

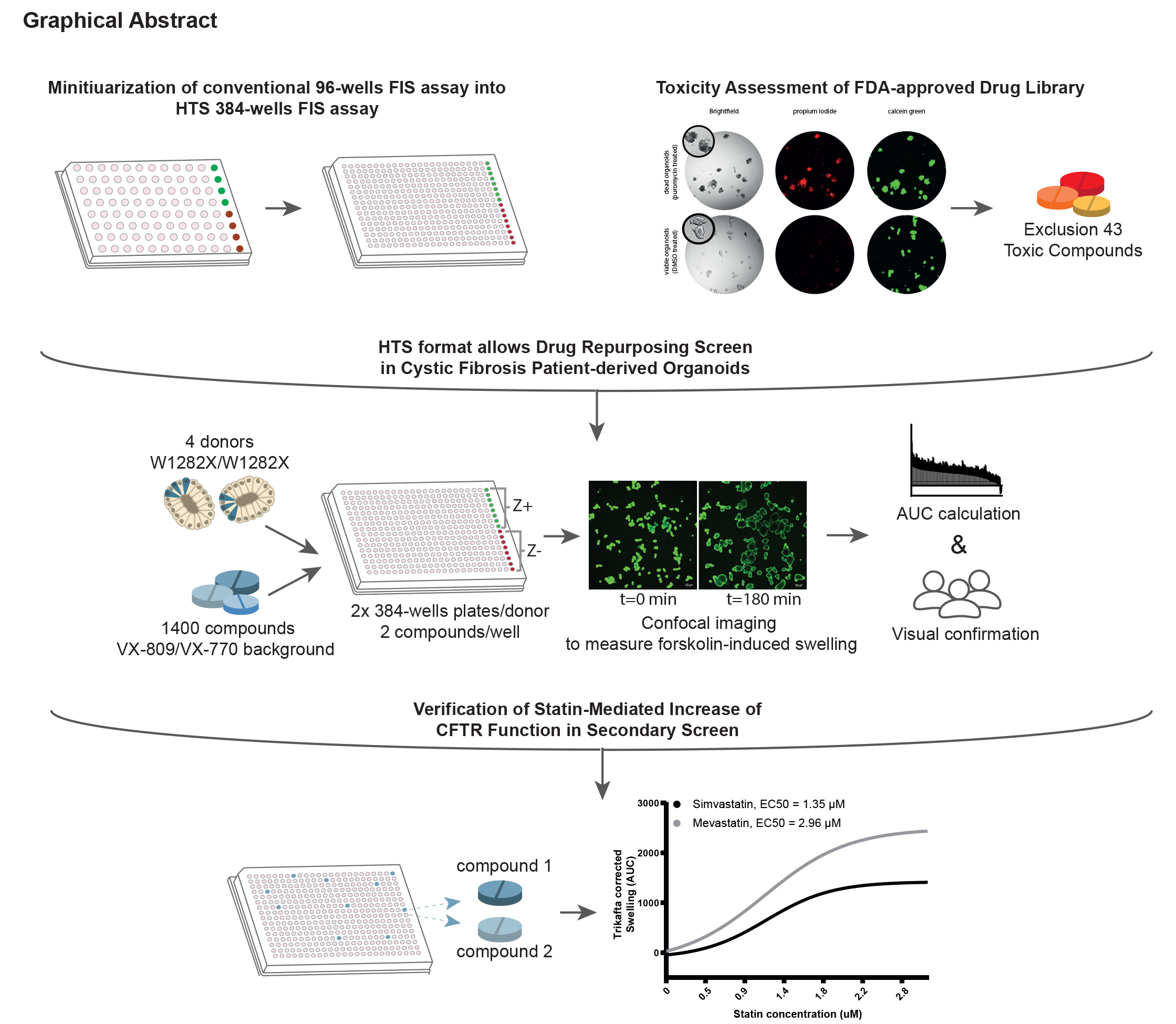
Abstract
Background
Cystic fibrosis (CF) is a rare hereditary disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Recent therapies enable effective restoration of CFTR function of the most common F508del CFTR mutation. This shifts the unmet clinical need towards people with rare CFTR mutations such as nonsense mutations, of which G542X and W1282X are most prevalent. CFTR function measurements in patient-derived cell-based assays played a critical role in preclinical drug development for CF and may play an important role to identify new drugs for people with rare CFTR mutations.
Methods
Here, we miniaturised the previously described forskolin-induced swelling (FIS) assay in intestinal organoids from a 96-well to a 384-well plate screening format. Using this novel assay, we tested CFTR increasing potential of a 1400-compound Food and Drug Administration (FDA)-approved drug library in organoids from donors with W1282X/W1282X CFTR nonsense mutations.
Results
The 384-well FIS assay demonstrated uniformity and robustness based on coefficient of variation and Z’-factor calculations. In the primary screen, CFTR induction was limited overall, yet interestingly, the top five compound combinations that increased CFTR function all contained at least one statin. In the secondary screen, we indeed verified that four out of the five statins (mevastatin, lovastatin, simvastatin and fluvastatin) increased CFTR function when combined with CFTR modulators. Statin-induced CFTR rescue was concentration-dependent and W1282X-specific.
Conclusions
Future studies should focus on elucidating genotype specificity and mode-of-action of statins in more detail. This study exemplifies proof of principle of large-scale compound screening in a functional assay using patient-derived organoids.
Short abstract
This study established a high-throughput functional assay using CF patient-derived intestinal organoids. 1400 FDA-approved compounds were screened, and it was found that statins increased function of W1282X/W1282X CFTR when combined with CFTR modulators. https://bit.ly/3NPXvhf
Introduction
Cystic fibrosis (CF) is a rare, monogenic disease that is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Mutations in CFTR lead to loss of chloride secretion and deficient fluid transport across tissue epithelium. Subsequently, thick and sticky mucus secretions are produced resulting in chronic bacterial infections, progressive loss of pulmonary function and multiorgan failure [1]. More than 1700 distinct mutations have been characterised, of which 400 are estimated to be disease-causing [2]. The F508del mutation is mainly prevalent, occurring on approximately one allele in 85% of the CF patients. For those patients, increasingly effective treatments called CFTR modulators have been developed in the last decade aiding in proper CFTR protein folding and membrane potentiation [3]. Recently a potent combination of the three CFTR modulators VX-445, VX-661 and VX-770 has been approved by the Food and Drug Administration (FDA) and European Medicines Agency (EMA). However, an urgent need for effective restoration of CFTR function persists for the remaining CF patients that cannot benefit from CFTR modulation.
