

Abstract
Background: Alzheimer’s disease (AD) is a chronic neurodegenerative disease with no specific disease-modifying treatment. β-secretase (BACE1) is considered the potential and rationale target because it is involved in the rate-limiting step, which produces toxic Aβ42 peptides that leads to deposits in the form of amyloid plaques extracellularly, resulting in AD.
Objective: This study aims to discuss the role and implications of BACE1 and its inhibitors in the management of AD.
Methods: We have searched and collected the relevant quality work from PubMed using the following keywords “BACE1”, BACE2”, “inhibitors”, and “Alzheimer’s disease”. In addition, we included the work which discusses the role of BACE1 in AD and the recent work on its inhibitors.
Results: In this review, we have discussed the importance of BACE1 in regulating AD progression and the current development of BACE1 inhibitors. However, the development of a BACE1 inhibitor is very challenging due to the large active site of BACE1. Nevertheless, some of the BACE1 inhibitors have managed to enter advanced phases of clinical trials, such as MK-8931 (Verubecestat), E2609 (Elenbecestat), AZD3293 (Lanabecestat), and JNJ-54861911 (Atabecestat). This review also sheds light on the prospect of BACE1 inhibitors as the most effective therapeutic approach in delaying or preventing AD progression.
Conclusion: BACE1 is involved in the progression of AD. The current ongoing or failed clinical trials may help understand the role of BACE1 inhibition in regulating the Aβ load and cognitive status of AD patients.
Keywords: BACE1, Beta-secretase, Aβ42 peptide, inhibitors, Alzheimer’s disease, neurodegenerative disease
1. INTRODUCTION
Dementia is characterized by a long-term and gradual inability to think and remember. Alzheimer's disease (AD) is the most general form of dementia, covering nearly 50-70% among all the cases [1]. Approximately 5.8 million people from the United States were suffering from AD in 2019, and out of them, 5.6 million people were aged 65 years or higher [2]. About 121,404 people died due to AD in 2017. In 2018 AD was ranked as the sixth major cause of death in the US and the fifth for Americans aged > 65 years [3]. The two main pathological indications of AD are the deposition of intracellular neurofibrillary tangles (NFT) and extracellular amyloid plaques [4, 5]. The cleavage of amyloid precursor protein (APP) by β- secretase and γ-secretase enzymes at specific sites lead to the formation of Aβ peptides and amyloid precursor protein intracellular domain (AICD), which further aggregate to form Aβ plaques (Fig. 1) [6]. The current AD treatment includes acetylcholinesterase (AChE) inhibitors and NMDA receptor antagonists, which only provide symptomatic relief. Hence there is a need for disease-modifying treatments along with treatments that provide symptomatic benefits. There have been many potential therapeutic targets that have been explored to combat AD progression like β-secretase 1 (beta-site APP cleaving enzyme 1 (BACE1)), γ-secretase, nuclear factor E2 related factor 2 (Nrf2), protein tyrosine phosphatase 1B (PTP1B) and phosphodiesterases (PDE), etc. [7]. BACE1 inhibition is the most rational approach as it blocks the first rate-limiting step for the formation of Aβ peptides. The design and development of the BACE1 inhibitors have proven to be very challenging due to the large binding pocket and structural flexibility of the BACE1 enzyme [8].
Fig. (1).
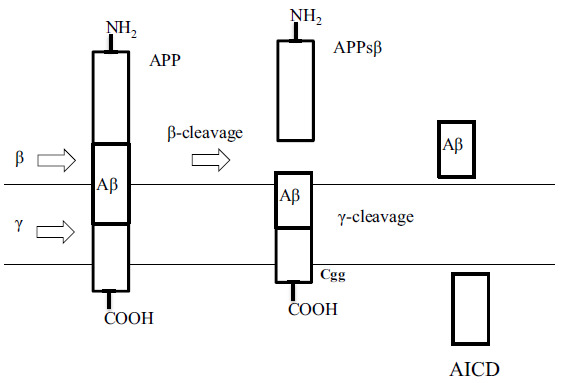
The amyloidogenic pathway of amyloid precursor protein (APP) proteolysis [5].
Decades of research have been done to find selective and safe BACE1 inhibitors. Some of the molecules showed promising results of cellular Aβ reduction in animal models and reached advanced human phase clinical trials. Unfortunately, most of them were terminated from the clinical trials due to inefficacy in providing cognitive benefit and exhibiting toxicity. The lack of efficacy of the BACE1 inhibitors in clinical trials may involve many factors, including being administered too late in the time course of the disease following irreversible synapse and neuron loss or due to the unresolved complicated pathophysiology of AD [9]. This review summarizes the importance of BACE1 in the regulation of AD progression and the current development of selective BACE1 inhibitors.
2. BACE1 / β SECRETASE
BACE1 is a type 1 transmembrane aspartyl protease consisting of 501 amino acids mainly expressed in the neurons. The extraneuronal portion of APP is cleaved by BACE1 to generate a large soluble extracellular fragment sAPPβ and an intracellular C-terminal fragment (βCTF) which is called C99 (Fig. 1). Intramembrane cleavage of C99 by γ secretase complex gives a cytosolic C57 or C59 AICD fragment and extracellular Aβ peptides of different lengths. The 42 amino acid form of Aβ (Aβ1-42) is vulnerable to oligomerization and leads to the formation of amyloid plaques [7].
2.1. Genomic and Proteomic Information About BACE1 and BACE2
The catalytic domain of BACE1 consists of two signature motif sequences called DTGS (93-96 amino acid residues) and DSGT (289–292 amino acid residues). They show activity at an acidic pH and are located within the lumen of the intracellular cavity, incorporating the trans-Golgi network and endosomes (Fig. 2) [10]. Other aspartyl proteases include BACE2, pepsin, cathepsin D, rennin, and cathepsin E [4, 11]. There are minimum structural differences between these enzymes, indicating that high selectivity is required for BACE1 inhibition and to avoid proteolysis of unintended substrates. BACE2 is a close homolog of BACE1, showing 64% amino acid similarity, but BACE2 is not present to a high level in neurons and does not form toxic Aβ peptides upon cleavage of APP [12-14].
Fig. (2).
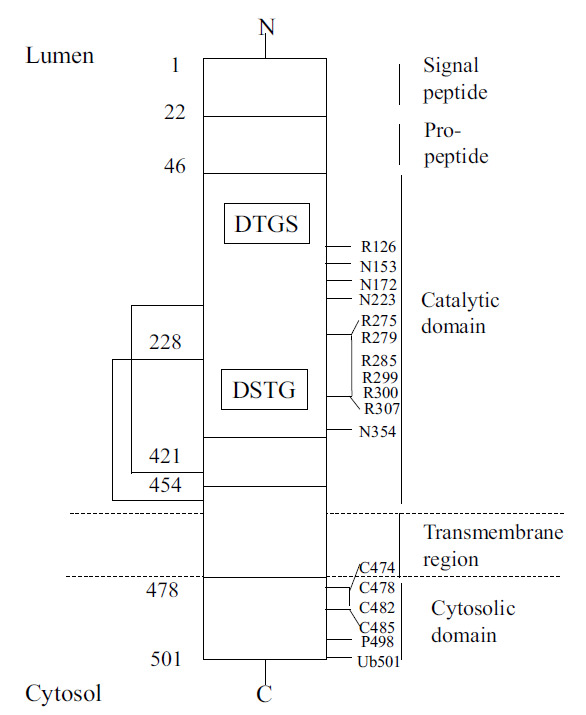
BACE1 primary sequence features [The complete primary structure (sequence) is not shown, only select features] [14].
Analysis of BACE1 and BACE2 protein sequences from some mammalian species utilizing the ET (Evolutionary trace sequence) method revealed the difference in ligand binding sites. Although BACE1 and BACE2 have a similar biological role in proteolyzing APP, they cleave at different sites producing various cleavage products, one which is amyloidogenic from BACE1 and others that are non-amyloidogenic from BACE2 [15]. The structural characteristics of BACE1 were further elucidated after discovering some early potent BACE1 inhibitors like OM99-2 containing eight residues whose interactions with the BACE1 sub-pockets are shown in Fig. (3) [16].
Fig. (3).
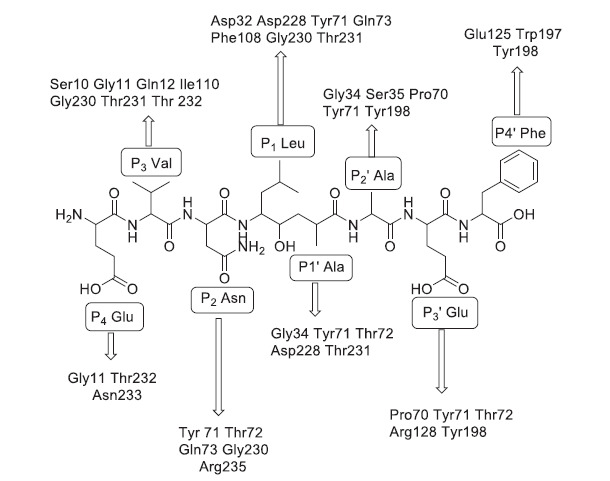
Binding interactions of the first BACE1 inhibitor OM99-2 with different sub-pockets of BACE1 [16].
Twenty-eight amino acid residues for BACE1 and twenty-four amino acid residues for BACE2 were recognized as a ligand-binding site for OM99-2. The surface of the ligand‐binding site consists of subpockets S1, S2, S3, S4, S1¢, S2¢, S3¢, and S4¢ [17]. Two catalytic aspartic acid residues were identified to be required for enzymatic activity Asp32 and Asp228 of BACE1 and Asp48 and Asp241 of BACE2. Three main entities that form the large ligand binding site are the catalytic dyad, the flap region, and the 10s loop. The 10s loop is positioned around Ser10 in the S3 pocket. It acquires an open conformation allowing maximum binding of the inhibitor to the S3 pocket [18]. The ligand-binding site comprises four group-specific residues, which are Pro70, Ile110, Ile126, and Asn233 of BACE1 and Lys86, Leu126, Leu142, and Leu246 of BACE2. Although amino acid residues like Ile and Leu are hydrophobic and equally small, the orientation of the alkyl group in the 3D protein structure accounts for selectivity in binding of the ligand to BACE1 and BACE2. Most of the aspartyl proteases possess a loop structure that covers the active site called the flap region. Tyr71 in the flap region of BACE1 plays a crucial role in determining conformational flexibility. In the flap region, Pro70 of BACE1 is replaced with Lys86 of BACE2. The cyclic nature of the proline residue may affect the flexibility of the flap region in BACE1 compared to the linear chain of lysine in BACE2, so ligand interaction with the proline residue might enhance the selectivity of the ligand towards BACE1. The last group-specific residue in the ligand-binding site is Asn233 of BACE1 and Leu246 of BACE2, which indicates that the BACE1 binding site in this region is more hydrophilic than BACE2. These differences can help in the design and development of selective inhibitors for either BACE1 or BACE2 [14].
2.2. Significance of Genetic Mutations in Alzheimer’s Disease
Genetic studies have revealed that over 200 autosomal dominant missense mutations have been observed in the amyloid precursor protein (APP) and presenilin 1 and 2 genes. These mutations increase the production of either total Aβ forms or only the toxic Aβ42 form causing familial Alzheimer’s disease (FAD). The FAD mutations are located very close to β-secretase and γ-secretase cleavage sites influencing APP processing by these enzymes, ultimately increasing the accumulation of Aβ peptides. Particularly, the Swedish mutation (K670N; M671L) [19] and the A67RV mutation [20] are specifically located C-terminal to the β-secretase cleavage site. Naturally, these mutations effectively increase the APP cleavage by BACE1, forming more C99 fragments and toxic Aβ peptides. However, there is the A673T mutation, which is protective against AD. The A673T mutation leads to nearly a 40% reduction in Aβ production due to reduced cleavage of APP by BACE1. Most carriers contain one copy of the A673T mutation, leading to a 20% reduction in Aβ production, which protects against AD [21-23]. This data provides strong evidence that BACE1 inhibition decreases the Aβ load in the brain and thus can be used as a disease-modifying treatment option for AD if employed before the accumulation of cellular Aβ plaques.
2.3. BACE1 Substrates and their Physiological Role
Proteomics analyses in cultured primary neurons have found BACE1 substrates with neuronal functions, excluding amyloid precursor protein (APP). Most of the substrates cleaved by BACE1 release an ectodomain fragment extracellularly, which might bind to the same (autocrine) or different (paracrine) cells to alter signal transduction and cell-cell interactions. Two BACE1 substrates are type III neuregulin 1 (NRG1), which triggers a myelination process in Schwann cells, and neuronal cell adhesion molecule close homolog of L1 (CHL1), which controls axonal functions. Thus, lack of BACE1 processing of type III NRG1 and CHL1 may lead to hypomyelination in BACE1- null mice and mistargeted axons in the hippocampus [24-26]. Analyzing the proteolysis of the different BACE1 substrates may be used to understand the underlying mechanism causing the toxicity following BACE1 inhibitor administration [27-29].
