

Abstract
Transcriptional heterogeneity among malignant cells of a tumor has been studied in individual cancer types and shown to be organized into cancer cell states; however, it remains unclear to what extent these states span tumor types, constituting general features of cancer. Here, we perform a pan-cancer single-cell RNA-Seq analysis across 15 cancer types and identify a catalog of gene modules whose expression defines recurrent cancer cell states including ‘stress’, ‘interferon response’, ‘epithelial-mesenchymal transition’, ‘metal response’, ‘basal’ and ‘ciliated’. Spatial transcriptomic analysis linked the interferon response in cancer cells to T cells and macrophages in the tumor microenvironment. Using mouse models, we further found that induction of the interferon response module varies by tumor location and is diminished upon elimination of lymphocytes. Our work provides a framework for studying how cancer cell states interact with the tumor microenvironment to form organized systems capable of immune evasion, drug resistance, and metastasis.
Introduction
Transcriptional heterogeneity in cancer is increasingly recognized as a driver of tumor progression, metastasis and treatment failure1–5. Single-cell RNA-Sequencing (scRNA-Seq) has enabled the unbiased transcriptomic profiling of individual tumor cells and has revealed a striking amount of heterogeneity among malignant cells of the same tumor6–12. Data from glioblastoma6,7, oligodendroglioma13, astrocytoma14, head and neck cancer10 and melanoma15,16 among others, indicates that, within a tumor, cancer cells are heterogeneous in their degree of differentiation, ranging from stem- or progenitor-like to fully differentiated. These studies performed in a variety of cancer types have also shown the existence of cancer cell states related to stress response, interferon response, and hypoxia6,7,9,10,17. Causative links have also been drawn between specific cell types of the tumor microenvironment (TME) and cancer cell states10,18,19. In one study, a population of partial epithelial-mesenchymal transition (pEMT) cancer cells at the leading edge of head and neck tumors was shown to interact with cancer-associated fibroblasts and mediate invasion10. These works motivated us to systematically study the relationships between cancer cell states and non-malignant cell types of the TME.
Results
Recurring gene modules across diverse cancer types
We collected 19 fresh primary untreated patient tumors spanning 9 cancer types immediately after surgery (Supplementary Table 1). We dissociated each tumor to obtain a single-cell suspension, and processed for scRNA-Seq without prior sorting to ensure an unbiased assessment of the tumor cellular composition. Our tumor collection included 9 cancer types: carcinoma of the ovary (OVCA), endometrium (UCEC), breast (BRCA), prostate (PRAD), kidney (KIRC), liver (LIHC), colon (COAD) and pancreas (PDAC), as well as one non-epithelial cancer type, gastrointestinal stromal tumor (GIST) (Fig. 1a). We first identified the malignant cells in our dataset by analyzing the transcriptomes using a combination of marker genes, singleR annotation20, and inferred copy number variation6 (Fig. 1b–c, Extended Data Fig. 1–2, see Methods). To extend our dataset, we also performed this analysis in tumors from prior publications, including additional PDAC21 and LIHC22, as well as additional tumor types: cholangiocarcinoma (CHCA)23, lung adenocarcinoma (LUAD)24, head and neck squamous cell carcinoma (HNSC)10, skin squamous cell carcinoma (SKSC)25, glioblastoma multiforme (GBM)7 and oligodendroglioma (OGD)13, resulting in a total of 19,942 malignant cells from 62 untreated primary tumors spanning 15 cancer types (Fig. 1a, Supplementary Table 2).
Figure 1: A catalog of recurrent cancer gene modules.
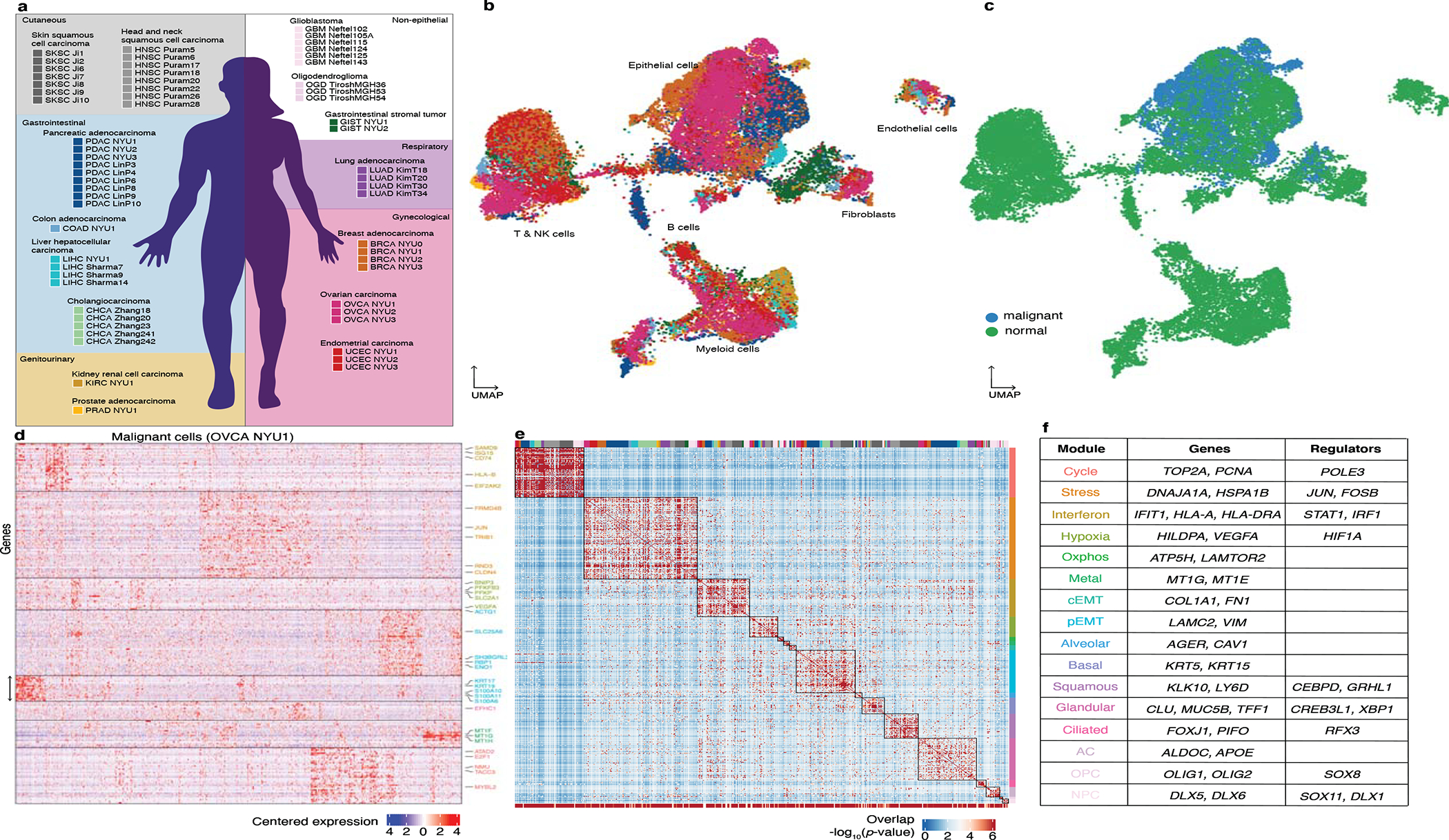
a. Schematic indicating the tumors collected in this study and tumors included from previous reports for joint analysis. The background color indicates the organ system of origin: cutaneous (grey), gastrointestinal (blue), gynecological (red) and genitourinary (yellow). White background indicates non-epithelial tumors.
b-c. UMAP embedding of cells from the 19 tumors collected in this study spanning a total of 9 cancer types, colored as in a (b) and colored by annotation as malignant or non-malignant (c).
d. Heatmap of expression levels for 241 genes in the malignant cells of the ovarian tumor OVCA NYU1. Genes are ordered by their module membership (horizontal lines) and the colors of the indicated genes correspond to their consensus module annotation.
e. Heatmap of the significance of the overlap between individual tumor modules (hypergeometric test p-value). The bottom bar indicates the significance of the overlap with consensus modules (hypergeometric test p-value). The top bar indicates the identity of the tumor samples, colored as in a. Extended Data Figure 3h indicates the significance of the overlap of each consensus module with each tumor specific gene module.
f. Table of consensus modules, selected genes and putative regulators identified using SCENIC (regulators identified in at least 2 tumors are shown), colored as in d. See also Extended Data Figure 3h and Supplementary Table 4.
Our approach to defining cell states involved first cataloging the underlying gene modules, following recent work that has identified gene modules as the defining features of cell states6,7,10,13. This is a flexible approach since it allows for cells expressing combinations of modules, and thus for the complexity of possible cell states. We analyzed the malignant cells using non-negative matrix factorization (NMF) to identify gene modules as sets of co-expressed genes (Fig. 1d, see Methods). Our method detects groups of genes that are co-expressed within the sample, i.e. that are expressed coherently in a subset of cells. To search for recurring gene modules across tumors, we then compared the gene composition of the identified modules (Fig. 1e, see Methods). By thus performing the integration at the level of gene modules rather than expression matrices, the impact of technical variation across the samples and studies is reduced. Despite the independent identification of these modules in a variety of cancer types – thereby not presuming recurrence – we found that modules obtained in different tumors overlap significantly (Fig. 1e, top bar).
The recurrence of the gene modules enabled us to construct a catalog of 16 consensus modules (henceforth ‘modules’; Fig. 1f, Extended Data Fig. 3f, Supplemental Table 3, see Methods), ranging from 9 to 297 genes. To establish whether a module is significantly present in the population of cancer cells in each tumor, we used the gene set overdispersion metric26 (Extended Data Fig. 4a, see Methods). Consistent with Figure 1d, some modules were enriched in specific organ systems (such as the brain or gynecological organs), while others spanned several organ systems and histologies. We also tested for the overdispersion of the modules in independent datasets representing normal epithelia from the fallopian tube27, breast28 and liver29, and found that most modules are present in normal tissues but with a lower dispersion (Extended Data Fig. 4b). This suggests that the modules are not specific to cancer, but rather are co-opted from existing ones and expressed more heterogeneously. Studying the gene composition and cancer type-specificity of the modules, we distinguished modules related to cellular processes (Extended Data Fig. 3a) from those related to cell identity (Extended Data Fig. 3b).
As expected, we recovered a highly recurrent module consisting of cell cycle genes (e.g., TOP2A, PCNA), capturing the subset of cancer cells in any tumor that is cycling at the time of sampling. Another process which recurred across tumor types was the stress response (e.g., JUN, FOS, HSPA1B), which has been previously described12,30,31 and was proposed by us to be a consistent feature of tumorigenesis6. We also present spatial transcriptomics data below (Fig. 4) that provides additional support for the in vivo existence of this state among cancer cells in the absence of dissociation for scRNA-Seq.
Figure 4. Mapping cancer cell states and their interactions with the TME.

a. Scoring ST spots for module expression. Top: Schematic of spots colored by their annotation as ‘Malignant’, ‘Both’ and ‘Normal’; ‘Malignant’ spots are scored for expression of each module. Bottom: BRCA NYU2 sample with ‘Malignant’ spots colored by their expression of the indicated modules. Scale bar represents 1mm.
b. Characterizing spots by cell type neighborhood. Top: Schematic with grey spots indicating the presence of the cell type of interest, orange indicating ‘Malignant’ spots, and dashed lines indicating their surrounding spots. Bottom: BRCA NYU2 sample with ‘Malignant’ spots colored by neighborhood macrophage score. Scale bar represents 1mm.
c. Characterizing ST spots by cell type proximity. Top: Schematic colored as in b with arrows indicating the distance to the closest spot containing the cell type of interest. Bottom: BRCA NYU2 sample with ‘Malignant’ spots colored by macrophage proximity score. Scale bar represents 1mm.
d. Heatmap of correlations (Pearson) between module scores and cell type neighborhood for BRCA NYU2 ‘Malignant’ spots. Boxes indicate clusters.
e-f. Plot of the relationship between the interferon response module score and macrophage neighborhood score (e) or proximity score (f) in the BRCA NYU2 ‘Malignant’ spots. Line and grey area represent linear regression and standard error.
g. Boxplot of correlation scores (±log10(p-value), Pearson) between module scores and macrophage neighborhood scores across 10 samples, colored as in Figure 1a. For each boxplot, the line indicates the median, the box indicates the 1st and 3rd quartile, the whiskers indicate the minimum and maximum values. Positive scores correspond to positive correlations. Dashed lines indicate p-value=0.05.
h-i. Map of correlations between module expression and cell type neighborhoods (h) and cell type proximity (i). Color represents the median ±log10(p-value) of the correlation (Pearson). Point size represents the fraction of samples in which the correlation is of the same sign as the median correlation. Black outlines indicate relationships with (1) median ±log10(p-value) greater than 0.75 and (2) fraction of samples greater than 0.5 - using both the neighborhood and proximity metrics.
An interferon response module12,32–34 was widely detected, containing both interferon-stimulated genes - such as STAT1 and IFIT1 - and components of antigen presentation - MHCI and MHCII, a well-characterized effect of interferon35–37. While MHC II expression is classically associated with professional antigen-presenting cells, this pathway has also been shown to be expressed in normal epithelial cells and in cancer cells38,39.
Two modules relating to metabolic processes were also found across a range of cancer types: a hypoxia module (e.g., VEGF, ADM)6 and an oxidative phosphorylation module (e.g., ATP5H, LAMTOR2)9. An additional gene module of metallothionein genes – which we refer to here as a metal-response module – may have a role in proliferation and drug resistance in several cancer types40–43.
