

Abstract
Artificial intelligence (AI) is a rapidly growing discipline in the field of chemical toxicology. Herein, we provide a broad overview of research presented at the Fall 2022 American Chemical Society meeting, highlighting how AI is being applied across various facets of drug design, development, and safety assessment.
The term artificial intelligence (AI) was coined in the 1950s and is defined as the ability of a computer or computer-controlled robot to perform tasks commonly associated with intelligent beings.1 AI programs are advanced machine learning (ML) methods which have evolved to solve complex tasks pertaining to image analysis, quantum chemistry, organic synthesis planning, and predicting molecular toxicity. Recent advances in AI architectures, such as neural networks, have dramatically increased their computational power and, in turn, their utility in complex systems.
Generally, sophisticated ML algorithms extract patterns from data sets to make predictions. This is accomplished by first training a ML model to prioritize and predict features within a curated data set, followed by testing the model’s accuracy with another known data set. Once validated, the model can be used to make predictions. Exciting progress in this area has sparked interest within the scientific community for the continued development of such tools for drug discovery and toxicology.
Current applications of AI within the field of chemical toxicology were presented at the 2022 American Chemical Society Fall meeting during the joint Chemical Toxicology Division/Medicinal Chemistry Division thematic session “Developing Role for Artificial Intelligence in Drug Discovery in Drug Design, Development, and Safety Assessment”. Some of the topics covered in the session are highlighted.
Patrick Walters from Relay Therapeutics discussed emerging areas where AI is contributing to drug discovery programs and highlighted open questions in the field.2 A major effort in drug discovery campaigns is to define a class of molecules’ structure— activity relationships (SAR). SAR information is critical in assessing a class of compounds’ therapeutic potential as well as potential toxicity. Typically, the greater selectivity a given compound has for its intended target, the lower the chances are for off-target side effects. To assess selectivity in silico, ML models have been developed to model the number of drug interactions within a protein target site in molecular dynamic simulations. Combining chemistry- and physics-based models with ML is a powerful approach to identify features driving drug selectivity which expedites molecular design and synthetic efforts to improve target specificity.
Another application of ML is the de novo generation of molecules through automated drug discovery. Advantages of this automated chemical design approach include reproducibility, scalability, 24/7 operation, and elimination of user bias. However, challenges remain in fully implementing the automated design of the new chemical matter without human participation. Full automation in drug discovery requires developing an increased trust in the ML process, overcoming technical challenges, and considering whether the resulting molecules are feasible to synthesize. In the current state of ML applications, human input contributes toward defining ML search goals, assay cascade design, assessing relevancy, and debugging artifacts in the learning process. In such methods, ML serves as an important component in the collaborative efforts between computational and experimental drug design.
The second speaker, Christina de Bruyn Kops from Vertex discussed how AI could be applied to study xenobiotic metabolism and identify potential sites for detoxification and targeted bioactivation of small molecules.3 Computational prediction of metabolism of small molecules falls into two categories: identification of sites of metabolism (SOM) and metabolic structure prediction. GLORYx is an ML approach to predict and rank the chemical structures of metabolites generated by oxidative, hydrolytic, and conjugative metabolism. First, the FAME3 software was implemented to predict SOMs. This was accomplished using an expertly annotated data set and a set of unique molecular circular atom-type descriptors as molecular fingerprints. GLORYx uses the SOM predictions to score and rank the predicted metabolites. Challenges in predicting conjugated metabolites include high false positive identification rates and difficulty in differentiating between reaction types that take place at the same SOM. Comparison of SOM prediction models with high-quality manually curated test sets revealed incorporation of SOM prediction improves prioritization of metabolites generated during metabolic processes.
