

Abstract
Huntington’s disease (HD) is a neurodegenerative genetic disorder caused by a CAG repeat expansion in the huntingtin gene. HD causes motor, cognitive, and behavioral dysfunction. Since no existing treatment affects the course of this disease, new treatments are needed. Inflammation is frequently observed in HD patients before symptom onset. Neuroinflammation, characterized by the presence of reactive microglia, astrocytes and inflammatory factors within the brain, is also detected early. However, in comparison to other neurodegenerative diseases, the role of neuroinflammation in HD is much less known. Work has been dedicated to altered microglial and astrocytic functions in the context of HD, but less attention has been given to glial participation in neuroinflammation. This review describes evidence of inflammation in HD patients and animal models. It also discusses recent knowledge on neuroinflammation in HD, highlighting astrocyte and microglia involvement in the disease and considering anti-inflammatory therapeutic approaches.
Keywords: Huntington’s disease, mutant huntingtin, neuroinflammation, astrocytes, microglia, neurodegeneration
1. INTRODUCTION
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder whose onset occurs in adulthood with a progressive course of motor, cognitive, and psychiatric symptoms. It is caused by CAG repeat expansion in the huntingtin (Htt) gene leading to an expanded polyglutamine region in exon 1 of the protein [1]. This region exhibits CAG repeat polymorphism below 35 CAG repeats; between 35-39 CAG repeats, HD penetrance is incomplete, and above 40 CAG repeats, penetrance is complete. Htt is found in all human and rodent tissues, although it is more abundant in the central nervous system (CNS), where it is found predominantly in the cytoplasm of brain cells [2]. There are numerous Htt functions that play a role in the development, axonal transport, and cell survival [3]. Mutant Htt (mHtt) forms soluble fragments and aggregates that appear to be toxic to neurons, particularly to medium spiny neurons in the striatum, subsequently reaching other areas such as the cerebral cortex, whereas the cerebellum is usually spared from the neurodegenerative process [4]. Although several mechanisms have been implicated in HD pathology, why mHtt causes neurodegeneration remains unsolved. These mechanisms include defects in axonal transport, such as vesicle transport and recycling. Indeed, impaired vesicular trafficking of brain-derived neurotrophic factor (BDNF) has been reported [5], and it may contribute to the decreased BDNF levels observed in HD patients [6]. Also, excitotoxicity induced by overactivation of N-methyl-D-aspartate (NMDA) receptors [7], transcriptional dysregulation [8, 9], altered proteostasis [10], and mitochondrial dysfunction [11], have all been associated with HD. In addition, HD may result from cell-autonomous and non-cell-autonomous mechanisms [12]. Neuroinflammation is another mechanism possibly involved in HD pathogenesis. Nevertheless, the exact contribution of the inflammatory process to this disease is not completely understood. Here, we review evidence of inflammation in HD in humans and in animal models to help understand the role of neuroinflammation in HD.
2. HUNTINGTON’S DISEASE
The age of onset of HD in patients is around 45 years; the higher the number of CAG repeats in the Htt gene, the earlier the symptoms appear. Between 4.4 and 11.5% of HD subjects have a late onset (over 60 years), which implies a slow and benign progression of the disease [13]. In rare cases, when symptoms begin before the age of 20, the disease is termed juvenile Huntington's disease, and subjects present behavioral disturbances, school learning difficulties, and cerebellar symptoms associated with motor, speech, and language delay [14]. Presymptomatic HD patients show no signs or symptoms of motor or cognitive disturbances but might exhibit some changes in neuroimaging. Prodromal HD subjects begin to have subtle motor and cognitive disturbances. Behavioral changes may occur often even prior to motor symptom onset. Presymptomatic and prodromal HD subjects are both premanifest HD, i.e., before the motor diagnosis of HD, and can be examined to find therapeutical targets that delay or modify the course of the disease [15]. A two-year clinical study involving a presymptomatic HD cohort approximately 24 years prior to anticipated clinical onset reported normal brain function but an increase in sensitive measures of neurodegeneration, such as smaller caudate and putamen volumes [16].
After the disease is diagnosed based on motor symptoms, the manifest or symptomatic HD patients go through early, moderate, and advanced disease stages. The symptoms that characterize HD when the disease becomes manifest include motor, cognitive, and psychiatric dysfunctions [14, 15]. Motor disturbances comprise the characteristic unwanted involuntary movements, which tend to be progressive. Early in the disease, the patients are mostly hyperkinetic with involuntary chorea. In later stages, however, hypokinesia with bradykinesia and dystonia predominate. Motor disturbances in daily activities progress over time and may lead to difficulties in walking and standing, as well as frequent falls. Behavioral and psychiatric symptoms are often consistent with frontal lobe dysfunction. Initially, patients may present poor attention, impulsivity, and irritability. Later in the disease, there is prominent apathy, loss of intuition, and creativity. Psychosis can appear at later stages. Cognitive decline is one of the main features of HD and may also be present before the onset of motor disturbances. The cognitive changes are more prominent for executive functions, i.e., patients have difficulty in organizing, multitasking, and planning. These symptoms then progress to dementia. It was recently reported that specific cortical changes can predict a decline in motor and cognitive performance, contributing to regional variability of cortical atrophy [17].
The mHtt inclusions are a key neuropathological hallmark of HD since these structures are detected in HD brains [18]. Nuclear inclusions can be detected before symptom onset in presymptomatic gene carriers [19]. Degeneration in HD brains initially involves the dorsal striatum where medium spiny neurons present nuclear inclusions and dystrophic neurites, whereas striatal interneurons are selectively preserved. Postmortem analysis indicates that 95% of HD brains present bilateral atrophy of the striatum [20]. In the cerebral cortex, the number of cortical pyramidal neurons in layers III, V, and VI of the prefrontal cortex of HD patients is reduced [21]. Indeed, it was shown that neuron cell loss in the cerebral cortex is heterogeneous, explaining different symptom profiles found in HD [22, 23]. There is also a remarkable white matter loss in HD patients even before symptoms develop [4]. Interestingly, increased density of oligodendrocytes in the striatum may precede symptom onset [20]. Reactive gliosis is consistently seen in the striatum of HD patients, where reactive fibrillary astrocytosis and reactive microglia are detected [20]. Importantly, reactive astrogliosis is not detected in the rest of the brain structures even when there is atrophy [20]. Astrocyte reactivity is an early event in HD since it can be detected in the striatum of presymptomatic carriers, increasing with disease progression [24]. Also, activated microglia are detected in the striatum and cerebral cortex, showing a disease grade-dependent increase [25]. By contrast, there are no infiltrating lymphocytes in HD brains [20].
3. INFLAMMATION IN HD PATIENTS
The innate immune system activation induces an immediate generic response to a wide range of pathogens or stressors, whereas the adaptive immune system generates a highly specific response largely mediated by T and B lymphocytes. In HD, there is considerable evidence of activation of the innate immune cells both in the CNS and in the periphery, but very little information regarding the role of the adaptive immune system is available. Inflammation in HD can be triggered by the response of glial cells to neuron-autonomous degeneration or by the activation of immune cells resulting from the expression of mHtt or both at the same time. Neither the mechanism nor the contribution of inflammation to HD pathogenesis has been elucidated.
3.1. Neuroinflammation
Neuroinflammation, characterized by reactive gliosis and the production of soluble inflammatory factors in the CNS, is a component of several neurodegenerative disorders [26, 27]. In contrast to other neurodegenerative diseases, HD shows no significant infiltration of peripheral immune cells into the brain [20, 25]; therefore, major inflammatory processes are performed by activated microglia and astrocytes. Reactive astrocytes are observed in presymptomatic HD and correlate with disease severity [24], whereas reactive T cells are not detected in HD brains [28]. Reactive microglia are present in the striatum and cortex of HD brains, where neuron loss is observed in postmortem studies [25]. Positron emission tomography (PET) imaging has the potential to detect changes in patients’ brains at the early stages of the disease. Neuroinflammation can be inferred from the detection of translocator protein (TSPO) expression, which is expressed at low levels in healthy CNS but upregulated by microglia and astrocytes in neuroinflammatory processes [29]. PET imaging shows increased levels of TSPO tracer in vivo in HD striatum correlative with HD severity, which was attributed to microglia activation [30]. Moreover, activation of microglia was also present in presymptomatic gene carriers [31], indicating that it is an early event in HD pathogenesis. However, these studies have been carried out using a second-generation TSPO tracer with a poor signal-to-background ratio. A recent study, involving another tracer with a high signal-to-background ratio, performed PET imaging on HD patients and found that microglial activation is significant in manifest HD but not in premanifest HD [32]. Nevertheless, this study had an important limitation because it included a small group of patients.
Inflammatory mediators are found in the CNS and the cerebral spinal fluid (CSF) of HD subjects (Table 1) [33-56]. In postmortem striatal tissue of HD patients, IL-6, IL-8, and tumor necrosis factor (TNF)-α levels have been found to be up-regulated [33]. Another report analyzed three brain regions: striatum, cerebral cortex, and cerebellum, with the last one usually less affected in HD patients. It showed significantly increased levels of IL-10 and CCL2 (also known as MCP-1) mRNA and a trend towards an increase in IL-1β and TNF-α mRNA, only in the striatum of HD patients; IL-8 mRNA was augmented in the striatum and cortex, whereas IL-6 and MMP9 exhibited increased mRNA levels in the cortex, striatum, and cerebellum [28]. IL-1β was found to be increased in cortical tissue of manifest HD subjects [42]. Surprisingly, only one study showed decreased levels of several cytokines, such as IL-1β, IL-8, and TNF-α [40].
Table 1.
Inflammation in HD patients.
| HD Patient Sample | Increased | Decreased | Unchanged | Refs. |
|---|---|---|---|---|
| CNS | - | - | - | - |
| Striatum mRNA | IL-6, IL-8, TNF-α IL-6, IL-8, MMP9, IL-10, CCL2/MCP1 IL-6, TLR2 C1q, C4, C3, clusterin, C5aR, C3aR C3, C4a, C4b, GFAP, YKL-40, Cx43 |
CX3CL1 | IL1-β, TNF-α | [33] [28] [34] [35] [8] [36] |
| Striatum protein | TLR4 (putamen) TGF-β1 (caudate) TGF-β1 (caudate) CCL5/RANTES (caudate) |
CX3CL1 (putamen) IL1-β, IL-8, TNF-α, CCL5, CCL4/MIP1β |
TREM2 (putamen) | [37] [38] [39] [36] [40] [41] |
| Cortex mRNA | IL-6, IL-8, MMP9 TLR2 Cx43, YKL-40 |
- | IL-6 | [28] [34] [8] |
| Cortex protein | C1q, C3, C4 CCL5/RANTES IL1-β |
TGF-β1 | TLR4, TREM2 | [37] [38] [35] [41] [42] |
| Cerebellum mRNA | IL-6, MMP9 C4b |
- | - | [28] [8] |
| CSF premanifest | YKL-40 | IL-6, IL-8 | [16] | |
| CSF manifest | IL-6, YLK-40, chitotriosidase Clusterin C1qc, C2, C3, YKL-40(CHI3L1) sCD27 MMP3, MMP9 IL-17, LT-α, VEGF |
IL-7, IL-16 | IL-8 YKL-40 i TIMP, MMP1 |
[43] [44] [45] [46] [47] [48] |
| Microglia differentiated from human PCSs | IL-6, TNF-α | - | IL1-β, IL-8, IL-10i | [49] |
| PERIPHERAL TISSUES | - | - | - | |
| Serum | C3, IgA, sTNF-R, sIL-2R, neopterin | TGF-β1 tryptophan |
TGF-β1 IgG, IgM, C4 |
[38] [50] |
| Plasma | IL-4, IL-5, IL-6, IL-8, TNF-α Clusterin TGF-β1, IL-6, VEGF, MMP8 CCL2/MCP1, MIP1β, eotaxin, eotaxin-3, MCP4 |
IL-18 C-reactive protein |
IgG, IgA, IgM IL-16, MMP3, MMP10, TIMP-2, MIP1α, MIP3β |
[33] [44] [51] [52] [53] |
| Plasma (premanifest HD) | IL-1β C-reactive protein in premanifest HD |
IL-6, IL-8, TNF-α i TNF-α |
[54] [55] |
|
| - | - | - | - | |
| Monocytes-macrophagess | IL-6 | - | - | [33] |
| Monocytes-macrophagess | IL-6, IL-8, TNF-α | - | - | [56] |
| Unstimulated monocytes | - | - | IL-6, TLR2 | [34] |
| Unstimulated lymphocytes and granulocytes | - | - | TGF-β1 expressing cells | [39] |
| Unstimulated monocytes | - | TGF-β1 expressing cells in premanifest HD | TGF-β1 expressing cells in manifest HD | [39] |
s=stimulated with LPS+IFN, i=increasing trend, PSC=pluripotent stem cells.
Examination of CSF from manifest HD showed elevated levels of IL-6, chitinase 3-like protein-1 (CHI3L1, also known as YKL-40), and chitotriosidase, no change in IL-8 levels, and no detectable IL-1β or TNF-α levels [43]. YKL-40 and chitotriosidase are glycosyl hydrolases secreted from immune cells, such as macrophages, astrocytes, and microglia, whose expression is increased in several CNS disorders [57, 58]. YKL-40 mRNA levels are markedly increased in human astrocytes and macrophages, and can also be induced by pro-inflammatory factors [59]. Remarkably, CSF levels of YKL-40 were higher in manifest HD subjects and correlated with the disease stage and motor score of HD patients [43]. Moreover, a premanifest HD cohort, estimated to be around 24 years before predicted clinical disease onset, was recently shown to have smaller caudate and putamen volumes together with high levels of YKL-40 in CSF [16].
Clusterin, whose expression is induced by inflammation, functions as a chaperone-like protein clearing aggregates, and it was suggested to prevent the Aβ deposition characteristic of Alzheimer’s disease [60]. Clusterin is involved in complement system regulation and its levels are increased in the caudate nucleus of HD brains [35]. Indeed, several complement pathway factors have been observed to be abundantly expressed in the HD striatum and absent in the cerebral cortex [35]. Clusterin was also increased in the CSF of HD subjects, whereas cytokines, such as IL-1β and TNF-α, were not detected [44]. Also, complement components were elevated in HD CSF [45].
Differential gene expression in the caudate, prefrontal cortex, motor cortex, and cerebellum of HD patients was examined by microarray analysis. Interestingly, HD caudate had the largest number of mRNA changes; also, the motor cortex exhibited changes, but the prefrontal cortex and cerebellum showed to be relatively unchanged [8]. Among the top 30 regulated genes increased in the striatum of HD subjects were inflammatory factors, such as complement proteins 3 (C3), C4a, and C4b, and glial fibrillary acidic protein (GFAP), whereas C4b was upregulated in the cerebellum [8].
Toll-like receptors (TLRs) are pattern recognition receptors that identify non-self-molecules and then activate immune system cells, resulting in cytokine production. TLR2 and 4 are expressed primarily by microglia and, to a lesser extent, by astrocytes, oligodendrocytes, and neurons [61]. HD brains show increased mRNA levels of IL-6 and TLR2 in the striatum, but only TLR2 is elevated in the cerebral cortex [34]. Recently, TLR4 protein was reported to be increased in the putamen of a small cohort of HD patients, whereas no differences were found in cortical tissue [37]. Interestingly, the authors found two TLR4 polymorphisms to be associated with the rate of motor decline in HD patients [37].
In human putamen, mRNA and protein levels of CX3CL1 chemokine (also known as fractalkine) were found to be decreased [36]. Transforming growth factor- β (TGF-β) is a multifunction cytokine with actions in the innate and adaptive immune responses and in neuroprotection [62]. TGF-β1 immunoreactivity is increased in the HD striatum and decreased in the HD cortex [38]. A more recent study showed TGF-β1 immunoreactive cells to be increased with disease severity and colocalized with a gradual increase in GFAP immunoreactive astrocytes, whereas microglial marker ionized calcium-binding adaptor molecule 1 (Iba1) showed no colocalization, suggesting that astrocytes upregulate TGF-β1 in the HD brain [39].
CCL5/RANTES, a chemokine with low levels of expression in the CNS, is also involved in neuroinflammation [63]. CCL5 immunoreactivity was found to be associated with GFAP in the caudate nucleus and frontal cortex of HD patients, whereas non-HD controls showed low or no reactivity [41].
Matrix metalloproteinases (MMP) are produced by glial cells in inflammatory processes and can induce neurotoxicity. MMPs were examined as potential modifiers of Htt proteolysis and toxicity in HD [64]. In fact, MMP9 expression was increased in the striatum, cortex, and cerebellum of HD patients [28]. MMP9 and MMP3 were found to be elevated in CSF of HD patients, which correlated with the subject’s motor score [47]. MMP3 is secreted by neurons and acts as an inflammatory signal, inducing cytokine release from microglia [65]. Hence, elevated MMPs in CSF support activation of microglia and astrocytes in the HD brain.
A recent study evaluated the expression of sCD27, a marker of adaptive immunity that reflects T-cell mediated neuroinflammation, but although its levels were increased in the CSF of HD subjects, they did not correlate with clinical scores [46]. The CSF of HD subjects was shown to have decreased IL-7 and IL-16 levels and increased IL-17 and lymphotoxin-α levels, together with a higher frequency of CD4-positive T cells and Th17.1 cells in premanifest HD, providing evidence of lymphocyte activation in HD [48]. Recently, human HD pluripotent stem cells were differentiated into microglial-like cells and showed increased IL-6 and TNF-α release when stimulated with bacterial lipopolysaccharide (LPS) plus interferon-γ (IFN-γ) [49]. Altogether, the results reflect innate immune activation in HD, indicating an inflammatory environment in the HD brain. This effect is far more evident in the striatum of HD subjects but is also present in the cerebellum, suggesting that inflammation may impact brain regions differently.
3.2. Peripheral Inflammation
The value of inflammatory factors as biomarkers of HD progression is still under investigation; nevertheless, several factors produced by innate immune cells are elevated in the plasma of HD patients, indicating that there is immune peripheral activation (Table 1). Cytokines IL-6, IL-8, and TNF-α [33], as well as clusterin [44], VEGF, and MMP8 [51] are higher in the plasma of HD patients. Plasma clusterin levels increase in parallel with CSF clusterin levels.
TGF-β1 has neuroprotective and anti-inflammatory properties, and its peripheral levels may depend on the production by monocytes/macrophages. TGF-β1 levels were reported to decrease in plasma of asymptomatic HD, whereas they did not change in symptomatic HD patients [38]. In another report, the same group demonstrated that lymphocytes and granulocytes expressing TGF-β1 were similar in control and HD patients, but macrophages expressing TGF-β1 in premanifest HD individuals were lower in comparison to control patients [39]. This decrease was not observed in manifest HD patients with more severe disease scores. However, the plasma of manifest HD subjects also showed increased levels of TGF-β1, whereas no change was detected in premanifest individuals [51]. Regarding chemokines, very little information is available, although chemokines, such as CCL4/MIP1β, CCL2/MCP1, CCL13/MCP4, CCL11/ eotaxin, and CCL26/eotaxin-3, were found to be elevated in HD plasma [52] while others showed no change [51]. However, it is still unknown how chemokine alterations might influence HD pathology.
Regarding peripheral immune cells in HD, IL-6, IL-8, and TNF-α were elevated in HD blood monocytes and macrophages from premanifest, early, and late manifest HD patients stimulated in vitro with LPS and IFN [33, 56]. Interestingly, in this study, human HD myeloid cells showed activation of the NF-κB pathway caused by mHtt interaction with IKKBγ, leading to degradation of the inhibitor IκB and consequent NF-κB activation [56]. Since NF-κB is a master regulator of the inflammatory response, its activation could result in pro-inflammatory cytokine production. Moreover, myeloid cells derived from HD patients treated with Htt-lowering siRNA showed decreased inflammatory cytokine levels and NF-κB translocation [56], thereby confirming that mHtt expression in immune cells induces the activation of these cells. In fact, mHtt levels in monocytes and T cells correlate with disease burden scores and caudate atrophy in HD patients [66]. Although HD individuals had increased mRNA levels of IL-6 and TLR2 in the striatum, no change was detected in unstimulated monocytes [34]. Regarding adaptive immune cells, T cell activation, proliferative response, and cytokine release upon stimulation [67], as well as plasma levels of immunoglobulins, were unchanged in patients [33]. However, one study showed higher serum levels of IgA than controls [50]. This study also examined soluble receptor levels of TNF-α and IL-2, neopterin, C3, which were higher in HD patients, whereas tryptophan levels were decreased [50]. In premanifest HD patients, although plasma levels of IL-6, IL-8, and TNF-α showed an increasing trend, only IL-1β levels were increased [54]. Another study showed C-reactive protein to be elevated in premanifest HD, although no changes in TNF-α were observed [55]. In manifest HD subjects, C-reactive protein plasma levels were found to be decreased, although several other factors did not change [53].
Among all the factors reported to increase in HD, IL-6 and TNF-α seem to be most consistently elevated in the plasma of manifest HD patients, which may be caused by activated innate immune cells. Further studies are needed to determine how the immune system responds to HD. Hopefully, more uniform methods to assay patients’ samples that yield more consistent results will contribute to the design of therapies targeting the immune system centrally or peripherally. In conclusion, inflammation is observed in the brain and peripheral tissues of HD patients. Whether it is the cause or the consequence of neuron death is still a matter of debate.
4. HD RODENT MODELS
Animal models of HD have been very helpful in unraveling some mechanisms of neurodegeneration. An animal model of human disease must recreate the symptoms, pathological changes, and mechanisms observed in human patients. This is very difficult to achieve with a single model because rodents do not develop HD as humans do. Therefore, our current knowledge of this disease has been built on diverse studies utilizing different types of HD models with different characteristics.
The first animal models for studying HD were the non-genetic or toxic models. Although they are unrelated to the presence of poli-Q expanded proteins, they generate massive neuron death, which enables the analysis of potential neuroprotective treatments for HD. Excitotoxicity induced using kainic acid causes striatal pathology, but since it has side effects, it was later replaced by quinolinic acid (QA), an NMDA receptor agonist. QA is a downstream metabolite of the kynurenine pathway, a major catabolic route of dietary tryptophan in mammals. In the brain, tryptophan is taken up by astrocytes, microglia, and dendritic cells, and converted into kynurenine, and after several steps, it is transformed into QA. Since QA cannot cross the blood-brain barrier (BBB), it is usually administered in the striatum. QA causes striatal neurodegeneration by excitotoxicity in rats [68], mice [69], and primates [70], like in human HD [71], and although it does not affect interneurons, it induces astrogliosis [72] and motor and cognitive symptoms [73] similar to those of HD.
