

Abstract
Gram-negative bacteria are intrinsically resistant to many classes of antibiotics, predominantly due to the impermeability of the outer membrane and the presence of efflux pumps. Small molecule adjuvants that circumvent these resistance mechanisms have the potential to expand therapeutic options for treating Gram-negative infections to encompass antibiotic classes that are otherwise limited to treating Gram-positive infections. Adjuvants that effect increased antibiotic permeation, either by physical disruption of the outer membrane or through interference with synthesis, transport, or assembly of membrane components, and adjuvants that limit efflux, are discussed as potential avenues to overcoming intrinsic resistance in Gram-negative bacteria.
Graphical abstract
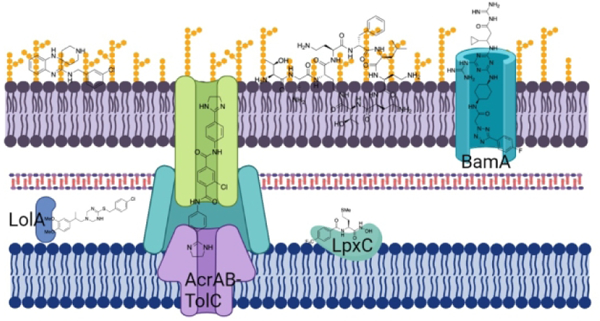
Introduction.
Infections caused by multidrug-resistant (MDR) bacteria, in some cases resistant to every clinically available antibiotic, are increasing in prevalence and represent a serious health threat.1 The use of antibiotics as the primary treatment for bacterial infections over the last 70 years has enabled significant medical advances; practices such as: surgery, premature infant care, cancer chemotherapy, and transplantation would not be feasible without effective antibiotics.2 The World Health Organization (WHO) states that “a post-antibiotic era - in which common infections and minor injuries can kill - is a very real possibility for the 21st century”3. If resistance continues to rise at the current rate it is projected that MDR bacterial infections will account for over 10 million deaths, and a global economic cost of 100 trillion USD annually by 2050.4, 5 The rise in MDR infections has coincided with a decrease in the number of novel antibiotics entering the clinic, due in part to significant divestment of pharmaceutical companies from antibiotic drug discovery.6, 7
Infections caused by MDR Gram-negative bacteria are of particular concern. Of the six bacterial species identified as the greatest threat to human health, the ESKAPE pathogens, four are Gram-negative: Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species.8 The last novel class of antibiotics with activity against Gram-negative bacteria introduced into the clinic were the fluoroquinolones in the 1960’s,9 and even last resort antibiotics such as polymyxins are experiencing increasing levels of resistance.10 Carbapenem resistant A. baumannii, P. aeruginosa, and K. pneumoniae were included in the highest category of a list of pathogens released by the WHO in 2017 as resistant to the majority of currently available antibiotics and urgently requiring new therapeutic options.11
In this Digest we give a brief background as to why infections caused by Gram-negative bacteria are so challenging to overcome, present an overview of traditional and current treatment approaches, and discuss recent advances in the use of small molecule antibiotic adjuvants to tackle this problem.
Challenges of treating Gram-negative infections.
The fundamental structural differences between the cell envelopes of Gram-positive and Gram-negative bacteria (Figure 1) play a significant role (along with the presence of efflux pump systems) in the response to many antibiotics. The Gram-positive cell envelope consists of a symmetrical phospholipid bilayer, the cytoplasmic membrane (CM), surrounded by a cell wall comprised of many layers of peptidoglycan 30 – 100 nm thick, with a narrow periplasmic space between the two.12 The Gram-negative cell envelope also contains a CM and cell wall, however the peptidoglycan layer is much thinner, typically only a few nanometers thick.13 The major difference, and that which plays the most significant role in the differing response to antibiotics, is the presence of an outer membrane (OM). The Gram-negative OM is an asymmetrical lipid bilayer, the inner leaflet of which contains phospholipids, while the outer leaflet is composed of glycolipids, principally lipopolysaccharide (LPS). LPS typically consists of a hydrophobic domain known as lipid A, a core oligosaccharide, and a distal polysaccharide or O-antigen,14 (in lipooligosaccharide (LOS) O-antigen is not present). Most OM proteins are either lipoproteins anchored in the inner leaflet of the OM, or integral transmembrane ß-barrel proteins, known as porins, that mediate the uptake of small molecules across the OM in addition to contributing to membrane stability.13, 15
Figure 1.
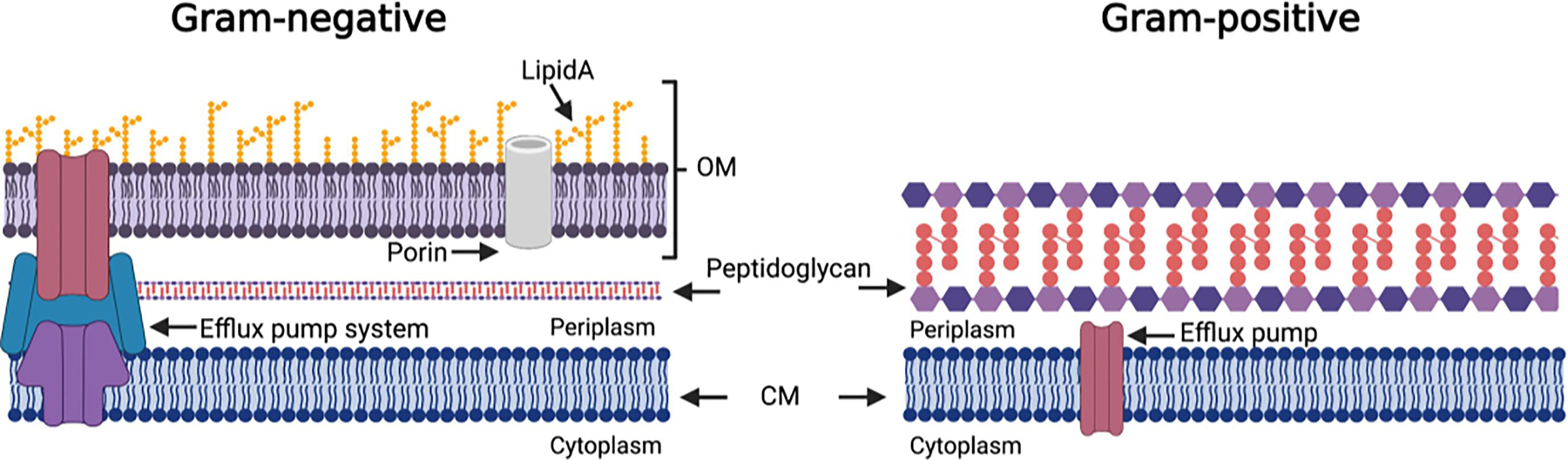
Schematic representation of major structural differences between the Gram-negative and Gram-positive cell envelopes.
Both LPS and porins play a critical role in the barrier function of the OM, LPS effectively prevents access of hydrophobic molecules, while diffusion of hydrophilic molecules through porins is limited to molecules below ~700 Da.13 This combination confers intrinsic resistance to many antibiotic classes, including macrolides, glycopeptides, and lipopeptides, despite the fact that the targets of most antibiotics are highly conserved across Gram-positive and Gram-negative species.16
Current treatment approaches for Gram-negative infections.
Treatment options for infections caused by Gram-negative bacteria have typically consisted of small broad-spectrum hydrophilic antibiotics such as ß-lactams, tetracyclines, and fluoroquinolones that enter the cell through porins.17 Other antibiotics used to treat infections caused by Gram-negative bacteria gain access via a self-promoted uptake pathway that involves the displacement of divalent cations and subsequent destabilization of the LPS; such antibiotics are usually cationic and include polymyxins, aminoglycosides, and the later generation macrolide azithromycin. Some compounds enter through a combination of both pathways.18, 19
Most antibiotics used to treat infections caused by Gram-negative bacteria (with the exception of polymyxins) are broad-spectrum, and acquired resistance is becoming widespread.20 This is in part due to the fact that many resistance determinants reside on mobile genetic elements and thus can disseminate rapidly, and multiple resistance genes are often co-harbored on the same genetic elements.21 As the pool of effective antibiotics for the treatment of Gram-negative infections is inherently smaller than that for infections caused by Gram-positive bacteria, increasing levels and prevalence of resistance to established therapeutic options represents a formidable predicament.
