

Abstract
Pluripotent human embryonic stem cell (hESC) lines are a valuable in vitro tool to differentiate specific cell lineages, including cells from all three germ layers, i.e., neuronal cells, myocytes, and hematopoietic cells, including progenitors (described here), lymphoid cells, and myeloid cells. However, dramatically different cell subtypes and functional properties of specific cells can arise depending on the differentiation technique used. We previously optimized hematopoietic stem cell differentiation from two different NIH-approved hESC lines to generate CD34+ hematopoietic progenitor cells (HPCs). Infection of these HPCs with a common herpesvirus (human cytomegalovirus) results in maintenance of viral latency, capability of viral reactivation, recapitulation of viral mutant phenotypes, and virus-induced myelosuppression of hematopoietic differentiation. However, different HPC subpopulations support different viral latency and reactivation phenotypes, and different hESC-to-HPC differentiation methods alter the ratio of stem cell subsets. In addition, differences in differentiation methods are dependent on both protocol/reagents and user techniques. Here, we report a simplified and optimized method to generate large numbers of CD34+ HPCs with consistent phenotypes and demonstrate a comparison of several common methods that can be used to control the ratio of available HPC subpopulations. A key aspect of this approach is that we achieve consistency in differentiation across users in different laboratories and, importantly, among newly trained individuals.
Keywords: cell biology, cell culture, differentiation, embryonic stem cells, hematopoietic progenitor cells
INTRODUCTION
Human embryonic stem cells (hESCs) were first described by Thomson et al. (1998), and these original lines and others have been a valuable in vitro tool to generate different cell lineages. These multipotent cells can be induced to differentiate into multiple lineages; in the hematopoietic lineage alone, hESCs can be differentiated into hematopoietic progenitor cells (HPCs, described below) and their downstream-lineage cells [myeloid (Choi, Vodyanik, & Slukvin, 2011; Li et al., 2012) and lymphoid (Galić et al., 2006)]. HPCs derived from hESCs share many of the characteristics of primary progenitor cells, including broad lymphoid-myeloid potential; directed, cytokine-mediated differentiation into mature myeloid, lymphoid, and endothelial cells; and recapitulation of specific disease states (reviewed by Rao & Greber, 2017; Vazin & Freed, 2010).
We assessed multiple differentiation methods including commercially available media (STEMCELL Technologies STEMdiff Hematopoietic Kit), modifications of long-term bone marrow cultures with stromal cell feeder layers (Choi et al., 2011), serum-free or feeder-free differentiation methods (Kim et al., 2017; Kovarova, Latour, Chason, Tilley, & Koller, 2010; Salvagiotto et al., 2011), and our own modifications. Using these methods, hESCs differentiate into hematopoietic cells through a process intended to mimic natural hematopoietic processes in early embryonic development through a hematopoietic-endothelial transition. This process begins with differentiation to an endothelial-like adherent layer within the first several days, followed by commitment to hematopoiesis, and finally further expansion of the hematopoietic population. HPCs generated with this protocol are a mixture of early hematopoietic stem cells (HSCs) and HPCs, all of which are CD34+, are not yet lineage committed, and have downstream differentiation capabilities. Although the specific timing for commitment, expansion, and optimal harvest varied between methods, all protocols tested yielded CD34+ HPCs. However, different differentiation methods gave rise to very different total HPC yields and to an HPC population with dramatically different cell subtypes and phenotypes. Selection of the correct method for the intended assay requires asking if the cells are already differentiating further toward specific lineages or are lacking diversity in early HSC versus HPC subpopulations. To avoid this issue, we selected two main methods that generated HPCs expressing only stem cell and early lineage commitment markers and lacking all mature lineage receptor expression (Crawford et al., 2021).
Stem cells and their essential stemness properties, however, cannot be defined by phenotype alone. One critical property is their ability to function, including maintenance of self-renewal ability and the capacity to differentiate to multiple lineages. Different HPC subsets, and therefore hESC differentiation methods that give rise to different proportions of HPC subsets, have dramatically different functional biological outcomes. This is exemplified by our recent work (Crawford et al., 2021), where we demonstrate that different HPC subsets respond very differently to viral infection. In this work, we further characterized the two best differentiation methods tested. These methods, namely commercial media (from the STEMCELL Technologies STEMdiff Hematopoietic Kit) and a feeder cell-supported culture system (Choi et al., 2011), were selected as best based on total yield of CD34+ HPCs per starting material with best reproducibility between batches in the first trials. We also previously demonstrated that the differentiation method can be used to control the ratio of available HPC subpopulations.
In addition, differences in differentiation success are dependent not only on the protocol and reagents used but also on user techniques. Here, we report a simplified and optimized method to generate large numbers of CD34+ HPCs with consistent phenotypes. This approach is based on a commercially available kit to maintain quality control, and this kit can be supplemented with additional cytokines or paired with downstream assays to customize HPCs for different uses. A key aspect of this approach is that we achieve consistency in differentiation across users in different laboratories and, importantly, among newly trained individuals. This article includes detailed description of the maintenance of hESCs (required for successful later differentiation; Basic Protocol 1), the differentiation of hESCs to HPCs (Basic Protocol 2), and their subsequent functional outcomes for long-term survival as HPCs and downstream differentiation to the myeloid lineage (Basic Protocol 3). Also included are support protocols for banking hESCs at low passage via freezing and subsequent optimization of each lot for use (Support Protocol 1); for counting aggregates for consistency in downstream assays, including differentiation (Support Protocol 2); and for analysis of HPC phenotype by flow cytometry (Support Protocol 3).
STRATEGIC PLANNING
Appropriate maintenance of hESCs in their undifferentiated state is essential for proper downstream differentiation and function. It is also recommended to obtain several hESC lines (we use WA01 and WA09, two comparable but genetically distinct lines) for all experiments, as there are differences in differentiation and phenotypes between cell lines. Banking low-passage hESCs for experiments and maintaining multiple lines are critical for reproducible and reliable results. An initial 3 to 6 weeks should be allocated at the beginning to make both a golden stock bank and a working bank for a small laboratory. A golden stock bank of hESCs frozen at as low a passage as possible (within 2 to 4 passages of the initial stock) creates a supply of consistent cells for long-term and future use. A second, working bank can then be created “on demand” from the golden stock bank at regular intervals to provide a consistent and reproducible supply of cells for experimental use. In addition, advance planning is recommended to time the start of differentiation appropriately to account for (1) hESCs from frozen stocks requiring 2 to 4 passages prior to the initiation of differentiation and (2) HPCs harvested fresh from differentiation having significantly higher viability and better function as compared with HPCs frozen between differentiation and use. Outlines of the main steps and differentiation experiments are shown in Figures 1A and 2E and described in more detail in the timeline.
Figure 1.
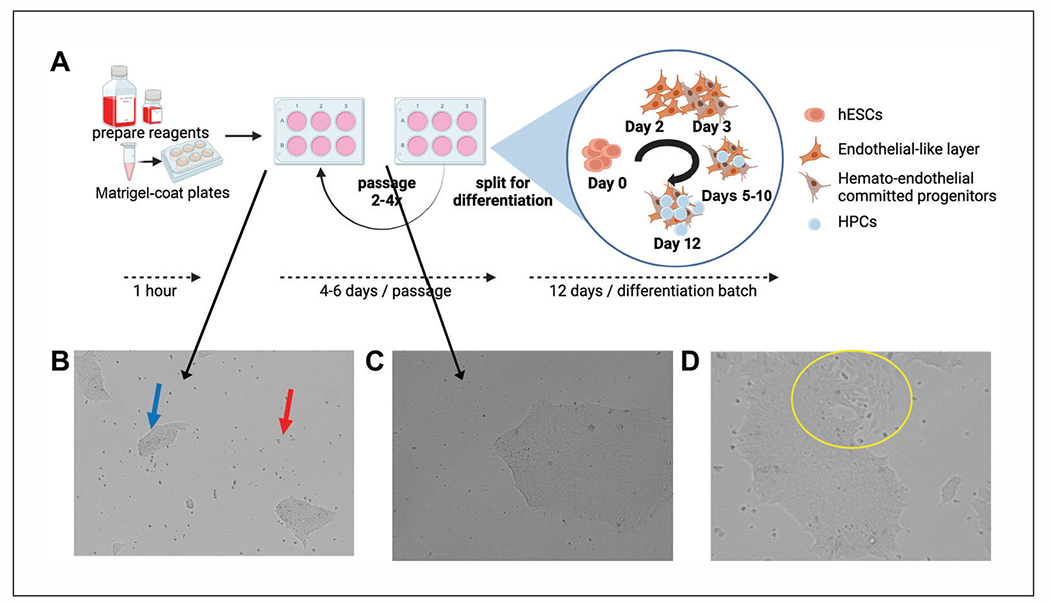
Maintenance of hESCs. (A) Overview of hESC maintenance and differentiation experiments with estimated timing. Reagents for maintenance and differentiation require thawing overnight at 4°C. hESC should be passaged at least twice after thawing and recovery prior to beginning a differentiation experiment. A normal passaging period for healthy WA01 or WA09 cells split at an average colony size with ~10% initial confluence equivalency should be passaged every 4 to 6 days. Note that aggregates grow exponentially faster at later days post-split and can easily become overcrowded. Differentiation of hESCs to HPCs requires 12 days using this method and begins with aggregate plating, as for passaging, followed by incubation in specific differentiation media that induce hemato-endothelial differentiation that mimics embryonic development from aggregate expansion and endothelial differentiation to hematopoietic development in layers on top of the adherent endothelial-like cells (more details in Fig. 2). Panel (A) was created with BioRender.com. (B) WA01 hESCs at passage 30 were cultured and maintained in mTeSR1. The image shows a representative view of hESCs at 1 day post-split where both adherent aggregates (blue arrow) and some floating cells (red arrow) are present prior to medium exchange. (C) A single aggregate of WA01 cells from a low-density split at 5 days and ready for passage. Note that although aggregates are not at 90% confluence equivalency, individual aggregates are large and dense. (D) WA09 (passage 34) hESCs, which were cultured as in (B), showing signs of spontaneous differentiation (yellow circle) based on morphological changes among individual cells at the edges of the aggregate and changes in overall aggregate-edge morphology.
Figure 2.
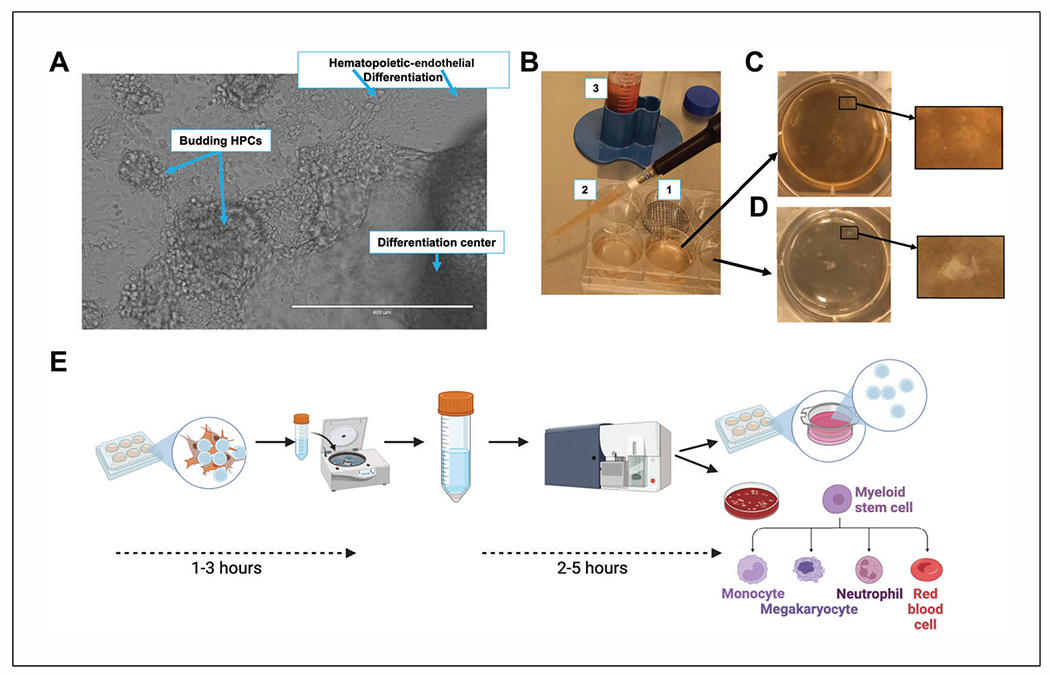
Differentiation of hESCs to HPCs and harvest for downstream analysis. (A) Expanded view of differentiating hESCs at 7 days post-initiation using the STEMdiff Hematopoietic Kit. Clockwise from top: endothelial layer from initial hematopoietic differentiation showing early hematopoietic cells (left) and endothelial cells (right); a further progressed aggregate demonstrating a dense differentiation center, with hematopoietic cells in a dome on top of the adherent endothelial layer; and HPCs that are budding off of the differentiation center. Scale bar is 400 μm. (B) Macro view of hematopoietic differentiation at day 12 post-initiation showing an example of a gridded well (1) for counting aggregates at day 0 and the density of budded HPCs (2, 3) that are floating in the culture medium at the round 1 (initial) harvest. Following initial harvest and replacement of medium with DMEM/F12, hematopoietic and endothelial cells remain in the well and aggregates appear normal (C), and following multiple rounds of trituration, only endothelial cells remain, and all HPCs have been collected, with some aggregates having rounded up edges or beginning to detach from the plate (D). Enlarged views of aggregates remaining on the dish in (C) and (D) are also shown. Images were selected to demonstrate the view of the aggregates in wells while working with the dish within the biosafety cabinet without a microscope. (E) An overview of timing for harvest and downstream analysis. A typical experiment uses HPCs harvested fresh on the day of experiment initiation, which requires 1 to 3 hr of harvest time depending on experience and the number of wells. An experiment using a whole kit of differentiation media (15 wells) performed by an experienced user takes ~2 hr for harvest, collection, and preparation of cells for downstream use. Harvested cells are typically 40% to 80% CD34+ HPCs, as collected straight from the culture. For experiments requiring a pure population of CD34+ HPCs or specific subsets, cells should be further separated using magnetic bead isolation and/or FACS (requiring an additional 2 to 4 hr). Post-separation, downstream plating for long-term liquid culture and myeloid colony-forming assays requires an additional ~1 hr. Panel (E) was created using BioRender.com.