Selective death of striatal neurons can also be induced by mitochondrial dysfunction. 3-nitropropionic acid (3NP) is a natural product found in fungi and plants that acts as an irreversible inhibitor of mitochondrial complex II succinate dehydrogenase [74]. Since it crosses the BBB, systemically administered 3NP produces selective striatal neuron loss and motor and cognitive dysfunction similar to HD in rodents and nonhuman primates [75]. Specifically, the intraperitoneal administration of 3NP in murine models causes selective damage to striatal neurons with atrophy of the striatum [76] and enables the reproduction of HD hyperkinetic and hypokinetic symptoms depending on the time of administration and dose used [77]. 3NP increases the sensibility to basal levels of glutamate induced by the relief of blockade at the NMDA receptor and raises the number of electrons released by mitochondria, generating a rise in reactive oxygen species (ROS) production and oxidative stress, thereby leading to cell death [78]. Mitochondrial ROS can activate innate immunity [79], thus creating a vicious circle. 3NP-generated excitotoxicity and oxidative stress induce neuronal death by both necrosis and apoptosis [80], and these mechanisms may be related to neuroinflammation. We demonstrated that 3NP reduces the viability of PC12 neurons while increasing ROS levels and inducing apoptosis [81]. Also, neuronal cell lines from rat striatum expressing a fragment of human Htt with 15 or human mHtt with 120 CAG repeats exhibit decreased viability in response to 3NP [82, 83], implicating mitochondrial dysfunction as a mechanism of neurodegeneration.
Genetic models of HD are transgenic animals developed to carry either N-terminal fragment of human mHtt or the full-length human mHtt randomly inserted in its genome. This implies that mHtt expression depends on variable promoters and that transgenic mice express three copies of the Htt gene: two murine wild-type Htt and one human mHtt. R6/1 and R6/2 were the first transgenic N-terminal models described [84], and R6/2 is the most widely used HD model. R6/2 model was generated by insertion of a fragment of exon 1 of human mHtt with 144 CAG repeats under the control of human Htt promoter. This model shows a very aggressive phenotype with neuron loss and atrophy in the striatum and early onset of behavioral symptoms [85]. R6/2 mice die after 10 to 13 weeks and show nuclear inclusions of Htt [86]. Nevertheless, R6/2 mice show moderate neuronal death in contrast to human brains, which exhibit profound loss of striatal and cortical neurons [87]. The N171-82Q mouse, generated by insertion of a 171, a fragment with 82 CAG repeats of human Htt [88], presents striatal neuron loss and inclusions [89]. In general, N-terminal fragment models exhibit a rapid onset of motor, cognitive, and behavioral symptoms, weight loss, and early death, as well as a widespread and generalized degenerative phenotype [90].
Full-length Htt models express human genomic mHtt transgene under the control of human Htt promoter and regulatory elements in a yeast artificial chromosome (YAC) or a bacterial artificial chromosome (BAC). YAC models include YAC18, YAC46, YAC72, and YAC128, designated for the number of CAG repeats [91, 92], whereas BACHD mice [93] express mHtt with 97 CAG repeats. However, YAC46 and YAC72 exhibit almost no behavioral deficit, whereas YAC128 and BACHD mice display selective atrophy of the striatum and cortex, together with slow progressive motor deficits. Thereby, transgenic mice must have higher CAG repeats than human patients to recapitulate some features of HD. Nevertheless, these full-length models exhibit milder motor dysfunction than the N-terminal model R6/2 [94]. BACHD mice expressing 225 CAG repeats show very early motor dysfunction, widespread Htt aggregates, and neuron loss [95]. Another full-length model was generated expressing 16, 48, or 89 CAG repeats in which only mice with 48 or 89 repeats showed neuron loss in the striatum and cerebral cortex and progressive behavioral and motor dysfunction [96]. A transgenic HD rat model expressing a fragment of Htt with 51 human-derived CAG repeats inserted in rat Htt showed slow disease progression and pathology restricted to the striatum [97]. Later, the BACHD rat expressing full-length human Htt with 97 CAG repeats, under the control of human Htt promoter and regulatory elements, showed early progressive motor impairment and Htt aggregates in the cortex and striatum [98].
Knock-in full-length models were developed for animals carrying a fragment of the human mHtt gene replacing its endogenous Htt gene. In these models, the endogenous promoter of mouse Htt and its regulatory elements drive mHtt expression. Therefore, knock-in mice expressing a hybrid Htt with only two copies of Htt have a genomic context more comparable to that of the human disease than transgenic models. Knock-in mice showed late onset of motor and behavioral symptoms and progressive mild pathology, with no reduction in life span. HdhQ111 [99], CAG140 [100], Hdh(CAG)150 [101], and zQ175 [102] are the most common knock-in models used to study HD. HdhQ111 mice show nuclear inclusions but lack striatal atrophy, and neuronal loss is observed at a late stage at 24 months, while CAG140 mice exhibit late neuropathology features at 24 months [103]. However, zQ175 animals show nuclear aggregates, early onset of motor symptoms, and a more severe phenotype compared to the others [102].
In conclusion, HD mice do not recapitulate all neuropathologic changes displayed by human HD brains in one animal model. HD models exhibit mild neuron loss occurring at a late stage, and knock-in mice show more subtle symptoms than transgenic mice [87]. Despite these observations, animal models are the best tool available for investigating HD pathogenesis.
4.1. Reactive Astrocytes in HD Models
Astrocyte reactivity occurs in response to a variety of pathological conditions and involves morphological changes since they become hypertrophic, as well as transcriptional changes, such as upregulation of GFAP expression [104]. Being the main constituent of intermediate filaments in astrocytes, GFAP is consequently regarded as a hallmark of reactive astrogliosis. In human HD patients, the brain reactivity of astrocytes is detected only in the striatum [20]. HD astrocytes have increased soma size, altered arborization, and GFAP expression that increases with disease severity [24]. Astrogliosis can be present in some rodent HD models as shown in Table 2. Non-genetic models reproduce increased GFAP expression in the striatum. Intrastriatal administration of QA induces astrogliosis in the striatum of rats [68, 72] and mice [69]. Likewise, the intraperitoneal administration of 3NP induces higher GFAP immunoreactivity in the striatum of rats [105-109] and mice [110, 111], although when the 3NP dose is high, it kills neurons and astrocytes so that the lesion zone lacks GFAP staining while the marginal area shows increased GFAP immunoreactivity [106]. Even though the expression of mHtt in transgenic models induces astrocyte activation, several models do not recapitulate astrogliosis, but they show dysfunction. Astrogliosis in the striatum is undetectable in R6/1 [112] and R6/2 mice [84, 113], although one study found increased GFAP levels in R6/2 mice 3 months of age [85]. In other studies, N171-82Q mice showed increased GFAP expression at 4 months [113, 114]. Full-length Htt transgenic mice do not show reactive astrogliosis in the striatum [115-117] except for HD89 [96] and BAC-225Q mice, which show increased GFAP levels at 12 months of age [95]. Knock-in mice exhibit striatal astrogliosis at later stages, such as 12-24 months [101, 118-122]. In nonhuman primates, two HD-monkeys were generated via lentiviral transfer of exon 1 of the mHtt transgene with 29Q or 70Q repeats, respectively. Both HD-monkeys showed striatal neuron death and increased GFAP expression in the striatum, which was higher in the monkey carrying mHtt with 70Q repeats [123]. Intriguingly, reactive astrocytes are not recapitulated in all HD models, thus caution must be taken in order to draw conclusions from these models.
Table 2.
Astrogliosis in rodent HD models.
| Toxic Models | Strain | Dose/Day |
Astrogliosis/
Brain Region |
Age or
Weight |
Refs. |
|---|---|---|---|---|---|
| Rats+QA | Wistar Sprague-Dawley |
75 nmol/µl c 50 µg/µl c |
+/Str +/Str |
1 m 1 m |
[68] [72] |
| Mice+QA | C129SvEv | 50 nmol/µl c | +/Str | 1 m | [69] |
| Rats+3NP | Sprague-Dawley Sprague-Dawley Sprague-Dawley Wistar Wistar |
7.5 mg/kg a 25 mg/kg a 60 mg/kg a 15 mg/kg b 10 mg/kg a |
+/Str +/Str +/Str +/Str +/Str |
3 m 250-280 g 3 m 250-300 g 200-250 g |
[105] [106] [107] [108] [109] |
| Mice+3NP | C57BL/6 C57BL/6J |
50 mg/kg a 15 mg/kg a |
+/Str +/Str, -/Cx |
4 m 18-22 g |
[110] [111] |
| Transgenic Mice |
Transgene/
promoter |
CAG repeats |
Astrogliosis/
brain region |
Age | Refs. |
| R6/1 | Human exon 1/ human |
116 | +/Cx, -/Str -/Str |
4, 6 m 9 m |
[112] [113] |
| R6/2 | Human exon1/ human |
144- variable |
-/Str +/Str |
1-3 m 3m |
[84], [113] [85] |
| N171-82Q | Human 171aa/ murine prion |
82 | +/Str +/Str, +/Cx |
4 m 4m |
[113] [114] |
| HD89 | Full length Htt/ CMV |
89 | +/Str, +/Cx | 6-12 m | [96] |
| YAC128 | Full length Htt/ human |
128 | +/Cx, -/Str -/Str |
15, 24 m 12 m |
[115] [116] |
| BACHD | Full length Htt/ human |
97 | -/Str | 12 m | [117] |
| BAC-225Q | Full length Htt /mouse |
225 | -/Str +/Str |
6 m 12 m |
[95] |
| Knock-in Mice | |||||
| HdhQ111 | Human exon 1/ mouse |
111 | +/Str | 24 m | [120] |
| Hdh(CAG)150 | Murine exon 1/ mouse |
150 | +/Str -/Str -/Str |
14 m 5-6 m 5-24 m |
[101] [113] [121] |
| Hdh200 | Murine exon 1/ mouse |
200 | +/Str, +/Cx | 40 and 80 w | [118] |
| zQ175 | Human-murine exon 1/ mouse |
188 | +/Str, +/Cx +/Str |
49-53 w 12 m |
[119] [122] |
Str: striatum, Cx: cerebral cortex, m: months, w: weeks, a: intraperitoneal, b: subcutaneous, c: intrastriatal.
4.2. Reactive Microglia in HD Models
Microgliosis is detected in the caudate putamen, cortex, and globus pallidus of HD brains [25]. The number of reactive microglia expressing increased levels of HLA class II was elevated in HD caudate [35]. Microglial activation correlates with disease severity in HD patients [30], and can be detected before symptoms appear [31]. Moreover, microglial activation in HD patients is present in brain regions related to cognitive functions [124]. Pro-inflammatory cytokines are elevated in plasma and striatum of HD patients [33]. Human monocytes, mHtt-expressing macrophages, and microglia are hyperreactive in response to LPS releasing higher levels of IL-6 than control cells [33], indicating that peripheral cells can mirror pathological changes in the HD brain.
Microgliosis is also seen in HD models. Iba1-positive cells are found to be increased in the cortex but not in the striatum of R6/2 mice [125]. However, microglia cell number is increased in the striatum of R6/2 mice and in HD brains [126], showing ferritin accumulation, and a few of them colocalizing with Htt [127]. Also, N171-82Q mice at 4 months of age show increased Iba1-positive cells in the cerebral cortex and striatum [114], and 12 month-old, but not 6-month-old, BAC-225Q mice exhibit higher microglial cell numbers in the cortex [95]. mHtt inclusions can be present in microglial cells of R6/2 and zQ175 mice, but its numbers are negligible in comparison to neuronal and astrocyte inclusions [128]. Microglial morphology and brain vasculature are altered in YAC128 mouse striatum [129], and proliferating microglia are observed near mHtt-expressing neurons [130]. More recently, it was shown that striatal microglia from R6/2 mice display a more mature morphological phenotype, increased phagocytosis, and fewer contacts with synapsis [131].
4.3. Inflammation in HD Models
Aside from the reactivity of astrocytes and microglia, inflammation in the brain and the periphery has been demonstrated in several HD animal models (Table 3). QA induces the release of cytokines TNF-α, IL-6, and IL-1β in the striatum of rats [132, 133] and mice [134]. Systemic treatment with 3NP exhibits increased pro-inflammatory cytokine levels, including TNF-α, IL-6, and IL-1β, in the striatum of rats [135] and mice [136, 137]. Interestingly, TLR4 [138], inducible nitric oxide synthase (iNOS), and cyclooxygenase 2 (COX2) [137], all inflammatory factors, were also found to be increased in 3NP-treated animals. Another study showed overexpression of IL-1α, TNF-α, C1q, and C3 in the striatum, hippocampus, and cerebellum of Wistar rats treated with 3NP [139]. Several reports on R6/2 mice showed increased levels of cytokines in the striatum, plasma, serum, and peripheral immune cells [125, 140-144]. In the N171-82Q mice, one study found increased plasma levels of IL-12 but not of IL-4 [145]. Regarding YAC128 mice, levels of IL-6 were increased in serum and macrophages [33, 144, 146], whereas BACHD mice exhibited increased cytokine levels in several peripheral organs but not in serum [147]. As for knock-in HD mice, HdhQ140 and HdhCAG150 animals had several pro-inflammatory cytokines levels elevated in plasma [145] and serum [33], respectively, whereas blood monocytes in HdhCAG150 mice also exhibited high levels of cytokines [144]. zQ175 mice had increased TNF-α levels in the striatum but not in unstimulated peritoneal macrophages [141].
Table 3.
Inflammation in HD models.
| HD Model | Samples | Increased | Decreased | Unchanged | Refs. |
|---|---|---|---|---|---|
| Rats +QA c | Str Str |
IL-6, TNF-α, IFN-γ IL-1β, IL-6, |
- | TNF-α | [132] [133] |
| Rats +3NP a | Str Str Str, Hip, Cer |
IL-1β, IL-6, TNF-α IL-6, TLR4 IL-1α, TNF-α, C1q, C3 |
IL-10 | - | [135] [138] [139] |
| Mice +QA c | Str | IL-1β, IL-6, TNF-α | - | - | [134] |
| Mice+3NP a | Str Str |
IL-1β, TNF-α IL-1β, IL-6, TNF-α, iNOS, COX-2 |
- | - | [136] [137] |
| R6/2 | Str, Cx Str |
IL-1β, TNF-α IL-12, TNF-α |
- | - | [140] [141] |
| - | Plasma | IL-6, MMP-9, TGF-β1 IL-1β, IL-2, IL-6, IL-10, TNF-α IL-1β, IL-2, IL-5, IL-6, IL-10, TNF-α |
IL-18 | - | [51] [141] [142] |
| - | Serum | IL-6, IL-10, IL-1β, IL-12 TNF-α IL-1β, IL-2, IL-6, IL-10, TNF-α |
- | IL-8, TNF-α, IFN-γ | [33] [125] [143] |
| - | peritoneal macrophages | IL-4, IL-6, IL-10, IL-12 | - | IL-1β, TNF-α | [141] |
| - | blood monocytes s | IL-6, TNF-α | - | IL-1β, IL-10, IL-12 | [144] |
| N171-82Q | Plasma | IL-12 | - | IL-4 | [145] |
| YAC128 | Serum Serum |
IL-6, IL-8 IL-6 |
- | IL-1β, IL-10, IL-12, TNF-α, IFN-γ | [33] [146] |
| - | alveolar macrophages s | IL-6 | - | - | [33] |
| - | peritoneal macrophages s | IL-6 | - | - | [144] |
| BACHD | Kidney | IL-6 | - | IL-4, IL-5, IL-12, TNF-α, IFN-γ | [147] |
| Heart | IL-6, IL-12 | - | IL-4, IL-5, TNF-α, IFN-γ | ||
| Liver | IL-12, TNF-α | - | IL-4, IL-5, IL-6, IFN-γ | ||
| Spleen | IL-4 | IL-5, IL-6 | IL-12, TNF-α, IFN-γ | ||
| Serum | - | - | IL-4, IL-5, IL-6, IL-12, TNF-α, IFN-γ | ||
| HdhQ140 | Plasma | IL-1A, IL-1-β, IL-2, IL-4, IL-6, IL-10, IL12, IL-17A, TNF-α, G-CSF, GM-CSF | - | IFN-γ | [145] |
| HdhQ150 | Serum | IL-6, IL-10, IL-12 | - | IL-1β, IL-8, TNF-α, IFN-γ | [33] |
| - | blood monocytes s | IL-1β, IL-12, TNF-α | - | IL-6 | [144] |
| zQ175 | Str | TNF-α | - | IL-1β, IL-6, IL-10, IL-12 | [141] |
| - | peritoneal macrophages | IL-6, IL-10, IL-12 | - | IL-1β, TNF-α | [141] |
Str: striatum, Cx: cerebral cortex, Hip: hippocampus, Cer: cerebellum, a: intraperitoneal, c: intrastriatal, s: stimulated.
Altogether, these data indicate that astrocytes and microglia are altered in HD models. A moderate immune response is present in HD models. However, little is still known about what triggers the immune response in HD. More data are needed to define the role of immune response in HD.
5. ASTROCYTES IN HD
Astrocytes participate in maintaining brain physiological functions [148] and the inflammatory response [149]. HD patients and models show astrocyte dysfunction as well as reactive astrocytes. Astrocyte dysfunction occurs when these cells fail to fulfill their normal functions. Thus, deficits in ion homeostasis, calcium signaling, and neurotransmitter clearance have been shown to contribute to HD pathology [150] (Fig. 1 ). Also, HD astrocytes become reactive cells producing pro-inflammatory and harmful molecules. For example, R6/2 astrocytes secreted higher levels of TNF-α than wild-type astrocytes when stimulated by LPS [125, 140], and HD astrocytes from R6/2 mice exhibited decreased CCL5 release, an important chemokine for neurite development and activity in cortical neurons [41]. Since astrocytes can also release anti-inflammatory, neurotrophic, or antioxidant molecules, they are interesting targets for neuroprotective approaches in HD [151].
Fig. (1).
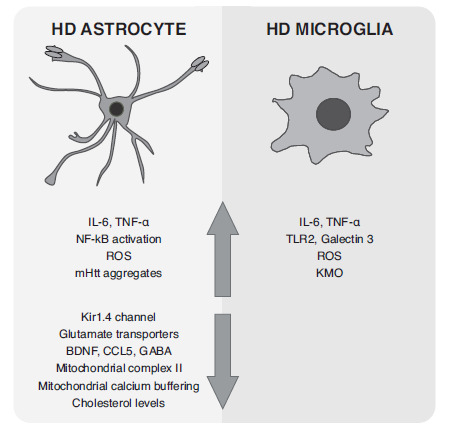
Changes exhibited by astrocytes and microglia in HD.
5.1. mHTT Expression in Astrocytes
mHtt inclusions have been found to occur in reactive astrocytes in HD patients [35, 152] and mouse models [152]. A detailed study on R6/2 and zQ175 mice showed that astrocytes exhibited both nuclear and cytoplasmic mHtt inclusions, whose number increased with age, and that these inclusions were less abundant than in neurons [128]. Astrocyte contribution to HD pathogenesis was demonstrated by selective overexpression of mHtt in astrocytes, which led to a motor and transcriptional dysfunction [153]. Also, astrocytic mHtt contributed to the BACHD phenotype as its reduction improved striatal and cortical volume loss as well as motor and psychiatric symptoms [154]. Moreover, a study showed that expressing miRNA directed to Htt in neurons alone was unable to reduce pathology of YAC128 mice and that reducing Htt in both neurons and astrocytes was required to achieve maximal benefit [155]. It was also demonstrated that increasing the expression of Hsp70-binding protein 1 (a protein that inhibits Hsp70 and is enriched in neurons) in astrocytes of HTT140Q knock-in mice increased levels of mHtt and induced exacerbation of pathology in these mice [156], further indicating that astrocyte dysfunction is involved in HD pathogenesis. Apart from contributing to pathology, HD astrocytes also lose their protective effects. When the striatum of immunodeficient mice was engrafted with mHtt-expressing human glial progenitor cells, mice showed impaired motor learning and hyperexcitability of striatal neurons. Conversely, transplanting mice with normal human glia improved motor function and life span and normalized neuron excitability in R6/2 mice [157]. Despite these data, the basis for glial pathology in HD is still poorly understood. Astrocytes expressing mHtt exhibit many changes, such as astrogliosis, secretion of inflammatory factors, impaired glutamate and potassium homeostasis, mitochondrial dysfunction, and decreased cholesterol synthesis. How these conditions contribute to neuroinflammation has not yet been established. Astrocyte dysfunction in HD may be a consequence of cell-autonomous and non-cell-autonomous mechanisms, such as neuronal dysfunction coexisting and contributing to HD pathology.
5.2. Potassium, Glutamate, and GABA Homeostasis
Maintenance of potassium (K+) homeostasis is an important function of astrocytes, and therefore, they express several K+ channels. Glia, mainly astrocytes, express Kir4.1 channels, which maintain membrane potential [158] and play a role in several neurodegenerative disorders [159]. Kir4.1 protein and function are reduced in astrocytes with mHtt inclusions of R6/2 and zQ175 mice, and when Kir4.1 expression in astrocytes is increased through adenovirus construction, the motor dysfunction is attenuated [160].
Excitotoxicity is induced by the overactivation of glutamate receptors in the synapsis. Glutamate uptake failure can contribute to increased extracellular glutamate levels and, consequently, induce excitotoxicity and neuronal death. Glutamate uptake is decreased in the prefrontal cortex of HD postmortem tissue [161]. Glutamate transporter 1 (GLT1 in rodents and EAAT2 in humans) and glutamate aspartate transporter (GLAST in rodents and EAAT1 in humans), mainly expressed by astrocytes to remove glutamate from the synaptic space, are downregulated in HD patients and HD mice [162]. Astrocytes expressing mHtt show decreased expression of only GLT1 [163] or both GLT1 and GLAST, and decreased EAAT2 immunoreactivity was observed in the caudate nucleus of HD subjects [24]. R6/2 mice brain and R6/2 mice cultured astrocytes also exhibit decreased GLT1 expression in the brain [152]. Moreover, when mHtt was expressed only in astrocytes, these animals presented HD behavioral phenotype and had reduced GLT1 levels and glutamate uptake [153]. Another report demonstrated that BACHD astrocytes release more glutamate than control astrocytes, likely due to increased glutamate de novo synthesis [164]. Ceftriaxone, a beta-lactam antibiotic, was shown to increase GLT1 expression and glutamate uptake and to improve the motor behavior of R6/2 mice [165]. Reduced GLT1 and GLAST levels could be a consequence of increased cytokine expression in the HD brain. TNF-α was shown to reduce GLAST levels in rat astrocytes [166, 167] and GLAST and GLT1 levels in human fetal astrocytes [168]. IL-1β reduced GLT1 protein levels in the spinal cord of an encephalomyelitis model [169], and was recently shown to downregulate GLT1 and GLAST proteins in human astrocytes derived from induced pluripotent stem cells [170]. Unfortunately, there are no data evaluating the effect of inflammatory cytokines on GLT1 or Kir4.1 expression in HD astrocytes or the effect of overexpressing GLT1 or Kir4.1 on the inflammatory response of HD astrocytes.
