

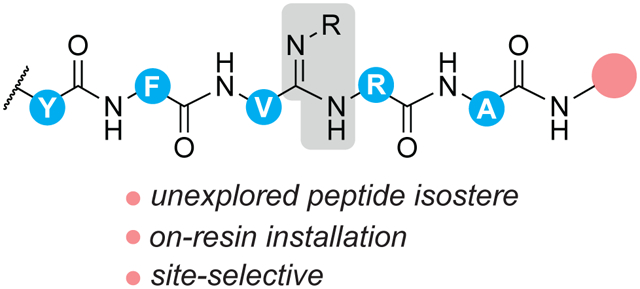
Abstract
Amidines are a structural surrogate for peptide bonds, yet have received considerably little attention in peptides due to limitations in existing methods to access them. The synthetic strategy developed in this study represents the first robust and general procedure for the introduction of amidines into the peptide backbone. We exploit and further develop the utility and efficiency of thioimidate protecting groups as a means to side-step reactivity that ultimately renders existing methods unsuitable for the installation of amidines along the main-chain of peptides. This work is significant because it describes a generally applicable path to access unexplored peptide designs and architectures for new therapeutics made possible by the unique properties of amidines.
Graphical Abstract
Numerous isosteres have been developed to mimic the shape and function of the native amide bond.1 One isostere in particular—the amidine—can occur naturally,2 but also contrasts with amides by displaying a dynamic hydrogen-bonding motif (Figure 1a). This unique ability of amidines to alter their hydrogen-bond donating and accepting character was exploited by Boger and coworkers to counter the resistance of bacteria towards vancomycin (1).3,4 Like wise, amidines are key features of other therapeutic enzyme/receptor inhibitors5 such as the anti-psychotic drug Clozapine,6 anti-infectives like Pafuramidine and Pentamidine,7 and the FDA approved anticoagulant Dabigatran.8 Further, beyond the ability of amidines to operate as hydrogen-bond shape shifters, the additional valence compared to the amide furnishes another site to append substituents (R, Figure 1a).
Figure 1.
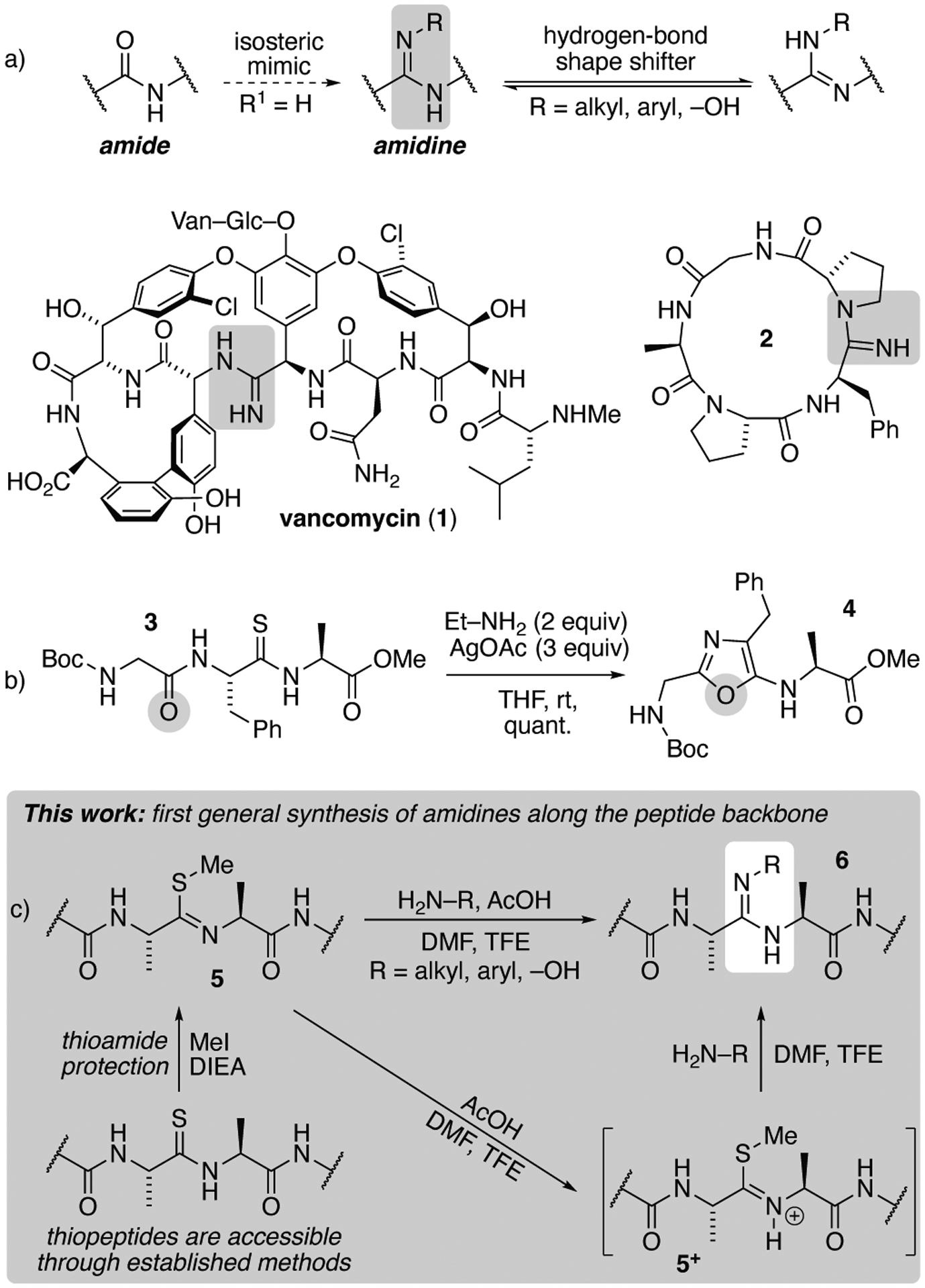
Amidines in peptidic molecules. (a) Malleable hydrogen-bonding motif of amidines and examples of conformationally constrained peptidic molecules that allows for application of the Ag(I)-promoted conversion of thioamide into amidine. (b) The Ag(I)-promoted method to convert thioamides into amidines is not applicable to linear peptides because of competing intramolecular attack by adjacent amides.24 (c) A new, general methods for amidine insertion into linear peptides.
While these distinct properties of amidines have served as design elements in numerous pharmaceutical compounds,7,9–11 polymeric materials,12 and even prebiotic building blocks,13,14 amidines have received relatively little attention in peptides due to a lack of compatibility and general methods for incorporation into peptides using standard Fmoc solid-phase peptide synthesis (SPPS) techniques.15,16
