

Abstract
Upon viral infection, cytoplasmic pattern recognition receptors detect viral nucleic acids and activate the adaptor protein VISA/MAVS- or MITA/STING-mediated innate antiviral response. Whether and how the innate antiviral response is regulated by neuronal endocrine functions is unclear. Here, we show that viral infection reduced the serum levels of the β-adrenergic hormones epinephrine and norepinephrine as well as the cellular levels of their receptors ADRB1 and ADRB2. We further show that an increase in epinephrine/norepinephrine level inhibited the innate antiviral response in an ADRB1-/2-dependent manner. Mechanistically, epinephrine/norepinephrine stimulation activated the downstream kinase PKA, which catalyzed the phosphorylation of MITA at S241, S243 and T263, inhibiting MITA activation and suppressing the innate immune response to DNA virus. In addition, phosphorylation of VISA at T54 by PKA antagonized the innate immune response to RNA virus. These findings reveal the regulatory mechanisms of innate antiviral responses by epinephrine/norepinephrine and provide a possible explanation for increased host susceptibility to viral infection in stressful and anxiety-promoting situations.
Keywords: beta-adrenoreceptor, PKA, innate immunity, virus, signal transduction
Subject terms: Innate immunity, Cell signalling
Introduction
The innate immune response is the first line of host defense against viral infection. The structurally conserved components of microbes called pathogen-associated molecular patterns (PAMPs) are recognized by host pattern-recognition receptors (PRRs), which leads to the induction of type I interferons (IFNs), proinflammatory cytokines and other downstream effector genes [1–3]. The most important viral PAMPs are viral nucleic acids, including viral DNA and/or RNA. During RNA virus infection, cytosolic viral RNA is sensed by cytoplasmic RIG-I-like receptor (RLR) family members, including retinoic acid-induced gene-I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5), which are recruited to the mitochondrion-associated adaptor protein called virus-induced signaling adaptor (VISA, also named MAVS, IPS-1 and Cardif), which transmits the signal [4–7]. VISA is a central platform and activates downstream TAK1-IKKβ and TBK1/IKKε kinases, leading to the activation of the transcription factors NF-κB and IRF3 and the induction of type I IFNs [3].
In mammalian cells, the most widely expressed and activated cytosolic viral DNA sensor is cGMP-AMP (cGAMP) synthase (cGAS) [8, 9]. The engagement of cGAS by DNA triggers a conformational change in the active site and catalyzes the synthesis of second messenger cGAMP from ATP and GTP, which in turn binds and activates the endoplasmic reticulum (ER) membrane-associated adapter protein mediator of IRF3 activation (MITA, also known as STING) [10, 11]. Activated MITA is translocated from the ER via the ER-Golgi intermediate compartment (ERGIC) to perinuclear punctate structures. In this process, MITA recruits TBK1 and IRF3, leading to the phosphorylation of IRF3 and induction of type I IFNs. Type I IFNs further induce the expression downstream antiviral genes via the JAK-STAT pathway, leading to innate antiviral responses [3, 12, 13].
Stress and anxiety increase human vulnerability to infection by viruses such as several different respiratory virus strains [14, 15]. However, the mechanisms underlying this susceptibility remain poorly understood. Under physiological conditions, acute stress or anxiety activates the hypothalamus-pituitary-adrenal cortex (HPA) axis and sympathetic nervous system (SNS), leading to the production of stress hormones, including glucocorticoids, epinephrine and norepinephrine [16–19]. Recently, the regulation of adaptive immunity and inflammation by stress hormones has been reported. For example, several studies have revealed the suppressive role of glucocorticoids in the lipopolysaccharide (LPS)-induced inflammatory response [20, 21] and T helper 1 (Th1) cell differentiation [22, 23]. Moreover, catecholamines have been shown to inhibit LPS-induced expression of cytokines and chemokines [24, 25]. Norepinephrine inhibited the production of IFN-γ and TNFα in mouse intestinal intraepithelial lymphocytes and macrophages via its receptors ADRB1/2 [25, 26]. Recently, ADRB2 signaling was reported to downregulate the production of IFN-γ in liver natural killer (NK) cells to reduce host resistance to murine cytomegalovirus (MCMV) infection [27]. However, whether stress hormones regulate innate antiviral responses is not clear.
In this study, we showed that the levels of the stress hormones epinephrine and norepinephrine and the mRNA levels of their receptors ADRB1/2 are downregulated in mice following viral infection. Furthermore, we found that the epinephrine/norepinephrine-ADRB1/2 axis activates protein kinase A (PKA), which in turn phosphorylates the innate immune signaling adaptor MITA or VISA, suppressing its activity and ultimately leading to the inhibition of innate antiviral responses. Our findings reveal a negative regulatory role of the stress hormones epinephrine and norepinephrine on the innate immune response to viral infection.
Materials and methods
Reagents, antibodies, cells and viruses
2′3′-cGAMP (tlrl-nacga23-02, Invitrogen); Lipofectamine 2000 (52887, Invitrogen); polybrene (TR-1003-G, Millipore); SYBR Green Mix (172-5274, Bio–Rad); a Dual-Specific Luciferase Assay Kit (E1980, Promega); puromycin (Thermo); geneticin (G-418) (11811-031, Gibco); RNAiso plus (9109, Takara Bio); ELISA kits for murine Ifn-β (42400, PBL), Tnf-α (430904, BioLegend), Cxcl10 (EMC121, NeoBioscience), Epinephrine (E-EL-0045c, Elabscience), Norepinephrine (E-EL-0047c, Elabscience); recombinant hIFN-β (300-02BC, PEPRO TECH); mIFN-β (8234-MB-010/CF, R&D); poly(I:C) (tlrl-pic-5, Invitrogen); herring testis (HT)-DNA (D6898, Sigma); L-epinephrine (HY-B0447B), norepinephrine (HY-13715), isoprenaline hydrochloride (HY-B0468), H89 (HY-15979), MK2206 (HY-108232), SB-203580 (HY-10256), and GDC-0994 (HY-15947) (MedChemExpress) were used in this study.
Antibodies against HA (TA180128, OriGene, 1:2000); Flag (F3165, 1:2000), β-actin (A2228, 1:10000) (Sigma); TBK1 (ab40676, 1:2000), pTBK1S172 (ab109272, 1:1000), and pIRF3S386 (ab76493, 1:1000) (Abcam); IRF3 (sc-33641, 1:1000) and STAT1 (SC-417, 1:1000) (Santa Cruz Biotechnology); STING (13647, 1:2000), pSTINGS366 (43499, 1:1000), pIRF3S396 (4947, 1:1000), pSTAT1Y701 (9167, 1:1000), ADRB1 (12271, 1:1000), and ADRB2 (8513, 1:1000) (CST); ADRB1 (AAR-023, 2.5 μg/sample) and ADRB2 (AAR-016, 2.5 μg/sample) (Alomone Labs); goat-anti-rabbit-PI (A-11012, Invitrogen, 1:1000) were used in this study. In addition, mouse anti-PKACΑ/Β antiserum against recombinant human full-length PKACB was derived as previously described [28]. Anti-phosphorylated (phospho)-MITAS241/S243 and anti-phospho-MITAT263 antibodies were prepared by the ABclonal Technology Company.
HEK293 cells and THP1 cells were obtained from ATCC.
Sendai virus (SeV), encephalomyocarditis virus (EMCV), vesicular stomatitis virus (VSV), herpes simplex virus-1 (HSV-1), and human cytomegalovirus (HCMV) were obtained previously described [29–32]. Herpes simplex virus-2 (HSV-2, ATCCVR-734) was obtained from CCTCC.
Plasmid constructs
Mammalian expression plasmids for Flag- or hemagglutinin (HA)-tagged cGAS, VISA, IRF3, PKACA/B, MITA and their mutants were constructed via standard molecular biology techniques. Guide RNA (gRNA) plasmids targeting ADRB1, ADRB2, PKACA, PKACB or MITA were constructed in a lenti-CRISPR-V2 vector, which was provided by Dr. Shu-Wen Wu (Wuhan University).
Mice
Adrb1−/−/Adrb2−/− (Adrb1- and Adrb2-double knockout (Adrb1/2-DKO) mice were purchased from GemPharmatech Co., Ltd., as previously described [33–36]. The mice were maintained in the specific-pathogen-free facility of the Medical Research Institute at Wuhan University. Eight- to ten-week-old mice were randomly allocated to each experimental group, and littermates were used as controls. Viral infection experiments were performed in animal biosafety level-2 (ABSL-2) laboratories. All animal experiments were performed in accordance with the Wuhan University Medical Research Institute Animal Care and Use Committee guidelines.
Preparation of primary mouse cells
Bone marrow-derived macrophages (BMDMs) were prepared as previously described [37–39]. Briefly, for the preparation of BMDMs, mouse bone marrow-derived monocytes (5 × 106) were cultured for 3–5 days in 100-mm dishes with 5 ml of 10% M-CSF-supplemented conditional medium from L929 cell culture.
