

Abstract
Translating our knowledge of the biological aging from animal models to humans may give rise to novel approaches of targeting multiple aging-related diseases simultaneously and increasing health span. Here, for the first time, we use transcriptomic signatures of monocytes to identify biological aging pathways underlying multiple aging-related diseases in humans. The ordinal logistic regression was used to cross-sectionally investigate transcriptomics of the comorbidity index in 1264 community-based Multi-Ethnic Study of Atherosclerosis (MESA) adults, 47% Caucasian, 32% Hispanic, 21% African American, and 51% female, aged 55–94 years. The comorbidity index was defined as the number of prevalent aging-related diseases including cardiovascular disease, type-2 diabetes, hypertension, cancer, dementia, chronic kidney disease, chronic obstructive pulmonary disease, and hip fracture. We identified 708 gene transcripts associated with the comorbidity index (FDR < 0.05) after adjusting for age, sex, ethnicity, and study site. In a weighted gene co-expression network analysis, as postulated, aging-related declines in apoptosis/autophagy (OR = 1.21 per SD increment, p = 0.0006) and ribosome/mitochondrion (OR = 0.90 per SD increment, p = 0.05) were positively associated with the comorbidity index. After adjusting for multiple comparisons, we identified 10 comorbidity-associated modules (FDR < 0.05), including the module of apoptosis/autophagy. There were three inter-correlated modules of these 10 involved in the complement subcomponent C1q, Fc gamma receptor I, and Fc gamma receptor III of the immune system, respectively. Aging-related upregulation of these three modules was positively associated with the comorbidity index. The odds of comorbidity increased with more of these modules acting together in a dose–response fashion. In conclusion, transcriptomic analysis of human immune cells may identify biomarker panels indicative of comprehensive biological mechanisms, especially immune signaling pathways, contributing to health aging.
Supplementary Information
The online version contains supplementary material available at 10.1007/s11357-022-00608-1.
Keywords: Monocyte, Transcriptomic, Aging, Comorbidity
Introduction
More than half of older adults have two or more chronic diseases [1]. With an expanding older population, there is a pressing need to improve health care for those with multiple chronic diseases [2]. The concept of geroscience holds that human health span and life span could be increased by directly targeting aging-related pathways as an alternative to targeting specific pathways for specific diseases. The hope is that by targeting the biology of aging, multiple age-related diseases/conditions could be affected simultaneously. Indeed, manipulations in energy and nutrient intake and at least five pharmacologic compounds approved for human use extend health span and life span in rodent models [3–7]. A growing body of findings from model systems supports the geroscience concept; however, evidence in humans is developing more slowly.
The pace of work in humans is slowed by the lack of appropriate biomarkers that can be used to evaluate the promise of geroscience-inspired interventions. There are many approaches being assessed including looking at changes in age-associated methylomic patterns, fluid biomarkers, and cellular markers of biologic age like telomere length [8]. Cellular transcriptomic patterns reflect genetic and internal and external environmental influences and may provide an integrated view of how these entities interact to influence the molecular mechanisms of the aging process. A key aspect of biomarker development is to show that candidate biomarkers broadly reflect organismal aging including associations with a variety of age-related diseases and health conditions.
Here, for the first time, we use transcriptomic profiles of immune cells (i.e., peripheral monocytes) to identify fundamental biological mechanisms of the aging process and related diseases in humans. The monocyte was chosen as the cell of interest both because of its relative accessibility and its key role in the pathogenesis of a number of age-related health conditions such as cardiovascular disease, cancer, neurodegenerative disease, type-2 diabetes, and osteoporosis [9]. We have previously reported on monocyte gene networks associated with chronologic age in 1264 participants (aged 55–94 years) in the Multi-Ethnic Study of Atherosclerosis (MESA). That analysis identified 2704 age-associated transcripts, which over-represented several modules including apoptosis/autophagy and ribosome/mitochondrion [10]. Alterations in autophagic flux [11] and mitochondrial bioenergetics have been recognized to be key contributors to the aging process and the development of aging-related diseases [12, 13].
In this paper, we test the hypothesis that the same age-related gene modules will be associated with the presence of multiple chronic health conditions both individually and the number of health conditions overall (i.e., multimorbidity). We also present exploratory analyses to identify additional gene modules associated with multimorbidity to provide additional clues underlying processes that may be in common to multiple age-related health conditions.
Methods
Study population
The Multi-Ethnic Study of Atherosclerosis was designed to investigate the progression of subclinical cardiovascular disease in a community-based cohort of 6814 participants. Since its inception in 2000, clinic visits (exams) and phone follow-up contacts collected extensive clinical, socio-demographic, lifestyle and behavior, and laboratory data [14]. The present analysis is based on analyses of purified monocyte samples from April 2010 to February 2012 examination (exam 5) of 1264 randomly selected MESA participants from four MESA field centers (Baltimore, MD; Forsyth County, NC; New York, NY; and St. Paul, MN). At exam 5, these participants also underwent assessment of cardiovascular disease, type-2 diabetes, and other aging-related diseases. The study protocol was approved by the Institutional Review Board at each site. All participants signed informed consent.