Recent syntheses of amidines in peptidic molecules have exploited thioamides17 as a site that can be activated with Ag(I) salts for conversion into amidines.3,18 Hutton and coworkers similarly demonstrated how other nucleophiles can be reacted with thioamides through activation with Ag(I).19–21 The installation of thioamides into peptides using standard SPPS procedures can be achieved with activated thioacyl amino acid precursors derived from commercially available Fmoc-amino acids.22,23 Thus, in principle, a thioamide-containing peptide should provide an avenue for the site-selective insertion of amidines into peptides. Unfortunately, examples of such chemistry within the context of polypeptides are noticeably scarce. The paucity of literature around the Ag(I)-promoted conversion of thioamides into amidines along the peptide backbone is likely due to a rapid intramolecular 5-exo-trig attack from an adjacent backbone amide onto the thioamide carbonyl (3→4, Figure 1b), crippling any chance for intermolecular attack by an amine nucleophile.
This type of ring closure has been observed previously for macrocycles in which both the N-terminal and/or C-terminal amides that bracket the thioamide along the backbone can cyclize,24 as well as for other nucleophiles that are in close proximity to the thioamide.21,25 These results likely explain why this seemingly facile method to access amidines has not been successful for linear peptides. Indeed, to our knowledge, only two examples of Ag(I)-mediated installation of amidines into peptide-like molecules have been reported (Figure 1, 1 and 2). The first was Boger’s amidinecontaining vancomycin synthesis mentioned above (1),3,4 and in the second, Yudin and coworkers demonstrated the conversion of a thioamide into an amidine within a small macrocyclic peptide (2).18 In both of these examples, the constrained nature of the macrocycles likely plays a role in preventing the 5-exo-trig cyclization, as formation of a planar oxazole within each small ring is conformationally disfavored.
We recently demonstrated how thioamides can be converted into thioimidates (5, Figure 1c) during SPPS to preserve the α-C stereochemistry of thioamides during subsequent elongation of the peptide on solid support.26,27 The basic conditions of SPPS generally maintain the thioimidate in its deprotonated state. The basicity of the thioimidate, however, means that it can be rendered more electrophilic upon protonation (5+).6 We therefore hypothesized that amidines could be introduced into peptides through the thioimidate rather than the thioamide (5→6, Figure 1c). In addition to being the first general approach to incorporate amidines into peptide backbones, we further note that this approach has several desirable features: (1) The synthesis would avoid the use of stoichiometric quantities3,4 of precious transition metals such as silver. (2) The amidine could be introduced on-resin prior to cleavage and deprotection of the peptide. The ability to introduce amidines while on resin would increase the generality of the method, as competing nucleophiles such as the reactive side-chain functional groups along the peptide would be necessarily capped. (3) Finally, we have already demonstrated how thiomidates are a preferred protection method for thioamides during SPPS,26,27 where subsequent exploitation of the thioimidate for installation of the amidine would provide enhanced utility and efficiency.
