

Abstract
Polyribonucleotide nucleotidyltransferase 1 (Pnpt1) plays critical roles in mitochondrial homeostasis by controlling mitochondrial RNA (mt-RNA) processing, trafficking and degradation. Pnpt1 deficiency results in mitochondrial dysfunction that triggers a type I interferon response, suggesting a role in inflammation. However, the role of Pnpt1 in inflammasome activation remains largely unknown. In this study, we generated myeloid-specific Pnpt1-knockout mice and demonstrated that Pnpt1 depletion enhanced interleukin-1 beta (IL-1β) and interleukin-18 (IL-18) secretion in a mouse sepsis model. Using cultured peritoneal and bone marrow-derived macrophages, we demonstrated that Pnpt1 regulated NLRP3 inflammasome-dependent IL-1β release in response to lipopolysaccharide (LPS), followed by nigericin, ATP or poly (I:C) treatment. Pnpt1 deficiency in macrophages increased glycolysis after LPS administration and mt-reactive oxygen species (mt-ROS) after NLRP3 inflammasome activation. Pnpt1 activation of the inflammasome was dependent on increased glycolysis and the expression of mitochondrial antiviral-signaling protein (MAVS) but not NF-κB signaling. Collectively, these data suggest that Pnpt1 is an important mediator of inflammation, as shown by activation of the NLRP3 inflammasome in murine sepsis and cultured macrophages.
Keywords: Pnpt1, Inflammasome, Mitochondria, Macrophage
Subject terms: Inflammasome, Peritoneal macrophages
Introduction
Mitochondrial homeostasis has attracted much attention in studies on immunity [1, 2]. Polyribonucleotide nucleotidyltransferase 1 (Pnpt1, previously PNPase or PNPase old-35) is a mitochondria-targeted, multidomain protein with 3′-5′ exoribonuclease activity. Pnpt1 localizes to the mitochondrial matrix and the intermembrane space [3]. In the matrix, it participates in tRNA processing, polyadenylation and degradation, while in the mitochondrial intermembrane space, it is involved in the import of different RNA species from the cytoplasm [3–6]. Recent studies have shown that Pnpt1 mutation or deficiency causes mitochondrial dysfunction that is pathogenic for several diseases [7–9]. Patients carrying hypomorphic mutations in the PNPT1 gene exhibited mitochondrial dsRNA (mt-dsRNA) and cytosolic dsRNA accumulation, which triggered Type I interferon (IFN) responses in cancer cells and fibroblasts [10, 11].
NLR family pyrin domain containing 3 (NLRP3) inflammasomes are multimolecular signaling complexes involved in the development of atherosclerosis, diabetes and several immune-mediated diseases [12–14]. NLRP3 inflammasome activation in macrophages depends on two functionally distinct steps: priming and activation. The priming step, which is classically triggered by microbe-derived LPS, stimulates the transcription of the proinflammatory cytokines pro-IL-1β and pro-IL-18 and the inflammasome component NLRP3 by activating the transcription factor nuclear factor-κB (NF-ĸB). Activation is triggered by adenosine triphosphate (ATP), nigericin, monosodium urate and poly I:C and leads to NLRP3 inflammasome assembly, which is followed by caspase-1 activation and cleavage of active proinflammatory IL-1β or IL-18 [12, 15, 16]. Gasdermin-D (GSDMD) was discovered to be a key effector that is downstream of caspase-1 activation. Active caspases cleave GSDMD to generate an N-terminal cleavage product that forms transmembrane pores to enable IL-1β release and drive pyroptosis, a lytic proinflammatory type of cell death [17–19].
Mitochondrial dysfunction plays a key role in inflammation through at least three mechanisms. (1) Release of several danger-associated molecular patterns (DAMPs). Under conditions of stress, mitochondria release DAMPs, such as DNA (mt-DNA), double-stranded RNA (mt-dsRNA), and reactive oxygen species (mt-ROS), that promote inflammation [2, 20–23]. The production of mt-ROS contributes to NLRP3 inflammasome activation and plays a critical role in the pathogenesis of exaggerated inflammation [24]. Highly unstable dsRNA derived from mitochondrial damage promotes the production of proinflammatory and antiviral cytokines through cytosolic sensors known as retinoic acid-inducible gene I. Cytosolic dsRNA also activates the NLRP3 inflammasome [25, 26], highlighting the important role of Pnpt1, which degrades dsRNA. (2) Function as an essential scaffold for signaling molecules. Mitochondrial antiviral-signaling protein (MAVS) is a critical protein for the production of IFNs [27]. MAVS acts as a scaffold molecule for NLRP3 oligomerization, which drives caspase-1 activation and IL-1β secretion [21]. The depletion of MAVS abrogates the Pnpt1-mediated IFN response, suggesting that the Pnpt1-induced IFN response is also regulated through MAVS signaling [10]. Taken together, these results suggest a role for the Pnpt1-MAVS axis in the regulation of the NLRP3 inflammasome. (3) Control of metabolic reprogramming. NLRP3 inflammasome activation and IL-1β release are regulated by glycolysis and mitochondrial metabolites [2, 28, 29]. For example, inhibiting glycolysis with 2-deoxy-d-glucose (2-DG) suppressed LPS-induced IL-1β gene expression and release in macrophages [30]. Inflammation is an energy- and redox-intensive process. LPS stimulation induces a glycolytic shift by downregulating mitochondrial respiration and increasing glycolytic gene expression. This glycolytic shift provides rapid ATP production and generates nicotinamide adenine dinucleotide phosphate and glutathione to protect cells from the production of excess ROS [31, 32]. NLRP3 and its critical binding partner NIMA-related kinase 7 (NEK7) are regulated by ROS [33, 34], suggesting that macrophage metabolic reprogramming could regulate NLRP3 inflammasome activation.
Although Pnpt1 depletion results in an IFN response and mitochondrial dysfunction, the role of Pnpt1 in NLRP3 inflammasome activation is unknown. We hypothesized that Pnpt1 is an important mediator of NLRP3 inflammasome activity by regulating mitochondrial function and signaling. To address this hypothesis, we studied inflammation in myeloid cell-specific Pnpt1-knockout and wild-type mice. These mice were challenged with LPS or LPS + ATP to induce a systemic inflammatory response. The major finding was significantly increased IL-1β production in Pnpt1-knockout mice compared to wild-type mice. Mechanistically, depleting Pnpt1 in macrophages increased NLRP3 inflammasome activation, as measured by IL-1β production, through a mechanism requiring MAVS signaling and metabolic reprogramming. These findings describe a novel Pnpt1-dependent pathway in which mitochondrial dysfunction promotes NLRP3 inflammasome activation due to decreased Pnpt1 expression.
Results
Pnpt1 deletion increases LPS-induced lung inflammation and NLRP3 inflammasome activation
To examine the effects of myeloid cell Pnpt1 depletion, we generated myeloid-specific Pnpt1-knockout mice (Pnpt1m−/−) by crossing Pnpt1flox/flox mice with LysM-Cre transgenic mice. Whole-body Pnpt1 knockout in mice is lethal, but Pnpt1m−/− mice have a normal body weight and phenotype. We first used an LPS sepsis mouse model to examine the role of Pnpt1 in lung injury and systemic inflammation [35]. Sepsis is a systemic inflammatory response that can cause life-threatening organ dysfunction. Sepsis-related multiple organ dysfunction syndrome and septic shock significantly increase patient mortality [36]. LPS, which is the major causative agent of gram-negative bacteria-induced sepsis, is capable of stimulating innate immune cells and triggering inflammatory signaling cascades [37]. To model sepsis, we intraperitoneally injected mice with LPS (i.p.). After 6 h, lung tissues and peritoneal lavage fluid were collected to assess the inflammatory response. At baseline, there was no difference in inflammation or lung morphology in wild-type mice (WT) compared to Pnpt1m−/− mice (Fig. 1A). In response to LPS, there was significant lung injury, as shown by increased pulmonary edema and inflammatory cell numbers (Fig. 1A). Macrophage infiltration was significantly increased in Pnpt1m−/– mice after LPS challenge, and immunohistochemistry (IHC) showed higher expression of MAC-2 in the lung sections of Pnpt1m−/− mice than in those of WT mice (Fig. 1A, B). However, the levels of myeloperoxidase (MPO), an abundant ROS-generating enzyme present in neutrophils and macrophages, were not different in Pnpt1m−/− mice compared to WT mice after LPS challenge (Fig. 1A, C).
Fig. 1.