Another set of modules correspond to cell identity, and appear to be related to the tissue and cell of origin (Extended Data Fig. 3b). The majority of the tumors profiled were of epithelial origin, and accordingly we identified modules overlapping with known epithelial cell type markers: an alveolar module (e.g., AGER, CAV1) which was particularly present as expected in LUAD44–46, as well as basal (e.g., KRT5 and KRT15), squamous (e.g., KLK10, LY6D), and glandular (e.g., CLU, MUC5B) cell modules (Extended Data Fig. 3f–g). A module composed of cilium-related genes (e.g., FOXJ1, PIFO) was present in endometrioid OVCA and UCEC (Supplementary Table 1), LUAD and GBM. The presence of the module in normal fallopian tube and lung epithelial tissues (Extended Data Fig. 4b–c)47,48 suggests that its differential expression in cancer mirrors the heterogeneity of the tissue of origin.
Two of the modules spanning multiple cancer types were related to epithelial-mesenchymal transition (EMT): a complete mesenchymal module (cEMT) (e.g., COL1A1, FN1) and a partial mesenchymal module (pEMT) (e.g., LAMC2, VIM) lacking canonical mesenchymal markers such as collagen genes10. The pEMT module has been recently characterized in HNSC10 and SKSC25 (Extended Data Fig. 3c–e), but is also found in GBM7, suggesting that cells from different lineages converge upon this identity in cancer. We detected the presence of the cEMT module in a minority of samples, but a range of cancer types: mainly PDAC, CHCA, LUAD, and GBM. These modules may represent two pathways converging on the phenotypic properties conferred by mesenchymal differentiation including migration and drug resistance11,49–51.
Finally, we identified three neurological cancer-specific modules that were analogous to those described by Tirosh et al.13 and Neftel et al.7 (Extended Data Fig. 3c): the astrocyte (AC)-like (e.g., APOE, ALDOC), oligodendrocyte progenitor cell (OPC)-like (e.g., OLIG1, OLIG2), and neural progenitor cell (NPC)-like (e.g., DLX1, DLX5) modules.
Defining cancer cell states by gene module expression
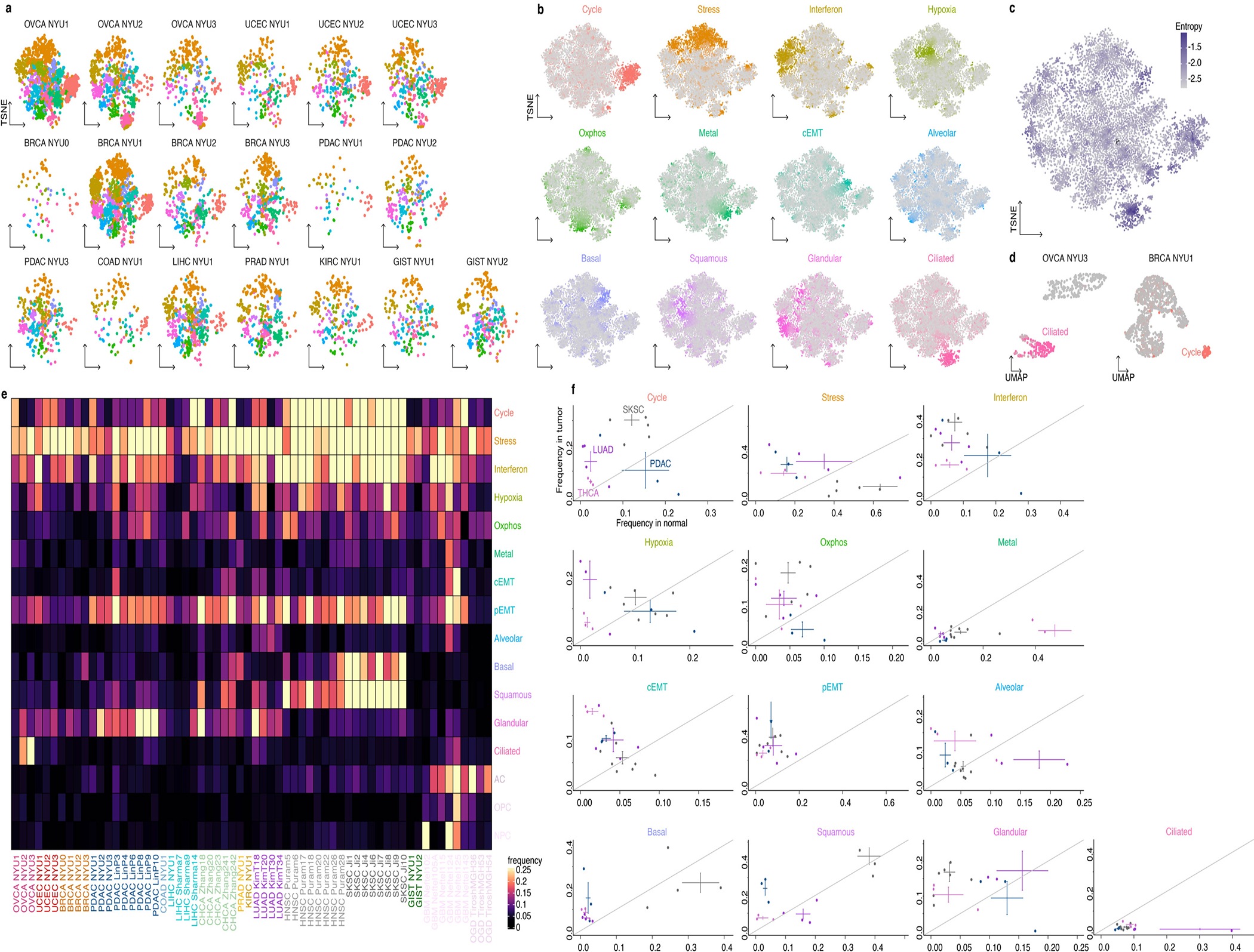
We next sought to understand how the gene modules are assembled at the level of individual cells, and scored each malignant cell for the expression of each of the modules (Fig. 2a, see Methods). Since the modules recur across cancer types, we reasoned that the cell module scores could serve as natural axes across which to compare cancer cells of different patients. Figure 2b represents a dimensionality reduction performed on the module scores of cells from 19 different tumors collected at NYU (see Methods). Most notably, the cancer cells in this space do not group by patient or cancer type, but rather by their most highly expressed module (Fig. 2b,c, Extended Data Fig. 5a–c). This is in sharp contrast with the finding that, in gene expression space, cancer cells cluster by patient30. We detected some co-expression between certain modules: for example pEMT is co-expressed with stress and interferon-response (Fig. 2a,d, Extended Data Fig. 5b). Together with the fact that cells do not form distinct clusters, this supports the view that cancer cell states do not generally represent discrete entities.
Figure 2: Expression of gene modules underlies cancer cell states.
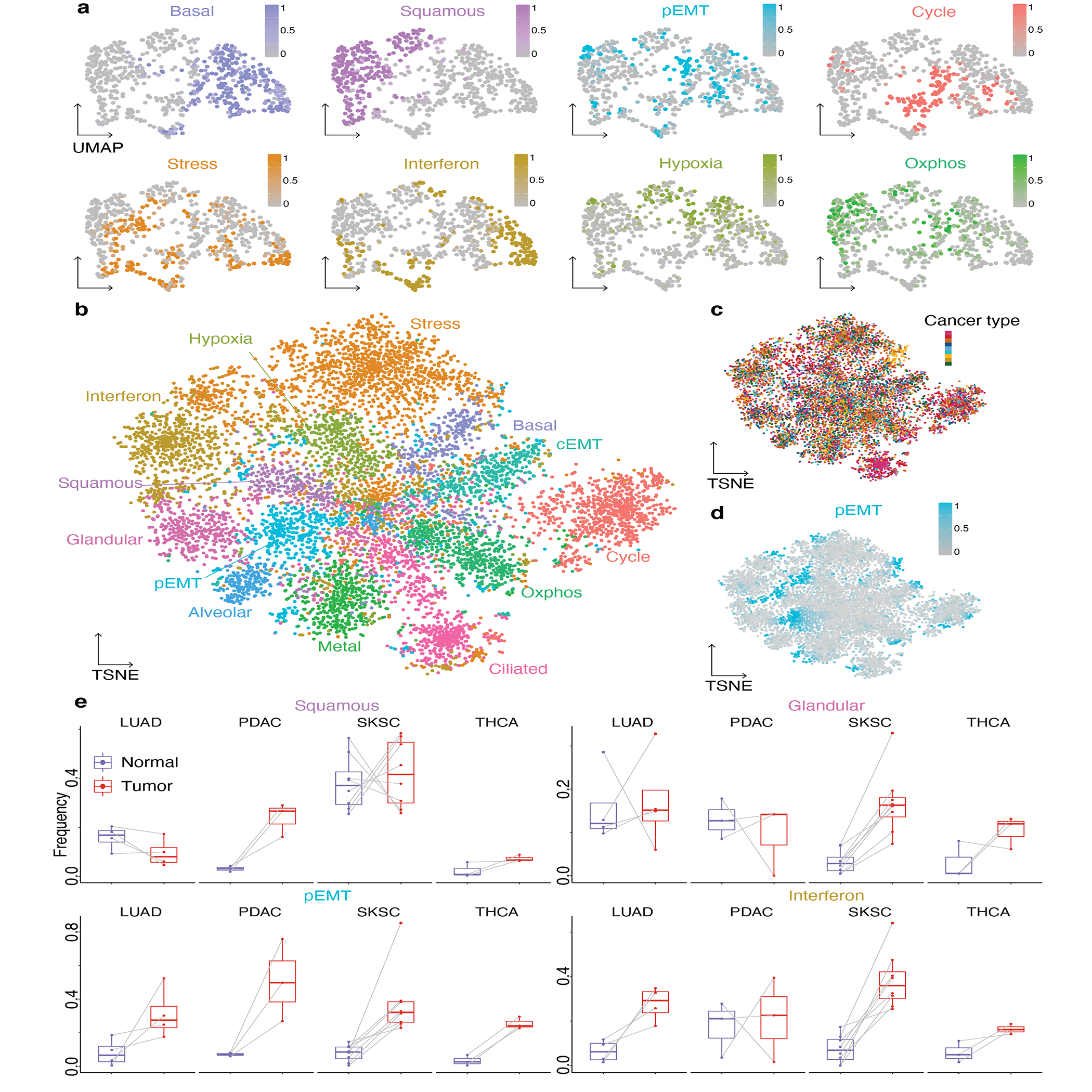
a. Gene expression UMAP embedding of malignant cells of SKSC Ji1, colored by module score for the 8 indicated modules.
b-d. Module score TSNE embedding of the 8613 malignant cells of all 19 tumors, colored by the most high scoring module (b), by cancer type as in Figure 1a (c) and by pEMT module score (d).
e. Boxplots of the expression frequency (fraction of cells with module score greater than 0.5) of the squamous, glandular, pEMT and interferon response modules in paired normal and tumor samples. For each boxplot, the line indicates the median, the box indicates the 1st and 3rd quartile, the whiskers indicate the minimum and maximum values. Gray lines connect data points from the same patient. LUAD: n=4; PDAC: n=3, SKSC: n=8; THCA: n=3.
Since certain modules are also present in non-cancer samples (Extended Data Fig. 4b–c), we asked whether the fraction of cells expressing each module varies between malignant and non-malignant epithelia across paired samples. The pEMT module was expressed at higher frequencies in all four cancer types relative to normal (Fig. 2e), in line with the common occurrence of EMT in tumor progression52. The interferon response module exhibited increased expression frequency in LUAD and SKSC relative to normal (Fig. 2e). Normal lung and skin have squamous components, and consistently we observed no difference in squamous expression in the tumor samples (Fig. 2e). In contrast, the squamous module was induced in the PDAC relative to normal ductal cells, indicating squamous differentiation in the malignant cell population. Several classifications of PDAC have been proposed based on bulk transcriptomics53–55, including a distinction between classical (high expression of glandular genes, including TFF1 and CEACAM6) and basal subtypes (high expression of squamous and basal genes, including LY6D and KRT15). Although squamous cell pancreatic cancer is rare56,57, the increase in squamous expression frequency in PDAC suggests that partial metaplasia towards a squamous program is common. Analogously, expression of the glandular module was unchanged in LUAD and PDAC relative to their normal counterparts, but increased in SKSC relative to normal skin. This pattern highlights both retention of modules expressed by the tissue of origin and redeployment of modules from other cell types.
Cancer cell state analysis of tumor cellular neighborhoods
To analyze the organization and interactions between cancer cell states and cells of the TME, we turned to sequencing-based spatial transcriptomics (ST)58. Since this technology is not at single-cell resolution, we performed non-negative linear least squares (NNLS) regression using the average expression profiles of cell types from the paired single cell data, and annotated spots as ‘Malignant’, containing only malignant cells, ‘Normal’, containing only immune and stromal cells, and ‘Both’, containing a combination (Fig. 3a, Extended Data Fig. 6–7, Supplementary Fig. 3, Supplementary File 2, see Methods).
Figure 3: Spatial organization of the tumor microenvironment.