Transfection and reporter assays
Transfection and reporter assays were performed as previously described [4, 32, 40–42]. HEK293 cells were transfected by the standard calcium phosphate precipitation method. THP1 cells were transfected with Lipofectamine 2000. To ensure that each cell received the same amount of total DNA, an empty control plasmid was included in each transfection procedure. To normalize transfection efficiency, a pRL-TK (Renilla luciferase) reporter plasmid (0.01 μg) was included in each transfection procedure. Luciferase assays were performed using a Dual-Specific Luciferase Assay Kit. Firefly luciferase activity was normalized on the basis of the Renilla luciferase activity.
Quantitative PCR
Total RNA was isolated for qPCR analysis to measure the mRNA abundance of the indicated genes. The data are shown as the relative abundance of the indicated mRNA derived from human or mouse cells and normalized to the level of GAPDH. Gene-specific primer sequences were as follows:
GAPDH: GAGTCAACGGATTTGGTCGT (forward) and
GACAAGCTTCCCGTTCTCAG (reverse);
IFNB1: TTGTTGAGAACCTCCTGGCT (forward) and
TGACTATGGTCCAGGCACAG (reverse);
CXCL10: GGTGAGAAGAGATGTCTGAATCC (forward) and
GTCCATCCTTGGAAGCACTGCA (reverse);
ISG56: GCCTTGCTGAAGTGTGGAGGAA (forward) and
ATCCAGGCGATAGGCAGAGATC (reverse);
TNFα: GCCGCATCGCCGTCTCCTAC (forward) and
CCTCAGCCCCCTCTGGGGTC (reverse);
ADRB1: GCCCTCGCCCTCGCCCTCGCC (forward) and
GCCGCTTACCCGCACGCCCG (reverse);
ADRB2: ATTGCCAAGTTCGAGCGTCTGC (forward) and
AAGAATATGGGCGGCCCCAAA (reverse);
Gapdh: ACGGCCGCATCTTCTTGTGCA (forward) and
ACGGCCAAATCCGTTCACACC (reverse);
Ifnb1: TCCTGCTGTGCTTCTCCACCACA (forward) and
AAGTCCGCCCTGTAGGTGAGGTT (reverse);
Cxcl10: ATCATCCCTGCGAGCCTATCCT (forward) and
GACCTTTTTTGGCTAAACGCTTTC (reverse);
Isg56: TACAGGCTGGAGTGTGCTGAGA (forward) and
CTCCACTTTCAGAGCCTTCGCA (reverse);
Tnfα: GGTGATCGGTCCCCAAAGGGATGA (forward) and
TGGTTTGCTACGACGTGGGCT (reverse);
Adrb1: TCATCGTGGTGGGTAACGTG (forward) and ACCAGCAATCCCATGACCAG (reverse);
Adrb2: GAGCGACTACAAACCGTCACCA (forward) and TGGAAGTCCAGAACTCGCACCA (reverse);
SeV genome: GCTCCGGATCGTTACCCATA (forward) and TCATTCCCTGTCTCAGCCTG (reverse);
EMCV genome: TCTTGGCCGCTTTGTCTAGA (forward) and TGGCTTGGTCTCGACTAGTG (reverse); and
HSV-1 genome: CGCACAGACGAAGACCTCAA (forward) and ACCACGTCTGGATTCACCAA (reverse).
CRISPR‒Cas9 knockout
The procedures for genome engineering using the CRISPR‒Cas9 system have been previously described [43, 44]. Briefly, double-stranded oligonucleotides corresponding to target sequences were cloned into s lenti-CRISPR-V2 vector and cotransfected with packaging plasmids into HEK293 cells. Two days after transfection, the viruses were harvested and used to infect THP1 cells. The infected cells were selected with puromycin (2 μg/ml) for at least 6 days. The following sequences in the indicated genes were targeted:
gNC (negative control): 5′-GTAGTCGGTACGTGACTCGT-3ʹ;
gADRB1 (#1): 5′-CCGCTTACCCGCACGCCCGT-3′;
gADRB2 (#1): 5′-TGACGTCGTGGTCCGGCGCA-3′;
gADRB1 (#2): 5′-TACGGACGAGGCGATGGCGT-3′;
gADRB2 (#2): 5′-CGTCTGCAGACGCTCGAACT-3′;
gPKACA (#1): 5′-TGAACCTTCCGATCCGCCGT-3′;
gPKACB (#1): 5′-CTGTAGTTGTACCTGTATAC-3′;
gPKACA (#2): 5′-CGATCTGGGCCGCGTAGAAA-3′;
gPKACB (#2): 5′-AAGCATACTCCAGTCGAACA-3′;
gMITA: 5′-GCTGGGACTGCTGTTAAACG-3′.
Coimmunoprecipitation and immunoblot analysis
Cells were lysed in NP-40 lysis buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mM phenylmethylsulfonyl fluoride). For each immunoprecipitation experiment, a 0.4-ml aliquot of lysate was incubated with the indicated antibody or control IgG (0.5 μg) and 30 μl of a 1:1 slurry of Protein G Sepharose beads (GE Healthcare) for 3 h. The sepharose beads were washed three times with 1 ml of lysis buffer containing 0.5 M NaCl. The precipitates were fractionated via sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‒PAGE), and immunoblot analysis was performed with the indicated antibodies.
Phosphorylation site identification by mass spectrometry
HEK293 cells were transfected with Flag-MITA and HA-PKACA/B or an empty vector. Flag-MITA was immunoprecipitated and desalted. Mass spectrometry analysis was performed as previously described by SpecAlly (Wuhan) Life Science and Technology Company [45].
Preparation of anti-phospho-MITAS241/S243 and anti-phospho-MITAT263 antibodies
Preparation of anti-phospho-MITAS241/S243 and anti-phospho-MITAT263 was performed by the ABclonal Technology Company. Briefly, peptides with C-DRVY(S-p)N(S-p)IYE or C-EYA(T-p)PLQ sequences were synthesized and conjugated to KLH for immunization of experimental Japanese white-eared rabbits. Rabbit antiserum was passed through affinity columns twice for conjugation with the synthesized control peptide (C-DRVYSNSIYE or C-EYATPLQ). The serum was then purified by affinity columns via conjugation with modified peptide C-DRVY(S-p)N(S-p)IYE or C-EYA(T-p)PLQ. The identity of the eluted anti-phospho-MITAS241/S243 and anti-phospho-MITAT263 antibodies were validated by dot blot analysis with both synthesized unmodified and modified peptides.
cGAMP affinity assay
HEK293 cells (1 × 108) were transfected with the indicated plasmids for 24 h and then lysed in NP-40 lysis buffer. After centrifugation at 13,000 rpm for 20 min, the supernatant was incubated with 2′3′-cGAMP and anti-Flag agarose beads at 4 °C for 12 h. The agarose beads were washed three times with 1 ml of ddH2O and resuspended in 0.4 ml of ddH2O before heating at 95 °C for 10 min. They were then centrifuged at 13,000 rpm for another 10 min to remove denatured proteins and agarose beads. The cGAMP level in the heat-resistant supernatants containing cGAMP were was measured with an Ultimate 3000 UHPLC Dionex (Sunnyvale, CA) coupled with a TSQ Quantiva mass spectrometer (Thermo Fisher, Waltham, MA). Chromatographic separation was performed on a Waters C18 column at 40 °C. The appropriate productions were chosen to quantify cGAMP by selective reaction monitoring (SRM).
Flow cytometry
To determine the protein levels of ADRB1 and ADRB2 in THP1 cells and BMDMs, the cells (1 × 106) were uninfected or infected with SeV or HSV-1 (MOI = 2) for 6 h. The cells were then subjected to staining with the indicated antibodies for 60 min followed by flow cytometry analysis. Data were acquired on the BD LSRFortessaX-20 instrument and analyzed with FlowJo software.
Digitonin permeabilization
cGAMP (1 μg/ml) was used to treat THP1 cells (1 × 106) pretreated with a digitonin permeabilization solution (50 mM HEPES pH 7.0, 100 mM KCl, 3 mM MgCl2, 0.1 mM DTT, 85 mM sucrose, 0.2% BSA, 1 mM ATP, 0.1 mM GTP and 2 μg/mL digitonin) at 37 °C for 20 min. Four hours later, the THP1 cells were collected and subjected to qPCR analysis.
Statistical analysis
GraphPad Prism and SPSS Statistics software was used for statistical analyses. Quantitative data in histograms are shown as the means ± SDs. Data were analyzed by the log-rank (Mantel‒Cox) test or Student’s unpaired t test. The number of asterisks represent the degree of significance with respect to the P value. Statistical significance was set to P < 0.05, * and P < 0.01, **.