2.4. Role of BACE1 in the Production of Toxic Aβ Peptides
Amyloid plaques are extracellular deposits of Aβ42 fragments in the brain parenchyma and cerebral blood vessels. These fragments are formed by the cleavage of APP by different secretase enzymes (β and γ-isoforms) in brain neurons, as discussed in the previous section. APP (APP695) is a transmembrane glycoprotein observed on the cell membrane of many cells like microglia, astrocytes, and neuronal cells in the CNS [30]. APP is important in neural growth, synaptic formation, and repair [31]. But highest activity levels were observed in neural tissue and neuronal cells [32, 33]. The α-secretase cleaves APP mainly outside the CNS in peripheral cells and tissues. The β and ɣ-secretase cleave APP mainly in the CNS neuronal cells and, with sequential reactions, lead to shorter insoluble peptide fragments called Aβ- fragments, which are ~39-43 amino acids long (Fig. 4). These Aβ-fragments form unusual insoluble complex structures around neuronal cells in the brain and comprise a histopathological characteristic of AD pathophysiology called senile plaques. The control of amyloid processing of APP protein can be carried out by modulation of β-secretase (BACE1) [21-23].
Fig. (4).
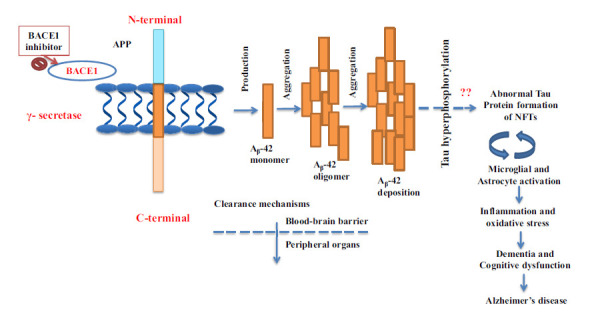
Schematic representation for the formation of toxic Aβ peptides and NFTs. APP is a type 1 transmembrane protein sequentially cleaved by two aspartic proteases to generate Aβ. First, the β-secretase enzyme cuts the APP to create the N-terminus of Aβ. Gamma-secretase cleavage next yields production of Aβ42 monomer, followed by aggregation of Aβ and deposition of Aβ. The amyloid plaques are extracellular deposits of Aβ42 fragments in the brain parenchyma.
3. BACE1 INHIBITORS IN CLINICAL TRIALS
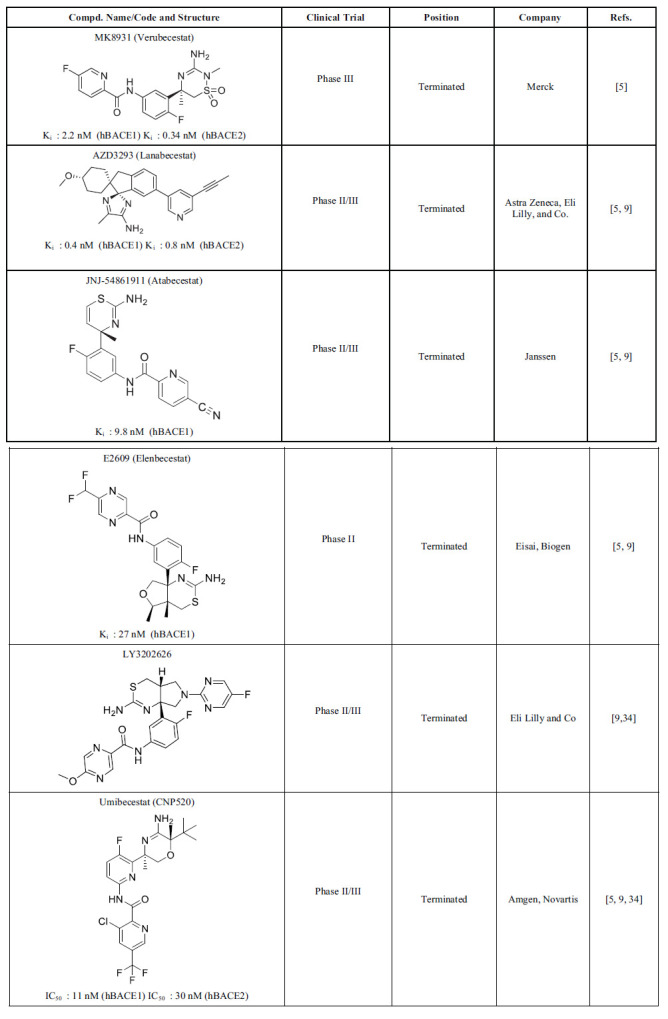
Some small molecule BACE1 inhibitors have been developed orally bioavailable and have entered into human clinical trials, as summarized below in Table 1 [5, 9, 34].
Table 1.
BACE1 inhibitors in different phases of clinical trials.
|
AstraZeneca developed AZD3293 (Lanabecestat), and it belongs to the acyl guanidine class of BACE1 inhibitors. The phase I trial results of Lanabecetat demonstrated excellent safety and metabolic profiles. The results of phase II/III trials showed improved cognition and decreased CSF Aβ42 levels. However, the phase II/III trials were discontinued because they caused psychiatric adverse events, weight loss, and depigmentation of the epidermis (outer layer of the skin) [3]. Merck pharmaceutical company developed one of the most promising BACE1 inhibitors, MK8931 (Verubecestat) (IC50 = 2.2 nM), which entered a phase III clinical trial. The high potency of Verubecestat is due to the interaction of the amidine moiety with the catalytic diad of BACE1. The lower pKa of the iminothiadiazinane core of Verubecestat is responsible for the low P-gp efflux [35]. Even though it showed remarkable pharmacokinetics and physiochemical properties, it was discontinued in the phase III trial. The major reasons to discontinue were the lack of efficacy of Verubecestat to produce cognitive benefits for AD patients and adverse effects like rashes [8]. Eisa developed E2609 (Elenbecestat) as a small molecule BACE1 inhibitor. The phase I trial of Elenbecestat demonstrated that a single oral ascending dose of 5, 10, 25, 50, 100, 200, 400, or 800 mg could reduce CSF Aβ42 levels by 92% at an 800 mg single dose. CSF Aβ42 levels are frequently inversely proportional to the brain, so decreased CSF levels likely indicate increased levels of brain Aβ42 levels. Decreased CSF Aβ42/ Aβ40 is a promising biomarker for AD [36]. These results were consistent with the phase II trial, and it was recently discontinued due to notable side effects like dizziness and nightmares. E2609 also caused elevation of liver enzymes and hippocampal atrophy [5]. JNJ-54861911 (Atabecestat) is a brain penetrable small molecule BACE1 inhibitor developed by Janssen. The phase I trial showed promising results, which allowed it to enter phase II trials. The results of the phase II trial demonstrated that 5, 25, and 50 mg doses of Atabecestat could reduce CSF Aβ levels by 50, 80, and 90%, respectively, indicating high potency for BACE1 inhibition. However, the phase II trials were terminated due to adverse effects like abnormal elevation in liver enzymes [34]. LY3202626 is a compound in the acylguanidine class of BACE1 inhibitors developed by Eli Lilly and Co. No clear effect on cognition was found; hence the phase II trial of LY3202626 was terminated. The phase II/III trial of Umibecestat (CNP520) was discontinued due to cognitive worsening in the active treatment group [37].
4. DESIGN AND DEVELOPMENT OF BACE1 INHIBITORS
The first group of BACE1 inhibitors developed was peptidomimetics based on the non-cleavable peptide transition state analogs with the amino acid sequence of the APP β site [38]. These peptidomimetic molecules are potent since they efficiently bind to the large active site of BACE1. But, unfortunately, these peptide inhibitors possess poor in-vivo pharmacological properties such as oral bioavailability, serum half-life, and BBB penetration [39].
The second group developed consists of small molecule BACE1 inhibitors that are large enough to bind to the active site and small enough to exhibit good pharmacokinetic properties and brain penetration. However, most of the second-generation molecules are substrates of P-glycoprotein, an ATP-dependent drug efflux pump for xenobiotics in the brain, hence cannot attain high concentrations in the brain. The third modern group of small molecule BACE1 inhibitors has been developed with adequate BBB penetration, pharmacokinetic properties, and Aβ level-reducing activity in the brain (Fig. 5).
Fig. (5).
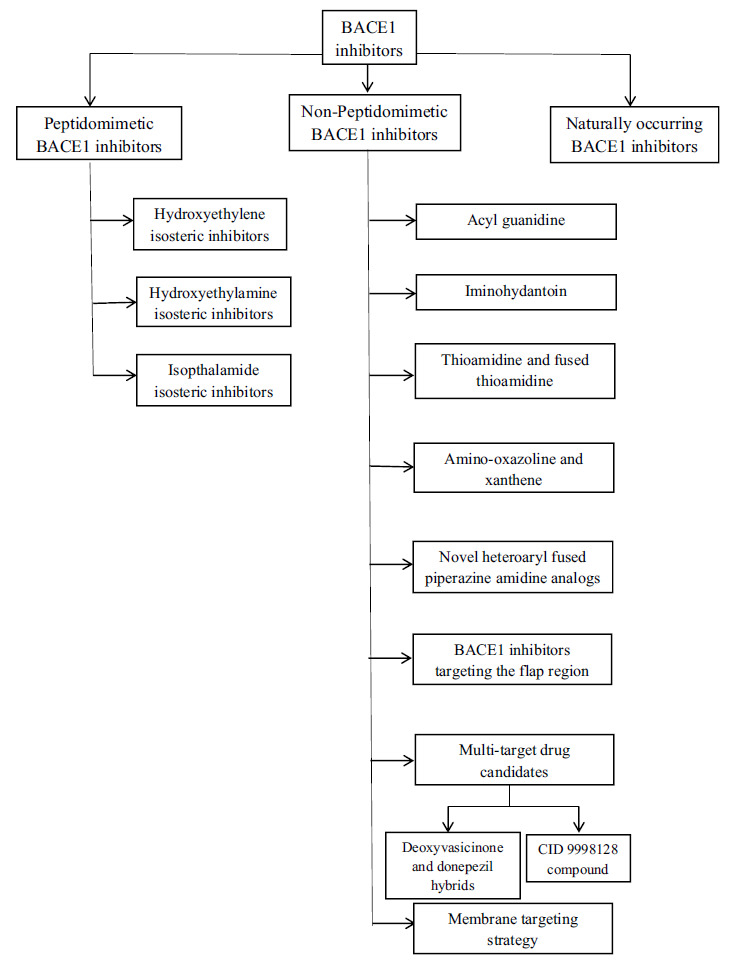
Schematic representation of different types of BACE1 inhibitors.
4.1. Peptidomimetic BACE1 Inhibitors
This class of compounds contains an amide bond or one of its isosteres. Transition-state bioisosteres like hydroxyl ethylene,-hydroxyl ethylamine-, isopthalamide-based inhibitors as well as reduced amide, statins, norstatins, and macrocyclic-based inhibitors are used for the design of aspartyl protease inhibitors [14, 39, 40].
Hydroxyethylene isosteric inhibitors are known to be the first class of BACE1 inhibitors discovered. Compound 1 (Fig. 6), which includes hydroxy ethylene along with an oxazolylmethyl substituent at the P3 position, is a more potent peptidomimetic BACE1 inhibitor (IC50= 0.12 nM) with high selectivity of >2500 over Cathepsin D (CatD) and >3800 fold over BACE2. On the other hand, Compound 2 (Fig. 6) consists of bulkier substituents like benzyl and isopropylamine on a hydroxy ethylene skeleton to accommodate P1 and P3 positions present on the binding site of the enzyme BACE1 (IC50= 1.1 nM) but gave poor in-vivo inhibition of Aβ [41, 42]. An example of a hydroxyl ethylamine inhibitor is Compound 3 (Fig. 6) with the 3-methoxybenzyl group at P1 with a phenylalanine side chain at the P1’ position. It showed an IC50 of 1.0 nM and a 39-fold selectivity for BACE1 over BACE2 and 23-fold over CatD. It showed a 65% reduction in Aβ levels in transgenic mice as well as improvement in cognitive function [43]. Another example consists of a six-membered ring sultam derivative Compound 4 (Fig. 5) having an IC50 of a 4 nM and 44-fold selectivity for BACE1 over BACE2 and 663-fold over CatD. The high potency of this compound was due to the presence of a meta ethylene moiety that fits the S3 subpocket of BACE1, which increases binding affinity to the active site by enhancing interaction with the catalytic dyad. Compound 4 exhibited low oral bioavailability within transgenic mice when co-administered with a P-gp inhibitor, and also a 55% reduction in Aβ42 levels was observed [44, 45]. Isopthalamide isostere-based inhibitors with a substituent like a hydroxyethyl amine, as demonstrated by Compounds 5 and 6), showed high potency and selectivity for BACE1 [46]. Compound 5 (Fig. 6) showed an IC50 of 2.5 nM, but low selectivity against CatD hence required modification. The main difference in S1’ subpockets of BACE1 and CatD was studied by structure-activity relationship (SAR) to increase the selectivity of the inhibitor toward BACE1. It was observed that the S1’ subpocket of BACE1 can accommodate large groups with polar interactions, while the S1’ subpocket of CatD can only form hydrophobic interactions with the ligand. This information led to adding a small polar group like methoxy at the P1’ position present in Compound 6 (Fig. 6) with increased potency (IC50= 0.3 nM) and 1,000-fold selectivity for BACE1 over CatD. Most of the peptidomimetic inhibitors exhibited poor oral bioavailability, limited BBB penetration, low serum half-life, and low reduction of Aβ levels, which shifted the research interest towards the next generation of BACE1 inhibitors called nonpeptidomimetic inhibitors [47].
