

Abstract
Mannose is a naturally occurring sugar widely consumed in the daily diet; however, mechanistic insights into how mannose metabolism affects intestinal inflammation remain lacking. Herein, we reported that mannose supplementation ameliorated colitis development and promoted colitis recovery. Macrophage-secreted inflammatory cytokines, particularly TNF-α, induced pathological endoplasmic reticulum stress (ERS) in intestinal epithelial cells (IECs), which was prevented by mannose via normalization of protein N-glycosylation. By preserving epithelial integrity, mannose reduced the inflammatory activation of colonic macrophages. On the other hand, mannose directly suppressed macrophage TNF-α production translationally by reducing the glyceraldehyde 3-phosphate level, thus promoting GAPDH binding to TNF-α mRNA. Additionally, we found dysregulated mannose metabolism in the colonic mucosa of patients with inflammatory bowel disease. Finally, we revealed that activating PMM2 activity with epalrestat, a clinically approved drug for the treatment of diabetic neuropathy, elicited further sensitization to the therapeutic effect of mannose. Therefore, mannose metabolism prevents TNF-α-mediated pathogenic crosstalk between IECs and intestinal macrophages, thereby normalizing aberrant immunometabolism in the gut.
Keywords: Colitis, Mannose, Macrophages, Intestinal epithelial cells, TNF-alpha, Endoplasmic reticulum stress
Subject terms: Chronic inflammation, Monocytes and macrophages
Introduction
Mannose is a natural aldohexose sugar found in many fruits and vegetables, such as cranberries, peaches, eggplants and green beans. The blood concentration of mannose ranges from 20 to 80 μM and can exceed 900 μM upon dietary supplementation without discernable side effects [1], proving its safety as a natural sugar. At present, mannose is commonly marketed as a dietary supplement for the treatment of urinary tract infections by inhibiting bacterial adherence to the urinary tract [2]. A recent report also revealed the antitumor function of mannose [3]. However, whether mannose has therapeutic potential in a broader range of diseases remains elusive.
Inflammatory bowel disease (IBD) is an inflammatory disorder with a continually increasing incidence worldwide. The etiology of IBD is complex and involves sophisticated bidirectional epithelial-immune interactions. Compromised intestinal epithelial function allows the invasion of gut microbes into the lamina propria, triggering the activation of gut immune cells, which produce large amounts of inflammatory cytokines that damage intestinal epithelial cells (IECs). How to prevent this vicious IEC-immune cell pathological cycle is of great importance for preventing IBD progression. Excessive endoplasmic reticulum stress (ERS) is a hallmark of IBD pathogenesis that leads to IEC death and thus disrupts epithelial integrity [4]. Some FDA-approved chaperone drugs that decrease ERS, such as tauroursodeoxycholate and 4-phenylbutyrate, have been utilized in the treatment of IBD [5]. However, the actual therapeutic efficacy of these ERS suppressors is unsatisfactory. This might be because hyperactivated colonic immune cells produce inflammatory mediators to continuously trigger ERS in IECs. However, the physiological inducers of ERS in the colitis microenvironment are largely unidentified.
In the present work, we revealed that TNF-α is a key ERS inducer during colitis development and that mannose prevents TNF-α-mediated pathogenic crosstalk between IECs and macrophages in intestinal inflammation.
Results
Mannose supplementation ameliorates intestinal inflammation
First, we hypothesized that mannose supplementation has an impact on the progression of colitis. To test this hypothesis, D-mannose was added to the drinking water of DSS-fed mice. Eight days after DSS administration, mice in the DSS-alone control group exhibited severe colitis symptoms, including massive body weight loss, colon shortening, and histological damage, whereas mice supplemented with mannose exhibited significant amelioration of these colitis markers (Fig. 1A–C). Separate from the DSS model, mannose also significantly ameliorated the pathogenesis of TNBS-induced colitis (Fig. 1D, E) as well as spontaneous colitis in IL-10 knockout (Il-10-/-) mice (Fig. 1F, G).
Fig. 1.

Mannose supplementation protects mice against colitis. A, B Mice were fed normal drinking water (n = 4) or 2.5% DSS (n = 7) for 8 days with or without 15% mannose treatment (dissolved in the drinking water). Body weight was measured daily (A), and colon length (B) was measured on Day 8. C Tissue damage was evaluated by H&E staining (n = 3/H2O group and n = 8/DSS group). D, E Mice were intrarectally injected with 2.5% TNBS with or without 15% mannose, and body weight (D) and colon length (E) were measured (n = 5–6). F, G Il-10−/− mice were administered 5% mannose for 10 weeks, and rectal prolapse (F) and body weight (G) were evaluated (n = 7). H Mice were fed 2.5% DSS for 6 days without mannose, followed by administration of 15% mannose until Day 16, and body weight was measured daily (n = 6). I, J Mice were pretreated with 15% mannose for 5 days, followed by treatment with 2.5% DSS in the absence of mannose (n = 5). Body weight was measured daily (I), and colon length was measured on Day 7 (J). The data are presented as the means ± SEMs. *p < 0.05; **p < 0.01; ***p < 0.001; two-tailed unpaired Student’s t test
Given the striking phenotypic difference with mannose supplementation, we further asked whether mannose could be involved in the recovery phase of colitis. Mice were fed DSS for 6 days prior to mannose administration in the regular drinking water. In this context, mannose significantly accelerated the recovery of mice against colitis relative to that in the control group (Fig. 1H). However, when mice were pretreated with mannose and given only regular water during DSS challenge, mannose did not exhibit any therapeutic effect on colitis (Fig. 1I, J), suggesting that the sustained administration of mannose is required for its anticolitic function.
Mannose has previously been reported to prevent diet-induced obesity by altering the gut microbiome [6]. Indeed, by microbiomics, we found that mannose supplementation altered the composition of the fecal microbiota and enhanced the α-diversity and Shannon diversity in colitic mice (Fig. S1A, B). However, in a fecal microbiota transplant (FMT) experiment, recipient mice transplanted with the fecal microbiota from control or mannose-fed colitic mice developed similar degrees of colitis (Fig. S1C), suggesting that the difference in the microbiome does not significantly contribute to the protective role of mannose.
Mannose suppresses pathological ERS in IECs by normalizing protein N-glycosylation
To understand the mechanisms of action of mannose, RNA sequencing was performed. We harvested colon tissues four days after DSS challenge, a time point when colitis progression is still mild, to investigate the early events regulated by mannose. The induction of endoplasmic reticulum stress (ERS) is a hallmark of colitis that drives the progression of inflammation by triggering IEC death [5, 7]. Notably, we found general upregulation of ERS marker genes in the colons of mice given DSS without mannose compared with healthy colons, and this upregulation was markedly prevented by mannose supplementation (Fig. 2A). In contrast, genes known to be inhibited by ERS were downregulated in the colons of mice given only DSS, whereas treatment with mannose reversed the downregulation of these genes (Fig. 2A). QPCR further confirmed that the levels of key ERS markers, such as GRP78, CHOP, and CASP12, were significantly reduced in IECs from mannose-treated mice compared with those from control mice (Fig. 2B). As expected, mannose treatment prevented the apoptosis of IECs, as evidenced by the reduced protein level of cleaved caspase 3 (Fig. 2C). We further treated colon explants from colitic mice with mannose and found that mannose markedly reduced GRP78 and CHOP expression, indicating that mannose could also alleviate established ERS in inflamed tissues (Fig. 2D).
Fig. 2.