Approximately 10% of the worldwide CF population carry premature termination codon (PTC) mutations, resulting in production of truncated CFTR protein. Previous preclinical research identified compounds with readthrough (RT) activity that introduce an amino acid at the PTC site and thereby results in full-length protein production [4, 5]. Clinical efficacy of RT approaches is however limited, as demonstrated by for example clinical studies with RT agent Ataluren/PTC-124 [6]. A novel recently developed RT agent, ELX-02 (NB124; Eloxx Pharmaceuticals, Watertown, MA, USA), resulted in production of full-length CFTR protein and restoration of CFTR function in G542X/G542X intestinal organoids [7]. However, in a study characterising the effect of ELX-02 in a larger set of patient-derived organoids, ELX-02 as single agent resulted in only limited restoration of CFTR function [8]. Altogether, this underlines the need for continuing the search for novel CFTR modulating molecular entities for people with CFTR PTC mutations.
Drug development in the context of rare diseases where the numbers of patients are low is challenging. For these patient populations, repurposing of clinically approved drugs could be beneficial. Drug repurposing is an attractive approach that aims to extend the indication of already marketed drugs [9]. A prerequisite for drug repurposing is that the exploited assay is robust and is associated with clinical features of disease, such as therapeutic response. CFTR function measurements in patient-derived intestinal organoids (PDIOs) are associated with clinical features of CF and may enable drug repurposing in a personalised setting. We previously developed a PDIO-based assay for CF based on forskolin-induced swelling (FIS). Forskolin induces fluid secretion into the organoid lumen resulting in CFTR-dependent swelling [10, 11]. CFTR function measurements in PDIOs are associated with 1) disease severity indicators of CF, 2) long-term disease progression and 3) therapeutic response, enabling preclinical drug efficacy and mode-of-action studies [12, 13]. As such, the FIS assay is well suited for identifying and prioritising drugs that influence CFTR function. However, the 96-well format of the assay limits its use when higher throughput screening is required.
Here, we miniaturised the 96-well FIS assay towards a 384-well plate format enabling drug screening in PDIOs at higher throughput. Important factors when validating assay quality are within and between experiment variability, drift and edge effects within plates and comparison of the outcomes of experiments performed with the original 96-well set-up to the novel 384-well set-up [14]. As these validations yielded positive results, Z’-factors were calculated to ultimately summarise assay performance [15]. The 384-well FIS assay was subsequently used to identify compounds from an FDA-approved, commercially available drug library for their capacity to increase CFTR function. The screen was performed in PDIOs harbouring W1282X/W1282X CFTR, as W1282X encompasses 18% of all PTC mutations, thereby being the most common PTC mutation in CF patients after G542X (CFTR2 database, 2021) [16]. Additionally, W1282X is one of the PTC sites that is closest to the normal termination codon (NTC). As such, W1282X protein is less severely truncated than in other PTC cases, and exploiting PDIOs with this genotype allows potential identification of hits that can improve organoid swelling through various mode of actions, ranging from nonsense-specific effects to more general CFTR-modulating effects.
In brief, we show proof of principle of a high-throughput screening (HTS) assay using PDIOs in a functional assay. By screening a library of FDA-approved compounds, we pave the way for drug repurposing for people with CF for whom no clinical options are available.
Materials and methods
Collection of primary epithelial cells of CF patients
All experimentation using human tissues described herein was approved by the medical ethical committee at University Medical Center Utrecht (UMCU; TcBio#19-831). Informed consent for intestinal tissue collection, generation, storage and use of the organoids was obtained from all participating patients. Biobanked intestinal organoids are stored and catalogued (https://huborganoids.nl/) at the foundation Hubrecht Organoid Technology (http://hub4organoids.eu) and can be requested at info@huborganoids.nl.
Human intestinal organoid culture
Human intestinal organoid culture was executed as described by Vonk et al. [11].
Compounds
The FDA library, purchased from Selleck Chemicals GmbH Europe (Z178323-100uL-L1300), was stored at −80°C. All other compounds described in this study were purchased at SelleckChem and dissolved in DMSO at a 20-mM concentration.
384-well FIS assay validation
Differences between the 96-well FIS assay as previously described by Vonk et al. [11] and the 384-well FIS assay described in this study are summarised in table 1 in the Results section. To characterise FIS in a 384-well format, a quality replicate experiment was performed. Three biological replicate experiments were performed on different days with organoids of different passages, with three technical replicates per condition. The F508del/S1251N organoids were submerged in 8 µL complete culture medium, the F508del/F508del organoids in 8 µL complete culture medium supplemented with 3 µM VX-809. 24 h later, FIS measurements were assessed in the presence of VX-770 (3 µM) and low (0.008 µM) and high (5 µM) forskolin (forskolin) concentrations, resulting in minimum (min) signal values and maximum (max) signal values. Organoid swelling was monitored for 60 min using a Zeiss LSM 710 confocal microscope. Total organoid surface area per well was quantified based on calcein green staining using Zen Blue Software and area under the curve over time was calculated as described by Vonk et al. [11]. CV values were calculated according to the following formula: % CV=(sd of means)/(mean of means)×100.
TABLE 1.
Adaptations in forskolin-induced swelling (FIS) protocol of original 96-well format to allow a 384-well format
| 96-well format, as described by Vonk et al. [11] | 384-well format | |
| 1 | Plates are prewarmed at 37°C prior to organoid-matrigel addition | Plates are precooled at −20°C, and kept on ice during organoid-matrigel plating |
| 2 | 4 μL 50% matrigel organoid suspension is added per well as drops | 10 μL 25% matrigel organoid suspension is added per well with an automatic multichannel, to cover the whole surface |
| 3 | – | Plate is spun down in a centrifuge to ensure matrigel coverage of the whole well surface |
| 4 | After 10 min at 37°C, 50 μL medium per well is added | After 10 min at 37°C, 8 μL medium per well is added |
| 5 | X/Y/Z location is manually set for each well | X/Y/Z is automatically set based on plate lay-out and autofocus of 12 reference points |
Min and max swelling enabled Z’-factor calculation of each 384-well plate according to the following formula: Z’-factor=1 − (3×(σp + σn)/(μp − μn)), where σp is the standard deviation of the max signal wells (n=128 per plate, +CFTR modulator(s) and 5 µM forskolin), σn is the standard deviation of the min signal wells (n=128 per plate, +CFTR modulator(s) and 0.008 µM forskolin), μp is the mean of the max signal wells and μn is the mean of the min signal wells.