CAUTION: All work should follow local biosafety and stem cell regulatory guidelines. Tissue culture techniques should be carried out using aseptic technique and sterile handling, including use of biosafety cabinets. Human cells should be considered BSL2. Personal protective equipment (PPE) should include a dedicated tissue culture gown and gloves, separate from that used for other tissue culture work.
NOTE: All solutions and equipment coming into contact with cells must be sterile, and proper sterile technique should be used accordingly. A biosafety cabinet/laminar flow hood (Baker SteriGARD Class II Type A2 Biosafety Cabinet or equivalent) and sterile filter tips must be used for cell culture.
NOTE: All culture incubations are performed in a humidified 37°C, 5% CO2 incubator, with minimal vibrations, unless otherwise specified.
BASIC PROTOCOL 1 MAINTENANCE OF HUMAN EMBRYONIC STEM CELLS (hESCs)
Maintenance of hESCs is essential to keep cells undifferentiated and provide a consistent source of cells for reproducible experiments. hESCs are cultured on a Matrigel substrate in defined medium with daily medium changes to prevent cell toxicity and maintain constant levels of nutrients and growth factors. Sterile technique is essential, as cells are typically cultured in media free of antibiotics and antifungals to improve cell growth and minimize spontaneous differentiation. Cultures should be checked at each medium change to monitor the health of the cells and the growth of cell aggregates. Quality-control testing for tissue culture of primary and/or sensitive cells, including regular mycoplasma, microbiological, and endotoxin testing and reagent validation, and regular training updates, among other measures, should be included if not already part of standard practice. Healthy cells have a high nuclear-to-cytoplasmic ratio; are adherent, with smooth edges; and exhibit rapidly increased growth after several days in culture. hESCs in this protocol are maintained as aggregates (Fig. 1B) to promote expansion and suppress spontaneous differentiation. Although hESCs can be grown either as aggregates or as single cells (easier for cell counting and essential for generating clonal cell lines), aggregate growth supports a consistent cell phenotype, prevents the development of abnormal clones, and is required for long-term maintenance and robust initiation of hematopoietic differentiation (see Basic Protocol 2).
Materials
mTeSR1 supplement and basal medium (STEMCELL Technologies, cat. no. 85852)
Matrigel, hESC qualified (Corning, cat. no. 35427; aliquot according to manufacturer’s instructions; store at −20°C)
DMEM/F12 medium (Gibco, cat. no. 11320-033), 4°C
hESCs [e.g., cells used in our study: WA01 (H1) NIH registry #0043 (WiCell, cat. no. WA01 [mTeSR Plus/Matrigel]) and WA09 (H9), NIH registry #0062 (WiCell, cat. no. WA09 [mTeSR Plus/Matrigel])]
Dry ice
Dulbecco’s phosphate-buffered saline (DPBS), without calcium and magnesium (Corning, cat. no. 21-031-CV; store at room temperature)
ReLeSR (STEMCELL Technologies, cat. no. 05873; store at room temperature)
15- and 50-ml conical tubes
6-well tissue culture plates
37°C water bath
5-, 10-, and 25-ml serological pipets
Tissue culture centrifuge (with buckets that can hold 15- and 50-ml conical tubes)
Tissue culture microscope (inverted microscope with 4×, 10×, and 20× objectives; Nikon Ti2 or equivalent)
Additional reagents and equipment for freezing cells (see Support Protocol 1, steps 1 to 11; optional)
Preparation
-
1
Thaw mTeSR1 supplement at 4°C overnight or at room temperature until just thawed.
-
2Aliquot appropriately in 50-ml conical tubes and store at −20°C.A useful aliquot size is 10 ml supplement aliquoted into a 50-ml conical tube.
-
3
Prepare complete mTeSR1 medium by combining cold mTeSR1 basal medium with supplement at a 4:1 ratio.
Coating plates with Matrigel
-
4Thaw a Matrigel aliquot on ice.Choose an aliquot size appropriate for the number of wells needed.When preparing to either thaw cells or split cells, coat plates with Matrigel. In this protocol, all hESC culture steps (passage, testing, differentiation) are performed in 6-well plates with 1 ml Matrigel coating solution per well.
-
5
Aliquot 1 ml cold DMEM/F12 medium per well into a 15- or 50-ml conical tube and place on ice.
-
6
Add 500 μl cold DMEM/F12 to the thawed Matrigel aliquot (of any size) and quickly but thoroughly mix by pipetting. Then, transfer to the conical tube containing the remaining DMEM/F12.
-
7
Mix the Matrigel + DMEM/F12 mixture by inverting the tube.
-
8
Plate 1 ml Matrigel + DMEM/F12 per well of a 6-well tissue culture plate.
-
9
Rock the dish to completely cover the wells.
-
10Incubate at room temperature for 1 hr to allow Matrigel to solidify on the well surface.Do not exceed 3 hr of incubation and do not let plates dry out.Proceed to step 11 during the incubation.
Thawing hESCs
-
11Before starting cell culture, warm complete mTeSR1 medium from step 3 to room temperature.Do not use a water bath to warm the medium.If feeding only 1 to 2 wells of cells, aliquot the complete medium into smaller volumes (~10 ml) to avoid repeated warming of larger medium volumes.
-
12Identify the batch of hESCs to be used and determine the optimal number of wells needed per vial of frozen cells.The optimal number of wells for each vial of frozen cells is based on aggregate number and is either provided in the information included with commercially obtained cells or determined experimentally after banking cell batches (see Support Protocol 1).
-
13
Remove Matrigel + DMEM/F12 solution from the pre-coated well(s) from step 10 by aspiration and proceed immediately to step 14.
-
14
Add 1 ml complete mTeSR1 medium to each Matrigel-coated well.
-
15
Remove needed number of hESC vials from liquid nitrogen using standard safety precautions. Place vials on dry ice and transfer to tissue culture room.
-
16
Thaw vial(s) by quick immersion in a 37°C water bath with constant swirling until only a small ice piece remains.
-
17Prepare 10 ml cold DMEM/F12 in a 15-ml conical tube (with a separate tube for each vial of cells) and use it to wash freezing medium off the hESCs:
- Using a 5-ml serological pipet, add 1 ml cold DMEM/F12 dropwise to the hESC vial.Here and in steps 17b to 17d, be careful not to break up aggregates.
- Gently draw entire contents of the vial into the pipet and add dropwise to the 15-ml conical tube containing the remaining 9 ml DMEM/F12.
- Draw up 2 ml new cold DMEM/F12 and rinse the vial. Transfer slowly (but faster than dropwise) to the 15-ml conical tube to resuspend the cells.
- Cap tube and invert gently once to mix.The final volume is now 14 ml (10 ml DMEM/F12 plus 2 ml cells in freezing medium plus 2 ml additional DMEM/F12).
-
18
Spin down cells for 5 min at 300 × g at room temperature and decant off medium.
-
19Resuspend cells gently in complete mTeSR1 medium (1 ml per well of a 6-well plate) using a 5-ml serological pipet.Pipet gently and carefully and use a 5-ml serological pipet (which has a wider bore than a micropipet tip) to avoid breaking up aggregates.
-
20
Gently add cells to the prepared Matrigel-coated plates (see step 14) by adding 1 ml cell aggregate/medium suspension to each well.
-
21Place plates in the incubator and distribute aggregates evenly across the surface and incubate for 24 hr.To distribute, leave the plate sitting flat on the incubator shelf and move the plate quickly using back-and-forth and side-to-side motions to minimize settling of cells in the center of the wells.IMPORTANT NOTE: Be sure not to disturb the cells for the next 24 hr. Minimize vibrations from nearby equipment and the force of opening/closing the incubator door.The following section assumes that day 1 is the day immediately following the day the cells were split or thawed.
Maintenance of hESC cultures
Day 1
-
22.
Warm complete mTeSR1 medium from step 3 to room temperature (2 ml per well of cells).
-
23.Remove cells from the incubator (see step 21).Do not remove more than two plates at a time to minimize exposure of the cells to ambient temperatures, as hESCs spontaneously differentiate if the temperature fluctuates.
-
24.Check cells on one plate for aggregate adherence, aggregate number, and aggregate dispersal using a tissue culture microscope.Aggregate assessment varies depending on the day post-thaw/post-split and the intended use. See Table 1 for specifics and Figure 1B for example plating density. The ideal number of aggregates depends on application (hESC maintenance vs. differentiation of hESCs to HPCs; see Basic Protocol 2 and Fig. 3). Maintenance of hESCs is more flexible than differentiation, and an initial aggregate density of about 100 to 500 aggregates per well is usable following the guidelines in steps 33 to 35.
-
25.
Aspirate medium from the plate, being careful not to disturb the cell aggregates or scratch the plate.
-
26.
Using a 10-ml or 25-ml serological pipet, immediately add 2 ml complete mTeSR1 medium to each well, being careful not to disturb cell aggregates.
-
27.
Repeat steps 24 to 26 for the other plates.
-
28.
Return plates to the incubator, being careful not to shake them.
Table 1.
Aggregate Analysis (Basic Protocol 1)
| Description | Day 1 | Day 2 | Day 3 and onward | |
|---|---|---|---|---|
| Aggregates present | Small aggregates (left two aggregates in Fig. 1B) | Continue with step 22 to feed the cells | Check that aggregates are still present and have maintained their size or enlarged slightly (Fig. 1B, right aggregate) and then continue with step 29 to feed the cells | Confirm that aggregates are appropriately attached to the plate and have normal morphology and edges (Fig. 1C) |
| No aggregates visible | Cells appear as single cells or very small clusters of cells (4-10 cells) attached to the plate | If the medium is clear and no “sheets” of cells are visible, continue with step 22 and check the wells again at day 2 (aggregates could be too small to see) | Check to see if aggregates are now visible (Fig. 1B). If some aggregates are present, continue with step 29 to feed cells. If no aggregates are present, discard and try again, handling the cells more gently and/or plating at a higher density. | |
| Cells are all in “sheets” or a film across the surface of the well | Cells appear as single cells floating in the medium, or medium is cloudy, with cells that are floating but stuck together (sheets) without contamination | Cells either 1) were plated too densely, and it is likely that aggregates will not form, or 2) are just in single-cell format and will not survive or form aggregates. Try continuing with step 22 and check again at day 2 for aggregates. | If small aggregates are now visible, continue with step 29 to feed the cells. If aggregates are still not visible, the cells were plated too densely, and aggregates will not form. Discard and try again, plating at a lower density. |
Figure 3.