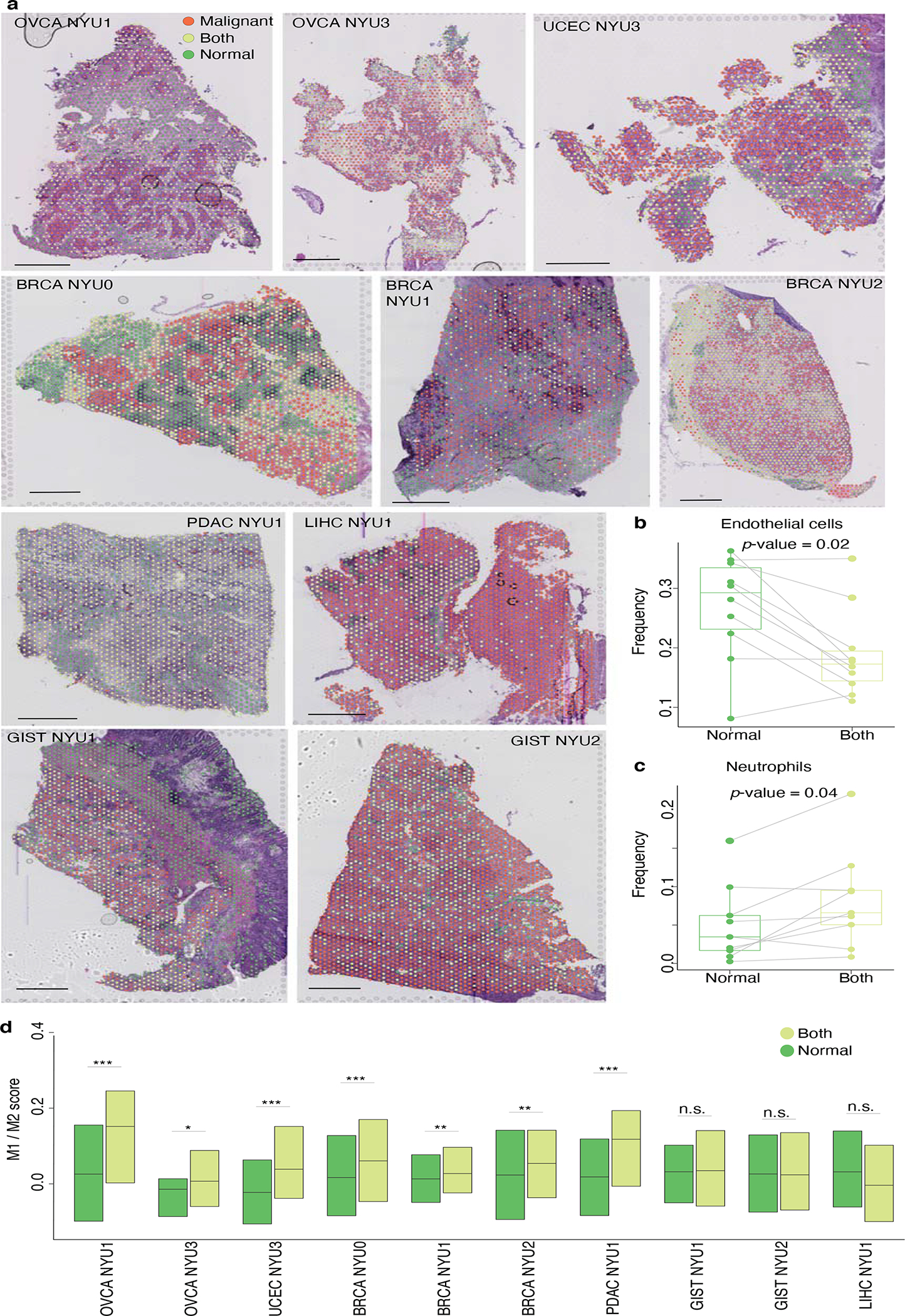
a. H&E images for the 10 indicated patient tumors overlaid with the locations of the spatial transcriptomic spots colored according to their annotation as ‘Malignant’, ‘Both’, or ‘Normal’. Bar plots indicate the fraction of non-malignant cell types in the ‘Normal’ and ‘Both’ spots for each sample. Scale bar represents 1mm.
b-d. Boxplots of the fractions of endothelial cells (b), neutrophils (c) and M1/M2 score (d) in ‘Normal’ and ‘Both’ spots for each sample (n=10; *, p-value<0.05; ***, p-value<0.001; two-sided Wilcoxon test). For each boxplot, the line indicates the median, the box indicates the 1st and 3rd quartile, the whiskers indicate the minimum and maximum values.
The presence of ‘Normal’ and ‘Both’ spots in each sample enabled us to ask how the cell type composition of the tissue changes in the presence of malignant cells. We found that the fraction of endothelial cells was lower in the spots also containing malignant cells (Fig. 3b), suggesting an incomplete vascularization of the tumor59,60. Conversely, neutrophils are present in higher numbers in the ‘Both’ spots (Fig. 3c). Tumor-associated macrophages are broadly defined as M1 - anti-tumor/pro-inflammatory - and M2 - pro-tumor/anti-inflammatory61–63. For each spot containing a macrophage, we scored its M1/M2 polarity (Fig. 3d, Supplementary Fig. 4, see Methods). In the six gynecological samples - ovarian, endometrial and breast cancer - we found that ‘Both’ spots are higher in M1/M2 than ‘Normal’ spots, suggesting a robust anti-tumor macrophage activity in proximity to cancer cells.
We then sought to query the composition of cellular neighborhoods in terms of cancer cell states. For this, we mapped cancer cell states within each ST sample, scoring each ‘Malignant’ spot for its expression of each module (Fig. 4a, see Methods). To characterize the cell type composition surrounding each ‘Malignant’ spot, we calculated, for each cell type, two score indices meant to capture their microenvironment. We defined the ‘neighborhood score’ as the fraction of surrounding spots containing that cell type (Fig. 4b, see Methods). This score thus directly measures the cell type composition in the adjacent spots. The proximity score measures how close the spot of interest is to each cell type, and is calculated as the inverse of the shortest distance to a cell of that type (Fig. 4c, see Methods).
Correlating the module scores and cell type neighborhood profiles across the ‘Malignant’ spots of each tumor revealed how cancer cell states and cell types of the TME co-localize to form ‘neighborhoods’ (Fig. 4d). For the BRCA NYU2 tumor shown in Fig. 4a–c, one grouping in the heatmap of relationships contained the interferon response module and macrophages. Studying this correlation more closely confirmed the significance of this positive relationship between the module score and the macrophage neighborhood score (Fig. 4e). A consistent relationship was also observed when computing the presence of macrophages using the proximity score (Fig. 4f). To explore this relationship across all samples, we calculated the correlation score (±log10(p-value)) of the macrophage neighborhood (Fig. 4g). The correlation with the interferon response was positive in all the samples, and significant for 8 of 10 samples.
Extending this analysis for all pairs of cell types and modules using both measures (Fig. 4h,i), we identified other consistent co-localizations of cell states with cell types of the TME. Cancer cells undergoing EMT were positively correlated with fibroblasts and endothelial cells, and negatively correlated with other malignant cells, consistent with the finding that they are enriched at the interface of the tumor (Extended Data Fig. 8) and interact with cancer-associated fibroblasts10. In addition to macrophages, the interferon response-expressing cancer cells co-localize with T cells (Fig. 4h,i), which we validated using CO-Detection by indEXing (CODEX)64 - a multiplexed protein staining assay - which enables spatial analysis at single-cell resolution (Extended Data Fig. 9). This suggests that at least a subset of cancer cell states interact with the TME, either being elicited by immune or stromal cells, or altering the cell type composition of their surroundings.
Modulation of the interferon response by the TME
In tumors, IFNα and IFNβ are secreted by cancer cells and DCs in response to DNA fragments activating the cGAS/STING pathway65–67, and result in T cell priming and antitumor activity33. IFNγ is mainly produced by adaptive immune cells upon activation34, and leads to up-regulation of MHC I genes, initially facilitating tumor rejection but ultimately leading to IFN-unresponsive tumors through immunoediting68. To further test the relationship between the interferon response and T cells in the TME, we used an established allograft mouse cancer model in which the TME can be readily perturbed (see Methods). We performed scRNA-Seq on four orthotopic pancreatic tumors to verify that gene modules could be recapitulated in the orthotopic model. Identifying gene modules using NMF as we did previously, we found that five were recapitulated in this system: cycling, stress response, interferon response, hypoxia, and glandular differentiation (Fig. 5a,b, Supplemental Table 4).
Figure 5: Cancer cell states in perturbed tumor microenvironments.
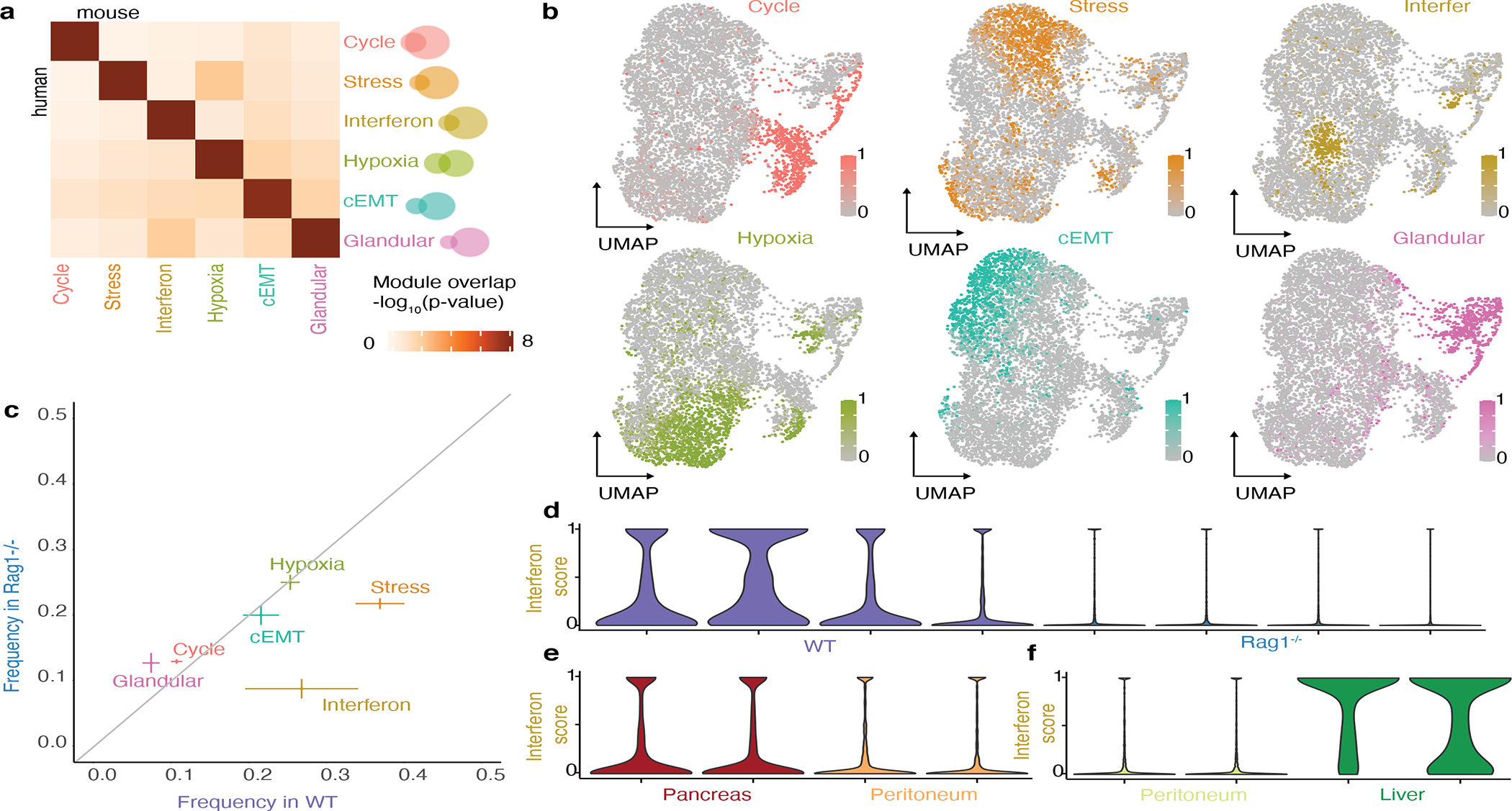
a. Heatmap of the significance of the overlap between modules obtained by NMF in an orthotopic model of pancreatic cancer and modules obtained in patient samples in Figure 1f (hypergeometric test p-value).
b. UMAP embedding of malignant cells from orthotopic pancreatic tumors, colored according to the expression score of the six modules shown in a.
c. Module expression frequencies in WT mice vs Rag1−/− mice (mean ± standard error obtained from 4 biological replicates per condition).
d-f. Violin plots of interferon module expression score in WT mice vs Rag1−/− mice (d), pancreas versus peritoneum (e), peritoneum versus liver (f).
In parallel, we collected scRNA-Seq data from four orthotopic tumors formed in Rag1−/− mice, which lack T and B cells. Analyzing the gene module expression in the malignant cells from these tumors we found that cycling, stress response, hypoxia and glandular differentiation were expressed at similar frequencies between the Rag1−/− and WT mice (Fig. 5c). In contrast, the interferon response module was expressed at lower frequencies in the tumors from the Rag1−/− mice (p<10−10, Kolmogorov-Smirnov test, Fig. 5d). Furthermore, all of the genes of the interferon response module were up-regulated in the interferon response-expressing cells relative to other cancer cells, suggesting that a coordinated response is maintained - albeit in fewer cells (Extended Data Fig. 10d). The MHC I genes of the interferon response module (B2m, H2-D1, H2-K1) have a lower overall expression in the Rag1−/− mice (although they remain relatively up-regulated in the interferon response-expressing cells), suggesting that lymphocyte depletion has an additional general effect on the expression of MHC I genes which is interferon response-independent. The remaining expression in the lymphocyte-depleted condition suggests other causes of interferon response in cancer cells, either cancer extrinsic, for example interferon secretion by NK cells, or cancer-intrinsic, consistent with reports of an interferon response module in vitro69. The frequency of interferon response cells also varied by tumor location (Fig. 5e,f). Notably, these findings do not discriminate between signaling mechanisms eliciting an interferon response and long term immunoediting leading to selection of the state within the tumor37,68.
Discussion
Cancer cell states cannot be defined as distinctly as cell types6,16,30. Our results indicate that this likely follows from the expression of combinations of modules: since these are not generally mutually exclusive (Fig. 2a, Supplementary Fig. 1), this leads to continuous variation rather than discrete clusters (Fig. 5b). Conversely, distinct states may be observed when the gene modules are mutually exclusive with others, as in the case of the cycling and cilium gene modules (Fig. 2b, Supplementary Fig. 1). Our analysis thus supports the notion that the basic underlying units of tumor transcriptional variability are the gene modules, whose combinatorial products define the cancer cell states.
Much of the heterogeneity observed across cancer cells appears to result from redeployment of modules typically expressed in other cellular and developmental contexts70. Indeed, our catalog of cancer gene modules includes general features of cell physiology (eg. cycling), specific processes and responses (eg. interferon response), and developmental programs (eg. basal). Relative to their normal counterparts, cancer cells exploit the existing gene modules, expressing them at different levels (Fig. 2e, Extended Data Fig. 4d) and more heterogeneously (Extended Data Fig. 4c).