Fig. (6).
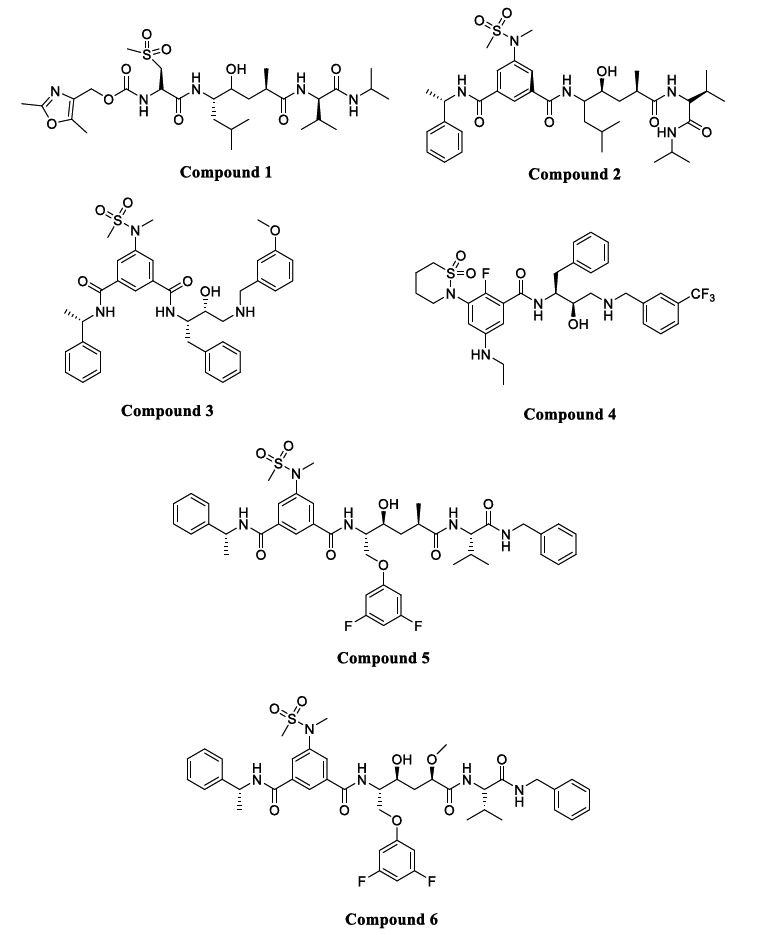
Peptidomimetic BACE1 inhibitors.
4.2. Non-peptidomimetic BACE1 Inhibitors
Development of non-peptidomimetic inhibitors was performed by high-throughput screening (HTS) campaigns followed by SAR studies of the best hit compounds. Non-peptidomimetic inhibitors showed better metabolic stability and oral bioavailability, improved BBB penetration, and reduced P-gp efflux ratio. The main feature of these compounds is the presence of an aminoazine residue in their structure. The different classes of these inhibitors include the compounds that contain acylguanidine, 2-aminopyridine, thioamidine, aminoimidazole-amino/iminohydantoin, dihydroquinazoline, and amino- thiazoline/oxazoline/quinoline/ pyrrolidine moieties [14, 48].
4.2.1. Acyl Guanidine
The acyl guanidine scaffold is the most promising class of inhibitors with high BACE1 potency, excellent pharmacokinetic characteristics, and lower P-gp efflux. Thus some molecules of this class were entered into human phase clinical trials [49, 50]. In Compound 7 (Fig. 7), adding a pyrrolidine moiety led to the development of a better and more potent compound with an IC50 of 0.25 µM against BACE1. Molecular modelling studies of this compound showed a unique U-shaped conformation that orients the aryl group to the S2 subpocket of BACE1. Moreover, SAR studies revealed that the addition of an ether linkage on the aryl group was directed toward macrocrystallzation through a rigidification strategy.
Fig. (7).
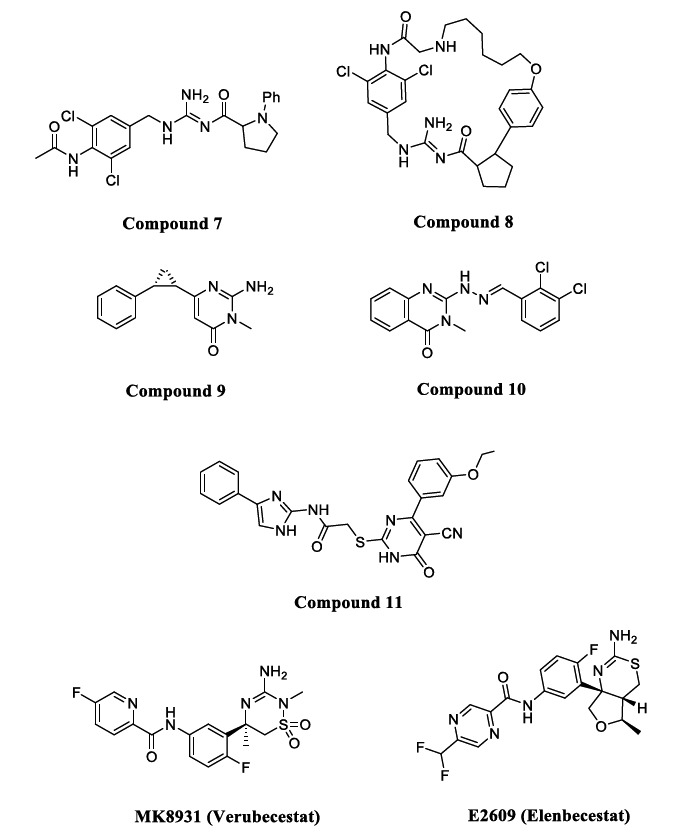
Acyl guanidine BACE1 inhibitors.
Compound 8 (Fig. 7) was developed by the same strategy and showed improved in-vitro and in-vivo potency with a significant reduction in Aβ levels. In addition, it showed BACE1 inhibition with an IC50 of 0.32 nM, a low P-gp efflux ratio of 2.3, and a 700-fold selectivity over CatD [51].
Acyl guanidine in the heterocyclic ring led to the design of 2-amino-pyrimidin-4-ones, aminopyrimidines, and quinazolinones to treat AD [52]. The binding affinity of Compound 9 (Fig. 6, IC50 = 154 µM) with BACE1 was enhanced using a rigidification approach with sp3 hybridized carbon. A CH-pi interaction was reported between the rigid cyclopropane and the Tyr71 residue of BACE1 [53]. As acyl guanidine is a very important scaffold for targeting BACE1, quinazoline-hydrazones (Compound 10, Fig. 7, IC50 = 3.7 µM) have been explored recently as novel BACE1 inhibitors [54]. Yan et al. discovered acyl guanidine molecules with a thioacetamide linkage (Compound 11, Fig. 6, IC50 = 4.6 µM) to be potent BACE1 inhibitors. Thioacetamide linkage also led to an increase in BBB permeability and low cellular cytotoxicity [55, 56]. One of the highly effective compounds containing the guanidine scaffold is MK8931 (Verubecestat) (Fig. 7), the first BACE1 inhibitor to reach a phase III clinical trial. It has 45000-fold selectivity for BACE1 over CatD and a potent IC50 of 2.2 nM due to strong hydrogen bonding between the amidine moiety and the catalytic dyad. The S3 subpocket of BACE1 is occupied by the pyridine fluoro substituent of verubecestat, which is also close to the Ala335 methyl side chain [35]. E2609 (Elenbecestat) (Fig. 7) is another guanidine-based inhibitor tested in phase III clinical trial with an IC50 of 27 nM [57-59].
4.2.2. Iminohydantoin
A compound with an iminohydantoin scaffold linked to a chloropyridine analog was developed (Compound 12) (Fig. 8), having a BACE1 IC50 of 21 nM and selectivity of 350-fold over CatD. A further modification was done to maximize the binding interaction with a subpocket of the enzyme, which was obtained by replacing the chloro group with the propenyl group. The linearity of the propenyl group led to strong interactions with the S3 pocket. The compounds with cyclopropyl substituent attained 6-fold more potency over compounds with phenyl substituent. Compound 13 (Fig. 8) was developed using the above modification and showed 5 fold increased potency (IC50 = 5.4 nM), improved selectivity for BACE1 (7500 fold over CatD), and showed excellent bioavailability [60].
Fig. (8).
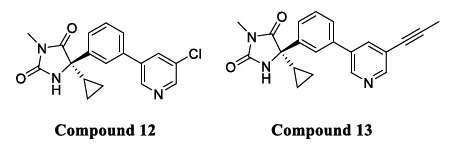
Iminohydantoin BACE1 inhibitors.
4.2.3. Thioamidine and Fused Thioamidine
Compounds with a benzothiazine (thioamidine based) scaffold were recognized as a different class of non-peptidomimetic BACE1 inhibitors. Compound 14 (Fig. 9) was the first orally bioavailable non-peptidomimetic inhibitor, which was later terminated from Phase 1 clinical trials due to toxic effects on the epithelial pigment of the eyes [61]. Structural modification of Compound 14 led to the discovery of another Compound 15 (Fig. 9) with potent BACE1 inhibitory activity, but clinical side effects were the main concern [62]. Fused thioamidine core-based structures were selective towards BACE1 over CatD with enhanced pharmacokinetic properties and improved BBB permeation. Compound 16 (Fig. 9) with a fused thioamidine ring was reported to lower amyloid-beta load in plasma and lumbar CSF in human subjects [56, 63].
Fig. (9).
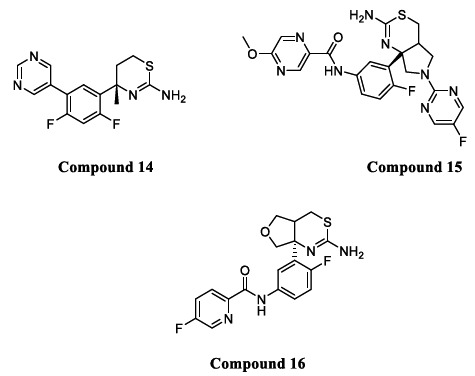
Thioamidine based BACE1 inhibitors.
4.2.4. Amino-oxazoline and Xanthene based β-secretase (BACE1) Inhibitors
Other aminoazine inhibitors, including amino-oxazoline and xanthene cores compounds, exhibited remarkable inhibitory potencies but a high P-gp efflux ratio. Compound 17 (Fig. 10), consisting of a functionalized 3-aza-2-fluoroxan- thene core, formed strong hydrogen bonds with Trp76. Other hydrogen bonds were formed between the oxygen of dihydropyran and Tyr198 and pyridyl nitrogen with Ser229 as observed in cocrystal structures of Compound 17 bound with BACE1 (IC50 = 0.3 nM and 2.2 P-gp efflux ratio) [64, 65].
Fig. (10).
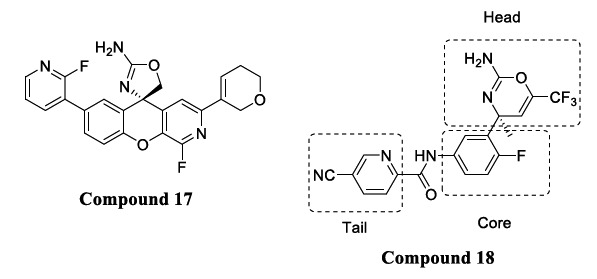
Xanthene and amino-oxazoline based derivatives.
An HTS campaign was performed on dihydrooxazine-based analogs to lower P-gp efflux and mitigate hERG inhibitory activity by controlling pKa [59, 60]. A lowering of the pKa of the amine or amidine moieties (which interact with the catalytic aspartates of BACE1) was obtained by incorporating the double bond in dihydrooxazine to convert it into an oxazine, led to reduced P-gp efflux. Such additional modification led to the development of Compound 18 (pKa = 7.0) [66].
Compound 18 (Fig. 10) consisted of substituted amino-oxazines with potent BACE1 activity (IC50 = 12 nM and 1.9 P-gp efflux ratio), which showed a dose-dependent reduction in Aβ levels in the brain as well as good (68%) oral bioavailability in rats. This class of inhibitors showed potent BACE1 inhibition, but there was a lack of detail regarding their selectivity profile. The X-ray structure of Compound 18 complexed with BACE1 shows the different interactions of amidine with the two catalytic aspartate residues 32 and 228 of the enzyme. Two aromatic rings like fluorophenyl and cyano pyridine accommodate the S1 and S3 pockets of the enzyme, and an amide NH forms a hydrogen bond with the backbone carbonyl oxygen of Gly230. Non-fluorine analogs were found to be 2-3 times less potent than the fluorine analog of Compound 18, indicating the importance of fluorine [67].
4.2.5. Novel Heteroaryl Fused Piperazineamidine Analogs as BACE1 InhibitorS
Ciordia et al. previously reported new candidates using computational approaches to include a series of cyclic amidines, acyl guanidines, and amino piperazinones [68-70]. Unfortunately, amino piperazinones (Compound 19, Fig. 11) showed poor brain penetration, preventing further development. Nevertheless, amino piperazinones inspired the development of the amino morpholine series having high pKa values (pKa< 9.6) but a strong P-gp efflux. Lowering of the pKa (pKa 6.5-7.5) was achieved by the addition of an electron-withdrawing group (trifluoromethyl) at the C-2 position of the morpholine ring seen in Compound 20 (Fig. 11).