Toxicity screen using 384-well plates
Organoids were plated into 384-well plates and were incubated for 24 h with 8 µL complete culture medium supplemented with a single FDA compound per well at a final concentration of 3 µM.1443 compounds were tested in total, divided over five 384-well plates, on three different PDIO lines (2× F508del/S1251N organoids, 1× F508del/F508del organoids). Bright field images were taken per well, and organoid viability was scored in a binary way (live/apoptotic) by three blinded investigators, based on comparison with positive control PDIOs treated with puromycin, which results in clear phenotypical differences such as cell blebbing. To assess cellular toxicity in a quantitative way, organoids were stimulated with calcein green (7 µM) for 30 min and propidium iodide (0.1 mg·mL−1) for 10 min prior to confocal imaging. Total organoid area per well was determined based on total calcein green staining (Zeiss, excitation at 488 nM) and amount of dead cells per well was determined by total area of PI staining (Zeiss, excitation at 564 nM) using Zen Image analysis software module (Zeiss, Oberkochen, Germany). The ratio of total area calcein green and total area PI (=T − score) was calculated to correct for varying number of organoids between wells. To further correct for the varying organoid sizes among plates the Z-score was calculated between the compound-treated organoids and control DMSO-treated organoids per plate. The Z-score was determined according to the following formula: Z-score = (x − μ)/σ, where x is the calcein/PI ratio of each single FDA compound, μ is the mean calcein/PI ratio of 16 control wells on each plate and σ is the standard deviation of the calcein/PI ratio of the same 16 control wells on each plate. Z-scores of the single FDA compounds were compared to Z-scores of wells treated (n=71) with a toxic concentration of puromycin (24 h, 3 mg·mL−1). Z-scores that were below Q1 − (3×IQR) of all the DMSO-treated wells (Z-score= −2.5) or Z-scores that were above Q3 + (3×IQR) of all puromycin-treated wells (Z-score = 14.8) were excluded. One biological replicate experiment was performed, with one technical replicate per condition.
FDA-screen using 384-well plates
Four W1282X/W1282X organoid lines were plated into 384-well plates and were incubated for 24 h with 8 µL complete culture medium supplemented with two FDA compounds per well (1400 compounds in total, divided over two 384-well plates) as well as VX-809, all tested at 3 µM. The day after plating, FIS measurements were assessed in the presence of VX-770 (3 µM) and 5 µM forskolin. No FDA compounds were added to eight wells per plate as negative control (= min signal), and F508del/S1251N organoids were added to eight wells per plate and treated with VX-770 to serve as positive control (= max signal). Three biological replicate experiments were performed on different days with organoids of different passages, with one technical replicate per condition. Organoid swelling was monitored as previously described for 180 min using a Zeiss LSM 710 confocal microscope. Additionally, bright field images were taken per well, and organoid morphology was scored in a binary way (no swelling/swelling) by three blinded investigators.
Conventional 96-well format FIS assay
96-well FIS assays were executed as previously described by Vonk et al. [11]. FIS of F508del/F508del and F508del/S1251N organoids with or without CFTR modulator(s) and increasing concentration of forskolin performed in 96-well plates was compared to FIS under similar conditions performed in 384-well plates. F508del/F508del organoids were treated with VX-809 (3 µM) for 24 h prior to FIS assays. VX-770 (3 µM) was added simultaneously with forskolin for 1 h. Z’-factors were calculated based on the minimal swelling signals induced with 0.008 or 0.02 µM forskolin + CFTR modulators and the maximum swelling signals induced with 5 µM forskolin + CFTR modulators. For the statin experiments in W1282X/W1282X, F508del/F508del and R334W/R334W donors, compounds were added 24 h prior to FIS measurements with different combinations of CFTR correctors (3 µM). Before FIS measurements, VX-770 (3 µM) and forskolin (5 µM) were added and organoid swelling was monitored during 180 min. Three biological replicate experiments were performed on different days with organoids of different passages, with three technical replicates per condition.
qPCR
PDIOs were treated with statins (3 µM) or nonsense-mediated decay (NMD) inhibitor SMG1i (0.3 µM) during 24 h prior to RNA isolation using a Qiagen RNA isolation kit (Qiagen, Valencia, CA, USA), according to the manufacturer's protocol. The RNA yield was measured by a NanoDrop spectrophotometer (ThermoFisher), and extracted mRNA was used for cDNA synthesis using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA) according to the manufacturer's protocol. A final cDNA concentration of 1000 ng·µL−1 was subjected to a two-step quantitative real-time PCR (qPCR) SYBR green reaction (CFX Connect Real time PCR, CFX-384 Real-time PCR, Bio-Rad) in a total assay volume of 10 μL. Primer sequences for GAPDH, YWHAZ, CFTR, PIAS1 and STAT1 are given in table 2. Expression levels of investigated genes were normalised against mRNA levels of housekeeping genes GAPDH and YWHAZ and thereafter to untreated control samples (ΔΔCT method). Melt peaks were analysed to confirm amplification of a single project. Two biological replicate experiments were performed on different days with organoids of different passages, with three technical replicates per condition.
TABLE 2.
Primer sequences for quantitative PCR reactions
| Gene | Forward primer | Reverse primer |
| CFTR | CAACATCTAGTGAGCAGTCAGG | CCCAGGTAAGGGATGTATTGTG |
| PIAS1 | GCGGACAGTGCGGAACTAAA | ATGCAGGGCTTTTGTAAGAAGT |
| STAT1 | ACTCAAAATTCCTGGAGCAG | ACGCTTGCTTTTCCTTATGTT |
| GAPDH | TGCACCACCAACTGCTTAGC | GGCATGGACTGTGGTCATGAG |
| YWHAZ | CTGGAACGGTGAAGGTGACA | AAGGGACTTCCTGTAACAATGCA |
Statistics
Statistical analysis was performed using GraphPad Prism 8.0 (GraphPad Software, La Jolla, CA, USA). Data on the graphs are presented as mean±sem, as experiments were performed in triplicate with three technical replicates per biological replicate. One-way analysis of variance (ANOVA) analyses with Dunnet's post hoc test were performed to analyse the differences in the secondary W1282X/W1282X screen, where a separate test was performed for each compound combination group to compare compound A/compound B or compound combination AB to the Trikafta background. Differences were considered significant at p<0.05. Statistical analysis of the primary W1282X/W1282X screen was performed in RStudio, and averages were calculated of the replicate experiments of the four donors combined for all compound combinations. To analyse whether these means increased FIS in comparison to the average of the plate, one-way t-tests were performed, and p-values were adjusted for multiple testing using the Benjamin–Hochberg FDR method.
Results
The 384-well FIS assay is reproducible, spatially uniform and has a comparable dynamic range to the 96-well FIS assay
First, we adapted several practical aspects of the 96-well FIS format which was previously described by Vonk et al. [11] to allow a higher throughput working method (table 1). In brief, 384-well plates were precooled prior to organoid addition, a higher volume of a lower matrigel-percentage was added to each well to cover the whole well surface and during image acquisition X/Y/Z locations were based on autofocus.