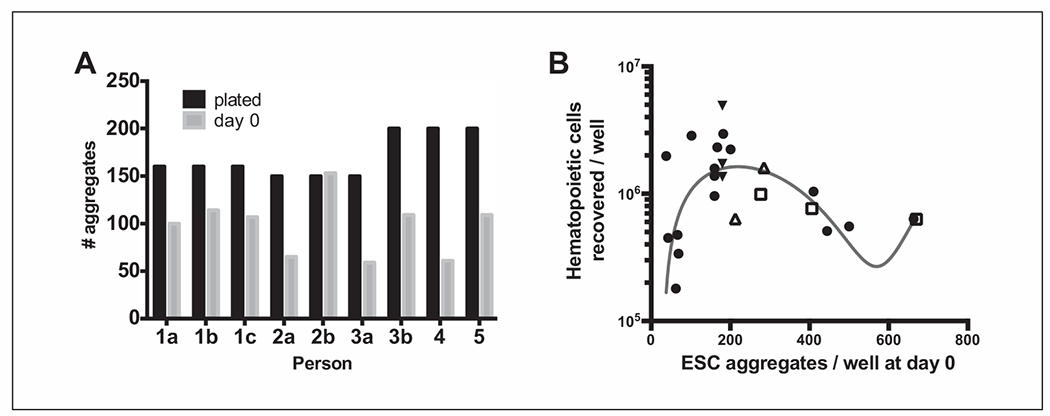
Aggregate plating variations (between batches and users) and impact on HPC yield. (A) A trial experiment using matched-passage WA01 aggregates (passaged by one user) that were then split and plated as if for differentiation by five different users. Three users (1, 2, 3) repeated this experiment with 1 to 2 additional batches of WA01 cells at different passages (replicates b and c). Aggregate counts are shown for the intended plating density (plated, black bars) and the number of aggregates present on the following day (day 0, gray bars). (B) Cumulative data from one user (after initial trial batches to gain experience) with multiple differentiation experiments using WA01 cells showing the relationship between plating density (aggregates per well at day 0) and the total number of HPCs recovered at 12 days post-differentiation (per well). WA01 cells from passage 29 to passage 39 were used for all experiments and were all maintained and differentiated by one user. Trend assessment (blue line) was determined by a third-order nonlinear polynomial curve fit using GraphPad Prism 9. Filled round symbols indicate individual differentiation experiments, different days, and different passages of cells. Open symbols indicate matched-passage cells intentionally plated at different densities (open triangle, p31; open square, p30). Filled triangles indicate different-passage cells all plated on the same day at the same density.
Days 2 through ~6
-
29.
Repeat steps 22 to 28 daily.
For determination of passaging time, see Table 1. For troubleshooting aggregate health, see Table 2.
-
30.Check cells for aggregate condition (adherence, number, and dispersal) and for signs of spontaneous differentiation.See Figures 1C and 1D for example images of large normal aggregates and spontaneous differentiation. Spontaneous differentiation is normal in hESC cultures at a rate of <2% of total aggregates and can be used as a quick surrogate for pluripotency. However, excess spontaneous differentiation (>5% of total aggregates or occurring every passage), visualized by the appearance of endothelial-like cells or spiky protrusions from the edges of aggregates (Fig. 1D, yellow circle), indicates an unhealthy culture.
-
31.Estimate aggregate number and size.The overall aggregate number will influence the passage date (more aggregates will require an earlier passage even though the aggregate size will be smaller, as a more crowded well will have aggregates that begin to merge earlier), and this will influence the passage timing and ratio. Aggregate size will also influence passage timing (a plate with few but very large aggregates will also require an earlier passage, as cells in the center of a large aggregate have less nutrient accessibility and reduced physical space for growth, resulting in senescence and cell death).
-
32.Estimate upcoming passage date based on aggregate density.Cells should be passaged when aggregates are densely packed but not completely merged. Merged aggregates essentially function as extra-large aggregates, with the same reduction in cell health (see step 31). Note that aggregates grow quicker at later dates from passaging and can rapidly expand from one day to the next near the end.
-
33.Feed or passage cells as appropriate.To feed, follow steps 22 to 28. To passage (or split), see steps 34 to 46.
Table 2.
Troubleshooting Guide for Aggregate Growth
| Problem | Possible cause | Solution |
|---|---|---|
| Aggregate loss or cell death | Aggregate size is suboptimal (too small or too large) | Re-plate cells using gentler technique (to reduce cell death) or check attachment conditions (to reduce cell loss; e.g., check Matrigel batch, cell density) |
| Poor recovery of aggregates post-thaw | Aggregate number per vial is suboptimal (too few or too many) | Re-check optimal thaw ratio |
| Aggregates lost at handling | Re-plate cells using gentler technique (to reduce cell death) or check attachment conditions (to reduce cell loss, e.g., check Matrigel batch, cell density) | |
| Too many aggregates post-thaw or split | Too many aggregates plated | Plate fewer aggregates at next split (and monitor for faster expansion and increased risk of spontaneous differentiation) |
| Sparse aggregates post-thaw or split | Too few aggregates plated | Increase density at initial plating. Split cells at 5-6 days post-split and use a lower split ratio (1:2-1:4) to reset and recover. |
| Excess spontaneous differentiation | Rough handling, suboptimal aggregate density, skipped medium changes | hESCs with spontaneous differentiation are more adherent than normal aggregates and can be removed at passage by shortening the ReLeSR exposure time. This will sacrifice some healthy aggregates (also remaining attached to the plate) but removes areas of differentiation from the passage. |
Passaging hESCs
-
34.Determine split ratio of the cells by estimating (or counting; see Support Protocol 2) the number of aggregates and the size of the aggregates present.The split ratio is determined both by the number of aggregates you obtain after splitting the cells (estimated based on aggregate size and density, the specifics of the handling technique, and the amount of spontaneous differentiation present and verified with the actual aggregate count post-split) and by the intended downstream use for the cells (see below). Specifics within the suggested range should be empirically determined.For expansion or banking of cells, use a 1:10 to 1:50 split ratio. For expanding cells for hematopoietic differentiation, use a 1:10 to 1:20 ratio. For freezing cells, use a 1:5 to 1:10 ratio.
-
35.
Prepare new Matrigel-coated plates for passaging as above (see steps 4 to 10) or prepare for freezing cells (see Support Protocol 1, steps 1 to 11).
-
36.
Warm complete mTeSR1 medium from step 3 to room temperature (1 ml per well to harvest plus enough medium for plating or freezing).
-
37.Remove one plate from the incubator and aspirate medium from the wells.Work with just one plate at a time, as it is critical to not let the plates dry out.
-
38.Wash wells with 1 ml DPBS and aspirate wash.Gently add PBS down the side of the wells without disturbing the aggregates.
-
39.Add 1 ml ReLeSR per well and aspirate all ReLeSR within 1 min.This leaves a thin (invisible) film of liquid over the cells.ReLeSR is an enzymatic solution used to detach aggregates or cells from Matrigel-coated plates. ReLeSR is stored and used at room temperature.A good time for ReLeSR incubation is the time it takes to add 1 ml liquid to five additional wells (i.e., if you are processing a whole 6-well plate, add ReLeSR to each well and then remove the ReLeSR from the wells in order, without rushing; if you are processing fewer than six wells, mentally mimic processing the other wells to space out the timing.
-
40.Incubate the plate after ReLeSR removal at 37°C for 5 to 7 min.The incubation time varies depending on (1) the individual user (even after training), (2) the incubator (even with frequent calibration), and (3) the aggregate size and density (larger and denser aggregates will take longer to remove). A good practice is to place the plate in the incubator for 4.5 min and then remove it and check under the microscope for loosening of aggregates (aggregate edges should be compacted, and cells should be retracted into the body of the aggregate). Incubate longer as needed, with continual monitoring under the microscope at room temperature. Reference images of aggregates at different times during ReLeSR incubation can be found in the ReLeSR manual from STEMCELL Technologies.
-
41.
Add 1 ml complete mTeSR1 medium per well to stop ReLeSR action.
-
42.Detach colonies by gentle but firm tapping on the sides of the plate.Hold the plate firmly in one hand and tap the side of the plate using the other hand. Then, rotate the plate and repeat the tapping on each of the remaining sides, distributing the force equally across the plate. Empirically determine the number of times to tap the plate. A good starting point is 8 to 13 times per side, resulting in release of ~95% of all aggregates without reducing individual aggregate size more than 50%.
-
43.If working with multiple wells, use a 5-ml serological pipet to transfer all cell aggregates to a 15- or 50-ml conical tube. Pipet up and down once when all cells are transferred to the conical to get even distribution of aggregates in solution and help homogenize aggregate size.Optional: Check that the cell aggregates are appropriately sized by counting the aggregates and/or individual cells as described in Support Protocol 2. If aggregates are too big, pipet gently to break up.
-
44.
Repeat steps 37 to 43 for all remaining plates to be harvested.
-
45.Plate cells at the desired density on the prepared plates from step 35 or freeze cells as in Support Protocol 1, steps 1 to 11.The desired density should be such that cultures can be split every 4 to 7 days and is usually a 1:10 to 1:50 split. If the cells were too dense or too sparse at the last split, adjust the split ratio accordingly.
-
46.
Place plate back in the incubator, evenly distribute cells (see step 21), and leave undisturbed for 24 hr.
BASIC PROTOCOL 2 DIFFERENTIATION OF hESCs TO HEMATOPOIETIC PROGENITOR CELLS (HPCs)
Differentiation of hESCs (Basic Protocol 1) to HPCs can be performed with a variety of methods (Akopian et al., 2010; Choi et al., 2011; Crawford et al., 2021; Kim et al., 2017; Salvagiotto et al., 2011). Here, we use defined commercial media and two distinct cytokine stimulation steps. First, hESCs are passaged, and early-stage (1-day post-passage) aggregates are induced to undergo a hematopoietic-endothelial transition beginning with differentiation to an endothelial-like adherent layer (days 1 to 3), followed by commitment to hematopoiesis (days 3 to 5) and finally further expansion of the hematopoietic population (days 6 to 12). After hematopoietic commitment and expansion, HPCs are harvested off the endothelial cell layer, collected, and washed, and then they can be used for downstream assays. This protocol is optimized to use one complete bottle of the STEMd-iff Hematopoietic Kit to differentiate 15 wells of hESCs and yields ~2 × 107 CD34+ HPCs. However, the entire protocol can easily be scaled down proportionally. An outline of the differentiation is shown in the last panel of Figure 1A.
NOTE: At all steps, be careful not to disturb the aggregates by scratching with the pipet tip or by pipetting liquid onto the aggregate surfaces directly, and watch out for drying of exposed well surfaces during handling steps.
Additional Materials (also see Basic Protocol 1)
STEMdiff Hematopoietic Kit, containing STEMdiff basal medium and Supplements A and B (STEMCELL Technologies, cat. no. 05310; store all at −20°C)
IMDM (Iscove’s Modification of DMEM; HyClone, cat. no. SH30228.01)
Trypan blue
Vortex
70-μm nylon cell strainers (Fisher, cat. no. 22363548, or equivalent)
Additional reagents and equipment for counting aggregates (see Support Protocol 2) and counting HPCs
Preparation
-
1.
Culture hESCs for at least two passages after thawing (see Basic Protocol 1).
-
2.Split cells, preparing two wells of hESCs for each batch to be used for differentiation (see Basic Protocol 1).This is a normal passage of hESCs as described in Basic Protocol 1 to maintain hESCs under normal growth conditions.
-
3.Feed and monitor hESCs as in Basic Protocol 1, steps 22 to 33, until ready for passaging again.hESCs should be split when aggregates are medium to large and ~75% confluent-equivalent within the well, although the cells perform better when split the day before you would normally split for passage so that cells are in exponential growth but not senescent. Therefore, it is possible to plan differentiation experiments within a ~2-day range but not exactly before the point of hESC split for differentiation.
Day -1: preparing and splitting cells
-
4.Prepare 15 wells in three separate 6-well tissue culture plates by coating with 1 ml prepared Matrigel solution (as in Basic Protocol 1, steps 4 to 10 and steps 13 and 14).Pre-grid the bottom of two wells for easier counting on day 0 by drawing lines ~4 mm apart with an ultra-fine-point permanent pen. Each grid square should be visible on all sides under a 4× objective under a microscope, and the squares are simply guides to keep track of your location in the well when counting. Grid these two wells on two separate plates for replicate counts so cells are exposed to outside-incubator conditions for minimal possible time to minimize spontaneous differentiation and cell death due to handling. An example grid is shown in Figure 2B, image 1.
-
5.Split hESCs using ReLeSR (see Basic Protocol 1, steps 36 to 43) and plate on 6-well plates for a final (on day 0) aggregate density of about 200 aggregates per well (Fig. 1B; see Support Protocol 2 for counting aggregates).This means that more aggregates are plated at this step to achieve an optimal concentration. For the author, 350 aggregates reliably result in 200 aggregates per well on the following day (day 0; see also Fig. 3A).IMPORTANT NOTE: Aggregates must number between 100 and 300 per well on the next day to get optimal differentiation. Aggregate counting can vary between users, as does the total number of aggregates that remain at day 0 (Fig. 3A), and each individual user should empirically determine the best plating density using repeated small-scale differentiation batches (see also Troubleshooting in the Commentary).
-
6.
Distribute aggregates evenly across the plate (see Basic Protocol 1, step 21) and culture overnight.
-
7.