Transcriptomic profiling of peripheral monocytes
Transcriptomic profiling of peripheral monocytes has been described previously [15]. Briefly, monocytes were isolated with anti-CD14 monoclonal antibody–coated magnetic beads, respectively, using AutoMACs automated magnetic separation unit (Miltenyi Biotec, Bergisch Gladbach, Germany). RNA was isolated from samples using the AllPrep DNA/RNA Mini Kit (Qiagen, Inc., Hilden, Germany). The Illumina HumanHT-12 v4 Expression BeadChip and Illumina Bead Array Reader were used to perform the genome-wide expression analysis, following the Illumina expression protocol. To avoid potential biases to chip effects, a random sampling was used to assign individual samples to specific BeadChips. These data have been deposited in the NCBI Gene Expression Omnibus and are accessible through GEO Series accession number (GSE56045).
Data pre-processing and QC analyses were performed in R (http://www.r-project.org/) using Bioconductor (http://www.bioconductor.org/) packages. The Illumina HumanHT-12 v4 Expression BeadChip included 48 K transcripts. Statistical analyses were limited to probes retained after applying the following QC elimination: non-detectable expression in ≥ 90% of MESA samples using a detection p-value cutoff of 0.0001, overlap with a repetitive element or region, low variance across the samples (< 10th percentile), or putative and/or not well-characterized genes, i.e., gene names starting with KIAA, FLJ, HS, Cxorf, MGC, or LOC. We included 14,619 gene transcripts (10,898 unique genes) for analysis. These pre-processing pipelines effectively removed large effects of sample position on chips; however, probe-specific chip effects were found for some gene transcripts. These probe-specific effects were included as covariates in all analyses.
Assessment of aging-related diseases and related variables
A comorbidity index was defined, similar to that used in the Multimorbidity Clinical Trials Consortium [16], as the number of prevalent aging-related diseases, including cardiovascular disease, type-2 diabetes, hypertension, cancer, dementia, chronic kidney disease, chronic obstructive pulmonary disease, and hip fracture. Prevalent cardiovascular disease was based on a past history of myocardial infarction, angina (which included definite angina and probable angina if coronary revascularization was performed at the same time or afterwards), resuscitated cardiac arrest, or stroke. Cardiovascular events are adjudicated using all death certificates, medical records for all hospitalizations, and selected outpatient cardiovascular diagnoses and procedures. Prevalent type-2 diabetes was defined as fasting glucose ≥ 7.0 mmol/L (≥ 126 mg/dL) or use of hypoglycemic medication, and impaired fasting glucose was defined as fasting glucose 5.6–6.9 mmol/L (100–125 mg/dL). Fasting serum glucose at each examination was measured by rate reflectance spectrophotometry using thin-film adaptation of the glucose oxidase method on the Vitros analyzer (Johnson & Johnson Clinical Diagnostics, Rochester, NY). Prevalent hypertension was defined as systolic blood pressure ≥ 130 mm Hg or diastolic blood pressure ≥ 85 or taking anti-hypertension medication. Blood pressure was measured in the right arm of the participant after 5 min in a sitting position with a Dinamap model Pro 100 automated oscillometric sphygmomanometer (Critikon, Tampa, FL). The second and third of three readings were averaged to obtain the blood pressure levels. Prevalent cancer, dementia, chronic kidney disease, chronic obstructive pulmonary disease, and hip fracture will be defined on the basis of the ICD codes abstracted directly from hospital records.
Detailed sociodemographic information including age, sex, ethnicity, education, smoking status, alcohol drinking, and statin use was obtained. Sedentary was defined as the total intentional moderate and vigorous exercise score < the median. Obese was defined as body mass index ≥ 30 kg/m2, and overweight was defined as body mass index 25.0–29.9 kg/m2. High triglyceride was defined as ≥ 150 mg/dL or taking cholesterol-lowering medication. Low HDL cholesterol was defined as < 40 mg/dL for male or < 50 for female or taking cholesterol-lowering medication. High IL6 was defined as plasma IL-6 > the median (1.19 pg/mL). High CRP was defined as plasma CRP > the median (1.94 mg/L).
Association analysis
Transcriptional profiles were analyzed to identify modules of highly correlated transcripts using the stability analysis–enhanced weighted gene co-expression network analysis (WGCNA) [17]. We first obtained a weighted network based on the pairwise correlations among all transcripts considered, with an adjustment to produce a scale-free topology. Then, using the topological overlap measure to estimate the network interconnectedness, the transcripts were hierarchically clustered. With respect to the default parameters of WGCNA, we changed only the correlation type from Pearson to biweight midcorrelation (which is more robust to outliers) and set the minimum size for module detection to 10. A total of 40 modules were generated. The eigengene score was defined as the first principal component of a transcriptional module. The Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics Resources 6.7 was used to examine enrichment of pathway genes in each module [18].
We first examined the relationship between the two previously identified aging-associated modules, involved in apoptosis/autophagy and ribosome/mitochondrion, respectively [10], and the comorbidity index using ordinal logistic regression at the significance level of p < 0.05. Covariates were age, sex, ethnicity, study site, technical covariates (express chip), and residual sample contamination with non-targeted cells (e.g., non-monocytes). The top aging-associated module was involved in apoptosis and autophagy. We also discussed another aging-associated module, which was enriched with ribosome/mitochondrion genes. Here, we further investigated these two aging-related modules in relation to aging-related diseases.