In the CNS, γ-gamma aminobutyric acid (GABA) is the main inhibitory neurotransmitter, and striatal MSNs are GABAergic neurons. A balance between the excitatory glutamatergic system and the inhibitory GABAergic system is essential to control movement. The CSF of premanifest HD subjects has low levels of GABA [171], and several studies describe a functional alteration of GABA activity in HD models and HD patients [172-174]. GABA transporters (GAT) are present in neurons and astrocytes. GAT-3 is predominantly expressed in astrocytes, and HD astrocytes have a reduced capacity to release GABA via GAT-3 [175]. The association between neuroinflammation and GABAergic tone has not been elucidated, but it may be an important mechanism in HD pathology. Indeed, increased membrane expression of astrocyte GAT-3 in the cerebellum was observed in neuroinflammation induced by hyperammonemia, which is associated with increased levels of GABA and motor and cognitive dysfunction [176]. More studies on HD astrocytes are needed to completely understand GABA and glutamate involvement in HD.
5.3. Mitochondrial Dysfunction
Mitochondrial dysfunction is observed in HD patients [177] and HD animal models [178]. The inhibition of succinate dehydrogenase from mitochondrial complex II by 3NP induced HD-like symptoms in animals and humans [75], and overexpression of complex II decreased lesion volume of HD animals, although it did not modify the number or size of mHtt aggregates [178]. Neurons have a decreased capacity to neutralize oxidative stress, and thus, astrocytes are known to provide neurons with antioxidant molecules, such as glutathione [179, 180], to prevent neuron death. In addition, dysfunctional astrocytes can contribute to pathology by producing lower levels of antioxidants in HD or by releasing more oxidant factors or both [181]. Astrocytes expressing mHtt induce oxidative stress in normal neurons [182]. mHtt can also influence astrocyte mitochondrial function since HD astrocytes from YAC128 mice show increased ROS levels and deficits in mitochondrial respiration [116]. We must also consider regional differences in mitochondrial function in the brain. Mitochondria from striatal neurons and astrocytes were reported to be more vulnerable to calcium loads than cortical neuronal and astrocyte mitochondria [183]. Striatal astrocytes were shown to be more vulnerable than cortical astrocytes to death by 3NP, which may be due to increased mitochondrial H2O2 levels [184]. Also, complex II succinate dehydrogenase activity loss and decreased protein expression were observed in striatal astrocytes from HDHQ150 mice but not in age-matched controls, thereby showing that HD astrocytes exhibit higher superoxide production and increased fatty acid oxidation [185]. Furthermore, treatment with XBJ-5-131, a mitochondrially targeted antioxidant compound, ameliorated HD phenotype in mice, reducing oxidative damage and neuron death and preventing motor dysfunction [186]. Expression of the transcription factor PGC-1α, which regulates mitochondrial biogenesis, is reduced in HD models [187, 188], though it is not known whether this factor is also diminished in astrocytes.
Systemic administration of 3NP increased levels of pro-inflammatory enzymes and cytokines [137, 139]. Astrocytes can respond to mitochondrial dysfunction by releasing inflammatory factors. We recently demonstrated that 3NP induces cell death of astrocytes, increases ROS levels, and induces TNF-α release into the culture medium of cortical and striatal astrocytes, whereas it decreases TGF-β release from cortical astrocytes [83]. We also observed regional differences in cytokine release induced by 3NP; striatal astrocytes released more TNF-α than cortical astrocytes and cortical astrocytes released less TGF-β than striatal astrocytes in response to 3NP [83]. Mitochondrial dysfunction can impact differently, inducing selective inflammation within the brain regions. This fact highlights the urgent need to improve our understanding of regional astrocyte heterogeneity.
5.4. Cellular Stress
Cellular stress, such as oxidative stress or alteration of proteostasis, requires the activation of pro-survival pathways driven by protective genes (vitagenes), resulting in the production of antioxidant and antiapoptotic molecules [189, 190]. Among these mechanisms are the Heat Shock Response (HSR) and the antioxidant response. HSR is an ancient protective mechanism that protects cells from stress by inducing the expression of heat shock proteins (Hsp) and repairing misfolded proteins and aggregations, thus avoiding cell death. Evidence shows that a variety of Hsps can inhibit mHtt aggregation [191]. Whole tissue homogenates from HD mice showed reduced levels of heat shock factor 1, Hsp70, and two Hsp40 members [192, 193]. Interestingly, overexpression of the small Hsp αB-crystallin in astrocytes was recently shown to ameliorate the neuropathology of BACHD mice by reducing Htt inclusions in the striatum and cortex [194]. Indeed, an inflammatory stimulus, such as LPS+IFN and nitric oxide, also induces the expression of Hsps in cultured astrocytes [195], indicating that inflammation can also cause cellular stress.
As mentioned above, oxidative stress may result from mitochondrial dysfunction; as neurons have low levels of antioxidant factors, astrocytes provide them with antioxidants, such as glutathione. Nuclear factor erythroid 2-related factor 2 (Nrf2) is the master regulator of the antioxidant response since it induces expression of antioxidant enzymes and is known to exert neuroprotection [196]. Indeed, Nrf2 over-expression is sufficient to provide neuroprotection in a toxic HD model induced by 3-NP [197]; astrocyte-specific overexpression of Nrf2 protected from malonate (another complex II inhibitor) toxicity in mice by decreasing the lesion size [198]. In HD mice, Nrf2 is not altered, and in striatal mouse cells expressing mHtt, there is evidence of impaired Nrf2 signaling [199]. Indeed, selective activation of Nrf2 signaling decreased IL-6 release in primary wild-type and YAC128 astrocytes and microglia and in human monocytes [200], showing an anti-inflammatory role for Nrf2. Data suggest that activation of astrocyte Nrf2 signaling can compensate for impaired Nrf2 response in HD neurons. It would be interesting to verify Nrf2 activation in HD patients.
5.5. BDNF
BDNF levels are decreased in HD patients [6]. Therefore, increasing BDNF levels ameliorates the HD phenotype in rodents [201, 202]. Crossing BDNF+/- mice with R6/1 mice resulted in earlier onset of motor dysfunctions, more severe uncoordinated movements, and striatal neuron loss, indicating that decreased BDNF levels play a key role in HD pathology [203]. BDNF release is impaired in mHtt-expressing astrocytes [204, 205], and BDNF release, induced in mouse mesencephalic astrocytes by glatiramer acetate treatment, increases BDNF/GFAP-positive cells and exerts neuroprotection in R6/2 and YAC128 mice [206]. Giralt et al. [207] reported that R6/2 mice overexpressing BDNF under GFAP promoter exhibited improved motor coordination and synaptic transmission, suggesting that expressing BDNF when GFAP is induced by HD pathology could be therapeutic. We recently demonstrated that BDNF increases expression of GLT1 and TGF-β release in striatal rat astrocytes [83], two effects that might contribute to BDNF protective effect on HD. We also showed that BDNF increased levels of the antioxidant glutathione, decreased production of ROS [81], and reduced TNF-α release [83] induced by 3NP in astrocytes, indicating that this neurotrophic factor also has antioxidant and anti-inflammatory actions in glial cells. BDNF increased Nrf2 expression in astrocytes, suggesting that it may promote cell survival by increasing glutathione levels [81], and that this transcription factor may mediate their antioxidant and possibly their anti-inflammatory actions. BDNF protective action may be exerted on HD astrocytes as well as HD neurons and, therefore, it could become an attractive therapeutic strategy.
5.6. Cholesterol Synthesis
Brain cholesterol is needed for biological membrane structures, myelin formation, neurite outgrowth, synaptogenesis, and neurotransmitter release. Cholesterol biosynthesis in the brain is a process that mainly occurs in astrocytes [208]. In the brain, cholesterol is oxidated to 24S-hydroxycholesterol (24OHC), whose levels are considered an indirect measure of cholesterol synthesis. The plasma of HD patients showed a gradual decrease in 24OHC levels, which correlated with HD progression [209-211]. Accordingly, several transgenic HD models exhibit decreased cholesterol levels in the brain [212], positioning 24OHC as a candidate biomarker for HD. Cholesterol dysregulation in astrocytes, due to mHtt action on transcription factor sterol regulatory-element binding protein (SREBP), produces less cholesterol available to neurons [213]. Other studies indicate that mHtt produces an accumulation of cholesterol within neural cells, contributing to excitotoxicity in HD neurons, and that in human caudate of postmortem HD brains, total cholesterol levels were found to be increased [214]. Recently, a gene therapy approach delivering CYP46A1, the rate-limiting enzyme for cholesterol degradation in the brain, prevented neuronal dysfunction, restored cholesterol homeostasis in zQ175 mice, and induced transcriptional changes in the striatum [215], suggesting that it may also modify inflammatory genes. Since dysfunctional cholesterol metabolism can induce a neuroinflammatory response in other neurodegenerative disorders by increasing the cholesterol content, this mechanism was also proposed to occur in HD [216]. Very recently, intrastriatal administration of adenovirus targeting astrocytes carrying the transcriptionally active N-terminal fragment of SREBP2 induced robust expression in astrocytes of R6/2 mice. This treatment activated cholesterol biosynthesis gene expression, restored synaptic transmission, decreased Htt aggregates, and attenuated behavioral deficits [217]. These data highlight the role of astrocytes as therapeutic targets for HD prevention.
5.7. Transcriptomic Studies
The transcriptome analysis of human HD post-mortem striatal tissue and striatal astrocytes isolated from zQ175 and R6/2 mice revealed a set of astrocyte genes whose expression at RNA and protein levels changed in HD mice and humans [122]. Altered genes were related to calcium signaling and neurotransmitter regulation and did not represent neurotoxic or protective phenotypes but rather astrocytes that have lost essential functions [122]. Interestingly, HD astrocytes from the cingulate cortex were analyzed by single nucleus RNA sequencing to examine single cell differences in gene expression. The study revealed that HD astrocytes exhibited great heterogeneity, which could be classified into five different states. In these HD human astrocytes, GFAP and inflammatory gene expressions were upregulated, whereas cholesterol biosynthesis and glutamate transporter 1 and 2 genes were downregulated [218]. More recently, another RNA sequencing study demonstrated that molecular changes in striatal astrocytes were context-dependent and that selective stimulation of the Gi-GPCR pathway corrected some HD phenotypes [219]. Finally, a study comparing RNA expression profiles of striatal astrocytes from HD models harboring truncated exon 1 or full-length mHtt, i.e., R6/2 and zQ175, or comparing human ESCs derived from HD subjects with fetal human striatal glia expressing truncated exon 1, revealed differences between cells expressing truncated versus full-length mHtt. R6/2 mice exhibited more prominent transcriptional changes than zQ175 mice since downregulation of cholesterol synthetic pathway was observed in truncated mHtt astrocytes, as derived from both R6/2 mice and human cells transduced with mHtt-73 CAG fragment [220]. Interestingly, the authors also identified a cohort of genes linked to the development and consolidation of cell morphology that was shared by all models analyzed, and found it to be in correlation with morphometric analysis showing aberrant astrocyte morphology [220]. The study also revealed the upregulation of several genes associated with inflammation in R6/2 microglia and, to a much lesser extent, in astrocytes. Although older astrocyte inflammatory signature was more similar to young microglia, zQ175 astrocytes did not exhibit such signature [220]. These results suggest that inflammatory phenotype in HD astrocytes is influenced by truncated rather than full-length mHtt. Nevertheless, all the studies described addressing the role of astrocytes are mainly based on RNA sequencing analysis, which needs confirmation by determining astrocyte protein expression in vivo.
All data on astrocytes suggest that they are dysfunctional and at the same time reactive in HD. However, what contributes to HD pathology remains to be elucidated.
6. MICROGLIA IN HD
Microglial cells derive from hematopoietic progenitors that enter the CNS early in development and proliferate to colonize the entire CNS [221]. Microglia are considered brain-resident macrophages representing 5-12% of total brain cells, and they are known to constantly survey the brain parenchyma. They provide immune surveillance and apoptotic clearance, modulate neurogenesis, and exert synaptic pruning functions [222]. They also provide trophic neuron support, contribute to the formation and maturation of developing neural circuits, and modulate synaptic plasticity and neurogenesis in the adult brain [221]. Reactive microglia are characterized by a change in cell shape, from a highly ramified morphology to an ameboid shape, and by molecular events that induce the production of inflammatory mediators.
HD microglia exhibit activation and inflammatory phenotypes (Fig. 1). Expression of mHtt in microglial cells induces a cell-autonomous increase in pro-inflammatory gene expression and neurotoxic effect on wild-type neurons. Untreated YAC128 microglia release higher levels of IL-6 than microglia from wild-type mice [47]. Crotti et al. demonstrated that mHtt-expressing microglia are hyperactivated and exhibit a transcriptionally pro-inflammatory response dependent on myeloid-determining transcription factors PU.1 and C/EBPs [34]. The same study showed that primary microglia from R6/2 and zQ175 mice and HD human striatum exhibit PU.1, IL-6, and TNF-α increased expression, an effect not observed in bone marrow macrophages from the same animals or in monocytes from HD subjects. Microglia from HD mice, but not from wild-type mice, increased the number of apoptotic neurons in culture [34]. Finally, they expressed mHtt only in microglial cells and injected LPS in mice striatum, indicating that mice expressing mHtt in microglia had higher neuron death [34]. Therefore, data point to a neurotoxic role for HD microglia, which also seems to be the case for human microglia. Concordantly, microglia depletion in R6/2 mice prevented motor and cognitive deficits and decreased astrogliosis [223]. However, mHtt selectively deleted or expressed only in microglial cells of BACHD mice did not modify the phenotype in either way [224]. Interestingly, a longevity-associated variant of BPIFB4 treatment of human immortalized microglial cells SV40 induced an M2 polarizing anti-inflammatory effect and prevented motor dysfunction and mHtt aggregation in R6/2 mice [225]. Altogether, these data indicate that expression of mHtt increases microglia reactivity, which may contribute to HD, although this effect alone is not sufficient to induce neuropathology in mice. More importantly, modulation of microglial activation might be beneficial for preventing HD progression.
The CX3CL1-CX3CR1 axis was recently found to be implicated in HD pathology. CX3CL1 is a chemokine expressed abundantly in the brain, particularly in neurons, whereas its receptor CX3CR1 is selectively expressed in microglial cells [226]. CXC3CL1-CX3CR1 signaling pathway mediates neuron-microglia interaction and microglia immunomodulatory and phagocytic activities [226]. CX3CL1, also known as fractalkine, is decreased in human putamen and in R6/1 mice striatum, suggesting that HD could affect neuron-microglia communication [36]. Although striatal microglia from R6/1 lack inflammatory morphological changes, they show increased engulfment of PSD-95 puncta, which was suggested to contribute to early striatal synaptic dysfunction in HD mice [36]. Therefore, aside from inflammatory microglial functions, the CX3CL1-CX3CR1 axis shows that physiological neuron-microglia interactions can be affected in HD.
Galectin 3 is the only galectin expressed in human and mouse brains [227]. It is upregulated in microglia and, when released, can act as a TRL4 agonist [228]. Increased levels of galectin 3 were found in the plasma and striatum of HD patients and R6/2 mice, but interestingly, galectin 3 expression was found only in microglia, and not in neurons or astrocytes of R6/2 mice [229]. Furthermore, knockdown of galectin 3 expression decreased microglia activation and IL-1β, while increasing IL-10 levels and improving the motor function of HD mice.
Microglia are implicated in the kynurenine pathway for tryptophan degradation, which is associated with excitotoxicity and mitochondrial damage. The rate-limiting enzyme of the pathway, indoleamine 2,3-dioxygenase, is located mainly in microglia [230] and catalyzes the oxidation of tryptophan to kynurenine, which counteracts the effect of QA and confers neuroprotection. Kynurenine accumulates by inhibition of the degrading enzyme kynurenine 3-monooxygenase (KMO), which is mainly found in microglia [231]. In the brains of HD patients and mice, QA and KMO levels are increased, and peripheral administration of a KMO inhibitor ameliorated HD pathology in R6/2 mice [232]. Nevertheless, since the KMO inhibitor cannot cross the BBB, the neuroprotective effect results from the inhibition of KMO in peripheral immune cells. Recently, KMO ablation in R6/2 mice resulted in reduced levels of inflammatory cytokines and increased levels of 3-hydroxykinurenine levels in the cortex and striatum, and despite modulating inflammatory and metabolic parameters, it did not improve motor function in HD mice [233]. Moreover, a comprehensive study focused on kynurenine metabolites in CSF and plasma of premanifest and manifest HD patients found no evidence to support the alteration of this pathway [234].
Mitochondrial dysfunction can also affect microglia. 3NP treatment of rat microglia can decrease the viability of these cells [82], whereas in mouse microglial cells, 3NP inhibits some IL-4 functions, such as induction of arginase activity and expression, inhibition of IL-6 and TNF-α, and production of IGF-1 [235]. In an in vivo study, rats showed microglial activation, ROS production, and cell death after 3NP treatment [236]. 3NP-treated rats exhibited microglial infiltration and striatal cell death in the lesion, as well as proliferation and morphological changes of NG2 glia, which may be involved in fibrotic scar formation in the lesion core [237].
Taken together, evidence points to a detrimental role for microglia in HD. Future identification of microglia phenotypes at different disease stages in HD models and subjects will help define their dysfunction or involvement in HD.
7. GLIAL CELLS DERIVED FROM HUMAN-INDUCED PLURIPOTENT STEM CELLS
Human astrocytes are different from rodent astrocytes in that the former are larger, more diverse, and more complex than the latter [238]. However, access to HD human astrocytes is very limited. Human data from HD patients derive mainly from post-mortem samples, taken mainly for histochemistry studies, thus representing the end stage of the disease. In recent years, it has become possible to generate human-induced pluripotent stem cells (hiPSC), and this capacity of reprogramming human fibroblast into neurons in vitro has aided in the study of HD [239]. Regarding glial cells, there are a few studies that have used hiPSC to generate these cells. The first study showed astrocytes generated from hiPSC, which used cells from an adult-onset HD subject with 50 CAG repeats and from his daughter with juvenile HD with 109 CAG repeats [240]. Both cell lines exhibited increased cytoplasmic vacuolation under basal conditions. Notably, autophagic vacuoles were found in primary lymphoblasts harvested from HD patients [241, 242], thus reproducing in hiPSC-derived astrocytes a phenotype observed in HD cells. Later, it was also shown that astrocytes generated from the iPSC of HD patients behaved similarly to mouse astrocytes from HD models [243]. Moreover, HD iPSC-derived astrocytes did not protect HD or control neurons from glutamate toxicity [243], emphasizing that HD astrocytes are dysfunctional. An interesting study showed that transplantation of normal cord blood-derived iPSC-derived neural progenitor cells on YAC128 mice induced an increased number of striatal neurons and improved motor and cognition function after five months [244]. The grafted cells predominantly differentiated into neurons, oligodendrocytes, and astrocytes. Most of the benefits observed seemed to derive from new astrocytes since transplanted animals exhibited increased expression of EAAT2, no loss of Kir4.1 channel expression, and remarkably human-GFAP positive cells co-localized with BDNF only in transplanted mice [244]. These data show that normal human iPSC can prevent astrocyte-mediated toxicity and dysfunction in HD mice. Regarding microglia, only one study assessed hiPSC-derived microglia. Human HD iPSCs differentiated into microglial-like cells showed increased IL-6 and TNF-α release when stimulated with LPS+IFN-γ [49]. Also, they released higher ROS levels and had increased levels of apoptosis [49]. The data confirm that microglia are activated in HD models as well as in HD hiPSC. Although little data regarding hiPSC is available, this promising approach can provide much information regarding HD glial cells.
8. NF-κB PATHWAY ACTIVATION
NF-κB is a master transcription factor that regulates the production of cytokines, neuroinflammation, and cell survival. NF-κB functions as homo or heterodimer of five Rel/NF-κB subunits: Rel A (p65), c-Rel, Rel B, p50, and p52. NF-κB is retained in the cytoplasm by the inhibitory proteins IκBs. Upon stimulus-inducing phosphorylation of IκB by IκB kinase (IKK), IκBs are ubiquitinated followed by proteasomal degradation, and free NF-κB can translocate to the nucleus. NF-κB is activated in neurodegenerative diseases, such as Alzheimer’s and Parkinson’s diseases [245]. In HD, there is also evidence of NF-κB activation. Normal Htt interacts with neuronal p50 and p65, while mHtt decreases the nuclear accumulation of NF-κB in neurons of HdhQ140 mice [246, 247]. The p65/p50 heterodimer activity is markedly increased in the striatum after 3NP administration in wild-type mice [248] and in the cortex and striatum of R6/2 mice [249]. NF-κB may regulate the pathogenesis of HD since, in PC12 neurons, mHtt binds to IKKγ, activates the IKK complex, and increases NF-κB-dependent gene expression. Furthermore, mHtt toxicity can also be reduced by inhibition of IKK and NF-κB activity [249]. In normal conditions, NF-κB helps maintain neuron health, but in pathological conditions, the activation of NF-κB is induced in glial cells as a physiological response to injury or stress. However, chronic or excessive glial NF-κB activation can be toxic [250]. Nuclear localization of p65 was not found in neurons or microglia but was evident in astrocytes from R6/2 mice and GFAP-positive cells of the caudate nucleus of HD patients [140]. Primary R6/2 astrocytes challenged with LPS exhibited higher IKK activity, leading to sustained NF-κB activation and the expression of TNF-α and IL-1β [140]. mHtt, through IKKβ, induces expression of IL-34 in human mesencephalic neuron cell line and brains of R6/2 and YAC128 mice [251]. IL-34, a major driver of microglia proliferation, induces a microglial response that exacerbates mHtt toxicity and may contribute to aberrant microglial activation in HD [251]. Although not found in microglia, p65 protein expression is increased in monocyte-derived macrophages from presymptomatic HD patients compared to symptomatic HD patients, whereas no differences were observed between controls and symptomatic HD subjects [39]. The same study demonstrated that mHTT binds IKKγ and causes increased NF-κB translocation in HD patients’ monocytes and increased TNF-α, IL-6, and IL-8 release, which was prevented with Htt-lowering by siRNA delivery. Cell-intrinsic mHtt expression induces hyperactivation of immune cells, such as macrophages in HD patients and microglia in HD mice, which may be a consequence of NF-κB direct or indirect activation by mHtt.