Pnpt1 deletion increased LPS-induced lung and systemic inflammation in vivo. Ten-week-old male Pnpt1m−/− and WT mice were sacrificed 6 h after i.p. injection of 40 mg/kg LPS or saline (control). Whole blood samples were collected, and the peritoneal cavities were washed with PBS. A Representative H&E, Mac-2, and MPO staining of lung sections. B Quantification of Mac-2 staining. C Quantification of MPO staining. ELISA analysis of IL-1β levels in (D) peritoneal lavage fluid and (E) plasma. Statistical analyses in (B–E) were performed using a 2-way ANOVA and Bonferroni’s post hoc test. N = 6 mice per group. ****P < 0.001 between the LPS-WT and LPS-Pnpt1m−/− groups. Bars represent the mean ± SEM. **p < 0.01, ***p < 0.001
To test whether Pnpt1 regulated cytokine secretion and systemic inflammation, we measured IL-1β, MCP-1 and IL-6 levels in peritoneal lavage fluid and plasma from Pnpt1m−/− and WT mice with or without LPS challenge. At baseline, there was no difference between WT and Pnpt1m−/− mice (Fig. 1D, E, Fig. S1A–D). In response to LPS, there were significantly higher levels of IL-1β and MCP-1 but not IL-6 in the peritoneal lavage fluid of Pnpt1m−/− mice than in WT mice (Fig. 1D, Fig. S1A and B). IL-1β levels in plasma were also higher in Pnpt1m−/− mice than in WT mice (Fig. 1E), but there was no difference in IL-6 or MCP-1 levels in plasma (Fig. S1C, D).
Intraperitoneal injection of LPS significantly increased the production and release of IL-6 and MCP-1 in mice. However, the secretion of IL-1β following inflammasome activation requires a second signal [38]. We an acute in vivo sepsis model that is clinically relevant to study the inflammasome pathway using a combination of LPS and ATP as a second signal [37]. To test the possibility that Pnpt1 deletion may increase inflammasome activation in vivo, Pnpt1m−/− and WT mice were injected with LPS and then ATP. Plasma and peritoneal fluid were harvested 1 h after ATP injection to measure IL-1β and IL-18 levels. In response to LPS + ATP, there were significantly increased levels of both cytokines in Pnpt1m−/− mice relative to WT mice (Fig. 2A–D). To further characterize in vivo inflammasome activation, peritoneal exudate cells were harvested for western blotting. We found that there was a twofold increase in GSDMD cleavage in Pnpt1-deficient mice in comparison to WT mice after LPS/ATP injection (Fig. S2). Furthermore, Pnpt1 deletion worsened survival after LPS/ATP challenge. (Fig. S3). Collectively, these data show that Pnpt1 deletion enhances inflammasome activation in sepsis models in vivo.
Fig. 2.

Pnpt1 deletion increased IL-1β and IL-18 production in a mouse sepsis model. Mice were injected with saline (control) or LPS (10 mg/kg) i.p. for 3 h followed by i.p. administration of ATP (100 mM in 100 µl, pH 7.4). Plasma and peritoneal fluid were harvested 1 h after ATP treatment for cytokine ELISA analysis. A IL-1β levels in plasma, (B) IL-1β levels in peritoneal fluid, (C) IL-18 levels in plasma, and (D) IL-18 levels in peritoneal fluid. (N = 5 mice from each control group, and N = 7 from each LPS + ATP group). Statistical analyses in (A–D) were performed using a 2-way ANOVA and Bonferroni’s post hoc test. *P < 0.05, **P < 0.01, ****P < 0.001 between the Pnpt1m−/− and WT groups after LPS + ATP treatment. Bars represent the mean ± SEM
Pnpt1 deficiency increases NLRP3 inflammasome activation independent of NF-κB signaling in macrophages
In vivo, we showed that Pnpt1 deficiency increased IL-1β in the peritoneal cavity after LPS + ATP injection. NLRP3 inflammasome activation requires a second signal, such as K+ efflux (stimulated by nigericin) or ATP, which induces NLRP3 oligomerization [12]. To study the role of Pnpt1 in NLRP3 inflammasome activation, we used peritoneal macrophages and mouse bone marrow-derived macrophages (BMDMs) from WT and Pnpt1m−/− mice stimulated with LPS and nigericin. IL-1β release was significantly increased in peritoneal macrophages and BMDMs from Pnpt1m−/− mice compared to WT mice (Fig. 3A, B). Similarly, in differentiated human THP-1 macrophages, in which Pnpt1 was depleted using siRNA, there was a twofold increase in IL-1β release (Fig. 3C). To further analyze the role of Pnpt1, we used ATP or poly (I:C) (a dsRNA mimic) as a second signal to induce inflammasome activation. Both stimuli significantly increased IL-1β secretion by Pnpt1m−/− BMDMs compared to WT BMDMs (Fig. 3D).
Fig. 3.

Pnpt1 deletion increased inflammasome activation independent of the NF-ĸB pathway. Macrophages from WT and Pnpt1m−/− mice were stimulated with or without LPS (100 ng/mL) for 3 h, followed by 2 µM nigericin (Nig) stimulation for 1 h. IL-1β release in the medium of (A) peritoneal macrophages, (B) BMDMs, and (C) THP-1 macrophages (control and Pnpt1 knockdown). BMDMs from WT and Pnpt1m−/− mice were stimulated with or without LPS (100 ng/mL) for 3 h and with (D) 2 µM ATP for 1 h. 10 µg/mL poly (I:C) for 24 h. IL-1β release in the medium was measured by ELISA. (Con: control). E Peritoneal macrophages were stimulated with LPS (100 ng/mL) for 1 h. NF-κB p65 translocation was measured by immunofluorescence (scale bar: 10 µm), DAPI (blue), and p65 (green). F Quantification was performed by ImageJ. N = 3 experiments. G Peritoneal macrophages were stimulated with or without LPS (100 ng/mL) for 3 h. The protein expression of NLRP3 and pro-IL-1β was analyzed by western blotting, and the images are representative of three independent experiments. H, I Peritoneal macrophages were stimulated with LPS (100 ng/mL) for 3 h and with 1 µM nigericin (Nig) for 30 min. The western blots are representative of three independent experiments. Statistical analyses in (A–D, F and I) were performed using a 2-way ANOVA and Bonferroni’s post hoc test. ****P < 0.001 between the Pnpt1m−/− and WT groups after stimulation. Bars represent the mean ± SEM
To determine the mechanism of Pnpt1-mediated inflammasome activation, we first focused on NF-κB signaling by assessing p65 nuclear translocation and the expression of Pnpt1, NLRP3 and pro-IL-1β in macrophages treated with LPS. In response to LPS (100 ng/mL), there was a 5-fold increase in the nuclear translocation of p65, which was not significantly different between Pnpt1m−/− and WT mice (Fig. 3E, F). In response to LPS, NLRP3 and pro-IL-1β expression were increased (Fig. 3G) in Pnpt1m−/− and WT macrophages, but there was no increase in Pnpt1 expression (Fig. 3G). Consistent with p65 nuclear translocation, LPS-induced NLRP3 and pro-IL-1β expression was not significantly different between Pnpt1m−/− and WT mice (Fig. 3G). Inflammasome activation induces the cleavage of pro-caspase-1 to form caspase-1. In comparison to WT cells, Pnpt1m−/− macrophages showed significantly increased cleavage of caspase-1 (Fig. 3H, I) and GSDMD (Fig. S4A, B), as well as LDH release in the medium (Fig. S4C). Furthermore, we performed experiments in which BMDMs were stimulated with nigericin after 10 min of LPS priming. Inflammasome activation was confirmed by the cleavage of GSDMD. We found that rapid priming followed by nigericin treatment induced GSDMD cleavage, and Pnpt1 deletion enhanced this effect (Fig. S5). To determine the specificity of Pnpt1 deletion on the NLRP3 inflammasome, BMDMs were stimulated with LPS for 3 h followed by transfection with 2 µg/mL flagellin (an activator of the NLRC4 inflammasome) or 2 µg/mL poly(dA:dT) (an activator of the AIM2 inflammasome) for another 3 h. We found that there were no differences between the Pnpt1WT and Pnpt1m−/− groups after NLRC4 or AIM2 inflammasome activation (Fig. S6A, B). Collectively, these findings show that Pnpt1-regulated NLRP3 inflammasome activation is independent of NF-κB signaling. Furthermore, Pnpt1 has a specific effect on the NLRP3 inflammasome but not on NLRC4 or AIM2.