The presence of an interferon response in cancer has been studied extensively, and attempts have been made to harness the response for therapy71. Here, we found that genes involved in interferon response are co-regulated and heterogeneously expressed across malignant cells of the tumors in all 15 cancer types studied here (Fig. 1), suggesting that the existence of this state is a necessary feature of tumorigenesis. Indeed, tumors lacking IFNγ receptors fail to develop in mouse models68. Paradoxically, however, in tumors containing both interferon responsive and unresponsive cells, the unresponsive cells increase in frequency68. Thus, the subset of cells expressing the interferon response module appears to support the growth of other cells within the tumor. This may be explained by the dual function of these genes - with MHCI and MHCII eliciting heightened immune detection of the cells, but CD274 (PDL1) leading to a generalized increase in immune tolerance71.
It remains unclear to what extent the heterogeneity among cancer cells results from heterogeneity in the signals they receive, or from intrinsic differences between the cells. Our observations of cancer states across a wide range of cancer types provide evidence that cancer cell states are not genetically defined, but rather represent cellular plasticity. In addition, in vitro and in vivo studies have shown that cancer cells exhibit a high degree of plasticity, and that populations seeded by a single state recover the same state proportions as the original tumor7,69. Thus, while the individual state identities are highly plastic, their overall distribution may be a stable property. Interestingly, in glioblastoma, tumors harboring different genetic drivers share the same set of states, but differ in the proportions of each state7. In this view, an early phase of tumorigenesis would generate the oncogenic background upon which later epigenetic changes would lead to heterogeneity among the malignant population72.
Several of the gene modules that we identify here may enable the hallmarks proposed by Hanahan and Weinberg73,74, raising the possibility that the cancer hallmarks do not need to be assembled by all individual cells. Rather, cell states may cooperate within the tumor ecosystem leading to higher fitness of the tumor as a whole75. For example, induction of angiogenesis or down-regulation of immune surveillance may be mediated by a subset of cancer cells to the benefit of the others. To understand these complex relationships, it is crucial to consider the physical constraints of the tumor, including signaling between neighboring cells, diffusion of oxygen and nutrients, and segregation into niches with distinct composition. The co-localization that we observed of interferon response-expressing cells with T cells and macrophages (Figs. 5,6) highlights that the functional role of cancer cell states may be understood by analyzing the tumor architecture.
Methods
Patient tumor scRNA-Seq:
Data collection and processing.
Tumors were collected post-operatively from patients who signed consent to use their biospecimen for research under IRB s16-00122. Each sample was washed in PBS and cut into 4–5-mm3 pieces, of which 2–3 were reserved for spatial transcriptomics (see below). The remainder was dissociated for scRNA-Seq using the Miltenyi human tumor dissociation kit according to manufacturer’s instructions. Red blood cell lysis was performed in ACK lysis buffer for 3 minutes. Cells were counted and viability was assessed by trypan blue on a hemocytometer. For samples with low viability (<50%), dead cells were removed using the Miltenyi dead cell separator. Single-cell encapsulation and library preparations were performed using the inDrop platform according to manufacturer’s instructions76. Libraries were sequenced on an Illumina NextSeq and reads aligned using the inDrop pipeline previously described77. To exclude cells with low quality transcriptomes from analysis, cells with fewer than 500 UMIs or more than 30% mitochondrial or ribosomal reads were filtered out. The Seurat v.4.0.3 single-cell transformation78,79 was used to normalize and center the data, and to identify variable genes.
Analysis of previously published data.
Published datasets were downloaded from GEO. For PDAC9, LIHC22, CHCA23, LUAD24, HNSC10, SKSC25, THCA80, and OGD13, raw counts were used and normalized as above. For GBM7, normalized data was used and centered, and variable genes were identified using the ‘vst’ method in the Seurat package78,79.
Cell type annotation and detection of malignant cells.
To annotate cell types and identify malignant cells, the following procedure was used.
Cell type identification.
Part 1: The Seurat package79 was used to select variable genes, reduce dimensionality, cluster the cells, and search for differentially expressed genes (using thresholds of p-value<0.01, percentage of cells expressing >10%, log fold-change >0.25, sorted by log fold-change). These genes were cross-referenced with the literature to identify immune (expressing PTPRC, CD19, CD4, CD8A, FOXP3, CD68, S100A8, MS4A2), stromal (expressing HBA1, PECAM1, COL4A1 or COL1A1), and epithelial (EPCAM, KRT) cell types. Part 2: The SingleR package v.1.4.120 was used with the Human Primary Cell Atlas database81 to annotate each cell as a cell type. Cells that received the following SingleR annotations were classified as non-malignant: B_cell, BM, BM & Prog., CMP, DC, Endothelial_cells Erythroblast, Gametocytes, GMP, HSC_-G-CSF, HSC_CD34+, Macrophage, MEP, Monocyte, MSC, Myelocyte, Neutrophils, NK_cell, Osteoblasts, Platelets, Pre-B_cell_CD34−, ProB_cell_CD34+, Pro-Myelocyte, and T_cells. Differential gene expression was performed to identify marker genes for each of the identified cell types and validate their annotation (Supplementary file 1). These annotations served as the basis for annotation of the spatial transcriptomic spots, and were used to validate the annotation of clusters identified by gene expression in Part 1.
Malignant cell identification.
Part 1: For epithelial and stromal tumors, the expression pattern of the epithelial and stromal cluster respectively was examined to distinguish malignant from non-malignant cells. Specifically, genes were identified which are overexpressed in malignant relative to normal tissue for each cancer type: WFDC282 for OVCA and UCEC; CLU83 and MGP83 for BRCA; LAMC284 TM4SF185 for PDAC; CEACAM586 for COAD; APOH87 for LIHC; TMPRSS288 and CLDN488 for PRAD; CA989 for KIRC; PDGFRA, KCNK390 and ANO191 for GIST. Part 2: RNA-based copy-number variation inference was performed on the putative set of malignant cells, as implemented in the inferCNV package v.1.6.06, using all other cells from the sample as a reference and searching for consistent patterns of copy-number variation. Part 3: Dimensionality reduction was performed on the putative sets of malignant cells from different tumors to validate that they form separate clusters, a known property of malignant cells30.
Non-negative matrix factorization (NMF) and module detection.
NMF was performed separately on the identified malignant cells of each sample (Fig. 1c). Starting from the normalized centered expression of variable genes, all negative values were set to 010. The “nsNMF” method was applied for ranks between 5 and 25 – as implemented in the NMF R package v0.23.010,92. To define non-overlapping gene modules, a gene ranking algorithm was implemented93. Beginning with the matrix of the contribution of genes (rows) to the factors (columns), two ranking matrices were constructed, (list 1) ranking the gene contributions to each factor and (list 2) ranking for each gene the factors to which it contributes. For each factor, genes were added in the order of their contribution (list 1), until a gene was reached which contributed more to another factor, i.e. its rank across factors (list 2) was not 1. Factors which yielded fewer than 5 genes were removed, and the procedure repeated. With this method, the number of modules was at most the rank of the NMF, and the modules were robust to the rank chosen. The highest rank for which the number of modules was equal to the rank was selected for downstream analysis.
Graph-based clustering and identification of consensus gene modules.
The full list of modules obtained for individual tumors was filtered to retain only those with at least 5% overlap (by Jaccard index) with at least 2 other modules. An adjacency matrix was then constructed connecting genes according to the number of individual tumor modules in which they co-occur. Gene-gene connections were filtered out if they occurred in fewer than 2 individual tumor modules, and genes with fewer than 3 connections were removed. The graph was clustered using infomap clustering implemented in the igraph package v.1.2.1194. Finally, modules with potential biological relevance were retained by filtering out those with fewer than 5 genes or without significant overlap with gene ontology terms. The final graph (Extended Data Fig. 3f) was visualized with the fruchterman-reingold layout.
SCENIC module identification and module comparison.
SCENIC regulon identification was performed using the SCENIC package v.1.2.295 implemented in R and Python. Genes were filtered to have at least 0.05 counts per cell on average and to be detected in at least 5% of the cells, and the transcription factor-binding databases used were 500bp-upstream and tss-centered-10kb. To compare modules obtained by NMF in individual tumors to each other (Fig. 1e, Extended Data Fig. 3g), the significance of the pairwise overlap was calculated using the hypergeometric distribution. SCENIC-derived regulons were similarly compared to the consensus modules (Extended Data Fig. 3h), and were considered to match a consensus module if the p-value of the overlap was <10−3. Transcription factors annotated to regulons matching each consensus module were then tabulated (Supplemental Table 4) and the top 1–2 factors found in multiple samples are shown in Figure 1f ‘Regulators’.
Module annotation.
Gene Ontology terms were accessed using the MSigDB package v.1.6.6.2 in R96 (Extended Data Fig. 2a). Cell type markers were downloaded from PanglaoDB97 (Extended Data Fig. 2b). Tumor-derived signatures were accessed from Neftel et al.7 (Extended Data Fig. 2c)” Puram et al.10 (Extended Data Fig. 2d) and Ji et al.25 (Extended Data Fig. 2e) The significance of the overlap between each consensus module and each downloaded gene set was calculated using the hypergeometric distribution.
Significance of module presence.
The previously described ‘overdispersion’ approach was used to quantify the differential expression of a particular module in a set of malignant cells (Extended Data Fig. 4a–c)26. For each module, PCA was performed on the expression of the module genes, and the variance explained by PC1 was calculated. For each module, 103 random lists of genes with similar expression levels were generated as has been done previously6, and the variance explained by PC1 in those genesets was calculated. The significance of the presence was calculated as −log10(p), where p is the fraction of random genesets that resulted in a higher PC1 variance than the module itself. This enabled the identification of tumors in which specific modules are differentially expressed in a statistically significant manner.
Module expression scoring.
The expression level of each module in individual cells (Fig. 2a,d, Extended Data Fig. 4b,d) was scored as follows. For each module, 103 random lists of genes with similar expression levels were generated as has been done previously6. For each cell, the average centered expression of these genesets was calculated, along with that of the module genes. p was defined as the fraction of random genesets with a higher average expression than the module itself. The score was defined as −log10(p) and rescaled linearly to [0,1]. A module was considered expressed in a given cell if its score was higher than 0.5, and these binary values were used to calculate the frequency of expression of each module in each sample (Fig. 2e, Extended Data Fig. 5e–g). The matrix of module expression scores was used to perform TSNE with 500 iterations and a perplexity of 100 (Fig. 2b–d, Extended Data Fig. 5a–c). The mixing of tumors in the TSNE was assessed by calculating the entropy at each point using its 20 nearest neighbors (Extended Data Fig. 5c).
Analysis of normal epithelial cells.
For normal fallopian tube27, breast28 epithelium and normal liver29, single-cell RNA-Seq data was downloaded from GEO. For LUAD24 and SKSC25, single-cell RNA-Seq of matched samples representing normal lung and skin were downloaded from GEO as for the tumor samples. Cells were then annotated according to the lines of evidence 1. and 2. (see ‘Cell type annotation and detection of malignant cells’) to identify epithelial cells. For pancreas, the single-cell RNA-Seq data collected from PDAC9 contained malignant as well as non-malignant epithelial cells, with non-malignant cells expressing high levels of epithelial cell markers (e.g., EPCAM) but low levels of cancer-specific genes (e.g., LAMC2, CDKN2A and TM4SF1) and displaying low CNV9. Further analysis of each normal cell dataset, including significance of module presence and scoring module expression, was performed as for the malignant cell datasets.
Patient tumor spatial transcriptomics:
Data collection and processing.
From the 10 tumors (OVCA NYU1, OVCA NYU3, UCEC NYU3, BRCA NYU0, BRCA NYU1, BRCA NYU2, PDAC NYU1, GIST NYU1, GIST NYU2, LIHC NYU1), 2–3 pieces were embedded in OCT by placing them cut side down into a plastic mold. The OCT-filled mold was then snap frozen in chilled isopentane and stored at −80°C until use. Cryosections were then cut at 10μm thickness and mounted onto Visium arrays. Tissue optimization and library preparation were performed according to manufacturer’s instructions, with 12 minutes of permeabilization. Libraries were sequenced on an Illumina NextSeq and aligned using the Visium SpaceRanger pipeline. As a quality control step, spots with fewer than 500 UMIs or more than 30% mitochondrial or ribosomal reads were filtered out. The Seurat single-cell transformation78,79 was used to normalize and center the data, and to identify variable genes.
Deconvolution of spatial transcriptomic spots.
Spots were annotated using three parallel methods. First, non-negative least squares (NNLS) regression was performed using the single-cell RNA-Seq expression profiles. Specifically, average profiles were calculated for each cell type (annotated using the SingleR package, see ‘Cell type annotation and detection of malignant cells’), using only the paired sample when possible (i.e. when at least 20 cells of that type were present) or the pooled expression profiles from all samples. These profiles were then used to perform linear regression on each spot using the NNLS98 package v.1.4 in R and obtain estimates for the coefficient of each cell type at each spot (Fig. 3, Extended Data Fig. 6, Supplementary Fig. 3). The genes used were the intersection of variable genes in the single cell data and spatially variable genes in the spatial transcriptomic data, obtained with ‘FindVariableFeatures’ and ‘FindSpatiallyVariableFeatures’ respectively79,98. Because the distributions of regression coefficients varied across cell types, and were not usually bimodal, thresholds for cell type presence/absence were set for each cell type individually using a null distribution of coefficients in the sample, as follows. First, spots were selected which had a predicted score of 0 for the cell type in question (see below for mutual nearest neighbor annotation prediction). The resulting gene expression matrix was shuffled and used for NNLS, in 100 independent iterations. The distribution of coefficients for the cell type in question was then used to set the threshold at the mean + 2 × standard deviations. Second, mutual nearest neighbor (MNN) integration was performed using the Seurat package79, using the same set of genes as for NNLS. Prediction coefficients were obtained using the ‘TransferData’ function, and were binarized using a threshold of 0.9. Finally, NMF was performed on each dataset as described for the scRNA-Seq data (see ‘NMF’). The output was processed as above (see ‘NMF’), and factors were named according to the gene with the highest coefficient (Supplementary Fig. 2).