Remove STEMdiff basal medium (from the STEMdiff Hematopoietic Kit) from the freezer and thaw at 4°C overnight.
Day 0: initial differentiation of aggregates
-
8.
Count aggregates from each gridded well independently, storing all other plates in the incubator throughout counting.
-
9.Confirm that about 200 aggregates are present per well.The number of aggregates present here at day 0 is generally reduced compared to the number plated at day -1 (see step 5 above), and each user should individually determine the survival rate to optimize to about 200 surviving aggregates per well at this step. It does not matter how many aggregates each user needs to plate at day 0 as long as the survival number is predictable (Fig. 3A).
-
10.Prepare Medium A:
- Retrieve STEMdiff basal medium from being thawed at 4°C overnight. If the medium is not completely thawed, place it temporarily at room temperature until all ice is gone. When the medium is thawed (do not let it warm up yet), proceed with steps 10b to 10g.
- Remove Supplement A (from the STEMdiff Hematopoietic Kit) from −20°C ~5 min before use.
- Aliquot 45.5 ml STEMdiff basal medium to a 50-ml conical tube.
- Vortex Supplement A and quick-spin.
- Add 225 μl Supplement A (all of it) to the STEMdiff basal medium aliquot to make Medium A.
- Return remaining STEMdiff basal medium to 4°C immediately.
- Allow Medium A aliquot to come to room temperature before proceeding.This protocol is for one batch (15 wells). If you are processing smaller batches, follow the manufacturer’s instructions to aliquot supplements and media.
-
11.Remove all plates from the incubator.This step involves a quick medium change only, so it is more practical to remove all the plates in one step to minimize opening/closing the incubator (and disturbing other cells), and the medium change is quick enough that this causes no harm to the current cells at this step.
-
12.
Aspirate medium from one plate.
-
13.
Immediately add 2 ml Medium A to each well.
-
14.
Repeat steps 12 and 13 for remaining plates.
-
15.
Return all plates to incubator and leave undisturbed.
-
16.
Store remaining Medium A at 4°C until day 2.
Day 2: differentiation of aggregates, part 1
-
17.
Warm Medium A to room temperature.
-
18.Remove plates from the incubator and check that aggregates are present and enlarged compared to day 0.If the user is comfortable with aggregate analysis, all three plates can be handled in one batch, as the medium change is quick. If desired or if still learning analysis, each plate can be handled individually.
-
19.
Remove 1 ml medium (half the well volume) from each well of one plate and discard.
-
20.
Immediately add 1 ml fresh Medium A to each well.
-
21.
Repeat steps 19 and 20 for all plates.
-
22.
Return all plates to incubator and leave undisturbed.
Day 3: differentiation of aggregates, part 2
-
23.Prepare Medium B:
- Remove Supplement B (from the STEMdiff Hematopoietic Kit) from −20°C ~5 min before use.
- Check that 75 ml STEMdiff basal medium remains.This volume should remain because the manufacturer slightly overfills the bottle. If 45.5 ml was used in step 10c (from a listed total of 120 ml), there should be enough left for 15 wells.You can keep the medium in the original bottle.
- Vortex Supplement B and quick-spin.
- Add 375 μl Supplement B (all of it) to STEMdiff basal medium to make Medium B.This protocol is for one batch (15 wells). If you are processing smaller batches, follow the manufacturer’s instructions to aliquot supplements and media.
-
24.
Remove all plates from incubator and check that aggregates are present and enlarged compared to day 2.
-
25.
Aspirate medium from each well of one plate.
-
26.
Immediately add 2 ml Medium B to each well.
-
27.
Repeat steps 25 and 26 for all plates.
-
28.
Return all plates to incubator and leave undisturbed.
-
29.
Store remaining Medium B at 4°C until needed.
Days 5, 7, and 10: differentiation of aggregates, part 3
-
30.
Warm Medium B to room temperature.
-
31.
Remove plates from the incubator and check that aggregates are differentiating.
-
32.Remove 1 ml medium (half the well volume) from each well of one plate and discard.At days 7 and 10, remove only 0.9 ml medium from each well to account for evaporation, so that the final well volume is 1 ml old medium plus 1 ml new medium after the medium change.A representative image of successful differentiation is shown in Figure 2A.
-
33.
Immediately add 1 ml fresh Medium B to each well.
-
34.
Repeat steps 32 and 33 for all plates.
-
35.
Return all plates to incubator and leave undisturbed.
-
36.
Store remaining Medium B at 4°C until needed.
Day 12: harvest of HPCs
-
37.
Warm DMEM/F12 medium to 37°C.
-
38.Set up 50-ml conical tubes to collect all harvested cells.Cells are collected by supernatant removal and multiple wash steps (with a minimum of two 50-ml conical tubes per batch). The number of wash steps varies at step 43 depending on the degree of HPC attachment and user variability, so additional 50-ml conical tubes for collection are likely to be needed. Have extra tubes available and use as many as needed to collect all HPCs. The number of tubes used in steps 40 to 43 does not matter, as all cells will be combined at step 46.
-
39.
Remove one plate from the incubator.
-
40.Harvest supernatant cells:
- Using a 1-ml pipet tip, gently pipet supernatant around the first well a couple of times and transfer all medium plus floating cells to a 50-ml conical tube.
- Add 2 ml warm DMEM/F12 to well.
- Repeat steps 40a and 40b for all remaining wells on the plate.
- Check that floaters and loosely attached cells are removed before proceeding.Some HPCs naturally release into the supernatant or are very loosely attached to the aggregate. These are visible as suspension cells at this step (Fig. 2B, image 2), and gentle, minimal pipetting force at this step removes these cells while protecting their viability before proceeding to dispersal of more firmly attached HPCs (see steps 41 to 43).HPCs are attached to the surface of the aggregate to different degrees (Fig. 2A) and must all be removed. This is achieved by balancing trituration of attached cells with frequent removal of detached cells to preserve viability in steps 41 and 42.
-
41.Harvest attached hematopoietic cells (first round of detachment):
- Pipet cells in one well gently but vigorously to break up (triturate; Fig. 2C).Go over every millimeter of the plate with the pipet ~2 mm away from the surface, but not touching.Minimize other wells/areas that are exposed to air by rotating the whole plate periodically.
- Transfer cell suspension to a 50-ml conical tube.
- Add 2 ml warm DMEM/F12 to the well.
- Repeat steps 41a to 41c for all remaining wells in plate.Check that attached hematopoietic cells are coming off but that the adherent layer remains intact (mature and budding HPCs vs. the hematopoietic-endothelial layer in Fig. 2A).
-
42.Harvest second set of attached hematopoietic cells (second round of detachment):
- Pipet cells in one well gently but vigorously to break up (triturate).Go over every millimeter of the plate with the pipet ~2 mm away from the surface, but not touching.Minimize other wells/areas that are exposed to air by rotating the whole plate periodically.
- Transfer cell suspension to a 50-ml conical tube.
- Add 2 ml DPBS to the well.
- Repeat steps 42a to 42c for all remaining wells in plate.Check that most of the attached hematopoietic cells are detached but that adherent cells are still attached (although likely lifting up).
-
43.Repeat step 42 (trituration and PBS coverage) as many additional times as needed until all hematopoietic cells are collected but minimal adherent cells are collected.See Figure 2D, demonstrating remaining aggregates (hemato-endothelial layer) and removal of budded and mature HPCs.
-
44.
Repeat steps 39 to 43 for all remaining plates.
-
45.
Spin down all collected cells for 5 min at 300 × g at room temperature.
-
46.
Pool all cells from the same batch (all three differentiation plates and all washes from steps 40 to 45) and resuspend in 25 ml DPBS.
-
47.
Pass cells through a 70-μm nylon cell strainer into a new 50-ml conical tube to break up clumps.
-
48.
Wash cell strainer with 5 ml DPBS to rinse all cells through.
-
49.Repeat wash in step 48 five times total.The final volume should be 50 ml.
-
50.
Spin down 5 min at 300 × g at room temperature.
-
51.
Remove and discard supernatant.
-
52.Resuspend as appropriate for downstream assays:
- For CD34 micro-bead isolation, resuspend all cells in 5 ml DPBS.
- For FACS isolation, resuspend all cells in 1 ml DPBS.
- For phenotyping all cells, resuspend in DPBS as in Support Protocol 3 or at 300 μl per original differentiation well.
- For myeloid colony assays, resuspend at 106 cells in 1 ml IMDM without additives (see Basic Protocol 3).
- For other assays, resuspend in medium appropriate for the assay, usually 1 ml assay-specific medium (see also Basic Protocol 3 for long-term culture).In steps 52a to 52e, the volume and type of medium used for resuspension depend on the number of wells harvested, the cell pellet size, and downstream applications. Some suggestions for a full batch of cells are described here.
-
53.Count using trypan blue to distinguish live versus dead cells.Full differentiation will yield a mixture of HPCs (majority population) that should be ≥80% live plus a minor fraction of dead cells (<5%), endothelial cells (lifted from the adherent layer during harvest), and erythroid differentiating cells (that due to heme production can easily be miscounted as trypan blue positive). Differentiation success (and viability) can be measured by flow cytometry–based phenotyping using Support Protocol 3. For troubleshooting the viability of differentiated cells, see Table 3.
-
54.
Plate cells for downstream assays (e.g., Basic Protocol 3) or store on ice.
Table 3.
Troubleshooting Guide for HPC Differentiation
| Problem | Possible cause | Solution |
|---|---|---|
| Minimal HPCs recovered at harvest | HPCs stuck on the adherent monolayer | Check plates and repeat trituration steps as needed |
| Poor differentiation of cells | Monitor differentiation at all major milestones (days 1, 2, 3, 5, 7, and 10 of differentiation) | |
| Too few aggregates (spaces remain between aggregates at harvest) | Re-plate or optimize plating density for each user (Fig. 3A and 3B). Check that aggregates are surviving split and are adherent at day 0 and optimize for survival rather than initial plating (Fig. 3A). | |
| Too many aggregates (aggregates are crowded by day 5) | Plate fewer aggregates. Re-plate or optimize plating density for each user (Fig. 3A and 3B). An optimal number of aggregates at day 0 for each user can be adjusted to optimize HPC yield (Fig. 3B). | |
| Individual aggregates are too small | Mid-sized aggregates are best. Re-plate or optimize plating density for each user (Fig. 3A and 3B). Reducing the ReLeSR incubation time by ~20% over normal may help. | |
| hESCs are already spontaneously differentiated | A small amount of spontaneous differentiation indicates cells have differentiation capability, but >5% differentiated cells in the culture prior to directed differentiation inhibits hematopoiesis. Small amounts of spontaneous differentiation can be removed at passage (Table 1, last row), but for excess differentiation, use a different batch of hESCs. | |
| Poor viability of HPCs at harvest | HPCs are dying during differentiation | Verify aggregate plating density and kinetics of differentiation (as above). See also troubleshooting for minimal recovery. |
| HPCs are lost/dying at harvest | Verify that HPCs are live and present in a differentiated well prior to harvest by sampling a small number of cells prior to trituration. Reduce pipetting force, handling steps, and waiting time (when handling large number of wells). See also troubleshooting for minimal recovery. | |
| Poor HPC survival or differentiation in downstream assays | Cells are “exhausted” by the time of harvest and have lost the ability to proliferate and/or differentiate | Verify differentiation kinetics and ensure aggregate density is not too crowded |
| Cells are stressed | Check aggregate size at initial plating. Larger aggregates become senescent in the center and have reduced longevity. Remove any antibiotics/antifungals from culture. Check for contamination (e.g., mycoplasma) by sampling medium prior to plating, collecting Medium A at day 3 instead of discarding and a sample of supernatant from day 12. | |
| Cells are not true HPCs | Expand phenotyping to ensure cells are HPCs and not already differentiating |
BASIC PROTOCOL 3 DOWNSTREAM FUNCTIONAL DIFFERENTIATION OF hESC-DERIVED HPCs TO MATURE LINEAGES
The main hallmarks of HPCs are long-term survival without differentiation (survival and self-renewal) and the ability to undergo directed, purposeful differentiation to their own specific downstream lineages (differentiation). This protocol outlines two different methods to test and validate HPC function following hESC-to-HPC differentiation (Basic Protocol 2). The cells discussed here are HPCs and no longer hESCs. Here, we first use stromal cells to maintain and expand HPCs in vitro (Walenda et al., 2010). Culturing HPCs on a feeder layer of bone marrow stromal cells for an additional 12 days allows measurement of long-term stem cell survival. Second, we use a classic method to assess the differentiation potential of HPCs to the myeloid lineage using a myeloid colony-forming assay (Bock, 1997), which is used extensively to assess stem cell function and predict transplant success. In this assay, single HPCs are plated in semi-solid medium supplemented with cytokines and expand to clonal colonies differentiating to myeloid and erythroid lineages. Colonies are counted at 7 days (total initial survival), 14 days (total colonies and early differentiation), and 21 days (complete differentiation for hESC-derived HPCs).