Pnpt1 controls metabolic reprogramming in peritoneal macrophages
The switch from mitochondrial respiration to glycolysis is crucial for the inflammatory response in monocytes, macrophages and dendritic cells [31, 39, 40]. Knocking out Pnpt1 induces metabolic reprogramming by decreasing oxidative phosphorylation and increasing glycolysis [41]. However, the effect of Pnpt1 knockout on metabolism in the context of bacterial stimulus, such as LPS, is not clear. To investigate the role of Pnpt1 in metabolic alterations in macrophages, LPS-activated and PBS-treated peritoneal macrophages were assessed using a Seahorse XF analyzer. Mitochondrial respiration was assessed by recording the oxygen consumption rate (OCR, an index of mitochondrial respiration) and extracellular acidification rate (ECAR, an index of glycolysis). Under basal conditions, there was no difference in the ECAR between Pnpt1m−/− and WT macrophages (Fig. 4A, B). LPS stimulation significantly increased the ECAR in Pnpt1m−/− and WT macrophages, but the effects were significantly greater in Pnpt1m−/− cells than in WT cells (Fig. 4A, B). Furthermore, the maximal ECAR was greater in Pnpt1m−/− macrophages than in WT macrophages after LPS stimulation. Consistent with the ECAR data, lactate levels were significantly increased in LPS- or LPS/nigericin-treated Pnpt1m−/− macrophages compared to WT macrophages (Fig. 4D) (Fig. S7). Although the OCR was decreased less in Pnpt1m−/− macrophages compared to WT macrophages (Fig. S8A, B), the maximal OCR was similar in Pnpt1m−/− macrophages and WT macrophages after LPS stimulation (Fig. S8C). Because activated macrophages in Pnpt1m−/− mice have greatly increased glycolysis compared to those in WT mice, it is possible that Pnpt1 deletion may alter the expression of genes related to glucose metabolism. Hypoxia-inducible factor-1 alpha (HIF-1α) and glucose transporter 1 (GLUT1) participate in the regulation of glucose metabolism in LPS-treated macrophages [28, 30, 42]. Following LPS stimulation, HIF-1α and GLUT1 mRNA levels were markedly increased, and there was a significantly greater increase in Pnpt1m−/− macrophages than in WT macrophages (Fig. 4E, F). These findings demonstrate that Pnpt1 regulates metabolic reprogramming in macrophages after LPS stimulation.
Fig. 4.

Pnpt1 deletion controls metabolic reprogramming and contributes to inflammasome activation. Untreated (Con) or LPS (100 ng/mL)-activated (3 h) peritoneal macrophages from Pnpt1m−/− and WT mice were subjected to a mitochondrial stress test using a Seahorse XF analyzer. A A representative Seahorse plot of the mitochondrial stress test, as assessed by the extracellular acidification rate (ECAR), an index of glycolysis after the injection of oligomycin (1 μg/ml), carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP, 1 μM), and rotenone (1 μM) plus antimycin (1 μM). B Quantification of the results in (A); ECAR was defined as the rate at time = 0. C Quantification of the results in (A). Maximal ECAR was defined as the highest rate at any time. D In separate experiments, peritoneal macrophages from Pnpt1m−/− and WT mice were stimulated with LPS for 3 h. Lactate production in the culture supernatants was measured. E The relative gene expression of HIF1α and (F) GLUT1 in response to 100 ng/mL LPS for 3 h was measured by RT‒PCR. G Peritoneal macrophages were incubated with PBS or 10 mM 2-deoxy-D-glucose (2DG) and stimulated with LPS (100 ng/mL) for 3 h, followed by (G) 2 µM nigericin (Nig) for 1 h or (H) 10 µg/mL poly(I:C) (iC) transfection for 12 h. IL-1β levels in the medium were measured by ELISA. Statistical analyses in (B–H) were performed using a 2-way ANOVA and Bonferroni’s post hoc test. Bars represent the mean ± SEM. *p < 0.05
To determine whether Pnpt1-mediated glycolysis contributes to IL-1β release, we used two glycolytic inhibitors: (1) 2-deoxy-D-glucose (2-DG), which is a competitive inhibitor of hexokinase in the glycolytic pathway [30], and (2) heptelidic acid, a GAPDH inhibitor. Inhibiting glycolysis with 10 mM 2-DG decreased IL-1β release after LPS-nigericin or LPS-poly (I:C) treatment in BMDMs. Similarly, heptelidic acid treatment decreased IL-1β release after LPS-nigericin treatment in Pnpt1WT and Pnpt1m-/- BMDMs (Fig. S9). Statistically, there was a significant interaction between Pnpt1 and 2-DG or heptelidic acid that inhibited IL-1β release (Fig. 4G, H). Interestingly, Pnpt1-mediated enhancement of caspase-1 cleavage was not affected by 2-DG treatment (Fig. S10). These results suggest that glycolysis has a minimal effect on Pnpt1-mediated NLRP3 inflammasome activation.
Pnpt1 deletion increases inflammasome activation independent of mitochondrial ROS
Pnpt1 is located in the mitochondrial intermembrane space and regulates the expression of electron transport chain components [6], and Pnpt1 mutations impair mitochondrial function [8, 9]. To determine the role of Pnpt1-mediated mt-ROS generation in inflammasome activation, we measured mitochondrial superoxide (MitoSOX Red™ Indicator) and hydrogen peroxide (Amplex® Red Hydrogen Peroxide/Peroxidase Assay). Under baseline conditions, there was no difference in mt-ROS between Pnpt1m−/− and WT peritoneal macrophages (Fig. 5A, B). However, in response to inflammasome activation (LPS + Nig), there was an ~4-fold increase in mt-ROS, which was significantly greater in Pnpt1m−/− cells than in WT cells (Fig. 5A, B). Hydrogen peroxide production showed similar results (Fig. 5C). Mitochondrial membrane potential (ΔΨm) was analyzed by measuring JC-1 fluorescence, was similar at baseline (Fig. 5D) and was decreased following LPS + Nig treatment. The decrease was significantly greater in Pnpt1m−/− cells than in WT cells (Fig. 5D). Next, we incubated macrophages with the mitochondrial superoxide antioxidant MnTBAP (10 µM) plus LPS followed by nigericin. Exogenous MnTBAP was unable to block IL-1β secretion in either Pnpt1WT or Pnpt1m−/− macrophages after LPS-nigericin treatment (Fig. 5E). Similarly, the exogenous antioxidant NAC (500 µM) was unable to block IL-1β secretion in either Pnpt1WT or Pnpt1m−/− macrophages after LPS-Poly(I:C) treatment (Fig. 5F). These results suggest that the increase in mitochondrial ROS in Pnpt1m−/− macrophages does not contribute to inflammasome activation.
Fig. 5.

Pnpt1 deletion increased inflammasome activation independent of mitochondrial ROS. Peritoneal macrophages from Pnpt1m−/− and WT mice were primed with 100 ng/ml LPS for 3 h, followed by nigericin (2 μM) stimulation for 1 h. A Fluorescence microscopy analysis of mtROS production. Nuclei were stained with DAPI (blue) and MitoSOX (red). Scale bar, 20 μm. B MitoSOX Red fluorescence intensity was normalized to the control. C Hydrogen peroxide production was measured in the culture supernatants of untreated (Control) or LPS + Nig (Nig) activated peritoneal macrophages from Pnpt1m−/− and WT mice (n = 3 experiments with duplicate samples from each group). D Quantitative analysis of mitochondrial membrane potential was determined by measuring JC1 fluorescence in peritoneal macrophages (n = 3 experiments with duplicate samples from each group). E Peritoneal macrophages were incubated with PBS or 10 µM MnTBAP (Mn) and stimulated with LPS (100 ng/mL) for 3 h, followed by 2 µM nigericin (Nig) stimulation for 1 h. F Macrophages were incubated with 500 µM N-acetylcysteine (NAC) and stimulated with LPS (100 ng/mL) for 3 h, followed by 10 µg/mL poly(I:C) (iC) transfection for 12 h. IL-1β levels in the medium were measured by ELISA. Statistical analyses in (B–F) were performed using a 2-way ANOVA and Bonferroni’s post hoc test. Bars represent the mean ± SEM. *p < 0.05, ****p < 0.001
Pnpt1-mediated IL-1β release requires the mitochondrial antiviral-signaling protein MAVS
To determine the role of MAVS in Pnpt1-mediated IL-1β secretion, we measured the dsRNA-mediated IFN response because MAVS activation is required for this response. To determine the role of Pnpt1 deletion in macrophages, we used a monoclonal antibody (J2) that specifically detects dsRNA. We observed a strong dsRNA signal in Pnpt1-deficient macrophages (Fig. 6A, B). Pnpt1 knockdown in 293 T cells significantly increased the mRNA levels of IFN-β and interferon-induced gene 15 (ISG-15) compared to those in the control group (Fig. 6C, D). siRNA knockdown of MAVS abrogated the IFN response in Pnpt1-deficient cells (Fig. 6C, D). These data suggest that MAVS is an essential effector of the Pnpt1-mediated IFN response. Next, we determined whether Pnpt1-mediated IL-1β release requires MAVS. MAVS expression in BMDMs (Fig. 6E) or THP-1 macrophages (Fig. S11) was depleted with siRNA followed by stimulation with LPS/Nig. We showed that reducing MAVS expression decreased inflammasome activation, as measured by IL-1β release, only in Pnpt1-depleted cells (Fig. 6E). Consistent with these results, MAVS knockdown decreased Pnpt1-mediated IL-1β release after poly (I:C) treatment (Fig. 6F). Zhang, W et al. (2019) found that exogenous lactate reduced the IFN response by inhibiting MAVS activation [43]. The researchers suggested that lactate was a natural metabolite inhibitor of MAVS. We found that exogenous 10 mM lactate significantly reduced IL-1β release in Pnpt1-depleted macrophages (Fig. S12). A previous study showed that MAVS oligomerization facilitates the recruitment of NLRP3 inflammasome components to mitochondria, enhancing inflammasome formation and activity [21]. Here, we found that Pnpt1m−/− cells exhibited significantly increased MAVS oligomerization compared to WT cells under baseline conditions, but there was no MAVS oligomerization after LPS/Nig treatment (Fig. S13). To determine whether Pnpt1 deficiency triggers the IFN response in macrophages, we measured the mRNA levels of ISG-15 after cytosolic poly (I:C) treatment in LPS-primed macrophages. We found that MAVS depletion decreased Pnpt1-mediated ISG-15 gene expression in THP-1 macrophages after cytosolic poly (I:C) treatment (Fig. 6G). Collectively, these results suggest that MAVS expression is required for Pnpt1-mediated NLRP3 inflammasome activation (IL-1β release) and the IFN response (IFN-β and ISG-15 expression).