Using these adjustments, we verified the quality and reliability of the 384-well FIS assay. In figure 1a we summarise the advised [14] steps of assay quality validation when miniaturising a cell-based assay. To demonstrate FIS assay reproducibility, we performed replicate experiments in two organoid lines with different genotypes. FIS was assessed in F508del/S1251N organoids treated with CFTR potentiator VX-770 and in F508del/F508del organoids treated with CFTR potentiator VX-770 and CFTR corrector VX-809. Minimal (min) and maximal (max) signal of swelling was induced with 0.008 and 5 µM forskolin, respectively. The mean as well as the spread of the min and max signals were comparable between the replicate experiments both for F508del/S1251N organoids (figure 1b) and F508del/F508del organoids (figure 1c). In order to further characterise precision and repeatability within and between these replicate experiments, coefficient of variation (CV) was calculated [17]. For the max signal, the mean intra-assay CV values were 16% and 13% for the F508del/F508del and F508del/S1251N organoids respectively, and the inter-assay CV values were respectively 4% and 8%. CV values should not exceed 20% and CV values under 10% are considered excellent, underlining the reproducibility of the 384-well FIS assay [14].
FIGURE 1.

The 384-well forskolin-induced swelling (FIS) assay is reproducible, spatially uniform and has a similar dynamic range to the 96-well FIS-assay. a) Summary of optimisation steps for assay development, specifically for miniaturising a cell-based assay from 96-well format to 384-well format. b) and c) Replicate experiments of three 384-well plates with respectively F508del/S1251N and F508del/F508del patient-derived intestinal organoids (PDIOs), performed on three different culturing days. VX-770 (3 µM) and forskolin (0.008 µM) were added to 128 wells of each 384-well plate to induce minimal swelling (=min signal wells). Maximum swelling was achieved by the addition of 5 µM forskolin and VX-770 (3 µM) to another 128 wells (=max signal wells). The mean±sd swelling of all min and max signal wells of each plate depicted in b and c are summarised in the bar graphs. d) Swelling of F508del/F508del PDIOs treated with VX-770/VX-809 and F508del/S1251N PDIOs treated with VX-770 and in presence of an increasing concentration of forskolin, measured in 96-well plates and 384-well plates (data points represent mean±sd, n=3 for the 96-well experiments, n=1 for the 384-well experiments). e) Z’-factors calculated for each replicate experiment plate included in b, c and d. CV: coefficient of variation; WP: well plate; fsk: forskolin; AUC: area under the curve.
Next, edge effect for both outer rows and columns as well as overall horizontal or vertical drift on the plates was characterised (supplementary figure S1). As R2 values reach 0 in both donors for both the row and the column-oriented data, no horizontal or vertical drift across the plates is present. Additionally, the max area under the curve (AUC) values of the outer rows and columns (indicated with red dots) do not differ significantly from the rest of the data in both donors.
We next compared the signal amplitude of the 384-well FIS assay with the conventional 96-well assay. F508del/F508del and F508del/S1251N organoids with or without CFTR modulator(s) were stimulated with an increasing concentration of forskolin (figure 1d). This allows characterisation of “medium” swelling levels in the FIS assay, which is crucial when characterising the capacity of the assay to capture hit compounds during a screen [14]. Medium signal needs to lie between the negative and positive controls, and we show that indeed at 0.128 and at 0.8 µM forskolin the mid-range of the assay is covered. Differences between the 96-well and 384-well assay are negligible, except for 0.128 µM forskolin in the F508del/F508del organoids. Lastly, Z’-factors were calculated as an indicator of assay quality. The Z’-factor is a parameter based on positive and negative control that ranges between 0 and 1, with 1 indicating a perfect assay and Z’-factors larger than 0.4 considered acceptable [14]. Whilst mean Z’-factors from the 96-well format experiments were higher compared to those of the 384-well format experiments (figure 1e), the mean Z’-factor of the F508del/S1251N organoids was 0.4 and the mean Z’-factor of the F508del/F508del organoids was 0.5, indicating adequate assay robustness.
43. Out of 1443 FDA-approved compounds induce toxicity in PDIOs
Prior to assessing the effect of compounds on CFTR function restoration, we assessed potential toxicity of the 1443 FDA-approved compounds. Organoid viability was determined by a dual live cell staining approach in which calcein was used to stain metabolically active cells and propidium iodide (PI) to visualise dead cells, a toxicity assay that has previously been performed on intestinal organoids [18, 19]. The ratio of total calcein green area and total PI area was calculated to correct for varying number of organoids between wells, after which values were normalised to Z-scores facilitating comparisons between plates (figure 2a). Z-scores beyond 2, indicating toxicity, were found for 41 compounds (figure 2b). Additionally, organoid morphology was verified by three blinded observers. This led to a further exclusion of two additional compounds. The majority of the toxic compounds (81%), listed in supplementary table S1, are described as anti-cancer drugs. All 43 compounds were excluded from further screening.
FIGURE 2.

43 out of 1443 Food and Drug Administration (FDA)-approved compounds induce toxicity in patient-derived intestinal organoids (PDIOs). a) Schematic of toxicity assessment pipeline. The ratio of total area calcein green and total area propidium iodide (PI) (T-score) was calculated to correct for varying number of organoid structures between wells. To further normalise to the negative control organoids, the Z-score was calculated between the compound-treated PDIOs and control (DMSO-treated) PDIOs. b) Z-scores of the compounds labelled as toxic based on brightfield image scoring, compared to Z-scores of dead PDIOs (treated for 24 h with puromycin 3 mg·mL−1) and viable PDIOs not exposed to compounds. Bars represent mean±sd, n=3 for toxic compounds, n=71 for dead and healthy controls.
FDA-approved drugs increase FIS in homozygous W1282X/W1282X organoid lines
We next set out to identify FIS-increasing compounds. A schematic of the workflow is shown in figure 3a. Four organoid lines homozygous for W1282X-CFTR were pretreated with VX-809 and VX-770 to increase baseline function of W1282X/W1282X, facilitating hit detection. Two FDA-approved compounds were combined in each well. F508del/S1251N organoids treated with VX-770 were used as positive control on each plate, allowing CV value and Z’-factor calculation for quality control purposes. Inter- and intra-assay CV values were 17% and 14% respectively, averaged over the four donors, highlighting robustness of the assay. For each donor two 384-well plates were measured in triplicate, resulting in six Z’-factors for donor 1 and five Z’-factors for donors 2–4 due to technical errors. Mean Z’-factors for all plates approximated 0.4, ranging between 0.3 and 0.5 (figure 3b).
FIGURE 3.