While attempts to identify novel antibiotic classes with activity against Gram-negative bacteria have proven fruitless in recent decades, synthetic modification of scaffolds within known classes has seen some success in (at least temporarily) overcoming acquired resistance. ß-Lactam antibiotics such as the later generation cephalosporins and newer carbapenems display increased stability toward various classes of ß-lactamases.22,23 Similarly, the novel ß-lactamase inhibitors avibactam and vaborbactam show increased inhibition of class A and C ß-lactamases compared to ß-lactam containing inhibitors.24, 25 Semi-synthetic aminoglycosides such as arbekacin and plazomicin are not as readily modified by aminoglycoside modifying enzymes,26 while the glycylcycline tigecycline circumvents many of the resistance mechanisms that befall the tetracyclines27.
Structural modifications to known antibiotic classes that overcome intrinsic resistance in Gram-negative bacteria, i.e., that convert a Gram-positive only antibiotic to a broad-spectrum antibiotic have been exploited for decades. The addition of an amine moiety to penicillin G brought about the first Gram-negative acting ß-lactam, ampicillin,28 while ring expansion of the erythromycin macrocycle with a basic nitrogen to form azithromycin results in improved activity against some Gram-negative species.29, 30 Richter and Hergenrother have developed a series of guiding principles for compound accumulation in Escherichia coli that they term “eNTRy Rules”19,31, which should aid in future structural modification approaches to expanding the spectrum of Gram-positive acting antibiotics to encompass Gram-negative species.
Combinations of two or more antibiotics have a long-term precedent in the management of infectious diseases,32 and this approach has been utilized to overcome the ineffectiveness of many antibiotics in monotherapy for treating Gram-negative infections.20 Polymyxins in particular have been utilized in combination with antibiotics from several other classes, including ß-lactams, tigecycline, fluoroquinolones, aminoglycosides, and rifampin.32 One mechanism by which polymyxins synergize with other antimicrobials is through a permeabilizing effect on the Gram-negative OM that facilitates entry of partner antibiotics.33 This has relevance both for antibiotics for which acquired resistance is a result of decreased uptake, and antibiotics to which Gram-negative bacteria are intrinsically resistant.32
Antibiotic adjuvants.
The combination of an antibiotic with a non-microbicidal adjuvant that potentiates antibiotic activity, termed an adjuvant,34 has long been successfully utilized clinically to overcome acquired resistance in the case of ß-lactamase inhibitors.35 The adjuvant approach also holds promise for intrinsic resistance in Gram-negative bacteria, and is receiving increasing attention. An adjuvant that circumvents intrinsic resistance to one or more of the several classes of antibiotics that are clinically employed to treat infections caused by Gram-positive bacteria would have significant implications for the treatment of infections caused by MDR Gram-negative bacteria, for which there are in some cases no available treatment options and very few likely to materialize in the near future.
OM disruptors.
Gram negative bacteria with a permeabilized OM exhibit increased susceptibility to otherwise Gram-positive selective antibiotics.19 The use of an adjuvant that permeabilizes the OM is therefore an attractive approach to sensitizing Gram-negative bacteria to Gram-positive selective antibiotics; however, indiscriminate membrane disruptors often exhibit mammalian toxicity and therefore require structural optimization for in vivo application.
As mentioned, polymyxin antibiotics are used clinically in combination with several other classes of antibiotics, and synergize through permeabilization of the OM to effect increased entry of the partner antibiotic to the cell.33 Polymyxin antibiotics have several distinct interactions beyond the initial encounter with LPS that play a role in the mechanism of bacterial killing.36 This phenomenon has been exploited to develop analogues that retain permeabilization but lack bactericidal activity. Polymyxin B nonapeptide (PMBN) 1 (Figure 2), generated by the enzymatic removal of the acyl group and N-terminal 2,4-diaminobutyryl (Dab) residue from polymyxin B (PMB), retains the OM permeabilizing activity of PMB but lacks microbicidal activity.37 PMBN increases the susceptibility of E. coli to the hydrophobic antibiotics erythromycin, clindamycin, rifampin, fusidic acid, novobiocin, and cloxacillin.37 However, as nephrotoxicity is a common adverse effect of polymyxin antibiotics, and PMBN retains this nephrotoxicity, the further development of polymyxin analogues to both improve potency and reduce toxicity has been explored.
Figure 2.
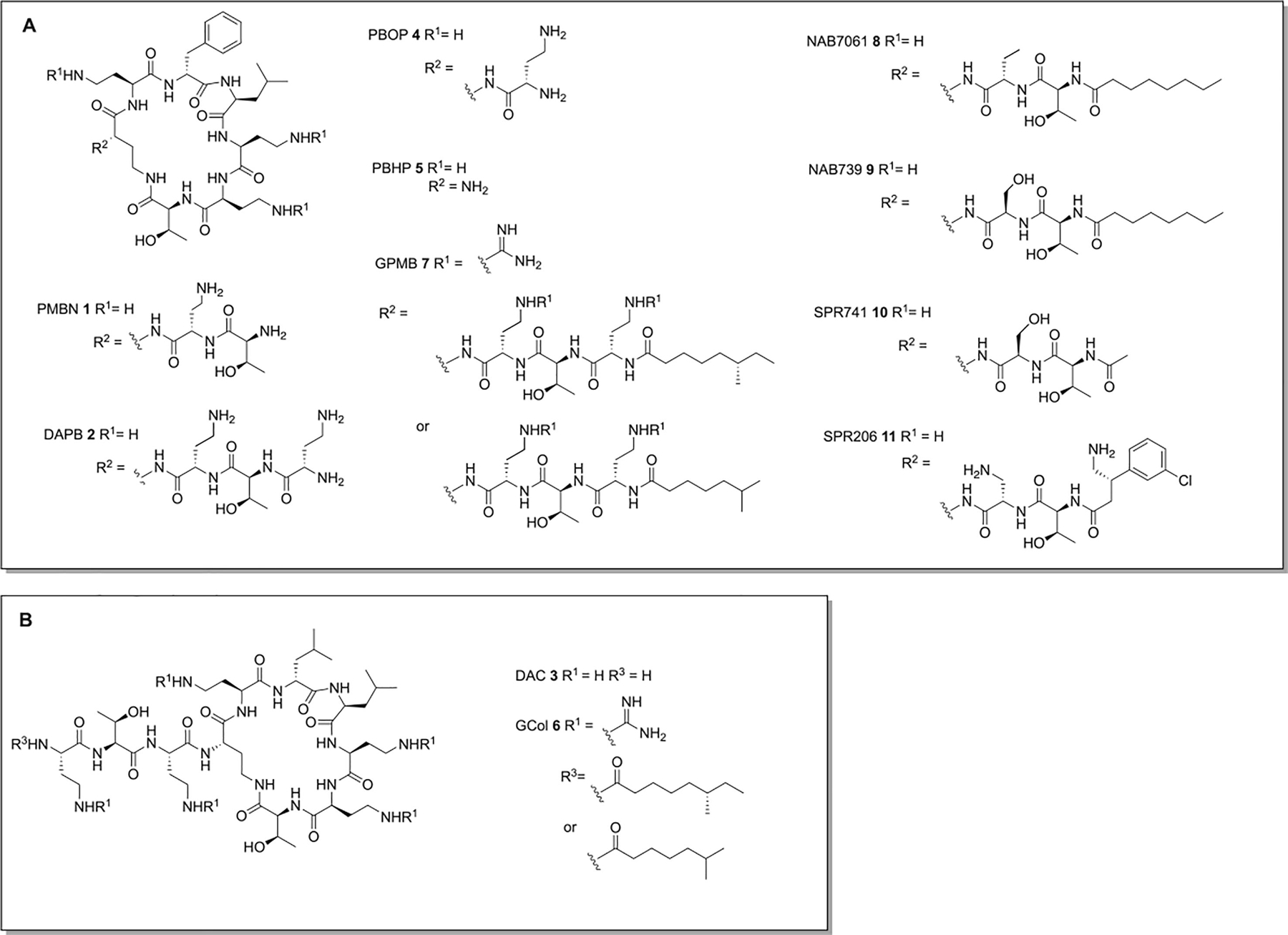
Polymyxin derived adjuvants. A: Adjuvants derived from the polymyxin B scaffold. B Adjuvants derived from the colistin scaffold.