Fig. 6.

MAVS is required for Pnpt1-regulated NLRP3 inflammasome activation. A Double-stranded RNA was measured in untreated BMDMs from WT and Pnpt1m−/− mice by immunofluorescence (J2 antibody, green) and DAPI (blue), scale bar: 5 µm (B) The ratio of total J2-positive cells/DAPI-positive cells was quantified by ImageJ. HEK-293T cells were transfected with scramble (control), Pnpt1 or MAVS siRNA for 48 h, and gene expression was measured by real-time PCR. C ISG-15 and (D) IFN-β mRNA levels in cell lysates. E BMDMs were treated with scrambled or MAVS siRNA for 48 h and stimulated with LPS (100 ng/mL) for 3 h, followed by nigericin (Nig, 2 µM) stimulation for 1 h. F THP-1 macrophages were treated with scrambled, Pnpt1 or MAVS siRNA for 48 h and stimulated with LPS (100 ng/mL) for 3 h, followed by 10 µg/mL poly(I:C) transfection (iC) for 12 h. The amount of IL-1β in the medium was measured. G The amount of ISG mRNA in cell lysates after stimulation with LPS plus poly (I:C) in cells treated with scrambled or MAVS siRNA was measured (n = 4 experiments, control: con; knockdown: KD). Statistical analyses in (B) were performed using an unpaired t-test. Statistical analyses in (C–G) were performed using a 2-way ANOVA and Bonferroni’s post hoc test. Bars represent the mean ± SEM. ***p < 0.001, ****p < 0.001
Discussion
The major finding of this study is that Pnpt1 deficiency enhances NLRP3 inflammasome activation in cultured macrophages and mouse sepsis models (Fig. 7). To study the role of Pnpt1 in inflammasome signaling, we generated conditional myeloid cell lineage-specific deletion of Pnpt1 in mice (Pnpt1m−/−). We showed that Pnpt1m-/- mice that were injected with LPS or LPS + ATP had excessive lung inflammation and increased IL-1β release in plasma and peritoneal fluid compared to WT mice. Using cultured peritoneal macrophages and BMDMs, we showed that Pnpt1 deletion increased mitochondrial dysfunction after LPS + nigericin treatment and LPS-induced glycolysis. Furthermore, inhibiting glycolysis using 2-DG and heptelidic acid, as well as depleting MAVS, significantly decreased the effect of Pnpt1 on IL-1β release after nigericin or cytosolic poly(I:C) treatment in LPS-primed macrophages. These results show that Pnpt1 is a novel mediator of the mitochondria-immune response. Specifically, we found that Pnpt1 acted as a negative regulator of the NLRP3 inflammasome in a mouse sepsis model and in cultured macrophages by controlling the MAVS pathway and glucose metabolism (Fig. 6).
Fig. 7.
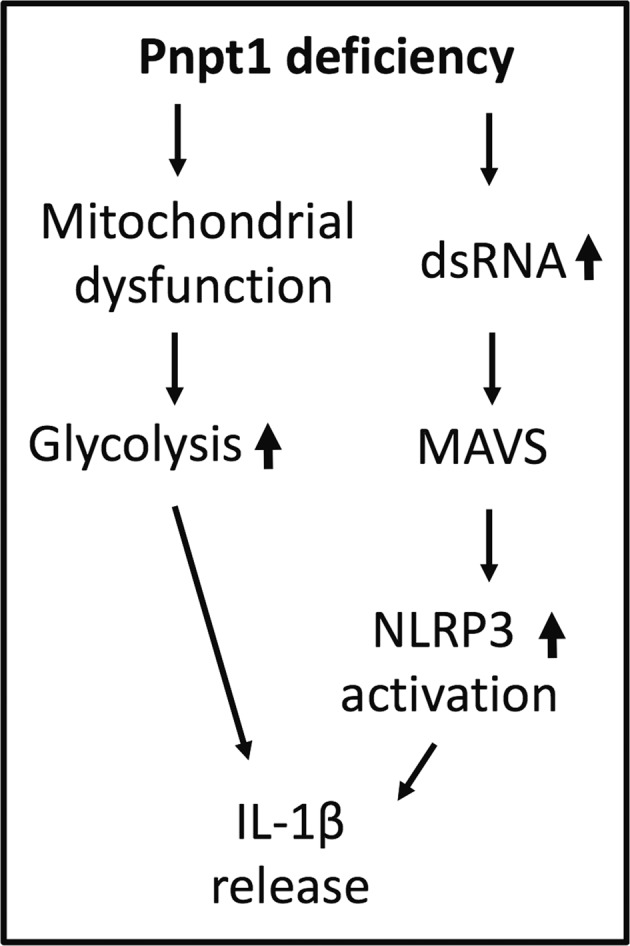
Model of Pnpt1-mediated NLRP3 inflammasome activation by MAVS and glycolysis. After stimulation with LPS and nigericin or poly(I:C), NLRP3 inflammasome activation and IL-1β release were significantly upregulated in Pnpt1-deficient macrophages. Inhibiting glycolysis or reducing MAVS expression ameliorated these effects
A novel finding was the requirement for MAVS to stimulate assembly of the NLRP3 inflammasome in the context of decreased Pnpt1. Our data are consistent with previous reports that decreased Pnpt1 stimulated the MAVS-dependent IFN response [10]. MAVS oligomerizes on the outer mitochondrial membrane to create a scaffold for the assembly of the NLRP3 inflammasome [21]. However, the role of the Pnpt1-MAVS pathway in the regulation of inflammasome activation is unknown. In this study, MAVS knockdown decreased Pnpt1-mediated IL-1β release after nigericin- or poly (I:C)-induced inflammasome activation, suggesting a Pnpt1-MAVS axis that regulates NLRP3 inflammasome activity. There may be other connections between Pnpt1 and MAVS, such as mt-dsRNA, which may be released due to increased mitochondrial membrane permeability in the context of Pnpt1 depletion [10, 44]. This could cause the accumulation of cytosolic dsRNA, triggering an MDA-5- and MAVS-dependent NLRP3 inflammasome response.
MAVS can also be activated by dsRNA-independent mechanisms. For example, the generation of mt-ROS can trigger MAVS oligomerization and mediate an inflammasome response [45, 46]. This mechanism was supported by findings that antioxidants could suppress ROS-induced MAVS oligomerization and inflammasome activation [45]. In our study, mt-ROS and mitochondrial membrane potential were further enhanced in Pnpt1-deficient cells compared to WT cells after inflammasome activation. It is possible that MAVS senses mt-ROS, which in turn promotes NLRP3 inflammasome activation.
Glycolysis is increased during inflammatory activation of immune cells, including macrophages [1, 42, 47]. LPS stimulation results in rapid transcriptional and posttranslational responses that involve genes regulating the metabolic shift from oxidative phosphorylation to glycolysis [42]. In this study, we showed that Pnpt1 deficiency increased LPS-induced lactate production and glycolysis, as well as HIF-1α gene expression and transcriptional activity, as measured by GLUT1 expression. Our observations are consistent with reports that Pnpt1 deficiency causes a glycolytic shift in stem cells [48] and fibroblasts [41]. In our study, inhibition of glycolysis with 2-DG decreased IL-1β release after LPS + nigericin or LPS + poly (I:C) treatment of BMDMs, suggesting that the hyperinflammatory phenotype in Pnpt1-deficient macrophages may involve metabolic reprogramming. HIF-1α signaling and glycolytic activity play critical roles in viral and innate immune responses. For example, HIF-1α is important for the full expression of many glycolytic genes, including GLUT1, hexokinase, and GAPDH. The increase in glycolysis and the accumulation of succinate enhance the production of IL-1β [30]. It is thus likely that the increase in IL-1β release following inflammasome activation in Pnpt1-deficient macrophages depends on a combination of glycolysis and mitochondrial signaling.