Food and Drug Administration (FDA)-approved drugs increase forskolin-induced swelling (FIS) in homozygous W1282X/W1282X patient-derived intestinal organoids (PDIOs). a) Schematic of workflow of FDA-approved drug library testing on W1282X/W1282X PDIOs. b) Z’-factor for each 384-well plate for the four PDIOs. F508del/S1251N PDIOs treated with VX-770 served as positive control. c) Area under the curve (AUC) values of the top 17 compound combinations, means of the triplicate experiments of all PDIOs are shown with the different PDIOs indicated by the four different coloured dots. d) Pictures of the PDIO line 1, of the primary screen, for the top five compound combinations and the DMSO control (t=0 h and t=3 h).
17 compound combinations resulted in a mean increase of at least 500 AUC (figure 3c). The top five compound combinations reaching the highest AUC values were verified by visual analysis (figure 3d and table 3). Subsequently, p-values were calculated for the difference between the mean of each plate and the FDA-compound treated wells. Prior to multiple testing correction of the p-values by the Benjamin–Hochberg false discovery rate (FDR) method, 40 compound combinations resulted in a significant increase (p<0.05) in AUC compared to the mean of the plate (supplementary table S2). Included in these 40 compound combinations were the top five hits based on AUC values and nine out of 12 of the rest of the top 17. However, after FDR correction, only the positive controls remained significantly different. Still, interestingly one of each of the compounds of the top five compound combinations is a statin, suggesting a statin-induced effect on organoid swelling. Altogether the results provided sufficient rationale to confirm whether these statins have indeed potential to increase CFTR function.
TABLE 3.
Hits based on mean area under the curve (AUC) over all donors (>500)
| Compound 1 | Classification compound 1 | Compound 2 | Classification compound 2 | Mean AUC | Based on visual analysis (max=36) | ||
| 1 | Pitavastatin | HMG Co-A reductase | Alibendol | Choleretic | 862.9 | 26 | |
| 2 | Simvastatin | HMG Co-A reductase | Tamoxifen citrate | Oestrogen antagonist | 712.1 | 9 | |
| 3 | Fenspiride HCl | PDE4 inhibitor | Mevastatin | HMG Co-A reductase | 671.8 | 25 | |
| 4 | Guaifenesin | Expectorant | Fluvastatin sodium | HMG Co-A reductase | 658.3 | 24 | |
| 5 | Valsartan | Angiotensin II antagonist | Lovastatin | HMG Co-A reductase | 599.2 | 33 | |
| 6 | Carvedilol | β2 antagonist | Formoterol hemifumarate | β2 agonist | 574.7 | 1 | |
| 7 | CO-1686 | TRK | Palbociclib | CDK4/6 inhibitor | 569.8 | 2 | |
| 8 | Tivantinib | c-Met inhibitor | Darifenacin HBr | Muscarinic antagonist | 566.2 | 1 | |
| 9 | Sofosbuvir | HCV polymerase inhibitor | Sertaconazole nitrate | Anti-fungal | 536.6 | 0 | |
| 10 | Deoxycholic acid | Cytolytic agent | Riociguat | GC stimulator | 532.8 | 0 | |
| 11 | Voxtalisib | mTor inhibitor | L-Glutamine | Amino acid | 527.6 | 3 | |
| 12 | Butoconazole nitrate | Inflammation blocker | Ketotifen fumarate | Antihistamine | 518.7 | 0 | |
| 13 | LEE011 | CDK4/6 inhibitor | Heparin sodium | Glycosaminoglycan | 510.5 | 0 | |
| 14 | Albendazole oxide | Anthelmintic | Diclazuril | Coccidiostaticum | 507.7 | 3 | |
| 15 | Levobupivacaine HCl | Neuronal Na channel inhibitor | Chlorquinaldol | Antiseptic | 504.8 | 0 | |
| 16 | Nilotinib | BCR-Abl inhibitor | Dienogest | Progesterone | 503.5 | 0 | |
| 17 | Dovitinib | RTK inhibitor | Letrozole | Aromatase inhibitor | 500.5 | 0 |
HCV: hepatitis C virus; GC: guanylate cyclase; RTK: receptor tyrosine kinase. Top hit compounds are indicated by bold type.
To assess whether the statins of the compound combinations were responsible for AUC increase, the top five different compound combinations were repurchased from an independent supplier and tested on W1282X/W1282X donors 1 and 3. Compounds were tested in combination with VX-445/VX-661/VX-770 to further maximise the window of opportunity to detect effects in comparison to the primary screen (figure 4b). The data of both donors combined confirmed that simvastatin, mevastatin, fluvastatin and lovastatin further induced organoid swelling, yet not significantly in both donors. Alibendol additionally resulted in a significant increase in AUC. In the absence of CFTR modulator treatment no simvastatin- or mevastatin-induced CFTR response was observed in both W1282X/W1282X donors (figure 4b). AUC values, however, do not reach the values of positive control-treated PDIOs which were treated with RT agent ELX-02, NMD inhibitor SMG1i and Trikafta. By assessing swelling induction with a concentration range of simvastatin and mevastatin, we observed a concentration-dependent increase in AUC levels (figure 4c). Concentrations above 3 µM resulted in decreased AUC values due to compound-induced toxicity (data not shown). Using 3 µM as maximal value, EC50s were respectively 1.35 and 2.96 µM for simvastatin and mevastatin, whereas a higher maximum swelling was induced with mevastatin than with simvastatin.
FIGURE 4.

Statins increase forskolin-induced swelling (FIS) in homozygous W1282X/W1282X patient-derived intestinal organoids (PDIOs) in a concentration-dependent manner. a) Area under the curve (AUC) values of the secondary FIS screen for two W1282X/W1282X PDIOs, in which compounds of the primary screen top five were tested separately and as original combination. Trikafta (CFTR modulators VX-445/VX-661/VX-770) was used as background. b) AUC values for two W1282X/W1282X PDIOs upon treatment with the statins and CFTR modulators separately (all at 3 µM), as well as the combination and a positive control consisting of 80 µM ELX-02, 0.3 µM SMG1i and 3 µM VX-445/VX-661/VX-770 (Trikafta). Means are shown of three biological replicate experiments with three technical replicates per condition; sem is indicated by error bars. c) Curve-fitting line for AUC values of W1282X/W1282X PDIO line 1 upon a mevastatin and simvastatin concentration gradient, corrected for Trikafta-induced baseline swelling. d) AUC values for R334W/R334W and F508del/F508del PDIOs upon simvastatin and mevastatin treatment, with respectively VX-770 and VX-770/VX-809 as background (all at 3 µM). Means are shown of three biological replicate experiments with three technical replicates per condition; sem is indicated by error bars.
To evaluate whether the statins increased FIS of specifically W1282X-CFTR, we assessed statin-induced swelling in organoid cultures homozygously expressing F508del/F508del and R334W/R334W in combination with VX-809/VX-770 and VX-770, respectively. For these genotypes, we did not observe a further increase of FIS by mevastatin and simvastatin (figure 4d).