Mannose inhibits ERS in IECs by normalizing protein N-glycosylation. A Colon tissues harvested from healthy and colitic mice (4 days after DSS challenge, n = 3/group) were subjected to RNA sequencing analysis. The levels of ERS-associated genes were analyzed. B The expression of GRP78, CHOP, and CASP12 in IECs from healthy (n = 4/group) and colitic (n = 7/group) mice was analyzed by QPCR. C The levels of cleaved caspase-3 in IECs were evaluated by immunoblotting. D Colon explants were isolated from colitic mice (n = 5/group) and cultured in the presence or absence of mannose (10 mM). GRP78 and CHOP expression was evaluated by immunoblotting. M = mannose. E The levels of the indicated ISC markers were analyzed by RNA sequencing. F The number of Ki67+ cells was determined by immunofluorescence staining using Lgr5EGFP reporter mice (n = 3/group). G Colon crypts were isolated from healthy (n = 4/group) or DSS-fed (n = 7/group) mice and cultured in vitro for 5 days. The viability of the organoids was analyzed by an MTT assay. H, I HCoEpiC cells (H) and colon organoids (I) were treated with CtrlSN or CoSN in the presence or absence of mannose (10 mM) for 24 h, and the levels of GRP78 and CHOP were measured by QPCR. J, K The viability of HCoEpiC cells (J) and colon organoids (K) was evaluated by CCK8 and MTT assays, respectively. L Schematic showing the mechanisms of ERS induction by 2DG and Tm. M IECs were treated with 2DG or Tm in the presence or absence of mannose for 24 h, and GPR78 and CHOP expression was evaluated by QPCR. N Cell viability was evaluated by a CCK8 assay. O IECs were stimulated with CoSN for 24 h, and protein glycosylation was evaluated by flow cytometry using conA staining. P The level of glycosylated proteins in colon tissues was measured by conA staining. The data are presented as the means ± SEMs. *p < 0.05; **p < 0.01; ***p < 0.001; two-tailed unpaired Student’s t test
ERS has been reported to cause the loss of intestinal stem cells (ISCs) [8]. We found that the expression of key ISC markers, such as Lgr5, Hopx, Sox9, Ascl2, Axin2, and Ki67, a proliferation marker expressed by IECs, were dramatically reduced in the inflamed colons. Notably, mannose treatment significantly reversed the decreases in the levels of these markers (Fig. 2E). Using Lgr5EGFP reporter mice, we discovered that the numbers of Lgr5+ crypts and Ki67+ IECs in mannose-treated colon sections were significantly higher than those in colon sections from control mice (Fig. 2F). In agreement with this finding, the growth potential of colon organoids isolated from mannose-treated mice was significantly maintained compared with that of colon organoids isolated from control mice (Fig. 2G), suggesting that mannose prevented colitis-induced ISC loss.
We then examined the impact of mannose on ERS-challenged IECs in vitro. To this end, a normal IEC cell line (HCoEpiC) and colon organoids were treated with the culture supernatant of colon explants from healthy mice (CtrlSN) or colitic mice (CoSN). The results indicated that compared with CtrlSN treatment, CoSN treatment significantly upregulated the expression of ERS markers and reduced the viability of IECs, while these effects were significantly rescued by mannose (Fig. 2H–K).
To understand how mannose alleviates ERS in IECs, we treated IECs with 2-deoxyglucose (2DG) and tunicamycin (Tm), which induce ERS through distinct mechanisms. 2DG acts as a mannose analog to antagonize the effect of mannose on N-glycosylation, while Tm prevents the addition of glucosamine to the sugar chain during N-glycosylation, thus interfering with the first step of N-linked glycan biosynthesis [9] (Fig. 2L). The results indicated that mannose treatment significantly reversed 2DG-induced but not Tm-induced GRP78 and CHOP expression (Fig. 2M). Consistent with this finding, mannose reversed the 2DG-induced but not the Tm-induced viability loss (Fig. 2N), suggesting that mannose might alleviate ERS by normalizing protein N-glycosylation. In contrast, blocking O-glycosylation using benzyl-α-GalNAc did not induce ERS or a reduction in viability in IECs (Fig. S2). We further performed concanavalin A (conA) staining to quantify the glycosylated protein levels in the groups. CoSN significantly reduced glycoprotein levels in IECs, and this effect was prevented by mannose (Fig. 2O). In agreement with this finding, the level of glycosylated proteins was higher in IECs from control colitic mice than in IECs from mannose-treated mice (Fig. 2P). Therefore, mannose corrects inflammation-induced abnormal protein N-glycosylation and thus reduces pathological ERS in the colonic epithelium.
Mannose prevents TNF-α-induced ERS and epithelial damage
Given the significant reduction in ERS by mannose treatment, we next sought to elucidate how the anti-ERS property of mannose contributes to its therapeutic function. Therefore, we treated mice with DSS-induced colitis with Tm to induce ERS in vivo. Consistent with a previous report, forced induction of ERS aggravated colitis development [7]. After Tm treatment, the amelioration of colitis by mannose was abrogated to some extent (Fig. 3A, B), suggesting that the normalization of N-glycosylation in IECs is partially attributed to the function of mannose.
Fig. 3.

Mannose prevents the pathological crosstalk between IECs and macrophages. A, B Mice were fed 2.5% DSS with or without intraperitoneal administration of Tm or cLipo (n = 6/group), and body weight (A) and colon length (B) were measured. C The percentages of CD11b+F4/80hi macrophages and CD11b+Ly6G+ neutrophils in the lamina propria were analyzed by flow cytometry (gated on CD45+ cells). D Bacterial translocation was evaluated in colon tissues via FISH and costaining with F4/80+ macrophages. E The bacterial load in the LP was determined by 16S rRNA QPCR (n = 3/group). F The levels of TNF-α, IL-6 and IL-1β in colon homogenates were evaluated by ELISA (n = 8/group). G, H Colon organoids were cultured in CtrlMSN or ManMSN (30% v/v) from healthy mice (H2O) or colitic mice (DSS) in the presence of IgG or an anti-TNFα antibody for 24 h. Organoid viability was assessed by an MTT assay (G), and the levels of GRP78 and CHOP were evaluated by QPCR (H). I, J Colon organoids were stimulated with TNFα in the presence or absence of Tm for 24 h, and organoid viability was assessed by an MTT assay (I). The levels of GRP78 and CHOP were evaluated by QPCR (J). K, L Mice were fed 2.5% DSS and then treated with a TNF-α neutralizing antibody, Tm or both (n = 5/group). Body weight was measured (K). The expression of ERS markers in IECs was evaluated by QPCR (L). The data are presented as the means ± SEMs. *p < 0.05; **p < 0.01; ***p < 0.001; two-tailed unpaired Student’s t test
Disruption of intestinal epithelial integrity allows the invasion of gut bacteria into the lamina propria (LP), leading to hyperactivation of LP resident immune cells. We and others have reported that macrophage-derived inflammatory cytokines are key driving factors of colitis [10, 11]. The flow cytometry results demonstrated that the infiltration of CD11b+F4/80hi macrophages and CD11b+Ly6G+ neutrophils in the LP was significantly reduced by mannose treatment (Fig. 3C). Consistent with the epithelial barrier preservation, mannose significantly prevented bacterial invasion into the LP, as evidenced by FISH and quantification of 16S ribosomal RNA (Fig. 3D, E). Furthermore, the colonic levels of TNF-α and IL-6, which are produced mainly by macrophages, were significantly decreased by mannose treatment (Fig. 3F). The levels of other inflammatory cytokines (IL-1β, CXCL10, IL-12, and IFN-γ) and anti-inflammatory cytokines (IL-10) were not obviously affected by mannose in colitic mice (Fig. S3). Notably, when we further depleted macrophages in Tm-treated mice using clodronate liposomes (cLipo), the combined treatment with Tm and cLipo completely abrogated the anticolitic role of mannose (Fig. 3A, B), suggesting that the anticolitic effect of mannose is also dependent on macrophage function. However, unlike in IECs, the expression of ERS markers in colitic macrophages was not affected by mannose administration (Fig. S4).
On the other hand, the levels of T-cell-derived cytokines were comparable between control and mannose-treated mice (Fig. S5A). Moreover, mannose retained its capacity to ameliorate colitis in severe combined immunodeficient (SCID) mice (Fig. S5B), confirming that the adaptive immune system is dispensable for the anticolitic effect of mannose.