Pnpt1 controls mt-dsRNA through two mechanism.: (1) As a component of the degradosome, Pnpt1 cooperates with SUV3 and degrades mt-dsRNA antisense transcripts in the mitochondrial matrix. (2) In the mitochondrial intermembrane space, Pnpt1 prevents the release of mt-dsRNA into the cytoplasm, independent of SUV3 helicase function [49]. Pnpt1 depletion in HeLa cells leads to the accumulation of dsRNAs in mitochondria and the cytoplasm to trigger a type I IFN response through MAVS signaling [10]. We observed a strong dsRNA signal (Fig. 6A, B) and MAVS oligomerization (Fig. S13) in Pnpt1-deficient macrophages. Interestingly, no MAVS oligomerization was detected after LPS + Nig treatment (Fig. S13). Furthermore, in response to inflammasome activation, there was a general decrease in MAVS expression. This inflammasome-mediated MAVS degradation has been reported in the literature, although the mechanism is unknown [50]. Reducing MAVS expression using siRNA in BMDMs (Fig. 6E) and THP-1 macrophages (Fig. 6F), as well as inhibiting MAVS activity with exogenous lactate (Fig. S11), reversed the effect of Pnpt1 on IL-1β release after NLRP3 inflammasome activation. Taken together, these data suggest that Pnpt1 deficiency enhanced inflammasome activation through a mechanism that was different from that of nigericin or ATP. More specifically, MAVS regulates NLRP3 activation only in response to stimuli that directly activate MAVS, such as the combination of mitochondrial dysfunction and the accumulation of dsRNA.
Pnpt1 deletion in peritoneal macrophages did not affect the NF-ĸB pathway (as measured by LPS-induced p65 nuclear translocation) or NLRP3 and pro-IL-1β protein expression. These results suggest that NF-ĸB was required for the expression of inflammasome components but was not important for the observed Pnpt1-mediated effects. LPS treatment triggers IL-6 and MCP-1 secretion, but mature IL-1β production is a multistep process that is dependent on NLRP3 inflammasome activation [51]. In our mouse sepsis model, IL-1β levels in plasma and peritoneal fluid were higher in Pnpt1m−/− mice than in WT mice (Fig. 1E), but there was no difference in IL-6 or MCP-1 levels, supporting a mechanism whereby Pnpt1 regulates NLRP3 inflammasome activation independent of the NF-ĸB pathway.
Pnpt1 is evolutionarily conserved and has several well-described mitochondrial functions [52]. Increases in mt-ROS and the OCR and ECAR were expected in Pnpt1-depleted cells. The magnitude of the change at baseline was small, but the effect of Pnpt1 deletion was obvious after stimulation with LPS alone or LPS + ATP. These effects were demonstrated in cultured macrophages and in an animal model of sepsis. Because the expression of Pnpt1 can be found in many different cells, this pathway is likely to be important not only in macrophages but also in most cells activated by DAMPs that stimulate the inflammatory response. Future studies will be required to characterize the specific role of Pnpt1 in other cell types involved in inflammation and the pathogenesis of disease. Furthermore, as Pnpt1 is a exoribonuclease, it would be informative to understand whether Pnpt1 RNase activity is required for enhancing glycolysis, mitochondrial dysfunction, or inflammasome activation.
Collectively, our results indicate that Pnpt1 is involved in the regulation of NLRP3 inflammasome activation by MAVS and a glycolytic shift and strengthen the importance of mitochondrial signaling and glucose metabolism in controlling inflammation. Further understanding of the interplay between Pnpt1, mitochondrial homeostasis, and immune responses is likely to yield new insights into the pathogenesis of inflammatory diseases.
Methods
Mice
Myeloid-specific Pnpt1-knockout mice (Pnpt1m−/−) were generated by crossing Pnpt1flox/flox mice with LysM-Cre transgenic mice. All mice were on a C3H/HeJ background, maintained under specific pathogen–free conditions with free access to pellet food and water, and kept on a 12 h light–dark cycle. Ten-week-old male Pnpt1m−/− mice and Pnpt1WT littermates were used. Animal welfare and experimental procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, USA) and ethical regulations of the University of Rochester.
Chemicals and reagents
LPS (Escherichia coli O127:B8, L3129), nigericin (n7143), PMA (phorbol 12-myristate 13-acetate) (P8139), ATP (A7699), poly(I:C) (P1530-25MG), oligomycin (O4876), carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP, C2920), rotenone (R8875), antimycin (A8674), 2-DG (D6134-5G), MnTBAP (475870), N-acetyl-cysteine (A7250) and anti-dsRNA clone rJ2 (MABE1134) were purchased from Sigma‒Aldrich (St. Louis, MO). Heptelidic acid (14079) was purchased from Cayman Chemical (Ann Arbor, MI). Lipofectamine 2000 (11668027) was purchased from Thermo Fisher Scientific (Waltham, MA). Enzyme-linked immunosorbent assay (ELISA) kits for mouse IL-6 (431304), mouse MCP-1 (432704), mouse IL-1β (432604) and human IL-1β (437004) were purchased from BioLegend. Mouse IL-1β (BMS6002TWO) and mouse IL-18 (88-50618-22) ELISA kits were obtained from Thermo Fisher Scientific. Anti-mouse/human Mac-2 (Galectin-3) (CL8942AP, 1:200 dilution) was purchased from Cedarlane Laboratories. Lab Vision™ Myeloperoxidase (MPO) and rabbit polyclonal antibodies (RB-373-A, 1:300 dilution) were purchased from Thermo Fisher Scientific. Anti-Pnpt1 (14487-1-AP) was purchased from Proteintech (Rosemont, IL). Anti-NLRP3 (AG-20B-0014-C100, 1:1000 dilution) and anti-Caspase-1 (p20) (mouse) (AG-20B-0042-C100, 1:1000 dilution) were purchased from AdipoGen. NF-ĸB p65 (D14E12) XP® Rabbit mAb (8242 S, 1: 1000 dilution), β-actin (4970, 1:2000 dilution), anti-mouse IgG, HRP-linked antibodies (7076), anti-rabbit IgG, HRP-linked antibodies (7074), and anti-rodent antibodies (4983 S, 1:1000 dilution) were purchased from Cell Signaling Technology (Danvers, MA). The IL1-β antibody (GTX10750, 1:1000 dilution for immunoblotting) was purchased from GeneTex (Irvine, CA). Anti-mouse GSDMD (ab209845, 1:1000 dilution) was purchased from Abcam (Cambridge, United Kingdom). Anti-goat IgG and HRP-linked antibodies were purchased (805-035-180) from Jackson ImmunoResearch (West Grove, PA), and DAPI-Fluoromount-G (0100-20) was purchased from Southern Biotech (Birmingham, AL).
Mouse sepsis model
LPS-induced sepsis
Sepsis was induced in Pnpt1WT and Pnpt1m–/– mice by intraperitoneal (IP) injection of 40 mg/kg bacterial endotoxin (E. coli O127:B8, LPS) 6 h before harvesting. LPS/ATP-induced sepsis: Sepsis was induced in mice by IP injection of LPS (10 mg/kg); 3 h later, 100 mM/100 µl ATP per mouse (pH 7.4) (Sigma‒Aldrich, A7699) was administered by IP injection as previously described [37]. Whole blood samples were collected in tubes with 10 µL of 500 mM EDTA and centrifuged at 1000 × g for 10 min at 4 °C. Plasma was collected and aliquoted for cytokine assays. Peritoneal fluids were harvested by washing the mouse peritoneal cavity with 2 mL of ice-cold PBS supplemented with 1 mM EDTA. The cell suspension was centrifuged at 500 × g for 5 min at 4 °C, and the supernatant was collected and aliquoted for cytokine assays. The survival rate was monitored every 6 h for a total of 60 h after ATP injection.
Histopathological analysis of lung tissues
Lung tissues of mice from each group were harvested aseptically 6 h after LPS/PBS injection and fixed in 10% formalin overnight. The fixed tissues were embedded in paraffin and then sliced into 5 μm-thick sections. The sections were subjected to hematoxylin and eosin (H&E) staining or immunohistochemical (IHC) staining according to a standard protocol. H&E and IHC images were observed by light microscopy (Nikon ECLIPSE 80i). IHC staining was independently assessed by three experienced investigators. The immunoreactive score was determined by multiplying the staining intensity in four gradations by the percentage of positive cells. Thus, a score from 0 to 12 points was obtained [53].