Results
Viral infection downregulates the expression of epinephrine/norepinephrine and their receptors
Previously, it had been shown that stress and anxiety increase human vulnerability to viral infection [14, 15]. We investigated whether stress hormones such as epinephrine and norepinephrine are involved in the regulation of the host defense against viruses. We first measured the serum levels of epinephrine and norepinephrine in mice before and after viral infection. The results showed that the infection of mice with the RNA virus SeV or the DNA virus HSV-1 for 6 h led to decreased serum levels of epinephrine and norepinephrine (Fig. 1A). Interestingly, infection with SeV or HSV-1 also reduced the serum levels of DOPA and dopamine, which are precursors of epinephrine and norepinephrine [46] (Fig. 1B). Furthermore, qPCR showed that infection with SeV or HSV-1 reduced the mRNA levels of the epinephrine/norepinephrine receptors Adrb1 and Adrb2 in the spleen and lung of the mice (Fig. 1C). In bone marrow-derived macrophages (BMDMs), both Adrb1 and Adrb2 mRNA levels were decreased following SeV or HSV-1 infection (Fig. 1D). In addition, the mRNA levels of the ADRB1 and ADRB2 genes were reduced following SeV or HSV-1 infection in human monocytic THP1 cells (Fig. S1A). The reduction in ADRB1/2 expression following HSV-1 infection was not blocked in MITA- or p65-deficient THP1 cells (Fig. S1B), suggesting that this effect was not mediated by the IRF3 or NF-κB pathway. Furthermore, by performing flow cytometry analyses, we confirmed that infection with SeV or HSV-1 reduced the protein levels of ADRB1 and ADRB2 in both the BMDMs and THP1 cells (Figs. 1E, S1C). These results suggest that the levels of the β-adrenoreceptors ADRB1 and ADRB2 are decreased following viral infection in various immune cells and tissues of mice.
Fig. 1.

Viral infection downregulates epinephrine/norepinephrine and their receptors. A Measurement of epinephrine and norepinephrine in the serum of virus-infected mice. C57BL/6 male mice (n = 6 in each group) were uninfected or intravenously infected with SeV (5 × 107 pfu) or HSV-1 (1 × 107 pfu) for 6 h before serum was collected for use in ELISAs. B Targeted metabolomic analysis of DOPA and dopamine in the serum of SeV- or HSV-1-infected mice. C57BL/6 male mice (n = 8 in each group) were uninfected or intravenously infected with SeV (5 × 107 pfu) or HSV-1 (1 × 107 pfu), and at the indicated times, the serum was collected and target metabolomic analysis was performed by liquid chromatography‒mass spectrometry (LC‒MS). C Measurement of virus-induced transcription of Adrb1 and Adrb2 genes in mouse tissues. C57BL/6 male mice were uninfected (n = 4) or intravenously infected with SeV (5 × 107 pfu, n = 5) or HSV-1 (1 × 107 pfu, n = 5), and 6 h later, the spleen and lung tissues were collected for qPCR analysis performed to determine the mRNA levels of Adrb1 and Adrb2 genes. D Measurement of the mRNA levels of the Adrb1 and Adrb2 genes in bone marrow-derived macrophages (BMDMs). BMDMs (1 × 106) were uninfected or infected SeV or HSV-1 (MOI = 1), and 6 h later, qPCR analysis to determine the mRNA levels of the Adrb2 gene was performed. E Measurement of the protein levels of ADRB1 and ADRB2 in BMDMs. BMDMs (1 × 106) were uninfected or infected with SeV or HSV-1 (MOI = 2), and 6 h later, flow cytometry analysis with the indicated antibodies and calculation of+ mean fluorescence intensity (MFI) (shown in a histogram) was performed. The data are shown as the mean ± SD of one representative experiment performed in triplicate. The experiments were repeated two times with similar results. ns, no significance, P > 0.05; *P < 0.05; **P < 0.01
β-adrenoreceptor signaling inhibits antiviral innate immune responses
Since viral infection causes the downregulated expression of epinephrine/norepinephrine and their receptors, we next investigated whether the stress hormones epinephrine (EPI) and norepinephrine (NE) regulate innate antiviral responses. We found that epinephrine or norepinephrine stimulation inhibited SeV- or HSV-1-induced transcription of downstream antiviral genes, including IFNB1, CXCL10 and TNFα (Fig. 2A), but exerted no marked effect on the IFN-β-induced transcription of the ISG56 gene in THP1 cells (Fig. 2B). Isoprenaline (ISO), an agonist of ADRB1 and ADRB2, with a function similar to that of epinephrine and norepinephrine [47, 48], also markedly inhibited SeV- or HSV-1-induced transcription of the IFNB1 and CXCL10 genes in THP1 cells (Fig. 2C). In similar experiments, epinephrine, norepinephrine or isoprenaline treatment profoundly inhibited the transcription of the IFNB1 and CXCL10 genes induced by other viruses, including the DNA viruses HSV-2 and HCMV as well as the RNA viruses EMCV and VSV, in THP1 cells (Figs. 2C, S2A, B). Consistently, ISO inhibited the phosphorylation of MITAS366, IRF3S386 and STAT1Y701 induced by HSV-1, HSV-2 and HCMV (Figs. 2D, S2C), as well as the phosphorylation of IRF3S386 and STAT1Y701 induced by SeV, EMCV and VSV, in THP1 cells (Figs. 2D, S2D). Notably, phosphorylation of these proteins is a hallmark of the activation of downstream components in innate antiviral signaling pathways [3, 49]. Interestingly, the phosphorylation of Ser172 in TBK1, an upstream kinase that phosphorylates MITA and IRF3, was not affected or was slightly increased following SeV or HSV-1 infection (Fig. 2D). These results suggest that epinephrine/norepinephrine negatively regulated innate antiviral responses by inhibiting the activation of downstream components in innate antiviral pathways.
Fig. 2.

The β-adrenergic hormones inhibit the virus-induced innate immune response. A Effects of epinephrine (EPI) and norepinephrine (NE) on virus-induced transcription of antiviral genes. THP1 cells (1 × 106) were treated with EPI or NE (10 μM) and then uninfected or infected with SeV or HSV-1 (MOI = 1), and at the indicated times, qPCR analysis was performed to determine the levels of the indicated mRNAs. B Effects of EPI and NE on IFN-β-induced transcription of downstream genes. THP1 cells (1 × 106) were untreated or treated with IFN-β (20 ng/ml) and EPI or NE (10 μM) for 6 h, and then, qPCR analysis was performed to measure the levels of the indicated mRNAs. C Effects of EPI/NE/isoprenaline (ISO) on virus-induced transcription of antiviral genes. THP1 cells (1 × 106) were treated with ISO (10 μM) and uninfected or infected with SeV or HSV-1 (MOI = 1) for 6 h or treated with EPI, NE or ISO (10 μM) and uninfected or infected with EMCV (MOI = 1). Ten hours later, qPCR analysis was performed to determine the levels of the indicated mRNAs. D Effects of ISO on virus-induced phosphorylation of TBK1, MITA, IRF3 and STAT1. THP1 cells (1 × 106) were treated with ISO (10 μM) and uninfected or infected with SeV or HSV-1 (MOI = 2) for the indicated times, and then, immunoblot analysis with the indicated antibodies was performed. The data in A–C are shown as the mean ± SD of one representative experiment performed in triplicate. All experiments were repeated at least two times with similar results. ns, no significance, P > 0.05; *P < 0.05; **P < 0.01
We next identified the roles of epinephrine/norepinephrine in the regulation of innate antiviral immunity in vivo. We found that the intraperitoneal administration of ISO inhibited the production of serum Ifn-β that had been induced by intravenous infection with SeV, EMCV or HSV-1 (Fig. 3A). Administration of ISO also promoted SeV, EMCV or HSV-1 replication in mice (Fig. 3B). qPCR experiments indicated that ISO inhibited SeV- or HSV-1-induced transcription of the Ifnb1, Cxcl10 and Tnfα genes in the spleen and lung of the mice (Fig. 3C). Furthermore, intraperitoneal administration of ISO rendered the mice more susceptible to EMCV- or HSV-1-induced death (Fig. 3D). These results suggest that ISO negatively regulated innate antiviral responses in the mice.
Fig. 3.