The NF-κB pathway is also activated by TLR signaling. TLR2 and TLR4 are abundantly expressed by microglial cells. However, little is known regarding TLRs and neuroinflammation in HD. N171-82Q mice lacking either TLR2, TRL3, or TLR4 had their lifespan extended [252], and administration of 3NP to rats increased TLR4 expression in the striatum [138]. Curiously, mast cells from R6/1 mice showed decreased TRL4-dependent TNF secretion [253]. Cells expressing TLR4 have been recently shown to be higher in postmortem HD striatum, whereas the TLR4 single nucleotide polymorphisms found were associated with changes in motor progression rather than cognition in manifest HD subjects [37]. The multiligand receptor for advanced glycation end products (RAGE) recognizes endogenous molecules in inflammatory processes [254]. RAGE levels are increased in neurons and, to a lesser extent, in astrocytes from the caudate nucleus of HD brains [255, 256]. RAGE levels are also elevated in medium spiny neurons from R6/2 mice [257]. Moreover, the staining pattern for RAGE immunoreactivity in the caudate nucleus appeared to correlate with the pattern of neuron degeneration in HD [255]. Since both RAGE and TLRs have several possible ligands, identification of their specific ligands in HD may help understand RAGE and TRL roles in HD pathogenesis.
High mobility group Box 1 (HMGB1) is a nuclear protein that stabilizes chromatin and regulates transcription and DNA repair. HMGB1 can be released from neurons, reactive astrocytes, and microglia. As an alarmin, it can drive inflammation by activating RAGE, TLR2, and TLR4 receptors and recruiting inflammatory cells. HMGB1 release is observed in neurons exposed to several toxic factors [258] and in astrocytes in response to cytokines [259]. mHtt induces HMGB1 exit from the nucleus to the cytoplasm, and high levels of HMGB1 can induce mHtt aggregation [260]. Rats treated with 3NP showed increased HMGB1 expression in the striatum and in cultured primary striatal neurons [261]. Therefore, HMGB1-TLR or HMGB1-RAGE signaling could be involved in neuroinflammation and neurodegeneration in HD. However, further studies must clarify this issue. HMGB1 can also directly interact with Htt, an interaction increased by oxidative stress [262]. HMGB1 may act as a chaperone to reduce the formation of mHtt aggregates in vitro [260], and HD mice show a reduction of HMGB1 nuclear levels in striatal neurons with Htt inclusions [263]. However, high levels of HMGB1 induce more mHtt aggregate formation in vitro [260]. Given that HMGB1 has a role in DNA repair, which can localize to nuclear inclusion bodies in neurons of HD models, and its reduction can activate genotoxic stress signals [263], nuclear HMGB1-Htt interaction could contribute to HD pathology. Nevertheless, more studies are needed to fully demonstrate this hypothesis. Although evidence shows NF-κB activation in HD, how and in which cells it is activated is far less clear.
9. THERAPIES TARGETING NEUROINFLAMMATION IN HD
9.1. Cytokines as Therapeutic Targets
Of all the cytokines, IL-6 and TNF-α seem to be those most consistently found to be elevated in the plasma and striatum of manifest HD patients. Both are multifunctional cytokines associated with several cellular functions. IL-6 triggers the acute phase response, and although it is often regarded as a pro-inflammatory cytokine, it also promotes the differentiation of oligodendrocytes and acts as a neurotrophic factor by regenerating peripheral nerves [264]. Systemic treatment with IL-6 neutralizing antibody partially rescues weight loss and motor deficit in R6/2 mice at 9 weeks [126], highlighting that modulating peripheral immune cells can ameliorate HD pathogenesis. However, a recent report demonstrated that IL-6 knockout in R6/2 mice led to more severe motor dysfunction due to the dysregulation of genes involved in synaptic transmission and glutamatergic synapses, whereas inflammatory genes were not altered [265]. Indeed, IL-6 expressing lentiviral vectors injected into the striatum of Wistar rats prevented the striatal damage induced by QA [266], suggesting that some cytokines, such as IL-6, elevated in HD striatum, may actually play a protective rather than a pathogenic role.
IL-1β is activated by cleavage to mature form by caspase 1 or caspase 8 after TLR4 activation [267]. Mature IL-1β was increased in cortical tissue of manifest HD subjects and in brains of R6/2 mice [42]. Interestingly, R6/2 mice expressing a dominant-negative mutant of caspase 1 under neuron-specific enolase promoter significantly decreased brain IL-1β levels and delayed progression of motor dysfunction as well as mortality in comparison to R6/2 mice [42]. However, when N171-82Q mice were crossed with IL-1 receptor 1 knockout mice, these animals presented exacerbated motor dysfunction, increased mHtt aggregates, and GFAP expression in the cortex and striatum in comparison to N171-82Q animals [268]. Similar results were obtained when IL-6 was deleted in R6/2 mice, indicating that cytokines also performed beneficial functions in the brain of HD animals and that complete inactivation of cytokines is, therefore, not a therapeutic strategy for HD. Recently, caspase 1 was described to cleave Htt, and inhibition of Htt cleavage by caspase 1 decreased mHtt accumulation [269]. Thus, the benefits observed in caspase 1 dominant negative mutant expression in R6/2 mice could be due to less cleavage of Htt as well as proIL-1β. Further studies are needed to elucidate this issue.
TNF-α is involved in tissue homeostasis, cell proliferation, and survival; it is also a key modulator of the inflammatory response [270]. TNF-α increased levels are a hallmark of neuroinflammation found in neurodegenerative diseases. TNF-α can potentiate glutamate-induced cytotoxicity by inhibiting glutamate transport in astrocytes, and it can induce glutamate release by astrocytes and microglia [271]. The rationale for treating neurodegenerative diseases by reducing brain TNF-α is that it can impact two pathological pathways in HD: excitotoxicity and inflammation. Decreasing TNF-α may ameliorate inflammation and increase glutamate transporter expression. However, no studies have investigated both conditions. Remarkably, although TNF-α levels are increased in HD patients and animal models, decreasing TNF-α in HD has not been entirely successful. Treatment of HD mice by intracerebroventricular infusion of a selective soluble inhibitor of TNF-α (XPro1595) decreased TNF-α levels in the cortex and striatum of R6/2 mice, increased neuron density, improved motor function, and decreased mHtt aggregates and gliosis [125]. However, adeno-associated virus (AAV)-vectors expressing dominant-negative-TNF-α injected in the striatum of adult YAC128 mice did not modify motor function, degeneration of medium spiny neurons, or neuroinflammation [272]. The authors explained that a single bilateral injection of AAV-DN-TNF, which may be expressed at 50% of the striatum at best, together with declined expression over time, may not have been sufficient to affect the course of HD in mice. Recently, it was shown that multiple systemic injections of another anti-TNF drug, etanercept, for three weeks lowered the plasma levels of several cytokines, such as IL-6, IL-1β, and TNF-α, which were elevated in R6/2 mice, while having no effect on striatal cytokine levels or motor dysfunction [143]. Therefore, lowering cytokine levels in the periphery is not sufficient to induce improvement of HD, suggesting that systemic administration may not be suitable for HD treatment. Indeed, it was reported that etanercept, which is a large molecule, does not cross the BBB, although other TNF inhibitors do [273]. It was proposed that perispinal injection allows larger molecules, such as etanercept, to enter the brain [274]. This route of administration as beneficial was evidenced in a very small clinical trial [275]. Therefore, studies on the route of administration of candidate drugs, such as TNF inhibitors, should be carefully designed to reliably reach the brain.
9.2. Clinical Trials Concerning Inflammation in HD
Since there is no disease-modifying therapy for HD, the current treatment is aimed at managing chorea and psychiatric symptoms. In the search for effective therapies, several drugs have been tested in clinical trials, including immunomodulators, but they failed to meet their efficacy endpoints [276]. Sativex is a mix of phytocannabinoids (delta-9-tetrahydrocannabinol and cannabidiol) that improves clasping behavior but had no effect on rotarod performance in R6/2 mice [277]. Although cannabinoids are immunomodulators within the brain, a clinical trial (NTC 01502046) showed no differences in motor, cognitive, or behavioral scores in HD patients [278]. Minocycline is a second-generation tetracycline with anti-inflammatory and anti-apoptotic properties [279]. Three clinical trials were performed with minocycline. In the last trial (NTC 00277355), 87 HD patients were treated for 18 months with minocycline, testing the hypothesis that its treatment would reduce the mean decline in total functionality score of subjects by at least 25%, but this goal was not achieved [280].
Laquinimod is an oral drug that decreases NF-κB/GFAP positive astrocytes in mice treated with cuprizone, a model of multiple sclerosis [281]. Laquinimod also upregulates BDNF in multiple sclerosis patients [282]. LPS-stimulated monocytes from HD patients treated with laquinimod reduced cytokine production, although NF-κB activation was not modified [283]. Laquinimod exerted a mild amelioration of motor dysfunction and striatal pathology in YAC128 and R6/2 mice [146, 284]. However, a clinical trial evaluating laquinimod in HD patients failed to meet the primary endpoint since there were no differences in the functional capacity of HD patients treated with laquinimod. Nevertheless, the secondary endpoint was met as patients showed a reduction in caudate atrophy (LEGATO-HD, NTC02215616).
Pepinemab (VX15/2503) is a humanized monoclonal antibody that blocks semaphorin 4D (SEMA4D), also known as CD100, a transmembrane protein that regulates the function of T lymphocytes [285], migration, and differentiation of oligodendrocytes [286], and it is a novel therapeutic for multiple sclerosis [287]. Anti-SEMA4D antibody ameliorates the pathological features of HD, such as striatal and cortical atrophy, and improves behavioral symptoms in YAC128 mice; however, it has no effect on motor dysfunction [288]. A phase 2 clinical trial (SIGNAL, NTC02481674), in which HD subjects were treated for 18 months with intravenous administration of the antibody, 20 mg/kg once a month, showed that the endpoint, finding a patient population that can benefit from treatment, was not achieved. Nevertheless, the company reported a trend towards cognitive improvement in two cognitive assessments [289].
Another therapeutic approach is to decrease mHtt expression in HD patients, which has proven beneficial in animal models. Several drugs that lower Htt RNA expression, such as antisense oligonucleotides (ASO) and miRNA, are under study. Encouraging results of a phase I/II trial showed that a particular ASO, which targeted both Htt and mHtt, significantly lowered mHtt CSF levels [276]. Unfortunately, the phase III trial was halted because ASO failed to demonstrate more efficacy than the placebo [290]. Since normal Htt has several functions, loss of normal Htt may also contribute to HD pathogenesis. Thus, selective silencing of mHtt without disturbing normal Htt is a big challenge. Finally, some preclinical studies support a therapeutic role for passive immunization via targeted mHtt-specific intrabodies [291]. It remains to be investigated if this strategy can be translated into future clinical trials.
CONCLUSION
The precise knowledge of HD pathology mechanisms is lacking, which hinders the development of effective therapeutics for HD. Preclinical research on animal models of HD has led to the identification of diverse protective agents showing an effective reduction in pathology; however, most of the studies have been performed using the toxic HD models [292]. One observation of these studies is that treating HD animals with a protective agent before causing HD by toxic drug administration may not be the best way to prove its benefits in a neurodegenerative disease, which takes years to develop. This limitation can be overcome by using genetic HD models where neurodegeneration is in progress. Moreover, studies show that loss of normal Htt functions can also contribute to the pathology. Depletion of normal Htt in medium spiny neurons was shown to lead to motor dysfunction, loss of striatal neurons with aging, and reactive gliosis, similarly to HD pathology [293]. Although several compounds have been found to be protective in different transgenic HD models, their clinical trials have not been very successful. This may be due to the fact that the studies failed to detect changes in disease parameters or that targeting a single pathological pathway may not be sufficient to prevent disease progression. This may also reflect the limitations of animal models in recapitulating a complex human disease, such as HD. Also, the drugs examined can exert a hormetic response. Hormesis is a dose-response phenomenon characterized by low-dose stimulation and high-dose inhibition [190], meaning that every drug has a dual effect on cells depending on its dose. For example, mitochondrial ROS at high levels are toxic to cells, but at low levels, they perform physiological cellular functions [189]. The same may be true for protective compounds and inflammatory factors, as they can be beneficial at low levels but may contribute to neurodegeneration at high levels.
Immune activation and brain inflammation are important characteristics of HD. Since they can be detected in premanifest HD, they can be targeted early in disease therapy. In recent years, astrocyte and microglia participation in neuroinflammation and HD pathogenesis has been investigated, and studies reflect glial involvement in the course of the disease. Glial dysfunction as well as glial reactivity can contribute to the neurodegenerative process. However, the exact mechanisms whereby neuroinflammation contributes to HD are still unclear. It is also unclear whether immune activation is a pathological mechanism of HD in itself or the consequence of neuronal/glial dysfunction. Although we now know much more about neuroinflammation in HD, we still lack information to help us direct efforts to slow or prevent this fatal disease. Nevertheless, targeting detrimental effects of neuroinflammation could have a beneficial outcome for HD. In addition, clarifying the role of immune factors as biomarkers of the disease is another challenge for the scientific community. Several issues should be addressed in the future to collaborate in the pursuit of effective treatments. Can glial cells become therapeutic targets for HD? Can we aim to modify one single pathway in HD? Would this be sufficient to modify the course of the disease? Could a combination of therapeutic strategies increase the success of HD treatments? Hopefully, in the near future, we will find answers to some of these questions.
ACKNOWLEDGEMENTS
Declared none.
LIST OF ABBREVIATIONS
- GABA
γ-gamma aminobutyric acid
- 24OHC
24S-hydroxycholesterol
- 3NP
3-nitropropionic acid
- AAV
Adeno-associated virus
- ASO
Antisense oligonucleotides
- BAC
Bacterial artificial chromosome
- BBB
Blood-brain barrier
- BDNF
Brain-derived neurotrophic factor
- CNS
Central nervous system
- CSF
Cerebral spinal fluid
- YKL-40
Chitinase 3-like protein-1
- C3
Complement proteins 3
- COX2
Cyclooxygenase 2
- GAT
GABA transporters
- GFAP
Glial fibrillary acidic protein
- GLAST
Glutamate aspartate transporter
- GLT1
Glutamate transporter 1
- HMGB1
High mobility group Box 1
- Htt
Huntingtin
- HD
Huntington’s disease
- Hsp
Heat shock proteins
- HSR
Heat shock response
- hiPSC
Human-induced pluripotent stem cells
- IKK
IκB kinase
- iNOS
Inducible nitric oxide synthase
- IFN-γ
Interferon-γ
- KMO
Kynurenine 3-monooxygenase
- LPS
Lipopolysaccharide
- Iba1
Marker ionized calcium-binding adaptor molecule 1
- MMP
Matrix metalloproteinases
- mHtt
Mutant Htt
- NMDA
N-methyl-D-aspartate
- Nrf2
Nuclear factor erythroid 2-related factor 2
- PET
Positron emission tomography
- QA
Quinolinic acid
- ROS
Reactive oxygen species
- RAGE
Receptor for advanced glycation end products
- SEMA4D
Semaphorin 4D
- SREBP
Sterol regulatory-element binding protein
- TLRs
Toll-like receptors
- TGF-β
Transforming growth factor-β
- TSPO
Translocator protein
- TNF-α
Tumor necrosis factor
- YAC
Yeast artificial chromosome
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971–983. doi: 10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- 2.Sharp A.H., Loev S.J., Schilling G., Li S.H., Li X.J., Bao J., Wagster M.V., Kotzuk J.A., Steiner J.P., Lo A. Widespread expression of Huntington’s disease gene (IT15) protein product. Neuron. 1995;14(5):1065–1074. doi: 10.1016/0896-6273(95)90345-3. [DOI] [PubMed] [Google Scholar]
- 3.Saudou F., Humbert S. The Biology of Huntingtin. Neuron. 2016;89(5):910–926. doi: 10.1016/j.neuron.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 4.Ross C.A., Tabrizi S.J. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10(1):83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 5.Gauthier L.R., Charrin B.C., Borrell-Pagès M., Dompierre J.P., Rangone H., Cordelières F.P., De Mey J., MacDonald M.E., Lessmann V., Humbert S., Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118(1):127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 6.Zuccato C., Marullo M., Conforti P., MacDonald M.E., Tartari M., Cattaneo E. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington’s disease. Brain Pathol. 2008;18(2):225–238. doi: 10.1111/j.1750-3639.2007.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fan M.M., Raymond L.A. N-methyl-D-aspartate (NMDA) receptor function and excitotoxicity in Huntington’s disease. Prog. Neurobiol. 2007;81(5-6):272–293. doi: 10.1016/j.pneurobio.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 8.Hodges A., Strand A.D., Aragaki A.K., Kuhn A., Sengstag T., Hughes G., Elliston L.A., Hartog C., Goldstein D.R., Thu D., Hollingsworth Z.R., Collin F., Synek B., Holmans P.A., Young A.B., Wexler N.S., Delorenzi M., Kooperberg C., Augood S.J., Faull R.L., Olson J.M., Jones L., Luthi-Carter R. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 2006;15(6):965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- 9.Steffan J.S., Kazantsev A., Spasic-Boskovic O., Greenwald M., Zhu Y.Z., Gohler H., Wanker E.E., Bates G.P., Housman D.E., Thompson L.M. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA. 2000;97(12):6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soares T.R., Reis S.D., Pinho B.R., Duchen M.R., Oliveira J.M.A. Targeting the proteostasis network in Huntington’s disease. Ageing Res. Rev. 2019;49:92–103. doi: 10.1016/j.arr.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng J., Winderickx J., Franssens V., Liu B. A Mitochondria-Associated Oxidative Stress Perspective on Huntington’s Disease. Front. Mol. Neurosci. 2018;11:329. doi: 10.3389/fnmol.2018.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Creus-Muncunill J., Ehrlich M.E. Cell-Autonomous and non-cell-autonomous pathogenic mechanisms in huntington’s disease: Insights from in vitro and in vivo models. Neurotherapeutics. 2019;16(4):957–978. doi: 10.1007/s13311-019-00782-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaganti S.S., McCusker E.A., Loy C.T. What do we know about Late Onset Huntington’s Disease? J. Huntingtons Dis. 2017;6(2):95–103. doi: 10.3233/JHD-170247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roos R.A. Huntington’s disease: a clinical review. Orphanet J. Rare Dis. 2010;5:40. doi: 10.1186/1750-1172-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bates G.P., Dorsey R., Gusella J.F., Hayden M.R., Kay C., Leavitt B.R., Nance M., Ross C.A., Scahill R.I., Wetzel R., Wild E.J., Tabrizi S.J. Huntington disease. Nat. Rev. Dis. Primers. 2015;1:15005. doi: 10.1038/nrdp.2015.5. [DOI] [PubMed] [Google Scholar]