To investigate how mannose mediates the crosstalk between macrophages and IECs, we treated colonic organoids with the culture supernatant of colonic macrophages isolated from control (CtrlMSN) or mannose-treated (ManMSN) colitic mice or healthy mice (Fig. 3G). Compared with CtrlMSN, ManMSN had a significantly impaired capacity to induce ERS and organoid damage, and this protective effect was abrogated in the presence of Tm. Importantly, neutralization of TNF-α markedly reduced ERS and organoid damage in CtrlMSN cultured organoids and, albeit to a lesser extent, in ManMSN cultured organoids (Fig. 3G, H). On the other hand, mannose prevented exogenous TNF-α-induced organoid damage and expression of GRP78 and CHOP (Fig. 3I, J). Again, this effect was greatly abrogated by Tm. In contrast to TNF-α, IL-6 failed to induce organoid damage or the expression of ERS markers (Fig. S6).
To validate the role of N-glycosylation in TNF-α-induced colitis, we neutralized TNF-α in DSS-treated mice. Treatment with an anti-TNF-α antibody (αTNF-α) significantly mitigated colitis in mice and reduced the expression of ERS markers in IECs. Importantly, the anticolitic effect of αTNF-α was greatly impaired by coadministration of Tm (Fig. 3K, L), indicating that ERS suppression is a crucial mechanism for the therapeutic effect of TNF-α blockade.
Collectively, these findings indicate that macrophage-derived inflammatory cytokines play a pivotal role in inducing ERS and IEC damage. Disruption of the epithelial barrier in turn leads to inflammatory activation of LP macrophages. This pathological macrophage-IEC crosstalk can be prevented by mannose.
Mannose inhibits TNF-α production translationally by promoting GAPDH binding to TNF-α mRNA
The aforementioned results indicated that mannose suppressed TNF-α-induced IEC cytotoxicity; however, it remained unknown whether TNF-α production by macrophages is directly mitigated by mannose supplementation. Using flow cytometry and ELISA, we found that mannose significantly reduced the level of TNF-α in CD68+ colonic macrophages (Fig. 4A, B). Further in vitro experiments demonstrated that mannose but not other hexoses dose-dependently suppressed the secretion of TNF-α from colonic macrophages and peritoneal macrophages upon treatment with LPS, PGN and heat-killed E. coli (Fig. 4C, D). Disrupting the interaction between mannose and the mannose receptor (MR) with mannan or silencing MR expression failed to weaken the anti-inflammatory role of mannose, suggesting the receptor-independent mechanism of mannose (Fig. S7).
Fig. 4.

Mannose inhibits macrophage production of TNF-α. A The production of TNF-α in colonic CD68+ macrophages was evaluated by flow cytometry (n = 8/group). B Magnetically sorted colonic macrophages were cultured for 24 h, and the level of TNF-α in the culture supernatant was evaluated by ELISA (n = 4 or 7/group). C, D Peritoneal macrophages (C) and colonic macrophages (D) were treated with 1 μg/ml LPS, 20 μg/ml PGN or heat-killed E. coli (MOI = 10) in the presence of various sugars (20 mM), and TNF-α production was evaluated by ELISA. E, F Peritoneal macrophages were left unstimulated or stimulated with 1 μg/ml LPS in the absence or presence of mannose, and the changes in key metabolic intermediates in glycolysis, the PPP, the TCA, and FAO were analyzed by widely targeted metabolomics. G Peritoneal macrophages were left unstimulated or stimulated with 1 μg/ml LPS in the absence or presence of mannose for 6 h, and the ECAR was evaluated by a Seahorse assay. H Peritoneal macrophages were stimulated with 1 μg/ml LPS in the presence of 5 mM 2FDG and 5 mM 2DG, and TNF-α production was evaluated by ELISA. I, J Peritoneal macrophages were stimulated with 1 μg/ml LPS for the indicated times; the mRNA level of TNF-α was evaluated by QPCR (I), and the phosphorylation of p65, ERK, JNK and p38 was evaluated by immunoblotting (J). K The intracellular level of TNF-α was measured by flow cytometry after LPS stimulation. The data are presented as the means ± SEMs. *p < 0.05; **p < 0.01; ***p < 0.001; two-tailed unpaired Student’s t test
Given that mannose is a metabolizable sugar, we speculated that mannose might exert its anti-inflammatory effect by altering cellular metabolic processes. Therefore, widely targeted metabolomics was performed to determine which potential metabolic pathways might be altered. The results demonstrated that mannose treatment decreased the levels of several glycolytic metabolites downstream of fructose 1,6-bisphosphate (Fig. 4E, F). Consistent with this finding, mannose reduced the extracellular acidification rate (ECAR) in LPS-activated macrophages (Fig. 4G). The level of the pentose phosphate pathway (PPP) intermediate ribose-5-P was moderately reduced by mannose. In contrast, mannose did not obviously affect the levels of tricarboxylic acid cycle (TCA) intermediates (malic acid, citric acid, and cis-aconitic acid) or a fatty acid oxidation (FAO) intermediate (β-hydroxybutyrate) (Fig. 4F). Consistent with this finding, blocking the FAO, TCA and PPP pathways with etomoxir, malonic acid and 6-aminonicotinamide, respectively, did not reduce the capacity of mannose to suppress TNF-α production (Fig. S8), while blocking glycolysis using 2DG or 2-FDG completely abrogated the function of mannose (Fig. 4H).
Surprisingly, although enhanced glycolysis has been reported to transcriptionally promote TNF-α expression [12], we found that mannose failed to inhibit TNF-α mRNA expression in activated macrophages (Fig. 4I). Moreover, mannose did not affect the phosphorylation of inflammatory transcription factors, including NF-κB and MAPKs (ERK, JNK, and p38) (Fig. 4J). The flow cytometry results demonstrated that mannose obviously reduced the intracellular level of TNF-α (Fig. 4K), suggesting that mannose inhibits TNF-α protein translation in macrophages rather than only preventing its extracellular secretion.
Therefore, polysome profiling was performed to examine the effect of mannose on TNF-α protein translation [13]. The results indicated that LPS stimulation shifted the distribution of TNF-α mRNA from light polysomes to heavy polysomes in macrophages, whereas mannose treatment led to a reduction in the abundance of TNF-α mRNA in the heavy polysome fractions (Fig. 5A). Compared with that in control macrophages (MφCtrl), the most robustly downregulated glycolytic intermediate in mannose-treated macrophages (MφMan) was glyceraldehyde 3-phosphate (G3P) (Fig. 4E), which is used in a reaction catalyzed by GAPDH to generate 1,3-bisphosphoglycerate (1,3-BPG). Interestingly, GAPDH also performs a less-known function as a translational repressor by binding to AU-rich elements (AREs) in the 3’UTRs of mRNAs, including TNF-α mRNA [14, 15]. The RNA-binding capacity of GAPDH was competitively inhibited by G3P (Fig. 5B). To determine whether the reduced G3P level is responsible for the anti-inflammatory role of mannose via regulation of GAPDH activity, we treated macrophages with a GAPDH inhibitor—heptelidic acid (HA)—or with exogenous G3P. The results showed that HA significantly decreased TNF-α production in MφCtrl but not in MφMan, while G3P significantly increased TNF-α production in MφMan but not in MφCtrl. Under both conditions, the level of TNF-α was comparable between MφCtrl and MφMan (Fig. 5C, D), further confirming that mannose suppresses TNF-α production by reducing the intracellular G3P level.
Fig. 5.