Isoprenaline inhibits the virus-induced innate immune response in vivo. A Effects of ISO on virus-induced serum levels of Ifn-β. C57BL/6 male mice (n = 3 in each group) were intraperitoneally injected with PBS or ISO (5 mg/kg) and intravenously infected with SeV (5 × 107 pfu), EMCV (5 × 106 pfu) or HSV-1 (1 × 107 pfu). Four hours later, ELISAs were performed to measure serum cytokine levels. B Effects of ISO on viral replication in mice. C57BL/6 male mice (n = 5 in each group) were injected intraperitoneally with PBS or ISO (5 mg/kg) and intravenously infected with SeV (5 × 107 pfu), EMCV (5 × 106 pfu) or HSV-1 (1 × 107 pfu). Viral genomic copy numbers in the spleens of SeV-infected mice or the brains of EMCV- or HSV-1-infected mice were quantified by qPCR. C Effects of ISO on virus-induced transcription of antiviral genes in mouse tissues. C57BL/6 male mice were intraperitoneally injected with PBS or ISO (5 mg/kg) and intravenously infected with SeV (5 × 107 pfu, n = 5) or HSV-1 (1 × 107 pfu, n = 3). Four hours later, the spleen and lung tissues were collected and qPCR analysis was performed to determine the levels of the indicated mRNAs. D Effects of ISO on the virus-induced death of mice. C57BL/6 male mice were intraperitoneally injected with PBS or ISO (5 mg/kg) and intravenously infected with HSV-1 (1 × 107 pfu, n = 10) or EMCV (5 × 105 pfu, n = 14). The survival rates were recorded daily for 2 weeks. The data in (A–C) are presented as the mean ± SD. All experiments were repeated at least two times with similar results. *P < 0.05; **P < 0.01
ADRB1/2 signaling activates PKA to inhibit innate antiviral responses
We next investigated how activation of ADRB1/2 inhibits innate antiviral responses. These hormones have been shown to commonly trigger downstream signaling events via their receptors ADRB1/2, which are G-protein coupled receptors (GPCRs) [50, 51]. Ligand stimulation of these receptors triggers their recruitment of stimulating adenylate cyclase G protein (Gs), leading to the activation of multiple downstream effector proteins, such as guanine exchange protein activated by cAMP (EPAC) and several kinases, including cAMP-dependent protein kinase A (PKA), p38, extracellular signal-regulated kinase (ERK) and RAC serine/threonine-protein kinase (AKT) [52–57]. Therefore, we generated ADRB1 and ADRB2 double-deficient (gADRB1/2) human monocytic THP1 cells via the CRISPR/Cas9 method (Figs. 4A, S3A). We found that ISO treatment profoundly reduced the mRNA levels of the IFNB1, CXCL10 and TNFα genes that had been expressed by infection with SeV, EMCV or HSV-1 in control but not in ADRB1/2-double-knockout THP1 cells (Figs. 4B, S3B). In similar experiments, ISO or ADRB1/2 double knockout exerted no marked effect on the IFN-β-induced transcription of the ISG56 gene in THP1 cells (Fig. 4C). In addition, ISO inhibited the mRNA levels of the IFNB1 and CXCL10 genes induced by the transfection of the synthetic RNA analog poly(I:C) or dsDNA HT-DNA in control but not in ADRB1/2-double-knockout THP1 cells (Fig. 4D). Immunoblot experiments showed that SeV- or HSV-1-induced phosphorylation of MITAS366, IRF3S386 and STAT1Y701 but not TBK1S172 was inhibited following ISO treatment in control but not in ADRB1/2-double-knockout THP1 cells (Fig. 4E). Collectively, these results suggest that ISO induced signaling through ADRB1/2 to inhibit virus-induced innate antiviral responses.
Fig. 4.

Isoprenaline inhibits virus-induced immune response via ADRB1 and ADRB2 in THP1 cells. A The knockout efficiency of ADRB1 and ADRB2 in gNC and gADRB1/2(#1) THP1 cells was shown by immunoblotting. B Effects of ISO on virus-induced transcription of antiviral genes in gADRB1/2 (#1) and control (gNC) THP1 cells. The cells (1 × 106) were treated with ISO (10 μM) and uninfected or infected with SeV, EMCV or HSV-1 (MOI = 1) for the indicated times before qPCR analysis was performed to determine the levels of the indicated mRNAs. C Effects of ISO on IFN-β-induced transcription of downstream genes in gADRB1/2(#1) and control (gNC) THP1 cells. The cells (1 × 106) were untreated or treated with IFN-β (20 ng/ml) and ISO (10 μM), and 6 h later, qPCR analysis was performed to determine the levels of the indicated mRNAs. The data are reported as the mean ± SD, n = 3. ns not significant. D Effects of ISO on poly(I:C)- or HT-DNA-induced transcription of antiviral genes in gADRB1/2(#1) and control (gNC) THP1 cells. The cells (1 × 106) were treated with ISO (10 μM) and untransfected or transfected with poly(I:C) or HT-DNA (2 μg/ml) for the indicated times before qPCR analysis was performed to determine the levels of the indicated mRNAs. E Effects of ISO on virus-induced phosphorylation of TBK1, MITA, IRF3 and STAT1 in gADRB1/2(#1) and control (gNC) THP1 cells. The cells (1 × 106) were treated with ISO (10 μM) and uninfected or infected with SeV or HSV-1 (MOI = 2) for the indicated times before immunoblot analysis with the indicated antibodies was performed. All experiments were repeated at least two times with similar results
To determine which downstream pathways activated by ADRB1/2 are involved in the inhibition of innate antiviral responses, we examined the effects of the related kinase inhibitors on the ISO-triggered inhibition of innate antiviral responses. qPCR experiments indicated that H89, which is a specific PKA inhibitor but not inhibitors of other examined kinases, including p38, ERK or AKT, reversed ISO-triggered decreases in the mRNA levels of the IFNB1 gene, the expression of which had been induced by SeV infection, in THP1 cells (Fig. 5A). These results suggest that PKA mediated the inhibition of innate antiviral responses triggered by the epinephrine/norepinephrine-ADRB1/2 axis. The PKA catalytic subunits PKACA and PKACB are highly conserved and functionally redundant upon epinephrine/norepinephrine stimulation [58, 59]; therefore, we generated PKACA and PKACB double-deficient (gPKACA/B) human monocytic THP1 cells using the CRISPR/Cas9 method (Figs. 5B, S3C). Consistently, ISO stimulation reduced the mRNA levels of the IFNB1, CXCL10 and TNFα genes, the expression of which had been induced by SeV, EMCV or HSV-1 infection (Figs. 5B, S3D) as well as by poly(I:C) or HT-DNA transfection (Fig. 5C) in control but not in PKACA/B-double-knockout THP1 cells. In similar experiments, the level of IFN-β-induced transcription of the ISG56 gene was comparable between the control and PKACA/B-double-knockout THP1 cells (Fig. 5D). These results suggest that PKACA/B mediated the transcriptional inhibition of antiviral genes that had been triggered by either RNA or DNA virus.
Fig. 5.

Isoprenaline negatively regulates the innate antiviral response via the ADRB1/2-PKA axis. A Effects of kinase inhibitors on ISO-triggered inhibition of SeV-induced transcription of the IFNB1 gene. THP1 cells (1 × 106) were treated with the PKA inhibitor H89 (5 μM), AKT inhibitor MK2206 (2 μM), P38 inhibitor SB-203580 (1 μM) or ERK inhibitor GDC-0994 (1 μM) for 1 h and then were treated with ISO (10 μM) and infected with SeV (MOI = 1). Six hours later, a qPCR analysis to determine the mRNA levels of the IFNB1 gene was performed. The results are shown as the inhibition rate (%). B Effects of ISO on virus-induced transcription of antiviral genes in gPKACA/B (#1) and control (gNC) THP1 cells. The cells (1 × 106) were treated with ISO (10 μM) and uninfected or infected with SeV, EMCV or HSV-1 (MOI = 1) for the indicated times before qPCR analysis to determine the levels of the indicated mRNAs. The knockout efficiency of PKACA/B is shown in the immunoblots in the right panels. C Effects of ISO on poly(I:C)- or HT-DNA-induced transcription of antiviral genes in gPKACA/B(#1) and control (gNC) THP1 cells. The cells (1 × 106) were treated with ISO (10 μM) and untransfected or transfected with poly(I:C) or HT-DNA (2 μg/ml) for the indicated times before qPCR analysis was performed to determine the levels of the indicated mRNAs. D Effects of ISO on IFN-β-induced transcription of downstream genes in gPKACA/B(#1) and control (gNC) THP1 cells. The cells (1 × 106) were untreated or treated with IFN-β (20 ng/ml) and ISO (10 μM). Six hours later, qPCR analysis was performed to determine the levels of the indicated mRNAs. The data are shown as the mean ± SD (A, D) or the means (B, C) of one representative experiment performed in triplicate. All experiments were repeated at least two times with similar results. ns, not significant, **P < 0.01
PKACA/B phosphorylate MITA to inhibit its activation
We next determined the components in innate antiviral signaling pathways that are targeted by PKACA/B. Immunoblot experiments showed that ISO stimulation inhibited the RNA virus SeV-induced phosphorylation of IRF3S386 and STAT1Y701 in control but not in PKACA/B-double-knockout THP1 cells (Fig. 6A). Previously, it has been reported that PKACA/B negatively regulated RNA virus-triggered innate immune signaling by phosphorylating VISA or MAVS and priming it for MARCH5-mediated degradation [28]. Consistent with this report, we found that PKACA/B interacted with VISA (Fig. S4A) and inhibited VISA-mediated activation of ISRE (Fig. S4B). Immunoblot analysis indicated that PKACA/B catalyzed phosphorylation of VISA at T54 (Fig. S4C). Importantly, we found that phosphorylation of VISA at T54 was induced after SeV infection and was further enhanced by ISO treatment in control but not in PKACA/B-double-knockout THP1 cells (Fig. S4D). These results suggest that epinephrine/norepinephrine activated PKACA/B to phosphorylate VISA/MAVS, thereby inhibiting RNA virus-triggered innate immune responses.