To determine the feasibility of installing an amidine via the thioimidate in linear peptides, we began with compound 7 as a simple test substrate that was free of any other competing functional groups (Table 1). The thioimidate was found highly amenable to conversion into amidine 8 with weak acid in DMF, conditions that are compatible with SPPS procedures.28 It should be noted that under basic conditions related to SPPS, the thioimidate is nonreactive towards amine nucleophiles and no amidine formation has been observed.26,27 Presumably, the acetic acid creates a mildly basic buffer with the amine, where protonation of the thioimidate (pka = 7.5–8)29 thereby increases its electrophilicity towards nucleophilic substitution with the amine. Notably, less nucleophilic amines such as aniline (entry 5) were less efficient in forming amidine relative to primary amines (entries 1–4). Likewise, more sterically hindered secondary amines (entries 6 and 7) provided only modest to good yields of amidine, even after extended reaction times. Finally, primary amidines were accessed from ammonium salts in only modest yields (entries 8 and 9). The addition of acetic acid was not found to have any effect, likely due to the presence of the internal acid of the ammonium cation. While ammonia is a weaker nucleophile than primary amines, this result was at first surprising because ammonia appeared to be even less efficient than aniline (entry 8 and 9 versus 5). We hypothesized, however, that the lower yields were due primarily to low solubility of the ammonium salts in DMF.
Table 1.
Conversion of Thioimidate into Amidine
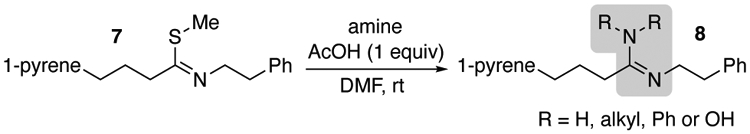
| ||||
|---|---|---|---|---|
| entry | amine | equivalents | time (h) | yield (%) |
| 1 | BnNH2 | 10 | 2 | 99 |
| 2 | BnNH2 | 5 | 2 | 98 |
| 3 | BnNH2 | 1 | 2 | 92 |
| 4 | H2N-OH | 10 | 0.5 | 95 |
| 5 | PhNH2 | 10 | 6 | 65 |
| 6 | Me2NH | 10 | 18 | 85 |
| 7 | piperidine | 10 | 24 | 54 |
| 8 | NH4Cla | 5 | 3 | 56 |
| 9 | NH4OAca | 2 | 3 | 34 |
Pyrene was used as internal standard.
No AcOH was added.
% Yield values are the average of two experiments.
To test this hypothesis, trifluoroethanol (TFE) was included in the solvent mixture (Table 2). The modified solvent conditions afforded increase solubility of ammonium salts, leading to homogenous solutions with significantly improved yields (entries 1 and 2). Moreover, the addition of TFE was found to improve the yields and reaction times of alkyl amines (entry 3), including amines with lower nucleophilicity due to both electronic (entry 4) or steric (entry 5) effects relative to neat DMF (Table 1). Other fluorinated alcohols like the more expensive hexafluoroisopropanol (HFIP) were found to have a similar results as TFE. The beneficial effects of TFE obviously extend beyond simple solubility, wherein the strong hydrogen-bond donor ability of fluoroalcohols may help to meditate the shuttling of protons during the reaction.30,31
Table 2.
Formation of Amidine with the Addition of Fluoroalcohol
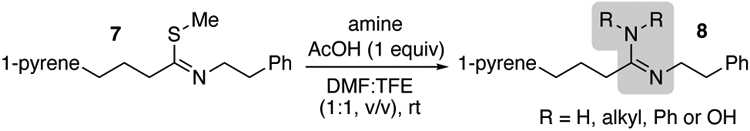
| ||||
|---|---|---|---|---|
| entry | amine | equivalents | time (h) | yield (%) |
| 1 | NH4Cla | 5 | 3 | 98 |
| 2 | NH4OAca | 2 | 3 | 99 |
| 3 | BnNH2 | 10 | 1 | 98 |
| 4 | PhNH2 | 10 | 3 | 99 |
| 5 | piperidine | 10 | 3 | 75 |
Pyrene was used as internal standard.
no AcOH was added.
% Yield values are the average of two experiments.