To identify other gene transcripts associated with comorbidity burden, we fit separate linear regression models using the linear model (lm) function in R with each transcript expression as a predictor of the comorbidity index. p-values were adjusted for multiple testing using the q-value false discovery rate (FDR) method [19]. To minimize false-positive results in the genome-wide analyses of the tested gene transcripts, we applied a commonly used FDR threshold of 0.05. We then associated the eigengene for each consensus module with the comorbidity index using ordinal logistic regression (FDR < 0.05).
Results
Characteristics of the study population
The study population comprised 1264 community-based adults, 47% Caucasian, 32% Hispanic, 21% African American, and 51% female, aged 55–94 years. The prevalence of aging-related diseases was 13%, 15%, 61%, 12%, 1%, 8%, 2%, and 0.6% for cardiovascular disease, type-2 diabetes, hypertension, cancer, dementia, chronic kidney disease, chronic obstructive pulmonary disease, and hip fracture, respectively. While only 30% of individuals were disease free, 43%, 16%, 7% (N = 92), 3% (N = 38), 0.6% (N = 7), and 0.2% (N = 3) had 1, 2, 3, 4, 5, and 6 diseases, respectively. Older persons, men, and non-Caucasians were more likely to have multiple aging-related diseases. These comorbidities often occurred in persons with lower education and less physical activities, alcohol abstainers, individuals with obesity, higher triglycerides, lower HDL cholesterol, higher IL6 and CRP, and statin users (Table 1).
Table 1.
Population characteristics, means (SD) or percent, by comorbidity, the Multi-Ethnic Study of Atherosclerosis, 2010–2012
| Comorbidity | p* | ||||
|---|---|---|---|---|---|
| 0 (N = 383) | 1 (N = 542) | 2 (N = 200) | 3–6 (N = 140) | ||
| Age, year | 65.28 (8.5) | 70.32 (8.89) | 72.21 (9.89) | 75.03 (7.9) | < 0.001 |
| Female | 49.61 | 54.98 | 54 | 38.57 | 0.005 |
| Caucasian | 53.79 | 44.46 | 41 | 43.57 | 5.42E − 06 |
| Hispanic | 33.68 | 29.34 | 30.5 | 37.86 | |
| African American | 12.53 | 26.2 | 28.5 | 18.57 | |
| Less than high school | 13.09 | 14.05 | 18 | 17.86 | 0.001 |
| High school or some college | 45.55 | 54.53 | 56.5 | 56.43 | |
| College degree or higher | 41.36 | 31.42 | 25.5 | 25.71 | |
| Sedentary | 37.44 | 52.97 | 57.79 | 63.31 | < 0.001 |
| Never smoker | 50.13 | 49.81 | 48.49 | 42.34 | 0.20 |
| Former smoker | 40.32 | 42.86 | 40.4 | 51.09 | |
| Current smoker | 9.55 | 7.33 | 11.11 | 6.57 | |
| Current drinker | 48.3 | 46.94 | 37.69 | 39.57 | 0.04 |
| Normal weight | 26.11 | 16.79 | 21.61 | 13.57 | < 0.001 |
| Over weight | 41.78 | 39.11 | 28.64 | 35 | |
| Obese | 32.11 | 44.1 | 49.75 | 51.43 | |
| High triglycerides | 44.39 | 51.29 | 50 | 58.57 | 0.03 |
| Low HDL cholesterol | 49.48 | 47.05 | 51.5 | 66.43 | 0.003 |
| High IL6 | 37.97 | 49.91 | 62.89 | 64.03 | < 0.001 |
| High CRP | 37.26 | 52.08 | 65.99 | 59.42 | < 0.001 |
| Statin use | 9.16 | 15.68 | 20.10 | 22.86 | < 0.001 |
*The trend analysis for continuous variables and the chi squared test for categorical variables
Hypothesized apoptosis/autophagy and ribosome/mitochondrion modules
We previously identified 24, out of 40, modules associated with chronologic age (FDR < 0.05). For convenience, we refer to each module by a color name. Steelblue was the top aging-associated module (FDR = 8.86E − 25), followed by darkmagenta (FDR = 9.10E − 11) and brown (FDR = 2.42E − 09). The steelblue module, enriched with the transcription factor activity (KLF6, TSC22D3, KLF9, CEBPD, CSRNP2, MXD1, and AHR), contained 16 transcripts. We previously described apoptosis and autophagy machinery in this module, including inhibitors of apoptosis and autophagy such as MCL1 (myeloid cell leukemia sequence 1) [10]. The positive association of steelblue with age indicated aging-related decline in apoptosis and autophagy. The brown module, enriched with the ribosome (N = 69) and mitochondrion (N = 147), contained 711 transcripts. Aging-related decline in ribosome and mitochondrion was also discussed previously [10].