- 16.Scahill R.I., Zeun P., Osborne-Crowley K., Johnson E.B., Gregory S., Parker C., Lowe J., Nair A., O’Callaghan C., Langley C., Papoutsi M., McColgan P., Estevez-Fraga C., Fayer K., Wellington H., Rodrigues F.B., Byrne L.M., Heselgrave A., Hyare H., Sampaio C., Zetterberg H., Zhang H., Wild E.J., Rees G., Robbins T.W., Sahakian B.J., Langbehn D., Tabrizi S.J. Biological and clinical characteristics of gene carriers far from predicted onset in the Huntington’s disease Young Adult Study (HD-YAS): a cross-sectional analysis. Lancet Neurol. 2020;19(6):502–512. doi: 10.1016/S1474-4422(20)30143-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson E.B., Ziegler G., Penny W., Rees G., Tabrizi S.J., Scahill R.I., Gregory S. dynamics of cortical degeneration over a decade in Huntington’s disease. Biol. Psychiatry. 2021;89(8):807–816. doi: 10.1016/j.biopsych.2020.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DiFiglia M., Sapp E., Chase K.O., Davies S.W., Bates G.P., Vonsattel J.P., Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277(5334):1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 19.Gómez-Tortosa E., MacDonald M.E., Friend J.C., Taylor S.A., Weiler L.J., Cupples L.A., Srinidhi J., Gusella J.F., Bird E.D., Vonsattel J.P., Myers R.H. Quantitative neuropathological changes in presymptomatic Huntington’s disease. Ann. Neurol. 2001;49(1):29–34. doi: 10.1002/1531-8249(200101)49:1<29:AID-ANA7>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 20.Vonsattel J.P., Keller C., Del Pilar Amaya M. Neuropathology of Huntington’s disease. Handb. Clin. Neurol. 2008;89:599–618. doi: 10.1016/S0072-9752(07)01256-0. [DOI] [PubMed] [Google Scholar]
- 21.Sotrel A., Paskevich P.A., Kiely D.K., Bird E.D., Williams R.S., Myers R.H. Morphometric analysis of the prefrontal cortex in Huntington’s disease. Neurology. 1991;41(7):1117–1123. doi: 10.1212/WNL.41.7.1117. [DOI] [PubMed] [Google Scholar]
- 22.Thu D.C., Oorschot D.E., Tippett L.J., Nana A.L., Hogg V.M., Synek B.J., Luthi-Carter R., Waldvogel H.J., Faull R.L. Cell loss in the motor and cingulate cortex correlates with symptomatology in Huntington’s disease. Brain. 2010;133(Pt 4):1094–1110. doi: 10.1093/brain/awq047. [DOI] [PubMed] [Google Scholar]
- 23.Mehrabi N.F., Waldvogel H.J., Tippett L.J., Hogg V.M., Synek B.J., Faull R.L. Symptom heterogeneity in Huntington’s disease correlates with neuronal degeneration in the cerebral cortex. Neurobiol. Dis. 2016;96:67–74. doi: 10.1016/j.nbd.2016.08.015. [DOI] [PubMed] [Google Scholar]
- 24.Faideau M., Kim J., Cormier K., Gilmore R., Welch M., Auregan G., Dufour N., Guillermier M., Brouillet E., Hantraye P., Déglon N., Ferrante R.J., Bonvento G. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington’s disease subjects. Hum. Mol. Genet. 2010;19(15):3053–3067. doi: 10.1093/hmg/ddq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sapp E., Kegel K.B., Aronin N., Hashikawa T., Uchiyama Y., Tohyama K., Bhide P.G., Vonsattel J.P., DiFiglia M. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J. Neuropathol. Exp. Neurol. 2001;60(2):161–172. doi: 10.1093/jnen/60.2.161. [DOI] [PubMed] [Google Scholar]
- 26.Nichols M.R., St-Pierre M.K., Wendeln A.C., Makoni N.J., Gouwens L.K., Garrad E.C., Sohrabi M., Neher J.J., Tremblay M.E., Combs C.K. Inflammatory mechanisms in neurodegeneration. J. Neurochem. 2019;149(5):562–581. doi: 10.1111/jnc.14674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marogianni C., Sokratous M., Dardiotis E., Hadjigeorgiou G.M., Bogdanos D., Xiromerisiou G. Neurodegeneration and Inflammation-An Interesting Interplay in Parkinson’s Disease. Int. J. Mol. Sci. 2020;21(22):E8421. doi: 10.3390/ijms21228421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Silvestroni A., Faull R.L., Strand A.D., Möller T. Distinct neuroinflammatory profile in post-mortem human Huntington’s disease. Neuroreport. 2009;20(12):1098–1103. doi: 10.1097/WNR.0b013e32832e34ee. [DOI] [PubMed] [Google Scholar]
- 29.Papadopoulos V., Baraldi M., Guilarte T.R., Knudsen T.B., Lacapère J.J., Lindemann P., Norenberg M.D., Nutt D., Weizman A., Zhang M.R., Gavish M. Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006;27(8):402–409. doi: 10.1016/j.tips.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 30.Pavese N., Gerhard A., Tai Y.F., Ho A.K., Turkheimer F., Barker R.A., Brooks D.J., Piccini P. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology. 2006;66(11):1638–1643. doi: 10.1212/01.wnl.0000222734.56412.17. [DOI] [PubMed] [Google Scholar]
- 31.Tai Y.F., Pavese N., Gerhard A., Tabrizi S.J., Barker R.A., Brooks D.J., Piccini P. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain. 2007;130(Pt 7):1759–1766. doi: 10.1093/brain/awm044. [DOI] [PubMed] [Google Scholar]
- 32.Rocha N.P., Charron O., Latham L.B., Colpo G.D., Zanotti-Fregonara P., Yu M., Freeman L., Furr Stimming E., Teixeira A.L. Microglia activation in basal ganglia is a late event in huntington disease pathophysiology. Neurol. Neuroimmunol. Neuroinflamm. 2021;8(3):e984. doi: 10.1212/NXI.0000000000000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Björkqvist M., Wild E.J., Thiele J., Silvestroni A., Andre R., Lahiri N., Raibon E., Lee R.V., Benn C.L., Soulet D., Magnusson A., Woodman B., Landles C., Pouladi M.A., Hayden M.R., Khalili-Shirazi A., Lowdell M.W., Brundin P., Bates G.P., Leavitt B.R., Möller T., Tabrizi S.J. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 2008;205(8):1869–1877. doi: 10.1084/jem.20080178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crotti A., Benner C., Kerman B.E., Gosselin D., Lagier-Tourenne C., Zuccato C., Cattaneo E., Gage F.H., Cleveland D.W., Glass C.K. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat. Neurosci. 2014;17(4):513–521. doi: 10.1038/nn.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singhrao S.K., Neal J.W., Morgan B.P., Gasque P. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington’s disease. Exp. Neurol. 1999;159(2):362–376. doi: 10.1006/exnr.1999.7170. [DOI] [PubMed] [Google Scholar]
- 36.Kim A., García-García E., Straccia M., Comella-Bolla A., Miguez A., Masana M., Alberch J., Canals J.M., Rodríguez M.J. Reduced fractalkine levels lead to striatal synaptic plasticity deficits in Huntington’s disease. Front. Cell. Neurosci. 2020;14:163. doi: 10.3389/fncel.2020.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vuono R., Kouli A., Legault E.M., Chagnon L., Allinson K.S., La Spada A., Biunno I., Barker R.A., Drouin-Ouellet J. Association between toll-like receptor 4 (TLR4) and triggering receptor expressed on myeloid cells 2 (TREM2) genetic variants and clinical progression of Huntington’s disease. Mov. Disord. 2020;35(3):401–408. doi: 10.1002/mds.27911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Battaglia G., Cannella M., Riozzi B., Orobello S., Maat-Schieman M.L., Aronica E., Busceti C.L., Ciarmiello A., Alberti S., Amico E., Sassone J., Sipione S., Bruno V., Frati L., Nicoletti F., Squitieri F. Early defect of transforming growth factor β1 formation in Huntington’s disease. J. Cell. Mol. Med. 2011;15(3):555–571. doi: 10.1111/j.1582-4934.2010.01011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Pardo A., Alberti S., Maglione V., Amico E., Cortes E.P., Elifani F., Battaglia G., Busceti C.L., Nicoletti F., Vonsattel J.P., Squitieri F. Changes of peripheral TGF-β1 depend on monocytes-derived macrophages in Huntington disease. Mol. Brain. 2013;6:55. doi: 10.1186/1756-6606-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laprairie R.B., Warford J.R., Hutchings S., Robertson G.S., Kelly M.E., Denovan-Wright E.M. The cytokine and endocannabinoid systems are co-regulated by NF-κB p65/RelA in cell culture and transgenic mouse models of Huntington’s disease and in striatal tissue from Huntington’s disease patients. J. Neuroimmunol. 2014;267(1-2):61–72. doi: 10.1016/j.jneuroim.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 41.Chou S.Y., Weng J.Y., Lai H.L., Liao F., Sun S.H., Tu P.H., Dickson D.W., Chern Y. Expanded-polyglutamine huntingtin protein suppresses the secretion and production of a chemokine (CCL5/RANTES) by astrocytes. J. Neurosci. 2008;28(13):3277–3290. doi: 10.1523/JNEUROSCI.0116-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ona V.O., Li M., Vonsattel J.P., Andrews L.J., Khan S.Q., Chung W.M., Frey A.S., Menon A.S., Li X.J., Stieg P.E., Yuan J., Penney J.B., Young A.B., Cha J.H., Friedlander R.M. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington’s disease. Nature. 1999;399(6733):263–267. doi: 10.1038/20446. [DOI] [PubMed] [Google Scholar]
- 43.Rodrigues F.B., Byrne L.M., McColgan P., Robertson N., Tabrizi S.J., Zetterberg H., Wild E.J. Cerebrospinal fluid inflammatory biomarkers reflect clinical severity in Huntington’s disease. PLoS One. 2016;11(9):e0163479. doi: 10.1371/journal.pone.0163479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dalrymple A., Wild E.J., Joubert R., Sathasivam K., Björkqvist M., Petersén A., Jackson G.S., Isaacs J.D., Kristiansen M., Bates G.P., Leavitt B.R., Keir G., Ward M., Tabrizi S.J. Proteomic profiling of plasma in Huntington’s disease reveals neuroinflammatory activation and biomarker candidates. J. Proteome Res. 2007;6(7):2833–2840. doi: 10.1021/pr0700753. [DOI] [PubMed] [Google Scholar]
- 45.Fang Q., Strand A., Law W., Faca V.M., Fitzgibbon M.P., Hamel N., Houle B., Liu X., May D.H., Poschmann G., Roy L., Stühler K., Ying W., Zhang J., Zheng Z., Bergeron J.J., Hanash S., He F., Leavitt B.R., Meyer H.E., Qian X., McIntosh M.W. Brain-specific proteins decline in the cerebrospinal fluid of humans with Huntington disease. Mol. Cell. Proteomics. 2009;8(3):451–466. doi: 10.1074/mcp.M800231-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niemelä V., Burman J., Blennow K., Zetterberg H., Larsson A., Sundblom J. Cerebrospinal fluid sCD27 levels indicate active T cell-mediated inflammation in premanifest Huntington’s disease. PLoS One. 2018;13(2):e0193492. doi: 10.1371/journal.pone.0193492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Connolly C., Magnusson-Lind A., Lu G., Wagner P.K., Southwell A.L., Hayden M.R., Björkqvist M., Leavitt B.R. Enhanced immune response to MMP3 stimulation in microglia expressing mutant huntingtin. Neuroscience. 2016;325:74–88. doi: 10.1016/j.neuroscience.2016.03.031. [DOI] [PubMed] [Google Scholar]
- 48.von Essen M.R., Hellem M.N.N., Vinther-Jensen T., Ammitzbøll C., Hansen R.H., Hjermind L.E., Nielsen T.T., Nielsen J.E., Sellebjerg F. Early intrathecal T helper 17.1 cell activity in huntington disease. Ann. Neurol. 2020;87(2):246–255. doi: 10.1002/ana.25647. [DOI] [PubMed] [Google Scholar]
- 49.O’Regan G.C., Farag S.H., Casey C.S., Wood-Kaczmar A., Pocock J.M., Tabrizi S.J., Andre R. Human Huntington’s disease pluripotent stem cell-derived microglia develop normally but are abnormally hyper-reactive and release elevated levels of reactive oxygen species. J. Neuroinflammation. 2021;18(1):94. doi: 10.1186/s12974-021-02147-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leblhuber F., Walli J., Jellinger K., Tilz G.P., Widner B., Laccone F., Fuchs D. Activated immune system in patients with Huntington’s disease. Clin. Chem. Lab. Med. 1998;36(10):747–750. doi: 10.1515/CCLM.1998.132. [DOI] [PubMed] [Google Scholar]
- 51.Chang K.H., Wu Y.R., Chen Y.C., Chen C.M. Plasma inflammatory biomarkers for Huntington’s disease patients and mouse model. Brain Behav. Immun. 2015;44:121–127. doi: 10.1016/j.bbi.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 52.Wild E., Magnusson A., Lahiri N., Krus U., Orth M., Tabrizi S.J., Björkqvist M. Abnormal peripheral chemokine profile in Huntington’s disease. PLoS Curr. 2011;3:RRN1231. doi: 10.1371/currents.RRN1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Silajdžić E., Rezeli M., Végvári Á., Lahiri N., Andre R., Magnusson-Lind A., Nambron R., Kalliolia E., Marko-Varga G., Warner T.T., Laurell T., Tabrizi S.J., Björkqvist M. A critical evaluation of inflammatory markers in Huntington’s Disease plasma. J. Huntingtons Dis. 2013;2(1):125–134. doi: 10.3233/JHD-130049. [DOI] [PubMed] [Google Scholar]
- 54.Politis M., Lahiri N., Niccolini F., Su P., Wu K., Giannetti P., Scahill R.I., Turkheimer F.E., Tabrizi S.J., Piccini P. Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington’s disease gene carriers. Neurobiol. Dis. 2015;83:115–121. doi: 10.1016/j.nbd.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 55.Wang R., Ross C.A., Cai H., Cong W.N., Daimon C.M., Carlson O.D., Egan J.M., Siddiqui S., Maudsley S., Martin B. Metabolic and hormonal signatures in pre-manifest and manifest Huntington’s disease patients. Front. Physiol. 2014;5:231. doi: 10.3389/fphys.2014.00231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Träger U., Andre R., Lahiri N., Magnusson-Lind A., Weiss A., Grueninger S., McKinnon C., Sirinathsinghji E., Kahlon S., Pfister E.L., Moser R., Hummerich H., Antoniou M., Bates G.P., Luthi-Carter R., Lowdell M.W., Björkqvist M., Ostroff G.R., Aronin N., Tabrizi S.J. HTT-lowering reverses Huntington’s disease immune dysfunction caused by NFκB pathway dysregulation. Brain. 2014;137(Pt 3):819–833. doi: 10.1093/brain/awt355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yeo I.J., Lee C.K., Han S.B., Yun J., Hong J.T. Roles of chitinase 3-like 1 in the development of cancer, neurodegenerative diseases, and inflammatory diseases. Pharmacol. Ther. 2019;203:107394. doi: 10.1016/j.pharmthera.2019.107394. [DOI] [PubMed] [Google Scholar]
- 58.Elmonem M.A., van den Heuvel L.P., Levtchenko E.N. Immunomodulatory effects of chitotriosidase enzyme. Enzyme Res. 2016;2016:2682680. doi: 10.1155/2016/2682680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bonneh-Barkay D., Bissel S.J., Kofler J., Starkey A., Wang G., Wiley C.A. Astrocyte and macrophage regulation of YKL-40 expression and cellular response in neuroinflammation. Brain Pathol. 2012;22(4):530–546. doi: 10.1111/j.1750-3639.2011.00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moon H.J., Herring S.K., Zhao L. Clusterin: a multifaceted protein in the brain. Neural Regen. Res. 2021;16(7):1438–1439. doi: 10.4103/1673-5374.301013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaur G., Behl T., Sehgal A., Singh S., Sharma N., Kumar A., Arora S., Bungau S. RETRACTED ARTICLE: Role of TLR2 and TLR4 signaling in Parkinson’s disease: an insight into associated therapeutic potential. J. Mol. Neurosci. 2021;71(11):2429. doi: 10.1007/s12031-021-01811-z. [DOI] [PubMed] [Google Scholar]
- 62.Hammond T.R., Marsh S.E., Stevens B. Immune signaling in neurodegeneration. Immunity. 2019;50(4):955–974. doi: 10.1016/j.immuni.2019.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pittaluga A. CCL5-Glutamate cross-talk in astrocyte-neuron communication in multiple sclerosis. Front. Immunol. 2017;8:1079. doi: 10.3389/fimmu.2017.01079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miller J.P., Holcomb J., Al-Ramahi I., de Haro M., Gafni J., Zhang N., Kim E., Sanhueza M., Torcassi C., Kwak S., Botas J., Hughes R.E., Ellerby L.M. Matrix metalloproteinases are modifiers of huntingtin proteolysis and toxicity in Huntington’s disease. Neuron. 2010;67(2):199–212. doi: 10.1016/j.neuron.2010.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim Y.S., Kim S.S., Cho J.J., Choi D.H., Hwang O., Shin D.H., Chun H.S., Beal M.F., Joh T.H. Matrix metalloproteinase-3: a novel signaling proteinase from apoptotic neuronal cells that activates microglia. J. Neurosci. 2005;25(14):3701–3711. doi: 10.1523/JNEUROSCI.4346-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weiss A., Träger U., Wild E.J., Grueninger S., Farmer R., Landles C., Scahill R.I., Lahiri N., Haider S., Macdonald D., Frost C., Bates G.P., Bilbe G., Kuhn R., Andre R., Tabrizi S.J. Mutant huntingtin fragmentation in immune cells tracks Huntington’s disease progression. J. Clin. Invest. 2012;122(10):3731–3736. doi: 10.1172/JCI64565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miller J.R., Träger U., Andre R., Tabrizi S.J. Mutant huntingtin does not affect the intrinsic phenotype of human huntington’s disease T lymphocytes. PLoS One. 2015;10(11):e0141793. doi: 10.1371/journal.pone.0141793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pierozan P., Gonçalves Fernandes C., Ferreira F., Pessoa-Pureur R. Acute intrastriatal injection of quinolinic acid provokes long-lasting misregulation of the cytoskeleton in the striatum, cerebral cortex and hippocampus of young rats. Brain Res. 2014;1577:1–10. doi: 10.1016/j.brainres.2014.06.024. [DOI] [PubMed] [Google Scholar]
- 69.Amaral A.U., Seminotti B., da Silva J.C., de Oliveira F.H., Ribeiro R.T., Vargas C.R., Leipnitz G., Santamaría A., Souza D.O., Wajner M. Induction of neuroinflammatory response and histopathological alterations caused by quinolinic acid administration in the striatum of glutaryl-CoA dehydrogenase deficient mice. Neurotox. Res. 2018;33(3):593–606. doi: 10.1007/s12640-017-9848-0. [DOI] [PubMed] [Google Scholar]
- 70.Emerich D.F., Thanos C.G., Goddard M., Skinner S.J., Geany M.S., Bell W.J., Bintz B., Schneider P., Chu Y., Babu R.S., Borlongan C.V., Boekelheide K., Hall S., Bryant B., Kordower J.H. Extensive neuroprotection by choroid plexus transplants in excitotoxin lesioned monkeys. Neurobiol. Dis. 2006;23(2):471–480. doi: 10.1016/j.nbd.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 71.Beal M.F., Ferrante R.J., Swartz K.J., Kowall N.W. Chronic quinolinic acid lesions in rats closely resemble Huntington’s disease. J. Neurosci. 1991;11(6):1649–1659. doi: 10.1523/JNEUROSCI.11-06-01649.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Björklund H., Olson L., Dahl D., Schwarcz R. Short- and long-term consequences of intracranial injections of the excitotoxin, quinolinic acid, as evidenced by GFA immunohistochemistry of astrocytes. Brain Res. 1986;371(2):267–277. doi: 10.1016/0006-8993(86)90362-8. [DOI] [PubMed] [Google Scholar]
- 73.Emerich D.F., Cain C.K., Greco C., Saydoff J.A., Hu Z.Y., Liu H., Lindner M.D. Cellular delivery of human CNTF prevents motor and cognitive dysfunction in a rodent model of Huntington’s disease. Cell Transplant. 1997;6(3):249–266. doi: 10.1177/096368979700600308. [DOI] [PubMed] [Google Scholar]
- 74.Ludolph A.C., He F., Spencer P.S., Hammerstad J., Sabri M. 3-Nitropropionic acid-exogenous animal neurotoxin and possible human striatal toxin. Can. J. Neurol. Sci. 1991;18(4):492–498. doi: 10.1017/S0317167100032212. [DOI] [PubMed] [Google Scholar]
- 75.Brouillet E., Jacquard C., Bizat N., Blum D. 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J. Neurochem. 2005;95(6):1521–1540. doi: 10.1111/j.1471-4159.2005.03515.x. [DOI] [PubMed] [Google Scholar]
- 76.Borlongan C.V., Nishino H., Sanberg P.R. Systemic, but not intraparenchymal, administration of 3-nitropropionic acid mimics the neuropathology of Huntington’s disease: a speculative explanation. Neurosci. Res. 1997;28(3):185–189. doi: 10.1016/S0168-0102(97)00045-X. [DOI] [PubMed] [Google Scholar]
- 77.Borlongan C.V., Koutouzis T.K., Freeman T.B., Hauser R.A., Cahill D.W., Sanberg P.R. Hyperactivity and hypoactivity in a rat model of Huntington’s disease: the systemic 3-nitropropionic acid model. Brain Res. Brain Res. Protoc. 1997;1(3):253–257. doi: 10.1016/S1385-299X(96)00037-2. [DOI] [PubMed] [Google Scholar]
- 78.Túnez I., Tasset I., Pérez-De La Cruz V., Santamaría A. 3-Nitropropionic acid as a tool to study the mechanisms involved in Huntington’s disease: past, present and future. Molecules. 2010;15(2):878–916. doi: 10.3390/molecules15020878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhong F., Liang S., Zhong Z. Emerging role of mitochondrial DNA as a major driver of inflammation and disease progression. Trends Immunol. 2019;40(12):1120–1133. doi: 10.1016/j.it.2019.10.008. [DOI] [PubMed] [Google Scholar]
- 80.Pang Z., Geddes J.W. Mechanisms of cell death induced by the mitochondrial toxin 3-nitropropionic acid: acute excitotoxic necrosis and delayed apoptosis. J. Neurosci. 1997;17(9):3064–3073. doi: 10.1523/JNEUROSCI.17-09-03064.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Saba J., Turati J., Ramírez D., Carniglia L., Durand D., Lasaga M., Caruso C. Astrocyte truncated tropomyosin receptor kinase B mediates brain-derived neurotrophic factor anti-apoptotic effect leading to neuroprotection. J. Neurochem. 2018;146(6):686–702. doi: 10.1111/jnc.14476. [DOI] [PubMed] [Google Scholar]
- 82.Saba J., Carniglia L., Ramírez D., Turati J., Imsen M., Durand D., Lasaga M., Caruso C. Melanocortin 4 receptor activation protects striatal neurons and glial cells from 3-nitropropionic acid toxicity. Mol. Cell. Neurosci. 2019;94:41–51. doi: 10.1016/j.mcn.2018.12.002. [DOI] [PubMed] [Google Scholar]
- 83.Saba J., López Couselo F., Turati J., Carniglia L., Durand D., de Laurentiis A., Lasaga M., Caruso C. Astrocytes from cortex and striatum show differential responses to mitochondrial toxin and BDNF: implications for protection of striatal neurons expressing mutant huntingtin. J. Neuroinflammation. 2020;17(1):290. doi: 10.1186/s12974-020-01965-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mangiarini L., Sathasivam K., Seller M., Cozens B., Harper A., Hetherington C., Lawton M., Trottier Y., Lehrach H., Davies S.W., Bates G.P. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87(3):493–506. doi: 10.1016/S0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 85.Stack E.C., Kubilus J.K., Smith K., Cormier K., Del Signore S.J., Guelin E., Ryu H., Hersch S.M., Ferrante R.J. Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington’s disease transgenic mice. J. Comp. Neurol. 2005;490(4):354–370. doi: 10.1002/cne.20680. [DOI] [PubMed] [Google Scholar]