Deacylation of polymyxin B or colistin to generate deacylpolymyxin B (DAPB) 2 and deacylcolistin (DAC) 3 (Figure 2) respectively, results in more potent activity with several antibiotics against E. coli and Salmonella typhimurium than PMBN (300 fold reduction in MIC compared to a 10-fold reduction effected by PMBN at 3 μg/mL).38 However, these two compounds retain antibacterial activity, likely as a result of the six positive charges.38 Further truncation of the polymyxin core in polymyxin B octapeptide (PBOP) 4 and polymyxin B heptapeptide (PBHP) 5 (Figure 2), results in low inherent toxicity toward E. coli and S. typhimurium. Both compounds sensitize E. coli and S. typhimurium to rifampin, erythromycin, clindamycin, and novobiocin, with the more positively charged PBOP exhibiting more potent activity.39 PMBN, PBOP, and PBHP also act as potent synergists with erythromycin, lincomycin, linezolid, nisin, and vancomycin against P. aeruginosa PA14.40
Guanidylated-colistin (GCol) 6 and guanidylated-polymyxin B (GPMB) 7 (Figure 2) possess guanidine moieties in place of the 4-amino groups of the Dab residues of colistin and polymyxin respectively.41 Both compounds synergize with erythromycin and rifampin against multiple strains of P. aeruginosa, A. baumannii, E. coli, E. cloacae, and K. pneumoniae, displaying superior activity than PMBN, however GCol and GPMB exhibit similar cytotoxicity in comparison to PMB toward HEK293, and HepG2 cell lines.41
Reducing the number of positive charges carried by the polymyxin scaffold as a means to reduce nephrotoxicity has been investigated, and several analogues with only three positive charges (compared to the five of polymyxin antibiotics and PMBN) retain potentiation activity.42,43 NAB7061 8 and NAB739 9 (Figure 2), in which the Dab residue of the linear portion of the polymyxin is replaced with an aminobutyryl (Abu) or D-Ser residue respectively, both synergize potently with rifampin, clarithromycin, and vancomycin.42 Compared to PMB, both compounds exhibit lower affinity for the brush border membrane (BBM) of the renal cortex, and exhibit no cytotoxicity against V79 Chinese hamster lung fibroblast cells at the highest concentration tested (128 μg/mL).42
SPR741 10 (Figure 2), previously known as NAB741, also carries three positive charges, and has an acetyl group in place of the hydrophobic octanoyl residue.43 SPR741 was initially reported to sensitize E. coli, K. pneumoniae, E. cloacae, and A. baumannii to rifampin, clarithromycin, azithromycin, mupirocin, fusidic acid, and vancomycin at 4 μg/mL,43 and later reported to additionally sensitize E. coli and K. pneumoniae to erythromycin, retapamulin, and telithromycin, and A. baumannii to erythromycin, and retapamulin.44 The combination of 4 μg/mL SPR741 and 1 μg/mL rifampin inhibits bacterial growth of all but one of a panel of 28 extensively drug resistant A. baumannii clinical isolates, and the combination is bactericidal and synergistic in vitro. In a murine pulmonary model of A. baumannii infection, 60 mg/kg SPR741 and 5 mg/kg rifampin effected a survival rate of 90% compared to 50% for rifampin alone, and decreased the bacterial burden in lung tissue by 6 log10 CFU/g compared to the untreated control, an additional 2 log10 CFU/g compared to rifampin alone.45
SPR741 acts predominantly on the OM, effecting little depolarization the E. coli CM compared to PMB and PMBN while causing 10–15 nm undulations in the cell surface. Genetic truncation of the LPS core selectively increases sensitivity to SPR741 over PMB and PMBN, indicating heptose residues in the LPS core prevent access of SPR741 to the CM.46 SPR741 exhibits no cytotoxicity against V79 Chinese hamster lung fibroblast cells at the highest concentration tested (128 μg/mL), while renal clearance was 400-fold higher than that of colistin.43 SPR741 entered phase 1 clinical trials in 2016 but was discontinued in 2020 and replaced with SPR206 11 (Figure 2), a polymyxin with standalone antibacterial activity.47
Several other peptide membrane disrupters have been reported. The cathelicidin peptides L-11 12 (RIVQRIKKWLR-NH2) and its D-amino acid counterpart D-11 13 sensitize E. coli, P. aeruginosa, K. pneumoniae, and A. baumannii to vancomycin,48 while D-11 also sensitizes K. pneumoniae to rifamycins, aminocoumarins, and macrolides.49 Activity is lost upon addition of exogenous LPS or Mg2+, indicating that the peptide binds LPS.48 Linear lipopeptide paenipeptin analogues 14 and 15 (Figure 3) potentiate rifampin and clarithromycin against A. baumannii and K. pneumoniae to nanomolar concentrations, and additionally potentiate erythromycin and vancomycin against A. baumannii. Once again, addition of exogenous LPS neutralizes activity indicating LPS binding is driving the increase in permeability.50 Short linear antibacterial peptide (SLAP)-S25 16 (Figure 3), increases the efficacy of rifampin and vancomycin against E. coli. Exogenous LPS or addition of divalent cations reduces activity and SLAP-S25 binds LPS, even when phosphoethanolamine (PetN) modified, with high affinity in vitro. Exogenous CM phospholipids also reduce activity, and phosphatidylglycerol (PG) abolishes activity, with SLAP-S25 binding PG with higher affinity than LPS, suggesting the adjuvant activity of SLAP-S25 is a result of triggering membrane damage through binding both the OM and CM.51
Figure 3.
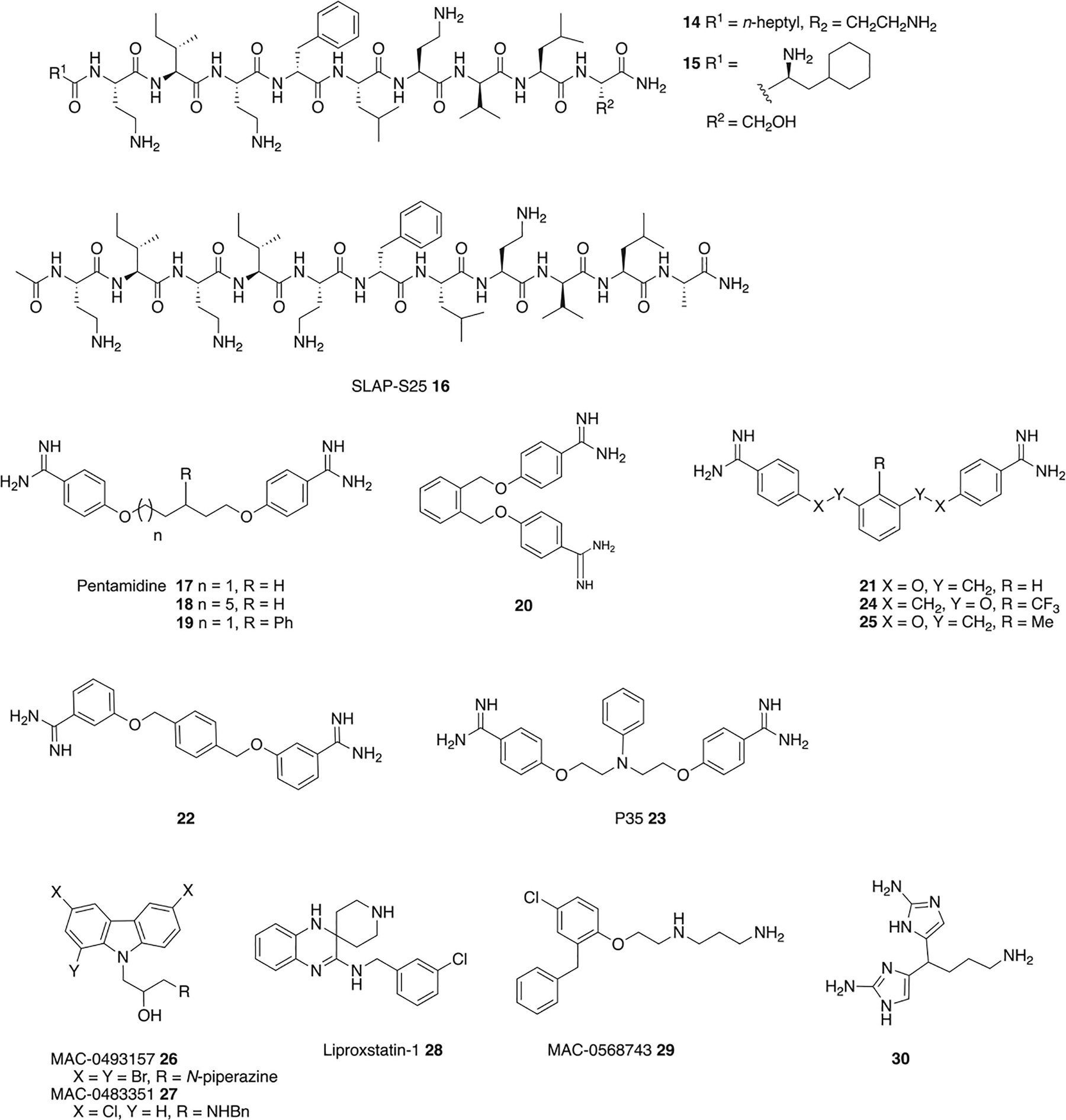
Adjuvants that act via OM disruption.