Fig. (11).
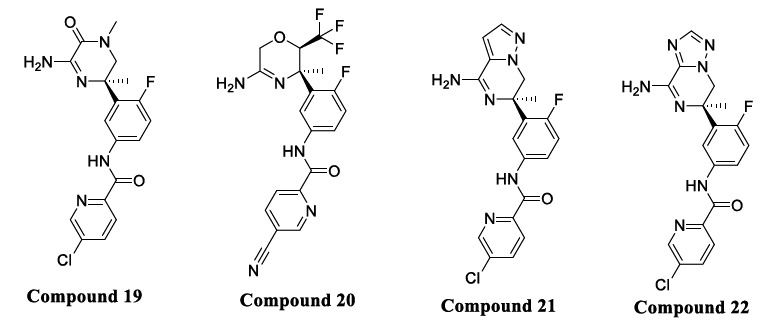
Fused piperazineamidine analogs.
A recent study focused on the bioisosteric replacement of an amide functional group of amino piperazinones with nitrogen fused to a five-membered heteroaromatic ring where the bridgehead nitrogen was maintained. Imidazopiperazine was first synthesized and compared with amino piperazinone and found to have similar potency in BACE1 cellular and enzymatic assays. Furthermore, the imidazopiperazine enabled deeper exploration of the synthesis of different bicycles based upon ring-opening of versatile cyclic sulphamidate. Cyclic sulphamidates are important and versatile intermediates used in organic synthesis to undergo highly selective reactions towards O-, S-, N-, C-, P-, and F- based nucleophiles [71].
Out of several compounds, Compounds 20, 21, and 22 (Fig. 11) exhibited the necessary in vitro profile merging potency with good intrinsic permeability, a reduced P-gp efflux, and hERG liability, and the heteroaryl fused aminopiperazine have been adopted for further profiling and optimization [72].
4.2.6. Structure-based Design of BACE1 Inhibitors and Targeting the Flap Region to Achieve Selectivity Over BACE2
Comparative analyses between inhibitor-bound BACE1 and BACE2 structures revealed differences in conformational flexibility in the flap and 10s loop region, where BACE1 shows a stabilized open conformation with the large binding site (Fig. 12) [73, 74], and BACE2 has a stabilized closed conformation [75, 76]. The reported selective BACE1 inhibitor (Compound 23) utilized an interaction of the ethyl group on the pyridine ring with the flap region to gain selectivity over BACE2 (> 227 fold and IC50 of 44 nM) [77]. In addition, a distinct conformation was observed between BACE1 and BACE2 in the 10s loop S3 pocket, where BACE2 has a smaller pocket than BACE1. Such differences offered another approach to design a selective BACE1 inhibitor (Fig. 11). Hilpert et al. at Roche explored amide substituents in their oxazine series directed in the S3 pocket, which inspired the author to optimize the amide substituent by incorporating larger substituents to increase selectivity and potency [65]. Compound 24 has a thiazine ring as the main nucleus. Spirocycles were added to the 5th position of thiazine, allowing for high selectivity over BACE2. This strategy was discovered by leveraging cocrystal structures of Compounds 23 and 24 (Fig. 12).
Fig. (12).
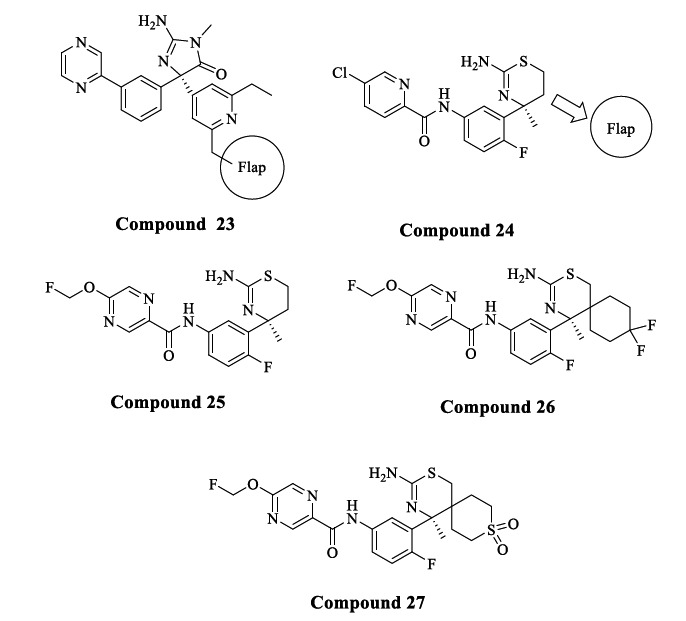
BACE1 inhibitors that target the flap region.
A balanced profile of cellular potency and BACE1 selectivity was seen by the introduction of a flouromethoxy group on the pyrazine ring as an amide substituent of Compound 24, resulting in Compound 25 (IC50 = 4.9 nM, and 16-fold selectivity over BACE2). Compound 25 served as a starting point to explore substituents at the 5th position of the thiazine ring. Previously the authors tried a phenyl ring substituent on the 5th position of the thiazine with both R and S configurations, but this resulted in increased potency on both BACE1 and BACE2 and minimal selectivity. A cocrystal structure revealed that replacing the phenyl ring with bulky substituents like saturated cycle/spirocycle led to a favourable interaction with the flap region to gain selectivity.
Compound 26, having difluorocyclohexyl as a spirocycle, showed an IC50 of 16 nM and 104-fold selectivity over BACE2. In contrast, Compound 27 with a sulphonyl group replacing the difluoromethylene of Compound 26 showed an increased potency of IC50 of 3.8 nM and 550-fold selectivity over BACE2. However, Compound 27 reduced cellular potency because of poor permeability and high P-gp efflux ratio, whereas Compound 26 displayed good cellular potency due to optimum lipophilicity and low P-gp efflux. High P-gp efflux in the case of Compound 27 may be due to the interaction of the polar sulfonyl group with the P-gp. Crystal structures of Compound 27 with BACE1 and BACE2 revealed that the flap of BACE1 forms an open conformation where the hydrogen bond between the phenolic OH of Tyr71 and the NH of the indole of Tyr76 was disrupted. It was also observed that the spirocycle interacts with the side chain of Tyr71, supporting the design hypothesis. The selectivity observed in Compound 27 is because BACE1 has a much larger movement of the flap compared to BACE2, which indicates a destabilization, hence resulting in the increased selectivity towards BACE1 and loss of BACE2 potency [78].
4.2.7. Multi-target Drug Candidates
(a) Deoxyvasicinone and Donepezil Hybrids
Du et al. selected acetylcholinesterase (AChE), Aβ1-42 peptide, and BACE1 as targets for symptomatic relief and to halt the chronic progression of AD. Deoxyvasicinone is a naturally occurring alkaloid composed of a quinazolinone fused with pyrrolidine. Its derivatives were suggested to be novel AChE inhibitors by the author's previous study [79-81]. Donepezil is an FDA-approved drug used as an AChE inhibitor to treat AD [82]. The pharmacophore benzyl piperidine of Donepezil binds to the active catalytic site of AChE [83, 84]. According to a literature study, the aryl piperazine class of molecules showed good BACE1 inhibition potency [85, 86]. Hence, the piperidine ring of donepezil was replaced with a piperazine ring to enhance the interaction with the active site of BACE1. According to a research study, some amount of reduction in the aggregation of Aβ can be achieved by the interaction of the ligand with the peripheral anionic site (PAS) of AChE. Deoxyvasicinone-donepezil hybrid compounds were synthesized (Fig. 13), out of which Compounds 28 and 29 exhibited remarkable inhibitory activities against BACE1 (IC50= 0.834 nM, 0.129 nM), hAChE (IC50= 56.14 nM, 3.29 nM) and Aβ1-42 aggregation (IC50 = 13.26 nM, 9.26 nM). Molecular docking studies of the most potent Compound 29 revealed binding with CAS and PAS of hAChE and important cationic-π interaction between piperazine and the Tyr71 residue in the active site of BACE1. Furthermore, both Compounds 28 and 29 exhibited lower cytotoxicity and excellent neuroprotective activity against Aβ1-42-induced damage in SH-SY5Ycells. Therefore, this strategy of pharmacophore combination can be used to design multifunctional agents for treating AD [87].
Fig. (13).
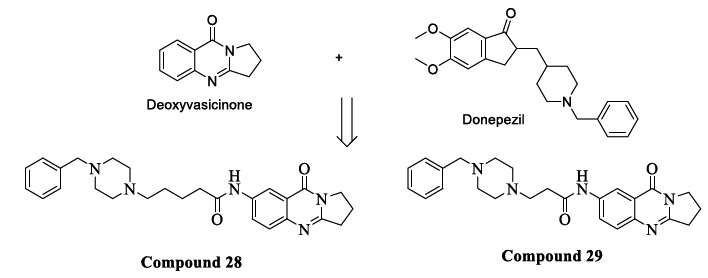
Deoxyvasicinone and donepezil hybrids.
(b) CID 9998128 Compound
CID 9998128 is a multi-target drug candidate obtained from the large PubChem database [88] by a high-throughput screening protocol [89]. This compound exhibited a high binding affinity with Aβ fibrils, BACE1, γ-secretase, PPAR-γ, and AChE. CID 9998128 has three structural parts – a quinazoline (Part 1), an indazole (Part 2), and a methylpyridine (Part 3) (Fig. 14). This compound showed potent inhibitory activity against Aβ fibrils formation and β-secretase (BACE1) as confirmed by in-silico and in vitro experiments. Molecular docking simulation was carried out between the BACE1 crystal structure (pdb: 1M4H) [38] with CID 9998128, and nine nonbonded interactions were observed (Van der Waal's interactions) along with three hydrogen bonds. The binding energy was found to be -9.8 kcal/mol, which was measured using a molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) method. This measurement showed that Van der Waal's interactions were more important than electrostatic interactions. Furthermore, the compound's negatively charged indazole (Part 3) destabilizes the interaction with 1M4H and Aβ1-42 peptides due to repulsive coulombic forces. These results suggested the indazole moiety should not be used as a pharmacophore in a multi-target drug candidate for AD. Furthermore, CID 9998128 was shown to have an in vitro EC50 of 15 μM against BACE1 using a FRET assay as well as a dose-dependent inhibitory activity against Aβ1-42 fibrils formation recorded as an IC50 = 43.6 µM and a DC50 (half-maximal dissociate activity) = 22.7 µM. Hence, these measurements suggested that CID 9998128 could be potential multi-target drug treatment for AD [90].
Fig. (14).
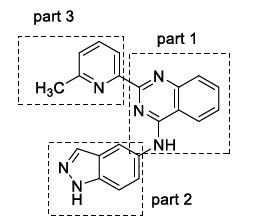
CID 9998128 compound.
4.2.8. Membrane Targeting Strategy for Effective BACE1 Inhibition
Amyloid precursor protein (APP) is an amyloidogenic substrate cleaved by the BACE1 enzyme active at acidic pH in the endosomes. So, to specifically inhibit endosomal β-secretase/BACE1, an endocytosis-dependent BACE1 inhibitor is needed. This strategy will eliminate the off-target side effects associated with endocytosis independent non-amyloidogenic substrates. It has been shown that endosome-targeted inhibitors are more potent and efficient than free inhibitors [91]. It is reported that membrane targeting can be achieved by using the sterol group as the membrane anchor linked to the C terminus of the inhibitor, produced by solid-phase synthesis. A sterol moiety will direct the inhibitor towards the lipophilic endosomes. The above results were confirmed by in-vivo treatment of a Drosophila model with sterol-linked BACE1 inhibitor. A high survival rate was observed compared to the control model [92]. Another study demonstrated that treatment of APPs/PSDE9 mice hippocampus with sterol-linked BACE1 inhibitor or free BACE1 inhibitor confirmed that the sterol-linked inhibitor has more Aβ reducing activity than the free inhibitor [93].
4.3. Naturally Occurring BACE1 Inhibitors
In recent years, many drugs of natural origin have been studied. It is because of the ability of the phytoconstituents to bind with multiple therapeutic targets rather than a single target. This multi-target-based strategy is useful in treating complex diseases like AD [94].
Flavonoids like polymethoxy flavones obtained from the extract of black ginger, i.e., Kaemferia parviflora (KP), were found to have BACE1 inhibitory activity. KP plants also exhibit antioxidant and anticarcinogenic properties [95]. The extract of the KP plant consists of mainly three polymethoxyflavone components, namely 3,5,7,3’,4’-pentamethoxy- flavone (PMF), 5,7-trimethoxyflavone (TMF), and 5,7,4’-dimethoxyflavone (DMF), which inhibits BACE1 and shows high selectivity towards BACE1 over α-secretase and other serine proteases. An in-silico study was carried out by Youn et al., which highlighted the potent BACE1 inhibitory activity of poly methoxy flavones and strong H-bond interactions between poly methoxy flavones and amino acid residues of BACE1. Among the three components, TMF (Compound 30) exhibited the most potent activity (IC50 = 36.9 µM), followed by DMF (Compound 31) (IC50 = 49.5 µM) and PMF (Compound 32) displayed the lowest potency with an IC50 = 59.8 µM [96]. Structure-activity relationship (SAR) studies revealed that methoxy groups at the C5 and C7 positions of ring A are necessary for BACE1 inhibition. Additionally, the methoxy group at C4’ position on ring B enhanced BACE1 activity, while adding the methoxy groups at the C3 and C3’ positions reduced BACE1 activity (Fig. 15). These compounds were reported to be non-competitive BACE1 inhibitors, which means they will bind to allosteric sites of the BACE1 enzyme [97]. They can also pass the BBB, which was confirmed by their detection in the rat brain [98].