- 86.Davies S.W., Turmaine M., Cozens B.A., DiFiglia M., Sharp A.H., Ross C.A., Scherzinger E., Wanker E.E., Mangiarini L., Bates G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90(3):537–548. doi: 10.1016/S0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 87.Vonsattel J.P. Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115(1):55–69. doi: 10.1007/s00401-007-0306-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schilling G., Becher M.W., Sharp A.H., Jinnah H.A., Duan K., Kotzuk J.A., Slunt H.H., Ratovitski T., Cooper J.K., Jenkins N.A., Copeland N.G., Price D.L., Ross C.A., Borchelt D.R. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 1999;8(3):397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- 89.McBride J.L., Ramaswamy S., Gasmi M., Bartus R.T., Herzog C.D., Brandon E.P., Zhou L., Pitzer M.R., Berry-Kravis E.M., Kordower J.H. Viral delivery of glial cell line-derived neurotrophic factor improves behavior and protects striatal neurons in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA. 2006;103(24):9345–9350. doi: 10.1073/pnas.0508875103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee C.Y., Cantle J.P., Yang X.W. Genetic manipulations of mutant huntingtin in mice: new insights into Huntington’s disease pathogenesis. FEBS J. 2013;280(18):4382–4394. doi: 10.1111/febs.12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Slow E.J., van Raamsdonk J., Rogers D., Coleman S.H., Graham R.K., Deng Y., Oh R., Bissada N., Hossain S.M., Yang Y.Z., Li X.J., Simpson E.M., Gutekunst C.A., Leavitt B.R., Hayden M.R. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 2003;12(13):1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- 92.Hodgson J.G., Agopyan N., Gutekunst C.A., Leavitt B.R., LePiane F., Singaraja R., Smith D.J., Bissada N., McCutcheon K., Nasir J., Jamot L., Li X.J., Stevens M.E., Rosemond E., Roder J.C., Phillips A.G., Rubin E.M., Hersch S.M., Hayden M.R. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23(1):181–192. doi: 10.1016/S0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 93.Gray M., Shirasaki D.I., Cepeda C., André V.M., Wilburn B., Lu X.H., Tao J., Yamazaki I., Li S.H., Sun Y.E., Li X.J., Levine M.S., Yang X.W. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci. 2008;28(24):6182–6195. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Menalled L., El-Khodor B.F., Patry M., Suárez-Fariñas M., Orenstein S.J., Zahasky B., Leahy C., Wheeler V., Yang X.W., MacDonald M., Morton A.J., Bates G., Leeds J., Park L., Howland D., Signer E., Tobin A., Brunner D. Systematic behavioral evaluation of Huntington’s disease transgenic and knock-in mouse models. Neurobiol. Dis. 2009;35(3):319–336. doi: 10.1016/j.nbd.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wegrzynowicz M., Bichell T.J., Soares B.D., Loth M.K., McGlothan J.S., Mori S., Alikhan F.S., Hua K., Coughlin J.M., Holt H.K., Jetter C.S., Pomper M.G., Osmand A.P., Guilarte T.R., Bowman A.B. Novel BAC mouse model of Huntington’s disease with 225 CAG repeats exhibits an early widespread and stable degenerative phenotype. J. Huntingtons Dis. 2015;4(1):17–36. doi: 10.3233/JHD-140116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Reddy P.H., Williams M., Charles V., Garrett L., Pike-Buchanan L., Whetsell W.O., Jr, Miller G., Tagle D.A. Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat. Genet. 1998;20(2):198–202. doi: 10.1038/2510. [DOI] [PubMed] [Google Scholar]
- 97.von Hörsten S., Schmitt I., Nguyen H.P., Holzmann C., Schmidt T., Walther T., Bader M., Pabst R., Kobbe P., Krotova J., Stiller D., Kask A., Vaarmann A., Rathke-Hartlieb S., Schulz J.B., Grasshoff U., Bauer I., Vieira-Saecker A.M., Paul M., Jones L., Lindenberg K.S., Landwehrmeyer B., Bauer A., Li X.J., Riess O. Transgenic rat model of Huntington’s disease. Hum. Mol. Genet. 2003;12(6):617–624. doi: 10.1093/hmg/ddg075. [DOI] [PubMed] [Google Scholar]
- 98.Yu-Taeger L., Petrasch-Parwez E., Osmand A.P., Redensek A., Metzger S., Clemens L.E., Park L., Howland D., Calaminus C., Gu X., Pichler B., Yang X.W., Riess O., Nguyen H.P. A novel BACHD transgenic rat exhibits characteristic neuropathological features of Huntington disease. J. Neurosci. 2012;32(44):15426–15438. doi: 10.1523/JNEUROSCI.1148-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wheeler V.C., Gutekunst C.A., Vrbanac V., Lebel L.A., Schilling G., Hersch S., Friedlander R.M., Gusella J.F., Vonsattel J.P., Borchelt D.R., MacDonald M.E. Early phenotypes that presage late-onset neurodegenerative disease allow testing of modifiers in Hdh CAG knock-in mice. Hum. Mol. Genet. 2002;11(6):633–640. doi: 10.1093/hmg/11.6.633. [DOI] [PubMed] [Google Scholar]
- 100.Menalled L.B., Sison J.D., Dragatsis I., Zeitlin S., Chesselet M.F. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J. Comp. Neurol. 2003;465(1):11–26. doi: 10.1002/cne.10776. [DOI] [PubMed] [Google Scholar]
- 101.Lin C.H., Tallaksen-Greene S., Chien W.M., Cearley J.A., Jackson W.S., Crouse A.B., Ren S., Li X.J., Albin R.L., Detloff P.J. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum. Mol. Genet. 2001;10(2):137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- 102.Menalled L.B., Kudwa A.E., Miller S., Fitzpatrick J., Watson-Johnson J., Keating N., Ruiz M., Mushlin R., Alosio W., McConnell K., Connor D., Murphy C., Oakeshott S., Kwan M., Beltran J., Ghavami A., Brunner D., Park L.C., Ramboz S., Howland D. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington’s disease: zQ175. PLoS One. 2012;7(12):e49838. doi: 10.1371/journal.pone.0049838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stricker-Shaver J., Novati A., Yu-Taeger L., Nguyen H.P. Genetic rodent models of huntington disease. Adv. Exp. Med. Biol. 2018;1049:29–57. doi: 10.1007/978-3-319-71779-1_2. [DOI] [PubMed] [Google Scholar]
- 104.Escartin C., Galea E., Lakatos A., O’Callaghan J.P., Petzold G.C., Serrano-Pozo A., Steinhäuser C., Volterra A., Carmignoto G., Agarwal A., Allen N.J., Araque A., Barbeito L., Barzilai A., Bergles D.E., Bonvento G., Butt A.M., Chen W.T., Cohen-Salmon M., Cunningham C., Deneen B., De Strooper B., Díaz-Castro B., Farina C., Freeman M., Gallo V., Goldman J.E., Goldman S.A., Götz M., Gutiérrez A., Haydon P.G., Heiland D.H., Hol E.M., Holt M.G., Iino M., Kastanenka K.V., Kettenmann H., Khakh B.S., Koizumi S., Lee C.J., Liddelow S.A., MacVicar B.A., Magistretti P., Messing A., Mishra A., Molofsky A.V., Murai K.K., Norris C.M., Okada S., Oliet S.H.R., Oliveira J.F., Panatier A., Parpura V., Pekna M., Pekny M., Pellerin L., Perea G., Pérez-Nievas B.G., Pfrieger F.W., Poskanzer K.E., Quintana F.J., Ransohoff R.M., Riquelme-Perez M., Robel S., Rose C.R., Rothstein J.D., Rouach N., Rowitch D.H., Semyanov A., Sirko S., Sontheimer H., Swanson R.A., Vitorica J., Wanner I.B., Wood L.B., Wu J., Zheng B., Zimmer E.R., Zorec R., Sofroniew M.V., Verkhratsky A. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021;24(3):312–325. doi: 10.1038/s41593-020-00783-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cirillo G., Maggio N., Bianco M.R., Vollono C., Sellitti S., Papa M. Discriminative behavioral assessment unveils remarkable reactive astrocytosis and early molecular correlates in basal ganglia of 3-nitropropionic acid subchronic treated rats. Neurochem. Int. 2010;56(1):152–160. doi: 10.1016/j.neuint.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 106.Mu S., Liu B., Ouyang L., Zhan M., Chen S., Wu J., Chen J., Wei X., Wang W., Zhang J., Lei W. Characteristic changes of astrocyte and microglia in rat striatum induced by 3-NP and MCAO. Neurochem. Res. 2016;41(4):707–714. doi: 10.1007/s11064-015-1739-2. [DOI] [PubMed] [Google Scholar]
- 107.Cirillo G., Cirillo M., Panetsos F., Virtuoso A., Papa M. Selective vulnerability of basal ganglia: Insights into the mechanisms of bilateral striatal necrosis. J. Neuropathol. Exp. Neurol. 2019;78(2):123–129. doi: 10.1093/jnen/nly123. [DOI] [PubMed] [Google Scholar]
- 108.Nishino H., Fujimoto I., Shimano Y., Hida H., Kumazaki M., Fukuda A. 3-Nitropropionic acid produces striatum selective lesions accompanied by iNOS expression. J. Chem. Neuroanat. 1996;10(3-4):209–212. doi: 10.1016/0891-0618(96)00134-2. [DOI] [PubMed] [Google Scholar]
- 109.Suganya S.N., Sumathi T. Effect of rutin against a mitochondrial toxin, 3-nitropropionicacid induced biochemical, behavioral and histological alterations-a pilot study on Huntington’s disease model in rats. Metab. Brain Dis. 2017;32(2):471–481. doi: 10.1007/s11011-016-9929-4. [DOI] [PubMed] [Google Scholar]
- 110.Valdeolivas S., Navarrete C., Cantarero I., Bellido M.L., Muñoz E., Sagredo O. Neuroprotective properties of cannabigerol in Huntington’s disease: studies in R6/2 mice and 3-nitropropionate-lesioned mice. Neurotherapeutics. 2015;12(1):185–199. doi: 10.1007/s13311-014-0304-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ranju V., Sathiya S., Kalaivani P., Priya R.J., Saravana Babu C. Memantine exerts functional recovery by improving BDNF and GDNF expression in 3-nitropropionic acid intoxicated mice. Neurosci. Lett. 2015;586:1–7. doi: 10.1016/j.neulet.2014.11.036. [DOI] [PubMed] [Google Scholar]
- 112.Bayram-Weston Z., Jones L., Dunnett S.B., Brooks S.P. Light and electron microscopic characterization of the evolution of cellular pathology in the R6/1 Huntington’s disease transgenic mice. Brain Res. Bull. 2012;88(2-3):104–112. doi: 10.1016/j.brainresbull.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 113.Yu Z.X., Li S.H., Evans J., Pillarisetti A., Li H., Li X.J. Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington’s disease. J. Neurosci. 2003;23(6):2193–2202. doi: 10.1523/JNEUROSCI.23-06-02193.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dutta D., Majumder M., Paidi R.K., Pahan K. Alleviation of Huntington pathology in mice by oral administration of food additive glyceryl tribenzoate. Neurobiol. Dis. 2021;153:105318. doi: 10.1016/j.nbd.2021.105318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bayram-Weston Z., Jones L., Dunnett S.B., Brooks S.P. Light and electron microscopic characterization of the evolution of cellular pathology in YAC128 Huntington’s disease transgenic mice. Brain Res. Bull. 2012;88(2-3):137–147. doi: 10.1016/j.brainresbull.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 116.Ehrnhoefer D.E., Southwell A.L., Sivasubramanian M., Qiu X., Villanueva E.B., Xie Y., Waltl S., Anderson L., Fazeli A., Casal L., Felczak B., Tsang M., Hayden M.R. HACE1 is essential for astrocyte mitochondrial function and influences Huntington disease phenotypes in vivo. Hum. Mol. Genet. 2018;27(2):239–253. doi: 10.1093/hmg/ddx394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mantovani S., Gordon R., Li R., Christie D.C., Kumar V., Woodruff T.M. Motor deficits associated with Huntington’s disease occur in the absence of striatal degeneration in BACHD transgenic mice. Hum. Mol. Genet. 2016;25(9):1780–1791. doi: 10.1093/hmg/ddw050. [DOI] [PubMed] [Google Scholar]
- 118.Heng M.Y., Duong D.K., Albin R.L., Tallaksen-Greene S.J., Hunter J.M., Lesort M.J., Osmand A., Paulson H.L., Detloff P.J. Early autophagic response in a novel knock-in model of Huntington disease. Hum. Mol. Genet. 2010;19(19):3702–3720. doi: 10.1093/hmg/ddq285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vagner T., Dvorzhak A., Wójtowicz A.M., Harms C., Grantyn R. Systemic application of AAV vectors targeting GFAP-expressing astrocytes in Z-Q175-KI Huntington’s disease mice. Mol. Cell. Neurosci. 2016;77:76–86. doi: 10.1016/j.mcn.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 120.Agostoni E., Michelazzi S., Maurutto M., Carnemolla A., Ciani Y., Vatta P., Roncaglia P., Zucchelli S., Leanza G., Mantovani F., Gustincich S., Santoro C., Piazza S., Del Sal G., Persichetti F. Effects of Pin1 loss in hdh(Q111) knock-in mice. Front. Cell. Neurosci. 2016;10:110. doi: 10.3389/fncel.2016.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bayram-Weston Z., Torres E.M., Jones L., Dunnett S.B., Brooks S.P. Light and electron microscopic characterization of the evolution of cellular pathology in the Hdh(CAG)150 Huntington’s disease knock-in mouse. Brain Res. Bull. 2012;88(2-3):189–198. doi: 10.1016/j.brainresbull.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 122.Diaz-Castro B., Gangwani M.R., Yu X., Coppola G., Khakh B.S. Astrocyte molecular signatures in Huntington’s disease. Sci. Transl. Med. 2019;11(514):eaaw8546. doi: 10.1126/scitranslmed.aaw8546. [DOI] [PubMed] [Google Scholar]
- 123.Lallani S.B., Villalba R.M., Chen Y., Smith Y., Chan A.W.S. Striatal interneurons in transgenic nonhuman primate model of Huntington’s disease. Sci. Rep. 2019;9(1):3528. doi: 10.1038/s41598-019-40165-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Politis M., Pavese N., Tai Y.F., Kiferle L., Mason S.L., Brooks D.J., Tabrizi S.J., Barker R.A., Piccini P. Microglial activation in regions related to cognitive function predicts disease onset in Huntington’s disease: a multimodal imaging study. Hum. Brain Mapp. 2011;32(2):258–270. doi: 10.1002/hbm.21008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hsiao H.Y., Chiu F.L., Chen C.M., Wu Y.R., Chen H.M., Chen Y.C., Kuo H.C., Chern Y. Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum. Mol. Genet. 2014;23(16):4328–4344. doi: 10.1093/hmg/ddu151. [DOI] [PubMed] [Google Scholar]
- 126.Bouchard J., Truong J., Bouchard K., Dunkelberger D., Desrayaud S., Moussaoui S., Tabrizi S.J., Stella N., Muchowski P.J. Cannabinoid receptor 2 signaling in peripheral immune cells modulates disease onset and severity in mouse models of Huntington’s disease. J. Neurosci. 2012;32(50):18259–18268. doi: 10.1523/JNEUROSCI.4008-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Simmons D.A., Casale M., Alcon B., Pham N., Narayan N., Lynch G. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia. 2007;55(10):1074–1084. doi: 10.1002/glia.20526. [DOI] [PubMed] [Google Scholar]
- 128.Jansen A.H., van Hal M., Op den Kelder I.C., Meier R.T., de Ruiter A.A., Schut M.H., Smith D.L., Grit C., Brouwer N., Kamphuis W., Boddeke H.W., den Dunnen W.F., van Roon W.M., Bates G.P., Hol E.M., Reits E.A. Frequency of nuclear mutant huntingtin inclusion formation in neurons and glia is cell-type-specific. Glia. 2017;65(1):50–61. doi: 10.1002/glia.23050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Franciosi S., Ryu J.K., Shim Y., Hill A., Connolly C., Hayden M.R., McLarnon J.G., Leavitt B.R. Age-dependent neurovascular abnormalities and altered microglial morphology in the YAC128 mouse model of Huntington disease. Neurobiol. Dis. 2012;45(1):438–449. doi: 10.1016/j.nbd.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 130.Kraft A.D., Kaltenbach L.S., Lo D.C., Harry G.J. Activated microglia proliferate at neurites of mutant huntingtin-expressing neurons. Neurobiol. Aging. 2012;33(3):621.e17–e633. doi: 10.1016/j.neurobiolaging.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Savage J.C., St-Pierre M.K., Carrier M., El Hajj H., Novak S.W., Sanchez M.G., Cicchetti F., Tremblay M.E. Microglial physiological properties and interactions with synapses are altered at presymptomatic stages in a mouse model of Huntington’s disease pathology. J. Neuroinflammation. 2020;17(1):98. doi: 10.1186/s12974-020-01782-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gill J.S., Jamwal S., Kumar P., Deshmukh R. Sertraline and venlafaxine improves motor performance and neurobehavioral deficit in quinolinic acid induced Huntington’s like symptoms in rats: Possible neurotransmitters modulation. Pharmacol. Rep. 2017;69(2):306–313. doi: 10.1016/j.pharep.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 133.Ferreira F.S., Schmitz F., Marques E.P., Siebert C., Wyse A.T.S. Intrastriatal quinolinic acid administration impairs redox homeostasis and induces inflammatory changes: Prevention by kynurenic acid. Neurotox. Res. 2020;38(1):50–58. doi: 10.1007/s12640-020-00192-2. [DOI] [PubMed] [Google Scholar]
- 134.Saliba S.W., Vieira E.L., Santos R.P., Candelario-Jalil E., Fiebich B.L., Vieira L.B., Teixeira A.L., de Oliveira A.C. Neuroprotective effects of intrastriatal injection of rapamycin in a mouse model of excitotoxicity induced by quinolinic acid. J. Neuroinflammation. 2017;14(1):25. doi: 10.1186/s12974-017-0793-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kaur N., Jamwal S., Deshmukh R., Gauttam V., Kumar P. Beneficial effect of rice bran extract against 3-nitropropionic acid induced experimental Huntington’s disease in rats. Toxicol. Rep. 2015;2:1222–1232. doi: 10.1016/j.toxrep.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Abdelfattah M.S., Badr S.E.A., Lotfy S.A., Attia G.H., Aref A.M., Abdel Moneim A.E., Kassab R.B. Rutin and selenium co-administration reverse 3-nitropropionic acid-induced neurochemical and molecular impairments in a mouse model of Huntington’s disease. Neurotox. Res. 2020;37(1):77–92. doi: 10.1007/s12640-019-00086-y. [DOI] [PubMed] [Google Scholar]
- 137.Jang M., Choi J.H., Chang Y., Lee S.J., Nah S.Y., Cho I.H. Gintonin, a ginseng-derived ingredient, as a novel therapeutic strategy for Huntington’s disease: Activation of the Nrf2 pathway through lysophosphatidic acid receptors. Brain Behav. Immun. 2019;80:146–162. doi: 10.1016/j.bbi.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 138.El-Abhar H., Abd El Fattah M.A., Wadie W., El-Tanbouly D.M. Cilostazol disrupts TLR-4, Akt/GSK-3β/CREB, and IL-6/JAK-2/STAT-3/SOCS-3 crosstalk in a rat model of Huntington’s disease. PLoS One. 2018;13(9):e0203837. doi: 10.1371/journal.pone.0203837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lopez-Sanchez C., Garcia-Martinez V., Poejo J., Garcia-Lopez V., Salazar J., Gutierrez-Merino C. Early reactive A1 astrocytes induction by the neurotoxin 3-nitropropionic acid in rat brain. Int. J. Mol. Sci. 2020;21(10):E3609. doi: 10.3390/ijms21103609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hsiao H.Y., Chen Y.C., Chen H.M., Tu P.H., Chern Y. A critical role of astrocyte-mediated nuclear factor-κB-dependent inflammation in Huntington’s disease. Hum. Mol. Genet. 2013;22(9):1826–1842. doi: 10.1093/hmg/ddt036. [DOI] [PubMed] [Google Scholar]
- 141.Pido-Lopez J., Andre R., Benjamin A.C., Ali N., Farag S., Tabrizi S.J., Bates G.P. In vivo neutralization of the protagonist role of macrophages during the chronic inflammatory stage of Huntington’s disease. Sci. Rep. 2018;8(1):11447. doi: 10.1038/s41598-018-29792-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Simmons D.A., Mills B.D., Butler Iii R.R., Kuan J., McHugh T.L.M., Akers C., Zhou J., Syriani W., Grouban M., Zeineh M., Longo F.M. Neuroimaging, urinary, and plasma biomarkers of treatment response in Huntington’s disease: Preclinical evidence with the p75NTR ligand LM11A-31. Neurotherapeutics. 2021;18(2):1039–1063. doi: 10.1007/s13311-021-01023-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pido-Lopez J., Tanudjojo B., Farag S., Bondulich M.K., Andre R., Tabrizi S.J., Bates G.P. Inhibition of tumour necrosis factor alpha in the R6/2 mouse model of Huntington’s disease by etanercept treatment. Sci. Rep. 2019;9(1):7202. doi: 10.1038/s41598-019-43627-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Träger U., Andre R., Magnusson-Lind A., Miller J.R., Connolly C., Weiss A., Grueninger S., Silajdžić E., Smith D.L., Leavitt B.R., Bates G.P., Björkqvist M., Tabrizi S.J. Characterisation of immune cell function in fragment and full-length Huntington’s disease mouse models. Neurobiol. Dis. 2015;73:388–398. doi: 10.1016/j.nbd.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Corey-Bloom J., Aikin A.M., Gutierrez A.M., Nadhem J.S., Howell T.L., Thomas E.A. Beneficial effects of glatiramer acetate in Huntington’s disease mouse models: Evidence for BDNF-elevating and immunomodulatory mechanisms. Brain Res. 2017;1673:102–110. doi: 10.1016/j.brainres.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Garcia-Miralles M., Hong X., Tan L.J., Caron N.S., Huang Y., To X.V., Lin R.Y., Franciosi S., Papapetropoulos S., Hayardeny L., Hayden M.R., Chuang K.H., Pouladi M.A. Laquinimod rescues striatal, cortical and white matter pathology and results in modest behavioural improvements in the YAC128 model of Huntington disease. Sci. Rep. 2016;6:31652. doi: 10.1038/srep31652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Valadão P.A.C., Oliveira B.D.S., Joviano-Santos J.V., Vieira E.L.M., Rocha N.P., Teixeira A.L., Guatimosim C., de Miranda A.S. Inflammatory changes in peripheral organs in the BACHD murine model of Huntington’s disease. Life Sci. 2019;232:116653. doi: 10.1016/j.lfs.2019.116653. [DOI] [PubMed] [Google Scholar]