Statins have been described to interact with many pathways additional to cholesterol synthesis, including STAT1/3, p38, MAPK and Akt phosphorylation. Firstly, we investigated whether 6-fluoromevalonate (6F), a compound that inhibits cholesterol synthesis further down the synthesis pathway than statins do, also results in rescue of CFTR function. However, 6F did not result in further rescue of CFTR in the W1282/W1282X PDIOs, indicating that the observed CFTR rescue upon statin treatment is not cholesterol mediated (supplementary figure S2A). We subsequently investigated whether statins inhibit NMD, the process in which PTC harbouring mRNAs are recognised and degraded. As statins were combined with CFTR modulators, NMD inhibition could result in a pool of truncated CFTR protein that could be partially rescued by CFTR modulators. Additional to CFTR mRNA levels, we investigated PIAS1 and STAT1 mRNA levels, as reduction of PIAS1 upon statin treatment has been linked to normalised STAT1 activation and CFTR function [20]. However, whilst NMD inhibitor SMG1i indeed resulted in an increase of CFTR mRNA levels, CFTR, PIAS1 and STAT1 mRNA levels did not increase upon statin treatment (supplementary figure S2B).
Discussion
Whilst potent compounds have been developed for the most common F508del-CFTR mutation, there's a lack of medication for other genotypes and an urgent need to improve therapy for these CF patients. Approximately 10% of the worldwide CF population carry PTC mutations that result in production of truncated CFTR protein with severe loss-of-function. Developing new drugs is a time-consuming and expensive process. In this regard, drug repurposing is an attractive solution providing economical and time-wise benefits. In this study, we developed a 384-well version of the FIS assay, exemplifying the feasibility of large-scale compound screening with a functional read-out using patient-derived organoids.
We adapted several steps compared to the previously described 96-well FIS assay [11]. Importantly, medium replacement steps are not needed in this experimental pipeline, enabling a straightforward working scheme. To ensure successful screening, quality and reliability of the 384-well FIS assay was characterised. We show that replicate experiments yield similar results with low intra- and inter-assay CV values and without outer row- or column-associated effects. Overall, assay performance of the 384-well FIS assay was adequate with Z’-factors reaching 0.4 or higher [14]. Z’-factors were robust in the W1282X/W1282X screen as well; however, the positive control here consisted of F508del/S1251N organoids treated with VX-770 as a more suitable positive control specifically for W1282X/W1282X organoids was not available. Potentially in future studies the recently described RT agent 2,6-diaminopurine (DAP) can be used for this [21] or a combination of compounds that together result in higher levels of CFTR rescue [8]. The gain of throughput did result in lower Z’-factors for 384-well plates than obtained for 96-well plates in the FIS assay. In the future, additional automation such as automated organoid dispensers, drug printers and centrifugal washers might further reduce technical variability, whereas biological robustness could be improved by incorporating a proper positive control for the PDIO's genotype of interest. Such improvements could aid in increasing the Z’-factor. Yet, so far, mainly small-scale screenings using PDIOs have been performed, e.g. <100 compounds [22, 23]. Although recent work showed the implementation of organoid cultures in 3D matrix in a 384- and 1536-well format for HTS, exploited read-outs such as fluorescence-based viability analyses were relatively simple and exhibited lower Z’-factors [24]. Jiang et al. [25] performed high-throughput experiments in the context of CFTR, using induced pluripotent stem cells (IPSCs) differentiated into early-stage lung progenitor cells in a fluorescence-based assay of CFTR channel activity, yet also with a lower Z’-factor (0.34). Berg et al. [26] describe a 96-well screening assay using 3D-cultured primary airway cells with a disease-relevant read-out consisting of liquid secretion quantification, yet Z’-factors were not described in this study. The clinical relevance of the exploited FIS assay in our study in combination with the robust Z’-factors in a 384-well plate format, underline the strength of this study.
Whilst toxicity testing was not the main focus in our study, we exploited the 384-well screening pipeline to analyse potential toxicity of the FDA library. Indeed, 43 compounds induced toxicity and were excluded for further characterisation. This approach could be translated to different PDIO systems, for example when comparing compound-induced toxicity in tumour and wild-type organoids derived from the same patient. Notably, we could miss potential efficacies of compounds that act at a nanomolar range that were excluded based on toxic effects observed at a micromolar range testing. However, we expect that compound efficacies are not completely abolished at higher concentrations. As such, we assumed that this risk was acceptable in the context of workload.
The effect of the 1400 FDA-approved compounds on CFTR-mediated fluid secretion was assessed in W1282X/W1282X PDIOs. Using a statistical approach, after multiple testing correction, only the positive controls remained significantly different. However, 17 compound combinations resulted in an mean increase of at least 500 AUC. An important remark is that in this work, we push towards highest sensitivity by measurement of swelling at a high forskolin concentration (5 µM)in a 3-h measurement, as we anticipated that such assay conditions were needed to be able to detect the marginal effects that are needed for detection of responses of single drugs in the context of stop codons. Organoid assay conditions that are associated with disease severity indicators are mostly observed at 0.8 µM forskolin in 1 h swell conditions [27], whereas therapeutic responses are mostly analysed at 0.128 µM forskolin and 1 h swell conditions [13, 28]. The amount of rescue we observe under the assay conditions in this study is below levels associated with the studies cited above, and thus are anticipated to have less clinical impact. However, we previously found that disease severity in a clinical setting for patients with F508del/F508del CFTR could be linked to variation in organoid function under conditions where organoids were stimulated with 5 µM forskolin and 3 h stimulation [27]. We therefore cannot rule out that these small changes we observe have no impact on long-term disease progression. The AUC analysis of the FIS experiments, the results based on visual analysis of PDIO swelling and the statistical analysis in which statins merely reached statistical significance, all pointed in the same direction concerning the top five compound combinations. Interestingly, one of each of the compounds of this top five compound combinations was a statin, suggesting a statin-induced effect on organoid swelling. Indeed, in the subsequent confirmation screen four out of five statins significantly increased AUC values as separate compound as well as in the original combination in which the statins were tested. Importantly, in this secondary screen CFTR modulators VX-445/VX-661/VX-770 were used as a background instead of VX-809/VX-770 due to its higher potency to increase CFTR function, thereby further maximising the window of opportunity to detect effects, whilst additionally possessing a more favourable safety profile and a more clinically robust benefit [29].
In order to assess whether the statins increased CFTR function only in W1282X/W1282X organoid cultures, we assessed the effect of simvastatin and mevastatin on R334W/R334W and F508del/F508del organoid cultures, in combination with VX-770 and VX-809/VX-770 respectively. However, here no further increase in CFTR function was observed. This points in the direction that the effect is W1282X/W1282X or PTC specific. Interestingly, a HTS study using Fischer rat thyroid (FRT) cells stably expressing F508del-CFTR with a yellow fluorescent protein flux assay as read-out, did reveal atorvastatin calcium and fluvastatin as hits [30]. In future studies, assessing the effect of statins on a large panel of organoid with different genotypes will be valuable to draw firm conclusions on this.
