

Abstract
Chronic stimulation of cardiac α1A-adrenergic receptors (α1A-ARs) improves symptoms in multiple preclinical models of heart failure. However, the translational significance remains unclear. Human engineered heart tissues (EHTs) provide a means of quantifying the effects of chronic α1A-AR stimulation on human cardiomyocyte physiology. EHTs were created from thin slices of decellularized pig myocardium seeded with human induced pluripotent stem cell (iPSC)-derived cardiomyocytes and fibroblasts. With a paired experimental design, EHTs were cultured for 3 wk, mechanically tested, cultured again for 2 wk with α1A-AR agonist A61603 (10 nM) or vehicle control, and retested after drug washout for 24 h. Separate control experiments determined the effects of EHT age (3–5 wk) or repeat mechanical testing. We found that chronic A61603 treatment caused a 25% increase of length-dependent activation (LDA) of contraction compared with vehicle treatment (n = 7/group, P = 0.035). EHT force was not increased after chronic A61603 treatment. However, after vehicle treatment, EHT force was increased by 35% relative to baseline testing (n = 7/group, P = 0.022), suggesting EHT maturation. Control experiments suggested that increased EHT force resulted from repeat mechanical testing, not from EHT aging. RNA-seq analysis confirmed that the α1A-AR is expressed in human EHTs and found chronic A61603 treatment affected gene expression in biological pathways known to be activated by α1A-ARs, including the MAP kinase signaling pathway. In conclusion, increased LDA in human EHT after chronic A61603 treatment raises the possibility that chronic stimulation of the α1A-AR might be beneficial for increasing LDA in human myocardium and might be beneficial for treating human heart failure by restoring LDA.
NEW & NOTEWORTHY Chronic stimulation of α1A-adrenergic receptors (α1A-ARs) is known to mediate therapeutic effects in animal heart failure models. To investigate the effects of chronic α1A-AR stimulation in human cardiomyocytes, we tested engineered heart tissue (EHT) created with iPSC-derived cardiomyocytes. RNA-seq analysis confirmed human EHT expressed α1A-ARs. Chronic (2 wk) α1A-AR stimulation with A61603 (10 nM) increased length-dependent activation (LDA) of contraction. Chronic α1A-AR stimulation might be beneficial for treating human heart failure by restoring LDA.
Keywords: α1A-adrenergic receptor, engineered heart tissue, heart failure, human, induced pluripotent stem cell
INTRODUCTION
Multiple preclinical animal models of heart failure show significantly improved cardiac function following chronic treatment with an α1A-adrenergic receptor (α1A-AR) agonist (1–4). Consistent with this, in humans, the ALLHAT clinical trial found worse heart failure outcomes in patients treated with an α1-adrenergic receptor (α1-AR) antagonist, suggesting that α1-ARs mediate protective effects in human heart failure patients [reviewed in Zhang et al. (5)]. However, there has been no direct test of α1A-AR agonist therapy in human. Engineered heart tissue (EHT) created using human induced pluripotent stem cell iPSC-derived cardiomyocytes (iPSC-CMs) is an experimental approach to study the in vitro properties of human cardiac tissue (6–11). EHT can be maintained long term in culture and thus can be used as a testing platform to evaluate the effect of chronic treatment with candidate therapeutics in human cardiomyocytes.
We used a paired experimental design to mechanically test human EHT, then treated for 2 wk with α1A-AR agonist A61603 (10 nM) or vehicle control, and mechanically retested after drug washout for 24 h.
We found that chronic treatment for 2 wk with A61603 caused increased length-dependent activation (LDA) of contraction compared with before treatment. LDA was not increased in vehicle-treated controls. LDA is an intrinsic myocardial property that underlies the organ-level Frank–Starling relationship where increased ventricular filling (preload) results in increased ejection volume during the next beat (12). LDA is blunted in heart failure, which might contribute to impaired pump function (13, 14). Thus, increased LDA mediated by chronic stimulation of the α1A-AR might be beneficial for increasing LDA in the human myocardium and might be beneficial for treating human heart failure by restoring LDA.
In contrast, average EHT force development was unchanged after chronic A61603 treatment but was increased after vehicle treatment, suggesting that EHT maturation occurred with vehicle treatment but not with A61603 treatment.
In summary, in human iPSC-CMs, chronic treatment with the α1A-AR agonist A61603 had a positive effect to increase LDA but did not change EHT force. These findings might be relevant for efforts to develop new heart failure treatments that target the α1A-AR.
METHODS
Creation of EHT
EHTs were formed with one million human iPSC-CMs (PGP1 cell line; Coriell Institute, male) and 50,000 human cardiac fibroblasts per tissue, using decellularized porcine myocardial scaffolds (dimensions, 150 mm thick, 2.75 mm wide, and 6.34 mm long) that were incorporated into a polymer cassette (MyoPod) (Fig. 1A). EHTs were cultured at 37°C for 3–5 wk in DMEM, 5% CO2, and B27 supplement. EHTs were created in two separate iPSC-CM differentiation batches (two biological replicates).
Figure 1.

A: schematic of engineered heart tissue (EHT) mounted in MyoPod cassette. EHT length was varied using an adjustable force transducer claw. B: schematic of the experimental protocol.
Mechanical Testing
After 3-wk culture, EHTs were mounted at culture length (0% stretch) in the MyoLab mechanical testing system. EHTs were superfused at 0.3 mL/min with DMEM media at 37°C.
EHTs were preconditioned with three slow stretch cycles from −5 to +5% of culture length. EHTs were returned to culture length and electrically paced with square wave stimuli of 5-ms duration, 9–12-V amplitude, and 1–1.4-Hz frequency. The contraction force and timing of contraction and relaxation were calculated using custom software and the values reported were obtained at culture length (unless another EHT length is indicated).
After a stable baseline force was attained, length-dependent activation (LDA) was determined by measuring isometric twitch force at 11 different tissue lengths (−5 to +5% of initial length, at 1% increments). A unitless parameter for LDA was defined as the slope of the relationship of EHT force versus normalized EHT length, divided by the EHT force obtained at 0% stretch. After the LDA procedure, EHTs were returned to 0% stretch for 10 min. Finally, the stimulation rate was increased from baseline in steps of 0.2 Hz to determine the maximum capture rate (MCR, the maximum stimulus rate that still caused a synchronous contractile response of the EHT). The EHT testing protocol was completed in 60–90 min.
Effect of Chronic A61603 Treatment
A61603 is a potent and highly selective agonist for the α1A-AR subtype (3). Previous dose-finding studies reported that the EC50 for activation of ERK signaling (a known effect of α1A-AR stimulation) was 5 nM A61603 in human cardiomyocytes (15), 6 nM in neonatal rat cardiomyocytes (3), and 23 nM in cultured mouse cardiomyocytes (3). Moreover, the EC50 for the protection of cultured mouse cardiomyocytes against doxorubicin-induced toxicity was 14 nM A61603 (3). For the current study, a dose of 10 nM A61603 was used.