- 148.Verkhratsky A., Nedergaard M. Physiology of astroglia. Physiol. Rev. 2018;98(1):239–389. doi: 10.1152/physrev.00042.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sofroniew M.V. Astrocyte reactivity: subtypes, states, and functions in CNS innate immunity. Trends Immunol. 2020;41(9):758–770. doi: 10.1016/j.it.2020.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Khakh B.S., Beaumont V., Cachope R., Munoz-Sanjuan I., Goldman S.A., Grantyn R. Unravelling and Exploiting Astrocyte Dysfunction in Huntington’s Disease. Trends Neurosci. 2017;40(7):422–437. doi: 10.1016/j.tins.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015;16(5):249–263. doi: 10.1038/nrn3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shin, J.Y.; Fang, Z.H.; Yu, Z.X.; Wang, C.E.; Li, S.H.; Li, X.J. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol. 2005;171(6):1001–1012. doi: 10.1083/jcb.200508072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bradford, J.; Shin, J.Y.; Roberts, M.; Wang, C.E.; Li, X.J.; Li, S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc. Natl. Acad. Sci. USA. 2009;106(52):22480–22485. doi: 10.1073/pnas.0911503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wood T.E., Barry J., Yang Z., Cepeda C., Levine M.S., Gray M. Mutant huntingtin reduction in astrocytes slows disease progression in the BACHD conditional Huntington’s disease mouse model. Hum. Mol. Genet. 2019;28(3):487–500. doi: 10.1093/hmg/ddy363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Stanek L.M., Bu J., Shihabuddin L.S. Astrocyte transduction is required for rescue of behavioral phenotypes in the YAC128 mouse model with AAV-RNAi mediated HTT lowering therapeutics. Neurobiol. Dis. 2019;129:29–37. doi: 10.1016/j.nbd.2019.04.015. [DOI] [PubMed] [Google Scholar]
- 156.Jing L., Cheng S., Pan Y., Liu Q., Yang W., Li S., Li X.J. Accumulation of endogenous mutant huntingtin in astrocytes exacerbates neuropathology of Huntington disease in mice. Mol. Neurobiol. 2021;58(10):5112–5126. doi: 10.1007/s12035-021-02451-5. [DOI] [PubMed] [Google Scholar]
- 157.Benraiss A., Wang S., Herrlinger S., Li X., Chandler-Militello D., Mauceri J., Burm H.B., Toner M., Osipovitch M., Jim Xu Q., Ding F., Wang F., Kang N., Kang J., Curtin P.C., Brunner D., Windrem M.S., Munoz-Sanjuan I., Nedergaard M., Goldman S.A. Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nat. Commun. 2016;7:11758. doi: 10.1038/ncomms11758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Djukic B., Casper K.B., Philpot B.D., Chin L.S., McCarthy K.D. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci. 2007;27(42):11354–11365. doi: 10.1523/JNEUROSCI.0723-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhang X., Wan J.Q., Tong X.P. Potassium channel dysfunction in neurons and astrocytes in Huntington’s disease. CNS Neurosci. Ther. 2018;24(4):311–318. doi: 10.1111/cns.12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tong X., Ao Y., Faas G.C., Nwaobi S.E., Xu J., Haustein M.D., Anderson M.A., Mody I., Olsen M.L., Sofroniew M.V., Khakh B.S. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci. 2014;17(5):694–703. doi: 10.1038/nn.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hassel B., Tessler S., Faull R.L., Emson P.C. Glutamate uptake is reduced in prefrontal cortex in Huntington’s disease. Neurochem. Res. 2008;33(2):232–237. doi: 10.1007/s11064-007-9463-1. [DOI] [PubMed] [Google Scholar]
- 162.Estrada-Sánchez A.M., Rebec G.V. Corticostriatal dysfunction and glutamate transporter 1 (GLT1) in Huntington’s disease: interactions between neurons and astrocytes. Basal Ganglia. 2012;2(2):57–66. doi: 10.1016/j.baga.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chen L.L., Wu J.C., Wang L.H., Wang J., Qin Z.H., Difiglia M., Lin F. Rapamycin prevents the mutant huntingtin-suppressed GLT-1 expression in cultured astrocytes. Acta Pharmacol. Sin. 2012;33(3):385–392. doi: 10.1038/aps.2011.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lee W., Reyes R.C., Gottipati M.K., Lewis K., Lesort M., Parpura V., Gray M. Enhanced Ca(2+)-dependent glutamate release from astrocytes of the BACHD Huntington’s disease mouse model. Neurobiol. Dis. 2013;58:192–199. doi: 10.1016/j.nbd.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Miller B.R., Dorner J.L., Shou M., Sari Y., Barton S.J., Sengelaub D.R., Kennedy R.T., Rebec G.V. Up-regulation of GLT1 expression increases glutamate uptake and attenuates the Huntington’s disease phenotype in the R6/2 mouse. Neuroscience. 2008;153(1):329–337. doi: 10.1016/j.neuroscience.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Korn T., Magnus T., Jung S. Autoantigen specific T cells inhibit glutamate uptake in astrocytes by decreasing expression of astrocytic glutamate transporter GLAST: a mechanism mediated by tumor necrosis factor-alpha. FASEB J. 2005;19(13):1878–1880. doi: 10.1096/fj.05-3748fje. [DOI] [PubMed] [Google Scholar]
- 167.Tilleux S., Hermans E. Down-regulation of astrocytic GLAST by microglia-related inflammation is abrogated in dibutyryl cAMP-differentiated cultures. J. Neurochem. 2008;105(6):2224–2236. doi: 10.1111/j.1471-4159.2008.05305.x. [DOI] [PubMed] [Google Scholar]
- 168.Wang Z., Pekarskaya O., Bencheikh M., Chao W., Gelbard H.A., Ghorpade A., Rothstein J.D., Volsky D.J. Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology. 2003;312(1):60–73. doi: 10.1016/S0042-6822(03)00181-8. [DOI] [PubMed] [Google Scholar]
- 169.Prow N.A., Irani D.N. The inflammatory cytokine, interleukin-1 beta, mediates loss of astroglial glutamate transport and drives excitotoxic motor neuron injury in the spinal cord during acute viral encephalomyelitis. J. Neurochem. 2008;105(4):1276–1286. doi: 10.1111/j.1471-4159.2008.05230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Colombo E., Pascente R., Triolo D., Bassani C., De Angelis A., Ruffini F., Ottoboni L., Comi G., Martino G., Farina C. Laquinimod modulates human astrocyte function and dampens astrocyte-induced neurotoxicity during inflammation. Molecules. 2020;25(22):E5403. doi: 10.3390/molecules25225403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Manyam N.V., Hare T.A., Katz L., Glaeser B.S. Huntington’s disease. Cerebrospinal fluid GABA levels in at-risk individuals. Arch. Neurol. 1978;35(11):728–730. doi: 10.1001/archneur.1978.00500350032006. [DOI] [PubMed] [Google Scholar]
- 172.Hsu Y.T., Chang Y.G., Chern Y. Insights into GABAAergic system alteration in Huntington’s disease. Open Biol. 2018;8(12):180165. doi: 10.1098/rsob.180165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Cepeda C., Galvan L., Holley S.M., Rao S.P., André V.M., Botelho E.P., Chen J.Y., Watson J.B., Deisseroth K., Levine M.S. Multiple sources of striatal inhibition are differentially affected in Huntington’s disease mouse models. J. Neurosci. 2013;33(17):7393–7406. doi: 10.1523/JNEUROSCI.2137-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rosas-Arellano A., Tejeda-Guzmán C., Lorca-Ponce E., Palma-Tirado L., Mantellero C.A., Rojas P., Missirlis F., Castro M.A. Huntington’s disease leads to decrease of GABA-A tonic subunits in the D2 neostriatal pathway and their relocalization into the synaptic cleft. Neurobiol. Dis. 2018;110:142–153. doi: 10.1016/j.nbd.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 175.Wójtowicz A.M., Dvorzhak A., Semtner M., Grantyn R. Reduced tonic inhibition in striatal output neurons from Huntington mice due to loss of astrocytic GABA release through GAT-3. Front. Neural Circuits. 2013;7:188. doi: 10.3389/fncir.2013.00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hernandez-Rabaza V., Cabrera-Pastor A., Taoro-Gonzalez L., Gonzalez-Usano A., Agusti A., Balzano T., Llansola M., Felipo V. Neuroinflammation increases GABAergic tone and impairs cognitive and motor function in hyperammonemia by increasing GAT-3 membrane expression. Reversal by sulforaphane by promoting M2 polarization of microglia. J. Neuroinflammation. 2016;13(1):83. doi: 10.1186/s12974-016-0549-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Browne S.E., Bowling A.C., MacGarvey U., Baik M.J., Berger S.C., Muqit M.M., Bird E.D., Beal M.F. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann. Neurol. 1997;41(5):646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 178.Damiano M., Diguet E., Malgorn C., D’Aurelio M., Galvan L., Petit F., Benhaim L., Guillermier M., Houitte D., Dufour N., Hantraye P., Canals J.M., Alberch J., Delzescaux T., Déglon N., Beal M.F., Brouillet E. A role of mitochondrial complex II defects in genetic models of Huntington’s disease expressing N-terminal fragments of mutant huntingtin. Hum. Mol. Genet. 2013;22(19):3869–3882. doi: 10.1093/hmg/ddt242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Dringen R., Gutterer J.M., Hirrlinger J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur. J. Biochem. 2000;267(16):4912–4916. doi: 10.1046/j.1432-1327.2000.01597.x. [DOI] [PubMed] [Google Scholar]
- 180.Shih A.Y., Johnson D.A., Wong G., Kraft A.D., Jiang L., Erb H., Johnson J.A., Murphy T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003;23(8):3394–3406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ben Haim L., Carrillo-de Sauvage M.A., Ceyzériat K., Escartin C. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front. Cell. Neurosci. 2015;9:278. doi: 10.3389/fncel.2015.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Boussicault L., Hérard A.S., Calingasan N., Petit F., Malgorn C., Merienne N., Jan C., Gaillard M.C., Lerchundi R., Barros L.F., Escartin C., Delzescaux T., Mariani J., Hantraye P., Beal M.F., Brouillet E., Véga C., Bonvento G. Impaired brain energy metabolism in the BACHD mouse model of Huntington’s disease: critical role of astrocyte-neuron interactions. J. Cereb. Blood Flow Metab. 2014;34(9):1500–1510. doi: 10.1038/jcbfm.2014.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Oliveira J.M., Gonçalves J. In situ mitochondrial Ca2+ buffering differences of intact neurons and astrocytes from cortex and striatum. J. Biol. Chem. 2009;284(8):5010–5020. doi: 10.1074/jbc.M807459200. [DOI] [PubMed] [Google Scholar]
- 184.Misiak M., Singh S., Drewlo S., Beyer C., Arnold S. Brain region-specific vulnerability of astrocytes in response to 3-nitropropionic acid is mediated by cytochrome c oxidase isoform expression. Cell Tissue Res. 2010;341(1):83–93. doi: 10.1007/s00441-010-0995-3. [DOI] [PubMed] [Google Scholar]
- 185.Polyzos A.A., Lee D.Y., Datta R., Hauser M., Budworth H., Holt A., Mihalik S., Goldschmidt P., Frankel K., Trego K., Bennett M.J., Vockley J., Xu K., Gratton E., McMurray C.T. Metabolic reprogramming in astrocytes distinguishes region-specific neuronal susceptibility in huntington mice. Cell Metab. 2019;29(6):1258–1273.e11. doi: 10.1016/j.cmet.2019.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Polyzos A., Holt A., Brown C., Cosme C., Wipf P., Gomez-Marin A., Castro M.R., Ayala-Peña S., McMurray C.T. Mitochondrial targeting of XJB-5-131 attenuates or improves pathophysiology in HdhQ150 animals with well-developed disease phenotypes. Hum. Mol. Genet. 2016;25(9):1792–1802. doi: 10.1093/hmg/ddw051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cui L., Jeong H., Borovecki F., Parkhurst C.N., Tanese N., Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127(1):59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 188.Jin J., Albertz J., Guo Z., Peng Q., Rudow G., Troncoso J.C., Ross C.A., Duan W. Neuroprotective effects of PPAR-γ agonist rosiglitazone in N171-82Q mouse model of Huntington’s disease. J. Neurochem. 2013;125(3):410–419. doi: 10.1111/jnc.12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Calabrese V., Cornelius C., Dinkova-Kostova A.T., Calabrese E.J., Mattson M.P. Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid. Redox Signal. 2010;13(11):1763–1811. doi: 10.1089/ars.2009.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Dattilo S., Mancuso C., Koverech G., Di Mauro P., Ontario M.L., Petralia C.C., Petralia A., Maiolino L., Serra A., Calabrese E.J., Calabrese V. Heat shock proteins and hormesis in the diagnosis and treatment of neurodegenerative diseases. Immun. Ageing. 2015;12:20. doi: 10.1186/s12979-015-0046-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.San Gil R., Ooi L., Yerbury J.J., Ecroyd H. The heat shock response in neurons and astroglia and its role in neurodegenerative diseases. Mol. Neurodegener. 2017;12(1):65. doi: 10.1186/s13024-017-0208-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Chafekar S.M., Duennwald M.L. Impaired heat shock response in cells expressing full-length polyglutamine-expanded huntingtin. PLoS One. 2012;7(5):e37929. doi: 10.1371/journal.pone.0037929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hay D.G., Sathasivam K., Tobaben S., Stahl B., Marber M., Mestril R., Mahal A., Smith D.L., Woodman B., Bates G.P. Progressive decrease in chaperone protein levels in a mouse model of Huntington’s disease and induction of stress proteins as a therapeutic approach. Hum. Mol. Genet. 2004;13(13):1389–1405. doi: 10.1093/hmg/ddh144. [DOI] [PubMed] [Google Scholar]
- 194.Oliveira A.O., Osmand A., Outeiro T.F., Muchowski P.J., Finkbeiner S. αB-Crystallin overexpression in astrocytes modulates the phenotype of the BACHD mouse model of Huntington’s disease. Hum. Mol. Genet. 2016;25(9):1677–1689. doi: 10.1093/hmg/ddw028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Calabrese V., Copani A., Testa D., Ravagna A., Spadaro F., Tendi E., Nicoletti V.G., Giuffrida Stella A.M. Nitric oxide synthase induction in astroglial cell cultures: effect on heat shock protein 70 synthesis and oxidant/antioxidant balance. J. Neurosci. Res. 2000;60(5):613–622. doi: 10.1002/(SICI)1097-4547(20000601)60:5<613:AID-JNR6>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 196.Esteras N., Dinkova-Kostova A.T., Abramov A.Y. Nrf2 activation in the treatment of neurodegenerative diseases: a focus on its role in mitochondrial bioenergetics and function. Biol. Chem. 2016;397(5):383–400. doi: 10.1515/hsz-2015-0295. [DOI] [PubMed] [Google Scholar]
- 197.Shih A.Y., Imbeault S., Barakauskas V., Erb H., Jiang L., Li P., Murphy T.H. Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo. J. Biol. Chem. 2005;280(24):22925–22936. doi: 10.1074/jbc.M414635200. [DOI] [PubMed] [Google Scholar]
- 198.Calkins M.J., Vargas M.R., Johnson D.A., Johnson J.A. Astrocyte-specific overexpression of Nrf2 protects striatal neurons from mitochondrial complex II inhibition. Toxicol. Sci. 2010;115(2):557–568. doi: 10.1093/toxsci/kfq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Jin Y.N., Yu Y.V., Gundemir S., Jo C., Cui M., Tieu K., Johnson G.V. Impaired mitochondrial dynamics and Nrf2 signaling contribute to compromised responses to oxidative stress in striatal cells expressing full-length mutant huntingtin. PLoS One. 2013;8(3):e57932. doi: 10.1371/journal.pone.0057932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Quinti L., Dayalan Naidu S., Träger U., Chen X., Kegel-Gleason K., Llères D., Connolly C., Chopra V., Low C., Moniot S., Sapp E., Tousley A.R., Vodicka P., Van Kanegan M.J., Kaltenbach L.S., Crawford L.A., Fuszard M., Higgins M., Miller J.R.C., Farmer R.E., Potluri V., Samajdar S., Meisel L., Zhang N., Snyder A., Stein R., Hersch S.M., Ellerby L.M., Weerapana E., Schwarzschild M.A., Steegborn C., Leavitt B.R., Degterev A., Tabrizi S.J., Lo D.C., DiFiglia M., Thompson L.M., Dinkova-Kostova A.T., Kazantsev A.G. KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington’s disease patients. Proc. Natl. Acad. Sci. USA. 2017;114(23):E4676–E4685. doi: 10.1073/pnas.1614943114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Xie Y., Hayden M.R., Xu B. BDNF overexpression in the forebrain rescues Huntington’s disease phenotypes in YAC128 mice. J. Neurosci. 2010;30(44):14708–14718. doi: 10.1523/JNEUROSCI.1637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Gharami K., Xie Y., An J.J., Tonegawa S., Xu B. Brain-derived neurotrophic factor over-expression in the forebrain ameliorates Huntington’s disease phenotypes in mice. J. Neurochem. 2008;105(2):369–379. doi: 10.1111/j.1471-4159.2007.05137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Canals J.M., Pineda J.R., Torres-Peraza J.F., Bosch M., Martín-Ibañez R., Muñoz M.T., Mengod G., Ernfors P., Alberch J. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease. J. Neurosci. 2004;24(35):7727–7739. doi: 10.1523/JNEUROSCI.1197-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wang L., Lin F., Wang J., Wu J., Han R., Zhu L., Difiglia M., Qin Z. Expression of mutant N-terminal huntingtin fragment (htt552-100Q) in astrocytes suppresses the secretion of BDNF. Brain Res. 2012;1449:69–82. doi: 10.1016/j.brainres.2012.01.077. [DOI] [PubMed] [Google Scholar]
- 205.Hong Y., Zhao T., Li X.J., Li S. Mutant Huntingtin Impairs BDNF Release from Astrocytes by Disrupting Conversion of Rab3a-GTP into Rab3a-GDP. J. Neurosci. 2016;36(34):8790–8801. doi: 10.1523/JNEUROSCI.0168-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Reick C., Ellrichmann G., Tsai T., Lee D.H., Wiese S., Gold R., Saft C., Linker R.A. Expression of brain-derived neurotrophic factor in astrocytes - Beneficial effects of glatiramer acetate in the R6/2 and YAC128 mouse models of Huntington's disease. Exp. Neurol. 2016;285(Pt A):12–23. doi: 10.1016/j.expneurol.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 207.Giralt A., Carretón O., Lao-Peregrin C., Martín E.D., Alberch J. Conditional BDNF release under pathological conditions improves Huntington’s disease pathology by delaying neuronal dysfunction. Mol. Neurodegener. 2011;6(1):71. doi: 10.1186/1750-1326-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sodero A.O. 24S-hydroxycholesterol: Cellular effects and variations in brain diseases. J. Neurochem. 2021;157(4):899–918. doi: 10.1111/jnc.15228. [DOI] [PubMed] [Google Scholar]
- 209.Leoni V., Caccia C. 24S-hydroxycholesterol in plasma: a marker of cholesterol turnover in neurodegenerative diseases. Biochimie. 2013;95(3):595–612. doi: 10.1016/j.biochi.2012.09.025. [DOI] [PubMed] [Google Scholar]
- 210.Leoni V., Mariotti C., Nanetti L., Salvatore E., Squitieri F., Bentivoglio A.R., Bandettini di Poggio M., Piacentini S., Monza D., Valenza M., Cattaneo E., Di Donato S. Whole body cholesterol metabolism is impaired in Huntington’s disease. Neurosci. Lett. 2011;494(3):245–249. doi: 10.1016/j.neulet.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 211.Leoni V., Mariotti C., Tabrizi S.J., Valenza M., Wild E.J., Henley S.M., Hobbs N.Z., Mandelli M.L., Grisoli M., Björkhem I., Cattaneo E., Di Donato S. Plasma 24S-hydroxycholesterol and caudate MRI in pre-manifest and early Huntington’s disease. Brain. 2008;131(Pt 11):2851–2859. doi: 10.1093/brain/awn212. [DOI] [PubMed] [Google Scholar]
- 212.Valenza M., Leoni V., Karasinska J.M., Petricca L., Fan J., Carroll J., Pouladi M.A., Fossale E., Nguyen H.P., Riess O., MacDonald M., Wellington C., DiDonato S., Hayden M., Cattaneo E. Cholesterol defect is marked across multiple rodent models of Huntington’s disease and is manifest in astrocytes. J. Neurosci. 2010;30(32):10844–10850. doi: 10.1523/JNEUROSCI.0917-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Valenza M., Marullo M., Di Paolo E., Cesana E., Zuccato C., Biella G., Cattaneo E. Disruption of astrocyte-neuron cholesterol cross talk affects neuronal function in Huntington’s disease. Cell Death Differ. 2015;22(4):690–702. doi: 10.1038/cdd.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.del Toro D., Xifró X., Pol A., Humbert S., Saudou F., Canals J.M., Alberch J. Altered cholesterol homeostasis contributes to enhanced excitotoxicity in Huntington’s disease. J. Neurochem. 2010;115(1):153–167. doi: 10.1111/j.1471-4159.2010.06912.x. [DOI] [PubMed] [Google Scholar]
- 215.Kacher R., Lamazière A., Heck N., Kappes V., Mounier C., Despres G., Dembitskaya Y., Perrin E., Christaller W., Sasidharan Nair S., Messent V., Cartier N., Vanhoutte P., Venance L., Saudou F., Néri C., Caboche J., Betuing S. CYP46A1 gene therapy deciphers the role of brain cholesterol metabolism in Huntington’s disease. Brain. 2019;142(8):2432–2450. doi: 10.1093/brain/awz174. [DOI] [PubMed] [Google Scholar]
- 216.González-Guevara E., Cárdenas G., Pérez-Severiano F., Martínez-Lazcano J.C. Dysregulated brain cholesterol metabolism is linked to neuroinflammation in Huntington’s disease. Mov. Disord. 2020;35(7):1113–1127. doi: 10.1002/mds.28089. [DOI] [PubMed] [Google Scholar]
- 217.Birolini G., Verlengia G., Talpo F., Maniezzi C., Zentilin L., Giacca M., Conforti P., Cordiglieri C., Caccia C., Leoni V., Taroni F., Biella G., Simonato M., Cattaneo E., Valenza M. Singlenucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol. Commun. 2020;8(1):19. doi: 10.1186/s40478-020-0880-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Al-Dalahmah O., Sosunov A.A., Shaik A., Ofori K., Liu Y., Vonsattel J.P., Adorjan I., Menon V., Goldman J.E. Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol. Commun. 2020;8(1):19. doi: 10.1186/s40478-020-0880-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Yu X., Nagai J., Marti-Solano M., Soto J.S., Coppola G., Babu M.M., Khakh B.S. Context-specific striatal astrocyte molecular responses are phenotypically exploitable. Neuron. 2020;108(6):1146–1162.e10. doi: 10.1016/j.neuron.2020.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Benraiss A., Mariani J.N., Osipovitch M., Cornwell A., Windrem M.S., Villanueva C.B., Chandler-Militello D., Goldman S.A. Cell-intrinsic glial pathology is conserved across human and murine models of Huntington’s disease. Cell Rep. 2021;36(1):109308. doi: 10.1016/j.celrep.2021.109308. [DOI] [PubMed] [Google Scholar]