Materials
Stromal cells [e.g., M2-10B and S1/S1 stromal cells obtained from STEMCELL Technologies on behalf of Dr. Donna Hogge at Terry Fox Laboratories, BC Cancer Agency; STEMCELL Technologies, 00301 and 00302 special request, not catalog listed; maintained and passaged according to Umashankar & Goodrum (2014)]
1 mg/ml collagen solution (see recipe)
DPBS, without calcium and magnesium (Corning, cat. no. 21-031-CV)
Stromal cell medium
HPCs differentiated from hESCs (see Basic Protocol 2)
Complete MyeloCult (MyeloCult H5100, STEMCELL Technologies, cat. no. 05150) containing 1 μM hydrocortisone (STEMCELL Technologies, cat. no. 74142; pre-filtered with a 0.22-μm syringe filter)
MethoCult SF H4436 (STEMCELL Technologies, cat. no. 04436)
IMDM (Iscove’s Modification of DMEM; HyClone, cat. no. SH30228.01)
Sterile water
6-well tissue culture plates
Appropriately sized tubes (e.g., 1.5- or 2.0-ml snap-cap microcentrifuge tubes or
5- or 15-ml conical tubes)
Secondary containers (for transport of cells for irradiation, as needed and appropriate for local biosafety regulations, and to contain colony assay plates/dishes, e.g., 245-mm square tissue culture dishes)
Radiation source appropriate for cell lines (e.g., 137Cs γ-irradiation source or X-ray source capable of delivery of 4 Gy)
Tissue culture centrifuge Transwells (Corning, cat. no 3412)
6-well flat-bottom dishes (SmartDishes, STEMCELL Technologies, cat. no. 27371, are required for STEMvision; these can be replaced with equivalent flat-bottom
6-well plates or 35-mm dishes for manual counting)
Vortex
35-mm dishes
STEMvision System (with human 14-day assay; STEMCELL Technologies, cat. no. 22005) or tissue culture microscope (inverted microscope with 4× and 20× objectives; Nikon Ti2 or equivalent)
Long-term bone marrow culture
Preparation
-
1.Obtain appropriate stromal cells for use in a stem cell feeder layer.We use a 1:1 ratio of M2-10B4 and S1/S1 mouse stromal cells expressing human G-CSF and IL-3 maintained and passaged according to prior work (Umashankar & Goodrum, 2014).
-
2.Coat one well of a 6-well tissue culture plate with 1 ml collagen solution for each group to be tested and incubate at room temperature for 2 min.For smaller cell numbers for analysis, scale down the cell culture plate and cell numbers proportionally.
-
3.
Remove collagen and allow plate to dry in the tissue culture hood for 1 hr.
-
4.Transfer stromal cells for irradiation in a secondary container and irradiate using a radiation source appropriate for cell lines.For M2-10B4 and S1/S1, irradiate at 4 Gy.
-
5.
Spin down cells in an appropriately sized tube for 5 min at 500 × g at room temperature. Aspirate supernatant and wash once with DPBS.
-
6.
Wash collagen-coated wells from step 3 with 2 ml DPBS per well three times and aspirate off wash solution.
-
7.
Add 1 ml stromal cell medium to each well.
-
8.Resuspend cells in 1 ml stromal cell medium per well and add to the plate.For a 6-well plate containing M2-10B4 and S1/S1 at a 1:1 ratio, we use 1.5 × 105 cells of each cell type per well.
-
9.
Incubate at 37°C overnight.
HPC culture
-
10.
Obtain HPCs differentiated from hESCs, either immediately from harvest or further sorted (via FACS for specific phenotypic properties as desired), and resuspend in 2 ml/well complete MyeloCult containing 1 μM hydrocortisone. Plate at 5 × 104 to 2 × 105 cells/well.
-
11.
Remove medium from the stromal cells and replace with 3 ml fresh complete MyeloCult containing 1 μM hydrocortisone.
-
12.
Add transwells to the wells.
-
13.
Add resuspended HPCs from step 10 (in 2 ml complete MyeloCult containing 1 μM hydrocortisone) to the top of the transwell.
-
14.
Incubate at 37°C with 5% CO2.
-
15.Change medium every 4 days by removing 2 ml medium from beneath the transwell and replacing with fresh complete MyeloCult containing 1 μM hydrocortisone.The medium on top of the transwell contains HPCs in suspension and should not be disturbed. The innate properties of a transwell will allow mixing of the fresh medium below the transwell with the medium on top of the transwell, providing new nutrients without disturbing the HPCs during culture.
HPC analysis
-
16.After 12 days in culture, pipet HPCs up and down to remove from the surface of the transwell and transfer to a 5- or 15-ml conical tube for analysis.Analysis may include 1) counting the cells using trypan blue to assess viability and cell growth; 2) analyzing the cells by flow cytometry using Support Protocol 3 to assess phenotype, including stem cell markers; or 3) plating the cells for downstream differentiation (i.e., myeloid colony-forming assays, as in steps 17 to 24).Cells can also be collected at desired time points (between plating and harvesting at 12 days) to track growth, survival, and stem cell properties over time. For cells that fail to survive during long-term culture, see Table 3 for troubleshooting.
Myeloid colony-forming assays
Preparation
-
17.Thaw MethoCult SF H4436 at 4°C overnight.Only MethoCult SF H4436 recapitulates viral (human cytomegalovirus, or HCMV)-induced myelosuppression with hESC-derived HPCs, a well-characterized and clinically relevant phenotype in HPCs, although multiple other media support myeloid colony formation alone from these cells (Crawford et al., 2021).There are multiple techniques for plating Methocult medium and cells to yield even dispersal of single cells throughout the well for accurate analysis. Descriptions and protocols can be found on the manufacturers’ websites, but our preferred methods (two options) are described in steps 20a and 20b.
-
18.
Shake MethoCult vigorously to mix and allow bubbles to disperse.
-
19.
Warm IMDM and DPBS to room temperature.
HPC plating
-
20.a. Plating option 1: Prepare three replicate wells per condition:
- Plate 1 ml MethoCult per well in a 6-well flat-bottom plate.SmartDishes (specific 6-well flat-bottom plates) are required if using the STEMvision Analysis System. If counting manually, 6-well tissue culture dishes or individual 35-mm dishes can be used.
- Resuspend HPCs in 100 μl IMDM per intended well in a 1.5- or 2.0-ml snap-cap microcentrifuge tube.HPCs from different sources have different differentiation potential, as measured by the number of colonies formed per initial number of HPCs plated in this assay (i.e., 1 colony to X initial HPCs). hESC-derived HPCs plated initially after harvest (without subsequent sorting for CD34+ cells) usually support colony formation at a plating density of 1:1000 to 1:5000 (or 104 cells per 1 ml Methocult). This is a reduction in colony-forming ability compared to that in cells from bone marrow (about 1:20 to 1:500 cells), cord blood (about 1:20 cells), and fetal liver (about 1:5 cells).
- Add 100 μl IMDM + cells to each well by evenly dispensing cells in a circle through the well. Start by dispensing cells with the pipet halfway between the edge and the center of the well and with the pipet halfway in the depth of the medium. Dispense cells evenly while moving the pipet in a circle within the well.
- Use the side of the pipet tip to mix the cells throughout the medium until all cells are spread equally throughout the well.Plating option 1 requires only the number of cells needed for each replicate but requires more practice for even dispersal of single cells throughout the medium.
-
20.b. Plating option 2: Prepare three replicate wells per condition:
- Obtain N+1 volume of cells in IMDM for all replicates (e.g., for three replicates, 4× cells, or 400 μl cells, is needed).HPCs from different sources have different differentiation potential, as measured by the number of colonies formed per initial number of HPCs plated in this assay (i.e., 1 colony to X initial HPCs). hESC-derived HPCs plated initially after harvest (without subsequent sorting for CD34+ cells) usually support colony formation at a plating density of 1:1000 to 1:5000 (or 104 cells per 1 ml Methocult). This is a reduction in colony-forming ability compared to that in cells from bone marrow (about 1:20 to 1:500 cells), cord blood (about 1:20 cells), and fetal liver (about 1:5 cells).
- Add N+1 ml MethoCult to a 5- or 15-ml tube (depending on replicate number and availability; e.g., for three replicates, 4 ml MethoCult is needed).
- Add cells to the medium.
- Mix well by vortexing and allow bubbles to settle.
- Plate 1.1 ml MethoCult + cells in each well of a 6-well flat-bottom plate.SmartDishes (specific 6-well flat-bottom plates) are required if using the STEMvision Analysis System. If counting manually, 6-well tissue culture dishes or individual 35-mm dishes can be used.Plating option 2 requires more cells and Methocult (to make a master mix) but less practice for even dispersal of single cells throughout the medium.
-
21.
Check each well for bubbles and complete coverage with medium.
-
22.
Place plates in a secondary container with open 35-mm dishes containing sterile water to maintain humidity and prevent drying of MethoCult.
-
23.
Incubate at 37°C with 5% CO2.
Colony counting
-
24.Count colonies at 7, 14, and 21 days post-plating using the STEMvision System or a tissue culture microscope.A valuable reference for colony types and counting can be found in the manuals from the main vendors (including STEMCELL Technologies), but note that colonies from hESCs are usually less abundant, more diffuse, and not quite typical in morphology compared to colonies from cord blood and bone marrow (and therefore do differ somewhat from the manuals’ images). Individual colony types from hESC-derived HPCs can be seen in Figure 4A from our original publication (Crawford et al., 2021). For cells that fail to differentiate into myeloid cells, confirm proliferation ability using long-term culture (above) and/or see Table 3 for troubleshooting.
SUPPORT PROTOCOL 1 FREEZING AND TESTING FROZEN BATCHES OF hESCs
Banked hESCs are essential for maintaining low-passage cells (required for appropriate differentiation) and a consistent lab stock of uniform cells. Cells currently (as of 2022) available from WiCell are shipped at passage 22 (WA01) or passage 26 (WA09). Although hESCs can be maintained in culture through more than 70 passages, pluripotency markers and gene expression stability change over time (Rosler et al., 2004). This is especially relevant for downstream differentiation to hematopoietic lineages, as Choi et al. (2011) recommend using cells no later than passage 50, and we have successfully used these hESCs for viral infection studies through passage 42 with no loss of function. This protocol outlines a robust serum-based freezing method to store cells where hESC aggregates are harvested from ideal growth conditions and cryogenically stored. Regardless of method, however, each batch of frozen hESCs varies slightly in recovery. The second part of this protocol outlines a process to empirically test frozen cell batches for viability, aggregate recovery, and aggregate condition using a plating dilution scheme and daily assessment.
Additional Materials (also see Basic Protocol 1)
Complete mTeSR1 medium (see Basic Protocol 1, step 3)
Freezing medium (see recipe)
Liquid nitrogen (in cryogenic storage system of choice)
Cryovials (1.8 or 2.0 ml, rated for liquid-phase storage appropriate for cryogenic storage system)
Controlled-rate freezing container (Corning, cat. no. 432138, or equivalent)
Freezing hESCs
-
1.Determine number of cryovials needed for freezing and pre-label.Post-thaw recovery varies due to a number of factors, so each batch should be pre-tested for optimal plating density (see steps 12 to 33):
-
1)Controllable factors: freezing medium used, handling and processing time and conditions, time stored at −80°C, long-term storage conditions (liquid nitrogen storage).
-
2)Partially controllable factors (may vary between users): aggregate density, aggregate size, minor culture condition variations (e.g., temperature fluctuations or vibrations from others using the same culture incubator).
-
3)Partially controllable factors due to specific cell line, batch, and culture conditions: time in culture since last passage, passage number.
-
4)Uncontrollable factors: cell density within aggregates, overall cellular survival in culture.
Note that these factors are not necessarily problematic but should be considered when testing batches for reproducibility. -
1)
-
2.
Warm complete mTeSR1 medium (0.5 ml per final vial plus 1 ml per well to harvest) to room temperature.
-
3.Passage hESCs as described in Basic Protocol 1, steps 34 to 44, and collect all cells for this batch into one conical tube.Use optimal and actively growing aggregates for freezing. Optimal aggregates are medium to large in size (500 to 1000 μm); are 70% to 80% confluent in the well, but with minimal aggregates touching each other; have even morphology, with minimal spontaneous differentiation and dense centers in the aggregate; and are at about 4 to 5 days after the previous split or 1 day before you would normally passage cells for maintenance growth.A good starting point is to freeze one well of a 6-well plate of large aggregates that are 70% to 80% confluent in one freezing vial. This is very approximately 750 to 1000 aggregates per vial, although we routinely split by confluence equivalence rather than counted aggregates, as the time and additional handling required to count aggregates reduce frozen aggregate survival. More details of the thawing ratio and adjustments are provided in the testing section of this protocol (below).