Cell culture
Peritoneal macrophage isolation
Peritoneal macrophages were stimulated by intraperitoneally injecting mice with 1 mL of sterile 2% Bio-Gel® P Polyacrylamide Beads (Bio-Rad, 150-4174, Hercules, CA); the cells were subsequently harvested 4 days later by flushing the peritoneal cavity with 5 ml of ice-cold phosphate-buffered saline (PBS) and were plated in 6-well plates (5 × 105 cells/well) in RPMI 1640 medium (Thermo Fisher Scientific, 11875-093, Waltham, MA) supplemented with 10% fetal bovine serum (Gibco, 10437-028, Waltham, MA), 100 U/mL penicillin, 100 μg/mL streptomycin, and 1% pyruvate (Thermo Fisher Scientific, 11360070). The next day, the cells were washed with serum-free medium twice before use [54]. Bone marrow progenitor cell isolation and BMDM differentiation: BMDM preparation was performed as previously described [55]. L929 conditioned media, which contains the macrophage growth factor M-CSF, was prepared by culturing L929 cells (ATCC) in complete DMEM (Thermo Fisher Scientific, MT10013CV) supplemented with 10% FBS and 1% penicillin and streptomycin for 10 days at 37 °C in 5% CO2. The L929 conditioned media was collected, filtered (Vacuum Filter/Storage Bottle System, Corning, 431153, Corning, NY), and stored at −80 °C until needed. To isolate BMDMs, tibias and femurs were removed from male and female mice and flushed with media using a 26-gauge needle. Bone marrow was collected and centrifuged at 500 × g for 2 min at 4 °C, resuspended in complete DMEM and filtered through a 70 μm cell strainer (VWR international, 10199-657, Radnor, PA). Bone marrow progenitor cells were cultured in 100 mm dishes for 6–7 days in 70% complete DMEM and 30% L929-conditioned medium. Fresh medium (5 mL) was added on Day 3. BMDMs were collected by scraping in cold EDTA (1 mM). After centrifugation, BMDMs were seeded on 12-well plates at a density of 1.6 × 105 cells/well in DMEM and incubated overnight before use. THP-1 differentiated macrophages: Human THP-1 monocytes were differentiated into macrophages by 24 h of incubation with 100 nM PMA (Sigma‒Aldrich) in complete RPMI medium at a density of 1.6 × 105 cells/well in 12-well plates. Cells were washed twice with 1× PBS and incubated with complete RPMI medium without PMA for 24 h prior to any experiment.
LPS stimulation
Macrophage cultures were rinsed twice with medium without serum or antibiotics (RPMI 1640 for peritoneal macrophages, THP-1 macrophages, and DMEM for BMDMs). Cells were exposed to LPS (100 ng/mL) for 3 h in appropriate growth media without serum or antibiotics.
Inflammasome stimulation
To activate NLRP3 inflammasomes, cells were primed for 3 h with LPS (100 ng/mL) followed by the addition of 2 µM nigericin or 2 mM ATP for 30–60 min. To activate the NLRP3 inflammasome with cytosolic poly(I:C), macrophages were stimulated with LPS (100 ng/mL) for 3 h followed by intracellular poly(I:C) administration (10 µg/mL) by transfection using Lipofectamine 2000 (Invitrogen, 11668027) according to the manufacturer’s protocol in OptiMEM (Gibco, 31985-070) for 12–24 h. To activate AIM2 inflammasomes, BMDMs were primed with LPS (100 ng/mL) for 3 h followed by (Poly(dA:dT), 2 µg/mL) for 3 h using LyoVecTM (InvivoGen, tlrl-patc) according to the manufacturer’s protocol. To activate NLRC4 inflammasomes, BMDMs were primed with LPS (100 ng/mL) for 3 h followed by stimulation with flagellin isolated from P. aeruginosa (2 µg/mL) for 3 h using FLA-PA Ultrapure (InvivoGen, tlrl-pafla) according to the manufacturer’s protocol.
Western blotting
Cell lysates were prepared using Cell Lysis Buffer (Cell Signaling Technology, 9803 S) containing protease and phosphatase inhibitors (Sigma Aldrich, St. Louis, MO, USA), followed by centrifugation at 12,000 rpm for 10 min. Protein concentrations were measured by BCA protein assay (Pierce™ BCA Protein Assay Kit, Thermo Fisher Scientific, 23225). Cell lysates were separated by 10% SDS‒PAGE and transferred onto nitrocellulose transfer membranes. After being blocked using 5% BSA, the membranes were incubated with primary antibodies at 4 °C overnight. Reactive proteins were detected using appropriate secondary antibodies followed by chemiluminescent detection and visualization. The protein signals were quantified by ImageJ software.
Analysis of mRNA expression
RNA isolation was performed using an RNeasy mini kit (Qiagen GmbH, Hilden, Germany). A total of 1 µg of RNA was reverse transcribed using an iScript™ cDNA Synthesis Kit (Bio-Rad, 1708891, Hercules, CA, USA). Quantitative PCR amplification reactions contained a target specific fraction of cDNA and 1 μM forward and reverse primers in iQ™ SYBR® Green Supermix (Bio-Rad, 1708882) and were analyzed using real-time qPCR. β-actin was used as a control for normalization. The ΔΔCT method was used to quantify mRNA levels. The primers used were as follows: mouse β-actin (forward: TTCAACACCCCAGCCATGT, reverse: GTAGATGGGCACAGTGTGGGT), mouse GLUT1 (forward: GGTTGTGCCATACTCATGACC, reverse: CAGATAGGACATCCAGGGTAGC), mouse HIF-1α (forward: CCCATTCCTCATCCGTCA AATA, reverse: GGCTCATAACCCATCAACTCA), mouse ISG15 (forward: CATCCTGGTGAGGAACGAAAGG, reverse: CTCAGCCAGAACTGGTCTTCGT), mouse IFN-β (forward: CTCAGCCAGAACTGGTCTTCGT, reverse: CTCAGCCAGAACTGGTCTTCGT), human ISG15 (forward: CTCAGCCAGAACTGGTCTTCGT, reverse: CTCAGCCAGAACTGGTCTTCGT), human IFN-β1 (forward: ATGACCAACAAGTGTCTCCTCC, reverse: GCTCATGGAAAGAGCTGTAGTG), and human β-actin (forward: TGTCCCCCAACTTGAGATGT, reverse: TGTGCACTTTTATTCAACTGGTC).
RNA interference
Differentiated THP-1 monocytes were treated with 1 µL/mL Lipofectamine RNAiMax (Invitrogen, 13778-075) and 3 nM siRNAs in OptiMEM (Gibco, 31985-070). Human siPnpt1 (HSS131759), Human Silencer® Select MAVS (s33179), Mouse Silencer® Select MAVS (s105944), and control negative siRNA (4390844) were purchased from Thermo Fisher Scientific.
Metabolism analysis
The glycolytic and mitochondrial activity of macrophages were measured using a Seahorse extracellular flux 96 analyzer (Seahorse Bioscience). Peritoneal macrophages were seeded at a density of 1 × 105 cells/well in 100 µl RPMI 1640 medium and allowed to adhere for 2 h. After the cells were washed with PBS, 200 µl of RPMI 1640 was added, and the cells were incubated overnight. The next day, the cells were washed twice and incubated with Seahorse XF base medium (Agilent, 103334-100) (Santa Clara, CA, USA) containing 2 g/L glucose, 1X GlutaMAX (Thermo Fisher Scientific, 1798324) and 1X sodium pyruvate (Thermo Fisher Scientific, 11160070) with or without LPS for 3 h in a CO2-free incubator. The OCR and ECAR were measured following the injection of oligomycin (1 μg/ml), carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP, 1 μM), and rotenone (1 μM) plus antimycin (1 μM).
Lactate measurement
Mouse peritoneal macrophages were seeded in six-well plates (5 × 105 cells/well) and treated with or without LPS for 3 h. L-Lactate levels in the supernatant were measured using an L-Lactate Assay Kit (1200011002, Eton Bioscience, San Diego, CA, USA) according to the manufacturer’s instructions.