Fig. 6.

PKACA/B targets MITA and mediates its phosphorylation. A Effects of ISO on virus-induced phosphorylation of TBK1, MITA, IRF3 and STAT1 in gPKACA/B(#1) and control (gNC) THP1 cells. The cells (1 × 106) were treated with ISO (10 μM) and uninfected or infected with SeV or HSV-1 (MOI = 2) for the indicated times before immunoblot analysis with the indicated antibodies was performed. B Effects of ISO on endogenous association of MITA with PKACA/B. THP1 cells (5 × 107) reconstituted with human Flag-PKACA/B were treated with ISO (10 μM) and uninfected or infected with HSV-1 (MOI = 2) for the indicated times before coimmunoprecipitation and immunoblot analysis with the indicated antibodies were performed. C HEK293 cells (1 × 106) were transfected with the indicated plasmids for 24 h before immunoblot analysis was performed with the indicated antibodies. D Effects of wild-type MITA and its mutants on ISRE activation. HEK293 cells (2 × 105) were transfected with wild-type MITA and its mutants. Twenty-four hours later, luciferase assays were performed. The data are shown as the mean ± SD, n = 3. E Effects of wild-type MITA and its mutants on HSV-1-induced phosphorylation of TBK1, MITA, IRF3 and STAT1. MITA-knockout (MITA-KO) THP1 cells (1 × 106) reconstituted with human MITA or its mutants were uninfected or infected with HSV-1 (MOI = 2). Six hours later, immunoblot analysis was performed with the indicated antibodies. F Effects of PKACA/B on the binding affinity of cGAMP for MITA and its mutants. HEK293 cells (1 × 108) were transfected with the indicated plasmids for 24 h. The cell lysates were then used for cGAMP affinity assays. The concentration of cGAMP (ng/ml) is shown in the histogram. The data are shown as the mean ± SD of one representative experiment performed in triplicate. **P < 0.01. The data shown in (A–E) are based on experiments repeated at least two times with similar results
Previously, upon sensing DNA by cGAS, activated TBK1 was demonstrated to phosphorylate MITAS366 and then IRF3S386, leading to the activation of IRF3 and the induction of downstream antiviral gene expression [12]. As shown by immunoblotting, ISO inhibited the DNA virus HSV-1-induced phosphorylation of MITAS366, IRF3S386 and STAT1Y701 in control but not in PKACA/B-double-knockout THP1 cells (Fig. 6A), suggesting that PKACA/B targeted MITA or a component upstream of MITA. Furthermore, we examined the effects of ISO on the synthesis of cGAMP in response to HT-DNA treatment as well as cGAMP-triggered signaling. As shown in Fig. S4E, F, ISO markedly inhibited cGAMP-induced transcription of the downstream IFNB1 and CXCL10 genes (Fig. S4F) but exerted little effect on HT-DNA-induced cGAMP production in THP1 cells (Fig. S4E), suggesting that ISO signaling exerted no marked effects on cGAMP synthesis (cGAS activity) but affected a component downstream of cGAMP (presumably MITA). Transient transfection and coimmunoprecipitation experiments indicated that PKACA/B interacted with MITA (Fig. S4A, G). Endogenous coimmunoprecipitation experiments confirmed that PKACA/B were associated with MITA (Fig. 6B). PKACA/B were constitutively associated with MITA in resting cells, and ISO stimulation further enhanced their association, but HSV-1 infection decreased their association (Fig. 6B). Luciferase assays showed that overexpression of PKACA or PKACB, but not their kinase-inactive mutants PKACA K73A or PKACB K73A, inhibited cGAS-MITA-mediated activation of ISRE (Fig. S4B). These data suggest that MITA is a target of PKACA/B in the ISO signaling pathway.
We next examined whether PKACA/B phosphorylate MITA. We cotransfected MITA and PKACA/B into HEK293 cells and immunoprecipitated MITA for follow-up mass spectrometry analysis. These experiments led to the identification of three phosphorylation sites at the cGAMP-binding domain (CBP) of MITA, namely, S241, S243 and T263 (Fig. S5A). To further explore the phosphorylation of endogenous MITA at these serine/threonine residues, we generated site-specific antibodies by using synthetic peptides that bear phosphorylation modifications at S241/S243 or T263 of human MITA (Fig. S5B). With these antibodies, we detected a signal of p-MITAS241/S243 or p-MITAT263 when wild-type human MITA but not MITAS241/S243D or MITAT263D was cotransfected with PKACA/B (Fig. 6C). In addition, we reconstituted human wild-type MITA and its mutants S241/243D and T263D into MITA-deficient THP1 cells via a pseudotyped retroviral-mediated gene transfer approach, and these cells were then infected with control or PKACA/B-encoding pseudotyped retrovirus. The results showed that overexpression of PKACA/B led to the phosphorylation of wild-type MITA but not that of the S241/S243D and T263D mutants, and these effects were enhanced after ISO treatment (Fig. S5C). Taken together, these results suggest that PKACA/B directly target MITA and catalyze the phosphorylation of MITA at multiple residues, including S241, S243 and T263.
Phosphorylation of MITA at S241, S243 and T263 impairs its ability to activate downstream signaling
We next investigated the biological functions of MITA phosphorylation at S241, S243 and T263. The replacement of S241/S243 or T263 of MITA with an alanine (A) residue had little effect on its ability to activate ISRE, whereas replacement of MITA S241/S243 with an aspartic acid (D) residue, which mimics phosphorylation at these residues, reduced its ability to activate downstream signaling, and the replacement of MITA T263 to an aspartic acid residue abolished its activity in ISRE reporter assays (Fig. 6D). Reconstitution experiments indicated that both MITAS241/243D and MITAT263D showed no ability to mediate HSV-1- or cGAMP-triggered induction of the downstream IFNB1 gene (Fig. S5D). In a consistent finding, reconstitution of wild-type MITA, MITAS241/S243A and MITAT263A but not MITAS241/S243D or MITAT263D rescued HSV-1-induced phosphorylation of MITAS366 and IRF3S386 (Fig. 6E), which is a hallmark of their activation [60, 61]. It had been previously shown that MITA S241, S243 and T263 are important for its cGAMP-binding ability [62–64] (Fig. S5E). We analyzed the structure of the MITA C-terminal domain (CTD) complexed with c-di-GMP (PDB ID: 4EF4) (Fig. S5E). We found that MITA S241, S243 and T263 in the cGAMP-binding domain (CBP) were structurally adjacent to c-di-GMP. Additionally, we performed affinity assays to examine the binding between cGAMP and MITA or mutant MITA in which all three serine/threonine residues (S241/S243/T263) were replaced with aspartic acid (3D). The results showed that overexpressed MITA(3D) led to a markedly reduced ability to bind cGAMP compared to wild-type MITA. Furthermore, PKACA/B markedly inhibited the binding of cGAMP to MITA but not MITA(3D) (Fig. 6F). Taken together, these results suggest that PKACA/B-mediated phosphorylation of MITA S241, S243 and T263 impaired its binding to cGAMP and its activation.
ADRB1/2 mediate the negative regulation of innate antiviral responses in mice
To investigate the roles of ADRB1/2-mediated signaling in the innate antiviral response in vivo, we generated Adrb1/2-double knockout (Adrb1/2-DKO) mice (Fig. S6A, B). qPCR experiments indicated that ISO, EPI or NE treatment inhibited the transcription of the Ifnb1, Cxcl10 and Tnfα genes that had been induced by SeV, EMCV, HSV-1 or HSV-2 in wild-type but not in Adrb1/2-DKO BMDMs (Figs. 7A, S6C, D). In similar experiments, ISO, EPI or NE treatment exerted no marked effect on Ifn-β-induced transcription of the Isg56 gene in either wild-type or Adrb1/2-DKO BMDMs (Figs. 7B, S6E). Immunoblot experiments showed that SeV- or HSV-1-induced phosphorylation of MitaS366, Irf3S386 and Stat1Y701 but not Tbk1S172 was inhibited following ISO treatment in wild-type but not in Adrb1/2-DKO BMDMs (Fig. S6F). To evaluate the roles of the epinephrine/norepinephrine-ADRB1/2 axis in innate antiviral responses in vivo, age- and sex-matched Adrb1+/−/2+/− and Adrb1−/−/2−/− mice were intraperitoneally administered ISO and then immediately intravenously infected with SeV, EMCV or HSV-1. As shown in Fig. 7C, double knockout of Adrb1 and Adrb2 reversed the inhibitory effects of ISO on SeV-, EMCV- or HSV-1-induced production of serum Ifn-β, Cxcl10 and Tnf-α⊡ In addition, Adrb1+/−/2+/− mice administered ISO were more susceptible to EMCV- or HSV-1-induced death than the PBS group, but ISO exerted no marked effect on HSV-1- or EMCV-induced death of Adrb1−/−/2−/− mice (Fig. 7D). Moreover, we found that Adrb1−/−/2−/− mice were more susceptible to HSV-1–induced death than Adrb1+/−/2+/− mice, and the fatality rate was similar to that of the ISO group Adrb1+/−/2+/− mice. It is possible that ISO regulates multiple physiological metabolic pathways through the receptors ADRB1 and ADRB2 and exerts other effects that modulate natural antiviral immune responses to DNA viruses. Collectively, these results suggest that the ISO-ADRB1/2 axis specifically inhibits innate antiviral signaling in both human and mouse immune cells and mice.