Fig. (15).
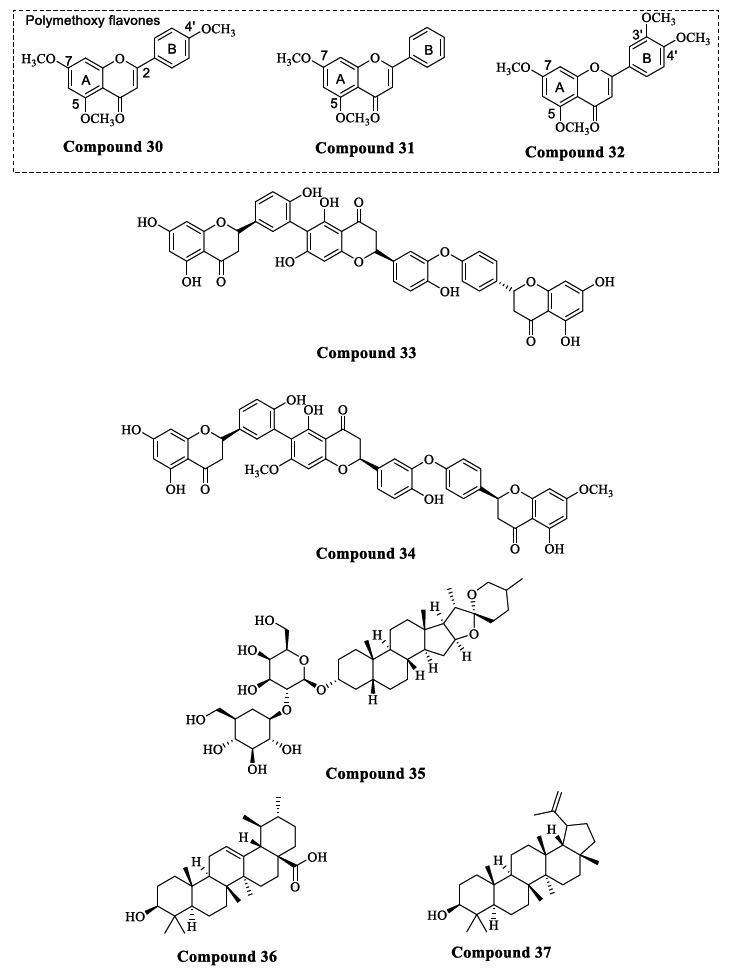
Naturally occurring BACE1 inhibitors.
The Selaginella doederleinii (SD) plant is used in Chinese herbal medicine as an anticancer, cardioprotective, and anti-inflammatory agent. Zou et al. isolated several triflavonoids like selagin triflavonoids from this plant, which have displayed potent BACE1 inhibitory activity. The results obtained in an in-vitro FRET assay suggested that Compound 33 was the most potent BACE1 inhibitor with an IC50 of 0.75 µM and Compound 34 was the weakest inhibitor (IC50 = 46.99 µM) (Fig. 15). Furthermore, SAR studies showed that selagin triflavonoids with a naringenin unit are preferred over those with an apigenin unit for potent BACE1 activity [99].
In traditional Chinese medicine, Rhizoma anemarrhenae (RA) and its related compounds are used to treat depression and dementia. Multiple bioactive constituents are present in RA, such as steroidal saponins, alkaloids, and flavonoids, out of which saponins are the main constituents [100]. Cell extraction techniques were used to screen potent active compounds from RA, and the pharmacological targets were predicted using a chemogenomics target knowledgebase and docking studies. As a result, Timosaponin A-III (TA-III) (Compound 35) was identified as the most potent active compound to inhibit BACE1. TA-III inhibited BACE1 in a dose-dependent manner with an IC50 of 7.45 µM. Furthermore, the pharmacological mechanism of action of TA-III was identified as reduction of Aβ aggregation by binding with BACE1 and prevention of the neuronal impairment by downregulation of NMDAR/ERK signaling pathway [101].
Hosen et al. isolated 40 compounds from different parts of the Leea indica plant [102], a folk medicinal plant and often used as an antispasmodic, antidiarrheal and anticancer agent [103]. Out of all the isolated compounds, two triterpene molecules, ursolic acid (Compound 36) and lupeol (Compound 37) displayed potent BACE1 inhibitory activity. SAR studies revealed the binding interaction of these triterpenes with the BACE1 active site. Hydrogen bonds were found between ursolic acid and the Asn233 and Thr232 residues. Also, there were hydrophobic interactions. In contrast, lupeol forms a hydrogen bond with Gly11 residue and displays strong hydrophobic interactions with the active site of BACE1. Lupeol was a more promising compound, as it was able to pass the BBB more efficiently than ursolic acid (based on their theoretical pharmacokinetics values) [102]. Lupeol is also neuroprotective and inhibits BACE1 with an IC50 of 5.12 µM [102, 104].
Most of the synthetic BACE1 inhibitors were terminated from clinical trials due to toxicity problems. Naturally derived BACE1 inhibitors are safe and display potent anti-AD effects. Further advanced in vivo studies are required to understand these natural compounds' mechanism of action and clinical benefits.
5. FUTURE DIRECTIONS
Since the active site of BACE1 (β-secretase) is large and has few structural similarities with other aspartyl proteases, it was very challenging to develop a potent, selective, and safe BACE1 inhibitor. Some compounds (e.g., verubecestat, and atabecestat) successfully entered the advanced phases of clinical trials but were unfortunately terminated due to lack of clinical benefits or toxicity problems [8]. Accumulation of toxic Aβ peptides in AD patients starts 10-15 years before the onset of AD symptoms which suggests that BACE1 inhibition would not be effective if patients are symptomatic [105, 106]. The pathophysiology and genetics of AD are very complex, so BACE1 inhibition or, in general, any anti-amyloid therapy might not be alone sufficient to reverse the progression of AD. The level of BACE1 inhibition required for reduction in Aβ load was revealed by the A637T APP mutation [21]. It was shown that 50% inhibition of BACE1 is sufficient to provide a 20% lowering of the cellular Aβ load in transgenic mice. Reducing cellular Aβ peptides by around 20% is preventative for AD if commenced before significant Aβ accumulation [107, 108]. However, the relationship between BACE1 inhibition and Aβ reduction level is not fully understood in humans at present. In the future, AD prevention clinical trials should be carried out by enrolling asymptomatic patients that carry FAD mutations and are at high risk for acquiring AD/prodromal AD. Recently, amyloid positron emission tomography (PET) and CSF Aβ42 measurement have been developed as primary diagnostic tools for prodromal AD identification [109, 110]. A particular dose range needs to be defined for BACE1 inhibitors to provide clinical benefit and offer better safety without hampering the synaptic functions of other BACE1 substrates. Treatments with BACE1 inhibitors are somewhat like cholesterol-lowering statin drugs. In both cases, if the treatment does not generally begin before the deposition of cholesterol in coronary arteries or accumulation of toxic Aβ in the brain, there will be no reversal in the progression of these diseases. The informational data obtained from current clinical trials or even failed clinical trials of BACE1 inhibitors is useful in understanding the level of BACE1 inhibition required to lower the level of Aβ peptides to delay the onset of AD or provide cognitive benefits in AD patients.
CONCLUSION
Alzheimer’s disease (AD) is a chronic neurodegenerative disorder associated with cognitive deficits. The current FDA-approved treatment for AD only provides symptomatic relief without any modification in disease progression. Some potential targets have been discovered as disease-modifying treatments for AD; out of them, BACE1 is the most rational approach as it is possibly involved in the rate-limiting step to produce Aβ peptides. The active site of BACE1 is large and shares 64% sequence similarity with its close homolog BACE2. Hence, developing potent, selective, and orally bioavailable small molecule BACE1 inhibitors is very challenging. The BACE1 inhibitors are characterized as peptidomimetic and non-peptidomimetic inhibitors, further classified into different structural classes. The peptidomimetic inhibitors mainly consist of hydroxy ethylene, hydroxyethyl amine, and isopthalamide-based inhibitors. These inhibitors exhibited good potency for BACE1 inhibition, but development was terminated due to poor oral bioavailability and stability problems. The nonpeptidomimetic inhibitors were structurally classified into acylguanidine, thioamidine, iminohydantoin, aminooxazoline, and xanthene. The acylguanidine class of inhibitors has been the most promising class of BACE1 inhibitors. Verubecestat, lanabecestat, elenbesestat, and atabecestat are some of the molecules from the acylguanidine class which successfully reached to late stages of clinical trials. However, they all failed to enter the market because of toxicity or a lack of clinical efficacy.
The literature suggests that the most effective BACE1 inhibitor is a non-peptidomimetic with substantial binding affinity and blood-brain barrier (BBB) permeability. Also, there should be low or no affinity to P-gp.
Looking at the bioactivity profile peptidomimetic Compound 1 turns out to be the most potent with hydroxy ethylene isosteric and oxazolylmethyl substituent (IC50= 0.12 nM) as well as higher selectivity of >3800 fold over BACE2 [41, 42]. But several limitations of the peptidomimetics like metabolic instability, poor oral bioavailability, and low BBB penetration led to the development of non-peptidomimetics with better pharmacokinetics and stability. In the case of non- peptidomimetics, the acyl guanidine Compound 7 with pyrrolidine moiety (IC50 of 0.25 µM against BACE1) is the most promising inhibitor with high BACE1 potency, excellent pharmacokinetic characteristics, and lower P-gp efflux [49, 50].
The multi-target drug ligands (MTDLs) approach was used to delay the progression of AD and provide cognitive benefit. A membrane targeting strategy was used to specifically deliver BACE1 inhibitors to the membrane where the cleavage process of APP is performed. With such a strategy, side effects caused by the deficiency of other non-amyloidogenic BACE1 substrates are reduced. Amyloid PET and CSF Aβ42 measurements have been developed recently to identify patients who are in the prodromal stage of AD and are genetically at high risk of developing AD. The A673T FAD mutation is protective against AD. Studies showed that 50% BACE1 inhibition is sufficient to lower the Aβ load by around 20%, which can delay the onset of AD if the treatment is started before the accumulation of significant levels of amyloid peptides. Deoxyvasicinone-donepezil hybrid compounds Compounds 28 and 29 were developed as multitargeted small molecules having inhibitory activity for BACE1 (IC50= 0.834 nM, 0.129 nM), hAChE (IC50= 56.14 nM, 3.29 nM) and Aβ1-42 aggregation (IC50 = 13.26 nM, 9.26 nM). Thus such a pharmacophore may be beneficial in the design of multifunctional agents for treating AD [87].
Results of the current clinical trials may be helpful in the future for understanding the connection between the level of BACE1 inhibition, amyloid load, and cognitive status. There is a need for the optimization of the correct dose of BACE1 inhibitors. The dose must be sufficient to provide clinical benefit without causing side effects. BACE1 inhibitors could be the best potential approach to combat AD despite their past failure in advanced clinical trial studies. The data from these current ongoing or failed clinical trials can help understand the relationship between BACE1 inhibition and amyloid load and the complexity of AD progression. A safe and selective BACE1 inhibitor is needed, which can be used as a treatment option in delaying or preventing the onset of AD.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
ACKNOWLEDGEMENTS
SP and BAV acknowledge the Department of Pharmaceuticals, Ministry of Chemical & Fertilizers, Govt of India for fellowship.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1. Dementia. Available from: https://www.who.int/news-room/fact-sheets/detail/dementia.