Detection of mitochondrial ROS
A total of 2.5 × 105 peritoneal macrophages/well were seeded in a 35 mm diameter cover glass dish (MatTek Corp. Ashland, MA). After 24 h, the cells were treated with 100 ng/ml LPS for 3 h. The culture medium was replaced with Hank’s balanced salt solution with calcium and magnesium (HBSS/Ca/Mg) containing 5 µM MitoSOX Red™ Mitochondrial Superoxide Indicator (Molecular Probes; Thermo Fisher Scientific M36008, Inc., Waltham, MA, USA), and the cells were incubated for 10 min at 37 °C. Following three washes, the cells were stimulated with nigericin (2 µM) for 30 min. The cells were fixed in 4% paraformaldehyde for 10 min. After three washes with 1× PBS, the cells were mounted using Fluoromount-G-DAPI. The cellular fluorescence (510/580 nm) intensity of 10 randomly selected cells in each sample was detected using a confocal inverted fluorescence microscope (Olympus Corporation, Tokyo, Japan) under ambient temperature. Fluorescence intensity profiles were obtained using the plot profile plugin of ImageJ software. Data were normalized against maximum intensity values in each channel. The results represent pooled data from three independent experiments, and the data were analyzed independently by two investigators to assure accuracy in the quantification process.
Determination of extracellular hydrogen peroxide production
Extracellular hydrogen peroxide was quantified using an Amplex® Red Hydrogen Peroxide/Peroxidase Assay Kit (Thermo Fisher Scientific, A22188) according to the manufacturer’s instructions. Briefly, macrophages were seeded in 6-well plates (5 × 105 cells/well) and activated with LPS for 3 h, and the supernatants were collected. Standard curve samples, controls, and experimental samples (50 µl of each) were pipetted into individual wells of a microplate, along with 100 µM Amplex® Red reagent and 0.2 U/mL HRP in a total volume of 100 µL. The samples were incubated at room temperature for 30 min in the dark. Fluorescence was measured with a microplate reader (BMG LabTech) at excitation wavelengths of 530–560 nm and an emission wavelength of 590 nm.
Mitochondrial membrane potential assay (JC-1)
Assays were performed using the JC-1 - Mitochondrial Membrane Potential Assay Kit (Abcam, ab113850, Cambridge, United Kingdom) according to the manufacturer’s instructions. After 24 h of pre‑incubation, the cells were treated with 0 or 100 ng/ml LPS for 3 h followed by nigericin (2 μM) for 1 h. The cells were washed once in dilution buffer and then stained with 20 µM JC-1 in 1X dilution buffer for 10 min at 37 °C. The cells were washed twice in 1× dilution buffer, and the fluorescence was measured (excitation at 570 nm/emission at 620 nm).
Nuclear translocation of p65
Peritoneal macrophages were seeded on glass coverslips in six-well plates and treated with LPS for 3 h. The cells were washed three times with PBS and fixed with 4% formaldehyde in PBS for 10 min. After being washed with PBS, the cells were permeabilized with 0.1% Triton X-100 in PBS for 1.5 min and incubated with blocking solution (3% BSA in PBS) for 1 h followed by incubation with the anti-p65 antibody overnight. The cells were washed three times with PBS and incubated with anti-rabbit secondary antibodies (Alexa Fluor 488; Molecular Probes) for 1 h in PBS in the dark. After being washed three times with PBS, the samples were mounted using antifade Gold reagent with DAPI (Invitrogen, P36935, Waltham, MA, USA). Immunofluorescence was detected by confocal microscopy. The colocalization of p65 and DAPI was quantified by ImageJ software. p65 translocation was quantified by comparing the ratio of the fluorescence pixel intensity in the nucleus (colocalization with DAPI) to the total cellular pixel intensity. At least 6 images were quantified for each experimental group [56].
Cytokine assays
IL-6, MCP-1, IL-1 β and IL-18 levels in plasma and peritoneal fluids were determined by ELISA according to the manufacturer’s instructions. For in vitro experiments, the culture media was cleared by centrifugation at 16,000 × g for 5 min and stored at −20 °C.
LDH assay
Culture media were collected and centrifuged at 500 × g for 5 min to remove cellular debris. LDH measurement was performed using the CyQUANT™ LDH cytotoxicity assay kit (Thermo Fisher Scientific, C20301, Waltham, MA) according to the manufacturer’s instructions. The data were plotted by normalizing the O.D. value obtained with wells treated with Triton X-100 (0.1%) as 100%.
MAVS oligomerization
For MAVS oligomer cross-linking, cells were lysed in buffer (0.5% Triton × 100, 20 mM HEPES-KOH, pH 7.5, 150 mM KCl, and complete protease and phosphatase inhibitor cocktail) on ice by passage 10 times through a G26 needle. The cell lysates were centrifuged at 6000 rpm at 4 °C for 10 min. The pellets were resuspended in PBS and crosslinked with 2 mM disuccinimidyl suberate (Thermo Fisher Scientific, 21655). The crosslinked pellets were centrifuged at 15,000 rpm for 15 min and dissolved directly in 1× SDS sample buffer.
Statistical analysis
Unless otherwise noted, in vitro experiments were repeated 3–4 times independently, with duplicate or triplicate wells averaged prior to statistical analysis. All data are presented as the mean ± SEM. GraphPad Prism 9.0 (GraphPad Software Inc., San Diego, CA, USA) was used for statistical analysis. Comparisons between two groups after stimulation were analyzed by two-way ANOVA. Some cell culture experiments were analyzed by one-way ANOVA followed by post hoc T-tests using the Bonferroni correction for multiple comparisons. P values are indicated as follows: * < 0.05, ** < 0.01, *** < 0.001, and **** < 0.0001.
Supplementary information
Acknowledgements
We thank Amanda Pereira and Sharon Senchanthisai for assistance with the maintenance and breeding of mice.
Author contributions
CGH, WJL, MS, and BCB designed the research. CGH, WJL, CLC, and MS performed the research. BCB contributed new reagents/analytical tools. CGH and WJL analyzed the data. CGH, WJL, and BCB wrote the paper.
Funding
This work was financially supported by the National Institute of Health (HL140958 to BCB), the Department of Defense (DM190884 to BCB), New York State Department of Health (C34726GG to BCB and CGH), University of Rochester Environmental Health Sciences Center (P30 ES001247 to BCB), and the National Natural Science Foundation of China (No. 82200268 to WJL).
Data availability
All data generated or analyzed during this study are included in this published paper and in its supplementary file.
Competing interests
The authors declare no competing interests.
Ethics approval
All of the experiments were approved by the University Committee on Animal Use for Research (UCAR) at the University of Rochester and followed the National Institutes of Health guidelines for experimental procedures on mice.
Footnotes
These authors contributed equally: Chia George Hsu, Wenjia Li.
Supplementary information
The online version contains supplementary material available at 10.1038/s41423-022-00962-2.