- 221.Kierdorf K., Prinz M. Microglia in steady state. J. Clin. Invest. 2017;127(9):3201–3209. doi: 10.1172/JCI90602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Casano A.M., Peri F. Microglia: multitasking specialists of the brain. Dev. Cell. 2015;32(4):469–477. doi: 10.1016/j.devcel.2015.01.018. [DOI] [PubMed] [Google Scholar]
- 223.Crapser J.D., Ochaba J., Soni N., Reidling J.C., Thompson L.M., Green K.N. Microglial depletion prevents extracellular matrix changes and striatal volume reduction in a model of Huntington’s disease. Brain. 2020;143(1):266–288. doi: 10.1093/brain/awz363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Petkau T.L., Hill A., Connolly C., Lu G., Wagner P., Kosior N., Blanco J., Leavitt B.R. Mutant huntingtin expression in microglia is neither required nor sufficient to cause the Huntington’s disease-like phenotype in BACHD mice. Hum. Mol. Genet. 2019;28(10):1661–1670. doi: 10.1093/hmg/ddz009. [DOI] [PubMed] [Google Scholar]
- 225.Di Pardo A., Ciaglia E., Cattaneo M., Maciag A., Montella F., Lopardo V., Ferrario A., Villa F., Madonna M., Amico E., Carrizzo A., Damato A., Pepe G., Marracino F., Auricchio A., Vecchione C., Maglione V., Puca A.A. The longevity-associated variant of BPIFB4 improves a CXCR4-mediated striatum-microglia crosstalk preventing disease progression in a mouse model of Huntington’s disease. Cell Death Dis. 2020;11(7):546. doi: 10.1038/s41419-020-02754-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Pawelec P., Ziemka-Nalecz M., Sypecka J., Zalewska T. The impact of the CX3CL1/CX3CR1 axis in neurological disorders. Cells. 2020;9(10):E2277. doi: 10.3390/cells9102277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Liu F.T., Rabinovich G.A. Galectins as modulators of tumour progression. Nat. Rev. Cancer. 2005;5(1):29–41. doi: 10.1038/nrc1527. [DOI] [PubMed] [Google Scholar]
- 228.Burguillos M.A., Svensson M., Schulte T., Boza-Serrano A., Garcia-Quintanilla A., Kavanagh E., Santiago M., Viceconte N., Oliva-Martin M.J., Osman A.M., Salomonsson E., Amar L., Persson A., Blomgren K., Achour A., Englund E., Leffler H., Venero J.L., Joseph B., Deierborg T. Microglia-secreted galectin-3 acts as a toll-like receptor 4 ligand and contributes to microglial activation. Cell Rep. 2015;10(9):1626–1638. doi: 10.1016/j.celrep.2015.02.012. [DOI] [PubMed] [Google Scholar]
- 229.Siew J.J., Chen H.M., Chen H.Y., Chen H.L., Chen C.M., Soong B.W., Wu Y.R., Chang C.P., Chan Y.C., Lin C.H., Liu F.T., Chern Y. Galectin-3 is required for the microglia-mediated brain inflammation in a model of Huntington’s disease. Nat. Commun. 2019;10(1):3473. doi: 10.1038/s41467-019-11441-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Heyes M.P., Achim C.L., Wiley C.A., Major E.O., Saito K., Markey S.P. Human microglia convert l-tryptophan into the neurotoxin quinolinic acid. Biochem. J. 1996;320(Pt 2):595–597. doi: 10.1042/bj3200595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Lu Y., Shao M., Wu T. Kynurenine-3-monooxygenase: A new direction for the treatment in different diseases. Food Sci. Nutr. 2020;8(2):711–719. doi: 10.1002/fsn3.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Zwilling D., Huang S.Y., Sathyasaikumar K.V., Notarangelo F.M., Guidetti P., Wu H.Q., Lee J., Truong J., Andrews-Zwilling Y., Hsieh E.W., Louie J.Y., Wu T., Scearce-Levie K., Patrick C., Adame A., Giorgini F., Moussaoui S., Laue G., Rassoulpour A., Flik G., Huang Y., Muchowski J.M., Masliah E., Schwarcz R., Muchowski P.J. Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell. 2011;145(6):863–874. doi: 10.1016/j.cell.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Bondulich M.K., Fan Y., Song Y., Giorgini F., Bates G.P. Ablation of kynurenine 3-monooxygenase rescues plasma inflammatory cytokine levels in the R6/2 mouse model of Huntington’s disease. Sci. Rep. 2021;11(1):5484. doi: 10.1038/s41598-021-84858-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Rodrigues F.B., Byrne L.M., Lowe A.J., Tortelli R., Heins M., Flik G., Johnson E.B., De Vita E., Scahill R.I., Giorgini F., Wild E.J. Kynurenine pathway metabolites in cerebrospinal fluid and blood as potential biomarkers in Huntington’s disease. J. Neurochem. 2021;158(2):539–553. doi: 10.1111/jnc.15360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Ferger A.I., Campanelli L., Reimer V., Muth K.N., Merdian I., Ludolph A.C., Witting A. Effects of mitochondrial dysfunction on the immunological properties of microglia. J. Neuroinflammation. 2010;7:45. doi: 10.1186/1742-2094-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Ryu J.K., Nagai A., Kim J., Lee M.C., McLarnon J.G., Kim S.U. Microglial activation and cell death induced by the mitochondrial toxin 3-nitropropionic acid: in vitro and in vivo studies. Neurobiol. Dis. 2003;12(2):121–132. doi: 10.1016/S0969-9961(03)00002-0. [DOI] [PubMed] [Google Scholar]
- 237.Jin X., Riew T.R., Kim H.L., Choi J.H., Lee M.Y. Morphological characterization of NG2 glia and their association with neuroglial cells in the 3-nitropropionic acid-lesioned striatum of rat. Sci. Rep. 2018;8(1):5942. doi: 10.1038/s41598-018-24385-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Oberheim N.A., Takano T., Han X., He W., Lin J.H., Wang F., Xu Q., Wyatt J.D., Pilcher W., Ojemann J.G., Ransom B.R., Goldman S.A., Nedergaard M. Uniquely hominid features of adult human astrocytes. J. Neurosci. 2009;29(10):3276–3287. doi: 10.1523/JNEUROSCI.4707-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Monk R., Connor B. Cell reprogramming to model Huntington’s disease: A comprehensive review. Cells. 2021;10(7):1565. doi: 10.3390/cells10071565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Juopperi T.A., Kim W.R., Chiang C.H., Yu H., Margolis R.L., Ross C.A., Ming G.L., Song H. Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington’s disease patient cells. Mol. Brain. 2012;5:17. doi: 10.1186/1756-6606-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Nagata E., Sawa A., Ross C.A., Snyder S.H. Autophagosome-like vacuole formation in Huntington’s disease lymphoblasts. Neuroreport. 2004;15(8):1325–1328. doi: 10.1097/01.wnr.0000127073.66692.8f. [DOI] [PubMed] [Google Scholar]
- 242.Martinez-Vicente M., Talloczy Z., Wong E., Tang G., Koga H., Kaushik S., de Vries R., Arias E., Harris S., Sulzer D., Cuervo A.M. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010;13(5):567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Garcia V.J., Rushton D.J., Tom C.M., Allen N.D., Kemp P.J., Svendsen C.N., Mattis V.B. Huntington’s disease patient-derived astrocytes display electrophysiological impairments and reduced neuronal support. Front. Neurosci. 2019;13:669. doi: 10.3389/fnins.2019.00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Park H.J., Jeon J., Choi J., Kim J.Y., Kim H.S., Huh J.Y., Goldman S.A., Song J. Human iPSC-derived neural precursor cells differentiate into multiple cell types to delay disease progression following transplantation into YAC128 Huntington’s disease mouse model. Cell Prolif. 2021;54(8):e13082. doi: 10.1111/cpr.13082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Mattson M.P., Camandola S. NF-kappaB in neuronal plasticity and neurodegenerative disorders. J. Clin. Invest. 2001;107(3):247–254. doi: 10.1172/JCI11916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Takano H., Gusella J.F. The predominantly HEAT-like motif structure of huntingtin and its association and coincident nuclear entry with dorsal, an NF-kB/Rel/dorsal family transcription factor. BMC Neurosci. 2002;3:15. doi: 10.1186/1471-2202-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Marcora E., Kennedy M.B. The Huntington’s disease mutation impairs Huntingtin’s role in the transport of NF-κB from the synapse to the nucleus. Hum. Mol. Genet. 2010;19(22):4373–4384. doi: 10.1093/hmg/ddq358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Yu Z., Zhou D., Cheng G., Mattson M.P. Neuroprotective role for the p50 subunit of NF-kappaB in an experimental model of Huntington’s disease. J. Mol. Neurosci. 2000;15(1):31–44. doi: 10.1385/JMN:15:1:31. [DOI] [PubMed] [Google Scholar]
- 249.Khoshnan A., Ko J., Watkin E.E., Paige L.A., Reinhart P.H., Patterson P.H. Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity. J. Neurosci. 2004;24(37):7999–8008. doi: 10.1523/JNEUROSCI.2675-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Dresselhaus E.C., Meffert M.K. Cellular specificity of NF-κB function in the nervous system. Front. Immunol. 2019;10:1043. doi: 10.3389/fimmu.2019.01043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Khoshnan A., Sabbaugh A., Calamini B., Marinero S.A., Dunn D.E., Yoo J.H., Ko J., Lo D.C., Patterson P.H. IKKβ and mutant huntingtin interactions regulate the expression of IL-34: implications for microglial-mediated neurodegeneration in HD. Hum. Mol. Genet. 2017;26(21):4267–4277. doi: 10.1093/hmg/ddx315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Griffioen K., Mattson M.P., Okun E. Deficiency of Toll-like receptors 2, 3 or 4 extends life expectancy in Huntington’s disease mice. Heliyon. 2018;4(1):e00508. doi: 10.1016/j.heliyon.2018.e00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Pérez-Rodríguez M.J., Ibarra-Sánchez A., Román-Figueroa A., Pérez-Severiano F., González-Espinosa C. Mutant Huntingtin affects toll-like receptor 4 intracellular trafficking and cytokine production in mast cells. J. Neuroinflammation. 2020;17(1):95. doi: 10.1186/s12974-020-01758-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Gonzalez-Reyes R.E., Rubiano M.G. Astrocyte’s RAGE: More than just a question of mood. Cent. Nerv. Syst. Agents Med. Chem. 2018;18(1):39–48. doi: 10.2174/1871524916999160505105121. [DOI] [PubMed] [Google Scholar]
- 255.Ma L., Nicholson L.F. Expression of the receptor for advanced glycation end products in Huntington’s disease caudate nucleus. Brain Res. 2004;1018(1):10–17. doi: 10.1016/j.brainres.2004.05.052. [DOI] [PubMed] [Google Scholar]
- 256.Kim J., Waldvogel H.J., Faull R.L., Curtis M.A., Nicholson L.F. The RAGE receptor and its ligands are highly expressed in astrocytes in a grade-dependant manner in the striatum and subependymal layer in Huntington’s disease. J. Neurochem. 2015;134(5):927–942. doi: 10.1111/jnc.13178. [DOI] [PubMed] [Google Scholar]
- 257.Anzilotti S., Giampà C., Laurenti D., Perrone L., Bernardi G., Melone M.A., Fusco F.R. Immunohistochemical localization of receptor for advanced glycation end (RAGE) products in the R6/2 mouse model of Huntington’s disease. Brain Res. Bull. 2012;87(2-3):350–358. doi: 10.1016/j.brainresbull.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 258.Faraco G., Fossati S., Bianchi M.E., Patrone M., Pedrazzi M., Sparatore B., Moroni F., Chiarugi A. High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J. Neurochem. 2007;103(2):590–603. doi: 10.1111/j.1471-4159.2007.04788.x. [DOI] [PubMed] [Google Scholar]
- 259.Hayakawa K., Arai K., Lo E.H. Role of ERK map kinase and CRM1 in IL-1beta-stimulated release of HMGB1 from cortical astrocytes. Glia. 2010;58(8):1007–1015. doi: 10.1002/glia.20982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Min H.J., Ko E.A., Wu J., Kim E.S., Kwon M.K., Kwak M.S., Choi J.E., Lee J.E., Shin J.S. Chaperone-like activity of high-mobility group box 1 protein and its role in reducing the formation of polyglutamine aggregates. J. Immunol. 2013;190(4):1797–1806. doi: 10.4049/jimmunol.1202472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Qi L., Sun X., Li F.E., Zhu B.S., Braun F.K., Liu Z.Q., Tang J.L., Wu C., Xu F., Wang H.H., Velasquez L.A., Zhao K., Lei F.R., Zhang J.G., Shen Y.T., Zou J.X., Meng H.M., An G.L., Yang L., Zhang X.D. HMGB1 Promotes mitochondrial dysfunction-triggered striatal neurodegeneration via autophagy and apoptosis activation. PLoS One. 2015;10(11):e0142901. doi: 10.1371/journal.pone.0142901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Son S., Bowie L.E., Maiuri T., Hung C.L.K., Desmond C.R., Xia J., Truant R. High-mobility group box 1 links sensing of reactive oxygen species by huntingtin to its nuclear entry. J. Biol. Chem. 2019;294(6):1915–1923. doi: 10.1074/jbc.RA117.001440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Qi M.L., Tagawa K., Enokido Y., Yoshimura N., Wada Y., Watase K., Ishiura S., Kanazawa I., Botas J., Saitoe M., Wanker E.E., Okazawa H. Proteome analysis of soluble nuclear proteins reveals that HMGB1/2 suppress genotoxic stress in polyglutamine diseases. Nat. Cell Biol. 2007;9(4):402–414. doi: 10.1038/ncb1553. [DOI] [PubMed] [Google Scholar]
- 264.Rothaug M., Becker-Pauly C., Rose-John S. The role of interleukin-6 signaling in nervous tissue. Biochim. Biophys. Acta. 2016;1863(6 Pt A):1218–1227. doi: 10.1016/j.bbamcr.2016.03.018. [DOI] [PubMed] [Google Scholar]
- 265.Wertz M.H., Pineda S.S., Lee H., Kulicke R., Kellis M., Heiman M. Interleukin-6 deficiency exacerbates Huntington’s disease model phenotypes. Mol. Neurodegener. 2020;15(1):29. doi: 10.1186/s13024-020-00379-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Bensadoun J.C., de Almeida L.P., Dréano M., Aebischer P., Déglon N. Neuroprotective effect of interleukin-6 and IL6/IL6R chimera in the quinolinic acid rat model of Huntington’s syndrome. Eur. J. Neurosci. 2001;14(11):1753–1761. doi: 10.1046/j.0953-816x.2001.01802.x. [DOI] [PubMed] [Google Scholar]
- 267.Gaidt M.M., Ebert T.S., Chauhan D., Schmidt T., Schmid-Burgk J.L., Rapino F., Robertson A.A., Cooper M.A., Graf T., Hornung V. Human monocytes engage an alternative inflammasome pathway. Immunity. 2016;44(4):833–846. doi: 10.1016/j.immuni.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 268.Wang C.E., Li S., Li X.J. Lack of interleukin-1 type 1 receptor enhances the accumulation of mutant huntingtin in the striatum and exacerbates the neurological phenotypes of Huntington’s disease mice. Mol. Brain. 2010;3:33. doi: 10.1186/1756-6606-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Martin D.D.O., Schmidt M.E., Nguyen Y.T., Lazic N., Hayden M.R. Identification of a novel caspase cleavage site in huntingtin that regulates mutant huntingtin clearance. FASEB J. 2019;33(3):3190–3197. doi: 10.1096/fj.201701510RRR. [DOI] [PubMed] [Google Scholar]
- 270.Clark I.A., Vissel B. A neurologist’s guide to TNF biology and to the principles behind the therapeutic removal of excess TNF in disease. Neural Plast. 2015;2015:358263. doi: 10.1155/2015/358263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Olmos G., Lladó J. Tumor necrosis factor alpha: a link between neuroinflammation and excitotoxicity. Mediators Inflamm. 2014;2014:861231. doi: 10.1155/2014/861231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Alto L.T., Chen X., Ruhn K.A., Treviño I., Tansey M.G. AAV-dominant negative tumor necrosis factor (DN-TNF) gene transfer to the striatum does not rescue medium spiny neurons in the YAC128 mouse model of Huntington’s disease. PLoS One. 2014;9(5):e96544. doi: 10.1371/journal.pone.0096544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Majumder S., Sreedhara S.R., Banerjee S., Chatterjee S. TNF α signaling beholds thalidomide saga: a review of mechanistic role of TNF-α signaling under thalidomide. Curr. Top. Med. Chem. 2012;12(13):1456–1467. doi: 10.2174/156802612801784443. [DOI] [PubMed] [Google Scholar]
- 274.Tobinick E. Rapid improvement of chronic stroke deficits after perispinal etanercept: three consecutive cases. CNS Drugs. 2011;25(2):145–155. doi: 10.2165/11588400-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 275.Ralph S.J., Weissenberger A., Bonev V., King L.D., Bonham M.D., Ferguson S., Smith A.D., Goodman-Jones A.A., Espinet A.J. Phase I/II parallel double-blind randomized controlled clinical trial of perispinal etanercept for chronic stroke: improved mobility and pain alleviation. Expert Opin. Investig. Drugs. 2020;29(3):311–326. doi: 10.1080/13543784.2020.1709822. [DOI] [PubMed] [Google Scholar]
- 276.Tabrizi S.J., Flower M.D., Ross C.A., Wild E.J. Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020;16(10):529–546. doi: 10.1038/s41582-020-0389-4. [DOI] [PubMed] [Google Scholar]
- 277.Valdeolivas S., Sagredo O., Delgado M., Pozo M.A., Fernández-Ruiz J. Effects of a sativex-like combination of phytocannabinoids on disease progression in R6/2 mice, an experimental model of Huntington’s disease. Int. J. Mol. Sci. 2017;18(4):E684. doi: 10.3390/ijms18040684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.López-Sendón Moreno J.L., García Caldentey J., Trigo Cubillo P., Ruiz Romero C., García Ribas G., Alonso Arias M.A., García de Yébenes M.J., Tolón R.M., Galve-Roperh I., Sagredo O., Valdeolivas S., Resel E., Ortega-Gutierrez S., García-Bermejo M.L., Fernández Ruiz J., Guzmán M., García de Yébenes Prous J. A double-blind, randomized, cross-over, placebo-controlled, pilot trial with Sativex in Huntington’s disease. J. Neurol. 2016;263(7):1390–1400. doi: 10.1007/s00415-016-8145-9. [DOI] [PubMed] [Google Scholar]
- 279.Garrido-Mesa N., Zarzuelo A., Gálvez J. Minocycline: far beyond an antibiotic. Br. J. Pharmacol. 2013;169(2):337–352. doi: 10.1111/bph.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.A futility study of minocycline in Huntington’s disease. Mov. Disord. 2010;25(13):2219–2224. doi: 10.1002/mds.23236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Brück W., Pförtner R., Pham T., Zhang J., Hayardeny L., Piryatinsky V., Hanisch U.K., Regen T., van Rossum D., Brakelmann L., Hagemeier K., Kuhlmann T., Stadelmann C., John G.R., Kramann N., Wegner C. Reduced astrocytic NF-κB activation by laquinimod protects from cuprizone-induced demyelination. Acta Neuropathol. 2012;124(3):411–424. doi: 10.1007/s00401-012-1009-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Thöne J., Ellrichmann G., Seubert S., Peruga I., Lee D.H., Conrad R., Hayardeny L., Comi G., Wiese S., Linker R.A., Gold R. Modulation of autoimmune demyelination by laquinimod via induction of brain-derived neurotrophic factor. Am. J. Pathol. 2012;180(1):267–274. doi: 10.1016/j.ajpath.2011.09.037. [DOI] [PubMed] [Google Scholar]
- 283.Dobson L., Träger U., Farmer R., Hayardeny L., Loupe P., Hayden M.R., Tabrizi S.J. Laquinimod dampens hyperactive cytokine production in Huntington’s disease patient myeloid cells. J. Neurochem. 2016;137(5):782–794. doi: 10.1111/jnc.13553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Ellrichmann G., Blusch A., Fatoba O., Brunner J., Reick C., Hayardeny L., Hayden M., Sehr D., Winklhofer K.F., Saft C., Gold R. Laquinimod treatment in the R6/2 mouse model. Sci. Rep. 2017;7(1):4947. doi: 10.1038/s41598-017-04990-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Giraudon P., Vincent P., Vuaillat C., Verlaeten O., Cartier L., Marie-Cardine A., Mutin M., Bensussan A., Belin M.F., Boumsell L. Semaphorin CD100 from activated T lymphocytes induces process extension collapse in oligodendrocytes and death of immature neural cells. J. Immunol. 2004;172(2):1246–1255. doi: 10.4049/jimmunol.172.2.1246. [DOI] [PubMed] [Google Scholar]
- 286.Yamaguchi W., Tamai R., Kageura M., Furuyama T., Inagaki S. Sema4D as an inhibitory regulator in oligodendrocyte development. Mol. Cell. Neurosci. 2012;49(3):290–299. doi: 10.1016/j.mcn.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 287.Carulli D., de Winter F., Verhaagen J. Semaphorins in adult nervous system plasticity and disease. Front. Synaptic Neurosci. 2021;13:672891. doi: 10.3389/fnsyn.2021.672891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Southwell A.L., Franciosi S., Villanueva E.B., Xie Y., Winter L.A., Veeraraghavan J., Jonason A., Felczak B., Zhang W., Kovalik V., Waltl S., Hall G., Pouladi M.A., Smith E.S., Bowers W.J., Zauderer M., Hayden M.R. Anti-semaphorin 4D immunotherapy ameliorates neuropathology and some cognitive impairment in the YAC128 mouse model of Huntington disease. Neurobiol. Dis. 2015;76:46–56. doi: 10.1016/j.nbd.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 289.2021. Vaccinex. Available from: https://www.vaccinex.com/pipeline/
- 290.Kwon D. Failure of genetic therapies for Huntington’s devastates community. Nature. 2021;593(7858):180. doi: 10.1038/d41586-021-01177-7. [DOI] [PubMed] [Google Scholar]
- 291.Fatoba O., Ohtake Y., Itokazu T., Yamashita T. Immunotherapies in Huntington’s disease and α-Synucleinopathies. Front. Immunol. 2020;11:337. doi: 10.3389/fimmu.2020.00337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Calabrese E.J., Bhatia T.N., Calabrese V., Dhawan G., Giordano J., Hanekamp Y.N., Kapoor R., Kozumbo W.J., Leak R.K. Cytotoxicity models of Huntington’s disease and relevance of hormetic mechanisms: A critical assessment of experimental approaches and strategies. Pharmacol. Res. 2019;150:104371. doi: 10.1016/j.phrs.2019.104371. [DOI] [PubMed] [Google Scholar]
- 293.Burrus C.J., McKinstry S.U., Kim N., Ozlu M.I., Santoki A.V., Fang F.Y., Ma A., Karadeniz Y.B., Worthington A.K., Dragatsis I., Zeitlin S., Yin H.H., Eroglu C. Striatal projection neurons require huntingtin for synaptic connectivity and survival. Cell Rep. 2020;30(3):642–657.e6. doi: 10.1016/j.celrep.2019.12.069. [DOI] [PMC free article] [PubMed] [Google Scholar]