- 2.Alzheimer’s Association 2019 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2019;15:321–387. doi: 10.1016/j.jalz.2019.01.010. [DOI] [Google Scholar]
- 3.Alzheimer Association Early signs and symptoms of Alzheimer’s. Alzheimers Dement. 2019:1–88. [Google Scholar]
- 4.Coimbra J.R.M., Marques D.F.F., Baptista S.J., Pereira C.M.F., Moreira P.I., Dinis T.C.P., Santos A.E., Salvador J.A.R. Highlights in BACE1 inhibitors for Alzheimer’s disease treatment. Front Chem. 2018;6:178. doi: 10.3389/fchem.2018.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das B., Yan R. A close look at BACE1 inhibitors for Alzheimer’s disease treatment. CNS Drugs. 2019;33(3):251–263. doi: 10.1007/s40263-019-00613-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amiri H., Saeidi K., Borhani P., Manafirad A., Ghavami M., Zerbi V. Alzheimer’s disease: Pathophysiology and applications of magnetic nanoparticles as MRI theranostic agents. ACS Chem. Neurosci. 2013;4(11):1417–1429. doi: 10.1021/cn4001582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaudhary A., Maurya P.K., Yadav B.S., Singh S., Mani A. Current therapeutic targets for Alzheimer’s disease. J. Biomed. (Syd.) 2018;3:74–84. doi: 10.7150/jbm.26783. [DOI] [Google Scholar]
- 8.Hall A., Gijsen H.J.M. Targeting β-secretase (BACE) for the treatment of Alzheimer’s disease. Compr. Med. Chem. III. 2017;7:326–383. doi: 10.1016/B978-0-12-409547-2.13809-0. [DOI] [Google Scholar]
- 9.Vassar R. BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimers Res. Ther. 2014;6(9):89. doi: 10.1186/s13195-014-0089-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan J., Venkatraman S., Zheng Y., McKeever B.M., Dillard L.W., Singh S.B. Structure-based design of β-site APP cleaving enzyme 1 (BACE1) inhibitors for the treatment of Alzheimer’s disease. J. Med. Chem. 2013;56(11):4156–4180. doi: 10.1021/jm301659n. [DOI] [PubMed] [Google Scholar]
- 11.Al-Tel T.H., Semreen M.H., Al-Qawasmeh R.A., Schmidt M.F., El-Awadi R., Ardah M., Zaarour R., Rao S.N., El-Agnaf O. Design, synthesis, and qualitative structure-activity evaluations of novel β-secretase inhibitors as potential Alzheimer’s drug leads. J. Med. Chem. 2011;54(24):8373–8385. doi: 10.1021/jm201181f. [DOI] [PubMed] [Google Scholar]
- 12.Solans A., Estivill X., de La Luna S. A new aspartyl protease on 21q22.3, BACE2, is highly similar to Alzheimer’s amyloid precursor protein β-secretase. Cytogenet. Cell Genet. 2000;89(3-4):177–184. doi: 10.1159/000015608. [DOI] [PubMed] [Google Scholar]
- 13.Bennett B.D., Babu-Khan S., Loeloff R., Louis J.C., Curran E., Citron M., Vassar R. Expression analysis of BACE2 in brain and peripheral tissues. J. Biol. Chem. 2000;275(27):20647–20651. doi: 10.1074/jbc.M002688200. [DOI] [PubMed] [Google Scholar]
- 14.Moussa-Pacha N.M., Abdin S.M., Omar H.A., Alniss H., Al-Tel T.H. BACE1 inhibitors: Current status and future directions in treating Alzheimer’s disease. Med. Res. Rev. 2020;40(1):339–384. doi: 10.1002/med.21622. [DOI] [PubMed] [Google Scholar]
- 15.Mirsafian H., Mat Ripen A., Merican A.F., Bin Mohamad S. Amino acid sequence and structural comparison of BACE1 and BACE2 using evolutionary trace method. ScientificWorldJournal. 2014;2014:482463. doi: 10.1155/2014/482463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu B., Xiong B., Qiu B.Y., Li X., Yu H.P., Xiao K., Wang X., Li J., Shen J.K. Construction of a small peptide library related to inhibitor OM99-2 and its structure-activity relationship to β-secretase. Acta Pharmacol. Sin. 2006;27(12):1586–1593. doi: 10.1111/j.1745-7254.2006.00432.x. [DOI] [PubMed] [Google Scholar]
- 17.Rombouts F.J.R., Alexander R., Cleiren E., De Groot A., Carpentier M., Dijkmans J., Fierens K., Masure S., Moechars D., Palomino-Schätzlein M., Pineda-Lucena A., Trabanco A.A., Van Glabbeek D., Vos A., Tresadern G. Fragment binding to β-secretase 1 without catalytic aspartate interactions identified via orthogonal screening approaches. ACS Omega. 2017;2(2):685–697. doi: 10.1021/acsomega.6b00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu Y., Li M.J., Greenblatt H., Chen W., Paz A., Dym O., Peleg Y., Chen T., Shen X., He J., Jiang H., Silman I., Sussman J.L. Flexibility of the flap in the active site of BACE1 as revealed by crystal structures and molecular dynamics simulations. Acta Crystallogr. D Biol. Crystallogr. 2012;68(Pt 1):13–25. doi: 10.1107/S0907444911047251. [DOI] [PubMed] [Google Scholar]
- 19.Mullan M., Crawford F., Axelman K., Houlden H., Lilius L., Winblad B., Lannfelt L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of β-amyloid. Nat. Genet. 1992;1(5):345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- 20.Di Fede G., Catania M., Morbin M., Rossi G., Suardi S., Mazzoleni G., Merlin M., Giovagnoli A.R., Prioni S., Erbetta A., Falcone C., Gobbi M., Colombo L., Bastone A., Beeg M., Manzoni C., Francescucci B., Spagnoli A., Cantù L., Del Favero E., Levy E., Salmona M., Tagliavini F. A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science. 2009;323(5920):1473–1477. doi: 10.1126/science.1168979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jonsson T., Atwal J.K., Steinberg S., Snaedal J., Jonsson P.V., Bjornsson S., Stefansson H., Sulem P., Gudbjartsson D., Maloney J., Hoyte K., Gustafson A., Liu Y., Lu Y., Bhangale T., Graham R.R., Huttenlocher J., Bjornsdottir G., Andreassen O.A., Jönsson E.G., Palotie A., Behrens T.W., Magnusson O.T., Kong A., Thorsteinsdottir U., Watts R.J., Stefansson K. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488(7409):96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 22.Maloney J.A., Bainbridge T., Gustafson A., Zhang S., Kyauk R., Steiner P., van der Brug M., Liu Y., Ernst J.A., Watts R.J., Atwal J.K. Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J. Biol. Chem. 2014;289(45):30990–31000. doi: 10.1074/jbc.M114.589069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benilova I., Gallardo R., Ungureanu A.A., Castillo Cano V., Snellinx A., Ramakers M., Bartic C., Rousseau F., Schymkowitz J., De Strooper B. The Alzheimer disease protective mutation A2T modulates kinetic and thermodynamic properties of amyloid-β (Aβ) aggregation. J. Biol. Chem. 2014;289(45):30977–30989. doi: 10.1074/jbc.M114.599027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo X., Prior M., He W., Hu X., Tang X., Shen W., Yadav S., Kiryu-Seo S., Miller R., Trapp B.D., Yan R. Cleavage of neuregulin-1 by BACE1 or ADAM10 protein produces differential effects on myelination. J. Biol. Chem. 2011;286(27):23967–23974. doi: 10.1074/jbc.M111.251538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hitt B., Riordan S.M., Kukreja L., Eimer W.A., Rajapaksha T.W., Vassar R. β-Site amyloid precursor protein (APP)-cleaving enzyme 1 (BACE1)-deficient mice exhibit a close homolog of L1 (CHL1) loss-of-function phenotype involving axon guidance defects. J. Biol. Chem. 2012;287(46):38408–38425. doi: 10.1074/jbc.M112.415505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salzer J.L. Axonal regulation of Schwann cell ensheathment and myelination. J. Peripher. Nerv. Syst. 2012;17(Suppl. 3):14–19. doi: 10.1111/j.1529-8027.2012.00425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou L., Barão S., Laga M., Bockstael K., Borgers M., Gijsen H., Annaert W., Moechars D., Mercken M., Gevaert K., De Strooper B. The neural cell adhesion molecules L1 and CHL1 are cleaved by BACE1 protease in vivo. J. Biol. Chem. 2012;287(31):25927–25940. doi: 10.1074/jbc.M112.377465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Montag-Sallaz M., Schachner M., Montag D. Misguided axonal projections, neural cell adhesion molecule 180 mRNA upregulation, and altered behavior in mice deficient for the close homolog of L1. Mol. Cell. Biol. 2002;22(22):7967–7981. doi: 10.1128/MCB.22.22.7967-7981.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pratte M., Rougon G., Schachner M., Jamon M. Mice deficient for the close homologue of the neural adhesion cell L1 (CHL1) display alterations in emotional reactivity and motor coordination. Behav. Brain Res. 2003;147(1-2):31–39. doi: 10.1016/S0166-4328(03)00114-1. [DOI] [PubMed] [Google Scholar]
- 30.Pimplikar S.W., Ghosal K. Amyloid precursor protein: More than just neurodegeneration. Stem Cell Res. Ther. 2011;2(5):39. doi: 10.1186/scrt80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Priller C., Bauer T., Mitteregger G., Krebs B., Kretzschmar H.A., Herms J. Synapse formation and function is modulated by the amyloid precursor protein. J. Neurosci. 2006;26(27):7212–7221. doi: 10.1523/JNEUROSCI.1450-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seubert P., Oltersdorf T., Lee M.G., Barbour R., Blomquist C., Davis D.L., Bryant K., Fritz L.C., Galasko D., Thal L.J., Lieberburg I., Schenk D.B. Secretion of β-amyloid precursor protein cleaved at the amino terminus of the β-amyloid peptide. Nature. 1993;361(6409):260–263. doi: 10.1038/361260a0. [DOI] [PubMed] [Google Scholar]
- 33.Hrabinova M., Pejchal J., Kucera T., Jun D., Schmidt M., Soukup O. Is It the Twilight of BACE1 Inhibitors? Curr. Neuropharmacol. 2021;19(1):61–77. doi: 10.2174/1570159X18666200503023323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Evin G., Lessene G., Wilkins S. BACE inhibitors as potential drugs for the treatment of Alzheimer’s disease: Focus on bioactivity. Recent Patents CNS Drug Discov. 2011;6(2):91–106. doi: 10.2174/157488911795933938. [DOI] [PubMed] [Google Scholar]
- 35.Scott J.D., Li S.W., Brunskill A.P.J., Chen X., Cox K., Cumming J.N., Forman M., Gilbert E.J., Hodgson R.A., Hyde L.A., Jiang Q., Iserloh U., Kazakevich I., Kuvelkar R., Mei H., Meredith J., Misiaszek J., Orth P., Rossiter L.M., Slater M., Stone J., Strickland C.O., Voigt J.H., Wang G., Wang H., Wu Y., Greenlee W.J., Parker E.M., Kennedy M.E., Stamford A.W. Discovery of the 3-Imino-1,2,4-thiadiazinane 1,1-dioxide derivative verubecestat (MK-8931)-A β-site amyloid precursor protein cleaving enzyme 1 inhibitor for the treatment of Alzheimer’s disease. J. Med. Chem. 2016;59(23):10435–10450. doi: 10.1021/acs.jmedchem.6b00307. [DOI] [PubMed] [Google Scholar]
- 36.Janelidze S., Stomrud E., Brix B., Hansson O. Towards a unified protocol for handling of CSF before β-amyloid measurements. Alzheimers Res. Ther. 2019;11(1):63. doi: 10.1186/s13195-019-0517-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hampel H., Vassar R., De Strooper B., Hardy J., Willem M., Singh N., Zhou J., Yan R., Vanmechelen E., De Vos A., Nisticò R., Corbo M., Imbimbo B.P., Streffer J., Voytyuk I., Timmers M., Tahami Monfared A.A., Irizarry M., Albala B., Koyama A., Watanabe N., Kimura T., Yarenis L., Lista S., Kramer L., Vergallo A. The β-secretase BACE1 in Alzheimer’s disease. Biol. Psychiatry. 2021;89(8):745–756. doi: 10.1016/j.biopsych.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hong L., Turner R.T., III, Koelsch G., Shin D., Ghosh A.K., Tang J. Crystal structure of memapsin 2 (β-secretase) in complex with an inhibitor OM00-3. Biochemistry. 2002;41(36):10963–10967. doi: 10.1021/bi026232n. [DOI] [PubMed] [Google Scholar]
- 39.Hong L., Koelsch G., Lin X., Wu S., Terzyan S., Ghosh A.K., Zhang X.C., Tang J. Structure of the protease domain of memapsin 2 (β-Secretase) complexed with inhibitor. Science (80-. ). 2000;290:150-153. doi: 10.1126/science.290.5489.150. [DOI] [PubMed] [Google Scholar]
- 40.Dash C., Kulkarni A., Dunn B., Rao M. Aspartic peptidase inhibitors: Implications in drug development. Crit. Rev. Biochem. Mol. Biol. 2003;38(2):89–119. doi: 10.1080/713609213. [DOI] [PubMed] [Google Scholar]