Mannose suppresses TNF-α translation by enhancing GAPDH binding to TNF-α mRNA. Peritoneal macrophages were treated with 1 μg/ml LPS in the presence or absence of mannose for 6 h. A Polysome profiling was performed. The relative abundances of free ribosomes, light ribosomes, and heavy ribosomes were determined. B Schematic showing the competitive binding of G3P and TNF-α mRNA to GAPDH. C, D Peritoneal macrophages were stimulated with 1 μg/ml LPS for 12 h in the presence or absence of HA (C) or exogenous G3P (D), and TNF-α production was evaluated by ELISA. E Macrophages were isolated magnetically from the colon, lung and liver of mice and were then stimulated with 1 μg/ml LPS for 12 h. TNF-α production was evaluated by ELISA. F Peritoneal macrophages were stimulated with 1 μg/ml LPS for different times, and the levels of the indicated proteins were evaluated by immunoblotting. G The level of p-eEF2 was evaluated by immunofluorescence staining 1 h after stimulation with 1 μg/ml LPS. H Peritoneal macrophages were stimulated with 1 μg/ml LPS for 1 h, and the amount of eEF2-bound TNF-α mRNA was evaluated by a RIP assay. I Peritoneal macrophages were stimulated with 1 μg/ml LPS in the presence or absence of HA for 30 min, and the level of p-eEF2 was evaluated by immunoblotting. J Peritoneal macrophages were stimulated with 1 μg/ml LPS for 1 h, and the amount of GAPDH-bound TNF-α mRNA was evaluated by a RIP assay. K The interaction between GAPDH and eEF2 was evaluated by CoIP. L Peritoneal macrophages were transfected with eEF2 siRNA for 48 h and stimulated with 1 μg/ml LPS, and TNF-α production was evaluated by ELISA. M Colonic macrophages were isolated from healthy or colitic mice, and the p-eEF2 level was evaluated by immunoblotting. O, P Mice were fed 2.5% DSS with or without intraperitoneal administration of HA (n = 5/group). Body weight was measured (O), and TNF-α production in colon tissues was evaluated by ELISA (P). The data are presented as the means ± SEMs. *p < 0.05; **p < 0.01; ***p < 0.001; two-tailed unpaired Student’s t test
We then isolated macrophages from mouse colons, lungs and livers and stimulated them with LPS. Similar to the observations with peritoneal macrophages, treatment of macrophages of various origins with mannose inhibited TNF-α protein production, but this effect was abrogated by HA (Fig. 5E), suggesting that the inhibitory mechanism of mannose is conserved among macrophages of different tissue origins, therefore indicating that mannose is a global anti-inflammatory agent.
We next sought to identify the signaling pathways involved in protein translation. The phosphorylation of eukaryotic translation initiation factor 4E (eIF4E), which mediates translation initiation, was not affected by mannose. On the other hand, the phosphorylation of eukaryotic translation elongation factor 2 (eEF2), which indicates repression of translational elongation [16, 17], was dramatically increased by mannose treatment, suggesting that mannose specifically interferes with translational elongation (Fig. 5F). Through immunofluorescence staining, we confirmed that LPS stimulation resulted in an obvious decrease in the p-eEF2 intensity in macrophages, which was then found to be largely rescued by mannose treatment (Fig. 5G).
We then performed RNA immunoprecipitation (RIP) to examine eEF2-binding to TNF-α mRNA. As shown in Fig. 5H, LPS challenge resulted in a marked increase in the amount of TNF-α mRNA immunoprecipitated with the anti-eEF2 antibody, indicating active translational elongation. However, mannose treatment significantly decreased the abundance of eEF2-binding TNF-α mRNA. In addition, HA treatment markedly increased the level of p-eEF2 in macrophages, indicating that GAPDH inhibition leads to translational repression (Fig. 5I). The RIP assay further demonstrated that LPS stimulation dramatically reduced the amount of GAPDH-binding TNF-α mRNA and that this decrease was significantly reversed by mannose (Fig. 5J). The coimmunoprecipitation (CoIP) assay showed that GAPDH can interact with eEF2 in resting macrophages. LPS stimulation resulted in a decreased GAPDH-eEF2 association, which was partially rescued by mannose (Fig. 5K). These results indicate that GAPDH is present in the translational regulation complex and that optimal TNF-α mRNA translation requires the dissociation of GAPDH from eEF2, which could be prevented by mannose.
To validate whether mannose reduces TNF-α production by suppressing eEF2-mediated translational elongation, we silenced eEF2 expression in macrophages with siRNA. Knockdown of eEF2 significantly reduced TNF-α production. Importantly, in eEF2-silenced macrophages, the inhibitory effect of mannose on TNF-α production was largely abrogated (Fig. 5L). In agreement with this finding, macrophages isolated from mannose-treated colitic mice had a higher level of p-eEF2 than those from control colitic mice (Fig. 5M).
In an animal experiment, we treated control and mannose-fed colitic mice with HA. Similar to mannose administration, HA administration exerted a prominent anticolitic effect and reduced TNF-α production in the colons of control mice. In contrast, HA only moderately alleviated colitis in mannose-fed mice and did not have a synergistic effect with mannose to inhibit TNF-α production (Fig. 5O, P), suggesting that the functions of HA and mannose in TNF-α inhibition are largely redundant.
Hence, mannose can specifically translationally repress TNF-α production by promoting GAPDH binding to TNF-α mRNA.
IBD patients have elevated serum mannose concentrations and mucosal PMM2 levels
We next sought to determine whether endogenous mannose metabolism is dysregulated in IBD patients. Through ultra-performance liquid chromatography (UPLC), we discovered that serum mannose concentrations were significantly higher in IBD patients than in healthy controls (Fig. 6A), indicating that IBD patients might have impaired mannose absorption or metabolism. Indeed, the expression of phosphomannomutase 2 (PMM2) and phosphomannose isomerase (MPI), two key enzymes that control the metabolic flux of mannose [18], showed significant upregulation and downregulation, respectively, in IBD mucosa compared to healthy mucosa (Fig. 6B). Moreover, the expression of PMM2 and MPI exhibited a significantly positive and negative correlation, respectively, with the serum mannose concentration (Fig. 6C). High PMM2 expression has been reported to promote the flux of available mannose toward N-glycosylation, while low MPI expression is known to enhance the antiglycolytic function of mannose [18, 19] (Fig. 6D). Interestingly, by analyzing a single-cell sequencing dataset, we found that PMM2 upregulation occurred mainly in the IEC cluster, suggesting that IECs require high PMM2 expression to alleviate ERS (Fig. 6E). In agreement with this finding, the serum mannose concentration was significantly positively correlated with mucosal GRP78 and CHOP expression (Fig. 6F). On the other hand, MPI downregulation was observed in the immune cell cluster (Fig. 6E), potentially indicating that LP immune cells such as macrophages need to downregulate glycolysis to suppress inflammatory activation. Compared with healthy mice, colitic mice showed significantly enhanced PMM2 expression in IECs and significantly reduced MPI expression in macrophages (Fig. 6G), further validating the cell type-specific changes in mannose metabolism. Through reanalysis of an RNA-Seq dataset (GSE16879), it was found that 20 of the 33 genes enriched in the “Fructose and Mannose Metabolism” KEGG pathway were significantly altered in mucosal biopsies from IBD patients compared with tissues from healthy controls (Fig. 6H), again indicating dysregulated mannose metabolism in IBD patients.
Fig. 6.