Additionally, in combination with genotype specificity studies, more research is needed to elucidate the mode-of-action (MoA) of statins with respect to CFTR elevating function. Statins are generally known for their potential to inhibit HMG-CoA reductase, thereby inhibiting the production of cholesterol and isoprenoids [31, 32]. As such their potential to increase CFTR function is in fact contradictory to previous studies. By contributing to the creation and maintenance of the lipid rafts in which CFTR resides, cholesterol has been described to positively impact CFTR levels and function [33]. However, in this study more fundamental techniques were exploited, and results were not verified in primary patient-derived cells. Statins additionally have previously been described to have nanomolar median inhibitory concentration (IC50) as HMG-CoA reductase inhibitors in human cell lines [34]. The fact that we observe IC50 values that are higher could have two potential explanations: 1) the difference in model system and read-out – primary patient-derived cells used in a 3D Matrigel context versus cell background and the 2D systems in which statins’ IC50 was previously assessed; or 2) the effect is not mediated via HMG-CoA reductase inhibition but via another MoA. To assess whether the MoA of statins could be linked to their influence on cholesterol synthesis, we investigated the effect of 6F, a compound that also inhibits cholesterol synthesis. As no effect was observed, however, it is unlikely that the effect of the statins can be attributed to their inhibitory effect on cholesterol synthesis. Statins additionally have been described to interfere with PIAS1 and STAT1, which both have been linked to CFTR function [20], and Akt phosphorylation [35], which has recently been linked to NMD inhibition [36]. However, we did not observe differences in mRNA levels of PIAS1/STAT1 and CFTR upon statin treatment, indicating that the statin-mediated CFTR rescue is not achieved by NMD inhibition nor via statin-mediated correction of the STAT1 pathway. More elaborate studies should be performed to understand the exact MoA.
In conclusion, we have miniaturised our FIS assay into a robust 384-well plate high-throughput screen and used it to assess toxicity as well as CFTR increasing potential of an FDA-approved drug library. We found that statins increased CFTR function in CF PDIOs harbouring nonsense mutations. This finding can serve as an important starting point for developing novel treatment regimens for those CF patients that are left behind without a treatment regimen at present. Altogether, the developed pipeline in this study shows the potential of performing high-throughput, functional screenings assays on primary, patient-derived organoids.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00495-2022.SUPPLEMENT (399.2KB, pdf)
Graphical abstract 00495-2022.GRAPHICAL_ABSTRACT (892.2KB, jpg)
{kind=link}
Acknowledgements
We would like to thank the people with cystic fibrosis (CF) who gave informed consent for generating and testing their individual organoids; all members of the research teams of the Dutch CF clinics that contributed to this work; all colleagues of the HUB Organoid Technology for their help with generating intestinal organoids; and we thank Prof. Rene Eijkemans (Department of Biostatistics and Research Support, Utrecht University) for his valuable statistical advice and help.
Provenance: Submitted article, peer reviewed.
Author contributions: S. Spelier and E. de Poel contributed to the design of the study, the acquisition, verification, analysis and interpretation of the data, and drafted the manuscript. G.N. Ithakisiou, S.W.F. Suen, A.M. Vonk, J.E. Brunsveld, E. Kruisselbrink, D. Muilwijk and M.C. Hagemeijer contributed to the acquisition of study data and revised the manuscript. C.K. van der Ent and J.M. Beekman made substantial contributions to the conception and design of the study, and interpretation of data, and revised the manuscript.
Conflict of interest: C.K. van der Ent reports grants from GSK, Nutricia, TEVA, Gilead, Vertex, ProQR, Proteostasis, Galapagos NV and Eloxx, outside the submitted work; in addition, he has a patent (number 10006904) with royalties paid. J.M. Beekman reports personal fees from Vertex Pharmaceuticals, Proteostasis Therapeutics, Eloxx Pharmaceuticals, Teva Pharmaceutical Industries and Galapagos, outside the submitted work; in addition, he has a patent related to the FIS assay with royalties paid. All other authors have nothing to disclose.
Support statement: This work was funded by grants of the Dutch Cystic Fibrosis Foundation (NCFS) as part of the HIT-CF Program and by ZonMW grant number 91214103. Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Ehre C, Ridley C, Thornton DJ. Cystic fibrosis: an inherited disease affecting mucin-producing organs. Int J Biochem Cell Biol 2014; 52: 136–145. doi: 10.1016/j.biocel.2014.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sosnay PR, Siklosi KR, Van Goor F, et al. . Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet 2013; 45: 1160–1167. doi: 10.1038/ng.2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lopes-Pacheco M, Pedemonte N, Veit G. Discovery of CFTR modulators for the treatment of cystic fibrosis. Expert Opin Drug Discov 2021; 16: 897–913. doi: 10.1080/17460441.2021.1912732 [DOI] [PubMed] [Google Scholar]
- 4.Nudelman I, Rebibo-Sabbah A, Cherniavsky M, et al. . Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J Med Chem 2009; 52: 2836–2845. doi: 10.1021/jm801640k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Welch EM, Barton ER, Zhuo J, et al. . PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007; 447: 87–91. doi: 10.1038/nature05756 [DOI] [PubMed] [Google Scholar]
- 6.Kerem E, Konstan MW, De Boeck K, et al. . Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med 2014; 2: 539–547. doi: 10.1016/S2213-2600(14)70100-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crawford DK, Mullenders J, Pott J, et al. . Targeting G542X CFTR nonsense alleles with ELX-02 restores CFTR function in human-derived intestinal organoids. J Cyst Fibros 2021; 20: 436–442. doi: 10.1016/j.jcf.2021.01.009 [DOI] [PubMed] [Google Scholar]
- 8.de Poel E, Spelier S, Suen SWF, et al. . Functional restoration of CFTR nonsense mutations in intestinal organoids. J Cyst Fibros 2022; 21: 246–253. doi: 10.1016/j.jcf.2021.09.020 [DOI] [PubMed] [Google Scholar]
- 9.Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov 2004; 3: 673–683. doi: 10.1038/nrd1468 [DOI] [PubMed] [Google Scholar]
- 10.Dekkers JF, Wiegerinck CL, De Jonge HR, et al. . A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med 2013; 19: 939–945. doi: 10.1038/nm.3201 [DOI] [PubMed] [Google Scholar]
- 11.Vonk AM, van Mourik P, Ramalho AS, et al. . Protocol for application, standardization and validation of the forskolin-induced swelling assay in cystic fibrosis human colon organoids. STAR Protoc 2020; 1: 100019. doi: 10.1016/j.xpro.2020.100019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berkers G, van Mourik P, Vonk AM, et al. . Rectal organoids enable personalized treatment of cystic fibrosis. Cell Rep 2019; 26: 1701–1708.e3. doi: 10.1016/j.celrep.2019.01.068 [DOI] [PubMed] [Google Scholar]
- 13.Muilwijk D, de Poel E, van Mourik P, et al. . Forskolin-induced organoid swelling is associated with long-term CF disease progression. Eur Respir J 2022; 60: 2100508. doi: 10.1183/13993003.00508-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chai SC, Goktug AN, Chen T. Assay validation in high throughput screening: from concept to application. In: Vallisuta O, Olimat S, eds. Drug Discovery and Development – From Molecules to Medicine. London, IntechOpen, 2015; pp. 221–239. [Google Scholar]
- 15.Zhang JH, Chung TDY, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 1999; 4: 67–73. doi: 10.1177/108705719900400206 [DOI] [PubMed] [Google Scholar]
- 16.CFTR2. Database UCF. The Clinical and Functional TRanslation of CFTR (CFTR2). http://cftr2.org Date last updated: 29 April 2022. Date last accessed: 24 September 2022.