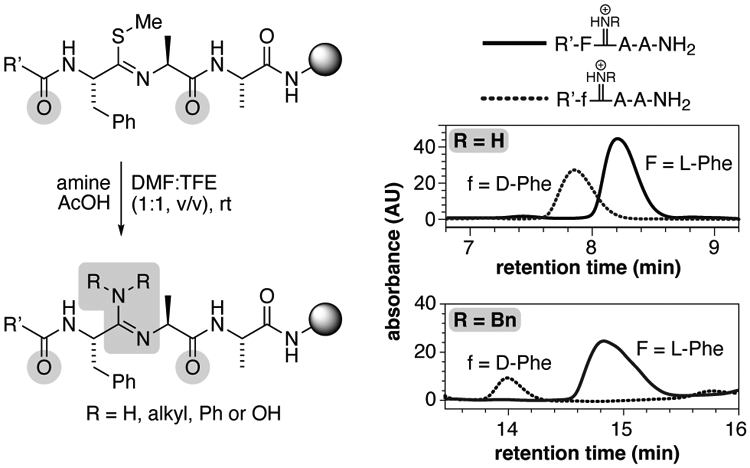
We next sought to explore the feasibility of this reaction along a peptide backbone and on solid support. We used the trimer PheSMe–Ala–Ala as a test sequence, where the thioimidate was placed at the linkage between the Phe and Ala residues (Table 3). Notably, this test trimer positioned an oxoamide to either side of the thioimidate linkage. Thus, the test trimer allowed us to explore the possibility that the oxoamide to either the N-terminal or C-terminal side of the thioimidate might react and cyclize onto the thioimidate to form oxazole or other products (akin to the side reaction observed with thiopeptides activated with Ag(I), Figure 1b).24,25
Table 3.
Amidine Formation from Thioimidate during Solid-Phase Peptide Synthesis
| ||||
|---|---|---|---|---|
| entry | amine | [amine] | [AcOH] | yield (%)b |
| 1 | NH4OAca | 5 | - | 93 |
| 2 | BnNH2 | 5 | 1 | 48 |
| 3 | BnNH2 | 1 | 1 | 17 |
| 4 | BnNH2 | 2 | 0.5 | 22 |
| 5 | PhNH2 | 5 | 1 | 44 |
| 6 | piperidine | 5 | 1 | trace |
R’ = 1-pyrenebutyric acid.
no AcOH was added.
% Yield of amidine determined relative to a R’-FAA-NH2 internal resin standard using LC-HRMS and monitoring at 330 nm (See SI for details).
Identity of the amidine confirmed via HRMS.
Using conditions that were derived from the results in Table 2, amidines were found to incorporate at the thioimidate site in serviceable yields after 24 h at room temperature (Table 3, entries 1–5). We found a 5:1 ratio of molarity of the alkyl amine to AcOH to be a generally applicable condition for amidine formation (entries 2 and 5). Higher concentrations of AcOH relative to benzyl amine (e.g. 5:5 or 5:2.5 M for [BnNH2]:[AcOH]) led to a salting out of benzylammonium acetate from solution. Lower concentrations of amine and AcOH produced lower yields (entries 3 and 4). Higher concentrations of acid may provide optimal yields for other amine nucleophiles, which certainly appears to be the case for the 1:1 molar equivalents of ammonia and acetic acid in NH4OAc (entry 1), but the general ratio of 5:1 M amine:AcOH was used to prepare the peptides described below.
To evaluate the possibility for epimerization of the α-C stereochemistry of the Phe residue during the reaction, we compared the results in entry 1 and entry 3 to analogous trimers prepared with authentic D-Phe (Table 3, inset). These results did not indicate epimerization to be a major concern as the two epimers were distinctly separated by chromatography. The results reported in Table 1 and 2 indicated that more sterically hindered secondary amines were more sluggish nucleophiles in the reaction. This behavior appears to be exacerbated on-resin as piperidine failed to give productive yields of corresponding amidine (entry 6). Importantly, in all cases, we did not observe products associated with oxazole-cyclized side product (Figure 1b). In fact, in all cases, no other unexpected species were observed besides the expected products and any unreacted starting material, indicating a clean conversion process. Finally, the ability to introduce amidines directly on-resin, while side-chain functional groups remain in protected form, increases the sequence applicability of the synthetic approach, with the important caveat that installation of an amidine at a site that precedes proline in the sequence (i.e. Xaa–Pro) would not be amenable to these methods because thioimidates cannot be formed at these positions.26
We next prepared a range of biologically relevant peptides to showcase the generality of using the thioimidate as a site for selective insertion of amidines (Scheme 1). Peptides were synthesized by insertion of the thioimidate following established procedures,26,27 followed by subsequent elongation to complete the sequence. Conversion to the amidine took place prior to final cleavage from the resin following a general procedure described in the SI.