- 41.Ghosh A.K., Kumaragurubaran N., Hong L., Lei H., Hussain K.A., Liu C.F., Devasamudram T., Weerasena V., Turner R., Koelsch G., Bilcer G., Tang J. Design, synthesis and X-ray structure of protein-ligand complexes: Important insight into selectivity of memapsin 2 (β-secretase) inhibitors. J. Am. Chem. Soc. 2006;128(16):5310–5311. doi: 10.1021/ja058636j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sandgren V., Bäck M., Kvarnström I., Dahlgren A. Design and synthesis of hydroxyethylene-based BACE-1 inhibitors incorporating extended P1 substituents. Open Med. Chem. J. 2013;7:1–15. doi: 10.2174/1874104501307010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang W.P., Huang X., Downs D., Cirrito J.R., Koelsch G., Holtzman D.M., Ghosh A.K., Tang J. Beta-secretase inhibitor GRL-8234 rescues age-related cognitive decline in APP transgenic mice. FASEB J. 2011;25(2):775–784. doi: 10.1096/fj.10-167213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beswick P., Charrier N., Clarke B., Demont E., Dingwall C., Dunsdon R., Faller A., Gleave R., Hawkins J., Hussain I., Johnson C.N., MacPherson D., Maile G., Matico R., Milner P., Mosley J., Naylor A., O’Brien A., Redshaw S., Riddell D., Rowland P., Skidmore J., Soleil V., Smith K.J., Stanway S., Stemp G., Stuart A., Sweitzer S., Theobald P., Vesey D., Walter D.S., Ward J., Wayne G. BACE-1 inhibitors part 3: Identification of hydroxy ethylamines (HEAs) with nanomolar potency in cells. Bioorg. Med. Chem. Lett. 2008;18(3):1022–1026. doi: 10.1016/j.bmcl.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 45.Rueeger H., Lueoend R., Machauer R., Veenstra S.J., Jacobson L.H., Staufenbiel M., Desrayaud S., Rondeau J.M., Möbitz H., Neumann U. Discovery of cyclic sulfoxide hydroxyethylamines as potent and selective β-site APP-cleaving enzyme 1 (BACE1) inhibitors: Structure based design and in vivo reduction of amyloid β-peptides. Bioorg. Med. Chem. Lett. 2013;23(19):5300–5306. doi: 10.1016/j.bmcl.2013.07.071. [DOI] [PubMed] [Google Scholar]
- 46.Kortum S.W., Benson T.E., Bienkowski M.J., Emmons T.L., Prince D.B., Paddock D.J., Tomasselli A.G., Moon J.B., LaBorde A., TenBrink R.E. Potent and selective isophthalamide S2 hydroxyethylamine inhibitors of BACE1. Bioorg. Med. Chem. Lett. 2007;17(12):3378–3383. doi: 10.1016/j.bmcl.2007.03.096. [DOI] [PubMed] [Google Scholar]
- 47.Björklund C., Oscarson S., Benkestock K., Borkakoti N., Jansson K., Lindberg J., Vrang L., Hallberg A., Rosenquist A., Samuelsson B. Design and synthesis of potent and selective BACE-1 inhibitors. J. Med. Chem. 2010;53(4):1458–1464. doi: 10.1021/jm901168f. [DOI] [PubMed] [Google Scholar]
- 48.Ghosh A.K., Osswald H.L. BACE1 (β-secretase) inhibitors for the treatment of Alzheimer’s disease. Chem. Soc. Rev. 2014;43(19):6765–6813. doi: 10.1039/C3CS60460H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gerritz S.W., Zhai W., Shi S., Zhu S., Toyn J.H., Meredith J.E., Jr, Iben L.G., Burton C.R., Albright C.F., Good A.C., Tebben A.J., Muckelbauer J.K., Camac D.M., Metzler W., Cook L.S., Padmanabha R., Lentz K.A., Sofia M.J., Poss M.A., Macor J.E., Thompson L.A. III Acyl guanidine inhibitors of β-secretase (BACE-1): Optimization of a micromolar hit to a nanomolar lead via iterative solid- and solution-phase library synthesis. J. Med. Chem. 2012;55(21):9208–9223. doi: 10.1021/jm300931y. [DOI] [PubMed] [Google Scholar]
- 50.Gu T., Wu W.Y., Dong Z.X., Yu S.P., Sun Y., Zhong Y., Lu Y.T., Li N.G. Development and structural modification of BACE1 inhibitors. Molecules. 2016;22(1):22. doi: 10.3390/molecules22010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boy K.M., Guernon J.M., Wu Y.J., Zhang Y., Shi J., Zhai W., Zhu S., Gerritz S.W., Toyn J.H., Meredith J.E., Barten D.M., Burton C.R., Albright C.F., Good A.C., Grace J.E., Lentz K.A., Olson R.E., Macor J.E., Thompson L.A. III Macrocyclic prolinyl acyl guanidines as inhibitors of β-secretase (BACE). Bioorg. Med. Chem. Lett. 2015;25(22):5040–5047. doi: 10.1016/j.bmcl.2015.10.031. [DOI] [PubMed] [Google Scholar]
- 52.Brown T. Design thinking. Harv. Bus. Rev. 2008;86(6):84–92, 141. [PubMed] [Google Scholar]
- 53.Yonezawa S., Yamamoto T., Yamakawa H., Muto C., Hosono M., Hattori K., Higashino K., Yutsudo T., Iwamoto H., Kondo Y., Sakagami M., Togame H., Tanaka Y., Nakano T., Takemoto H., Arisawa M., Shuto S. Conformational restriction approach to β-secretase (BACE1) inhibitors: Effect of a cyclopropane ring to induce an alternative binding mode. J. Med. Chem. 2012;55(20):8838–8858. doi: 10.1021/jm3011405. [DOI] [PubMed] [Google Scholar]
- 54.Haghighijoo Z., Firuzi O., Hemmateenejad B., Emami S., Edraki N., Miri R. Synthesis and biological evaluation of quinazolinone-based hydrazones with potential use in Alzheimer’s disease. Bioorg. Chem. 2017;74:126–133. doi: 10.1016/j.bioorg.2017.07.014. [DOI] [PubMed] [Google Scholar]
- 55.Yan G., Hao L., Niu Y., Huang W., Wang W., Xu F., Liang L., Wang C., Jin H., Xu P. 2-Substituted-thio-N-(4-substituted-thiazol/1H-imidazol-2-yl)acetamides as BACE1 inhibitors: Synthesis, biological evaluation and docking studies. Eur. J. Med. Chem. 2017;137:462–475. doi: 10.1016/j.ejmech.2017.06.020. [DOI] [PubMed] [Google Scholar]
- 56.Iraji A., Khoshneviszadeh M., Firuzi O., Khoshneviszadeh M., Edraki N. Novel small molecule therapeutic agents for Alzheimer disease: Focusing on BACE1 and multi-target directed ligands. Bioorg. Chem. 2020;97:103649. doi: 10.1016/j.bioorg.2020.103649. [DOI] [PubMed] [Google Scholar]
- 57.Kennedy M.E., Stamford A.W., Chen X., Cox K., Cumming J.N., Dockendorf M.F., Egan M., Ereshefsky L., Hodgson R.A., Hyde L.A., Jhee S., Kleijn H.J., Kuvelkar R., Li W., Mattson B.A., Mei H., Palcza J., Scott J.D., Tanen M., Troyer M.D., Tseng J.L., Stone J.A., Parker E.M., Forman M.S. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer’s disease patients. Sci. Transl. Med. 2016;8(363):363ra150. doi: 10.1126/scitranslmed.aad9704. [DOI] [PubMed] [Google Scholar]
- 58.Egan M.F., Kost J., Tariot P.N., Aisen P.S., Cummings J.L., Vellas B., Sur C., Mukai Y., Voss T., Furtek C., Mahoney E., Harper Mozley L., Vandenberghe R., Mo Y., Michelson D. Randomized trial of verubecestat for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2018;378(18):1691–1703. doi: 10.1056/NEJMoa1706441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Piton M., Hirtz C., Desmetz C., Milhau J., Lajoix A.D., Bennys K., Lehmann S., Gabelle A. Alzheimer’s disease: Advances in drug development. J. Alzheimers Dis. 2018;65(1):3–13. doi: 10.3233/JAD-180145. [DOI] [PubMed] [Google Scholar]
- 60.Cumming J.N., Smith E.M., Wang L., Misiaszek J., Durkin J., Pan J., Iserloh U., Wu Y., Zhu Z., Strickland C., Voigt J., Chen X., Kennedy M.E., Kuvelkar R., Hyde L.A., Cox K., Favreau L., Czarniecki M.F., Greenlee W.J., McKittrick B.A., Parker E.M., Stamford A.W. Structure based design of iminohydantoin BACE1 inhibitors: Identification of an orally available, centrally active BACE1 inhibitor. Bioorg. Med. Chem. Lett. 2012;22(7):2444–2449. doi: 10.1016/j.bmcl.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 61.May P.C., Dean R.A., Lowe S.L., Martenyi F., Sheehan S.M., Boggs L.N., Monk S.A., Mathes B.M., Mergott D.J., Watson B.M., Stout S.L., Timm D.E., Smith Labell E., Gonzales C.R., Nakano M., Jhee S.S., Yen M., Ereshefsky L., Lindstrom T.D., Calligaro D.O., Cocke P.J., Greg Hall D., Friedrich S., Citron M., Audia J.E. Robust central reduction of amyloid-β in humans with an orally available, non-peptidic β-secretase inhibitor. J. Neurosci. 2011;31(46):16507–16516. doi: 10.1523/JNEUROSCI.3647-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boggs L.N., May P.C., Yang Z., Brier R.A., Monk S.A., Borders A.R., Winneroski L.L., Green S.J., Mergott D.J., McKinzie D.L. P3-035: A correlational analysis of exposure and pharmacodynamic effects of the Bace1 inhibitor LY3202626 in PDAPP mice following acute oral dosing. Alzheimers Dement. 2016;12:831–P831. doi: 10.1016/j.jalz.2016.06.1693. [DOI] [Google Scholar]
- 63.May P.C., Willis B.A., Lowe S.L., Dean R.A., Monk S.A., Cocke P.J., Audia J.E., Boggs L.N., Borders A.R., Brier R.A., Calligaro D.O., Day T.A., Ereshefsky L., Erickson J.A., Gevorkyan H., Gonzales C.R., James D.E., Jhee S.S., Komjathy S.F., Li L., Lindstrom T.D., Mathes B.M., Martényi F., Sheehan S.M., Stout S.L., Timm D.E., Vaught G.M., Watson B.M., Winneroski L.L., Yang Z., Mergott D.J. The potent BACE1 inhibitor LY2886721 elicits robust central Aβ pharmacodynamic responses in mice, dogs, and humans. J. Neurosci. 2015;35(3):1199–1210. doi: 10.1523/JNEUROSCI.4129-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cheng Y., Brown J., Judd T.C., Lopez P., Qian W., Powers T.S., Chen J.J., Bartberger M.D., Chen K., Dunn R.T., II, Epstein O., Fremeau R.T., Jr, Harried S., Hickman D., Hitchcock S.A., Luo Y., Minatti A.E., Patel V.F., Vargas H.M., Wahl R.C., Weiss M.M., Wen P.H., White R.D., Whittington D.A., Zheng X.M., Wood S. An orally available BACE1 inhibitor that affords robust CNS Aβ reduction without cardiovascular liabilities. ACS Med. Chem. Lett. 2014;6(2):210–215. doi: 10.1021/ml500458t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hilpert H., Guba W., Woltering T.J., Wostl W., Pinard E., Mauser H., Mayweg A.V., Rogers-Evans M., Humm R., Krummenacher D., Muser T., Schnider C., Jacobsen H., Ozmen L., Bergadano A., Banner D.W., Hochstrasser R., Kuglstatter A., David-Pierson P., Fischer H., Polara A., Narquizian R. β-Secretase (BACE1) inhibitors with high in vivo efficacy suitable for clinical evaluation in Alzheimer’s disease. J. Med. Chem. 2013;56(10):3980–3995. doi: 10.1021/jm400225m. [DOI] [PubMed] [Google Scholar]
- 66.Rankovic Z. CNS drug design: Balancing physicochemical properties for optimal brain exposure. J. Med. Chem. 2015;58(6):2584–2608. doi: 10.1021/jm501535r. [DOI] [PubMed] [Google Scholar]
- 67.Fuchino K., Mitsuoka Y., Masui M., Kurose N., Yoshida S., Komano K., Yamamoto T., Ogawa M., Unemura C., Hosono M., Ito H., Sakaguchi G., Ando S., Ohnishi S., Kido Y., Fukushima T., Miyajima H., Hiroyama S., Koyabu K., Dhuyvetter D., Borghys H., Gijsen H.J.M., Yamano Y., Iso Y., Kusakabe K.I. Rational design of novel 1,3-oxazine based β-secretase (BACE1) inhibitors: Incorporation of a double bond to reduce p-gp efflux leading to robust Aβ reduction in the brain. J. Med. Chem. 2018;61(12):5122–5137. doi: 10.1021/acs.jmedchem.8b00002. [DOI] [PubMed] [Google Scholar]
- 68.Ciordia M., Pérez-Benito L., Delgado F., Trabanco A.A., Tresadern G. Application of free energy perturbation for the design of BACE1 inhibitors. J. Chem. Inf. Model. 2016;56(9):1856–1871. doi: 10.1021/acs.jcim.6b00220. [DOI] [PubMed] [Google Scholar]
- 69.Keränen H., Pérez-Benito L., Ciordia M., Delgado F., Steinbrecher T.B., Oehlrich D., van Vlijmen H.W.T., Trabanco A.A., Tresadern G. Acylguanidine beta secretase 1 inhibitors: A combined experimental and free energy perturbation study. J. Chem. Theory Comput. 2017;13(3):1439–1453. doi: 10.1021/acs.jctc.6b01141. [DOI] [PubMed] [Google Scholar]
- 70.Tresadern G., Delgado F., Delgado O., Gijsen H., Macdonald G.J., Moechars D., Rombouts F., Alexander R., Spurlino J., Van Gool M., Vega J.A., Trabanco A.A. Rational design and synthesis of aminopiperazinones as β-secretase (BACE) inhibitors. Bioorg. Med. Chem. Lett. 2011;21(24):7255–7260. doi: 10.1016/j.bmcl.2011.10.050. [DOI] [PubMed] [Google Scholar]
- 71.Bower J.F., Rujirawanich J., Gallagher T. N-heterocycle construction via cyclic sulfamidates. Applications in synthesis. Org. Biomol. Chem. 2010;8(7):1505–1519. doi: 10.1039/b921842d. [DOI] [PubMed] [Google Scholar]