- 17.Reed GF, Lynn F, Meade BD. Use of coefficient of variation in assessing variability of quantitative assays. Clin Vaccine Immunol 2002; 9: 1235–1239. doi: 10.1128/CDLI.9.6.1235-1239.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grabinger T, Luks L, Kostadinova F, et al. . Ex vivo culture of intestinal crypt organoids as a model system for assessing cell death induction in intestinal epithelial cells and enteropathy. Cell Death Dis 2014; 5: e1228. doi: 10.1038/cddis.2014.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Rijn JM, Ardy RC, Kuloğlu Z, et al. . Intestinal failure and aberrant lipid metabolism in patients with DGAT1 deficiency. Gastroenterology 2018; 155: 130–143.e15. doi: 10.1053/j.gastro.2018.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kreiselmeier NE, Kraynack NC, Corey DA, et al. . Statin-mediated correction of STAT1 signaling and inducible nitric oxide synthase expression in cystic fibrosis epithelial cells. Am J Physiol Cell Mol Physiol 2003; 285: L1286–L1295. doi: 10.1152/ajplung.00127.2003 [DOI] [PubMed] [Google Scholar]
- 21.Trzaska C, Amand S, Bailly C, et al. . 2, 6–Diaminopurine as a highly potent corrector of UGA nonsense mutations. Nat Commun 2020; 11: 1. doi: 10.1038/s41467-020-15140-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sachs N, de Ligt J, Kopper O, et al. . A living biobank of breast cancer organoids captures disease heterogeneity. Cell 2018; 172: 373–386.e10. doi: 10.1016/j.cell.2017.11.010 [DOI] [PubMed] [Google Scholar]
- 23.Schütte M, Risch T, Abdavi-Azar N, et al. . Molecular dissection of colorectal cancer in pre-clinical models identifies biomarkers predicting sensitivity to EGFR inhibitors. Nat Commun 2017; 8: 1. doi: 10.1038/ncomms14262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du Y, Li X, Niu Q, et al. . Development of a miniaturized 3D organoid culture platform for ultra-high-throughput screening. J Mol Cell Biol 2020; 12: 630–643. doi: 10.1093/jmcb/mjaa036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang JX, Wellhauser L, Laselva O, et al. . A new platform for high-throughput therapy testing on iPSC-derived lung progenitor cells from cystic fibrosis patients. Stem Cell Rep 2021; 16: 2825–2837. doi: 10.1016/j.stemcr.2021.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berg A, Hallowell S, Tibbetts M, et al. . High-throughput surface liquid absorption and secretion assays to identify F508del CFTR correctors using patient primary airway epithelial cultures. SLAS Discov 2019; 24: 724–737. doi: 10.1177/2472555219849375 [DOI] [PubMed] [Google Scholar]
- 27.de Winter-de Groot KM, Janssens HM, van Uum RT, et al. . Stratifying infants with cystic fibrosis for disease severity using intestinal organoid swelling as a biomarker of CFTR function. Eur Respir J 2018; 52: 1702529. doi: 10.1183/13993003.02529-2017 [DOI] [PubMed] [Google Scholar]
- 28.Dekkers JF, Berkers G, Kruisselbrink E, et al. . Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci Transl Med 2016; 8: 344ra84. doi: 10.1126/scitranslmed.aad8278 [DOI] [PubMed] [Google Scholar]
- 29.Heijerman HGM, McKone EF, Downey DG, et al. . Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet 2019; 394: 1940–1948. doi: 10.1016/S0140-6736(19)32597-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin S, Sui J, Cotard S, et al. . Identification of synergistic combinations of F508del cystic fibrosis transmembrane conductance regulator (CFTR) modulators. Assay Drug Dev Technol 2010; 8: 669–684. doi: 10.1089/adt.2010.0313 [DOI] [PubMed] [Google Scholar]
- 31.Bansal AB, Cassagnol M. HMG-CoA Reductase Inhibitors. In: StatPearls [Internet]. Treasure Island, FL, StatPearls Publishing. Available from: www.ncbi.nlm.nih.gov/books/NBK542212/ Date last updated: 4 July 2022. [PubMed] [Google Scholar]
- 32.Zhao W, Zhao SP. Different effects of statins on induction of diabetes mellitus: an experimental study. Drug Des Devel Ther 2015; 9: 6211–6223. doi: 10.2147/DDDT.S87979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chin S, Ramjeesingh M, Hung M, et al. . Cholesterol interaction directly enhances intrinsic activity of the cystic fibrosis transmembrane conductance regulator (CFTR). Cells 2019; 8: 804. doi: 10.3390/cells8080804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slater EE, MacDonald JS. Mechanism of action and biological profile of HMG CoA reductase inhibitors. Drugs 1988; 36: Suppl 3, 72–82. doi: 10.2165/00003495-198800363-00016 [DOI] [PubMed] [Google Scholar]
- 35.Bonifacio A, Sanvee GM, Bouitbir J, et al. . The AKT/mTOR signaling pathway plays a key role in statin-induced myotoxicity. Biochim Biophys Acta 2015; 1853: 1841–1849. doi: 10.1016/j.bbamcr.2015.04.010 [DOI] [PubMed] [Google Scholar]
- 36.Palma M, Leroy C, Salomé-Desnoulez S, et al. . A role for AKT1 in nonsense-mediated mRNA decay. Nucleic Acids Res 2021; 49: 11022–11037. doi: 10.1093/nar/gkab882 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00495-2022.SUPPLEMENT (399.2KB, pdf)
Graphical abstract 00495-2022.GRAPHICAL_ABSTRACT (892.2KB, jpg)