As hypothesized, in ordinal logistic regression analyses, steelblue was positively associated with the comorbidity index while brown was inversely associated with the comorbidity index after adjusting for age, sex, ethnicity, study site, expression/chip, and residual sample contamination with non-monocytes (unless otherwise noted, the following analyses were conducted with the same adjustment) (Table 2). Due to small numbers of persons with more than 3 diseases, we combined persons with 3–6 diseases into one comorbidity group in all ordinal logistic regression analyses. After further adjusting for education, sedentary lifestyle, smoking status, alcohol drinking, body mass index, HDL cholesterol, and triglycerides, these associations were slightly attenuated (Table 3). In general, directions of these relationships between steelblue/brown and the comorbidity index were consistent across sex and ethnicity subgroups, even though p values for interactions of steelblue and sex, brown and sex, and brown and ethnicity were less than 0.05 (Table S1). Figure 1 shows the associations between steelblue and individual diseases assessed. We found that steelblue was positively associated with type-2 diabetes, chronic kidney disease, cancer, hip fracture, and hypertension. Brown was inversely associated with type-2 diabetes and was marginally inversely associated with chronic kidney disease and cancer. The associations of these two modules with the comorbidity index support deleterious roles of aging-related declines in apoptosis/autophagy and ribosome/mitochondrion.
Table 2.
Steelblue, brown, and other comorbidity-associated transcriptional modules, and enriched gene ontology (GO) terms (Benjamini p-value < 0.05) within each module
| Comorbidity* | Enriched gene sets | ||||
|---|---|---|---|---|---|
| OR (95% CI) | p value | FDR | GO terms | Benjamini p-value | |
| A priori modules | |||||
| Steelblue | 1.21 (1.09, 1.36) | 6.20E − 04 | 0.003 | Transcription factor activity | 0.01 |
| Brown | 0.90 (0.81, 1.00) | 0.05 | 0.14 |
Ribosome Mitochondrion |
1.75E − 29 5.66E − 29 |
| Other modules at FDR < 0.05 | |||||
| Darkgrey | 1.40 (1.25, 1.56) | 1.75E − 09 | 7.00E − 08 | ||
| Darkred | 1.41 (1.25, 1.59) | 1.07E − 08 | 2.14E − 07 | ||
| Sienna3 | 1.33 (1.19, 1.47) | 1.97E − 07 | 2.63E − 06 | Sterol metabolic process | 6.92E − 13 |
| Darkmagenta | 1.32 (1.29, 1.47) | 3.85E − 07 | 3.85E − 06 | ||
| Orangered4 | 1.31 (1.17, 1.46) | 1.15E − 06 | 9.18E − 06 | Fc gamma receptor I | 0.01 |
| Lightcyan | 1.27 (1.14, 1.42) | 2.59E − 05 | 1.73E − 04 | ||
| Mediumpurple3 | 1.22 (1.09, 1.35) | 2.85E − 04 | 0.002 | Complement subcomponent C1q chain A | 0.001 |
| Lightgreen | 1.18 (1.06, 1.32) | 0.003 | 0.01 | ||
| Yellowgreen | 1.16 (1.04, 1.29) | 0.008 | 0.03 | Chromatin assembly | 5.10E − 20 |
*Per SD increment in the eigengenes of these modules adjusting for age, sex, ethnicity, study site, expression/chip, and residual sample contamination with non-monocytes
Table 3.
Steelblue, brown, and other comorbidity-associated transcriptional modules with and without adjusting for demographics and other factors
| Comorbidity* (model 1) | Comorbidity* (model 2) | |||||
|---|---|---|---|---|---|---|
| OR (95% CI) | p value | FDR | OR (95% CI) | p value | FDR | |
| A priori modules | ||||||
| Steelblue | 1.45 (1.30, 1.60) | 2.17E − 12 | 8.70E − 11 | 1.26 (1.13, 1.42) | 6.18E − 05 | 8.24E − 04 |
| Brown | 0.80 (0.72, 0.88) | 1.33E − 05 | 5.31E − 05 | 0.89 (0.79, 0.99) | 0.03 | 0.10 |
| Other modules at FDR < 0.05 | ||||||
| Darkgrey | 1.35 (1.22, 1.50) | 9.33E − 09 | 7.47E − 08 | 1.25 (1.12, 1.40) | 1.04E − 04 | 8.30E − 04 |
| Darkred | 1.42 (1.28, 1.58) | 1.43E − 11 | 2.86E − 10 | 1.32 (1.17, 1.49) | 9.59E − 06 | 1.92E − 04 |
| Sienna3 | 1.27 (1.14, 1.40) | 5.43E − 06 | 2.41E − 05 | 1.16 1.04, 1.30) | 0.009 | 0.04 |
| Darkmagenta | 1.39 (1.26, 1.54) | 2.71E − 10 | 2.71 − 09 | 1.36 (1.22, 1.52) | 3.19E − 08 | 1.28E − 06 |
| Orangered4 | 1.33 (1.19, 1.47) | 1.11E − 07 | 7.43E − 07 | 1.20 (1.07, 1.35) | 0.001 | 0.01 |
| Lightcyan | 1.41 (1.27, 1.57) | 1.16E − 10 | 1.55E − 09 | 1.26 (1.12, 1.41) | 9.27E − 05 | 8.30E − 04 |
| Mediumpurple3 | 1.30 (1.17, 1.44) | 6.81E − 07 | 3.89E − 06 | 1.18 (1.06, 1.32) | 0.004 | 0.02 |
| Lightgreen | 1.27 (1.15, 1.41) | 3.54E − 06 | 1.77E − 05 | 1.15 (1.02, 1.29) | 0.02 | 0.07 |
| Yellowgreen | 1.12 (1.01, 1.23) | 0.03 | 0.08 | 1.13 (1.01, 1.26) | 0.03 | 0.10 |
*Per SD increment in the eigengenes of these modules: model 1: adjusting for expression/chip and residual sample contamination with non-monocyte and model 2: adjusting for age, sex, ethnicity, study site, education, sedentary lifestyle, smoking status, alcohol drinking, body mass index, HDL cholesterol, triglycerides, expression/chip, and residual sample contamination with non-monocytes
Fig. 1.