EHTs from a single iPSC-CM differentiation batch (one biological replicate, n = 14 technical replicates) were used to test the effects of A61603- or vehicle-treatment using a paired (before vs. after) testing protocol (Fig. 1B). EHTs were mechanically tested in the MyoLab after 3 wk of culture, and EHTs were returned to culture for a further 2 wk and chronically treated with either 10 nM A61603 or vehicle control (n = 7/group) with media changed every other day.
After 2 wk, plus a 24-h washout of drug, EHT mechanical testing was repeated. The washout period was to minimize the acute (inotropic) effects of α1A-AR activation and determine if residual (noninotropic) effects remained. Previous studies found that acute physiological effects of α1-AR stimulation were reversed 20 min after drug washout (16, 17).
Solutions
Vitamin C and HCl were used to stabilize A61603 solutions. A stock solution of 10 mM A61603 (Tocris) was made in 1 mM HCl and stored at −20°C. A working solution of 0.1 mM A61603 was made with 50 μL stock added to 4.7 mL culture medium, and 0.25 mL of vitamin-C (Sigma) stock solution (100 mM). For chronic treatment of EHT with 10 nM A61603, 100 μL of working solution was added per liter of culture medium. Vehicle treatment used the same quantities of vitamin C and HCl.
Effects of EHT Age and Prior Mechanical Testing
EHTs from a second iPSC-CM differentiation batch (one biological replicate, n = 13 technical replicates) were used. To determine the effect of EHT age, EHTs were tested on a single occasion after 3, 4, or 5 wk of culture (n = 3 to 4 per age group). To assess the effect of prior mechanical testing, the EHTs tested at 3 wk were retested 1 wk later (n = 4). Moreover, additional EHTs tested at 3 wk were retested 1 day later (n = 2).
RNA Extraction from EHT
To preserve RNA from EHTs treated with A61603 or vehicle, after final physiological testing, EHTs were preserved individually with RNA stabilization solution (RNAlater, ThermoFisher), stored at −80°C for ≈2 wk, and shipped on dry ice to the University of California Davis for RNA-seq analysis. For RNA extraction, individual EHTs were transferred to a 1.5-mL Eppendorf tube on dry ice and homogenized using a cold pestle followed by the addition of 0.25 mL of TRIzol (Cat. No. 15596026; Life Technologies), vortexed for 1 min, and incubated at room temperature (RT) for 10 min. Samples were transferred to a QIAshredder tube (Qiagen LLC, 79654), and centrifuged at 4°C, at 14,000 g, for 2 min. Eluate was returned to the original 1.5-mL Eppendorf tube, and an additional 0.75 mL of TRIzol was added, vortexed for 1 min, and incubated at RT for 10 min. Chloroform (0.2 mL) was added, briefly vortexed, and centrifuged at 4°C, at 14,500 g, for 15 min. The clear aqueous phase was transferred to a new 1.5-mL Eppendorf tube, avoiding the phenol-chloroform phase. RNA was precipitated from the aqueous phase by adding 0.6 mL of isopropyl alcohol, incubating for 5 min at RT, and centrifuging at 4°C, at 14,500 g, for 15 min. The supernatant was carefully discarded without disturbing the pellet. Three ethanol washes were performed using 0.75 mL of 70% ETOH preceding RNA cleanup. After the last wash, the tubes were left to air dry for 30 min at RT. RNA was dissolved in 50 µL of nuclease-free water and incubated at 55°C for 5 min. Purification of RNA was followed immediately after extraction using an RNA Clean and Concentrator kit (Cat. No. R1017, Zymo Research). Briefly, two volumes of RNA binding buffer were added to the eluate and mixed, followed by an equal amount of 100% of ETOH, which was then mixed and transferred to a Zymo-spin column. Sample was centrifuged at 13,000 g for 30 s and the supernatant discarded. DNase treatment was performed in column by adding 75 µL of DNA digestion buffer and 5 µL of Dnase I (Zymo Research, E1010) and incubating for 15 min at RT. Following incubation, 0.4 mL prep buffer was added, centrifuged at RT, at 13,000 g, for 30 s and the supernatant discarded; 0.7 mL of RNA wash buffer was then added, centrifuged at RT, at 13,000 g, for 30 s, and the supernatant discarded. A second round of 0.4 mL of RNA wash buffer was added, centrifuged at RT, at 13,000 g, for 30 s and the supernatant discarded. RNA was eluted by adding 30 µL of RNase-free water, incubating for 1 min, and centrifuging at RT, at 13,000 g, for 30 s. Samples required an RNA integrity number (RIN) > 7.0 and an A260/280 ratio of absorbance > 1.7 to proceed.
RNA Sequencing
Strand-specific and barcode-indexed RNA sequencing (RNA-seq) libraries were generated from 100–200 ng total RNA from each EHT and after poly-A enrichment using the Kapa mRNA-seq Hyper kit (Kapa Biosystems, Cape Town, South Africa) following the manufacturer instructions (unfortunately, because of a technical issue, RNA from one A61603-treated sample was lost at the time of library making). The fragment size distribution of the libraries was verified via microcapillary gel electrophoresis on a LabChip GX system (PerkinElmer, Waltham, MA). The libraries were quantified by fluorometry on a Qubit fluorometer (LifeTechnologies, Carlsbad, CA) and pooled in equimolar ratios. The pool was quantified by qPCR with a Kapa Library Quant kit (Kapa Biosystems) and sequenced on an Illumina NovaSeq 6000 system (Illumina, San Diego, CA) run with paired-end 150-bp reads.
RNA-Seq Data Processing and Analysis
Transcript abundance was quantified from the raw sequencing data using the program kallisto (18) and the Homo_sapiens.GRCh38.96.gtf genome build (19). RNA-Seq data for vehicle-treated and A61603-treated EHTs have been uploaded to the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) and can be retrieved using the series accession number GSE216439.
Gene-level quantification of expression levels, as well as statistical tests of differential expression with respect to A61603 versus vehicle, were performed using the package sleuth (20, 21) within the statistical computing environment R (22). The sleuth package performed likelihood ratio tests of differential expression at the transcript (isoform) level and then aggregated the P values, using the Lancaster method (23), to obtain gene-level P values. The package also provided the computations for principal components analysis (PCA), through standard R routines. Adrenergic receptors and selected contraction-related genes (i.e., calcium handling, myofilament, and collagen-related genes) with raw P values of <0.05 were considered statistically significant for an individual gene.
Functional pathways altered with A61603 treatment were identified using prioritized genes with raw P values of <0.1 among the genes at the transcriptome level and the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway database (24) (a collection of maps for biological pathways showing networks of molecular interactions and links to genes). The pathways were tested for overrepresentation using the hypergeometric test (one-sided Fisher’s exact test), producing nominal P values as the probability of observing as many (or more) prioritized genes in the pathway, under the null hypothesis that any such common genes are due purely to chance. Significantly altered pathways were defined as having P < 0.05 for the hypergeometric test.
Statistical Analysis
Data are presented as means ± SE. Statistical tests for RNA-seq data were described in the previous section. All other statistical tests (paired or unpaired t test, two-way ANOVA, F test of equality of group variances, linear regression, and coefficients of variation) were performed using Prism 9 software (GraphPad Software, La Jolla, CA) with a significance level set at P < 0.05.