-
4.
Spin down for 5 min at 300 × g at room temperature.
-
5.Gently remove supernatant without disturbing the cell pellet.Decant the supernatant and, keeping the tube upside down, blot on a Kimwipe/paper towel to drain remaining wash solution.
-
6.
Gently resuspend cells in 0.5 ml complete mTeSR1 medium per final vial of frozen cells, taking care to avoid breaking up aggregates.
-
7.Slowly add an equivalent volume (0.5 ml per final vial) of freezing medium.Freezing medium is used at a 1:1 ratio with normal growth medium (for hESCs, use complete mTeSR1) for final concentrations of 50% growth medium, 40% (v/v) serum, and 10% (v/v) dimethylsulfoxide (DMSO).
-
8.
Invert tube gently to mix.
-
9.
Aliquot 1 ml per cryovial.
-
10.
Freeze in a controlled-rate freezing container at —80°C for 24 hr.
-
11.Transfer to liquid nitrogen for long-term storage.hESCs are more sensitive than many cell types to long-term storage at −80°C and perform best if transferred to liquid nitrogen between 24 and 48 hr post-freezing.
Testing frozen batches of hESCs
-
12.
For each vial of cells to be tested, coat four wells of a 6-well tissue culture plate with Matrigel as described in Basic Protocol 1, steps 4 to 10.
-
13.
Warm 9 ml complete mTeSR1 medium to room temperature.
-
14.
Remove Matrigel solution from the plates and add 1 ml complete mTeSR1 medium to each of the four wells coated with Matrigel.
-
15.
Label the wells as 1:2, 1:5, 1:10, and 1:100.
-
16.Add additional complete mTeSR1 medium to each well so that final volume will be 2 ml at the end of the protocol:
- For 1:2 well, add 500 μl medium.
- For 1:5 well, add 800 μl medium.
- For 1:10 well, add 900 μl medium.
- For 1:100 well, add 990 μl medium.
-
17.
Thaw one vial of cells to be tested (see step 11) and wash with DMEM/F12 as described in Basic Protocol 1, steps 15 to 18.
-
18.
Resuspend in 1 ml complete mTeSR1 medium, mixing gently by pipetting.
-
19.Plate cells in the four pre-coated wells at different dilutions, starting with the lowest cell number (to avoid aggregate degradation from excessive mixing and to minimize inaccuracies due to settling of cells):
- For 1:100 well, add 10 μl cells.
- For 1:10 well, add 100 μl cells.
- For 1:5 well, add 200 μl cells.
- For 1:2 well, add 500 μl cells.
-
20.
Place the plate back in the incubator, distribute aggregates evenly (see Basic Protocol 1, step 21), and leave undisturbed for 24 hr.
-
21.
Check cells the following day for attachment.
-
22.
Change medium in all wells as described in Basic Protocol 1, steps 22 to 28, and culture for an additional 24 hr.
-
23.
Check cells the following day (day 2).
-
24.Determine well with the optimal aggregate conditions by assessing aggregate condition, aggregate size, and aggregate density as described in the Basic Protocol 1 and Table 1.A representative image of good aggregate density, size, and condition is shown in Figure 1B.
-
25.
Double-check all wells the following day (day 3) and confirm optimal thaw ratio as in step 24 for this batch.
-
26.Change medium in all wells that contain reasonable numbers of aggregates.Not all wells will be continued at this point. It is reasonable that, for example, the 1:2 dilution may well be too dense at this point (i.e., 50+% confluence) and/or that the 1:100 well may have no visible aggregates.
-
27.Culture cells for an additional 1 to 2 days and make a final determination of optimal thaw ratio depending on the aggregate density.If the 1:100 well is optimal, then this batch should be thawed into 100 wells. If the 1:10 well is optimal, then this batch should be thawed into 10 wells. If the 1:5 well is optimal, then this batch should be thawed into 5 wells. If the 1:2 well is optimal, then this batch should be thawed into two wells. It is rare that the optimal density is outside of this range, but for specifics, see steps 29 to 33.
-
28.
Optional: Continue culture (day 3 and onward) for optimal wells only, if desired, for additional expansion for banking or for an experiment. Change the medium on optimal well(s) daily as per normal culture protocols.
-
29.If the optimal density is close to ideal, but not ideal, then thaw future batches to the closest estimate.For example, if the 1:5 well is only slightly under the optimal density, thaw this batch at a 1:3 to 1:4 ratio.
-
30.If the optimal density is between the larger values (e.g., somewhere between 1:10 and 1:100, where 1:10 is too crowded but 1:100 is too low), then re-test within this range.Perform this step if the aggregate density does not match any of those mentioned in step 27.
-
31.If no aggregates are present in any well:
- Incubate for an additional day and check again.
- If no aggregates are present after 3 days, continue only 1:2 well for an additional 2 days to confirm.
- If no aggregates are present, re-test batch at a 1:1 thaw (i.e., one vial to one well).If no aggregates are present in a 1:1 thaw, this batch is not viable. Although it may be possible to recover this batch using a single-cell thaw protocol, this changes the cell properties, including phenotype and hematopoietic function. Thus, this batch should be discarded and a different batch used if at all possible.
-
32.If all wells are too dense, then estimate optimal thaw ratio as greater than 1:100 and update on next thaw or re-test using a 1:50, 1:100, 1:200, and 1:400 ratio.The 1:50 is added and the 1:100 is repeated because it is very rare to have a 1:100 well that is too dense and because it is possible that either there was an error in diluting the cells to the plate or the vial used for the initial test thaw had an unusual clump of cells, giving it a higher aggregate number than the average vial in the batch. This method increases the chance of success on the second test.
-
33.As needed, if the ideal thawing ratio is routinely outside of one vial plated to between 2 to 20 wells, then adjust the freezing ratio for vials that are intended for experimental use, as relatively few wells are needed for scaling up cells for hematopoietic differentiation:
- If recovery is routinely robust (e.g., one vial thaws into more than 20 wells), then increase freezing ratio to up to 2 to 3 vials per well to have a larger bank of lower-passage cells available.
- If recovery of these cells is routinely low (e.g., if one vial consistently provides only one well of sparse aggregates, and handling and other conditions are correct), then scale up to freezing two wells per vial.
SUPPORT PROTOCOL 2 COUNTING hESCs
The optimal number of aggregates in a single well varies depending on the technique used and individual variations in maintenance, expansion, banking, and differentiation. Counting the number of aggregates is a method to more precisely control this variability and also provides an opportunity to more accurately estimate aggregate size, the number of cells per well, and aggregate condition. For example, aggregates at ≥50 μm in diameter are most likely to adhere and grow after splitting (or after freezing or during differentiation). However, aggregates at >100 μm at initial plating are more likely to have increased spontaneous differentiation during routine passage, may be less likely to thaw properly, and may have reduced differentiation capabilities. Aggregate count accuracy varies between individuals, in some instances dramatically; however, person-to-person variation is less of an issue if the following is true: intra-person variation is low (your own counts are consistent) and you have optimized the protocols (splitting and differentiation) for what works with your counts (or you have calibrated your counts to the standard). Here, we describe counting hESCs both by aggregate count, through simple harvest of aggregates (as performed for passaging cells) and manual counting in a gridded dish using a standard microscope, and via single-cell counting using enzymatic dissociation of aggregates and standard counting using a hemocytometer.
Materials
DMEM/F12 medium (Gibco, cat. no. 11320-033)
hESC aggregates released using ReLeSR (see Basic Protocol 1, step 42)
5% trypsin/EDTA (Gibco, cat. no. 11320-033; store at 4°C)
DPBS, without calcium and magnesium (Corning, cat. no. 21-031-CV; store at room temperature)
Trypan blue
96-well flat-bottom plate
Ultra-fine-point pen
Tissue culture microscope (inverted microscope with 4×, 10×, and 20× objectives; Nikon Ti2 or equivalent)
1.5-ml microcentrifuge tubes
Microcentrifuge
Standard hemocytometer
Additional reagents and equipment for plating cells (see Basic Protocol 1, steps 34 to 46)
Counting hESC aggregates
-
1.Prepare three wells of a 96-well flat-bottom plate:
- Grid the bottom of each well using an ultra-fine-point pen to draw guidelines ~4 mm apart (or as needed for microscope) for counting.
- Add 40 μl DMEM/F12 to each well.
-
2.Place 5 μl hESC aggregates released using ReLeSR in each of the three wells containing DMEM/F12.Use a pipet tip no smaller than a P20 to avoid shearing aggregates.
-
3.
Mix gently by tapping and allow aggregates to settle (about 3 to 5 min).
-
4.Count the total number of aggregates in each well that are ≥50 μm in diameter under a tissue culture microscope.Optional: Count all aggregates of any size greater than four cells in the well. Keep small versus large aggregate counts separate.
-
5.Calculate total concentration of cell aggregates:
-
6.Calculate total number of cell aggregates in the sample:
-
7.Determine target number of cell aggregates to plate.Routine plating is between 350 and 1000 aggregates per well of a 6-well plate.Plating for hematopoietic differentiation is less than for passaging, typically between 300 and 600 aggregates per well of a 6-well plate.Total aggregates should be empirically determined for each user and assay (see Fig. 3). Note that conditions are optimized for aggregate culture, so single cells and 2-to 5-cell aggregates, although viable at the time of passaging, do not survive.
-
8.Calculate total volume of cell aggregates to plate for each well of the experiment:
-
9.
Continue with plating aggregates as desired (see Basic Protocol 1, steps 34 to 46).
Counting single hESCs
-
10.
Warm 0.5% trypsin/EDTA to room temperature.
-
11.Beginning with released hESCs, remove 50 μl aggregates and place in a 1.5-ml microcentrifuge tube.Use a pipet tip no smaller than a P20 to avoid shearing aggregates.
-
12.
Add 450 μl of 0.5% trypsin/EDTA solution to the tube and incubate at room temperature for 5 min to dissociate aggregates into single cells.
-
13.
Add 1 ml DPBS and spin down 5 min at 400 × g at room temperature in a microcentrifuge.
-
14.
Carefully remove all supernatant and resuspend cells in 50 μl DPBS.
-
15.
Remove 20 μl resuspended cells and mix with 20 μl trypan blue.
-
16.
Add 10 μl to each side of a standard hemocytometer.
-
17.
Count total number of cells on each side of the hemocytometer under a tissue culture microscope and average the counts.
-
18.Calculate total concentration of cells:
-
19.Calculate total number of cells in the sample:
-
20.
Determine the target number of cells to plate depending on the downstream application.
-
21.Calculate the total volume of cells to plate for each well of the experiment:
-
22.
Optional: To determine the average number of cells per aggregate, divide total number of cells from step 19 above by the total number of aggregates from step 6.
-
23.
Gently mix remaining aggregate mixture (from step 11) by pipetting and plate as desired (see Basic Protocol 1, steps 34 to 46).
SUPPORT PROTOCOL 3 PHENOTYPING BY FLOW CYTOMETRY
Flow cytometry is a well-established method to phenotype cell populations based on their expression of various cell surface receptors. Here, we describe a standard protocol to stain HPCs with a viability dye and with fluorescently conjugated antibodies for progenitor cell markers and to fix the cells to accommodate biosafety concerns with human progenitors in shared flow cytometry facilities. These data can then be used to assess the differentiation success of each batch of cells in a short (~3 hr) process.
NOTE: All centrifugation steps can be performed for 3 min at 500 × g at room temperature in 96-well plates (or for 5 min at 500 × g at room temperature in 5-ml polypropylene flow tubes). All supernatant is removed by decanting.
Materials
HPCs (see Basic Protocol 2)
DPBS, without calcium and magnesium (Corning, cat. no. 21-031-CV; store at room temperature)
Viability dye master mix [100 μl DPBS per sample plus viability dye at 1:500 dilution; for live-cell sorting, PI (BioLegend, cat. no. 421301); for fixed-cell analysis, ZombieAqua (BioLegend, cat. no. 423102)]
1 × FACS buffer (from 10× FACS buffer; see recipe), 4°C
FACS blocking buffer (see recipe), 4°C
Pre-titered antibodies (store at 4°C):
Anti-human CD29 (clone TS2/16, AlexaFluor700, BioLegend, cat. no. 303020)
Anti-human CD33 (clone P67.6, APC-Cy7, BioLegend, cat. no. 366614)
Anti-human CD34 (clone 561, APC, BioLegend, cat. no. 343608)
Anti-human CD38 (clone HIT2, Brilliant Violet 605, BioLegend, cat. no. 303532)
Anti-human CD43 (clone CD43-10G7, PE-Cy7, BioLegend, cat. no. 343208)
Anti-human CD45 (clone HI30, Brilliant Violet 785, BioLegend, cat. no. 304048)
Anti-human CD90 (clone 5E10, Brilliant Violet 421, BioLegend, cat. no. 328121)
Anti-human CD117 (clone 104D2, Brilliant Violet 650, BioLegend, cat. no. 313222)
FACS fixing buffer: 2% (v/v) neutral buffered formalin in deionized water (store at room temperature)
96-well plates (or 5-ml polypropylene flow tubes appropriate for flow cytometer)
Tissue culture centrifuge
Mini flow tubes (Fisher, cat. no. 07-200-319, or equivalent)
Flow cytometer (BD LSRII or equivalent)
NOTE: Antibodies for flow cytometry should be chosen with appropriate fluorophores for the flow cytometer available and the assay used. Suggested antibodies are shown for basic HPC phenotyping or equivalent.