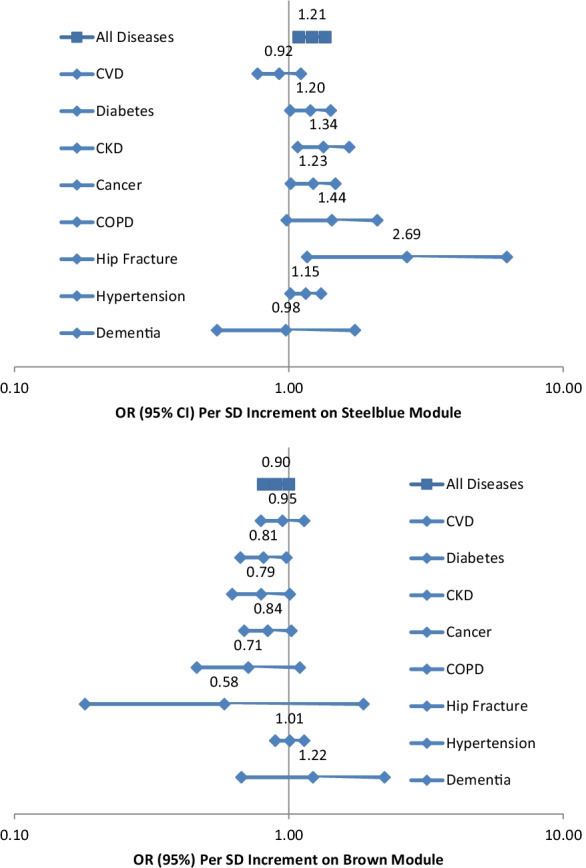
Associations of the steelblue and brown modules with comorbidity and individual aging-related diseases after adjusting for age, sex, ethnicity, study site, expression/chip, and residual sample contamination with non-monocytes
Transcripts associated with aging and related diseases
In a genome-wide analysis of peripheral monocytes using linear regression (Fig. 2), we identified 708 gene transcripts associated with the comorbidity index (FDR < 0.05). Table S2 shows the associations of individual transcripts within the steelblue and brown modules with the index. Among these 708 comorbidity-associated transcripts, 398 were also associated with aging (FDR < 0.05). Of the top 10 index-associated transcripts, FAM129A, PTGER2, CACNA2D3, and TEX2 were associated with aging (FDR < 0.05). Overall, the majority of comorbidity-associated transcripts were associated with chronologic age.
Fig. 2.

Manhattan plot showing the significance of mRNA expression association with comorbidity after adjusting for age, sex, ethnicity, study site, expression/chip, and residual sample contamination with non-monocytes, with the negative logarithm of the association p-values displayed on the Y-axis and transcripts along the X-axis. The dotted line represents the association significance of FDR = 0.05 (N = 708). The top 10 comorbidity-associated transcripts are also included
Modules related to immune function
Then, we conducted a comprehensive search for the comorbidity-associated modules after adjusting for multiple comparisons. Among the 40 modules, 10 including steelblue were positively associated with the comorbidity index in ordinal logistic regression analyses (FDR < 0.05) (Table 2). The metabolic, immune, transcription, and chromatin gene sets, which may reveal mechanistic pathways, were enriched in these modules. Out of these 10 modules, darkmagenta, lightcyan, mediumpurple3, steelblue, and lightgreen increased with aging (FDR < 0.05); in contrast, sienna3 and yellowgreen decreased with aging (FDR < 0.05) and darkgrey was unchanged with aging (FDR = 0.44) (Table S3). The increment with aging reached marginal significance for orangered4 (FDR = 0.06) and darkred (FDR = 0.06).
For monocytes, modules related to immune function such as mediumpurple3 implicate biologically plausible pathways leading to aging-related diseases. The mediumpurple3 module, enriched with the complement subcomponent C1q of the immune system (C1QB and C1QC), contained 5 transcripts. In fact, most transcripts within this module were positively (unless noted otherwise) associated with circulating measures of inflammatory biomarkers at baseline (about 10 years before transcriptomic profiling), IL6 (p = 0.002, < 0.001, 0.007, < 0.001 inverse association, and 0.81 for C1QC, LHFPL2, C1QB, ADAMTS5, and TIFA, respectively), and CRP (p = 0.002, 0.003, < 0.001, < 0.001 inverse association, and 0.07 for these transcripts, respectively).
As expected, the mediumpurple3 module was strongly correlated with another module related to immune function, the orangered4 module (Pearson correlation coefficient: 0.63, p < 0.001). The orangered4 module, enriched with the Fc gamma receptor I of the immune system (FCGR1C, FCGR1A, and FCGR1B), contained 10 transcripts. The mediumpurple3 module was also correlated with the lightgreen module (Pearson correlation coefficient: 0.33, p < 0.001). The lightgreen module contained 42 transcripts. There were two transcripts (FCGR3A and FCGR3B) in the lightgreen module, involved in the Fc gamma receptor III of the immune system.