RESULTS
Baseline contractions
Figure 2A shows typical data records of electrically stimulated twitch contractions of EHT measured at various EHT lengths both above and below the culture length. Figure 2B shows there was a close positive linear relationship between peak twitch contraction force versus EHT length. The linear regression R2 values were not different for EHT after vehicle treatment (0.99 ± 0.003) versus after A61603 treatment (0.98 ± 0.01) (P = 0.37, n = 7/group).
Figure 2.
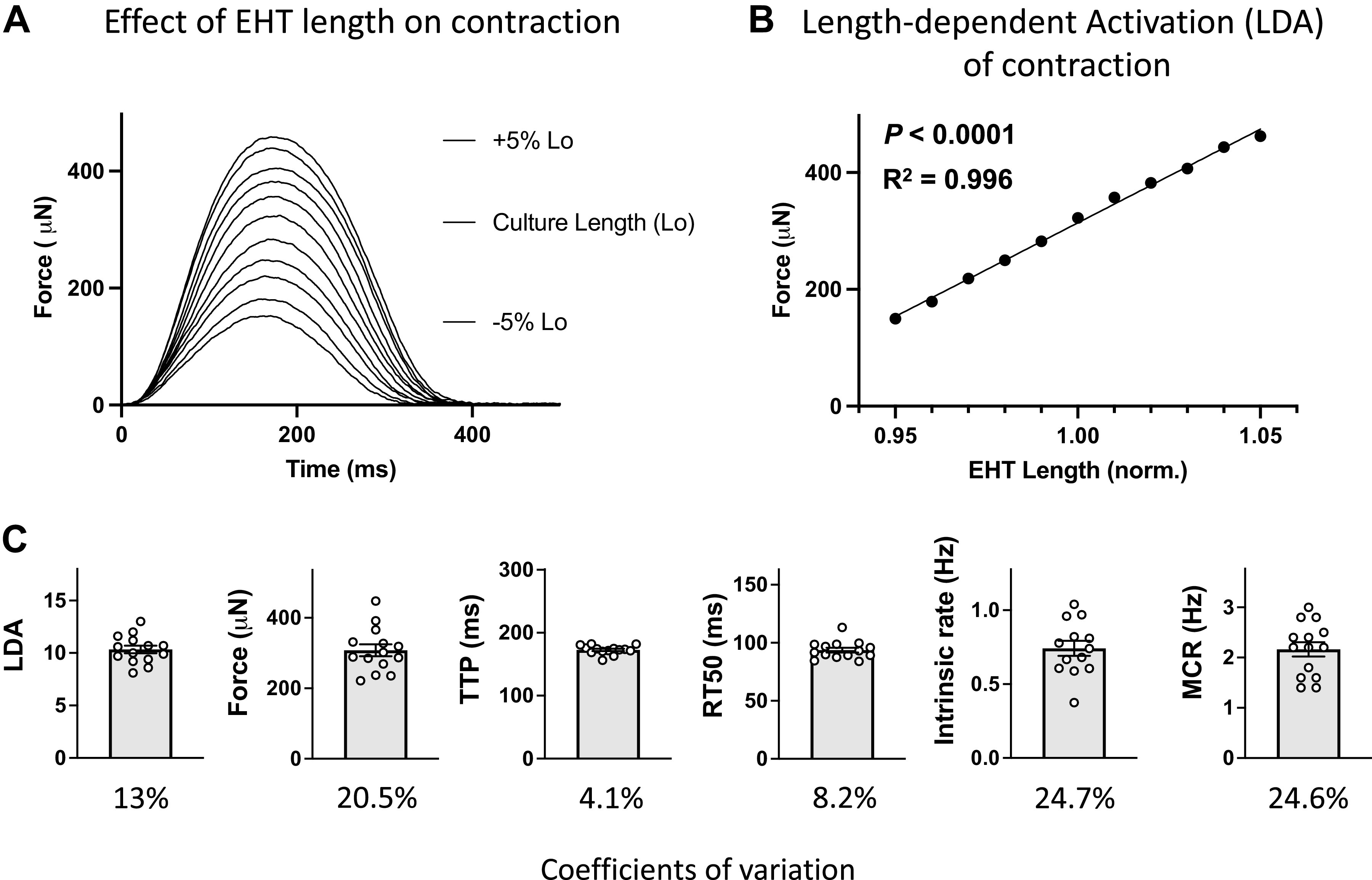
Representative records and pooled data for contractions of engineered heart tissue (EHT) after 3 wk culture, before drug/vehicle treatment. A: superimposed twitch contractions for EHT at various lengths above and below culture length (Lo). B: relationship between force vs. EHT length: same experiment as in A (means ± SE, n = 3 contractions, errors are smaller than symbols). C: baseline parameters before drug/vehicle treatment for all EHTs in cohort (individual and mean values, n = 14, and coefficients of variation).
Figure 2C shows values for parameters derived from contractions of EHT after 3 wk in culture before treatment with either A61603 or vehicle (n = 14). These baseline values show good reproducibility for EHT created from the same iPSC-CM differentiation batch. However, there were some differences noted between batches (see Chronic A61603 Treatment Did Not Increase EHT Force).
After 3 wk of culture, the average EHT force was ≈300 μN. Based on EHT width at 3 wk ≈1.5 mm, and EHT thickness ≈0.25 mm (7), the force per cross-sectional area was ≈1 mN/mm2.
Values for the twitch time to peak and time to 50% relaxation (173 ms, 94 ms) were similar to values previously measured in human cardiac trabeculae (203 ms, 125 ms) (17).
Intrinsic Contraction Rate
Figure 3 shows the intrinsic contraction rate measured from video recordings of EHT. Mean intrinsic rate before treatment was ≈0.75 Hz and the intrinsic rate was not statistically different after treatment with either vehicle or A61603. There was a trend toward greater variability in intrinsic rate after A61603 treatment versus baseline (coefficient of variation = 41%, n = 6 vs. 25%, n = 13, P = 0.051, F test).
Figure 3.

Chronic A61603 treatment did not change engineered heart tissue (EHT) intrinsic contraction rate. A: intrinsic rate of spontaneous EHT contractions measured before and after chronic (2 wk) treatment with vehicle or A61603 (means ± SE, n = 5–7/group, P values for mixed effects analysis). B: no change in intrinsic rate after treatment relative to before treatment (P value for unpaired t test).
Chronic A61603 Treatment Increased LDA
Figure 4A shows LDA values both before and after chronic treatment with vehicle control or with A61603. After chronic A61603 treatment, there was a trend toward a modest increase in LDA compared with before treatment (P = 0.06, n = 7/group, paired t test). However, LDA was not changed after vehicle control treatment. The experimental design allows a comparison of the before versus after treatment effect for each muscle. Figure 4B shows LDA after treatment as a percentage of LDA before treatment. LDA was ≈25% higher (P = 0.035) after A61603 treatment (117% of initial) than after vehicle treatment (93% of initial).
Figure 4.

Chronic A61603 treatment increased length-dependent activation (LDA). A: LDA measured before and after chronic (2 wk) treatment with vehicle or A61603 (means ± SE, n = 7/group, P values for paired t tests). B: LDA (%initial) was higher after A61603 treatment vs. after vehicle treatment (P value for unpaired t test).