Recommended controls
-
1.Plan for recommended controls:
- Negative controls: unstained samples, isotype-stained samples, and “true negative” stained samples (cells known to be negative for the selected marker).
- Background fluorescence controls: unstained sample (one well) of each cell type.
- FMO (fluorescent minus one) controls: one tube for each fluorophore used.
- Viability controls: negative and positive viability dye controls.
- Compensation controls as needed for the fluorophores used.The recommended flow cytometry controls are for the specific protocol described below. Specific details on how to prepare and use these controls and flow cytometry best practices are outside the scope of this protocol but can be found in detail elsewhere, including basic techniques (Maecker & Trotter, 2006), use of controls [including compensation (see Current Protocols article: Roederer, 2002a) and FMO controls (see Current Protocols article: Roederer, 2002b)], and large-panel optimization (see Current Protocols article: Ferrer-Font et al., 2021).
Viability staining and blocking
-
2.
Plate HPCs and controls in 96-well plates.
-
3.
Spin down and wash twice with DPBS.
-
4.Add 100 μl viability dye master mix to each sample and to positive viability dye control (see step 1).Proteins (including serum) neutralize viability dye activity, so samples must be washed and stained in plain DPBS.
-
5.
Incubate samples for 10 min at room temperature in the dark.
-
6.
Set up compensation controls (see step 1).
-
7.
Spin down cells and decant supernatant.
-
8.
Wash cells twice with cold 1 × FACS buffer (200 μl per well).
-
9.
Decant off supernatant and resuspend cells in cold FACS blocking buffer (50 μl per well).
-
10.
Incubate for 30 to 60 min at 4°C.
Surface antibody staining
-
11.
Spin down cells and decant off supernatant.
-
12.Resuspend cells in cold staining solution (cold FACS blocking buffer + pre-titered antibodies; 50 μl per sample) or cold FACS blocking buffer alone as appropriate:
- Staining solution: samples and stained controls (see step 1).
- Blocking buffer only: unstained controls and background fluorescence controls (see step 1).
- Other staining panels: isotype control and compensation (single-color) mixes (if compensating with stained cells instead of beads) (see step 1).
-
13.
Stain cells for 30 to 60 min at 4°C in the dark.
-
14.
Spin down cells and decant off supernatant.
Fixation
-
15.
Resuspend cells in room-temperature FACS fixing buffer (100 μl/well).
-
16.Fix cells for 10 to 15 min at room temperature.IMPORTANT NOTE: Do not reduce the fixing step to <10 min or increase it to >15 min.
-
17.
Spin down cells and decant supernatant.
-
18.
Wash cells twice in cold 1 × FACS buffer (200 μl/well).
-
19.
Resuspend in cold 1 × FACS buffer (120 μl/well) and transfer to mini flow tubes.
-
20.
Analyze samples using a flow cytometer.
REAGENTS AND SOLUTIONS
Collagen solution, 1 mg/ml
Resuspend collagen (Sigma, cat. no. C7661-5MG) in acetic acid at 3 mg/ml and dilute to 1 mg/ml in sterile water. Store ≤1 year at 4°C.
FACS blocking buffer
1% (v/v) human serum
1% (v/v) mouse serum
1 × FACS buffer (from 10×; see recipe)
Store ≤2 months at 4°C
FACS buffer, 10 ×
0.2M KH2PO4
1.5M NaCl
2.5% (w/v) bovine serum albumin (BSA)
0.5% (w/v) sodium azide
dH2O
Store ≤1 year at 4°C
Freezing medium
80% (v/v) fetal bovine serum (FBS; HyClone characterized; Fisher, cat. no. SH3007003, or equivalent; store at —20°C)
20% (v/v) DMSO (Sigma, cat. no. 472301, or equivalent; store at room temperature)
Filter-sterilize with 0.22-μm Steriflip conical filter (Millipore-Sigma, cat. no. SCNY00020)
Store ≤1 month at 4°C
COMMENTARY
Background Information
hESCs provide a valuable model to generate a consistent and reproducible source of CD34+ HPCs. Using this system, genetically identical batches of cells can be generated with a consistent phenotype, in contrast to genetically and phenotypically diverse primary HPCs. Since the original derivation of five hESC lines in 1998, thousands of different cell lines have been generated, including normal and disease models and a variety of modified lines (Ludwig et al., 2018). Many groups, including commercial vendors, have since developed methods for maintenance and differentiation to specific downstream lineages. However, variations in maintenance and differentiation methods result in different cells, function, and outcomes, and few comparative studies exist. There are two main components of differentiating HPCs from hESCs: 1) maintenance of hESCs and 2) directed differentiation.
First, we wanted to choose a maintenance method that maintains multiple hESC lines for future utility and supports their undifferentiated state without accumulation of mutations or chromosomal abnormalities. Akopian et al. (2010) directly compared eight different hESC lines in 10 different maintenance culture systems and found that only two commercial medium formulations supported all cell lines tested for 10 passages across five different laboratories. We chose one of these commercial medium platforms (mTeSR1) for our testing previously (Crawford et al., 2021) because it uses only two components, has relatively long storage stability, is used by stem cell-banking organizations (e.g., WiCell), and does not require maintenance of additional feeder cell lines, thus simplifying use for more users.
Second, we previously assessed eight different differentiation methods to derive HPCs from WA01 or WA09 for the number of HPCs derived per well of starting hESCs, the overall cost of materials, ease of use, the phenotype of CD34+ HPCs generated, and their long-term survival. The top two methods were compared in detail previously (Crawford et al., 2021). We chose to continue with the method described here, as HPCs generated with this commercial method (using the STEMdiff Hematopoietic Kit) had a reproducible yield and phenotype across both hESC lines tested (WA01 and WA09) and multiple passages (WA01 cells were tested between passages 29 and 39). Differentiation success was assessed by number of HPCs per well recovered at 12 days post-differentiation compared to the initial number of aggregates used (Fig. 3B) and the phenotype and function of the HPCs (Crawford et al., 2021). This method was chosen for ease of use for new users, simplicity (only three medium components are required), and reproducibility. Although the cost per batch is higher with this method than some of the others tested, this was mitigated by the batch-to-batch reproducibility and reduction in failed differentiation experiments with multiple users. This method also standardizes differentiation across users at different laboratories.
Importantly, we previously assessed these methods for function with both WA01 and WA09 hESCs. Many methods assess HPC phenotype by cell surface markers but include limited or no functional assessment. For studies assessing hESC function and downstream differentiation or using HPC differentiation as a control for other pathways, this is often sufficient. However, for using hESCs as a platform to generate consistent and reproducible pools of HPCs for studies on HPC function, further testing of downstream assays is essential. We previously assessed both hESC lines for biologically relevant outcomes following HCMV infection (Crawford et al., 2021). HCMV is a prototypical beta-herpesvirus that infects 40% to 90% of the world (Howley, Knipe, Cohen, & Damania, 2021), and although most infection is asymptomatic, the virus is a leading cause of congenital abnormalities and a significant cause of morbidity and mortality following transplant (Annaloro et al., 2021; Nogalski, Collins-McMillen, & Yurochko, 2014; Ramanan & Razonable, 2013; Stern et al., 2019; Voigt, Schaffrath Rosario, & Mankertz, 2016; Xia et al., 2021). HCMV infection in WA01 and WA09 hESC-derived HPCs reproduces phenotypes seen during infection of primary HPCs, which themselves reproduce biological phenotypes in vivo, including establishment of viral latency and reactivation, myelosuppression, and the function of specific viral genes, as determined with recombinant knockout viruses (Crawford et al., 2021). Cumulatively, these data demonstrate that reproducible maintenance of hESCs (Basic Protocol 1) and differentiation of hESCs to HPCs (Basic Protocol 2) provide a common platform for multiple groups to generate large numbers of HPCs from a common genetic background and with comparable phenotypes and functions to primary cells that are usable for downstream differentiation and analysis (Basic Protocol 3 and Support Protocol 3).
Critical Parameters
HPC differentiation from hESCs varies by protocol, timing, and user and often results in variability. We found that this method (Basic Protocol 2) is optimal for standardizing between users and yields the most HPCs with the most consistent phenotype. However, individual users should still optimize specific steps and perform quality control on all HPCs prior to interpretation of downstream assay results. Inefficient HPC differentiation or failure of HPCs to function downstream is likely indicative of poor-quality hESCs, spontaneous differentiation, or non-optimal differentiation techniques.
Quality of hESC culture
The quality of the initial culture (Basic Protocol 1) is essential for maintaining a usable bank of cells and for efficient HPC differentiation. All steps should be performed based on good laboratory practices and sterile technique. It is advisable to avoid antibiotics and antifungals in hESC culture when at all possible, as these induce cell stress and reduce HPC differentiation and survival. Cultures should be monitored regularly for quality and spontaneous differentiation (Fig. 1D) and appropriate growth (see Table 2 for troubleshooting). Morphological changes in hESC maintenance can indicate spontaneous differentiation (Fig. 1D) or the induction of senescence (indicated by slowing of aggregate growth or loss of differentiation potential). Cells should also regularly be screened for pluripotency markers, e.g., NANOG, OCT4, SOX2, SSEA3, and SSEA4 (Thomson et al., 1998), and checked for genetic abnormalities by karyotyping.
Seeding density
Aggregate density at the time of differentiation induction is critical to differentiation success. Too few aggregates reduce the overall yield and too many aggregates suppress differentiation and can induce cell death. The optimal plating density varies by hESC line and by user (Fig. 3A). Each user should optimize the ideal plating density for their cell line and standardize their counting procedures prior to large-batch production.
Maintenance culture conditions
Early-passage hESCs should be banked as soon as possible, as although hESCs can be maintained and passaged for greater than 100 passages, older lines have altered mitochondrial function and differentiation potential (Xie et al., 2011). Banking for long-term “golden” stocks (early-passage cells to be used for working-bank regeneration only) and a working bank for experimental use is important to enhance reproducibility. Thawed cells should be tested for viability and chromosomal and phenotype stability and observed for consistent morphology (Fig. 1B and 1C). Cells should be treated gently (minimal environmental vibrations, careful pipetting, and handling) and consistently (minimizing changes in the environment, including temperature, humidity, and atmospheric composition, and in timing for handling to avoid nutrient stress). Spontaneous differentiation (Fig. 1D) is common, and small amounts demonstrate that hESCs retain differentiation potential. However, excess spontaneous differentiation (>5% of cells in more than one well of a multiwell expansion) is a sign of stress. Undifferentiated cells can, in some cases, be removed at passaging (see Table 2); however, this can also select for a different subpopulation of hESCs and should be avoided if possible.
Differentiation culture conditions
Induction of HPC differentiation and progenitor cell expansion vary depending on the method used, including the timing of medium changes within a specific protocol. Protocol differences are key to differentiation differences (Crawford et al., 2021), which is a central premise of this article, but variations in timing within a protocol can also influence outcome. hESCs during differentiation and maintenance are sensitive to small fluctuations in conditions. Care should be taken to minimize handling (time out of the incubator, movement of the plates), external vibrations (incubator fans, external equipment, other use of the same incubator/shelf), and stress to the cells. Consistency in timing of medium changes (daily medium changes should be consistently at every 24 hr, rather than 16 hr followed by 32 hr, for example) also improves reproducibility.
Downstream differentiation
HPCs should be assessed for stem cell phenotype (expression of stem cell–related markers, e.g., CD34, CD33, CD90, CD117), hematopoietic commitment (e.g., CD43, CD45), minimal but present early erythroid lineage commitment (CD71), minimal but diverse early lineage commitment (e.g., CD29, CD33, CD5, CD7), and no mature lineage commitment (e.g., CD3, CD19, CD339). HPCs that do not pass quality control, i.e., have a suboptimal phenotype, poor harvest yield, low population viability, or morphological changes, are unlikely to survive through downstream assays.