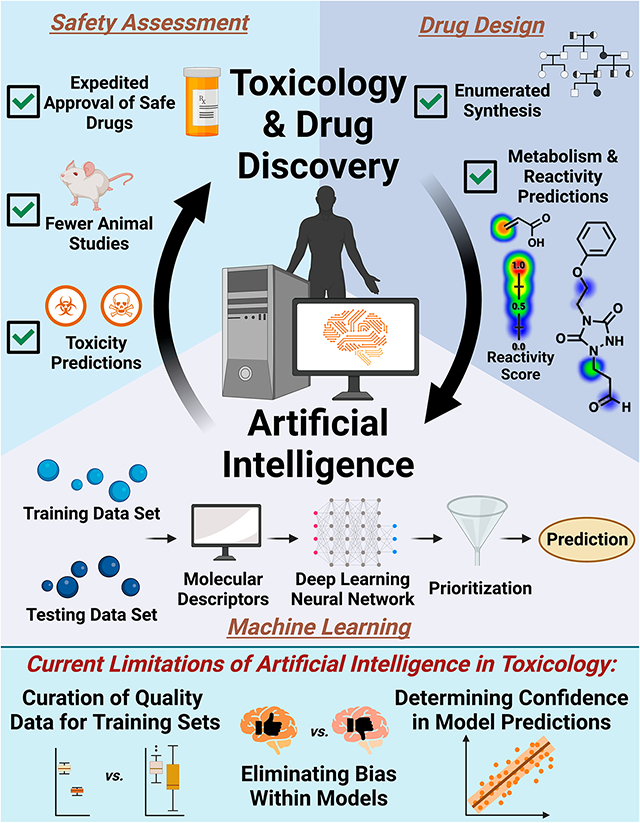
The third speaker, Joshua Swamidass from Washington University in St. Louis, MO, described how AI is utilized to identify reactive chemical substructures that are prone to metabolic bioactivation to toxic intermediates.4 In this capacity, ML was applied to bridge metabolism and toxicity models to understand how the bioactivation of metabolites leads to hypersensitivity and hepatotoxicity. In one example, an ML model was developed to predict the formation of quinone species in drug metabolism. This ML model successfully improved the ability to rationally modify drugs to prevent quinone formation. Additionally, this ML process was compared to general structural alerts used to identify potential chemical sites susceptible to bioactivation. When identified retrospectively, structural alerts are known to misclassify molecular toxicity. ML has been shown to provide more accurate toxicity predictions than broad structural alert approaches. This modeling framework has been applied to epoxidation, nitro-aromatic reduction, and thiophene sulfur-oxidation reactions and can be further expanded to other metabolic pathways in the future. Additionally, this approach could be coupled with ML platforms that infer the intermediate metabolites which may be useful for identifying bioactivation of drugs with unintended side effects that lead to the withdrawal of drugs used in clinical settings.
Jonathan Goodman from University of Cambridge discussed how AI can be applied in predictive toxicology5. Here, the complexity of designing useful AI predictive platforms was contextualized within the vastness of chemical space which is approximately 1024 drug-like compounds. This problem is further complicated when considering chemical reactivity and biological outcomes. Within this sea of complexity, there are approximately 108 known chemical entities deposited in various databases (e.g., PubChem). Thus, our exploration in this area is largely incomplete, and these missing data should be considered in the design of ML platforms. Other considerations for developing AI models were also discussed, including the importance of data quality and the need for defining similarity between compounds to make predictions.
To conclude the session, Zhichao Liu from the United States Food and Drug Administration (FDA) described the agency’s work to implement AI based workflows into the new drug approval process. SafetAI was introduced as a framework to assist in providing a safety profile for investigational new drug (IND) applications to allow drug candidates to enter Phase 1 clinical trials (https://www.fda.gov/about-fda/nctr-research-focus-areas/safetai-initiative). SafetAI facilitates drug safety research with a deep learning architecture which seeks to improve toxicity assessment and is currently being expanded to multiple organ systems. This tool may provide critical safety information during the IND review process and negate the need for intensive animal studies in accordance with the FDA Innovative Science and Technology Approaches for New Drugs program.
Examples of AI tools developed for assessing drug safety profiles include the deep learning-powered platforms drug-induced liver injury (DeepDILI) and DeepCarc. These tools were designed and evaluated as preclinical screening methods for DILI and cardiac toxicity of potential drug compounds, respectively.6 These tools use ML algorithms trained on drugs approved before 1997 to predict the DILI and cardiac toxicity of those approved thereafter. The DeepDILI model was also applied to predicting any DILI concerns from drug repurposing candidates. Such tools are publicly available through https://github.com/TingLi2016/DeepDILI and https://github.com/TingLi2016/DeepCarc.