Scheme 1.
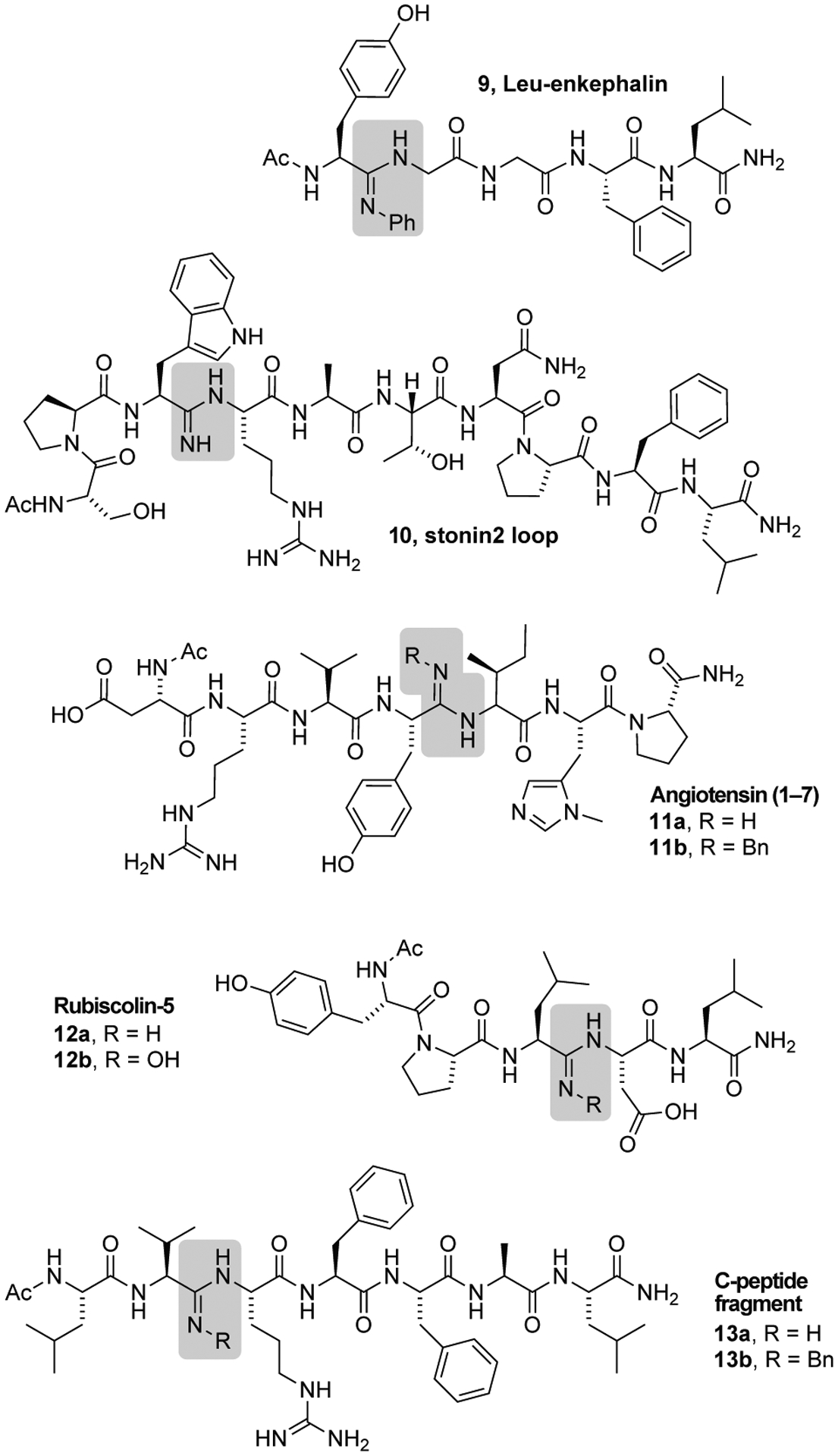
First General Synthesis of Amidine-Containing Peptides
The Leu-enkephalin pentapeptide (9) is a selective agonist for δ-opioid receptors, with potential to treat chronic pain and inflammation without eliciting respiratory depression and addiction. Despite these promising attributes, Leu-enkephalin has a poor pharmacokinetic profile due to rapid proteolysis of the Tyr-Gly linkage by aminopeptidase N in human plasma.32 Installation of an aniline amidine at this key metabolic site was readily achieved using our approach.