The anti-protozoal drug pentamidine 17 (Figure 3) is an OM permeabilizer that synergizes potently with rifampin, novobiocin, and erythromycin, but not vancomycin, against several Gram-negative species and, unlike for PMBN, activity is retained against polymyxin resistant isolates (harboring mcr-1).52 In a mouse model of colistin-susceptible A. baumannii infection, 10 mg/kg pentamidine and 5 mg/kg novobiocin effected 100% survival, while in a colistin-resistant A. baumannii model, 10 mg/kg pentamidine and 50 mg/kg novobiocin rescued 90% of the mice.52 Pentamidine causes 40 nm undulations on the surface of treated E. coli, cells, binds purified lipid A in vitro with high affinity (Kd ∼120 nM), results in enhanced LPS release similar to polymyxins, and its activity is impeded by both exogenous LPS and Mg2+, all of which indicate direct association with LPS.52 However, pentamidine is beset by side effects including: nephrotoxicity, hypotension, hypoglycemia, and QT prolongation, limiting its clinical potential.53
Preliminary structure functions studies on pentamidine revealed that each of the amidine groups is necessary for activity, and increased hydrophobicity, inter-amidine distance, and rigidity leads to increased rifampin potentiation, with compounds 18 and 19 exhibiting increased potency compared to pentamidine.52 From a series of bis-amidines with reduced linker flexibility, 20, 21 and 22 exhibited the greatest activity, synergizing to a greater degree than pentamidine with erythromycin, rifampin, and novobiocin against E. coli, K. pneumoniae, A. baumannii, and P. aeruginosa, although exhibiting lower activity than PMBN in most cases.54 P35 23, in which an N-phenyl group replaces the central carbon of pentamidine, synergizes with novobiocin to a much greater degree than pentamidine, and also potentiates rifampin, rifaximin, fusdic acid, roxithromycin, erythromycin, clarithromycin thiostrepton, trimethoprim, and linezolid at 50 μg/mL against A. baumannii, the latter three of which pentamidine does not potentiate.53 P35 has an improved toxicity profile compared to pentamidine, with lower cytotoxicity against HEK293 cells, a reduced impact on HepG2 cell cycle arrest, and lower hERG trafficking inhibition.53
The diamidine MD-124 24 was developed following an SAR study on the MD-100 (25) scaffold, which was identified from a screen of cationic compounds for rifampin potentiation in E. coli. MD-124, sensitizes E. coli, A. baumannii, and K. pneumoniae to rifampin and clarithromycin, with strain specific activity. Activity of MD-124 is reduced upon addition of divalent cations or exogenous LPS, and in an E. coli strain with a compromised OM and. A dansyl-PMBN fluorescent indicator suggested MD-124 binds to LPS, and molecular dynamics simulations more specifically indicate binding at the hydrophilic portion of LPS.55 The addition of phosphatidylcholine has a much lower effect on activity than addition of exogenous LPS, indicating selectivity toward LPS over eukaryotic phospholipids, and MD-124 exhibits low cytotoxicity toward HEK293 and NIH3T3 cells.55
The carbazoles MAC-0493157 26 and MAC-0483351 27 (Figure 3) sensitize E. coli to linezolid, while MAC-0493157 also potentiates novobiocin. Both compounds physically disrupt OM integrity to a similar extent as SPR741, in addition to disrupting the CM; however, both are cytotoxic toward HEK293 cells.56 Liproxstatin-1 28, a ferroptosis inhibitor in human cells, and MAC-0568743 29, potentiate large hydrophobic antibiotics (rifampin, novobiocin, erythromycin) in addition to the small hydrophilic linezolid in E. coli, but do not potentiate large, hydrophilic antibiotics. Both compounds retain activity with rifampin in K. pneumoniae and A. baumannii, while MAC-0568743 also exhibits activity in P. aeruginosa. Both compounds disrupt the OM and increase cell membrane roughness but have limited impact on CM integrity. Again, addition of Mg2+ or exogenous LPS suppresses activity, indicating LPS binding, which was confirmed with a BODIPY-cadaverine assay. However, against an E. coli waaC deletion mutant, in which LPS is truncated, both compounds exhibited more pronounced synergy with rifampin than in wild type E. coli, suggesting that LPS binding is independent of the LPS core. The two compounds may act through distinct or multiple mechanisms, as MAC-0568743 has no interaction with PetN modified LPS, while liproxstatin-1 has comparatively lower affinity for LPS.57
The bis-2-aminoimidazole 30 potentiates azithromycin against P. aeruginosa, reducing the MIC against PA01 from 256 μg/mL to 0.25 μg/mL at 60 μM, and to 1 μg/mL at 40 μM. Activity is retained against several cystic fibrosis clinical isolates including two mucoid strains, and 30 displays activity in a Galleria mellonella infection model. Activity is correlated with an increase in cell membrane permeability though direct binding of LPS has not been established.58
Interference with LPS synthesis or OM assembly.
Although the use of membrane permeabilizing adjuvants may prove to be a viable approach for the treatment of infections caused by some bacteria, toxicity issues and a potential lack of efficacy against increasing numbers of polymyxin resistant strains necessitate alternative approaches.
LPS/LOS is essential in most Gram-negative bacteria, however some species, including A. baumannii, remain viable upon complete loss of LPS/LOS.59 In A. baumannii this phenotype imparts attenuated fitness and virulence, and reduced susceptibility to polymyxin antibiotics,60 but also results in increased susceptibility to other antibiotic classes including macrolides and glycopeptides.61 Inducing the LOS deficient phenotype therefore represents a novel approach to sensitizing A. baumannii to these antibiotics.
Several aryl 2-aminoimidazole adjuvants enhance the sensitivity of A. baumannii to macrolide and glycopeptide antibiotics. Compounds 31 and 32 (Figure 4), reduce the clarithromycin MIC against A. baumannii 5075 (AB5075) from 32 μg/mL to 0.25 μg/mL at 30 μM. Adjuvant activity (defined here as lowering the clarithromycin MIC to 2 μg/mL or lower) is retained down to 10 μM, and compound 31 is active in a G. mellonella model of infection.62 Both compounds also sensitize A. baumannii to vancomycin, reducing the MIC of AB5075 from 256 μg/mL to 1 μg/mL at 30 μM. Activity is conserved with both classes of antibiotics, against a panel of primary clinical A. baumannii isolates that encompasses nearly all clinically relevant A. baumannii clades.63, 64 Compound 31 is not hemolytic and has low cytotoxicity against HaCaT keratinocyte cells.62
Figure 4.