Fig. 7.
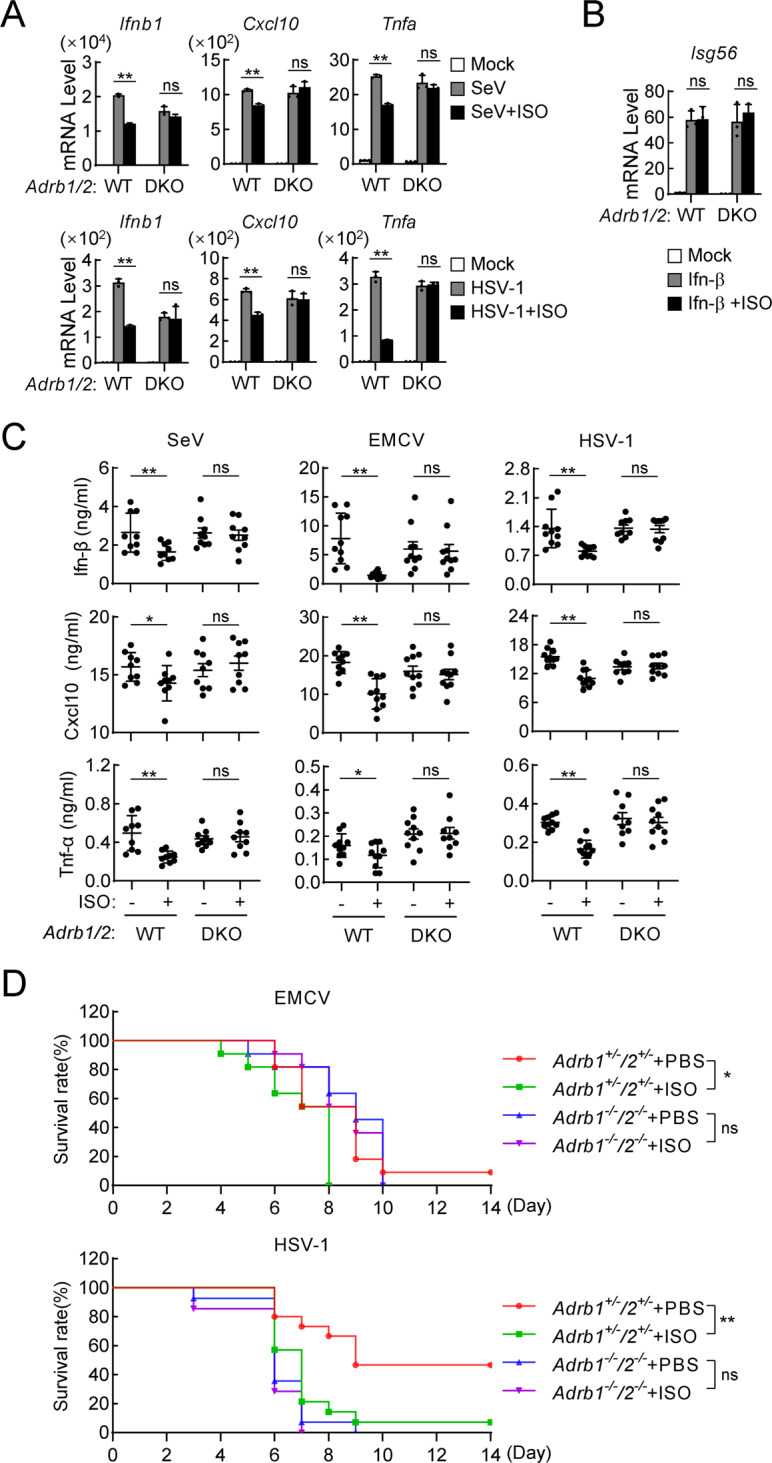
The isoprenaline-ADRB1/2 axis negatively regulates the innate antiviral response in mice. A Effects of ISO on virus-induced transcription of antiviral genes in wild-type and Adrb1/2-DKO BMDMs. Age- and sex-matched Adrb1+/+/Adrb2+/+ (WT) and Adrb1−/−/Adrb2−/− (DKO) mice were generated from Adrb1+/−/Adrb2+/− heterozygotes. BMDMs (1 × 106) from WT and Adrb1/2-DKO mice were treated with ISO (10 μM) and uninfected or infected with SeV (MOI = 1) for 6 h or HSV-1 (MOI = 1) for 3 h. Then, qPCR analysis was performed to determine the levels of the indicated mRNAs. The data are shown as the mean ± SD of one representative experiment performed in triplicate. The experiments were repeated three times with similar results. ns, not significant, **P < 0.01. B Effects of ISO on Ifn-β-induced transcription of downstream genes in WT and Adrb1/2-DKO BMDMs. BMDMs (1 × 106) from WT and Adrb1/2-DKO mice were untreated or treated with Ifn-β (100 ng/ml) and ISO (10 μM) for 6 h. Then, a qPCR analysis was performed to determine the levels of the indicated mRNAs. The data are shown as the mean ± SD of one representative experiment performed in triplicate. The experiments were repeated three times with similar results. ns, not significant. C Effects of ISO on virus-induced serum levels of Ifn-β, Cxcl10 and Tnf-α in WT and Adrb1/2-DKO mice. Age- and sex-matched WT and Adrb1/2-DKO mice (n = 9 or 10 in each group) were intraperitoneally injected with PBS or ISO (10 mg/kg) and intravenously infected with SeV (5 × 107 pfu), EMCV (5 × 106 pfu) or HSV-1 (1 × 107 pfu). Four hours later, ELISAs were performed to determine the serum cytokine levels. D Effects of ISO on the virus-induced death of mice. Age- and sex-matched Adrb1+/−/2+/− and Adrb1−/−/2−/− mice were intraperitoneally injected with PBS or ISO (10 mg/kg) and intravenously infected with HSV-1 (1 × 107 pfu, n = 14 or 15 in each group) or EMCV (5 × 106 pfu, n = 11 in each group). The survival rates were recorded daily for 2 weeks
Discussion
Stress and anxiety have been shown to increase human vulnerability to infection by viruses, including several different respiratory virus strains [14, 15], but the mechanisms of these effects remain poorly understood. In this study, we reveal a negative regulatory role of stress hormones, including epinephrine and norepinephrine, in innate antiviral responses.
Our results indicated that infection with either RNA or DNA virus reduced the serum levels of epinephrine/norepinephrine and the transcript levels of their receptors in mice. The reduction in these protein levels following viral infection was not mediated by the NF-kB or IRF3 activation pathway, and detailed mechanisms need to be investigated in future studies. We also showed that epinephrine, norepinephrine, and their clinical substitute isoprenaline inhibited the transcription of IFNB1 and other downstream antiviral genes in cells, as well as antiviral innate immune responses in mice. Furthermore, we found that these stress hormones inhibited virus-induced innate antiviral responses through the downstream kinase PKA. Knockout of PKACA/B reversed isoprenaline-triggered inhibition of virus-induced innate immune signaling and transcription of downstream antiviral genes. In our previous study, we demonstrated that PKA mediated the phosphorylation and proteasomal degradation of the adaptor protein VISA/MAVS after RNA virus-triggered innate immune signaling [28]. In this study, we demonstrated that PKA phosphorylates MITA/STING, a central adaptor protein in DNA virus-triggered innate immune signaling, at S241, S243 and T263. The phosphorylation of MITA at these residues led to the inhibition of its cGAMP binding activity and subsequent attenuation of the innate immune response to DNA virus. Our study provides a possible explanation for the higher vulnerability to viral infection due to stress and anxiety. Reporting a consistent finding, a prospective study in which the relationship of stress and recurrence of oral HSV lesions in women was assessed showed that epinephrine levels are elevated one week prior to oral lesion recurrence, providing an explanation for stress-induced reactivation of HSV [65]. It has also been shown that patients with severe enterovirus-71 (EV71) infection, involving both autonomic dysregulation and pulmonary edema, presented with significantly elevated levels of systemic catecholamines, including epinephrine and norepinephrine [66].
Our experiments suggest that viral infection downregulates Adrb1/2 expression in the lung. Since pulmonary epithelial cells can respond to viral infection, these cells may respond through stress hormone-mediated antiviral mechanisms. Recently, it has been reported that norepinephrine induced vasoconstriction and impaired leukocyte motility to suppress immune responses [67]. In light of these observations, in addition to the inhibition of innate antiviral pathways, epinephrine/norepinephrine may trigger vasoconstriction, contributing to suppression of antiviral responses in mice by impeding immune cell transmigration.