Annotation of spatial transcriptomic spots.
Signatures for M1 and M2 macrophages and for cytotoxic, helper and regulatory T cells were obtained by performing differential gene expression on the macrophage population of OVCA NYU1, keeping the top 100 genes by p-value for each cluster (Supplementary Fig. 4a). To ensure that the increase in M1/M2 score in the ‘Both’ spots relative to ‘Normal’ was not due to the presence of malignant cells themselves, we also scores the single-cell RNA-Seq data for the signatures, and confirmed that macrophages were the only cell type with wide bimodal distribution of M1/M2 scores (Supplementary Fig. 4b). Spatial transcriptomic spots containing macrophages were scored for the expression of the respective signatures using the ‘AddModuleScore’ function in Seurat79 (Fig. 3d). Distances were calculated using euclidean distance on the pixel coordinates, and scaled such that the unit is inter-spot distance (100μm). The depth of malignant spots was calculated as the shortest distance to a spot containing a non-malignant cell type. Proximity was defined as 1/(1+distance) (Fig. 4c). The neighborhood of a spot was defined as spots of distance ≤ 1 (including the spot itself), resulting in sets of ≤ 7 neighbors per spot. The neighborhood cell type fraction was then calculated from the binarized cell type annotations of this set (Fig. 4b). ‘Malignant’ spots were scored for the expression of each module using the Seurat function “AddModuleScore” (Fig. 4a)79.
Patient tumor CODEX:
Staining and image acquisition:
Four fresh frozen samples (OVCA NYU1, UCEC NYU3, LIHC NYU1, GIST NYU1) were cryosectioned at 10μm thickness and mounted onto a glass coverslip coated with poly-lysine. The tissue was stained and the CODEX multicycle reaction performed according to CODEX user manual Revision C. Briefly, the sample coverslip was placed on Drierite beads for 5 minutes, then incubated in Acetone for 10 minutes and set in a humidity chamber for 2 minutes. The sample was hydrated in CODEX Hydration Buffer for twice for 2 minutes, fixed in 1.6% Paraformaldehyde in CODEX Hydration Buffer for 10 minutes, then washed in CODEX Hydration Buffer twice for 2 minutes. The sample was then equilibrated in CODEX Staining Buffer for 30 minutes, then stained with a barcoded antibody cocktail in CODEX Blocking Buffer for 3 hours in a humidity chamber. Antibodies comprising the antibody cocktail are listed in Supplementary Table 7. The sample was washed three times for 2 minutes in CODEX Staining Buffer, fixed in 1.6% Paraformaldehyde in CODEX Storage Buffer for 10 minutes, and then washed 3 times in 1X PBS. The sample was incubated in 4°C methanol for 5 minutes, washed 3 times in 1X PBS, and then fixed using the CODEX Final Fixative Reagent Solution for 20 minutes in a humidity chamber. The sample was washed three times in PBS and then stored in CODEX Storage Buffer for 3 days until imaging. A commercial Akoya CODEX instrument and a Keyence BZ-X800 microscope with a 20x Nikon PlanApo NA 0.75 objective were used to treat and image tissue using complementary fluorescent reporters. The protocols from Akoya CODEX user manual revision C were followed. The four samples were imaged and processed in one CODEX run. Every sample was imaged in 64 (8 × 8) tiles.
Image processing:
Raw TIFF image files were processed using the CODEX Processor. ImageJ and its CODEX Multiplex Analysis Viewer (MAV) plugin were used to visualize, annotate and define cell populations. Supervised clustering was used to define populations for each tissue. Briefly, the gating function in CodexMAV was used to define cells as positive by setting a gate on log10 intensity vs frequency of the marker of interest, for the non-malignant cell populations. To define malignant cell populations in epithelial cancers log10 EpCAM versus log10 PanCK intensities were used to define a double positive population. For the GI stromal tumor, log10 Podoplanin vs. αSMA intensities were used, where double positive cells were annotated as muscle and Podoplanin+ αSMA- were annotated as malignant. Further characterization of the malignant interferon response cell state of the malignant cells, in both tumor types, was conducted by gating on the log10 HLA-DR intensity vs. frequency. All gating was performed on the segmented image of each sample separately, and was based on marker intensity value independent of the visualized image. The threshold dictating the gate was verified visually by validating overlap between the defined population and marker expression, and was set for each marker and for each sample independently. Gating for each population was done sequentially to avoid overlap between populations. The x and y coordinates and the annotation of each cell were exported for further analysis. Spatial analysis was done in R for each sample independently. For each tile of the image, a distance matrix was calculated and used to quantify the distribution of distances closest to populations of interest (e.g Macrophage) in interferon and non-interferon malignant cells, or the distribution of number of cells of interest in a distance of choice. Tiles with obvious bubbles, tissue folding issues, non-malignant cell dominance (more than 70%, mainly the blood vessel areas), or not containing any tissue, were excluded from the analysis. Tiles which passed quality control were used to derive statistics (Extended Data Fig. 9c–d).
Orthotopic pancreatic tumor mouse models and scRNA-Seq:
Tumor collection.
All experiments were approved by the New York University School of Medicine Institutional Animal Care and Use Committee (IACUC). Per protocol, tumors did not exceed 15% of normal body weight. Rag1−/− and WT C57BL/6 mice were obtained from Jackson Laboratories (Bar Harbor, ME). The KrasG12D;Tp53R172H;Pdx1Cre (KPC) derived cell line FC1242 was utilized for orthotopic injection of 100,000 cells into the tail of pancreata of 8–12 week old C57BL/6 or Rag−/− mice. To model liver and peritoneal metastases, mice received FC1242 via splenic (106 cells) and intraperitoneal (105 cells) injection, respectively. Tumors were harvested 2–3 weeks after injection and dissociated using Miltenyi mouse tumor dissociation kit enzymes D and R according to manufacturer’s instructions. Red blood cell lysis was performed for 3 minutes in ACK lysis buffer. Dead cells were removed using the Miltenyi dead cell separator. In order to hash and pool replicates, cells were then labeled with Biolegend oligonucleotide-conjugated antibodies according to manufacturer’s instructions. Single-cell encapsulation and library preparation were performed using the 10x Genomics Chromium. Libraries were sequenced on an Illumina NextSeq and reads aligned using the 10x Genomics CellRanger pipeline.
Transcriptomic analysis of mouse scRNA-Seq data.
Quality control and processing were performed separately for each mouse scRNA-Seq sample separately as for the human data (‘Patient tumor scRNA-Seq’). Samples from all 3 experiments were then combined for cell type annotation. NMF and module identification was performed for the 4 pancreatic WT tumors together. These modules were compared to the consensus modules obtained from patient tumors by orthology mapping using the biomart database99. Overlap between modules across species was tested using the hypergeometric distribution. Module expression was scored for each experiment separately as for the human data, and frequencies were compared across conditions using Kolmogorov-Smirnov tests on the distributions (maximum p-value of pairwise comparisons across conditions is reported).
Patient derived melanoma xenograft (PDX) models and scRNA-Seq:
Tumors collection:
PDX tumors were used to test our method for annotation of ST spots (Extended Data Fig. 7). Samples were obtained from the Hernando lab. Briefly, NSG (Jax 005557) mice were obtained from Jackson Laboratories (Bar Harbor, ME). Cells obtained from patient melanoma brain metastases were injected intradermally and collected after 82 days. For single-cell RNA-Seq, the sample was minced and incubated in 4mL HBSS buffer with 1mg Collagenase IV and 12.5uL DNAse I for 10min at 37C. Single-cell encapsulation and library preparation were performed using the 10x Genomics Chromium as above (‘Orthotopic pancreatic tumor mouse models and scRNA-Seq’). For spatial transcriptomics, samples were embedded in OCT and processed by Visium according to manufacturer’s instructions as above (‘Patient tumor spatial transcriptomics’). Libraries were sequenced on an Illumina NextSeq and reads aligned using the 10x Genomics pipelines.
Transcriptomic analysis of scRNA-Seq PDX data:
Quality control and processing were performed as for the human data (‘Patient tumor scRNA-Seq’). Cells with nCount_human>100nCount_mouse were annotated as human, cells with nCount_mouse>nCount_human were annotated as mouse, and other cells were discarded. The presence of modules in human cells was calculated as above (‘Significance of module presence’). Mouse cells were further clustered using Seurat and annotated using marker genes to obtain cell types for spatial transcriptomic deconvolution.
Transcriptomic analysis of ST PDX data:
Spatial transcriptomics analysis was performed as for the human data (‘Patient tumor spatial transcriptomics’). For NNLS deconvolution, the expression of human and mouse genes was used to generate a ‘ground truth’, and human orthologs of mouse genes were used to simulate patient tumors (Extended Data Fig. 7e–f).
Ethics and statistics:
Animal studies were performed in accordance with NYU IACUC protocol. Human samples were obtained according to NYU IRB. Statistics were derived using R, and the sample sizes, tests used and p-values are reported in the figure legends.
Extended Data
Extended Data Fig. 1. Quality control and cell type annotation in the single-cell RNA-Seq data.
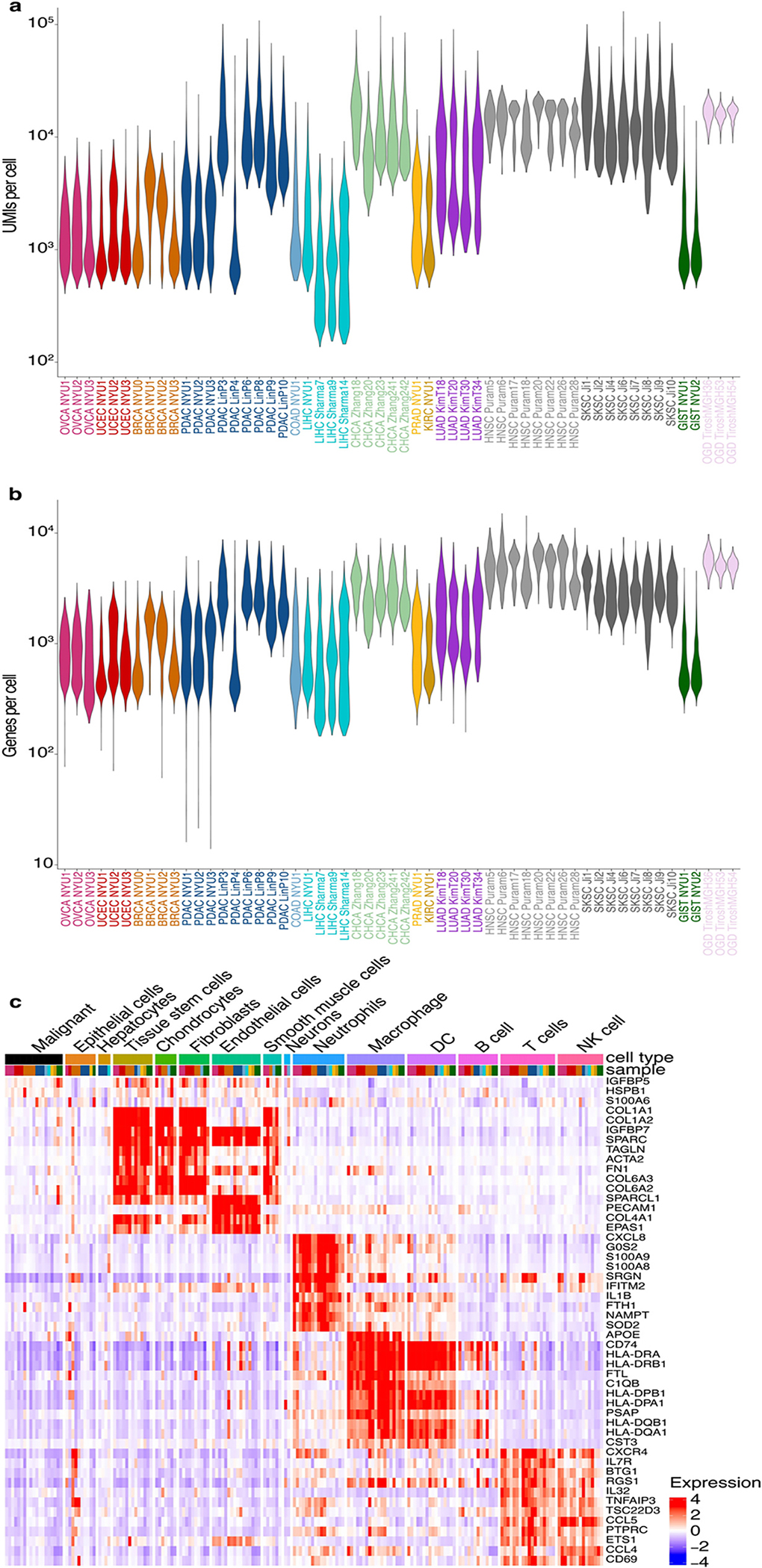
a. Violin plots of the number of UMIs per cell in each tumor sample.
b. Violin plots of the number of genes per cell in each tumor sample.
c. Heatmap of average scaled gene expression per cell type per sample. Top bar represents cell type (colored as indicated) and sample (colored as in Figure 1a).
Extended Data Fig. 2. Control analysis for purity of single-cell RNA-Seq data.

a. UMAP embeddings of cells annotated as malignant per cancer type or organ system, colored by sample.
b. Control analysis for annotations of cells as malignant, using the method described by Kim et al.1. Briefly, inferred CNV profiles (from the scRNA-Seq data) were scored as the sum of the squared values (shown as the x-axis). The cells with the top scores are assumed to be malignant and each cell is then correlated with their averaged profile (y-axis). In tumors with CNVs these two measures are consistent. Color indicates the annotation as malignant or normal cells, per sample.