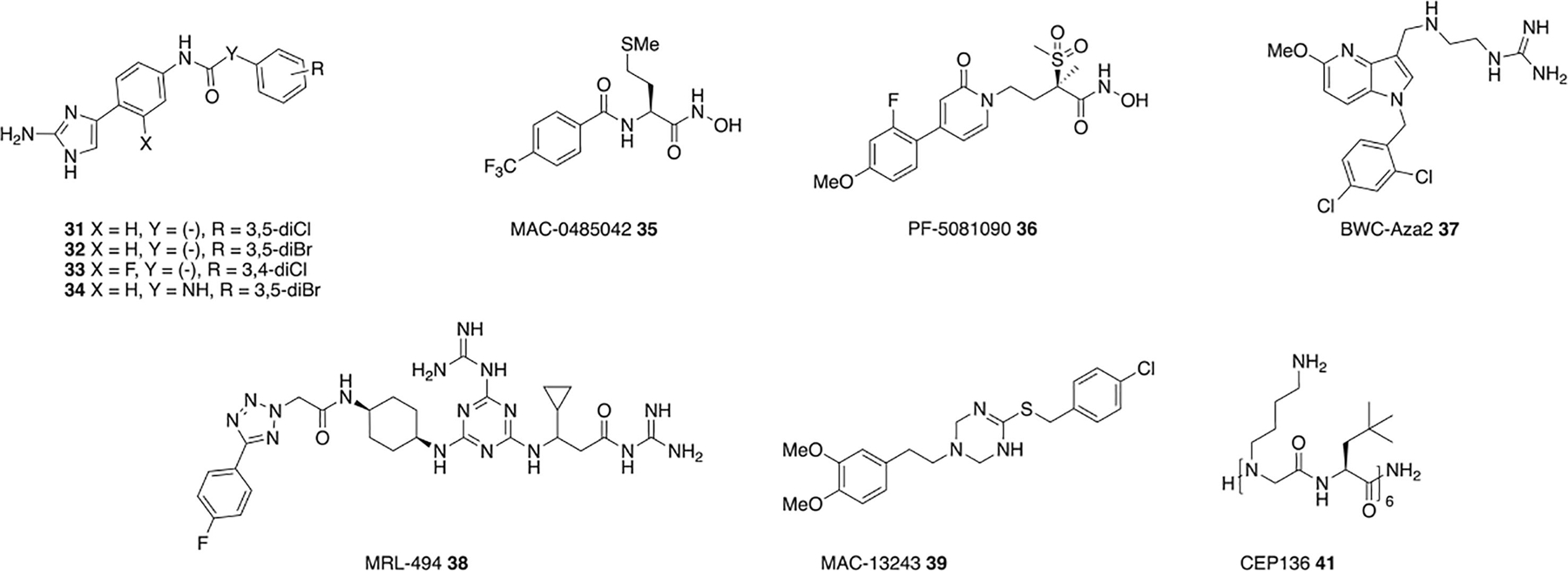
Adjuvants that interfere with OM component synthesis or assembly.
A structure activity relationship study on the aryl 2-aminoamidazole scaffold identified several compounds with increased macrolide potentiation activity, with 33 and 34 (Figure 4) reducing the clarithromycin MIC against AB5075 from 32 μg/mL to 1 μg/mL and 0.25 μg/mL respectively at 7.5 μM, and activity is retained across the same A. baumannii panel mentioned above.65 Although this series does cause membrane perturbation as determined by a BacLight assay, this appears to be decoupled from resistance suppression activity when compared to pentamidine.62 Instead compound 31 effects a shift toward palimitoylated lipid A species, an overall reduction of lipid A hydroxylation, and the absence of one lipid A species. This, along with the fact that the compound does not further sensitize a LOS-deficient strain to macrolides or vancomycin, indicates that this class of adjuvants is likely acting through modulation of LPS synthesis or assembly.62
Inhibitors of LpxC, which catalyzes the first committed step in lipid A biosynthesis, have potent antibacterial activity against several Gram-negative species, however inhibitors that weakly bind LpxC have potential as adjuvants. One weak LpxC inhibitor MAC-0485042 35 (Figure 4) potentiates rifampin and vancomycin without physical disruption of the OM. MAC-0485042 exhibits increased potency in an E. coli strain containing a H19Y mutation in LpxC (lpxC101), known to sensitize cells to LpxC inhibitors, which along with in vitro binding indicates that LpxC is a target of this compound.56 LpxC inhibitors exhibit poor antimicrobial activity against A. baumannii, likely because of non-essential nature of LOS in the species, but can increase susceptibility to other antibiotic classes as in the case of PF-5081090 36 with rifampin, azithromycin, and vancomycin. PF-5081090 36 significantly reduces lipid A, with a concomitant increase in OM permeability.66
The azaindole BWC-Aza2 37 potentiates rifampin, erythromycin, solithromycin, and novobiocin in P. aeruginosa, A. baumannii, Enterobacter cloacae, and Citrobacter freundii on par with PMBN. Activity is significantly impaired in a ΔLPS E. coli strain, and no change in activity is observed in the presence of exogenous LPS, indicating inhibition of LPS biosynthesis or assembly rather direct LPS binding, however the exact target is unknown as the ΔLPS strain harbors deletion of seven LPS synthesis genes.67 MRL-494 38 (Figure 4) inhibits assembly of proteins into the OM by binding directly or proximally to the essential exposed protein BamA of the β-barrel assembly machine, inhibiting the assembly of proteins into the OM to and sensitizing E. coli to rifampin.68 MAC13243 39 (Figure 4) is an inhibitor of LolA, a periplasmic chaperone that transports lipoproteins from the CM to the OM and partial depletion of which is enough to permeabilize the OM. MAC13243 increases membrane permeability and exhibits synergy against E. coli with erythromycin and novobiocin but not vancomycin or rifampin.69
The proline-hinged peptide KL-L9P (Ac-KLLKLLKKPLKLLK-NH2) 40 sensitizes MDR E. coli to linezolid, rifampin, and clarithromycin, MDR and non-MDR strains of K. pneumoniae and P. aeruginosa to erythromycin and rifampin and is active in a murine skin A. baumannii infection model. KL-L9P does not compromise membrane integrity but induces a morphological change that results in a large increase in fine surface roughness and fluidity. The authors hypothesize that KL-L9P binds to lipid A in a hinged helical formation, and creates a hard gel-like LPS domain, affecting arrangement of the OM.70
The peptidomimetic CEP-136 41 (Figure 4) sensitizes E. coli, K. pneumoniae, A. baumannii, and P. aeruginosa to rifampin, clarithromycin, and azithromycin in both lab strains and clinical isolates. While adjuvant activity is reduced in LPS-deficient E. coli, the MIC of CEP-136 is not affected indicating that the compound does not bind LPS. Synergy is particularly strong in a ΔrfaG mutant that has no outer core sugar, suggesting that CEP-136 facilitates antibiotic uptake via destabilization of the LPS inner core.71
Inhibition of efflux.
In addition to limited permeation of many antibiotics into Gram-negative cells, efflux pump systems contribute to intrinsic resistance in Gram-negative bacteria by keeping intracellular antibiotic concentrations low. Efflux pump inhibitors have long been investigated,72 and although none has yet made it to the clinic, this represents another adjuvant approach that has the potential to overcome intrinsic resistance in Gram-negative bacteria.
Anthracycline antibiotic 301A1 42 (Figure 5) is highly synergistic with rifampin and moderately synergistic with linezolid in E. coli and A. baumannii. Antibiotic 301A1 competes with Nile Red for efflux, and synergy with linezolid is lost in an E. coli strain overexpressing AdeIJK, pointing toward 301A1 competing with linezolid for efflux. Activity with rifampin is lost in E. coli rpoB mutants, indicating that 301A1 is directly involved in rifampin reaching RNA polymerase, and as there was no indication of membrane perturbation, the authors hypothesize 301A1 also competes with rifampin for efflux.73 A screen for inhibitors of AcrA of the AcrAB-TolC efflux pump system with sub-inhibitory concentrations of novobiocin in an OM permeable strain of E. coli identified NSC60339 43 (Figure 5), which potentiates novobiocin and erythromycin and dramatically inhibits the rate of efflux of the AcrAB-TolC substrate H33342. Proteolysis suggests NSC60339 binds AcrA and causes structural changes to the protein. Analogues 44 and 45 exhibit increased activity over NSC60339.74 The marine metabolite 3,4-dibromopyrrole-2,5-dione 46 (Figure 5) is synergistic with erythromycin in E. coli, inhibits RND pumps as shown by accumulation of H33342, and is inactive in a strain lacking the target RND pump (E. coli AG100A).75
Figure 5.