References
- 1.Timblin GA, Tharp KM, Ford B, Winchester JM, Wang J, Zhu S, et al. Mitohormesis reprogrammes macrophage metabolism to enforce tolerance. Nat Metab. 2021;3:618–35. doi: 10.1038/s42255-021-00392-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hooftman A, Angiari S, Hester S, Corcoran SE, Runtsch MC, Ling C, et al. The Immunomodulatory Metabolite Itaconate Modifies NLRP3 and Inhibits Inflammasome Activation. Cell Metab. 2020;32:468–78 e7. doi: 10.1016/j.cmet.2020.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen HW, Rainey RN, Balatoni CE, Dawson DW, Troke JJ, Wasiak S, et al. Mammalian polynucleotide phosphorylase is an intermembrane space RNase that maintains mitochondrial homeostasis. Mol Cell Biol. 2006;26:8475–87. doi: 10.1128/MCB.01002-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borowski LS, Dziembowski A, Hejnowicz MS, Stepien PP, Szczesny RJ. Human mitochondrial RNA decay mediated by PNPase-hSuv3 complex takes place in distinct foci. Nucleic Acids Res. 2013;41:1223–40. doi: 10.1093/nar/gks1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slomovic S, Schuster G. Stable PNPase RNAi silencing: its effect on the processing and adenylation of human mitochondrial RNA. Rna. 2008;14:310–23. doi: 10.1261/rna.697308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang G, Chen HW, Oktay Y, Zhang J, Allen EL, Smith GM, et al. PNPASE regulates RNA import into mitochondria. Cell. 2010;142:456–67. doi: 10.1016/j.cell.2010.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bamborschke D, Kreutzer M, Koy A, Koerber F, Lucas N, Huenseler C, et al. PNPT1 mutations may cause Aicardi-Goutieres-Syndrome. Brain Dev. 2021;43:320–4. doi: 10.1016/j.braindev.2020.10.005. [DOI] [PubMed] [Google Scholar]
- 8.von Ameln S, Wang G, Boulouiz R, Rutherford MA, Smith GM, Li Y, et al. A mutation in PNPT1, encoding mitochondrial-RNA-import protein PNPase, causes hereditary hearing loss. Am J Hum Genet. 2012;91:919–27. doi: 10.1016/j.ajhg.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alodaib A, Sobreira N, Gold WA, Riley LG, Van Bergen NJ, Wilson MJ, et al. Whole-exome sequencing identifies novel variants in PNPT1 causing oxidative phosphorylation defects and severe multisystem disease. Eur J Hum Genet. 2016;25:79–84. doi: 10.1038/ejhg.2016.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rötig A, et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature. 2018;560:238–42. doi: 10.1038/s41586-018-0363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghosh S, Guimaraes JC, Lanzafame M, Schmidt A, Syed AP, Dimitriades B, et al. Prevention of dsRNA-induced interferon signaling by AGO1x is linked to breast cancer cell proliferation. EMBO J. 2020;39:e103922. doi: 10.15252/embj.2019103922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018;17:688. [DOI] [PubMed]
- 13.Grebe A, Hoss F, Latz E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ Res. 2018;122:1722–40. doi: 10.1161/CIRCRESAHA.118.311362. [DOI] [PubMed] [Google Scholar]
- 14.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–61. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKee CM, Coll RC. NLRP3 inflammasome priming: A riddle wrapped in a mystery inside an enigma. J Leukoc Biol. 2020;108:937–52. doi: 10.1002/JLB.3MR0720-513R. [DOI] [PubMed] [Google Scholar]
- 16.Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol. 2021;18:2114–27. doi: 10.1038/s41423-021-00740-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25:1285–98. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 19.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 20.Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560:198–203. doi: 10.1038/s41586-018-0372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153:348–61. doi: 10.1016/j.cell.2013.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evavold CL, Hafner-Bratkovic I, Devant P, D’Andrea JM, Ngwa EM, Borsic E, et al. Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell. 2021;184:4495–511 e19. doi: 10.1016/j.cell.2021.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dominic A, Le NT, Takahashi M. Loop Between NLRP3 Inflammasome and Reactive Oxygen Species. Antioxid Redox Signal. 2022;36:784–96. [DOI] [PubMed]
- 24.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–5. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 25.Rajan JV, Warren SE, Miao EA, Aderem A. Activation of the NLRP3 inflammasome by intracellular poly I:C. FEBS Lett. 2010;584:4627–32. doi: 10.1016/j.febslet.2010.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem. 2006;281:36560–8. doi: 10.1074/jbc.M607594200. [DOI] [PubMed] [Google Scholar]
- 27.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–82. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 28.McGettrick AF, O’Neill LAJ. The Role of HIF in Immunity and Inflammation. Cell Metab. 2020;32:524–36. doi: 10.1016/j.cmet.2020.08.002. [DOI] [PubMed] [Google Scholar]
- 29.Hughes MM, O’Neill LAJ. Metabolic regulation of NLRP3. Immunol Rev. 2018;281:88–98. doi: 10.1111/imr.12608. [DOI] [PubMed] [Google Scholar]
- 30.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013;496:238–42. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wculek SK, Dunphy G, Heras-Murillo I, Mastrangelo A, Sancho D. Metabolism of tissue macrophages in homeostasis and pathology. Cell Mol Immunol. 2022;19:384–408. doi: 10.1038/s41423-021-00791-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Virag L, Jaen RI, Regdon Z, Bosca L, Prieto P. Self-defense of macrophages against oxidative injury: Fighting for their own survival. Redox Biol. 2019;26:101261. doi: 10.1016/j.redox.2019.101261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hughes MM, Hooftman A, Angiari S, Tummala P, Zaslona Z, Runtsch MC, et al. Glutathione Transferase Omega-1 Regulates NLRP3 Inflammasome Activation through NEK7 Deglutathionylation. Cell Rep. 2019;29:151–61 e5. doi: 10.1016/j.celrep.2019.08.072. [DOI] [PubMed] [Google Scholar]
- 34.Zhang T, Tsutsuki H, Islam W, Ono K, Takeda K, Akaike T, et al. ATP exposure stimulates glutathione efflux as a necessary switch for NLRP3 inflammasome activation. Redox Biol. 2021;41:101930. doi: 10.1016/j.redox.2021.101930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Z, Xu X, Ma J, Wu J, Wang Y, Zhou R, et al. Gene deletion of Gabarap enhances Nlrp3 inflammasome-dependent inflammatory responses. J Immunol. 2013;190:3517–24. doi: 10.4049/jimmunol.1202628. [DOI] [PubMed] [Google Scholar]
- 36.Fleischmann C, Scherag A, Adhikari NK, Hartog CS, Tsaganos T, Schlattmann P, et al. Assessment of Global Incidence and Mortality of Hospital-treated Sepsis. Current Estimates and Limitations. Am J Respir Crit Care Med. 2016;193:259–72. doi: 10.1164/rccm.201504-0781OC. [DOI] [PubMed] [Google Scholar]
- 37.Hsu CG, Chavez CL, Zhang C, Sowden M, Yan C, Berk BC. The lipid peroxidation product 4-hydroxynonenal inhibits NLRP3 inflammasome activation and macrophage pyroptosis. Cell Death Differ. 2022;29:1790–803. doi: 10.1038/s41418-022-00966-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Griffiths RJ, Stam EJ, Downs JT, Otterness IG. ATP induces the release of IL-1 from LPS-primed cells in vivo. J Immunol. 1995;154:2821–8. [PubMed] [Google Scholar]
- 39.Chi H. Immunometabolism at the intersection of metabolic signaling, cell fate, and systems immunology. Cell Mol Immunol. 2022;19:299–302. doi: 10.1038/s41423-022-00840-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paik S, Kim JK, Silwal P, Sasakawa C, Jo EK. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol. 2021;18:1141–60. doi: 10.1038/s41423-021-00670-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimada E, Ahsan FM, Nili M, Huang D, Atamdede S, TeSlaa T, et al. PNPase knockout results in mtDNA loss and an altered metabolic gene expression program. PLoS ONE. 2018;13:e0200925. doi: 10.1371/journal.pone.0200925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem. 2014;289:7884–96. doi: 10.1074/jbc.M113.522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang W, Wang G, Xu ZG, Tu H, Hu F, Dai J, et al. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell. 2019;178:176–89 e15. doi: 10.1016/j.cell.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lepelley A, Wai T, Crow YJ. Mitochondrial Nucleic Acid as a Driver of Pathogenic Type I Interferon Induction in Mendelian Disease. Front Immunol. 2021;12:729763. doi: 10.3389/fimmu.2021.729763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Franchi L, Eigenbrod T, Munoz-Planillo R, Ozkurede U, Kim YG, Arindam C, et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J Immunol. 2014;193:4214–22. doi: 10.4049/jimmunol.1400582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buskiewicz IA, Montgomery T, Yasewicz EC, Huber SA, Murphy MP, Hartley RC, et al. Reactive oxygen species induce virus-independent MAVS oligomerization in systemic lupus erythematosus. Sci Signal. 2016;9:ra115. doi: 10.1126/scisignal.aaf1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493:346–55. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- 48.Nishimura K, Aizawa S, Nugroho FL, Shiomitsu E, Tran YTH, Bui PL, et al. A Role for KLF4 in Promoting the Metabolic Shift via TCL1 during Induced Pluripotent Stem Cell Generation. Stem Cell Rep. 2017;8:787–801. doi: 10.1016/j.stemcr.2017.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grochowska J, Czerwinska J, Borowski LS, Szczesny RJ. Mitochondrial RNA, a new trigger of the innate immune system. Wiley Interdiscip Rev RNA. 2022;13:e1690. doi: 10.1002/wrna.1690. [DOI] [PubMed] [Google Scholar]
- 50.Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes-Alnemri T, et al. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol. 2013;191:4358–66. doi: 10.4049/jimmunol.1301170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jo EK, Kim JK, Shin DM, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. 2016;13:148–59. doi: 10.1038/cmi.2015.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Falchi FA, Pizzoccheri R, Briani F. Activity and Function in Human Cells of the Evolutionary Conserved Exonuclease Polynucleotide Phosphorylase. Int J Mol Sci. 2022;23:1652. [DOI] [PMC free article] [PubMed]
- 53.Yin L, Li W, Xu A, Shi H, Wang K, Yang H, et al. SH3BGRL2 inhibits growth and metastasis in clear cell renal cell carcinoma via activating hippo/TEAD1-Twist1 pathway. EBioMedicine. 2020;51:102596. doi: 10.1016/j.ebiom.2019.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hsu CG, Fazal F, Rahman A, Berk BC, Yan C. Phosphodiesterase 10A Is a Key Mediator of Lung Inflammation. J Immunol. 2021;206:3010–20. doi: 10.4049/jimmunol.2001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tweedell RE, Malireddi RKS, Kanneganti TD. A comprehensive guide to studying inflammasome activation and cell death. Nat Protoc. 2020;15:3284–333. doi: 10.1038/s41596-020-0374-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang C, Hsu CG, Mohan A, Shi H, Li D, Yan C. Vinpocetine protects against the development of experimental abdominal aortic aneurysms. Clin Sci (Lond) 2020;134:2959–76. doi: 10.1042/CS20201057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published paper and in its supplementary file.