Chronic A61603 Treatment Did Not Increase EHT Force
Interestingly, Fig. 5 shows that 2 wk after vehicle treatment, EHT contraction force was significantly increased by 35% compared with before vehicle treatment, suggesting EHT maturation. However, 2 wk after A61603 treatment average EHT force was not increased. These different responses did not involve EHT hypertrophy. EHT width at the start of culture (2.75 mm) was reduced to ≈1.5 mm after 3 wk of culture and was further decreased by ≈10% during the 2-wk treatment period (Fig. 5C). However, EHT width was not different between vehicle-treated versus A61603-treated EHT. The absence of hypertrophy suggests that the increased force in the vehicle-treated group was due to increased EHT intrinsic strength (force per cross-sectional area). Moreover, the findings suggest that A61303 treatment did not increase EHT intrinsic strength.
Figure 5.

Chronic A61603 treatment did not increase engineered heart tissue (EHT) force. A: twitch contraction force measured before and after chronic (2 wk) treatment with vehicle or A61603 (means ± SE, n = 7/group, P values for paired t tests). B: EHT force (%initial) was increased after vehicle treatment but not after A61603 treatment (P value for unpaired t test). C: EHT width before and after treatment with vehicle or A61603 (means ± SE, n = 7/group, P values for paired t tests).
To further investigate the increased force measured after 2 wk of vehicle treatment, we assessed the effect of EHT age. Figure 6 shows that there was not a significant change in the values of LDA, force, or twitch time to peak (TTP) with EHT age [though there was a decline in the time to 50% relaxation (RT50) with EHT age]. These results suggest that the increased EHT force after 2 wk of vehicle treatment was not due to the aging of EHT.
Figure 6.

Effect of engineered heart tissue (EHT) age on contraction parameters measured on a single occasion (n = 3–6 per age group; P values for linear regressions). A: length-dependent activation (LDA). B: force. C: time to peak force (TTP). D: time to 50% relaxation (RT50).
To determine the effect of repeat mechanical testing on EHT force, EHTs were mechanically tested after 3 wk in culture and then retested after 1 day or 1 wk (Fig. 7A). When compared with mechanical testing at 3 wk, EHT force was increased ≈30% when retested 1 day later and increased two- to threefold when retested 1 wk later.
Figure 7.

Repeat mechanical testing after 3 wk culture increased engineered heart tissue (EHT) force. A: relationship of EHT force (normalized) to EHT age. First, EHT mechanical testing was at 3 wk (n = 6, same EHT as Fig. 6B), with retesting after 1 day or 1 wk (P value for linear regression). B: effect of both EHT age and repeat testing on EHT force (data from Fig. 6B and Fig. 7A) (P values for unpaired t tests).
The effects of both EHT age and prior mechanical testing are summarized in Fig. 7B. For EHT mechanically tested at 4 wk, EHT force was much higher if EHTs were previously tested at 3 wk. Thus, some aspects of mechanical testing promoted increased EHT force development. Moreover, the EHT force on retest was higher when the interval between tests was 1 wk versus 1 day, suggesting that prior mechanical testing stimulated a maturation process that subsequently developed over the following week.
Taken together, the findings suggest that the increased EHT force after vehicle treatment was due to an effect of repeat mechanical testing. However, after A61603 treatment, EHT force was not significantly increased with repeat mechanical testing.
Separate iPSC-CM differentiation batches were used to create EHT for studies of A61603/vehicle versus studies of aging/retesting. EHT made from separate iPSC-CM differentiation had similar values for both twitch force and for time to peak. However, the EHT used for aging/retesting had a 25% higher LDA and a 16% longer relaxation time (RT50).
Contraction Kinetics
The twitch time to peak force (TTP) was slightly prolonged after treatment with either vehicle (6.7%) or A61603 (4.7%) (Fig. 8A). These increases were not statistically different from each other (Fig. 8B).
Figure 8.

Effect of chronic A61603 treatment on the timing of contraction and relaxation. A: twitch time to peak (TTP) measured before and after chronic (2 wk) treatment with vehicle or A61603 (means ± SE, n = 7/group, P values for paired t tests). B: TTP after treatment relative to before treatment (P value for unpaired t test). C: time to 50% relaxation (RT50) measured before and after chronic (2 wk) treatment with vehicle or A61603 (means ± SE, n = 7/group, P values for paired t tests). D: RT50 after treatment relative to before treatment (P value for unpaired t test).
The time to 50% relaxation (RT50) did not change significantly from baseline after treatment with either vehicle or A61603 (Fig. 8C). However, RT50 was more variable after A61603 treatment than after vehicle treatment (coefficient of variation = 11.5 vs. 3.3%, P = 0.008, F test).
There was a trend to a lower maximum capture rate (MCR) after vehicle treatment (P = 0.095) but not after A61603 treatment (Fig. 9A). Moreover, MCR values tended to be more variable after A61603 treatment than after vehicle treatment. But there was no difference in the percent change in MCR between treatment groups (Fig. 9B).
Figure 9.

Effect of chronic A61603 treatment on the response to rapid pacing. A: maximum capture rate (MCR) measured before and after chronic (2 wk) treatment with vehicle or A61603 (means ± SE, n = 7/group, P values for paired t tests). B: MCR after treatment relative to before treatment (P value for unpaired t test). C: relationship between engineered heart tissue (EHT) force (normalized) and stimulation frequency before treatment with vehicle or A61603 (n = 14) and after vehicle treatment (n = 5) (means ± SE, ****P < 0.0001, **P < 0.01, paired t tests). D: force-frequency relation after A61603 treatment (n = 6) (means ± SE, ns, not significant, paired t test).
Figure 9C shows the relationship between contraction force and stimulation frequency for contractions used to assess MCR. For all EHTs before treatment with either vehicle or A61603 (n = 14), there was a modest increase (≈3%) in force with increasing stimulation frequency. Figure 9C also shows that the force-frequency response after 2-wk vehicle treatment was not appreciably changed relative to baseline. Thus, prior mechanical testing, which prompts EHT force maturation, did not impact the force-frequency response.
Figure 9D shows that following chronic treatment with A61603, the force-frequency relationship was variable among EHT samples. For all stimulation frequencies above 1 Hz, there was a statistically significant greater variability of normalized force values after A61603 treatment than after vehicle treatment (P = 0.05, F test). There was a trend toward increased force with increased stimulation frequency, but this did not reach statistical significance.
Taken together, contraction kinetics were not different between vehicle versus A61603 treatment, but there was a trend toward greater variability after A61603 treatment.
RNA-Seq Analysis
To investigate the effects of treatment with A61603 or vehicle on gene expression, bulk RNA-seq was performed. The total RNA recovered from EHTs that were treated with A61603 (346.5 ± 63 ng) or vehicle (356.5 ± 125 ng) were similar (P = 0.8976, means ± SE, n = 7/group). The RIN values and A260/280 values for EHTs that were treated with A61603 (7.5 ± 0.15 and 1.87 ± 0.03) or vehicle (7.7 ± 0.21 and 1.9 ± 0.05) were similar (P = 0.4606 and 0.6312, respectively, n = 7/group). An average of 32.2 ± 0.5 million reads per sample were produced by the sequencer, of which an average of 23.5 million (73%) were mapped to transcripts.