Current progress in AI research has revealed several lessons and limitations. AI tool designers should be cognizant of whether their platform is properly “fit-for-purpose” and ensure that detailed data curation protocol is in-place to accurately reflect toxicity at a particular end point. Throughout ML development, care must be taken to properly evaluate the model to ensure accurate predictive power and to assess the adaptability of the model. Reproducibility is another key aspect to consider in ML design, as factors such as random seeding and the versions of software packages being used have been found to be sources of variability. Measures to improve reproducibility include using docker containers to maintain consistent workflows and ensuring all data and code management platforms are publicly accessible. It is expected that overcoming these challenges will advance AI platforms.
To conclude, AI is a potentially powerful tool in a chemical toxicologist’s toolkit. AI-based platforms have been developed to assist in designing drug-like molecules, predicting a compound’s propensity to form toxic metabolites, and expediting the FDA drug approval process. While key advances have been made in these areas, there is still a need for continued innovation. Noted themes of the session touched on the importance of data quality in AI training sets, eliminating bias from models, and applying multiple AI tools to provide confidence in predictive outcomes. Currently, no single AI model exists to predict everything well, but when a particular model is used appropriately, extremely useful information can be gleaned. Future applications of AI in chemical toxicology are expected to produce important contributions to the field. Currently, the journal of Chemical Research in Toxicology has an open call for papers at the interface of AI and toxicology where researchers are encouraged to contribute (https://pubs.acs.org/doi/10.1021/acs.chemrestox.2c00196).7
ACKNOWLEDGMENTS
Funding for A.K.H.’s ACS meeting attendance was provided by the NIH Chemistry and Biology Interface Training grant T32 GM132029, UMN Doctoral Dissertation Fellowship, and TOXI Travel Award. L.E. was supported by IRACDA program grant K12 GM119955–06 to attend this meeting. Funding was provided by NIH 1R13ES034642-01 for the authors’ ACS meeting registration. The authors would like to thank F. Peter Guengerich and Nicholas A. Meanwell for presiding over this ACS Chemical Toxicology Division thematic session as well as Natalia Y. Tretyakova for her editing of the manuscript.
Footnotes
Views expressed in this editorial are those of the authors and not necessarily the views of the ACS.
The authors declare no competing financial interest.
REFERENCES
- (1).Moor J The Dartmouth College Artificial Intelligence Conference: The Next Fifty Years. AI magazine 2006, 27 (4), 87–91. [Google Scholar]
- (2).Goldman B; Kearnes S; Kramer T; Riley P; Walters WP Defining Levels of Automated Chemical Design. Journal of medicinal chemistry 2022, 65 (10), 7073–7087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).De Bruyn Kops C; Šícho M; Mazzolari A; Kirchmair J GLORYx: Prediction of the Metabolites Resulting from Phase 1 and Phase 2 Biotransformations of Xenobiotics. Chemical research in toxicology 2021, 34 (2), 286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hughes TB; Flynn N; Dang NL; Swamidass SJ Modeling the Bioactivation and Subsequent Reactivity of Drugs. Chemical research in toxicology 2021, 34 (2), 584–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wang MWH; Goodman JM; Allen TEH Machine Learning in Predictive Toxicology: Recent Applications and Future Directions for Classification Models. Chemical research in toxicology 2021,34 (2), 217–239. [DOI] [PubMed] [Google Scholar]
- (6).(a) Li T; Tong W; Roberts R; Liu Z; Thakkar S DeepDILI: Deep Learning-Powered Drug-Induced Liver Injury Prediction Using Model-Level Representation. Chemical research in toxicology 2021, 34 (2), 550–565. [DOI] [PubMed] [Google Scholar]; (b) Li T; Tong W; Roberts R; Liu Z; Thakkar S DeepCarc: Deep Learning-Powered Carcinogenicity Prediction Using Model-Level Representation. Frontiers in artificial intelligence 2021, 4, 757780–757780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Tetko IV; Klambauer G; Clevert D-A; Shah I; Benfenati E Artificial Intelligence Meets Toxicology. Chemical research in toxicology 2022,35 (8), 1289–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]