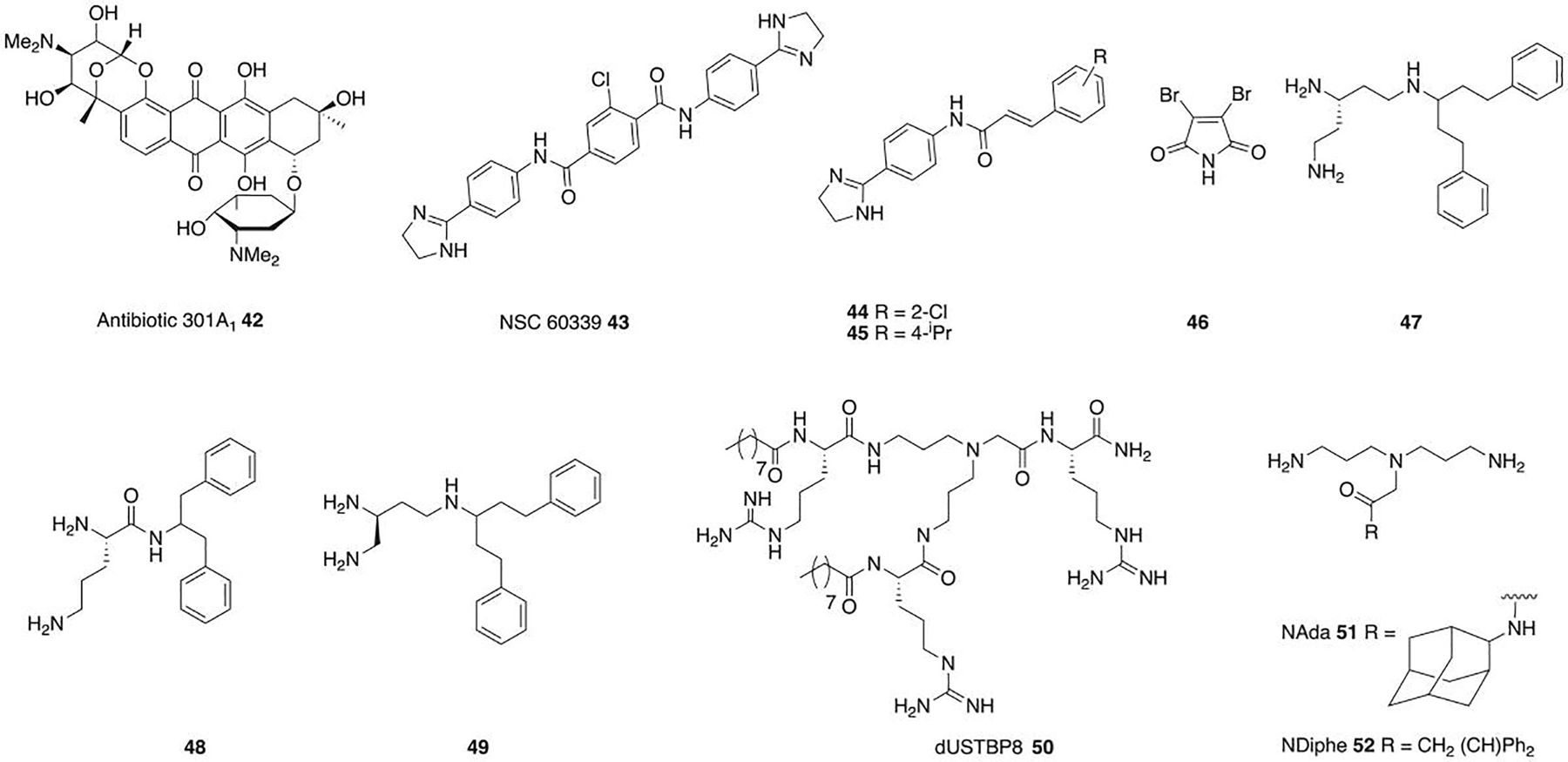
Adjuvants that inhibit efflux or act through multiple pathways.
Finally, some adjuvants likely act through more than one mode of action, typically affecting both the OM and efflux, either in concert to potentiate one antibiotic, or with different actions depending on the antibiotic. The triamine derivative of the known efflux inhibitor PAβN, compound 47 (Figure 5), potentiates clarithromycin against E. coli, reducing the MIC 32-fold to 2 μg/ml. The compound increases OM permeability and decreases efflux pump activity.76 Other PAβN analogues: 48 and 49, potentiate clarithromycin, azithromycin, and novobiocin in E. coli, with 48 having 128-fold greater potency than PAβN with clarithromycin. Both compounds affect efflux and also disrupt both the OM and CM.77
The dilipid ultrashort tetrabasic peptidomimetic dUSTBP8 50 (Figure 5) potentiates novobiocin and rifampin against MDR clinical isolates of P. aeruginosa, A. baumannii, K. pneumoniae, E. cloacae, and E. coli, with activity comparable to PMBN. Preliminary studies suggest OM permeabilization and/or disruption of efflux, potentially by sequestering lipids around transmembrane proteins of efflux pumps, or by altering the proton motive force required to power active efflux.78 Norspermidine derivatives with hydrophobic substituents show nonspecific, but weak membrane permeabilization and depolarization, and efflux inhibition in A. baumannii and P. aeruginosa. NAda 51 and NDiphe 52 (Figure 5) potentiate rifampin, erythromycin, and vancomycin in MDR K. pneumoniae, P. aeruginosa, and E. coli. Both compounds increase membrane permeability and effect small reductions in efflux, and the authors hypothesize that membrane depolarization may lead to an ion imbalance that indirectly slows efflux.79
In conclusion.
Infections caused by Gram-negative bacteria are becoming increasingly difficult, and in some cases impossible, to treat as a result of rising levels of resistance and a lack of novel Gram-negative acting antibiotics. Small molecule adjuvants that overcome intrinsic resistance in Gram-negative bacteria and sensitize the bacteria to approved antibiotics that are otherwise limited to treating Gram-positive infections have the potential to significantly expand therapeutic options for treating Gram-negative infections. Adjuvants that enhance antibiotic uptake through directly binding components of the OM are one approach, though many are beset with toxicity issues. Inhibitors of enzymes, chaperones, and assembly proteins involved in the synthesis, transport and assembly of the OM also show promise in overcoming intrinsic resistance, as do inhibitors of efflux.
Acknowledgments
The authors thank the US National Institutes of Health (R01AI136904 and R01AI167284) for funding. Graphical abstract and Figure 1 were created using BioRender.com
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brown ED, Wright GD. Antibacterial drug discovery in the resistance era. Nat 2016;529(7586): 336. [DOI] [PubMed] [Google Scholar]
- 2.Laxminarayan R, Duse A, Wattal C, et al. Antibiotic resistance-the need for global solutions. Lancet Infect Dis 2013;13(12): 1057. [DOI] [PubMed] [Google Scholar]
- 3.Organization WH. Antimicrobial Resistance - Global Report on Surveillance 2014.
- 4.O’Neill J Review on AMR. Antimicrobial resistance: tackling a crisis for the health and wealth of nations 2014.
- 5.Goff DA, Kullar R, Goldstein EJ, et al. A global call from five countries to collaborate in antibiotic stewardship: united we succeed, divided we might fail. Lancet Infect. Dis 2017;17(2): e56–e63. [DOI] [PubMed] [Google Scholar]
- 6.Tommasi R, Brown DG, Walkup GK, Manchester JI, Miller AA. ESKAPEing the labyrinth of antibacterial discovery. Nat Rev Drug Discov 2015;14(8): 529. [DOI] [PubMed] [Google Scholar]
- 7.Martens E, Demain AL. The antibiotic resistance crisis, with a focus on the United States. J Antibiot. (Tokyo) 2017;70(5): 520. [DOI] [PubMed] [Google Scholar]
- 8.Boucher HW, Talbot GH, Bradley JS, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 2009;48(1): 1. [DOI] [PubMed] [Google Scholar]
- 9.Lewis K Platforms for antibiotic discovery. Nat Rev Drug Discov 2013;12(5): 371. [DOI] [PubMed] [Google Scholar]
- 10.Khuntayaporn P, Thirapanmethee K, Chomnawang MT. An Update of Mobile Colistin Resistance in Non-Fermentative Gram-Negative Bacilli. Front Cell Infect Microbiol 2022;12: 882236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Organization WH. WHO priority pathogens list for R&D of new antibiotics 2017.