Extended Data Fig. 3. Additional analyses for the derivation of the cancer gene modules catalog.
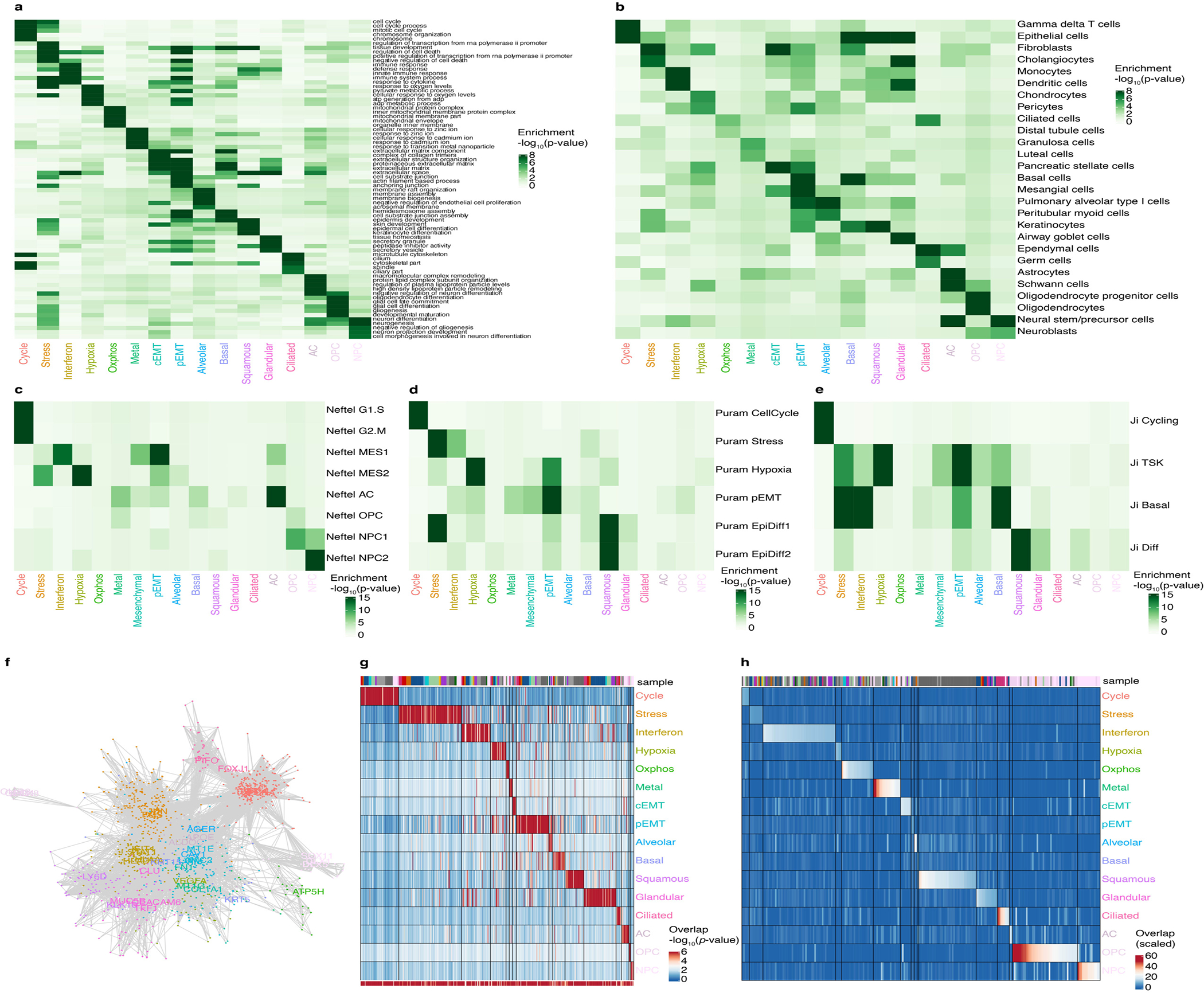
a-e. Heatmap of the significance of overlap (hypergeometric test) of the consensus modules across (a) the indicated Gene Ontology terms, (b) cell type markers, (c) signatures derived by Neftel et al.2, (d) signatures derived from Puram et al.3, and (e) signatures derived from Ji et al.4.
f. Network of genes belonging to the consensus modules, colored as in Figure 1f. Lines connect genes that are found together in at least 2 individual tumor modules (see Methods).
g. Heatmap of the significance of the overlap between consensus modules and individual tumor modules (hypergeometric test). The bottom bar indicates the significance of the overlap with consensus modules (hypergeometric test). The top bar indicates the identity of the tumor samples, colored as in Figure 1a.
h. Heatmap of the Jaccard similarity (intersect/union) between consensus modules and SCENIC regulons obtained for individual tumors. The bar indicates the identity of the tumor samples, colored as in Figure 1a. To test whether the catalog of 16 modules can also be detected using an independent approach, we used SCENIC5, a method that identifies genes that are both correlated in their expression and regulated by the same transcription factor. We found that each module of our catalog had significant overlap with several SCENIC regulons (Supplementary Table 4, see Methods). For instance, the interferon response module overlapped with several SCENIC regulons annotated with the transcription factors STAT1 and IRF1.
Extended Data Fig. 4. Pattern of presence and absence of the catalog gene modules across malignant and normal epithelial cells.
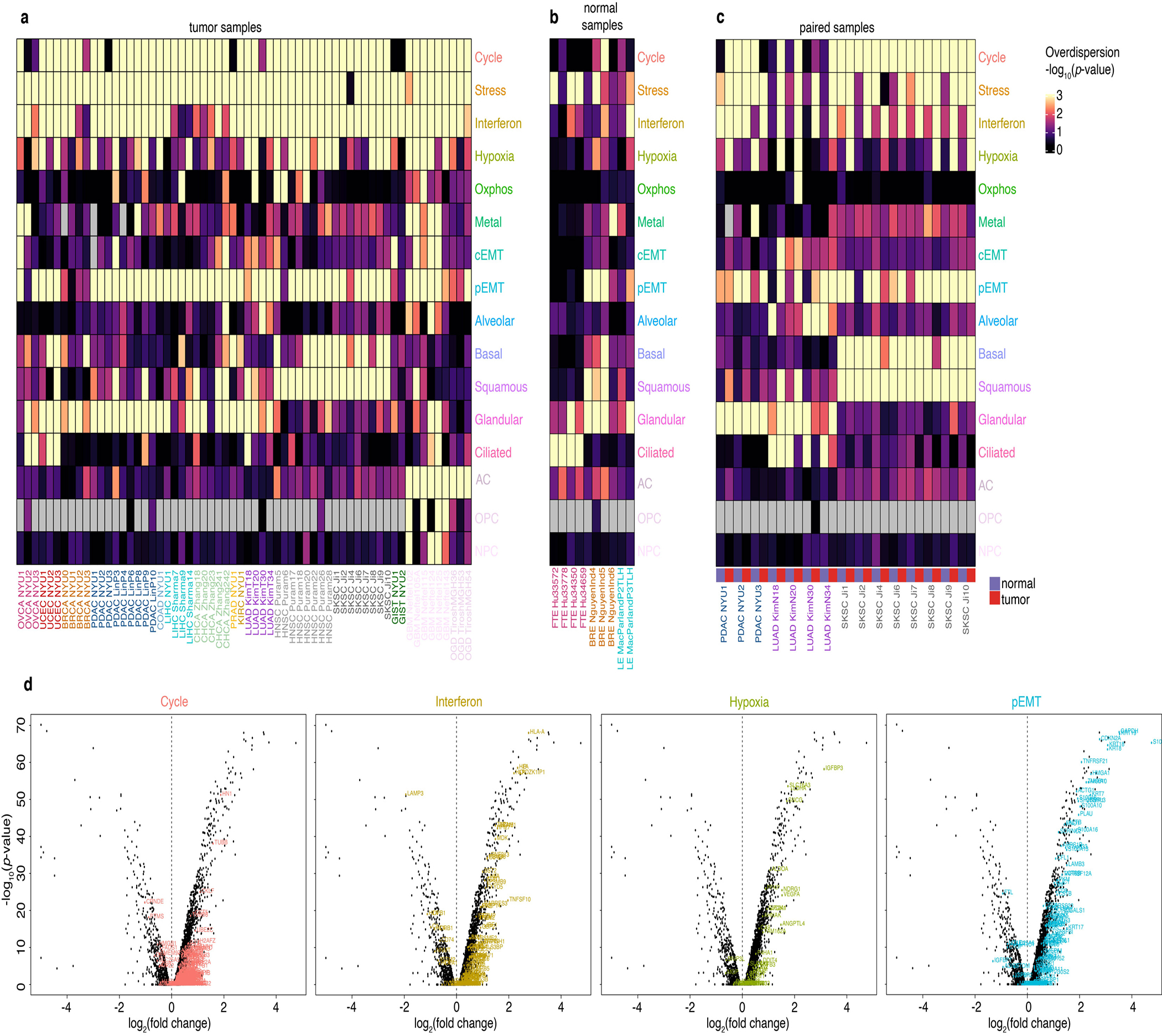
a-c. Heatmap of the significance of the presence of each module (see Methods) in the malignant cells of each tumor sample (a), in epithelial cells of normal samples 6–8 (b), and for malignant and epithelial cells from paired normal and tumor samples (c). Grey indicates a complete lack of gene expression of the module. FTE: Fallopian tube epithelium. BRE: Breast epithelium. LE: Liver epithelium.
d. Volcano plots of differential gene expression between malignant and normal epithelial cells from paired LUAD samples (Kim et al.1 T20 vs. N20). Each panel highlights genes from the indicated module.
Extended Data Fig. 5. Cell states across tumors.
a-c. Module score TSNE embedding of the cancer cells of all 19 tumors, colored by the most highly expressed module (a), by module score as in Figure 1f (b) and by entropy of tumor of origin (c).
d. Gene expression UMAP embedding of the cancer cells of OVCA NYU3 and BRCA NYU1, colored by module score for the Ciliated and Cycle module respectively. Unlike other modules, the cycle and cilium module were expressed by cells forming discrete clusters. These clusters are also identified when examining tumors individually in gene expression-based dimensionality reductions, and are therefore not artifacts of the module score dimensionality reduction (Extended Data Fig. 6d).
e. Heatmap of the frequency of expression of each module in the malignant cells of each sample.
f. Module expression frequencies in normal vs malignant epithelia. Points represent individual samples, bars indicate mean +/− standard error calculated across individual tumors of the same cancer type.
Extended Data Fig. 6. Spatial organization of the tumor microenviron ment.
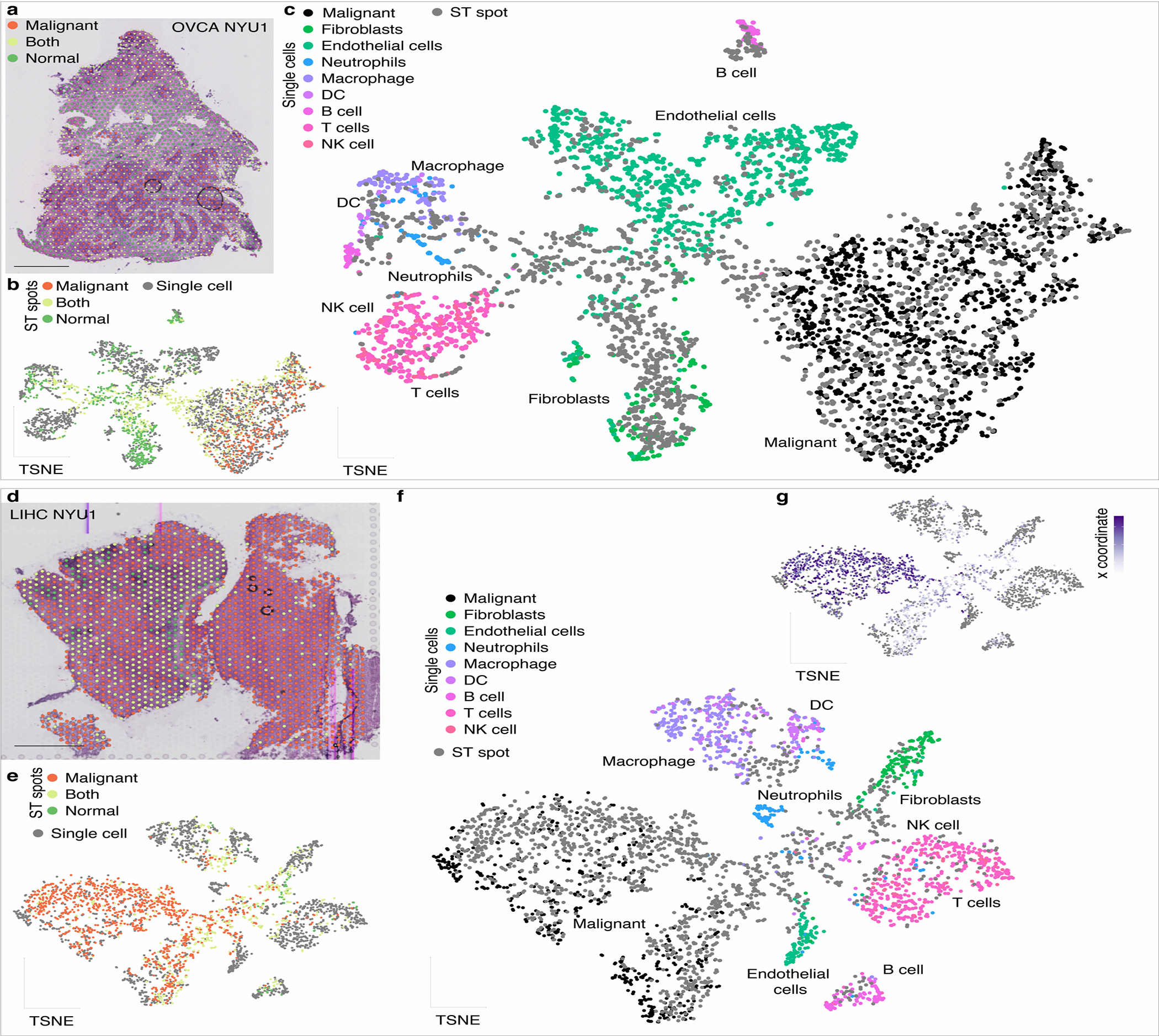
a. OVCA NYU1 H&E image with spots colored by annotation (scale bar represents 1mm).
b-c. Joint dimensionality reduction after mutual nearest neighbor integration (MNN) of single-cell and ST spots for the OVCA NYU1 sample, with (b) single-cell transcriptomes in gray and ST spots colored according to their annotation and (c) spots in gray and single-cell transcriptomes colored by their annotated cell type. The single cells form clusters at the periphery, indicating distinct cell types. The ST spots are either mixed with individual single-cell clusters, indicating a pure population, or bridge multiple clusters, indicating a combination of cell types. Specifically, ‘Malignant’ spots are mixed with the malignant cell cluster, ‘Normal’ spots are in the region of non-malignant cell types, and ‘Both’ spots span both malignant and non-malignant single-cell clusters.
d. LIHC NYU1 H&E image with spots colored by annotation as in a. (scale bar represents 1mm).
e-g. Joint dimensionality reduction of single-cell and ST spots for the LIHC NYU 1 sample, with (e) single-cell transcriptomes in gray and ST spots colored according to their annotation, (f) spots in gray and single-cell transcriptomes colored by their annotated cell type and (g) spots colored according to their coordinate along the x-axis. This sample has two spatially distinct tumor nodules, with the left having substantial mixing between malignant and nonmalignant cells and the right consisting of almost only malignant cells. The joint dimensionality reduction analysis reflects the two corresponding malignant clusters, which were not distinct when considering the single-cell dimensionality reduction alone.