Genes for all three α1-AR subtypes were expressed in EHT (Fig. 10A). Moreover, the expression level of the α1A-AR subtype was not significantly different between A61603-treated (n = 6) versus vehicle-treated EHT (n = 7, P = 0.36). The expression level of the α1A-AR subtype was less than the expression level of the β1-AR, consistent with previous studies (25). The α1D-AR subtype expression level was low, consistent with previous suggestions that the cardiac α1D-AR is localized to coronary vessels, not to cardiomyocytes (26, 27).
Figure 10.

Effects on chronic A61603 treatment on adrenergic receptors and contraction-related gene expression. A: expression of adrenergic receptors in engineered heart tissue (EHT) treated with A61603 (n = 6) or vehicle (n = 7). ****P < 0.0001. B: heat maps for selected contraction-related genes; values scaled from 0.4× (deep blue) to 1.6× (deep red) the average for the vehicle group. C: significantly changed biological pathways associated with prioritized genes in EHT treated with A61603 (n = 6) vs. vehicle (n = 7). Boldface indicates pathways with known links to α1-AR signaling. Origin value on the abscissa corresponds to P = 0.05.
A61603 treatment did not cause significant changes in the expression levels for several prespecified genes involved in Ca2+ handling, myofilament contraction, or collagen (Fig. 10B); this suggests that changes in the expression of these genes were not part of the response to chronic treatment with A61603 versus vehicle.
At the whole transcriptome level, 780 genes met the prespecified criteria of raw nominal P values of <0.1 and were pursued for additional pathway analysis. Under correction for multiple testing, no individual gene had a q value < 0.05 in A61603-treated versus vehicle-treated EHT. However, analysis of functional pathways for the 780 prioritized genes found several pathways in which genes with small nominal P values (P < 0.1) were significantly overrepresented in EHT treated with A61603 (Fig. 10C). Several of these pathways (5 out 18) have been previously implicated in α1-AR signaling, including calcium signaling (P = 0.00994), neuroactive ligand-receptor signaling (P = 0.01098), glycosaminoglycan biosynthesis (P = 0.01641), PI3K-AKT signaling (P = 0.0174165), and MAPK signaling (P = 0.02695).
DISCUSSION
Previous studies reported that chronic treatment with an α1A-AR agonist had beneficial effects in multiple animal models of heart failure (1–4). Here, human EHT was used to evaluate the effects of chronic α1A-AR agonist treatment on human cardiomyocyte physiology. EHTs were mechanically tested, then treated for 2 wk with A61603 or vehicle and retested after 24-h washout.
The major findings were 1) length-dependent activation (LDA) of EHT contraction was increased by 25% after chronic α1A-AR agonist (A61603) treatment, compared with after vehicle treatment; 2) average EHT force was not increased after A61603 treatment but was increased after vehicle treatment; and 3) RNA-seq analysis confirmed the expression of the α1A-AR in human EHT and found that chronic A61603 treatment caused changes in the expression of genes in multiple biological pathways that are linked to α1A-AR signaling.
In the context of preclinical testing, the current study found that chronic α1A-AR agonist treatment of human EHT had a positive effect to increase LDA. In heart failure where the effect of preload on cardiac output is reduced, increased myocardial LDA mediated by chronic α1A-AR stimulation might improve the function of the failing heart.
Increased LDA
For human EHT, chronic A61603 treatment increased LDA. However, LDA was not increased with EHT age. Moreover, LDA was not increased in the vehicle-treated group, indicating that EHT maturation did not involve increased LDA. Thus, increased LDA was specifically related to chronic A61603 treatment, but not to the components of the treatment protocol that involved aging (i.e., 2-wk treatment) or prior mechanical testing. Increased LDA is a novel effect of chronic α1A-AR stimulation and adds to multiple reported effects that may contribute to reversal of heart failure, including increased myofilament function, increased mitochondrial function, and decreased oxidative stress (1, 28).
LDA is a sarcomere-level correlate of the organ-level Frank–Starling relationship where increased filling of the ventricle (i.e., increased preload) results in greater ventricular ejection volume (13). LDA is attenuated in heart failure, which may contribute to the impaired function of the failing heart (13, 14). Increased LDA in human EHT after chronic A61603 treatment raises the possibility that chronic stimulation of the α1A-AR might be beneficial for increasing LDA in human myocardium and might be beneficial for treating human heart failure by restoring LDA.
The mechanisms underlying LDA are not fully resolved and involve multiple contributing effects on myofilament calcium sensitivity, cross-bridge recruitment, and posttranslational modifications (12, 29–31). Further study is needed to determine the role of these mechanisms in the increased LDA observed after chronic treatment of EHT with A61603. The current study did not find significant changes in the expression of specific genes that could account for increased LDA after A61603 treatment.
EHT Maturation
This study used a paired experimental design to mechanically test EHT both before and after treatment with A61603 or vehicle. Importantly, for the vehicle-treated group, we found that the first episode of mechanical testing prompted increased EHT contraction force, suggesting EHT maturation. As a cautionary note, future EHT studies using a paired experimental design with repeat mechanical testing would need to control for a potential EHT maturation effect.
EHT force was increased two- to threefold when mechanically retested after 1 wk. However, EHT force was increased by only 35% when vehicle-treated EHTs were retested after 2 wk. Possibly, the force maturation effect caused by mechanical testing might peak after 1 wk and then decline by 2 wk. However, the EHTs used at these two time points were created using different iPSC-CM differentiation batches, and EHT maturation may be affected by the iPSC-CM differentiation batch.
Currently, there is considerable interest in the factors promoting EHT maturation (32). Previous studies reported that chronic electrical pacing of EHT over several weeks resulted in improved Ca2+ handling and force development (33, 34). The current study found that just a single episode of mechanical testing involving electrical pacing for 60–90 min was sufficient to prompt subsequent EHT force maturation. What aspect of the testing protocol promoted EHT maturation is unknown (e.g., electrical pacing, faster pacing during force-frequency measures, mechanical stretch, etc.). Although electrically stimulated contractions can mediate EHT maturation (33, 34), the spontaneous contraction rate was not changed in the vehicle-treated group, suggesting that spontaneous contractions did not play a role in EHT maturation observed in the vehicle-treated group.
Chronic A61603 Treatment Did Not Increase EHT Force
Lower EHT force after chronic A61603 treatment versus after vehicle treatment might have occurred because A61603 treatment inhibited EHT maturation. After A6160 treatment, there were trends toward greater variability of intrinsic contraction rate, twitch timing, maximum capture rate (MCR), and force-frequency response. Variability in these parameters may reflect variability in EHT electrical properties (e.g., action potential duration, period of refractoriness, presence of arrhythmias), which might adversely affect EHT maturation. Nevertheless, it is unclear whether an absence of EHT maturation after A61603 treatment would be relevant in the context of heart failure and a therapeutic approach based on chronic α1A-AR stimulation.