The Eps15 protein is involved in a multiprotein complex with stonin2 to recruit machinery necessary for endocytosis.33 It was discovered that a truncated loop of stonin2, peptide (10), makes several key contacts with Eps15.34 We were successful in insertion of an amidine between two amino acids with (Trp–Arg) possessing larger side chains, one of them charged. The angiotensin (1–7) peptide binds and activates G-protein coupled Mas receptors with applications in pain management.35 New analogues of angiotensin (1–7) with unexplored biodistribution and pK a profiles may be possible through modification of the peptide backbone (11a and 11b).
Rubiscolin-5 is a naturally occurring peptide agonist with high selectivity against δ-opioid receptors over other signaling receptors.36 New classes of rubiscolin-5 compounds were accessed through modification of the polyamide backbone (12a and 12b).
Finally, amidine installation was also possible at bulky, β-branched amino acids such as the valine residue of a truncated sequence of insulin-related C-peptide (13a and 13b).37
The examples outlined in Scheme 1 demonstrates how different amidine scaffolds were able to withstand the strong acidic conditions (TFA) associated with both release of the peptide from the resin and cleavage of the side-chain protecting groups. The sequences also demonstrate how amidine formation was possible at different positions and between different residues along the backbone. Peptides were isolated in yields of 3–18% based on the reported loading of amino groups on the resin (Table S3). Isolated yields of the peptides are often low due to the linear nature of SPPS, and further complicated by the 3-phase sequence of (1) the problematic coupling of the thioamide residue, (2) conversion of thioamide into thioimidate, and (3) installation of the amidine that are not standard steps during SPPS. This procedure was not optimized for each peptide example (i.e. taking into account the sequence position). Instead, a standard procedure was applied for all peptides.
The results of Table 3 indicate that (3) installation of the amidine was efficient, or at least serviceable, to provide appropriate quantities of peptide for further study. Significant depression in isolated yield likely derives from the (1) coupling of the thioamide residue to the growing peptide. The thioamide coupling step is certainly not quantitative, requiring Ac2O capping in all cases to truncate unreacted chains (see SI Figure S12 for further discussion). Additionally, inclusion of significant quantities of oxoamide can occur at these sites despite pure thioacyl reagent.38 Thus, we are currently working on strategies to streamline the 3 nonstandard SPPS steps discussed above to improve the overall efficiency of the process.
The synthetic strategy developed in this study represents the first robust and general procedure for the introduction of amidines into the peptide backbone. Because amidines have remained largely unexplored in peptide science, this work is significant because it describes a generally applicable path to access unexplored peptide designs and architectures, enabled by the unique physicochemical properties and structure of the amidine (Figure 1a). The approach centers around exploiting the site-selective insertion of thioamides as a reactive handle for conversion into amidine, but avoids the detrimental side-reactivity associated with previous methods that ultimately prevents successful activation of thioamides with stoichiometric amounts of precious transition metals. Namely, we convert the thioamide into a thioimidate that not only protects the integrity of the α-C stereochemistry of the thioamide residue during normal peptide synthesis, but also provides an electrophilic site for downstream conversion into an amidine. Notably, the amidine is introduced while the peptide is still protected and on-resin, thereby preventing any potential reactivity with side-chain functional groups. Thus, this work increases the utility and efficiency of exploiting thioimidates for the solid-phase peptide synthesis of thiopeptides by introducing a whole new avenue for peptide diversification, as amidinopeptides. We anticipate the exploration of this new chemistry within the context of novel peptide scaffolds and therapeutic compounds.
Supplementary Material
Acknowledgement
The authors acknowledge the National Institutes of Health, National Institutes of General Medical Sciences under award number R35 GM142883. Luis Camacho acknowledges an award through the Alliances for Graduate Education and the Professoriate (AGEP) from the National Science Foundation (1848261). We also acknowledge Dr. Bryan Lampkin for helpful conversations regarding this work.
Footnotes
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website.