Troubleshooting
Understanding Results
During passage, hESCs should maintain normal morphology (Fig. 1B and 1C), with minimal spontaneous differentiation (Fig. 1D). Floating individual cells will be present following thawing or after initial split but should be reduced after 2 days in culture. If floating cells are continuous or reappear, this is a sign of cell death and culture stress. Spontaneous differentiation in minimal amounts is also normal, but >5%, in more than one well, or in back-to-back passages is detrimental and an indicator of culture stress. Visual assessment of morphology at medium change is critical to identifying problems early, but care should be taken to minimize the time that cells are outside of the incubator.
The initiation of differentiation is critical to overall HPC yield and function. Different users will evaluate the number of aggregates in solution differently as aggregates are more challenging to count than single cells. The specific counts or variations here are less important, assuming that counts are normalized between users (e.g., one person’s 100 aggregates/ml could be a second person’s 200 aggregates/ml) and that each user tests their optimal aggregate plating (plated number) for recovery on the following day (day 0 of differentiation; Fig. 3A) and optimizes this until it is reproducible. Figure 3A demonstrates a couple of these scenarios and outcomes. First, user 1 and user 3 have different counting methods from each other, but each user is internally reproducible for the proportion of aggregates that survive to day 0. In the second scenario, user 2 is comparable to user 1 in counts at plating but needs to standardize their individual methods to have consistent plating to day 0 survival between batches. Although these numbers can vary greatly, practice (10+ rounds of counting and plating aggregates at known numbers) and comparison with others improve user-to-user reproducibility. Each user should then confirm their ideal plating number to maximize HPC yield at the end of differentiation, although 200 aggregates per six wells present on day 0 is a good starting point for many (Fig. 3B).
During hematopoietic differentiation, handling should still be minimized, but morphology assessment at each medium change is an easy way to monitor the progress of differentiation. Differentiation of hESCs to HPCs follows natural differentiation along the hemato-endothelial lineage, beginning with flattening and expansion of the initial aggregates (first 2 days), followed by endothelial commitment and merging of some aggregates (day 3). After changing medium formulations with different cytokine support, differentiation proceeds along the hematopoietic lineage, and hematopoietic cells should be visible as small, rounded hematopoietic cells that begin to accumulate on top of the endothelial layer. As differentiation progresses, HPC differentiation and expansion will be visible using lower-power phase-contrast microscopy. A time course of this progression is shown in Figure 1A of our original publication (Crawford et al., 2021) and in the product information accompanying the STEMdiff Hematopoietic Kit. Figure 2A in the present article shows an enlarged image of day 7 differentiation, highlighting the transition along the hemato-endothelial lineage, with all major milestones visible at this midpoint.
Following differentiation, HPCs should be of a consistent phenotype and should be predominantly HPCs that are CD34+CD43+ and ~50% CD45+, with subsets expressing CD90, CD33, CD5, or CD71 and no cells expressing mature cell markers (i.e., CD3, CD19, CD339) (Crawford et al., 2021). HPCs should also be functional. In this method, we assessed function by both long-term culture with stromal cell support and myeloid differentiation (Basic Protocol 3). In longterm culture, cells should maintain viability, proliferate, and maintain stem cell function, including downstream differentiation potential. HPCs that do not survive in long-term culture are likely “exhausted” or lack further ability to proliferate and/or differentiate. This state correlates with hESC cultures (prior to differentiation) that are stressed or overly crowded and/or with crowded (too many initial aggregates) hematopoietic differentiation conditions. Functional cells, as assessed by myeloid differentiation, will have proliferative ability (based on colony formation and assessment of the ratio of colonies to initial cells plated) and multi-lineage differentiation capability (both myeloid and erythroid colonies form, and mature, heme-producing cells form by 21 days post-plating). Note that HPCs from hESCs have reduced colony-forming ability compared to primary cells (especially early, fetal liver-derived HPCs), and colonies are smaller (smaller colony diameter with fewer cells/colony) and slower to develop (21 days to heme production compared to 14). Overall, functional HPCs following differentiation will be able to survive and proliferate in supportive cultures and can further differentiate to downstream lineages compared to primary cells.
Time Considerations
Basic Protocol 1: 2 weeks per batch of cells, plus initial banking of 4 to 6 weeks depending on the desired size of the golden and working stock banks, which is performed once.
Basic Protocol 2: 13 days if starting from cells in culture, 3 weeks if thawing fresh cells. Note that for most experiments, harvesting HPCs (~3 hr), additional CD34 isolation (1 to 3 hr), and plating for downstream assays (1 to 3 hr) require a full day. Viably frozen hESC-derived HPCs are functional for phenotyping but appear to have less success in culture assays, as viability drops significantly upon freezing.
Basic Protocol 3: 12 days for minimal longterm culture, 21 days for myeloid colonyforming assays.
Support Protocol 1: 4 days.
Support Protocol 2: 30 min.
Support Protocol 3: 3 to 5 hr.
Acknowledgments
This publication was funded through a subaward to LBC from the Nebraska Center for Integrated Biomolecular Communication, a COBRE funded by P20 GM113126 (NIGMS). The author would also like to thank members of all the labs that contributed to development of these protocols.
Footnotes
Conflict of Interest
The funders had no role in protocol design, data collection, or analysis or interpretation of the data.
Data Availability Statement
All data are available within this publication or the primary paper referenced here (Crawford et al., 2021).
Literature Cited
- Akopian V, Andrews PW, Beil S, Benvenisty N, Brehm J, Christie M, … Suemori H (2010). Comparison of defined culture systems for feeder cell free propagation of human embryonic stem cells. In Vitro Cellular & Developmental Biology - Animal, 46(3), 247–258. doi: 10.1007/s11626-010-9297-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annaloro C, Serpenti F, Saporiti G, Galassi G, Cavallaro F, Grifoni F, … Onida F (2021). Viral infections in HSCT: Detection, monitoring, clinical management, and immunologic implications. Frontiers in Immunology, 11, 569381–569381. doi: 10.3389/fimmu.2020.569381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock TA (1997). Assay systems for hematopoietic stem and progenitor cells. Stem Cells, 15, Suppl 1, 185–195. doi: 10.1002/stem.5530150824 [DOI] [PubMed] [Google Scholar]
- Choi KD, Vodyanik M, & Slukvin II (2011). Hematopoietic differentiation and production of mature myeloid cells from human pluripotent stem cells. Nature Protocol, 6(3), 296–313. doi: 10.1038/nprot.2010.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford LB, Hancock MH, Struthers HM, Streblow DN, Yurochko AD, Caposio P, … Nelson JA (2021). CD34(+) Hematopoietic progenitor cell subsets exhibit differential ability to maintain human cytomegalovirus latency and persistence. Journal of Virology, 95(3), e02105–20. doi: 10.1128/jvi.02105-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Font L, Small SJ, Lewer B, Pilkington KR, Johnston LK, Park LM, … Price KM (2021). Panel optimization for highdimensional immunophenotyping assays using full-spectrum flow cytometry. Current Protocols, 1(9), e222. doi: 10.1002/cpz1.222 [DOI] [PubMed] [Google Scholar]
- Galić Z, Kitchen SG, Kacena A, Subramanian A, Burke B, Cortado R, & Zack JA (2006). T lineage differentiation from human embryonic stem cells. Proceedings of the National Academy of Sciences, 103(31), 11742–11747. doi: 10.1073/pnas.0604244103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howley PM, Knipe DM, Cohen JL, & Damania BA (Eds.) (2021). Fields Virology: DNA Viruses (Vol. 2). Wolters Kluwer. [Google Scholar]
- Kim S-J, Jung J-W, Ha H-Y, Koo SK, Kim E-G, & Kim J-H (2017). Generation of hematopoietic stem cells from human embryonic stem cells using a defined, stepwise, serum-free, and serum replacement-free monolayer culture method. Blood Research, 52(1), 37–43. doi: 10.5045/br.2017.52.1.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarova M, Latour AM, Chason KD, Tilley SL, & Koller BH (2010). Human embryonic stem cells: A source of mast cells for the study of allergic and inflammatory diseases. Blood, 115(18), 3695–3703. doi: 10.1182/blood-200908-237206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Yang H, Nan H, Liu P, Pang S, Zhao Q, … Wang F (2012). Identification of key regulatory pathways of myeloid differentiation using an mESC-based karyotypically normal cell model. Blood, 120(24), 4712–4719. doi: 10.1182/blood-2012-03-414979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig TE, Kujak A, Rauti A, Andrzejewski S, Langbehn S, Mayfield J, … Bhattacharyya A (2018). 20 years of human pluripotent stem cell research: It all started with five lines. Cell Stem Cell, 23(5), 644–648. doi: 10.1016/j.stem.2018.10.009 [DOI] [PubMed] [Google Scholar]
- Maecker HT, & Trotter J (2006). Flow cytometry controls, instrument setup, and the determination of positivity. Cytometry Part A, 69A(9), 1037–1042. doi: 10.1002/cyto.a.20333 [DOI] [PubMed] [Google Scholar]
- Nogalski MT, Collins-McMillen D, & Yurochko AD (2014). Overview of human cytomegalovirus pathogenesis. Methods in Molecular Biology, 1119, 15–28. doi: 10.1007/978-1-62703-788-4_2 [DOI] [PubMed] [Google Scholar]
- Ramanan P, & Razonable RR (2013). Cytomegalovirus infections in solid organ transplantation: A review. Infection & Chemotherapy, 45(3), 260–271. doi: 10.3947/ic.2013.45.3.260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao J, & Greber B (2017). Concise review: Signaling control of early fate decisions around the human pluripotent stem cell state. Stem Cells, 35(2), 277–283. doi: 10.1002/stem.2527 [DOI] [PubMed] [Google Scholar]
- Roederer M (2002a). Compensation in flow cytometry. Current Protocols in Cytometry, 22, 1.14.1–1.14.20. doi: 10.1002/0471142956.cy0114s22 [DOI] [PubMed] [Google Scholar]
- Roederer M (2002b). Multiparameter FACS analysis. Current Protocols in Immunology, 49(1), 5.8.1–5.8.10. doi: 10.1002/0471142735.im0508s49 [DOI] [PubMed] [Google Scholar]
- Rosler ES, Fisk GJ, Ares X, Irving J, Miura T, Rao MS, & Carpenter MK (2004). Long-term culture of human embryonic stem cells in feeder-free conditions. Developmental Dynamics, 229(2), 259–274. doi: 10.1002/dvdy.10430 [DOI] [PubMed] [Google Scholar]
- Salvagiotto G, Burton S, Daigh CA, Rajesh D, Slukvin II, & Seay NJ (2011). A defined, feeder-free, serum-free system to generate in vitro hematopoietic progenitors and differentiated blood cells from hESCs and hiPSCs. PLOS One, 6(3), e17829. doi: 10.1371/journal.pone.0017829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern L, Withers B, Avdic S, Gottlieb D, Abendroth A, Blyth E, & Slobedman B (2019). Human cytomegalovirus latency and reactivation in allogeneic hematopoietic stem cell transplant recipients. Frontiers in Microbiology, 10, 1186. doi: 10.3389/fmicb.2019.01186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, & Jones JM (1998). Embryonic stem cell lines derived from human blastocysts. Science, 282(5391), 1145–1147. doi: 10.1126/science.282.5391.1145 [DOI] [PubMed] [Google Scholar]
- Umashankar M, & Goodrum F (2014). Hematopoietic long-term culture (hLTC) for human cytomegalovirus latency and reactivation. Methods in Molecular Biology, 1119, 99–112. doi: 10.1007/978-1-62703-788-4_7 [DOI] [PubMed] [Google Scholar]
- Vazin T, & Freed WJ (2010). Human embryonic stem cells: Derivation, culture, and differentiation: A review. Restorative Neurology and Neuroscience, 28(4), 589–603. doi: 10.3233/rnn-2010-0543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt S, Schaffrath Rosario A, & Mankertz A (2016). Cytomegalovirus seroprevalence among children and adolescents in Germany: Data from the German health interview and examination survey for children and adolescents (KiGGS), 2003-2006. Open Forum Infectious Diseases, 3(1), ofv193. doi: 10.1093/ofid/ofv193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walenda T, Bork S, Horn P, Wein F, Saffrich R, Diehlmann A, … Wagner W (2010). Co-culture with mesenchymal stromal cells increases proliferation and maintenance of haematopoietic progenitor cells. Journal of Cellular and Molecular Medicine, 14(1-2), 337–350. doi: 10.1111/j.1582-4934.2009.00776.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, Yan H, Zhang Y, Wang C, Gao W, Lv C, … Liu Z (2021). Congenital human cytomegalovirus infection inducing sensorineural hearing loss. Frontiers in Microbiology, 12, 649690–649690. doi: 10.3389/fmicb.2021.649690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Hiona A, Lee AS, Cao F, Huang M, Li Z, … Wu JC (2011). Effects of long-term culture on human embryonic stem cell aging. Stem Cells and Development, 20(1), 127–138. doi: 10.1089/scd.2009.0475 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are available within this publication or the primary paper referenced here (Crawford et al., 2021).