Interestingly, the mediumpuple3 module was correlated with modules related to other biological functions, including yellowgreen (Pearson correlation coefficient: 0.28, p < 0.001) and sienna3 (0.21, < 0.001). The yellowgreen modules, enriched with the chromatin assembly (HIST1H2AC, HIST2H2AB, HIST2H2AA3, HIST2H2AA4, HIST1H2BD, HIST1H1C, HIST1H2BK, HIST2H2BE, HIST2H2AC, H2AFJ, and HIST2H4A), contained 14 transcripts. The sienna3 module, enriched with the sterol metabolic process (LDLR, CYP51A1, SQLE, HMGCS1, ABCA1, ABCG1, SC5DL, FDFT1, and SC4MOL), contained 15 transcripts.
Additive effects of multiple modules
We then focused on the 5 modules, which were positively associated with both the comorbidity index (FDR < 0.05) and age (FDR < 0.05), including the darkmagenta, lightcyan, mediumpurple3, steelblue, and lightgreen modules, as they may reflect fundamental mechanisms of the aging process underlying aging-related diseases. The darkmagenta module contained 16 transcripts, including TEX2 which was one of the top 10 cormorbidity-associated transcripts. The lightcyan module contained 46 transcripts, including PTGER2 which is one of the top 10 index-associated transcripts. The relationship between each of these 5 modules and the comorbidity index was consistent across sex and ethnicity subgroups (p for interactions > 0.05). Each of these 5 modules was associated with multiple aging-related diseases. To understand additive effects of these 5 modules, we created a combined risk score summarizing the score from each module (score = 1 if the eigengene greater than the module median, score = 0 if equal to or less than the median). The odds of comorbidity increased with a greater risk score in a dose–response fashion, with odds ratio of 5.5 comparing the most extreme risk groups (5 vs 0) (Fig. 3).
Fig. 3.
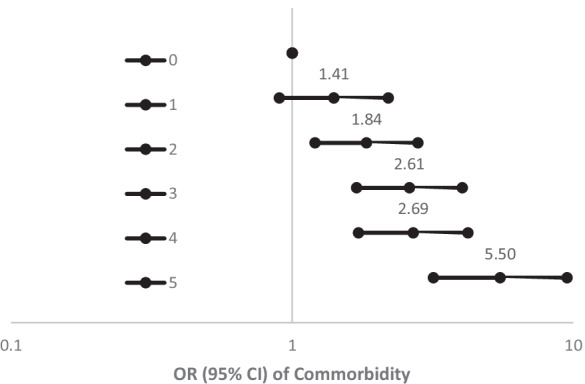
Association of the combined risk score (score = 1 if the eigengene greater than the module median, score = 0 if equal to or less than the median) from darkmagenta, lightcyan, mediumpurple3, steelblue, and lightgreen gene modules with comorbidity after adjusting for age, sex, ethnicity, study site, expression/chip, and residual sample contamination with non-monocytes
Discussion
The novel use of human peripheral monocytes enabled us to find that both the apoptosis/autophagy (steelblue) and ribosome/mitochondrion (brown) transcriptional modules in these immune cells were associated with aging-related diseases independent of chronological age. The transcriptomic analysis then identified 708 gene transcripts associated with aging-related diseases. In addition, after adjusting for multiple comparisons, a total of 10 transcriptional modules including the apoptosis/autophagy module were associated with aging-related diseases. These modules, especially those associated with chronological age, further implicate immune response and other biological pathways.
We demonstrated inverse relationships of apoptosis/autophagy and ribosome/mitochondrion with aging-related diseases. We previously reported that the top aging-associated module may be involved in decline of apoptosis and autophagy with aging in human peripheral monocytes [10]. Apoptosis may increase in some cell types with aging and decrease in other cell types [20]. Abundant evidence supports that autophagy declines with aging [21]. The aging-related disruption of apoptosis and autophagy has important pathophysiological consequences; however, human studies of their clinical implications are still lacking [22]. Now our data further linked inhibition of apoptosis and autophagy to aging-related diseases in humans. Another one of the top aging-associated modules, as we previously reported, may be involved in aging-related decline of ribosome and mitochondrion machinery (e.g., oxidative phosphorylation) in human peripheral monocytes [10]. Downregulation of mitochondrial genes is a common feature of aging in various human tissues [23, 24]. Increasingly, the role of mitochondrial dysfunction in the aging process has been studied, especially in animal experiments [13]. We extended these findings to demonstrate an association of downregulation of ribosome and mitochondrion genes with aging-related diseases in humans. Collectively, our findings provided initial epidemiological evidence that decline in apoptosis/autophagy and ribosome/mitochondrion may be fundamental mechanisms of the aging process underlying aging-related diseases.