Alternatively, lower force after A61603 treatment might reflect a rundown phenomenon. Specifically, in vitro studies of cardiac muscle preparations commonly find contraction force is reduced after inotropic stimulation and drug washout (i.e., rundown). In the current study, although EHTs were not electrically stimulated during A61603 treatment, inotropic stimulation by A61603 may have occurred during spontaneous contractions, and the lower force observed after A61603 washout might reflect postinotropic rundown.
Previous studies found that in mouse heart failure models, chronic A61603 treatment increased maximum force development of demembranated cardiac muscle fibers and increased fractional shortening in vivo (1–3). However, in the current study, chronic A61603 treatment did not increase EHT force. These discordant findings might reflect different experimental phenotypes and species (nonfailing human EHT vs. mouse heart failure models). Moreover, in the current study, A61603 was washed out before mechanical testing, but in the mouse heart failure models, A61603 was still present during testing.
Effect of Chronic A61603 Treatment on Gene Expression
RNA-seq analysis showed that the α1A-AR was present in human EHT (Fig. 10A), and the lower expression level relative to the β1-AR was consistent with a previous report (25). The presence of the α1A-AR in EHT is consistent with the idea that the observed mechanical effects of A61603 treatment occurred via α1A-AR signaling. When prespecified contraction-related genes were tested, no significant differences were seen in response to A61603 treatment, suggesting that these genes were not transcriptionally affected by A61603 treatment. This study was not powered to identify changes in the expression of individual genes with a significant q value that could explain the differences noted in LDA or force for EHT treated with either A61603 or vehicle.
However, RNA-seq analysis of prioritized genes ranked by the raw P values found significant changes in multiple biological pathways; five pathways were already known to be affected by α1-AR signaling, whereas thirteen others were not previously linked (Fig. 10C). There were significant changes in the MAPK signaling pathway, which is known to be activated by α1-AR stimulation and to mediate effects on myocyte hypertrophy, cell contraction, and cell survival (35–37). There were significant changes in the PI3K-Akt signaling pathway, which was previously reported to contribute to the hypertrophic response of A61603 stimulation (38). Although A61603 treatment did not change the expression level of a few prespecified major Ca2+ handling proteins (Fig. 10B), there were significant changes in other genes in the Ca2+ signaling pathway (Fig. 10C), which has been implicated in the regulation of contraction by α1A-ARs (39). There were also significant changes in the neuroactive ligand-receptor interaction pathway, which contains a large array of signaling receptors, including adrenergic receptors. There were significant changes in the glycosaminoglycan biosynthesis pathway, which involves numerous extracellular matrix molecules and is also known to be affected by α1-AR activation (40).
Finally, other pathways (13) not previously linked to α1-AR signaling were also significantly implicated. For example, there were significant changes in the glycosphingolipid biosynthesis pathway, which include sphingolipids that are known to mediate protective effects on myocyte survival (41). The other pathways identified in this analysis may suggest the existence of new molecular targets for the discovery of protective mechanisms involved in α1-AR signaling. Future studies powered for individual gene discovery will be needed to characterize potential new targets.
Strengths and Limitations of Human EHT
Human EHT has advantages as an experimental platform for translational studies. The geometrically simple form is ideal for mechanical testing. EHTs can be constructed using cells from heart failure human patients to evaluate patient-specific responses to drug treatments (42) and to investigate disease mechanisms (8). EHT can be maintained long term in culture to evaluate responses to chronic interventions.
There are additional advantages associated with the experimental system used in the current study. EHTs were constructed using standardized scaffolds consisting of cryosectioned and laser-cut pig myocardium (7). The resulting scaffolds were thin, which allows diffusional exchange between the EHT and culture medium. Moreover, the scaffolds had precisely defined dimensions with the longitudinal alignment of iPSC-CMs. The MyoLab system allows EHT to be automatically mounted at culture length for mechanical testing. Standardizing both the scaffold dimensions and length contributes to reproducible contractions measured between different EHTs. The MyoPod EHT mounting system allows EHT to be readily removed from culture for mechanical testing and then returned to culture. This nondestructive testing allows for repeated measurements in a single EHT, enabling the collection of longitudinal function data. Moreover, the MyoPod movable force transducer mount allows changes in the EHT length during mechanical testing. With the current experimental system, human EHTs have been used to investigate systolic and diastolic function, response to drugs, and the effects of cardiomyopathy mutations (6–11).
However, human EHTs have important limitations as a model system for human myocardium. EHT contains fewer cell types than human myocardium. Human iPSC-CMs are structurally and functionally immature relative to normal adult cardiomyocytes (11). Thus, signaling and metabolic pathways may differ from adult cells. Consistent with an immature phenotype, we found EHT force development (≈1 mN/mm2) was similar to a previous report (7) but well below that of adult human myocardium (16 mN/mm2) (17). Likewise, the maximum capture rate (≈2 Hz) was consistent with a previous report (43), but below that of human myocardium (≈3 Hz). Finally, the increase of EHT force (≈3%) with increased stimulation frequency was consistent with a recent study of EHTs (11), but well below that of human myocardium (≈35%) (44). Given the relative immaturity of human iPSC-CMs and the EHTs formed from them, results from studies performed in EHTs should be considered together with human and animal data and not as the sole tool to uncover the mechanistic effects of therapies.
Study Limitations
EHTs were constructed from healthy iPSC-CMs. Therefore, the findings of the current study show only the functional impact of A61603 on nonfailing myocardium and cannot be directly extrapolated to failing myocardium. The stability of A61603 in solution is unknown, and any significant degradation of A61603 during the 2-day period between media exchanges might attenuate the apparent treatment effects. After 2 wk of A61603 treatment, a 24-h washout was used. However, a previous study suggested that treatment with high doses (up to 1 mM) of phenylephrine may require >1 day for agonist to be eliminated from cells (45). Thus, although the current study used a very low dose (10 nM) of A61603, it remains possible that some agonist was retained within EHTs after washout. Finally, it is unclear if A61603 treatment inhibited EHT maturation, or if postinotropic rundown obscured monitoring of EHT force maturation.
Conclusions
Chronic stimulation of α1A-ARs resulted in increased length-dependent activation (LDA) of human EHT compared with treatment with vehicle control. This result raises the possibility that chronic stimulation of the α1A-AR might be beneficial for increasing LDA in human myocardium and might be beneficial for treating human heart failure by restoring LDA and thereby increasing the effect of preload on cardiac output.
DATA AVAILABILITY
Data will be made available upon reasonable request.
GRANTS
This work was supported by Department of Veterans Affairs Merit Review Award I01BX000740 (to A.J.B.); National Heart, Lung, and Blood Institute Grants HL154624 (to A.J.B. and N.C.C.) and HL31113 (to P.C.S.), and Harold S. Geneen Charitable Trust Awards Program (to J.E.L.). RNA sequencing was carried out at the University of California Davis Genome Center DNA Technologies and Expression Analysis Core, supported by National Institutes of Health Shared Instrumentation Grant 1S10OD010786-01.