PDF file describing synthetic procedures and molecular characterization/purity (NMR, LC-MS traces)
References
- (1).Choudhary A; Raines RT An Evaluation of Peptide-Bond Isosteres. ChemBioChem 2011, 12, 1801–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Clark KA; Seyedsayamdost MR Bioinformatic atlas of radical SAM enzyme-modified RiPP natural products reveals an isoleucine-tryptophan crosslink. J. Am. Chem. Soc 2022, 144, 17876–17888. [DOI] [PubMed] [Google Scholar]
- (3).Okano A; James RC; Pierce JG; Xie J; Boger DL Silver(I)-Promoted Conversion of Thioamides to Amidines: Divergent Synthesis of a Key Series of Vancomycin Aglycon Residue 4 Amidines That Clarify Binding Behavior to Model Ligands. J. Am. Chem. Soc 2012, 134, 8790–8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Xie J; Okano A; Pierce JG; James RC; Stamm S; Crane CM; Boger DL Total synthesis of [Ψ[C(S)NH]Tpg4]vancomycin aglycon, [Ψ[C(NH)NH]Tpg4]vancomycin aglycon, and related key compounds: reengineering vancomycin for dual D-Ala-D-Ala and D-Ala-D-Lac binding. J. Am. Chem. Soc 2012, 134, 1284–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Luo Y; Knuckley B; Lee Y-H; Stallcup MR; Thompson PR A fluoroacetamidine-based inactivator of protein arginine deiminase 4: design, synthesis, and in vitro and in vivo evaluation. J. Am. Chem. Soc 2006, 128, 1092–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hunziker F; Fischer E; Schmutz J 11-Amino-5H-dibenzo[b,e]-1, 4-diazepine. 10. Mitteilung über siebengliedrige Heterocyclen. Helv. Chim. Acta 1967, 50, 1588–1599. [Google Scholar]
- (7).Soeiro MNC; Werbovetz K; Boykin DW; Wilson WD; Wang MZ; Hemphill A Novel amidines and analogues as promising agents against intracellular parasites: a systematic review. Parasitology 2013, 140, 929–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lowenstern A; Al-Khatib SM; Sharan L; Chatterjee R; Allen LaPointe NM; Shah B; Borre ED; Raitz G; Goode A; Yapa R; Davis JK; Lallinger K; Schmidt R; Kosinski AS; Sanders GD Interventions for preventing thromboembolic events in patients with atrial fibrillation: A systematic review. Ann. Intern. Med 2018, 169, 774–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wang M-J et al. Design, synthesis, mechanisms of action, and toxicity of novel 20(s)-sulfonylamidine derivatives of camptothecin as potent antitumor agents. J. Med. Chem 2014, 57, 6008–6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Greenhill JV; Lue P Amidines and guanidines in medicinal chemistry. Prog. Med. Chem 1993, 30, 203–326. [DOI] [PubMed] [Google Scholar]
- (11).Diana GD Synthesis of noformycin. Journal of Medicinal Chemistry 1973, 16, 857–859. [DOI] [PubMed] [Google Scholar]
- (12).Quek JY; Davis TP; Lowe AB Amidine functionality as a stimulus-responsive building block. Chem. Soc. Rev 2013, 42, 7326–7334. [DOI] [PubMed] [Google Scholar]
- (13).Foden CS; Islam S; Fernández-García C; Maugeri L; Sheppard TD; Powner MW Prebiotic synthesis of cysteine peptides that catalyze peptide ligation in neutral water. Science 2020, 370, 865–869. [DOI] [PubMed] [Google Scholar]
- (14).Singh J; Whitaker D; Thoma B; Islam S; Foden CS; Aliev AE; Sheppard TD; Powner MW Prebiotic catalytic peptide ligation yields proteinogenic peptides by intramolecular amide catalyzed hydrolysis facilitating regioselective lysine ligation in neutral water. J. Am. Chem. Soc 2022, 144, 10151–10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Vastl J; Kartika R; Park K; Cho AE; Spiegel DA Peptidines: glycine-amidine-based oligomers for solution- and solid-phase synthesis. Chem. Sci 2016, 7, 3317–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Inokuchi E; Yamada A; Hozumi K; Tomita K; Oishi S; Ohno H; Nomizu M; Fujii N Design and synthesis of amidine-type peptide bond isosteres: application of nitrile oxide derivatives as active ester equivalents in peptide and peptidomimetics synthesis. Org. Biomol. Chem 2011, 9, 3421–3427. [DOI] [PubMed] [Google Scholar]
- (17).Lampkin BJ; VanVeller B Hydrogen Bond and Geometry Effects of Thioamide Backbone Modifications. J. Org. Chem 2021, 86, 18287–18291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Huh S; Appavoo SD; Yudin AK Amidine Functionality As a Conformational Probe of Cyclic Peptides. Org. Lett 2020, 22, 9210–9214. [DOI] [PubMed] [Google Scholar]