A number of other transcriptional modules, involved in multiple biological pathways of the aging process, were also linked to aging-related diseases. Particularly, there were at least three modules involved in the complement subcomponent C1q (mediumpurple3), Fc gamma receptor I (orangered4), and Fc gamma receptor III (lightgreen) of the immune system. While Fc gamma receptor I and III stimulate phagocytes to clear antigens and release cytokines through binding to IgG antibodies [25], C1q’s binding to IgG antibodies further enhances these phagocytes’ functions [26]. In addition, Fc gamma receptor III (CD16) along with CD14 has been used to classify monocyte subsets and CD16 + monocytes are pro-inflammatory [27]. Our findings of the correlations among these three modules are consistent with the synergistic nature of their biological functions. The correlations of the immune module with the modules of sterol metabolic process and chromatin assembly further indicate the interplay among various biological pathways of the aging process. Their associations with serum inflammatory markers reveal the crosstalk between immune cells and circulating cytokines. Immunosenescence, characterized by a loss in the naïve T cell population and chronic inflammation, has been increasingly recognized as an important player in the aging process, but the role of innate immune cells is less studied [28]. Although transcriptomics of whole blood demonstrated upregulation of immune system with aging [24], our monocyte data fill the knowledge gap by delineating multiple innate immune signaling pathways in relation to aging-related diseases. While aging-related changes in abovementioned immune functions may be deleterious, other changes could be compensatory responses. Both sterol metabolic process (sienna3) and chromatin assembly (yellowgreen) decreased with aging, but were positively associated with aging-related diseases. The module of sterol metabolic process was the top obesity-associated module in one of our previous reports [15]. The downregulation of this module with aging may reflect aging-related decrease in fat mass in older adults [29]. Aging-associated histone loss has been observed in animal models and humans [30], but its clinical implications are not yet clear. Here, histone loss was linked with beneficial effects in the aging process. Taken together, transcriptomic analysis enables us to dissect complex innate immune pathways and other molecular mechanisms underlying aging-related diseases.
A distinction of this study lies in that the use of human peripheral monocytes provides unique perspective on the contribution of the innate immunity to the aging process. Another strength is to assess transcriptomic profiles in relatively homogeneous cells, as opposed to mixed cell types (e.g., whole blood) which may produce type I and type II errors [31, 32]. The large sample size enhances our abilities to examine biological pathways across the strength spectrum in relation to aging-related diseases. We did correlate a number of transcriptional biomarker sets indicative of biological plausible pathways with aging-related diseases. However, some caution in interpreting the results is warranted as this is a cross-sectional study. So, it is possible that some of the signals detected reflect a response to the presence of chronic disease pathology. We plan to explore whether these modules predict the incidence of multiple diseases as the next logical step. Future intervention studies are also needed to assess responsiveness of these biomarkers.
Here, we identified in human monocytes a number of coordinated sets of transcriptional biomarkers reflecting molecular mechanisms of the aging process including apoptosis/autophagy, ribosome/mitochondrion, immune function, and chromatin assembly. The findings of apoptosis/autophagy and ribosome/mitochondrion corroborate previously proposed aging hallmarks of loss of proteostasis and mitochondrial dysfunction, respectively [33]. Other findings on interconnected innate immune pathways add new pieces in the puzzle for the aging hallmark of altered intercellular communication. Still, some findings such as the positive association of histone levels and aging-related diseases pose new questions on the aging hallmark of epigenetic alteration. There is a critical need for biomarkers reflecting the aging process to investigate aging biology in human studies and evaluate efficacy of interventions in clinical trials [8]. Omics approaches allow us to depict multiple aging hallmarks in human research by examining millions of molecules simultaneously [34, 35]. Particularly, omics profiling of disease-relevant human cells is likely to generate a number of biomarker panels reflecting comprehensive molecular mechanisms including cell-specific pathways of the aging process and to facilitate the geroscience-based therapeutics to improve health span.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank the other MEA investigators, the staff, and the participants of the MESA study for their valuable contributions. A full list of participating MESA investigators and institutions can be found at http://www.mesa-nhlbi.org.
Funding
The MESA project is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support for MESA is provided by contracts 75N92020D00001, HHSN268201500003I, N01-HC-95159, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92020D00003, N01-HC-95162, 75N92020D00006, N01-HC-95163, 75N92020D00004, N01-HC-95164, 75N92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, UL1-TR-001420, UL1TR001881, DK063491, and R01HL105756. The MESA Transcriptomics Studies were funded by NIH grants R01HL101250, R01HL119962, R01DK101921, R01HL135009, RF1AG054474, and U01AG060897. This work was partially supported by P30 AG21332.