IBD patients have dysregulated mannose metabolism. A The concentrations of mannose in serum from healthy controls (n = 30) and IBD patients (n = 20) were measured by UPLC. B The expression levels of PMM2 and MPI in the colonic mucosa were evaluated by QPCR. C Correlations between serum mannose concentrations and mucosal PMM2 and MPI expression were evaluated by Spearman rank correlation analysis. D Schematic showing MPI- and PMM2-mediated intracellular mannose metabolism. E The expression of PMM2 and MPI in the colonic mucosa was analyzed using Single Cell Portal (https://singlecell.broadinstitute.org/single_cell, accession SCP259). F Correlations between serum mannose concentrations and mucosal GRP78 and CHOP expression were evaluated by Spearman rank correlation analysis. G The expression of PMM2 and MPI in IECs and macrophages was evaluated by QPCR (n = 4 or 7/group). H The expression of genes involved in the “Fructose and Mannose Metabolism” KEGG pathway was analyzed using a GEO dataset (GSE16879). I–N The expression of PMM2 (I, L), its correlation with patient survival (J, M), and its correlation with the expression of GRP78 (K, N) in COAD (I–K, n = 290) and READ (L–N, n = 167) patients were analyzed using the TCGA database. The data are presented as the means ± SEMs. *p < 0.05; **p < 0.01; ***p < 0.001; two-tailed unpaired Student’s t test
Recurrent chronic colitis often leads to the development of colorectal cancer (CRC). Similar to IBD patients, CRC patients showed significantly increased PMM2 expression in tumor tissues compared with tumor-adjacent normal tissues (Fig. 6I, L). High PMM2 expression predicted more favorable patient survival (Fig. 6J, M). Additionally, the tumor expression of PMM2 was significantly positively correlated with GRP78 expression (Fig. 6K, N), indicating that PMM2 might also play a role in the colitis-CRC transition.
PMM2 activation improves the therapeutic potential of mannose
The aforementioned results were highly suggestive that PMM2 upregulation might serve as a compensatory mechanism in response to ERS. Therefore, we investigated whether activation of PMM2 enzymatic activity can further potentiate the ERS-suppressive role of mannose. Accordingly, we employed epalrestat, a clinically approved drug for the treatment of diabetic neuropathy [20] that has also been reported to function as a PMM2 activator [21, 22]. Epalrestat treatment significantly potentiated the capacity of mannose to protect organoids against CoSN-induced death (Fig. 7A). Then, we treated mice with epalrestat and mannose alone or in combination. Intriguingly, mice treated with both epalrestat and mannose gained greater protection from colitis than mice treated with mannose alone, as assessed by weight loss, colon length and histological score, suggesting that epalrestat can elicit sensitization to the anticolitic function of mannose (Fig. 7B–D). In addition, mice treated with both epalrestat and mannose had significantly lower expression of GRP78, CHOP and CASP12 in IECs than mice administered mannose alone (Fig. 7E). On the other hand, combined treatment with epalrestat and mannose significantly reduced colonic TNF-α production (Fig. 7F). Importantly, epalrestat failed to synergize with mannose to inhibit TNF-α production by macrophages in vitro, suggesting that epalrestat indirectly reduced TNF-α production in mannose-treated mice by preserving the IEC barrier (Fig. 7G).
Fig. 7.

Treatment with a PMM2 activator optimizes the therapeutic potential of mannose. A Colon organoids were treated with CoSN in the presence of epalrestat, mannose or both for 24 h. Organoid viability was evaluated by an MTT assay. B–D Mice were fed 2.5% DSS in combination with the administration of epalrestat, mannose or both. Body weight (B), colon length (C) and histological damage (D) were measured. E The levels of ERS markers in IECs were analyzed by QPCR. F TNF-α production in colon tissues was evaluated by ELISA. G Macrophages were treated with LPS in the presence of epalrestat, mannose or both for 12 h, and TNF-α production was evaluated by ELISA. The data are presented as the means ± SEMs. *Ctrl vs. Man; &Ctrl vs. Epal. #Man vs. Epal+Man. *, &p < 0.05; **, ##p < 0.01; ***p < 0.001; two-tailed unpaired Student’s t test
Taken together, these results indicated that mannose prevented the TNF-α-mediated pathogenic crosstalk between IECs and intestinal macrophages (Fig. 8).
Fig. 8.
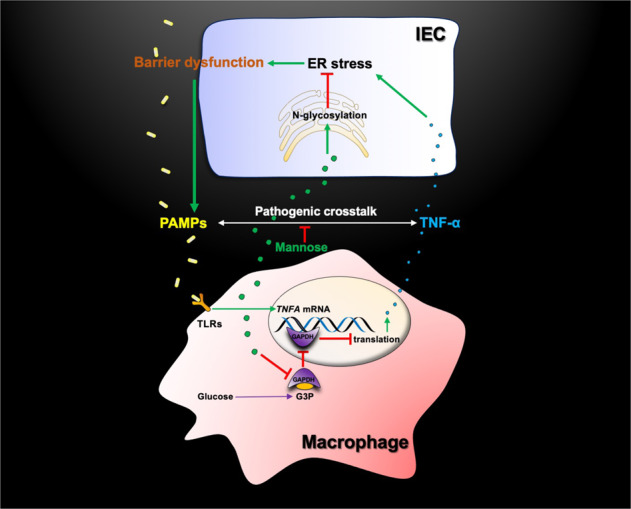
Model describing how mannose suppresses the pathogenic crosstalk between IECs and intestinal macrophages. PAMP pathogen-associated molecular pattern, PRR pattern recognition receptor
Discussion
In the present work, we reported that mannose, a simple sugar consumed daily in the average diet, possesses a unique anti-inflammatory capacity and provided key mechanistic insights into the dual function of mannose in IBD pathogenesis. ER stress, a hallmark of IBD, is a key driving factor in IBD etiology. Pharmacological intervention with ERS has been proven to be a promising strategy for IBD treatment [5, 23]. Notably, ongoing clinical trials are currently underway to assess the therapeutic efficacy and safety of ERS inhibitors in IBD patients. Herein, we identified mannose as a novel ERS suppressor, which means that ERS can be prevented by a naturally occurring substrate with clinical safety and efficacy. Additionally, we first reported that TNF-α, a predominant colitogenic cytokine, serves as a physiological ERS inducer in IECs. To further evaluate the ERS-inducing role of TNF-α in a clinically relevant context, we used αTNF-α to treat colitic mice and obtained two important findings. First, αTNF-α reduced the levels of ERS markers in IECs, confirming that TNF-α serves as an ERS inducer in vivo. In addition, in the presence of Tm, the therapeutic efficacy of infliximab was greatly blunted. Therefore, we present, to our knowledge, the first report indicating that preventing ERS is a crucial mechanism contributing to the function of αTNF-α. However, the exact mechanisms by which TNF-α induces ERS still need further investigation.
To further understand the mechanism underlying mannose-mediated amelioration of colitis, we sought to identify the target cell type on which mannose acts and found that mannose ameliorates colitis by targeting both IECs and macrophages. It is noteworthy that although macrophage-derived inflammatory cytokines are pivotal colitogenic factors, global macrophage depletion did not reduce disease severity. We rationalized that this was due to the importance of macrophages for mucosal repair and bacterial clearance in colitis [24, 25]. Regarding the effect of mannose on macrophages, although enhanced glycolysis is a prominent metabolic feature in inflammatory macrophages, the proinflammatory mechanisms of glycolysis remain poorly understood. In previous studies, glycolysis inhibition was reported to suppress the transcription of inflammatory cytokine genes by limiting the activation of upstream transcription factors, such as NF-κB [26, 27]. Surprisingly, here, mannose inhibited TNF-α production in a transcription-independent manner. Instead, the intracellular G3P level was decreased by mannose. Consequently, GAPDH was liberated from G3P to bind to TNF-α mRNA. In addition, eEF2 was found to be present in the GAPDH immunoprecipitate. Whether this interaction is direct or indirect and the exact mechanism by which GAPDH inhibits eEF2-mediated translational elongation still need further investigation and constitute a limitation of this study. These findings also indicate that glycolytic suppression induced by varying substances might inhibit inflammation via distinct mechanisms. In contrast to the findings of a previous study, we did not observe a robust impact of mannose on IL-1β production [28].