Extended Data Fig. 7. Validation of spot annotation and module presence using PDX samples.

a. UMAP of single-cell RNA-Seq data for sample 1, colored by the number of UMIs corresponding to human (left) or mouse (right) genes.
b. Heatmap of module presence in malignant cells of sample 1 (see Extended Data Fig. 4a–c and Methods) c-d. Spatial transcriptomic spots colored by the number of UMIs corresponding to human (left) or mouse (right) genes for sample 1 (c) and sample 2 (d). Scale bar represents 1mm.
e-f. Spatial transcriptomic spots colored by annotation as ‘Malignant’, ‘Both’ or ‘Normal’ using the NNLS method on the full transcriptome (left) or on human orthologs (right) in sample 1 (e) and sample 2 (f). To test the accuracy of the NNLS method to annotate spots, we performed paired scRNA-Seq and ST on two patient-derived melanoma xenografts (PDX). In this setting, only malignant cells are of human origin and therefore express human genes, enabling us to reliably identify malignant cells or spots. Using the NNLS method on the full mouse and human transcriptomes, we first established a ‘ground truth’ for spot identities. We then simulated the patient samples by converting mouse genes to their human orthologs, thereby removing the species information. This resulted in specificity of 99% (sample 1) and 89% (sample 2) specificity.
g-h. ‘Malignant’ spatial transcriptomic spots colored by expression score for the cycle, stress, hypoxia and pEMT modules for sample 1 (g) and sample 2 (h). To establish the validity of scoring ‘Malignant’ spots for gene modules, we scored the ‘Malignant’ spots for the expression of modules. Since human malignant cells can unambiguously be distinguished from mouse TME cells in this system, we first used the single cell data to confirm that the modules are differentially expressed by malignant cells themselves and rule out the possibility of an artifact stemming from TME contamination. For example, the pEMT module includes genes normally expressed by fibroblasts, but we detected its presence in the malignant cells.
Extended Data Fig. 8. Relationship between pEMT and cancer spot depth.
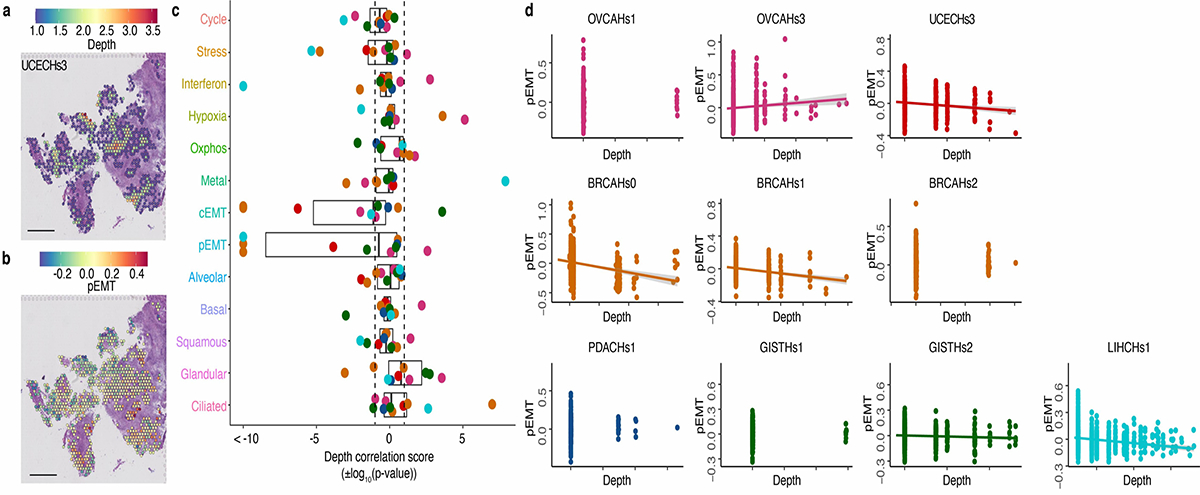
a. Sample UCEC NYU3, ‘Malignant’ only spots colored by their depth: the distance to the nearest spot containing non-malignant cells.
b. Sample UCEC NYU3, ‘Malignant’ only spots colored by pEMT module score.
c. Boxplots of correlation scores (±log10(p-value)) between module scores and depth of malignant spots across 10 samples, colored as in Figure 1a. For each boxplot, the line indicates the median, the box indicates the 1st and 3rd quartile, the whiskers indicate the minimum and maximum values. Positive scores correspond to positive correlations. Dashed lines indicate pvalue=0.05. Plots of the relationship between the pEMT module score and depth in the 10 ST samples, colored as in Figure 1a. Lines are drawn for correlations with p-value<0.05.
Extended Data Fig. 9. CODEX analysis of samples from four cancer types supporting a proximity of interferon responseexpressing malignant cells to macrophages and T cells.
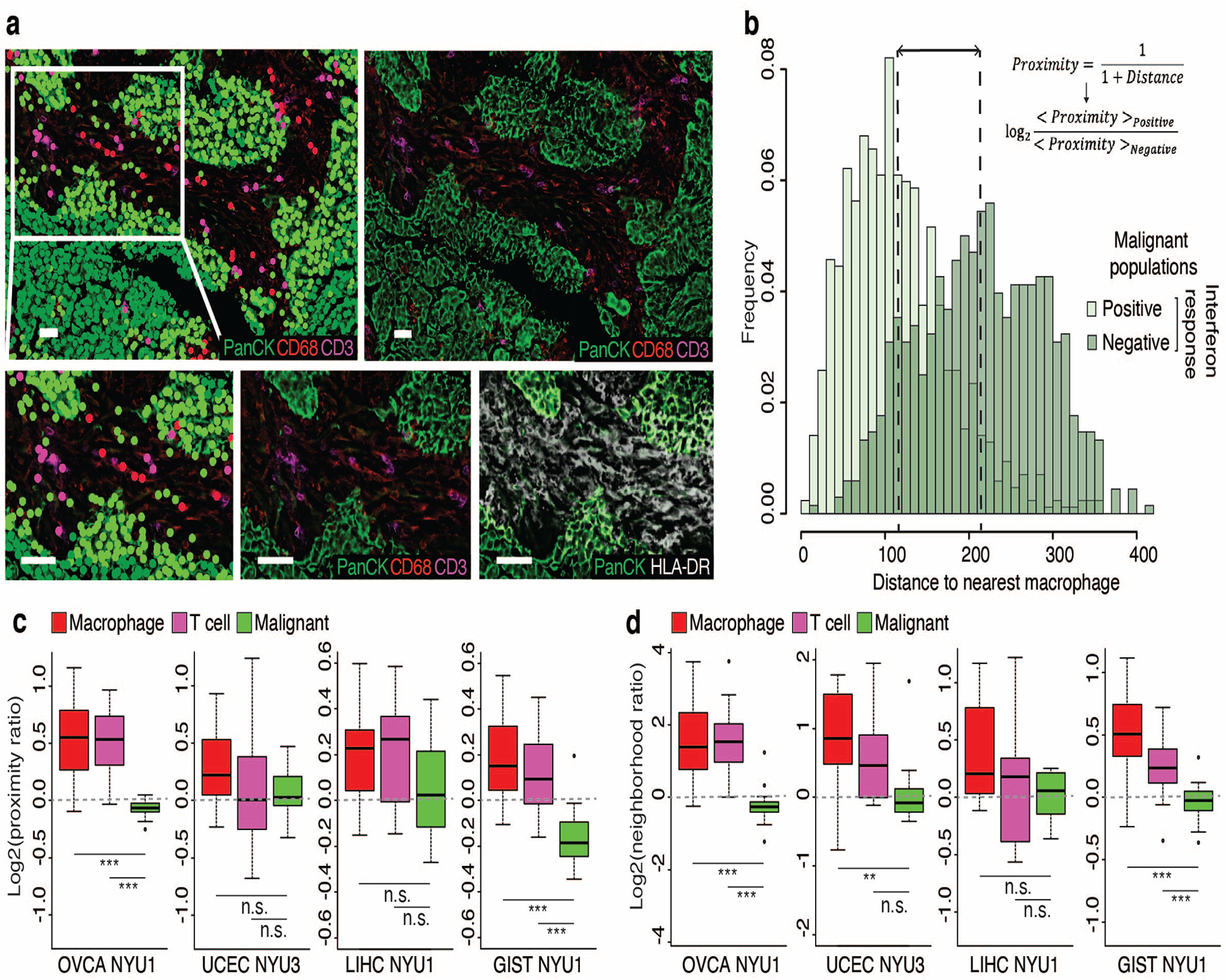
a. Cell populations and marker expression in a region of OVCA NYU1. Top row displays an entire tile, bottom row displays an enlargement. Top and bottom left: Colored by populations as defined in Extended Data Fig. 15. Top right and bottom center: Colored by expression of markers used to define cell types, as indicated. Bottom right: Colored by expression of PanCK and of HLA-DRA, used to define interferon response positive and negative malignant cells. Scale bar represents 50μm.
b. For the tile shown in a., histogram showing the distance between malignant cells and the nearest macrophage, for interferon-response positive (light green) and negative (dark green) malignant cells. Lines indicate the mean distance for each population, used to calculate the log2(proximity ratio). c-d. Boxplots of the distribution of log2(proximity ratio) (c) and log2(neighborhood ratio) (d) of macrophages, T cells and malignant cells across tiles of each sample (*, p-value<0.05; ***, p-value<0.001; two-sided t-test). For each boxplot, the line indicates the median, the box indicates the 1st and 3rd quartile, the whiskers indicate the minimum and maximum values.
Extended Data Fig. 10. Additional experiments.
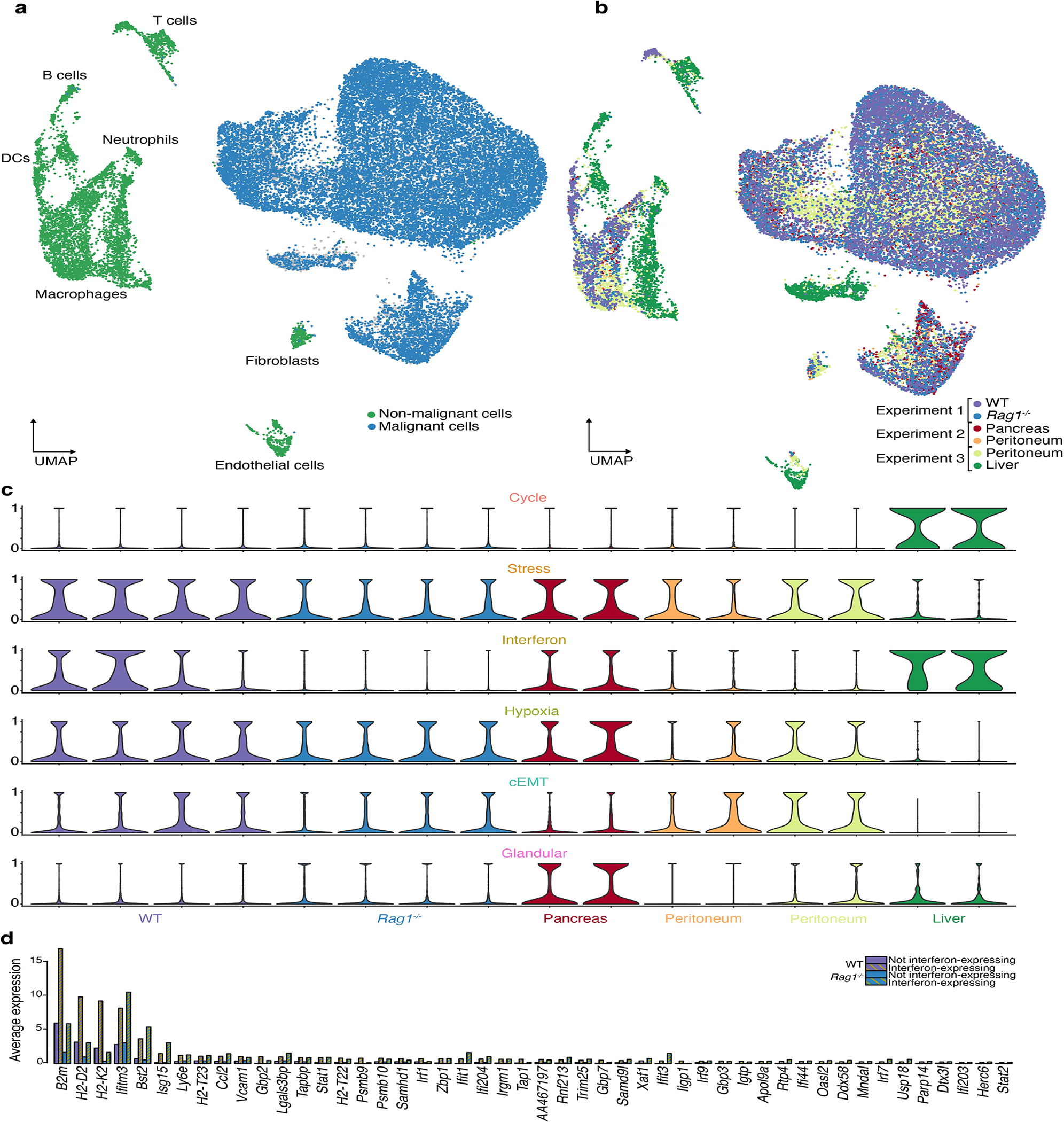
a. UMAP embedding of cells from 16 orthotopic pancreatic tumors across relating to the orthotopic and heterotopic mouse experiments. the 3 experiments, colored by annotation as malignant or non-malignant cells.
b. Same as a, colored by sample.
c. Violin plots of module expression scores in individual tumors across the 3 experiments. Barplots of the average expression of the interferon response module genes in cancer cells in the WT and Rag1−/− tumors according to their interferon response expression.