Considering our results, we propose a working model for the mechanisms for the negative regulation of innate antiviral responses by epinephrine and norepinephrine (Fig. S7). Epinephrine and norepinephrine secreted from adrenal glands and nerve terminals In an uninfected state bind to the receptors ADRB1/2 and activate the downstream kinase PKA. PKA in turn phosphorylates the innate immune signaling adaptor protein VISA at T54 after RNA virus infection or MITA at S241, S243 and T263 after DNA virus infection. This phosphorylation response suppresses the ability of VISA or MITA to trigger downstream TBK1 or IRF3 activation antiviral gene expression. Upon viral infection, the levels of epinephrine and norepinephrine as well as the levels of their receptors are markedly decreased, which drives the inhibitory effect of epinephrine and norepinephrine on VISA-/MITA-mediated antiviral innate immune signaling. How viral infection causes the decreased secretion of epinephrine and norepinephrine and the downregulation of their receptors needs to be further investigated in future studies. Nevertheless, our study reveals a conserved and common regulatory mechanism of innate antiviral responses by humoral epinephrine and norepinephrine. This mechanism might explain how type I IFNs are maintained at basal levels in an uninfected state and provide a checkpoint that licenses innate antiviral immunity in after viral infection. Manipulation of this mechanism may help to develop novel therapeutics for infectious and inflammatory diseases.
Supplementary information
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (32188101, 31830024, 31922021 and 32170713) and the CAMS Innovation Fund for Medical Sciences (2019-I2M-5-071).
Author contributions
MMH, YG, and HBS conceived and designed the study. YG, ZLR, XNZ, and SS performed the experiments. MMH, YG, and HBS analyzed all the data and wrote the manuscript.
Data availability
All the data are available in the main text or supplementary materials.
Competing interests
The authors declare no competing interests.
Contributor Information
Ming-Ming Hu, Email: mmhu@whu.edu.cn.
Hong-Bing Shu, Email: shuh@whu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41423-022-00967-x.
References
- 1.Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annu Rev Immunol. 2011;29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- 2.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 3.Hu MM, Shu HB. Cytoplasmic mechanisms of recognition and defense of microbial nucleic acids. Annu Rev Cell Dev Biol. 2018;34:357–79. doi: 10.1146/annurev-cellbio-100617-062903. [DOI] [PubMed] [Google Scholar]
- 4.Xu LG, Wang YY, Han KJ, Li LY, Zhai ZH, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–40. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 5.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–82. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 6.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–8. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 7.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–72. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 8.Sun LJ, Wu JX, Du FH, Chen X, Chen ZJJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–91. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li XD, Wu JX, Gao DX, Wang H, Sun LJ, Chen ZJJ. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science. 2013;341:1390–4. doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao FC, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–50. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–U674.. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu MM, Shu HB. Innate immune response to cytoplasmic DNA: mechanisms and diseases. Annu Rev Immunol. 2020;38:79–98. doi: 10.1146/annurev-immunol-070119-115052. [DOI] [PubMed] [Google Scholar]
- 13.Cai X, Chiu YH, Chen ZJJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell. 2014;54:289–96. doi: 10.1016/j.molcel.2014.03.040. [DOI] [PubMed] [Google Scholar]
- 14.Cohen S, Williamson GM. Stress and infectious disease in humans. Psychol Bull. 1991;109:5–24. doi: 10.1037/0033-2909.109.1.5. [DOI] [PubMed] [Google Scholar]
- 15.Glaser R, Rabin B, Chesney M, Cohen S. Stress-induced immunomodulation - Implications for infectious diseases? Jama-J Am Med Assoc. 1999;281:2268–70. doi: 10.1001/jama.281.24.2268. [DOI] [PubMed] [Google Scholar]
- 16.Herman JP. The neuroendocrinology of stress: Glucocorticoid signaling mechanisms. Psychoneuroendocrinology. 2022;137:105641. doi: 10.1016/j.psyneuen.2021.105641. [DOI] [PubMed] [Google Scholar]
- 17.Sapolsky RM, Krey LC, Mcewen BS. The neuroendocrinology of stress and aging - the glucocorticoid cascade hypothesis. Endocr Rev. 1986;7:284–301. doi: 10.1210/edrv-7-3-284. [DOI] [PubMed] [Google Scholar]
- 18.Habib KE, Gold PW, Chrousos GP. Neuroendocrinology of stress. Endocrinol Metab Clin North Am. 2001;30:695. doi: 10.1016/s0889-8529(05)70208-5. [DOI] [PubMed] [Google Scholar]
- 19.Wallace DM, Magnuson DJ, Gray TS. Organization of amygdaloid projections to brainstem dopaminergic, noradrenergic, and adrenergic cell groups in the rat. Brain Res Bull. 1992;28:447–54. doi: 10.1016/0361-9230(92)90046-z. [DOI] [PubMed] [Google Scholar]
- 20.Bhattacharyya S, Brown DE, Brewer JA, Vogt SK, Muglia LJ. Macrophage glucocorticoid receptors regulate Toll-like receptor 4-mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood. 2007;109:4313–9. doi: 10.1182/blood-2006-10-048215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li CYC, Munitic I, Mittelstadt PR, Castro E, Ashwell JD. Suppression of dendritic cell-derived IL-12 by endogenous glucocorticoids is protective in LPS-induced sepsis. Plos Biol. 2015;13:e1002269. doi: 10.1371/journal.pbio.1002269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franchimont D, Galon J, Gadina M, Visconti R, Zhou YJ, Aringer M, et al. Inhibition of Th1 immune response by glucocorticoids, dexamethasone selectively inhibits IL-12-induced Stat4 phosphorylation in T lymphocytes. J Immunol. 2000;164:1768–74. doi: 10.4049/jimmunol.164.4.1768. [DOI] [PubMed] [Google Scholar]
- 23.Chen LY, Jondal M, Yakimchuk K. Regulatory effects of dexamethasone on NK and T cell immunity. Inflammopharmacology. 2018;26:1331–8. doi: 10.1007/s10787-017-0418-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andrade-Mena CE. Catecholamines inhibit alpha/beta interferon production induced by lipopolysaccharide. Regul Pept. 1996;65:219–23. doi: 10.1016/0167-0115(96)00078-x. [DOI] [PubMed] [Google Scholar]
- 25.Ding X, Wang H, Qian X, Han X, Yang L, Cao Y, et al. Panicle-shaped sympathetic architecture in the spleen parenchyma modulates antibacterial innate immunity. Cell Rep. 2019;27:3799–3807.e3793. doi: 10.1016/j.celrep.2019.05.082. [DOI] [PubMed] [Google Scholar]
- 26.Takayanagi Y, Osawa S, Ikuma M, Takagaki K, Zhang J, Hamaya Y, et al. Norepinephrine suppresses IFN-gamma and TNF-alpha production by murine intestinal intraepithelial lymphocytes via the beta(1) adrenoceptor. J Neuroimmunol. 2012;245:66–74. doi: 10.1016/j.jneuroim.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 27.Wieduwild E, Girard-Madoux MJ, Quatrini L, Laprie C, Chasson L, Rossignol R, et al. beta2-adrenergic signals downregulate the innate immune response and reduce host resistance to viral infection. J Exp Med. 2020;217:e20190554. doi: 10.1084/jem.20190554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan BR, Zhou L, Hu MM, Li M, Lin H, Yang Y, et al. PKACs attenuate innate antiviral response by phosphorylating VISA and priming it for MARCH5-mediated degradation. PLoS Pathog. 2017;13:e1006648. doi: 10.1371/journal.ppat.1006648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu MM, Yang Q, Xie XQ, Liao CY, Lin H, Liu TT, et al. Sumoylation promotes the stability of the DNA sensor cGAS and the adaptor STING to regulate the kinetics of response to DNA virus. Immunity. 2016;45:555–69. doi: 10.1016/j.immuni.2016.08.014. [DOI] [PubMed] [Google Scholar]