In IBD patients, the increased serum mannose concentration might reflect that the intracellular uptake, transport, or catabolism of endogenous mannose is defective. These defects possibly lead to impairment of protein N-glycosylation and a subsequent increase in glycolysis. As PMM2 and MPI are the two key mannose-metabolizing enzymes sharing the same substrate, a high PMM2/MPI ratio is reported to both facilitate protein N-glycosylation and inhibit glycolysis [18]. As we reported herein, mannose metabolism in IECs and macrophages regulates cell function via distinct mechanisms. This differential utilization of endogenous mannose might be reflected by the observation that PMM2 upregulation and MPI downregulation specifically occur in IECs and colon macrophages, respectively. Based on this idea, we tried combination therapy with both mannose and epalrestat. The marked synergistic effect suggested that the enzymatic activity of PMM2 is required for the optimal anticolitic function of mannose. Given that epalrestat is a safe, orally available drug that was approved in Japan in 1992 for the treatment of diabetic neuropathy [29], combined administration of mannose and epalrestat is a feasible approach for the clinical treatment of IBD.
Collectively, these findings indicate that the anti-ERS and anti-inflammatory properties of mannose are highly intertwined and jointly confer protection against colitis.
Materials and methods
Study design
Colitic mice were fed mannose, and disease progression was monitored. The impact of mannose on colonic mucosal gene expression was evaluated by RNA sequencing. The effect of mannose on IECs was investigated using a CCK8 assay, QPCR, and flow cytometry. The effect of mannose on macrophage TNF-α production was investigated via metabolomics, enzyme-linked immunosorbent assay (ELISA), flow cytometry, RNA immunoprecipitation (RIP), coimmunoprecipitation (CoIP), and immunoblotting. The serum mannose concentration in IBD patients was measured by ultra-performance liquid chromatography (UPLC). Epalrestat, a phosphomannomutase 2 activator, was used in combination with mannose to study the synergistic effects of these agents.
Mice and colitis models
Wild-type C57BL/6 mice were purchased from SLAC Laboratory Animal Center (Shanghai, China) and were housed under specific pathogen-free conditions. The dextran sulfate sodium (DSS)-induced colitis model, trinitrobenzene sulfonic acid (TNBS)-induced colitis model and IL-10-deficient colitis model were established as we previously described [10]. According to the experimental design, different concentrations of D-mannose (Sigma–Aldrich, St. Louis, MO, USA) were added to the drinking water.
For ERS induction, mice were injected i.p. with tunicamycin (1 mg/kg, Sigma–Aldrich, St. Louis, MO, USA) on Days 0, 2, 4, and 6 of DSS treatment.
For PMM2 activation, mice were injected i.p. with epalrestat (40 mg/kg, TsBiochem, Shanghai, China) on Days 1, 3, 5, and 7 of DSS treatment.
For macrophage depletion, mice were injected i.p. with 200 μL of clodronate liposomes (FormuMax Scientific, Sunnyvale, CA, USA) on Days −2, 0, 2, 4, and 6 of DSS treatment.
For TNF-α neutralization, mice were injected i.p. with an anti-mouse TNF-α antibody (200 μg/mouse, BioXcell, Lebanon, NH, USA) on Days 1, 3, and 5 of DSS treatment.
For HA treatment, mice were injected i.p. with HA (0.5 mg/kg, MedChemExpress, Monmouth Junction, NJ, USA) on Days 1, 3, and 5 of DSS treatment.
Animal experiments were performed according to protocols approved by the Zhejiang University Institutional Animal Care and Use Committee.
Histopathology
The distal portions of colons were fixed with 4% paraformaldehyde (PFA). Paraffin-embedded, 5-μm-thick sections were used for hematoxylin-eosin (H&E) staining.
IBD patient specimens
Clinical specimens from IBD patients and healthy donors were collected from Sir Run Run Shaw Hospital, Zhejiang University School of Medicine. Experiments involving human participants were approved by the Medical Ethics Committee of Sir Run Run Shaw Hospital, Zhejiang University School of Medicine (20220209-296).
IEC cell line culture
HCoEpiC cells were purchased from the American Type Culture Collection (Manassas, VA, USA) and were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). HCoEpiC cells were confirmed by STR DNA fingerprinting at Texas MD Anderson Cancer Center and tested for mycoplasma negativity.
Isolation of mouse peritoneal macrophages
A total of 1 ml of 3% β-thioglycollate (Merck, Kenilworth, NJ, USA) was injected into the peritoneal cavities of mice. Three to five days later, cells in the peritoneal exudate were collected, and adherent macrophages were cultured at 37 °C in complete Dulbecco’s modified Eagle’s medium/nutrient mixture F-12 (DMEM/F-12) supplemented with 10% heat-inactivated FBS.
Isolation of mouse colonic macrophages
Phosphate-buffered saline (PBS)-rinsed colons were opened longitudinally and cut into 5 mm pieces. Colon fragments were incubated with Hanks’ balanced salt solution (HBSS) containing 5 mM ethylene diamine tetraacetic acid (EDTA), 1 mM dithiothreitol (DTT) and 5% FBS for 30 min at 37 °C with gentle shaking to remove colonic epithelial cells. The remaining tissue was cut into small pieces and digested with HBSS containing 5% FBS and 300 U/ml collagenase IV at 37 °C for 50 min with gentle shaking. The product was filtered through a 70-μm-pore-size nylon mesh and washed twice with PBS to obtain a single-cell suspension. Macrophages were magnetically sorted by an EasySep™ Mouse F4/80 Positive Selection Kit (STEMCELL, Cambridge, MA, USA). A total of 1 × 106 isolated macrophages were cultured in DMEM/F-12 medium supplemented with 10% FBS and 1× penicillin/streptomycin. Twelve hours later, the macrophage supernatant (MSN) was passed through 0.22-μm syringe filters and stored at −80 °C.
Organoid culture and viability assay
Colons were flushed with ice-cold PBS and opened longitudinally. The luminal surface of the colon was scraped using a glass coverslip to remove the luminal contents and villi. The colons were then cut into pieces of approximately 2 mm, washed 5 times with PBS, and incubated with 5 mM EDTA (in PBS) on ice for 40 min. Colon crypts were released by vigorous shaking. After filtering through 70-μm cell strainers, the crypt fraction was collected by centrifugation at 150 × g for 3 min at 4 °C prior to two washes with Advanced DMEM/F-12. The crypts were mixed with Matrigel® Matrix (Corning, NY, USA) at a 1:1 ratio and cultured in IntestiCult™ Organoid Growth Medium (STEMCELL, Cambridge, MA, USA). The medium was replaced every three days. The viability of organoids was measured by an MTT assay as previously described [30].
Flow cytometry
Cells were washed twice with PBS and treated with permeabilization buffer (eBioscience, San Diego, CA, USA) for 40 min at room temperature. The cells were then stained with the indicated fluorescently labeled antibodies/dyes in the dark for 30 min at room temperature. Samples were collected in an ACEA NovoCyteTM flow cytometer, and data were analyzed using FlowJo software (Tree Star). Cells were stained with antibodies specific for the following proteins: Con A (MKBio, Shanghai, China), CD68 (Abcam, Cambridge, MA, USA), TNF-α (Biolegend, San Diego, CA, USA), CD11b (Biolegend), F4/80 (Biolegend), Ly6G (Biolegend).
Fluorescence in situ hybridization (FISH)
The distal colon was fixed with 4% PFA overnight, and 20 and 30% sucrose solutions were used for dehydration. Then, the colon tissue was embedded in Optimum Cutting Temperature Compound (OCT) and cut into 5-μm-thick frozen sections. The slides were equilibrated in hybridization buffer (0.9 M NaCl, 20 mM Tris-HCl, 0.01% sodium lauryl sulfate, and 10% formamide; pH 7.5) with the FISH probe (10 ng/µl; EUB338) and incubated in a humidified chamber at 46 °C for 14 h. After incubation, the slides were gently washed three times in washing buffer containing 0.9 M NaCl and 20 mM Tris-HCl (pH 7.5; preheated to 42 °C). The samples were incubated with DAPI in PBS for 10 min at room temperature and then mounted with an antiquenching agent. Images were acquired on a Nikon A1 confocal microscope (Nikon, Tokyo, Japan).