- 12.Malanovic N, Lohner K. Gram-positive bacterial cell envelopes: The impact on the activity of antimicrobial peptides. Biochim Biophys Acta 2016;1858(5): 936. [DOI] [PubMed] [Google Scholar]
- 13.Silhavy TJ, Kahne D, Walker S. The bacterial cell envelope. Cold Spring Harb Perspect Biol 2010;2(5): a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem 2002;71: 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vergalli J, Bodrenko IV, Masi M, et al. Porins and small-molecule translocation across the outer membrane of Gram-negative bacteria. Nat Rev Microbiol 2020;18(3): 164. [DOI] [PubMed] [Google Scholar]
- 16.Musk DJ, Banko DA, Hergenrother PJ. Iron salts perturb biofilm formation and disrupt existing biofilms of Pseudomonas aeruginosa. Chem Biol 2005;12(7): 789. [DOI] [PubMed] [Google Scholar]
- 17.Nikaido H Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 2003;67(4): 593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farmer S, Li ZS, Hancock RE. Influence of outer membrane mutations on susceptibility of Escherichia coli to the dibasic macrolide azithromycin. J Antimicrob Chemother 1992;29(1): 27. [DOI] [PubMed] [Google Scholar]
- 19.Richter MF, Hergenrother PJ. The challenge of converting Gram-positive-only compounds into broad-spectrum antibiotics. Ann N Y Acad Sci 2019;1435(1): 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamma PD, Cosgrove SE, Maragakis LL. Combination therapy for treatment of infections with gram-negative bacteria. Clin Microbiol Rev 2012;25(3): 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paterson DL, Bonomo RA. Multidrug-Resistant Gram-Negative Pathogens: The Urgent Need for ‘Old’ Polymyxins. Adv Exp Med Biol 2019;1145: 9. [DOI] [PubMed] [Google Scholar]
- 22.Structural Palzkill T. and Mechanistic Basis for Extended-Spectrum Drug-Resistance Mutations in Altering the Specificity of TEM, CTX-M, and KPC β-lactamases. Front Mol Biosci 2018;5: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Queenan AM, Shang W, Flamm R, Bush K. Hydrolysis and inhibition profiles of beta-lactamases from molecular classes A to D with doripenem, imipenem, and meropenem. Antimicrob Agents Chemother 2010;54(1): 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bush K, Bradford PA. Interplay between β-lactamases and new β-lactamase inhibitors. Nat Rev Microbiol 2019;17(5): 295. [DOI] [PubMed] [Google Scholar]
- 25.Lomovskaya O, Sun D, Rubio-Aparicio D, et al. Vaborbactam: Spectrum of Beta-Lactamase Inhibition and Impact of Resistance Mechanisms on Activity in Enterobacteriaceae. Antimicrob Agents Chemother 2017;61(11): e01443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cox G, Ejim L, Stogios PJ, et al. Plazomicin Retains Antibiotic Activity against Most Aminoglycoside Modifying Enzymes. ACS Infect Dis 2018;4(6): 980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhanel GG, Homenuik K, Nichol K, et al. The glycylcyclines: a comparative review with the tetracyclines. Drugs 2004;64(1): 63. [DOI] [PubMed] [Google Scholar]
- 28.ACRED P, BROWN DM, TURNER DH, WILSON MJ. Pharmacology and chemotherapy of ampicillin--a new broad-spectrum penicillin. Br J Pharmacol Chemother 1962;18: 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jelić D, Antolović R. From Erythromycin to Azithromycin and New Potential Ribosome-Binding Antimicrobials. Antibiotics (Basel) 2016;5(3): 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gomes C, Martínez-Puchol S, Palma N, et al. Macrolide resistance mechanisms in Enterobacteriaceae: Focus on azithromycin. Crit Rev Microbiol 2017;43(1): 1. [DOI] [PubMed] [Google Scholar]
- 31.Richter MF, Drown BS, Riley AP, et al. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017;545(7654): 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bergen PJ, Smith NM, Bedard TB, Bulman ZP, Cha R, Tsuji BT. Rational Combinations of Polymyxins with Other Antibiotics. Adv Exp Med Biol 2019;1145: 251. [DOI] [PubMed] [Google Scholar]
- 33.Gordon NC, Png K, Wareham DW. Potent synergy and sustained bactericidal activity of a vancomycin-colistin combination versus multidrug-resistant strains of Acinetobacter baumannii. Antimicrob Agents Chemother 2010;54(12): 5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melander RJ, Melander C. The Challenge of Overcoming Antibiotic Resistance: An Adjuvant Approach? ACS Infect Dis 2017;3(8): 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Past Bush K. and Present Perspectives on β-Lactamases. Antimicrob Agents Chemother 2018;62(10). e01076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zabawa TP, Pucci MJ, Parr TR Jr, Lister T. Treatment of Gram-negative bacterial infections by potentiation of antibiotics. Curr Opin Microbiol 2016;33: 7. [DOI] [PubMed] [Google Scholar]
- 37.Vaara M, Vaara T. Sensitization of Gram-negative bacteria to antibiotics and complement by a nontoxic oligopeptide. Nat 1983;303(5917): 526. [DOI] [PubMed] [Google Scholar]
- 38.Viljanen P, Matsunaga H, Kimura Y, Vaara M. The outer membrane permeability-increasing action of deacylpolymyxins. The Journal of Antibiotics 1991;44(5): 517. [DOI] [PubMed] [Google Scholar]
- 39.Kimura Y, Matsunaga H, Vaara M. Polymyxin B octapeptide and polymyxin B heptapeptide are potent outer membrane permeability-increasing agents. J. Antibiot 1992;45(5): 742. [DOI] [PubMed] [Google Scholar]
- 40.Huang T, Zeng M, Fu H, et al. A novel antibiotic combination of linezolid and polymyxin B octapeptide PBOP against clinical Pseudomonas aeruginosa strains. Ann Clin Microbiol Antimicrob 2022;21(1): 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramirez DM, Ramirez D, Arthur G, Zhanel G, Schweizer F. Guanidinylated Polymyxins as Outer Membrane Permeabilizers Capable of Potentiating Rifampicin, Erythromycin, Ceftazidime and Aztreonam against Gram-Negative Bacteria. Antibiotics 2022;11(10): 1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vaara M, Fox J, Loidl GN, et al. Novel Polymyxin Derivatives Carrying Only Three Positive Charges Are Effective Antibacterial Agents. Antimicrob Agents Chemother 2008;52(9): 3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vaara M, Siikanen O, Apajalahti J, et al. A Novel Polymyxin Derivative That Lacks the Fatty Acid Tail and Carries Only Three Positive Charges Has Strong Synergism with Agents Excluded by the Intact Outer Membrane. Antimicrob Agents Chemother 2010;54(8): 3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Corbett D, Wise A, Langley T, et al. Potentiation of Antibiotic Activity by a Novel Cationic Peptide: Potency and Spectrum of Activity of SPR741. Antimicrob Agents Chemother 2017;61(8): AAC.00200-00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zurawski DV, Reinhart AA, Alamneh YA, et al. SPR741, an Antibiotic Adjuvant, Potentiates the in Vitro and In Vivo Activity of Rifampin against Clinically Relevant Extensively Drug-Resistant Acinetobacter baumannii. Antimicrob Agents Chemother 2017;61(12): e01239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.French S, Farha M, Ellis MJ, et al. Potentiation of Antibiotics against Gram-Negative Bacteria by Polymyxin B Analogue SPR741 from Unique Perturbation of the Outer Membrane. ACS Infect Dis 2020;6(6): 1405. [DOI] [PubMed] [Google Scholar]