Supplementary Material
{kind=link}
Acknowledgements:
We thank Rich White, Felicia Kuperwaser, Rahul Satija, and Benjamin Neel for critical readings and helpful suggestions. We thank Eva Hernando for helpful discussions and for providing the PDX samples. This work was supported by the following NIH grants: P50 CA225450 (to I.O. and I.Y.), R01 LM013522 (to I.Y.), R21 CA264361 (to I.Y.), GM126573 and F30 CA257400 (to D.B.)
Footnotes
Conflict of interests statement: The authors declare that they have no conflict of interest relating to this work.
Code availability:
Code from this manuscript is available at https://github.com/yanailab, and also through Zenodo (https://doi.org/10.5281/zenodo.6611786)100.
Data availability:
Data from this manuscript has been submitted to GEO with accession number GSE203612.
REFERENCES
- 1.Easwaran H, Tsai H-C & Baylin SB Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 54, 716–727 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGranahan N & Swanton C Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 168, 613–628 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Marusyk A & Polyak K Tumor heterogeneity: Causes and consequences. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer vol. 1805 105–117 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heppner GH & Miller BE Tumor heterogeneity: biological implications and therapeutic consequences. Cancer Metastasis Rev. 2, 5–23 (1983). [DOI] [PubMed] [Google Scholar]
- 5.Alizadeh AA et al. Toward understanding and exploiting tumor heterogeneity. Nat. Med. 21, 846–853 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patel AP et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neftel C et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 178, 835–849.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baron M et al. A population of stress-like cancer cells in melanoma promotes tumorigenesis and confers drug resistance. doi: 10.1101/396622. [DOI]
- 9.Moncada R et al. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol. 38, 333–342 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Puram SV et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 171, 1611–1624.e24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim C et al. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 173, 879–893.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Izar B et al. A single-cell landscape of high-grade serous ovarian cancer. Nat. Med. (2020) doi: 10.1038/s41591-020-0926-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tirosh I et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reitman ZJ et al. Mitogenic and progenitor gene programmes in single pilocytic astrocytoma cells. Nat. Commun. 10, 3731 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rambow F et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 174, 843–855.e19 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Baron M et al. The Stress-Like Cancer Cell State Is a Consistent Component of Tumorigenesis. Cell Syst 11, 536–546.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baron M et al. The Stress-Like Cancer Cell State Is a Consistent Component of Tumorigenesis. Cell Systems vol. 11 536–546.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dirkse A et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 10, 1787 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cazet AS et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat. Commun. 9, 2897 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aran D et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin W et al. Single-cell transcriptome analysis of tumor and stromal compartments of pancreatic ductal adenocarcinoma primary tumors and metastatic lesions. Genome Med. 12, 80 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharma A et al. Onco-fetal Reprogramming of Endothelial Cells Drives Immunosuppressive Macrophages in Hepatocellular Carcinoma. Cell 183, 377–394.e21 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Zhang M et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J. Hepatol. 73, 1118–1130 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Kim N et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 11, 2285 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ji AL et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 182, 1661–1662 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cantini L et al. Classification of gene signatures for their information value and functional redundancy. NPJ Syst Biol Appl 4, 2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu Z et al. The Repertoire of Serous Ovarian Cancer Non-genetic Heterogeneity Revealed by Single-Cell Sequencing of Normal Fallopian Tube Epithelial Cells. Cancer Cell 37, 226–242.e7 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Nguyen QH et al. Profiling human breast epithelial cells using single cell RNA sequencing identifies cell diversity. Nat. Commun. 9, 2028 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacParland SA et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 9, 4383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brady SW et al. Combating subclonal evolution of resistant cancer phenotypes. Nat. Commun. 8, 1231 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borden EC Interferons α and β in cancer: therapeutic opportunities from new insights. Nat. Rev. Drug Discov. 18, 219–234 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Dunn GP et al. A critical function for type I interferons in cancer immunoediting. Nat. Immunol. 6, 722–729 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Parker BS, Rautela J & Hertzog PJ Antitumour actions of interferons: implications for cancer therapy. Nat. Rev. Cancer 16, 131–144 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Vilgelm AE & Richmond A Chemokines Modulate Immune Surveillance in Tumorigenesis, Metastasis, and Response to Immunotherapy. Frontiers in Immunology vol. 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wan S et al. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS One 7, e32542 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dunn GP, Koebel CM & Schreiber RD Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 6, 836–848 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Park IA et al. Expression of the MHC class II in triple-negative breast cancer is associated with tumor-infiltrating lymphocytes and interferon signaling. PLOS ONE vol. 12 e0182786 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Axelrod ML, Cook RS, Johnson DB & Balko JM Biological Consequences of MHC-II Expression by Tumor Cells in Cancer. Clin. Cancer Res. 25, 2392–2402 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cherian MG, Jayasurya A & Bay B-H Metallothioneins in human tumors and potential roles in carcinogenesis. Mutat. Res. 533, 201–209 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Jin R et al. Metallothionein 2A expression is associated with cell proliferation in breast cancer. Carcinogenesis 23, 81–86 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Pereira H et al. Metallothionein expression in human breast cancer. The Breast vol. 1 159–160 (1992). [Google Scholar]
- 43.Pedersen MØ, Larsen A, Stoltenberg M & Penkowa M The role of metallothionein in oncogenesis and cancer prognosis. Prog. Histochem. Cytochem. 44, 29–64 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Laughney AM et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat. Med. 26, 259–269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marjanovic ND et al. Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell 38, 229–246.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maynard A et al. Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing. Cell 182, 1232–1251.e22 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hao D et al. Integrated Analysis Reveals Tubal- and Ovarian-Originated Serous Ovarian Cancer and Predicts Differential Therapeutic Responses. Clin. Cancer Res. 23, 7400–7411 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Zhang S et al. Both fallopian tube and ovarian surface epithelium are cells-of-origin for high-grade serous ovarian carcinoma. Nat. Commun. 10, 5367 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fischer KR et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature vol. 527 472–476 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aiello NM et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 45, 681–695.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cook DP & Vanderhyden BC Transcriptional census of epithelial-mesenchymal plasticity in cancer. Sci Adv 8, eabi7640 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kalluri R & Weinberg RA The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hayashi A et al. A unifying paradigm for transcriptional heterogeneity and squamous features in pancreatic ductal adenocarcinoma. Nature Cancer vol. 1 59–74 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Collisson EA et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 17, 500–503 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moffitt RA et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 47, 1168–1178 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baylor SM & Berg JW Cross-classification and survival characteristics of 5,000 cases of cancer of the pancreas. J. Surg. Oncol. 5, 335–358 (1973). [DOI] [PubMed] [Google Scholar]
- 57.Al-Shehri A, Silverman S & King KM Squamous Cell Carcinoma of the Pancreas. Current Oncology vol. 15 293–297 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ståhl PL et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Weis SM & Cheresh DA Tumor angiogenesis: molecular pathways and therapeutic targets. Nature Medicine vol. 17 1359–1370 (2011). [DOI] [PubMed] [Google Scholar]
- 60.Viallard C & Larrivée B Tumor angiogenesis and vascular normalization: alternative therapeutic targets. Angiogenesis 20, 409–426 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Solinas G, Germano G, Mantovani A & Allavena P Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 86, 1065–1073 (2009). [DOI] [PubMed] [Google Scholar]
- 62.Zhang M et al. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian Res. 7, 19 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yuan A et al. Opposite Effects of M1 and M2 Macrophage Subtypes on Lung Cancer Progression. Sci. Rep. 5, 14273 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Black S et al. CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nat. Protoc. 16, 3802–3835 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Diamond MS et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 208, 1989–2003 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Deng L et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41, 843–852 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ng KW, Marshall EA, Bell JC & Lam WL cGAS-STING and Cancer: Dichotomous Roles in Tumor Immunity and Development. Trends Immunol. 39, 44–54 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Williams JB et al. Tumor heterogeneity and clonal cooperation influence the immune selection of IFN-γ-signaling mutant cancer cells. Nat. Commun. 11, 602 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kinker GS et al. Pan-cancer single cell RNA-seq uncovers recurring programs of cellular heterogeneity. doi: 10.1101/807552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weinberg RA The Biology of Cancer. (Garland Pub, 2007). [Google Scholar]
- 71.Zaidi MR & Merlino G The two faces of interferon-γ in cancer. Clin. Cancer Res. 17, 6118–6124 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sottoriva A et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 47, 209–216 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hanahan D & Weinberg RA The Hallmarks of Cancer. Cell vol. 100 57–70 (2000). [DOI] [PubMed] [Google Scholar]
- 74.Hanahan D & Weinberg RA Hallmarks of Cancer: The Next Generation. Cell vol. 144 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 75.Archetti M & Pienta KJ Cooperation among cancer cells: applying game theory to cancer. Nat. Rev. Cancer 19, 110–117 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Klein AM et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baron M et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst 3, 346–360.e4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hafemeister C & Satija R Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pu W et al. Single-cell transcriptomic analysis of the tumor ecosystems underlying initiation and progression of papillary thyroid carcinoma. Nat. Commun. 12, 6058 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mabbott NA, Baillie JK, Brown H, Freeman TC & Hume DA An expression atlas of human primary cells: inference of gene function from coexpression networks. BMC Genomics 14, 632 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Galgano MT, Hampton GM & Frierson HF Comprehensive analysis of HE4 expression in normal and malignant human tissues. Modern Pathology vol. 19 847–853 (2006). [DOI] [PubMed] [Google Scholar]
- 83.Chen L, O’Bryan JP, Smith HS & Liu E Overexpression of matrix Gla protein mRNA in malignant human breast cells: isolation by differential cDNA hybridization. Oncogene 5, 1391–1395 (1990). [PubMed] [Google Scholar]
- 84.Kosanam H et al. Laminin, gamma 2 (LAMC2): a promising new putative pancreatic cancer biomarker identified by proteomic analysis of pancreatic adenocarcinoma tissues. Mol. Cell. Proteomics 12, 2820–2832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zheng B et al. TM4SF1 as a prognostic marker of pancreatic ductal adenocarcinoma is involved in migration and invasion of cancer cells. Int. J. Oncol. 47, 490–498 (2015). [DOI] [PubMed] [Google Scholar]
- 86.Jothy S, Yuan SY & Shirota K Transcription of carcinoembryonic antigen in normal colon and colon carcinoma. In situ hybridization study and implication for a new in vivo functional model. Am. J. Pathol. 143, 250–257 (1993). [PMC free article] [PubMed] [Google Scholar]
- 87.Jing X, Piao Y-F, Liu Y & Gao P-J Beta2-GPI: a novel factor in the development of hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 136, 1671–1680 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Landers KA et al. Identification of claudin-4 as a marker highly overexpressed in both primary and metastatic prostate cancer. Br. J. Cancer 99, 491–501 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liao SY, Aurelio ON, Jan K, Zavada J & Stanbridge EJ Identification of the MN/CA9 protein as a reliable diagnostic biomarker of clear cell carcinoma of the kidney. Cancer Res. 57, 2827–2831 (1997). [PubMed] [Google Scholar]
- 90.Allander SV et al. Gastrointestinal stromal tumors with KIT mutations exhibit a remarkably homogeneous gene expression profile. Cancer Res. 61, 8624–8628 (2001). [PubMed] [Google Scholar]
- 91.West RB et al. The Novel Marker, DOG1, Is Expressed Ubiquitously in Gastrointestinal Stromal Tumors Irrespective of KIT or PDGFRA Mutation Status. The American Journal of Pathology vol. 165 107–113 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gaujoux R & Seoighe C A flexible R package for nonnegative matrix factorization. BMC Bioinformatics 11, 367 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Carmona-Saez P, Pascual-Marqui RD, Tirado F, Carazo JM & Pascual-Montano A Biclustering of gene expression data by Non-smooth Non-negative Matrix Factorization. BMC Bioinformatics 7, 78 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nepusz GCA The igraph software package for complex network research. InterJournal Complex Systems, 1695 (2006). [Google Scholar]
- 95.Aibar S et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Franzén O, Gan L-M & Björkegren JLM PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mullen KM, van Stokkum IHM & Mullen MK Package ‘nnls’. (2015).
- 99.Durinck S et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440 (2005). [DOI] [PubMed] [Google Scholar]
- 100.Barkley D, Code for the analyses described in Barkley et al. Nature Genetics. Zenodo 10.5281/zenodo.6611786 (2022) [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data from this manuscript has been submitted to GEO with accession number GSE203612.