Immunofluorescence
Cells were seeded on glass slides. After treatment, the cells were washed three times with PBS, fixed with 4% PFA for 15 min, lysed with 0.5% Triton X-100 for 15 min, and blocked with 5% BSA for 1 h at room temperature. An anti-p-eEF2 (Abcam, Cambridge, MA, USA) primary antibody in 5% BSA was added dropwise to the slides, which were incubated in a humidified chamber in the dark at 4 °C overnight. The next day, the cells were washed with PBS and incubated with Alexa Fluor 488-conjugated anti-rabbit IgG (Invitrogen, Carlsbad, CA, USA) at room temperature in the dark for 1.5 h, incubated with DAPI for 5 min, and finally mounted with the antiquenching agent.
Human and mouse tissues were fixed with 4% PFA. Immunofluorescence staining was performed on 5-μm-thick paraffin sections after heat-induced antigen retrieval or on 5-μm-thick frozen sections. Primary antibodies specific for the following proteins were used: Grp78 (Beyotime, Shanghai, China), Ki67 (Biolegend), F4/80 (Biolegend), and p-eEF2 (Abcam, Cambridge, MA, USA).
CCK8 assay
HCoEpiC cells were seeded at 5 × 103 cells/well in a 96-well plate and incubated at 37 °C in DMEM supplemented with 10% FBS prior to the indicated treatment. Ten microliters of CCK-8 solution was added to each well prior to further incubation for 1 h. The absorbance was measured at 450 nm using a Synergy Mx M5 microplate reader (Molecular Devices, SV, USA).
ELISA
The levels of TNFα, IL-6, IL-1β, IL-12, CXCL10, IFN-γ, and IL-10 in the cell culture supernatant or tissue homogenate were measured by cytokine ELISA kits (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
QPCR
Total RNA was extracted from cells or tissues with TRIzol reagent (Pufei Biology, Shanghai, China) and reverse transcribed into cDNA by a ReverTra Ace Kit (Toyobo, Osaka, Japan). cDNA was amplified with THUNDERBIRD SYBR qPCR Mix (Toyobo, Osaka, Japan) on a StepOnePlus Real-Time PCR System (Bio-Rad, Hercules, CA, USA). The mRNA levels were normalized to β-actin. The primer sequences are listed in Supplementary Table 1.
RNA sequencing
Colon tissues were cut and homogenized in TRIzol on ice. Tissue RNA was subjected to mRNA sequencing utilizing next-generation sequencing (NGS) by Novogene Bio Technology Co., Ltd. (Beijing, China).
Colon explant culture
Approximately 100 mg of colon tissue was collected from each mouse, and the colons were opened longitudinally and washed twice with ice-cold PBS. Colon explants were cultured in 0.5 ml of DMEM/F-12 (per 100 mg of tissue) containing 10% FBS and 5× penicillin/streptomycin. Twelve hours later, the culture supernatant was harvested and centrifuged at 1000 × g for 10 min to remove tissue debris. The supernatant was collected for organoid treatment.
Fecal microbiota transplant (FMT)
The recipient mice were treated with an antibiotic cocktail (1 g/L ampicillin, 0.5 g/L vancomycin, 1 g/L neomycin, and 1 g/L metronidazole in sterilized water) for 3 weeks. The feces of the donor mice were collected on Day 3 and Day 4 of DSS treatment. The feces were dissolved in sterile PBS, filtered and centrifuged to collect the supernatant. The recipient mice were gavaged with the supernatant for 7 days for bacterial colonization and were then treated with DSS to induce colitis.
Immunoblotting
All cell lysates and the Prestained Protein Ladder (Thermo Fisher Scientific, MA, USA) were separated by 10% SDS-polyacrylamide gel electrophoresis and then transferred onto nitrocellulose membranes. After blocking with 5% skim milk in Tris-buffered saline containing 0.1% Tween 20 for 1 h at room temperature, the membranes were incubated with a specific primary antibody at 4 °C overnight. Then, the membranes were incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody. Protein signals were detected by a ChemiScope imaging system (Clinx, Shanghai, China) using an Ultra-Sensitive ECL Chemiluminescence Kit (Beyotime, Shanghai, China). Antibodies specific for the following proteins were used: GRP78, CHOP, p-p65, p65, p-eEF2, eEF2, p-eIF4E, p-ERK, ERK, p-JNK, JNK, p-p38, p38, cleaved caspase-3, and caspase 3. All antibodies were purchased from Cell Signaling Technology (Boston, MA, USA).
Metabolomic analysis
Peritoneal macrophages were seeded in 10 cm dishes and treated with LPS (1 μg/ml) for 9 h in the presence or absence of mannose (20 mM). The cells were removed by scraping in 60% chromatographic grade methanol and placed in liquid nitrogen for 15 min for rapid freezing. The samples were sent to Novogene Bio Technology Co., Ltd. (Beijing, China) for metabolomic analysis.
Seahorse metabolic assay
A total of 4 × 104 peritoneal macrophages were seeded in an XFe 96-well cell culture microplate in 200 μl of DMEM/F-12 medium, incubated at room temperature for 1 h, and then cultured overnight at 37 °C. After treatment, the DMEM/F-12 was removed, the wells were washed twice, and fresh assay medium was added. The ECAR was measured under basal conditions and after the addition of 10 mM glucose, 2 μM oligomycin, and 50 mM 2-deoxy-D-glucose as indicated. The data were collected with Wave software version 2.4 (Agilent, Palo Alto, CA, USA).
Polysome profiling assay
Polysome fractionation analysis was performed as previously described [13]. Briefly, cells were incubated with 100 μg/ml cycloheximide at 37 °C for 10 min and then lysed in lysis buffer (DEPC-treated water with 0.5% NP40, 100 mM KCl, 10 mM MgCl2, 20 mM Tris-HCl, 100 µg/ml cycloheximide, 2 mM DTT, 40 U/ml RNase inhibitor and protease inhibitor) at 4 °C for 20 min. Cell lysates were pelleted, and the supernatant was placed on top of a 10–50% continuous sucrose gradient. The tubes were centrifuged at 30000 rpm and 4 °C for 3 h. After ultracentrifugation, a pipette was used to carefully aspirate 12 fractions of the liquid and extract RNA for analysis.
Ultra-performance liquid chromatography (UPLC)
A serum sample (12 μl) was mixed with 28 μl of ultrapure water, 40 μl of 0.3 mM NaOH and 60 μl of 0.5 mM 1-phenyl-3-methyl-5-pyrazolone (PMP). The mixture was incubated at 70 °C for 1 h. After cooling, 80 μl od 0.3 mM hydrochloric acid was added to the test tube. The mixture was vigorously vortexed, and 500 μl chloroform was then added. The mixture was centrifuged at 8000 rpm for 3 min at 4 °C for separation into 2 layers. The chloroform layer was discarded, and the chloroform washing steps were repeated 3 times. The remaining layer was centrifuged at 13,000 rpm for 10 min at 4 °C. Then, 120 μl of the upper aqueous layer was transferred into 150 μl inserts with a preinstalled plastic spring (Waters WAT094171). The inserts were placed into 2 ml amber glass vials (Waters 186000846), and the vials were sealed with a screw cap (Waters 186000274). Then, the samples were centrifuged at 21000 rpm for 30 min at 4 °C. After centrifugation, the samples were loaded into an Agilent 1290 UPLC. Samples were monitored by a UV detector at excitation and emission wavelengths of 254 and 350 nm, respectively. The mannose concentration was calculated by comparing the peak area of derived mannose with that of glucose.
RNA immunoprecipitation (RIP)
Peritoneal macrophages were plated in 10 cm dishes overnight prior to the indicated treatment. The cells were collected with RIP lysis buffer and lysed by ultrasonication on ice. After centrifugation, the supernatants were collected and incubated with magnetic beads conjugated to an anti-eEF2 antibody (Abcam, Cambridge, MA, USA) or an anti-GAPDH antibody (Abcam, Cambridge, MA, USA) at 4 °C overnight. The next day, the supernatant was discarded, and the pellets were resuspended in 50 μl of Tris-EDTA buffer containing 1% SDS at 65 °C for 20 min. Three microliters of 6× loading buffer was added to 15 μl of the supernatant for immunoblotting. Then, 8 μl of protein K (20 mg/ml) and 15 μl of NaCl (5 M) were added to the remaining 35 μl of the supernatant, decrosslinking was performed at 55 °C for 1 h, and RNA was extracted with TRIzol.