- (19).Thombare VJ; Hutton CA Rapid, Traceless, AgIPromoted Macrocyclization of Peptides Possessing an N-Terminal Thioamide. Angew. Chem., Int. Ed 2019, 58, 4998–5002. [DOI] [PubMed] [Google Scholar]
- (20).Shabani S; Hutton CA Depsipeptide synthesis using a latestage Ag(i)-promoted macrolactonisation of peptide thioamides. Chem. Commun 2021, 57, 2081–2084. [DOI] [PubMed] [Google Scholar]
- (21).Taresh AB; Hutton CA Backbone thioamide directed macrocyclisation: lactam stapling of peptides. Org. Biomol. Chem 2022, 20, 1488–1492. [DOI] [PubMed] [Google Scholar]
- (22).Khatri B; Bhat P; Chatterjee J Convenient synthesis of thioamidated peptides and proteins. J. Pep. Sci 2020, 26, e3248. [DOI] [PubMed] [Google Scholar]
- (23).Shalaby MA; Grote CW; Rapoport H Thiopeptide Synthesis. α-Amino Thionoacid Derivatives of Nitrobenzotriazole as Thioacylating Agents. J. Org. Chem 1996, 61, 9045–9048. [DOI] [PubMed] [Google Scholar]
- (24).Hitotsuyanagi Y; Sasaki S-I; Matsumoto Y; Yamaguchi K; Itokawa H; Takeya K Synthesis of [L-Ala-1]RA-VII, [D-Ala-2]RA-VII, and [D-Ala-4]RA-VII by epimerization of RA-VII, an antitumor bicyclic hexapeptide from Rubia plants, through oxazoles. J. Am. Chem. Soc 2003, 125, 7284–7290. [DOI] [PubMed] [Google Scholar]
- (25).Ibara M; Abe T; Sawada D Chemo- and Site-Selective Replacement of N-Terminal Carbamates in Peptides. Org. Lett 2022, 24, 2131–2136. [DOI] [PubMed] [Google Scholar]
- (26).Camacho LA; Lampkin BJ; VanVeller B A Bottom-Up Approach To Preserve Thioamide Residue Stereochemistry during Fmoc Solid-Phase Peptide Synthesis. Org. Lett 2019, 21, 7015–7018. [DOI] [PubMed] [Google Scholar]
- (27).Camacho LA; Nguyen YH; Turner J; VanVeller B Deprotection Strategies for Thioimidates during Fmoc Solid-Phase Peptide Synthesis: A Safe Route to Thioamides. J. Org. Chem 2019, 84, 15309–15314. [DOI] [PubMed] [Google Scholar]
- (28).We note that the conditions to generate the amidine may elicit cleavage from resins that use acid-sensitive linkages between the C-terminal amino acid and the resin (e.g., 2-chlorotrityl or Sieber amide), but are compatible with Wang or Rink amide linkages.
- (29).Chaturvedi RK; Schmir GL Hydrolysis of thioimidate esters. II. Evidence for the formation of three species of the tetrahedral intermediate. J. Am. Chem. Soc 1969, 91, 737–746. [Google Scholar]
- (30).Motiwala HF; Armaly AM; Cacioppo JG; Coombs TC; Koehn KRK; Norwood VM 4th; Aubé J HFIP in organic synthesis. Chem. Rev 2022, 122, 12544–12747. [DOI] [PubMed] [Google Scholar]
- (31).Shuklov IA; Dubrovina NV; Boerner A Fluorinated alcohols as solvents, cosolvents and additives in homogeneous catalysis. Synthesis 2007, 2007, 2925–2943. [Google Scholar]
- (32).Altman RA; Sharma KK; Rajewski LG; Toren PC; Baltezor MJ; Pal M; Karad SN Tyr1-[(Z)CFCH]-Gly2 Fluorinated Peptidomimetic Improves Distribution and Metabolism Properties of Leu-Enkephalin. ACS Chem. Neurosci 2018, 9, 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Carbone R; Fré S; Iannolo G; Belleudi F; Mancini P; Pelicci PG; Torrisi MR; Di Fiore PP eps15 and eps15R are essential components of the endocytic pathway. Cancer Res. 1997, 57, 5498–5504. [PubMed] [Google Scholar]
- (34).Siegert TR; Bird MJ; Makwana KM; Kritzer JA Analysis of loops that mediate protein-protein interactions and translation into submicromolar inhibitors. J. Am. Chem. Soc 2016, 138, 12876–12884. [DOI] [PubMed] [Google Scholar]
- (35).Karnik SS; Singh KD; Tirupula K; Unal H Significance of angiotensin 1–7 coupling with MAS1 receptor and other GPCRs to the renin-angiotensin system: IUPHAR Review 22. B. J. Pharm 2017, 174, 737–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Cassell RJ; Mores KL; Zerfas BL; Mahmoud AH;Lill MA; Trader DJ; van Rijn RM Rubiscolins are naturally occurring G protein-biased delta opioid receptor peptides. Eur. Neuropsych 2019, 29, 450–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Vejrazkova D; Vankova M; Lukasova P; Vcelak J; Bendlova B Insights into the physiology of C-peptide. Physiol. Res 2020, 69, S237–S243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Newberry RW; VanVeller B; Raines RT Thioamides in the collagen triple helix. Chem. Commun. (Camb.) 2015, 51, 9624–9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.