- 47.Neha KPaIBSaRTCaOSR. Leaks in the Pipeline: a Failure Analysis of Gram-Negative Antibiotic Development from 2010 to 2020. Antimicrob Agents Chemother 2022;66(5): e00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Q, Cebrián R, Montalbán-López M, Ren H, Wu W, Kuipers OP. Outer-membrane-acting peptides and lipid II-targeting antibiotics cooperatively kill Gram-negative pathogens. Commun Biol 2021;4(1): 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cebrián R, Xu C, Xia Y, Wu W, Kuipers OP. The cathelicidin-derived close-to-nature peptide D-11 sensitises Klebsiella pneumoniae to a range of antibiotics in vitro, ex vivo and in vivo. Int J Antimicrob Ag 2021;58(5): 106434. [DOI] [PubMed] [Google Scholar]
- 50.Moon SH, Zhang X, Zheng G, Meeker DG, Smeltzer MS, Huang E. Novel Linear Lipopeptide Paenipeptins with Potential for Eradicating Biofilms and Sensitizing Gram-Negative Bacteria to Rifampicin and Clarithromycin. J Med Chem 2017;60(23): 9630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Song M, Liu Y, Huang X, et al. A broad-spectrum antibiotic adjuvant reverses multidrug-resistant Gram-negative pathogens. Nat Microbiol 2020;5(8): 1040. [DOI] [PubMed] [Google Scholar]
- 52.Stokes JM, MacNair CR, Ilyas B, et al. Pentamidine sensitizes Gram-negative pathogens to antibiotics and overcomes acquired colistin resistance. Nat Microbiol 2017;2: 17028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Macnair CR, Farha MA, Serrano-Wu MH, et al. Preclinical Development of Pentamidine Analogs Identifies a Potent and Nontoxic Antibiotic Adjuvant. ACS Infect Dis 2022;8(4): 768. [DOI] [PubMed] [Google Scholar]
- 54.Wesseling CMJ, Slingerland CJ, Veraar S, Lok S, Martin NI. Structure–Activity Studies with Bis-Amidines That Potentiate Gram-Positive Specific Antibiotics against Gram-Negative Pathogens. ACS Infect Dis 2021;7(12): 3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu B, Roy Choudhury M, Yang X, et al. Restoring and Enhancing the Potency of Existing Antibiotics against Drug-Resistant Gram-Negative Bacteria through the Development of Potent Small-Molecule Adjuvants. ACS Infect Dis 2022;8(8): 1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klobucar K, Jardine E, Farha MA, et al. Genetic and Chemical Screening Reveals Targets and Compounds to Potentiate Gram-Positive Antibiotics against Gram-Negative Bacteria. ACS Infect Dis 2022; 8(10):2 187. [DOI] [PubMed] [Google Scholar]
- 57.Klobucar K, Cote JP, French S, et al. Chemical Screen for Vancomycin Antagonism Uncovers Probes of the Gram-Negative Outer Membrane. ACS Chem Biol 2021;16(5): 929. [DOI] [PubMed] [Google Scholar]
- 58.Hubble VB, Hubbard BA, Minrovic BM, Melander RJ, Melander C. Using Small-Molecule Adjuvants to Repurpose Azithromycin for Use against Pseudomonas aeruginosa. ACS Infect Dis 2019;5(1): 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Simpson BW, Nieckarz M, Pinedo V, Mclean AB, Cava F, Trent MS. Acinetobacter baumannii Can Survive with an Outer Membrane Lacking Lipooligosaccharide Due to Structural Support from Elongasome Peptidoglycan Synthesis. mBio 2021;12(6): e0309921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arroyo LA, Herrera CM, Fernandez L, Hankins JV, Trent MS, Hancock RE. The pmrCAB operon mediates polymyxin resistance in Acinetobacter baumannii ATCC 17978 and clinical isolates through phosphoethanolamine modification of lipid A. Antimicrob Agents Chemother 2011;55(8): 3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moffatt JH, Harper M, Harrison P, et al. Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production. Antimicrob Agents Chemother 2010;54(12): 4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martin SE, Melander RJ, Brackett CM, et al. Small Molecule Potentiation of Gram-Positive Selective Antibiotics against Acinetobacter baumannii. ACS Infect Dis 2019;5(7): 1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taitt CR, Leski TA, Stockelman MG, et al. Antimicrobial resistance determinants in Acinetobacter baumannii isolates taken from military treatment facilities. Antimicrob Agents Chemother 2014;58(2): 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zurawski DV, Thompson MG, McQueary CN, et al. Genome sequences of four divergent multidrug-resistant Acinetobacter baumannii strains isolated from patients with sepsis or osteomyelitis. J Bacteriol 2012;194(6): 1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hubble VB, Bartholomew KR, Weig AW, et al. Augmenting the Activity of Macrolide Adjuvants against Acinetobacter baumannii. ACS Med Chem Lett 2020;11(9): 1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.García-Quintanilla M, Caro-Vega JM, Pulido MR, Moreno-Martínez P, Pachón J, McConnell MJ. Inhibition of LpxC Increases Antibiotic Susceptibility in Acinetobacter baumannii. Antimicrob Agents Chemother 2016;60(8): 5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sharma S, Rao R, Reeve SM, et al. Azaindole Based Potentiator of Antibiotics against Gram-Negative Bacteria. ACS Infect Dis 2021;7(11): 3009-. [DOI] [PubMed] [Google Scholar]
- 68.Hart EM, Mitchell AM, Konovalova A, et al. A small-molecule inhibitor of BamA impervious to efflux and the outer membrane permeability barrier. Proc Natl Acad Sci U S A 2019;116(43): 21748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Muheim C, Gotzke H, Eriksson AU, et al. Increasing the permeability of Escherichia coli using MAC13243. Sci Rep 2017;7(1): 17629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hyun S, Choi Y, Jo D, et al. Proline Hinged Amphipathic α-Helical Peptide Sensitizes Gram-Negative Bacteria to Various Gram-Positive Antibiotics. J Med Chem 2020;63(23): 14937. [DOI] [PubMed] [Google Scholar]
- 71.Mood EH, Goltermann L, Brolin C, et al. Antibiotic Potentiation in Multidrug-Resistant Gram-Negative Pathogenic Bacteria by a Synthetic Peptidomimetic. ACS Infect Dis 2021;7(8): 2152. [DOI] [PubMed] [Google Scholar]
- 72.Lomovskaya O, Bostian KA. Practical applications and feasibility of efflux pump inhibitors in the clinic--a vision for applied use. Biochem Pharmacol 2006;71(7): 910. [DOI] [PubMed] [Google Scholar]
- 73.Cox G, Koteva K, Wright GD. An unusual class of anthracyclines potentiate Gram-positive antibiotics in intrinsically resistant Gram-negative bacteria. J Antimicrob Chemother 2014;69(7): 1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Haynes KM, Abdali N, Jhawar V, et al. Identification and Structure-Activity Relationships of Novel Compounds that Potentiate the Activities of Antibiotics in Escherichia coli. J Med Chem 2017;60(14): 6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Whalen KE, Poulson-Ellestad KL, Deering RW, Rowley DC, Mincer TJ. Enhancement of Antibiotic Activity against Multidrug-Resistant Bacteria by the Efflux Pump Inhibitor 3,4-Dibromopyrrole-2,5-dione Isolated from a Pseudoalteromonas sp. J Nat Prod 2015;78(3): 402. [DOI] [PubMed] [Google Scholar]
- 76.Blankson G, Parhi AK, Kaul M, Pilch DS, LaVoie EJ. Structure-activity relationships of potentiators of the antibiotic activity of clarithromycin against Escherichia coli. Eur J Med Chem 2019;178: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Blankson GA, Parhi AK, Kaul M, Pilch DS, LaVoie EJ. Advances in the structural studies of antibiotic potentiators against Escherichia coli. Bioorg Med Chem 2019;27(15): 3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ramirez D, Berry L, Domalaon R, Brizuela M, Schweizer F. Dilipid Ultrashort Tetrabasic Peptidomimetics Potentiate Novobiocin and Rifampicin Against Multidrug-Resistant Gram-Negative Bacteria. ACS Infect Dis 2020;6(6): 1413. [DOI] [PubMed] [Google Scholar]
- 79.Dhanda G, Mukherjee R, Basak D, Haldar J. Small-Molecular Adjuvants with Weak Membrane Perturbation Potentiate Antibiotics against Gram-Negative Superbugs. ACS Infect Dis 2022;8(5): 1086. [DOI] [PubMed] [Google Scholar]