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Tinetti ME, Fried TR, Boyd CM. Designing health care for the most common chronic condition–multimorbidity. JAMA. 2012;307(23):2493–2494. doi: 10.1001/jama.2012.5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pearson-Stuttard J, Ezzati M, Gregg EW. Multimorbidity-a defining challenge for health systems. Lancet Public Health. 2019;4(12):e599–e600. doi: 10.1016/S2468-2667(19)30222-1. [DOI] [PubMed] [Google Scholar]
- 3.Harrison DE, Strong R, Allison DB, Ames BN, Astle CM, Atamna H, et al. Acarbose, 17-alpha-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell. 2014;13(2):273–282. doi: 10.1111/acel.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4:2192. doi: 10.1038/ncomms3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011;66(2):191–201. doi: 10.1093/gerona/glq178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Satoh A, Brace CS, Rensing N, Cliften P, Wozniak DF, Herzog ED, et al. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013;18(3):416–430. doi: 10.1016/j.cmet.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hwangbo DS, Lee HY, Abozaid LS, Min KJ. Mechanisms of lifespan regulation by calorie restriction and intermittent fasting in model organisms. Nutrients. 2020;12(4):1194. doi: 10.3390/nu12041194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Justice JN, Ferrucci L, Newman AB, Aroda VR, Bahnson JL, Divers J, et al. A framework for selection of blood-based biomarkers for geroscience-guided clinical trials: report from the TAME Biomarkers Workgroup. Geroscience. 2018;40(5–6):419–436. doi: 10.1007/s11357-018-0042-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karlmark KR, Tacke F, Dunay IR. Monocytes in health and disease - minireview. Eur J Microbiol Immunol (Bp) 2012;2(2):97–102. doi: 10.1556/EuJMI.2.2012.2.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reynolds LM, Ding J, Taylor JR, Lohman K, Soranzo N, de la Fuente A, et al. Transcriptomic profiles of aging in purified human immune cells. BMC Genomics. 2015;16(1):333. doi: 10.1186/s12864-015-1522-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146(5):682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 12.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 13.Haas RH. Mitochondrial dysfunction in aging and diseases of aging. Biology (Basel). 2019;8(2):48. doi: 10.3390/biology8020048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bild DE, Bluemke DA, Burke GL, Detrano R, Diez Roux AV, Folsom AR, et al. Multi-ethnic study of atherosclerosis: objectives and design. Am J Epidemiol. 2002;156(9):871–881. doi: 10.1093/aje/kwf113. [DOI] [PubMed] [Google Scholar]
- 15.Ding J, Reynolds LM, Zeller T, Muller C, Lohman K, Nicklas BJ, et al. Alterations of a Cellular Cholesterol Metabolism Network Are a Molecular Feature of Obesity-Related Type 2 Diabetes and Cardiovascular Disease. Diabetes. 2015;64(10):3464–74. doi: 10.2337/db14-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Espeland MA, Crimmins EM, Grossardt BR, Crandall JP, Gelfond JA, Harris TB, et al. Clinical trials targeting aging and age-related multimorbidity. J Gerontol A Biol Sci Med Sci. 2017;72(3):355–361. doi: 10.1093/gerona/glw220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(5):3. doi: 10.1186/gb-2003-4-5-p3. [DOI] [PubMed] [Google Scholar]
- 19.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tower J. Programmed cell death in aging. Ageing Res Rev. 2015;23(Pt A):90–100. doi: 10.1016/j.arr.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leidal AM, Levine B, Debnath J. Autophagy and the cell biology of age-related disease. Nat Cell Biol. 2018;20(12):1338–1348. doi: 10.1038/s41556-018-0235-8. [DOI] [PubMed] [Google Scholar]
- 22.Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15(2):81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Magalhaes JP, Curado J, Church GM. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. 2009;25(7):875–881. doi: 10.1093/bioinformatics/btp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peters MJ, Joehanes R, Pilling LC, Schurmann C, Conneely KN, Powell J, et al. The transcriptional landscape of age in human peripheral blood. Nat Commun. 2015;6:8570. doi: 10.1038/ncomms9570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ben Mkaddem S, Benhamou M, Monteiro RC. Understanding Fc receptor involvement in inflammatory diseases: from mechanisms to new therapeutic tools. Front Immunol. 2019;10:811. doi: 10.3389/fimmu.2019.00811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Son M, Diamond B, Santiago-Schwarz F. Fundamental role of C1q in autoimmunity and inflammation. Immunol Res. 2015;63(1–3):101–106. doi: 10.1007/s12026-015-8705-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong KL, Yeap WH, Tai JJ, Ong SM, Dang TM, Wong SC. The three human monocyte subsets: implications for health and disease. Immunol Res. 2012;53(1–3):41–57. doi: 10.1007/s12026-012-8297-3. [DOI] [PubMed] [Google Scholar]
- 28.Aiello A, Farzaneh F, Candore G, Caruso C, Davinelli S, Gambino CM, et al. Immunosenescence and its hallmarks: how to oppose aging strategically? A review of potential options for therapeutic intervention. Front Immunol. 2019;10:2247. doi: 10.3389/fimmu.2019.02247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding J, Kritchevsky SB, Newman AB, Taaffe DR, Nicklas BJ, Visser M, et al. Effects of birth cohort and age on body composition in a sample of community-based elderly. Am J Clin Nutr. 2007;85(2):405–410. doi: 10.1093/ajcn/85.2.405. [DOI] [PubMed] [Google Scholar]
- 30.Yi SJ, Kim K. New insights into the role of histone changes in aging. Int J Mol Sci. 2020;21(21):8241. doi: 10.3390/ijms21218241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong H, Yang X, Kaplan LM, Molony C, Schadt EE. Integrating pathway analysis and genetics of gene expression for genome-wide association studies. Am J Hum Genet. 2010;86(4):581–591. doi: 10.1016/j.ajhg.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H, et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325(5945):1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lorusso JS, Sviderskiy OA, Labunskyy VM. Emerging omics approaches in aging research. Antioxid Redox Signal. 2018;29(10):985–1002. doi: 10.1089/ars.2017.7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frenk S, Houseley J. Gene expression hallmarks of cellular ageing. Biogerontology. 2018;19(6):547–566. doi: 10.1007/s10522-018-9750-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.