- 72.Oehlrich D., Peschiulli A., Tresadern G., Van Gool M., Vega J.A., De Lucas A.I., Alonso de Diego S.A., Prokopcova H., Austin N., Van Brandt S., Surkyn M., De Cleyn M., Vos A., Rombouts F.J.R., Macdonald G., Moechars D., Gijsen H.J.M., Trabanco A.A. Evaluation of a series of β-secretase 1 inhibitors containing novel heteroaryl-fused-piperazine amidine warheads. ACS Med. Chem. Lett. 2019;10(8):1159–1165. doi: 10.1021/acsmedchemlett.9b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Patel S., Vuillard L., Cleasby A., Murray C.W., Yon J. Apo and inhibitor complex structures of BACE (β-secretase). J. Mol. Biol. 2004;343(2):407–416. doi: 10.1016/j.jmb.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 74.Hong L., Tang J. Flap position of free memapsin 2 (β-secretase), a model for flap opening in aspartic protease catalysis. Biochemistry. 2004;43(16):4689–4695. doi: 10.1021/bi0498252. [DOI] [PubMed] [Google Scholar]
- 75.Ostermann N., Eder J., Eidhoff U., Zink F., Hassiepen U., Worpenberg S., Maibaum J., Simic O., Hommel U., Gerhartz B. Crystal structure of human BACE2 in complex with a hydroxyethylamine transition-state inhibitor. J. Mol. Biol. 2006;355(2):249–261. doi: 10.1016/j.jmb.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 76.Banner D.W., Gsell B., Benz J., Bertschinger J., Burger D., Brack S., Cuppuleri S., Debulpaep M., Gast A., Grabulovski D., Hennig M., Hilpert H., Huber W., Kuglstatter A., Kusznir E., Laeremans T., Matile H., Miscenic C., Rufer A.C., Schlatter D., Steyaert J., Stihle M., Thoma R., Weber M., Ruf A. Mapping the conformational space accessible to BACE2 using surface mutants and cocrystals with Fab fragments, fynomers and xaperones. Acta Crystallogr. D Biol. Crystallogr. 2013;69(Pt 6):1124–1137. doi: 10.1107/S0907444913006574. [DOI] [PubMed] [Google Scholar]
- 77.Malamas M.S., Barnes K., Johnson M., Hui Y., Zhou P., Turner J., Hu Y., Wagner E., Fan K., Chopra R., Olland A., Bard J., Pangalos M., Reinhart P., Robichaud A.J. Di-substituted pyridinyl aminohydantoins as potent and highly selective human β-secretase (BACE1) inhibitors. Bioorg. Med. Chem. 2010;18(2):630–639. doi: 10.1016/j.bmc.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 78.Fujimoto K., Matsuoka E., Asada N., Tadano G., Yamamoto T., Nakahara K., Fuchino K., Ito H., Kanegawa N., Moechars D., Gijsen H.J.M., Kusakabe K.I. Structure-based design of selective β-site amyloid precursor protein cleaving enzyme 1 (BACE1) inhibitors: Targeting the flap to gain selectivity over BACE2. J. Med. Chem. 2019;62(10):5080–5095. doi: 10.1021/acs.jmedchem.9b00309. [DOI] [PubMed] [Google Scholar]
- 79.Darras F.H., Pockes S., Huang G., Wehle S., Strasser A., Wittmann H.J., Nimczick M., Sotriffer C.A., Decker M. Synthesis, biological evaluation, and computational studies of Tri- and tetracyclic nitrogen-bridgehead compounds as potent dual-acting AChE inhibitors and hH3 receptor antagonists. ACS Chem. Neurosci. 2014;5(3):225–242. doi: 10.1021/cn4002126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Darras F.H., Wehle S., Huang G., Sotriffer C.A., Decker M. Amine substitution of quinazolinones leads to selective nanomolar AChE inhibitors with ‘inverted’ binding mode. Bioorg. Med. Chem. 2014;22(17):4867–4881. doi: 10.1016/j.bmc.2014.06.045. [DOI] [PubMed] [Google Scholar]
- 81.Ma F., Du H. Novel deoxyvasicinone derivatives as potent multitarget-directed ligands for the treatment of Alzheimer’s disease: Design, synthesis, and biological evaluation. Eur. J. Med. Chem. 2017;140:118–127. doi: 10.1016/j.ejmech.2017.09.008. [DOI] [PubMed] [Google Scholar]
- 82.Tan C.C., Yu J.T., Wang H.F., Tan M.S., Meng X.F., Wang C., Jiang T., Zhu X.C., Tan L. Efficacy and safety of donepezil, galantamine, rivastigmine, and memantine for the treatment of Alzheimer’s disease: A systematic review and meta-analysis. J. Alzheimers Dis. 2014;41(2):615–631. doi: 10.3233/JAD-132690. [DOI] [PubMed] [Google Scholar]
- 83.Shidore M., Machhi J., Shingala K., Murumkar P., Sharma M.K., Agrawal N., Tripathi A., Parikh Z., Pillai P., Yadav M.R. Benzylpiperidine-linked diarylthiazoles as potential anti-Alzheimer’s agents: synthesis and biological evaluation. J. Med. Chem. 2016;59(12):5823–5846. doi: 10.1021/acs.jmedchem.6b00426. [DOI] [PubMed] [Google Scholar]
- 84.Ismaili L., Refouvelet B., Benchekroun M., Brogi S., Brindisi M., Gemma S., Campiani G., Filipic S., Agbaba D., Esteban G., Unzeta M., Nikolic K., Butini S., Marco-Contelles J. Multitarget compounds bearing tacrine- and donepezil-like structural and functional motifs for the potential treatment of Alzheimer’s disease. Prog. Neurobiol. 2017;151:4–34. doi: 10.1016/j.pneurobio.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 85.Iraji A., Firuzi O., Khoshneviszadeh M., Tavakkoli M., Mahdavi M., Nadri H., Edraki N., Miri R. Multifunctional iminochromene-2H-carboxamide derivatives containing different aminomethylene triazole with BACE1 inhibitory, neuroprotective and metal chelating properties targeting Alzheimer’s disease. Eur. J. Med. Chem. 2017;141:690–702. doi: 10.1016/j.ejmech.2017.09.057. [DOI] [PubMed] [Google Scholar]
- 86.Edraki N., Firuzi O., Foroumadi A., Miri R., Madadkar-Sobhani A., Khoshneviszadeh M., Shafiee A. Phenylimino-2H-chromen-3-carboxamide derivatives as novel small molecule inhibitors of β-secretase (BACE1). Bioorg. Med. Chem. 2013;21(8):2396–2412. doi: 10.1016/j.bmc.2013.01.064. [DOI] [PubMed] [Google Scholar]
- 87.Du H., Liu X., Xie J., Ma F. Novel deoxyvasicinone-donepezil hybrids as potential multitarget drug candidates for Alzheimer’s disease. ACS Chem. Neurosci. 2019;10(5):2397–2407. doi: 10.1021/acschemneuro.8b00699. [DOI] [PubMed] [Google Scholar]
- 88. N-1H-Indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/9998128.
- 89.Thai N.Q., Nguyen H.L., Linh H.Q., Li M.S. Protocol for fast screening of multi-target drug candidates: Application to Alzheimer’s disease. J. Mol. Graph. Model. 2017;77:121–129. doi: 10.1016/j.jmgm.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 90.Thai N.Q., Bednarikova Z., Gancar M., Linh H.Q., Hu C.K., Li M.S., Gazova Z. Compound CID 9998128 is a potential multitarget drug for Alzheimer’s disease. ACS Chem. Neurosci. 2018;9(11):2588–2598. doi: 10.1021/acschemneuro.8b00091. [DOI] [PubMed] [Google Scholar]
- 91.Rajendran L., Schneider A., Schlechtingen G., Weidlich S., Ries J., Braxmeier T., Schwille P., Schulz J.B., Schroeder C., Simons M., Jennings G., Knölker H.J., Simons K. Efficient inhibition of the Alzheimer’s disease β-secretase by membrane targeting. Science. 2008;320(5875):520–523. doi: 10.1126/science.1156609. [DOI] [PubMed] [Google Scholar]
- 92.Gouras G.K., Tampellini D., Takahashi R.H., Capetillo-Zarate E. Intraneuronal β-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol. 2010;119(5):523–541. doi: 10.1007/s00401-010-0679-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ben Halima S., Mishra S., Raja K.M.P., Willem M., Baici A., Simons K., Brüstle O., Koch P., Haass C., Caflisch A., Rajendran L. Specific inhibition of β-secretase processing of the Alzheimer disease amyloid precursor protein. Cell Rep. 2016;14(9):2127–2141. doi: 10.1016/j.celrep.2016.01.076. [DOI] [PubMed] [Google Scholar]
- 94.Newman D.J., Cragg G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016;79(3):629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 95.Kobayashi S., Kato T., Azuma T., Kikuzaki H., Abe K. Anti-allergenic activity of polymethoxyflavones from Kaempferia parviflora. J. Funct. Foods. 2015;13:100–107. doi: 10.1016/j.jff.2014.12.029. [DOI] [Google Scholar]
- 96.Youn K., Lee J., Ho C.T., Jun M. Discovery of polymethoxyflavones from black ginger (Kaempferia parviflora) as potential β-secretase (BACE1) inhibitors. J. Funct. Foods. 2016;20:567–574. doi: 10.1016/j.jff.2015.10.036. [DOI] [Google Scholar]
- 97.Williams P., Sorribas A., Liang Z. New methods to explore marine resources for Alzheimer’s therapeutics. Curr. Alzheimer Res. 2010;7(3):210–213. doi: 10.2174/156720510791050812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mekjaruskul C., Jay M., Sripanidkulchai B. Modulatory effects of Kaempferia parviflora extract on mouse hepatic cytochrome P450 enzymes. J. Ethnopharmacol. 2012;141(3):831–839. doi: 10.1016/j.jep.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 99.Zou Z., Xu P., Zhang G., Cheng F., Chen K., Li J., Zhu W., Cao D., Xu K., Tan G. Selagintriflavonoids with BACE1 inhibitory activity from the fern Selaginella doederleinii. Phytochemistry. 2017;134:114–121. doi: 10.1016/j.phytochem.2016.11.011. [DOI] [PubMed] [Google Scholar]
- 100.Ji K.Y., Kim K.M., Kim Y.H., Im A.R., Lee J.Y., Park B., Na M., Chae S. The enhancing immune response and anti-inflammatory effects of Anemarrhena asphodeloides extract in RAW 264.7 cells. Phytomedicine. 2019;59:152789. doi: 10.1016/j.phymed.2018.12.012. [DOI] [PubMed] [Google Scholar]
- 101.Wang H.Q., Liu M., Wang L., Lan F., Zhang Y.H., Xia J.E., Xu Z.D., Zhang H. Identification of a novel BACE1 inhibitor, timosaponin A-III, for treatment of Alzheimer’s disease by a cell extraction and chemogenomics target knowledgebase-guided method. Phytomedicine. 2020;75:153244. doi: 10.1016/j.phymed.2020.153244. [DOI] [PubMed] [Google Scholar]
- 102.Hosen S.M.Z., Rubayed M., Dash R., Junaid M., Mitra S., Alam M.S., Dey R. Prospecting and structural insight into the binding of novel plant-derived molecules of Leea indica as inhibitors of BACE1. Curr. Pharm. Des. 2018;24(33):3972–3979. doi: 10.2174/1381612824666181106111020. [DOI] [PubMed] [Google Scholar]
- 103.Wong Y.H., Abdul Kadir H., Ling S.K. Bioassay-guided isolation of cytotoxic cycloartane triterpenoid glycosides from the traditionally used medicinal plant. Leea indica. Evidence-based Complement. Altern. Med. 2012;2012 doi: 10.1155/2012/164689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kaundal M., Akhtar M., Deshmukh R. Lupeol isolated from betula alnoides ameliorates amyloid beta induced neuronal damage via targeting various pathological events and alteration in neurotransmitter levels in rat’s brain. J. Neurol. Neurosci. 2017;8:3–10. doi: 10.21767/2171-6625.1000195. [DOI] [Google Scholar]
- 105.Jack C.R. Jr Alzheimer disease: New concepts on its neurobiology and the clinical role imaging will play. Radiology. 2012;263(2):344–361. doi: 10.1148/radiol.12110433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Holtzman D.M., Morris J.C., Goate A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011;3(77):77sr1. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Laird F.M., Cai H., Savonenko A.V., Farah M.H., He K., Melnikova T., Wen H., Chiang H.C., Xu G., Koliatsos V.E., Borchelt D.R., Price D.L., Lee H.K., Wong P.C. BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J. Neurosci. 2005;25(50):11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McConlogue L., Buttini M., Anderson J.P., Brigham E.F., Chen K.S., Freedman S.B., Games D., Johnson-Wood K., Lee M., Zeller M., Liu W., Motter R., Sinha S. Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP transgenic mice. J. Biol. Chem. 2007;282(36):26326–26334. doi: 10.1074/jbc.M611687200. [DOI] [PubMed] [Google Scholar]
- 109.Jack C.R., Jr, Holtzman D.M., Ad L. Biomarker modeling of Alzheimer’s disease. Neuron. 2013;80(6):1347–1358. doi: 10.1016/j.neuron.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rosén C., Hansson O., Blennow K., Zetterberg H. Fluid biomarkers in Alzheimer’s disease - current concepts. Mol. Neurodegener. 2013;8:20. doi: 10.1186/1750-1326-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]