DISCLOSURES
Propria, LLC, has a financial interest in MyoPod products/services presented herein. S.G.C. is the founder of, holds equity in, and has received consulting fees from Propria, LLC. The experiments reported in the submitted manuscript were performed at Propria LLC. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
P.C.S., S.G.C., and A.J.B. conceived and designed research; C.R. and E.C.-T. performed experiments; C.R., J.E.L., E.C.-T., and O.C.D.L.C. analyzed data; C.R., J.E.L., E.C.-T., O.C.D.L.C., N.C.C., P.C.S., and A.J.B. interpreted results of experiments; J.E.L. and A.J.B. prepared figures; A.J.B. drafted manuscript; J.E.L., O.C.D.L.C., N.C.C., P.C.S., S.G.C., and A.J.B. edited and revised manuscript; C.R., J.E.L., E.C.-T., O.C.D.L.C., N.C.C., P.C.S., S.G.C., and A.J.B. approved final version of manuscript.
REFERENCES
- 1. Cowley PM, Wang G, Swigart PM, Raghunathan A, Reddy N, Dulam P, Lovett DH, Simpson PC, Baker AJ. Reversal of right ventricular failure by chronic alpha1A-subtype adrenergic agonist therapy. Am J Physiol Heart Circ Physiol 316: H224–H232, 2019. doi: 10.1152/ajpheart.00507.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cowley PM, Wang G, Joshi S, Swigart PM, Lovett DH, Simpson PC, Baker AJ. alpha1A-Subtype adrenergic agonist therapy for the failing right ventricle. Am J Physiol Heart Circ Physiol 313: H1109–H1118, 2017. doi: 10.1152/ajpheart.00153.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Montgomery MD, Chan T, Swigart PM, Myagmar BE, Dash R, Simpson PC. An alpha-1A adrenergic receptor agonist prevents acute doxorubicin cardiomyopathy in male mice. PLoS One 12: e0168409, 2017. doi: 10.1371/journal.pone.0168409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beak J, Huang W, Parker JS, Hicks ST, Patterson C, Simpson PC, Ma A, Jin J, Jensen BC. An oral selective alpha-1A adrenergic receptor agonist prevents doxorubicin cardiotoxicity. JACC Basic Transl Sci 2: 39–53, 2017. doi: 10.1016/j.jacbts.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang J, Simpson PC, Jensen BC. Cardiac alpha1A-adrenergic receptors: emerging protective roles in cardiovascular diseases. Am J Physiol Heart Circ Physiol 320: H725–H733, 2021. doi: 10.1152/ajpheart.00621.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ng R, Sewanan LR, Brill AL, Stankey P, Li X, Qyang Y, Ehrlich BE, Campbell SG. Contractile work directly modulates mitochondrial protein levels in human engineered heart tissues. Am J Physiol Heart Circ Physiol 318: H1516–H1524, 2020. doi: 10.1152/ajpheart.00055.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schwan J, Kwaczala AT, Ryan TJ, Bartulos O, Ren Y, Sewanan LR, Morris AH, Jacoby DL, Qyang Y, Campbell SG. Anisotropic engineered heart tissue made from laser-cut decellularized myocardium. Sci Rep 6: 32068, 2016. doi: 10.1038/srep32068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sewanan LR, Park J, Rynkiewicz MJ, Racca AW, Papoutsidakis N, Schwan J, Jacoby DL, Moore JR, Lehman W, Qyang Y, Campbell SG. Loss of crossbridge inhibition drives pathological cardiac hypertrophy in patients harboring the TPM1 E192K mutation. J Gen Physiol 153: e202012640, 2021. doi: 10.1085/jgp.202012640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sewanan LR, Shen S, Campbell SG. Mavacamten preserves length-dependent contractility and improves diastolic function in human engineered heart tissue. Am J Physiol Heart Circ Physiol 320: H1112–H1123, 2021. doi: 10.1152/ajpheart.00325.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shen S, Sewanan LR, Jacoby DL, Campbell SG. Danicamtiv enhances systolic function and frank-starling behavior at minimal diastolic cost in engineered human myocardium. J Am Heart Assoc 10: e020860, 2021. doi: 10.1161/JAHA.121.020860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Lange WJ, Farrell ET, Kreitzer CR, Jacobs DR, Lang D, Glukhov AV, Ralphe JC. Human iPSC-engineered cardiac tissue platform faithfully models important cardiac physiology. Am J Physiol Heart Circ Physiol 320: H1670–H1686, 2021. doi: 10.1152/ajpheart.00941.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Tombe PP, Mateja RD, Tachampa K, Ait Mou Y, Farman GP, Irving TC. Myofilament length dependent activation. J Mol Cell Cardiol 48: 851–858, 2010. doi: 10.1016/j.yjmcc.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. ter Keurs HE. Heart failure and Starling's Law of the heart. Can J Cardiol 12: 1047–1057, 1996. [PubMed] [Google Scholar]
- 14. McDonald KS, Hanft LM, Robinett JC, Guglin M, Campbell KS. Regulation of myofilament contractile function in human donor and failing hearts. Front Physiol 11: 468, 2020. doi: 10.3389/fphys.2020.00468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thomas RC, Singh A, Cowley P, Myagmar BE, Montgomery MD, Swigart PM, De Marco T, Baker AJ, Simpson PC. A Myocardial slice culture model reveals alpha-1A-adrenergic receptor signaling in the human heart. JACC Basic Transl Sci 1: 155–167, 2016. doi: 10.1016/j.jacbts.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Karsten AJ, Eckert RE. Involvement of signal transducing GTP-binding proteins in renal artery alpha 1-adrenoceptor mediated smooth muscle contraction. BJU Int 93: 622–625, 2004. doi: 10.1111/j.1464-410x.2003.04685.x. [DOI] [PubMed] [Google Scholar]
- 17. Janssen PML, Canan BD, Kilic A, Whitson BA, Baker AJ. Human myocardium has a robust alpha1A-subtype adrenergic receptor inotropic response. J Cardiovasc Pharmacol 72: 136–142, 2018. doi: 10.1097/FJC.0000000000000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34: 525–527, 2016. [Erratum in Nat Biotechnol 34: 888, 2016]. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 19. Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen HC, Kitts PA, , et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res 27: 849–864, 2017. doi: 10.1101/gr.213611.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pimentel H, Bray NL, Puente S, Melsted P, Pachter L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods 14: 687–690, 2017. doi: 10.1038/nmeth.4324. [DOI] [PubMed] [Google Scholar]
- 21. Pimentel H, McGee W. Sleuth: Tools for Investigating RNA-Seq. 2022.