- 30.Hu MM, Liao CY, Yang Q, Xie XQ, Shu HB. Innate immunity to RNA virus is regulated by temporal and reversible sumoylation of RIG-I and MDA5. J Exp Med. 2017;214:973–89. doi: 10.1084/jem.20161015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aghajanian GK, Vandermaelen CP. Intracellular identification of central noradrenergic and serotonergic neurons by a new double labeling procedure. J Neurosci. 1982;2:1786–92. doi: 10.1523/JNEUROSCI.02-12-01786.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu YZ, Su S, Gao YQ, Wang PP, Huang ZF, Hu MM, et al. Human cytomegalovirus tegument protein UL82 inhibits STING-mediated signaling to evade antiviral immunity. Cell Host Microbe. 2017;21:231–43. doi: 10.1016/j.chom.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 33.Bernstein D. Cardiovascular and metabolic alterations in mice lacking beta 1- and beta 2-adrenergic receptors. Trends Cardiovascular Med. 2002;12:287–94. doi: 10.1016/s1050-1738(02)00176-7. [DOI] [PubMed] [Google Scholar]
- 34.Chruscinski AJ, Rohrer DK, Schauble E, Desai KH, Bernstein D, Kobilka BK. Targeted disruption of the beta2 adrenergic receptor gene. J Biol Chem. 1999;274:16694–700. doi: 10.1074/jbc.274.24.16694. [DOI] [PubMed] [Google Scholar]
- 35.Rohrer DK, Chruscinski A, Schauble EH, Bernstein D, Kobilka BK. Cardiovascular and metabolic alterations in mice lacking both beta 1-and beta 2-adrenergic receptors. J Biol Chem. 1999;274:16701–8. doi: 10.1074/jbc.274.24.16701. [DOI] [PubMed] [Google Scholar]
- 36.Rohrer DK, Desai KH, Jasper JR, Stevens ME, Regula DP, Jr., Barsh GS, et al. Targeted disruption of the mouse beta1-adrenergic receptor gene: developmental and cardiovascular effects. Proc Natl Acad Sci USA. 1996;93:7375–80. doi: 10.1073/pnas.93.14.7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo WW, Li S, Li C, Lian H, Yang Q, Zhong B, et al. iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat Immunol. 2016;17:1057–66. doi: 10.1038/ni.3510. [DOI] [PubMed] [Google Scholar]
- 38.Luo WW, Li S, Li C, Zheng ZQ, Cao P, Tong Z, et al. iRhom2 is essential for innate immunity to RNA virus by antagonizing ER- and mitochondria-associated degradation of VISA. Plos Pathogens. 2017;13:e1006693. doi: 10.1371/journal.ppat.1006693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang Q, Liu TT, Lin H, Zhang M, Wei J, Luo WW, et al. TRIM32-TAX1BP1-dependent selective autophagic degradation of TRIF negatively regulates TLR3/4-mediated innate immune responses. Plos Pathogens. 2017;13:e1006600. doi: 10.1371/journal.ppat.1006600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo WW, Lian H, Zhong B, Shu HB, Li S. Kruppel-like factor 4 negatively regulates cellular antiviral immune response. Cell Mol Immunol. 2016;13:65–72. doi: 10.1038/cmi.2014.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu TT, Yang Q, Li M, Zhong B, Ran Y, Liu LL, et al. LSm14A plays a critical role in antiviral immune responses by regulating MITA level in a cell-specific manner. J Immunol. 2016;196:5101–11. doi: 10.4049/jimmunol.1600212. [DOI] [PubMed] [Google Scholar]
- 42.Lei CQ, Zhong B, Zhang Y, Zhang J, Wang S, Shu HB. Glycogen synthase kinase 3beta regulates IRF3 transcription factor-mediated antiviral response via activation of the kinase TBK1. Immunity. 2010;33:878–89. doi: 10.1016/j.immuni.2010.11.021. [DOI] [PubMed] [Google Scholar]
- 43.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–4. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–7. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shang J, Xia T, Han QQ, Zhao X, Hu MM, Shu HB, et al. Quantitative proteomics identified TTC4 as a TBK1 interactor and a positive regulator of SeV-induced innate immunity. Proteomics. 2018;18:1700403. doi: 10.1002/pmic.201700403. [DOI] [PubMed] [Google Scholar]
- 46.Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharm Rev. 2004;56:331–49. doi: 10.1124/pr.56.3.1. [DOI] [PubMed] [Google Scholar]
- 47.Degerman E, Smith CJ, Tornqvist H, Vasta V, Belfrage P, Manganiello VC. Evidence that insulin and isoprenaline activate the Cgmp-Inhibited Low-Km Camp phosphodiesterase in rat fat-cells by phosphorylation. Proc Natl Acad Sci USA. 1990;87:533–7. doi: 10.1073/pnas.87.2.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delpy E, Coste H, Gouville AC. Effects of cyclic GMP elevation on isoprenaline-induced increase in cyclic AMP and relaxation in rat aortic smooth muscle: role of phosphodiesterase 3. Br J Pharm. 1996;119:471–8. doi: 10.1111/j.1476-5381.1996.tb15696.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang Q, Shu HB. Deciphering the pathways to antiviral innate immunity and inflammation. Adv Immunol. 2020;145:1–36. doi: 10.1016/bs.ai.2019.11.001. [DOI] [PubMed] [Google Scholar]
- 50.Neves SR, Ram PT, Iyengar R. G protein pathways. Science. 2002;296:1636–9. doi: 10.1126/science.1071550. [DOI] [PubMed] [Google Scholar]
- 51.Shenoy SK, Lefkowitz RJ. beta-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharm Sci. 2011;32:521–33. doi: 10.1016/j.tips.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walsh DA, Perkins JP, Krebs EG. An adenosine 3′,5′-monophosphate-dependant protein kinase from rabbit skeletal muscle. J Biol Chem. 1968;243:3763–5. [PubMed] [Google Scholar]
- 53.de Rooij J, Zwartkruis FJT, Verheijen MHG, Cool RH, Nijman SMB, Wittinghofer A, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–7. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 54.Zheng M, Zhang SJ, Zhu WZ, Ziman B, Kobilka BK, Xiao RP. beta(2)-adrenergic receptor-induced p38 MAPK activation is mediated by protein kinase A rather than by G(i) or G beta gamma in adult mouse cardiomyocytes. J Biol Chem. 2000;275:40635–40. doi: 10.1074/jbc.M006325200. [DOI] [PubMed] [Google Scholar]
- 55.Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 56.Yano N, Ianus V, Zhao TC, Tseng A, Padbury JF, Tseng YT. A novel signaling pathway for beta-adrenergic receptor-mediated activation of phosphoinositide 3-kinase in H9c2 cardiomyocytes. Am J Physiol Heart Circ Physiol. 2007;293:H385–393. doi: 10.1152/ajpheart.01318.2006. [DOI] [PubMed] [Google Scholar]
- 57.Alcantara-Hernandez R, Hernandez-Mendez A. Adrenergic signaling molecular complexes. Gac Med Mex. 2018;154:223–35. doi: 10.24875/GMM.18002390. [DOI] [PubMed] [Google Scholar]
- 58.Tao M, Salas ML, Lipmann F. Mechanism of activation by adenosine 3′:5′-cyclic monophosphate of a protein phosphokinase from rabbit reticulocytes. Proc Natl Acad Sci USA. 1970;67:408–14. doi: 10.1073/pnas.67.1.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gill GN, Garren LD. Role of the receptor in the mechanism of action of adenosine 3′:5′-cyclic monophosphate. Proc Natl Acad Sci USA. 1971;68:786–90. doi: 10.1073/pnas.68.4.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao B, Shu C, Gao X, Sankaran B, Du F, Shelton CL, et al. Structural basis for concerted recruitment and activation of IRF-3 by innate immune adaptor proteins. Proc Natl Acad Sci USA. 2016;113:E3403–3412. doi: 10.1073/pnas.1603269113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen W, Srinath H, Lam SS, Schiffer CA, Royer WE, Jr., Lin K. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. J Mol Biol. 2008;379:251–60. doi: 10.1016/j.jmb.2008.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang YH, Liu XY, Du XX, Jiang ZF, Su XD. The structural basis for the sensing and binding of cyclic di-GMP by STING. Nat Struct Mol Biol. 2012;19:728–30. doi: 10.1038/nsmb.2333. [DOI] [PubMed] [Google Scholar]
- 63.Hong Z, Mei J, Li C, Bai G, Maimaiti M, Hu H, et al. STING inhibitors target the cyclic dinucleotide binding pocket. Proc Natl Acad Sci USA. 2021;118:e2105465118. doi: 10.1073/pnas.2105465118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ding C, Song Z, Shen A, Chen T, Zhang A. Small molecules targeting the innate immune cGASSTINGTBK1 signaling pathway. Acta Pharm Sin B. 2020;10:2272–98. doi: 10.1016/j.apsb.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Logan HL, Lutgendorf S, Hartwig A, Lilly J, Berberich SL. Immune, stress, and mood markers related to recurrent oral herpes outbreaks. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;86:48–54. doi: 10.1016/s1079-2104(98)90149-4. [DOI] [PubMed] [Google Scholar]
- 66.Fu YC, Chi CS, Chiu YT, Hsu SL, Hwang B, Jan SL, et al. Cardiac complications of enterovirus rhombencephalitis. Arch Dis Child. 2004;89:368–73. doi: 10.1136/adc.2003.029645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Devi S, Alexandre YO, Loi JK, Gillis R, Ghazanfari N, Creed SJ, et al. Adrenergic regulation of the vasculature impairs leukocyte interstitial migration and suppresses immune responses. Immunity. 2021;54:1219–30.e1217. doi: 10.1016/j.immuni.2021.03.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data are available in the main text or supplementary materials.