- 22.R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2021. https://www.r-project.org. [Google Scholar]
- 23. Lancaster HO. The combination of probabilities - an application of orthonormal functions. Aust J Stat 3: 20–33, 1961. doi: 10.1111/j.1467-842X.1961.tb00058.x. [DOI] [Google Scholar]
- 24. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28: 27–30, 2000. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Myagmar BE, Flynn JM, Cowley PM, Swigart PM, Montgomery MD, Thai K, Nair D, Gupta R, Deng DX, Hosoda C, Melov S, Baker AJ, Simpson PC. Adrenergic receptors in individual ventricular myocytes: the beta-1 and alpha-1B are in all cells, the alpha-1A is in a subpopulation, and the beta-2 and beta-3 are mostly absent. Circ Res 120: 1103–1115, 2017. doi: 10.1161/CIRCRESAHA.117.310520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jensen B, Swigart PM, Laden M, DeMarco T, Hoopes C, Simpson PC. The alpha-1D Is the predominant alpha-1-adrenergic receptor subtype in human epicardial coronary arteries. J Am Coll Cardiol 54: 1137–1145, 2009. doi: 10.1016/j.jacc.2009.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Turnbull L, McCloskey DT, O'Connell TD, Simpson PC, Baker AJ. Alpha 1-adrenergic receptor responses in alpha 1AB-AR knockout mouse hearts suggest the presence of alpha 1D-AR. Am J Physiol Heart Circ Physiol 284: H1104–H1109, 2003. doi: 10.1152/ajpheart.00441.2002. [DOI] [PubMed] [Google Scholar]
- 28. Li OY, Wang GY, Swigart PM, Marzban B, Simpson PC, Chesler NC, Beard DA, Baker AJ. Reversal of mitochondrial dysfunction and right ventricular failure after chronic treatment with alpha-1A-adrenergic agonist. Biophys J 121: 398A, 2022. doi: 10.1016/j.bpj.2021.11.787. [DOI] [Google Scholar]
- 29. Sequeira V, van der Velden J. The Frank-Starling Law: a jigsaw of titin proportions. Biophys Rev 9: 259–267, 2017. doi: 10.1007/s12551-017-0272-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Campbell KS, Janssen PML, Campbell SG. Force-dependent recruitment from the myosin off state contributes to length-dependent activation. Biophys J 115: 543–553, 2018. doi: 10.1016/j.bpj.2018.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sequeira V, Wijnker PJ, Nijenkamp LL, Kuster DW, Najafi A, Witjas-Paalberends ER, Regan JA, Boontje N, Ten Cate FJ, Germans T, Carrier L, Sadayappan S, van Slegtenhorst MA, Zaremba R, Foster DB, Murphy AM, Poggesi C, Dos Remedios C, Stienen GJ, Ho CY, Michels M, van der Velden J. Perturbed length-dependent activation in human hypertrophic cardiomyopathy with missense sarcomeric gene mutations. Circ Res 112: 1491–1505, 2013. [Erratum in Circ Res 113: e87, 2013]. doi: 10.1161/CIRCRESAHA.111.300436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo Y, Pu WT. Cardiomyocyte maturation: new phase in development. Circ Res 126: 1086–1106, 2020. doi: 10.1161/CIRCRESAHA.119.315862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ronaldson-Bouchard K, Ma SP, Yeager K, Chen T, Song L, Sirabella D, Morikawa K, Teles D, Yazawa M, Vunjak-Novakovic G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556: 239–243, 2018. doi: 10.1038/s41586-018-0016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shen S, Sewanan LR, Shao S, Halder SS, Stankey P, Li X, Campbell SG. Physiological calcium combined with electrical pacing accelerates maturation of human engineered heart tissue. Stem Cell Reports 17: 2037–2049, 2022. doi: 10.1016/j.stemcr.2022.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang Y, Wright CD, Merkwan CL, Baye NL, Liang Q, Simpson PC, O'Connell TD. An alpha1A-adrenergic-extracellular signal-regulated kinase survival signaling pathway in cardiac myocytes. Circulation 115: 763–772, 2007. doi: 10.1161/CIRCULATIONAHA.106.664862. [DOI] [PubMed] [Google Scholar]
- 36. Rohde S, Sabri A, Kamasamudran R, Steinberg SF. The alpha(1)-adrenoceptor subtype- and protein kinase C isoform-dependence of Norepinephrine's actions in cardiomyocytes. J Mol Cell Cardiol 32: 1193–1209, 2000. doi: 10.1006/jmcc.2000.1153. [DOI] [PubMed] [Google Scholar]
- 37. Xiao L, Pimental DR, Amin JK, Singh K, Sawyer DB, Colucci WS. MEK1/2-ERK1/2 mediates alpha1-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J Mol Cell Cardiol 33: 779–787, 2001. doi: 10.1006/jmcc.2001.1348. [DOI] [PubMed] [Google Scholar]
- 38. Vettel C, Wittig K, Vogt A, Wuertz CM, El-Armouche A, Lutz S, Wieland T. A novel player in cellular hypertrophy: Gibetagamma/PI3K-dependent activation of the RacGEF TIAM-1 is required for alpha(1)-adrenoceptor induced hypertrophy in neonatal rat cardiomyocytes. J Mol Cell Cardiol 53: 165–175, 2012. doi: 10.1016/j.yjmcc.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 39. Mohl MC, Iismaa SE, Xiao XH, Friedrich O, Wagner S, Nikolova-Krstevski V, Wu J, Yu ZY, Feneley M, Fatkin D, Allen DG, Graham RM. Regulation of murine cardiac contractility by activation of alpha(1A)-adrenergic receptor-operated Ca(2+) entry. Cardiovasc Res 91: 310–319, 2011. doi: 10.1093/cvr/cvr081. [DOI] [PubMed] [Google Scholar]
- 40. Shi T, Duan ZH, Papay R, Pluskota E, Gaivin RJ, de la Motte CA, Plow EF, Perez DM. Novel alpha1-adrenergic receptor signaling pathways: secreted factors and interactions with the extracellular matrix. Mol Pharmacol 70: 129–142, 2006. doi: 10.1124/mol.105.020735. [DOI] [PubMed] [Google Scholar]
- 41. Jin ZQ, Zhou HZ, Zhu P, Honbo N, Mochly-Rosen D, Messing RO, Goetzl EJ, Karliner JS, Gray MO. Cardioprotection mediated by sphingosine-1-phosphate and ganglioside GM-1 in wild-type and PKC epsilon knockout mouse hearts. Am J Physiol Heart Circ Physiol 282: H1970–H1977, 2002. doi: 10.1152/ajpheart.01029.2001. [DOI] [PubMed] [Google Scholar]
- 42. Tzatzalos E, Abilez OJ, Shukla P, Wu JC. Engineered heart tissues and induced pluripotent stem cells: macro- and microstructures for disease modeling, drug screening, and translational studies. Adv Drug Deliv Rev 96: 234–244, 2016. doi: 10.1016/j.addr.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cashman TJ, Josowitz R, Johnson BV, Gelb BD, Costa KD. Human engineered cardiac tissues created using induced pluripotent stem cells reveal functional characteristics of BRAF-mediated hypertrophic cardiomyopathy. PLoS One 11: e0146697, 2016. doi: 10.1371/journal.pone.0146697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mashali MA, Saad NS, Canan BD, Elnakish MT, Milani-Nejad N, Chung JH, Schultz EJ, Kiduko SA, Huang AW, Hare AN, Peczkowski KK, Fazlollahi F, Martin BL, Murray JD, Campbell CM, Kilic A, Whitson BA, Mokadam NA, Mohler PJ, Janssen PML. Impact of etiology on force and kinetics of left ventricular end-stage failing human myocardium. J Mol Cell Cardiol 156: 7–19, 2021. doi: 10.1016/j.yjmcc.2021.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ryall KA, Saucerman JJ. Automated imaging reveals a concentration dependent delay in reversibility of cardiac myocyte hypertrophy. J Mol Cell Cardiol 53: 282–290, 2012. doi: 10.1016/j.yjmcc.2012.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available upon reasonable request.