Statistical analysis
The data are presented as the means ± standard errors of the mean (SEMs). For comparisons between two groups, two-tailed unpaired Student’s t test was performed using GraphPad Prism (version 8.0). P < 0.05 was considered statistically significant.
Supplementary information
Supplementary table 1 - Primer sequences
Author contributions
Conceptualization: PX, YK. Methodology: QC, ZH, HZ, PX, YK, HC, XZ. Investigation: ZH, HZ, JL, TP, RTM, KG, MS, PX. Visualization: ZH, HZ, JL, PX. Funding acquisition: TP, PX, QC, YK. Project administration: PX, YK, XZ, HC, QC. Supervision: PX, YK, XZ, QC. Writing—original draft: ZH, JL, PX, RTM, YK. Writing—review and editing: ZH, JL, PX, RTM, YK.
Funding
This work was supported by the National Natural Science Foundation of China (82271862 and 81873418 to YK, 82171730 to PX, 81970484 to QC), the Zhejiang Provincial Ten Thousand Program for Leading Talents of Science and Technology Innovation (2021R52015 to YK), and the Natural Science Foundation of Zhejiang Province (LY20H160032 to PX, LQ21H090064 to TP).
Data availability
All data associated with this study are presented in the paper or Supplementary Materials and are available from the corresponding author upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Peng Xiao, Ziwei Hu.
Contributor Information
Peng Xiao, Email: tulipxp@zju.edu.cn.
Qian Cao, Email: caoq@zju.edu.cn.
Yuehai Ke, Email: yke@zju.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41423-022-00955-1.
References
- 1.Sharma V, Ichikawa M, Freeze HH. Mannose metabolism: more than meets the eye. Biochem Biophys Res Commun. 2014;453:220–8. doi: 10.1016/j.bbrc.2014.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ala-Jaakkola R, Laitila A, Ouwehand AC, Lehtoranta L. Role of D-mannose in urinary tract infections - a narrative review. Nutr J. 2022;21:18. doi: 10.1186/s12937-022-00769-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gonzalez PS, O’Prey J, Cardaci S, Barthet VJA, Sakamaki JI, Beaumatin F, et al. Mannose impairs tumour growth and enhances chemotherapy. Nature. 2018;563:719–23. doi: 10.1038/s41586-018-0729-3. [DOI] [PubMed] [Google Scholar]
- 4.Eugene SP, Reddy VS, Trinath J. Endoplasmic Reticulum Stress and Intestinal Inflammation: A Perilous Union. Front Immunol. 2020;11:543022. doi: 10.3389/fimmu.2020.543022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao SS, Zimmermann EM, Chuang BM, Song B, Nwokoye A, Wilkinson JE, et al. The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology. 2013;144:989–1000.e1006. doi: 10.1053/j.gastro.2013.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma V, Smolin J, Nayak J, Ayala JE, Scott DA, Peterson SN, et al. Mannose Alters Gut Microbiome, Prevents Diet-Induced Obesity, and Improves Host Metabolism. Cell Rep. 2018;24:3087–98. doi: 10.1016/j.celrep.2018.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Powell N, Pantazi E, Pavlidis P, Tsakmaki A, Li K, Yang F, et al. Interleukin-22 orchestrates a pathological endoplasmic reticulum stress response transcriptional programme in colonic epithelial cells. Gut. 2020;69:578–90. doi: 10.1136/gutjnl-2019-318483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heijmans J, van Lidth de Jeude JF, Koo BK, Rosekrans SL, Wielenga MC, van de Wetering M, et al. ER stress causes rapid loss of intestinal epithelial stemness through activation of the unfolded protein response. Cell Rep. 2013;3:1128–39. doi: 10.1016/j.celrep.2013.02.031. [DOI] [PubMed] [Google Scholar]
- 9.Andresen L, Skovbakke SL, Persson G, Hagemann-Jensen M, Hansen KA, Jensen H, et al. 2-deoxy D-glucose prevents cell surface expression of NKG2D ligands through inhibition of N-linked glycosylation. J Immunol. 2012;188:1847–55. doi: 10.4049/jimmunol.1004085. [DOI] [PubMed] [Google Scholar]
- 10.Xiao P, Zhang H, Zhang Y, Zheng M, Liu R, Zhao Y, et al. Phosphatase Shp2 exacerbates intestinal inflammation by disrupting macrophage responsiveness to interleukin-10. J Exp Med. 2019;216:337–49. doi: 10.1084/jem.20181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Na YR, Stakenborg M, Seok SH, Matteoli G. Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD. Nat Rev Gastroenterol Hepatol. 2019;16:531–43. doi: 10.1038/s41575-019-0172-4. [DOI] [PubMed] [Google Scholar]
- 12.Cameron AM, Castoldi A, Sanin DE, Flachsmann LJ, Field CS, Puleston DJ, et al. Inflammatory macrophage dependence on NAD(+) salvage is a consequence of reactive oxygen species-mediated DNA damage. Nat Immunol. 2019;20:420–32. doi: 10.1038/s41590-019-0336-y. [DOI] [PubMed] [Google Scholar]
- 13.Panda AC, Martindale JL, Gorospe M. Polysome Fractionation to Analyze mRNA Distribution Profiles. Bio Protoc. 2017;7:e2126. [DOI] [PMC free article] [PubMed]
- 14.Millet P, Vachharajani V, McPhail L, Yoza B, McCall CE. GAPDH Binding to TNF-alpha mRNA Contributes to Posttranscriptional Repression in Monocytes: A Novel Mechanism of Communication between Inflammation and Metabolism. J Immunol. 2016;196:2541–51. doi: 10.4049/jimmunol.1501345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galvan-Pena S, Carroll RG, Newman C, Hinchy EC, Palsson-McDermott E, Robinson EK, et al. Malonylation of GAPDH is an inflammatory signal in macrophages. Nat Commun. 2019;10:338. doi: 10.1038/s41467-018-08187-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaul G, Pattan G, Rafeequi T. Eukaryotic elongation factor-2 (eEF2): its regulation and peptide chain elongation. Cell Biochem Funct. 2011;29:227–34. doi: 10.1002/cbf.1740. [DOI] [PubMed] [Google Scholar]
- 17.Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999;68:913–63. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- 18.Ichikawa M, Scott DA, Losfeld ME, Freeze HH. The metabolic origins of mannose in glycoproteins. J Biol Chem. 2014;289:6751–61. doi: 10.1074/jbc.M113.544064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freeze HH. Towards a therapy for phosphomannomutase 2 deficiency, the defect in CDG-Ia patients. Biochim Biophys Acta. 2009;1792:835–40. doi: 10.1016/j.bbadis.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramirez MA, Borja NL. Epalrestat: an aldose reductase inhibitor for the treatment of diabetic neuropathy. Pharmacotherapy. 2008;28:646–55. doi: 10.1592/phco.28.5.646. [DOI] [PubMed] [Google Scholar]
- 21.Iyer S, Sam FS, DiPrimio N, Preston G, Verheijen J, Murthy K, et al. Repurposing the aldose reductase inhibitor and diabetic neuropathy drug epalrestat for the congenital disorder of glycosylation PMM2-CDG. Dis Model Mech. 2019;12:dmm040584. [DOI] [PMC free article] [PubMed]
- 22.Ligezka AN, Radenkovic S, Saraswat M, Garapati K, Ranatunga W, Krzysciak W, et al. Sorbitol Is a Severity Biomarker for PMM2-CDG with Therapeutic Implications. Ann Neurol. 2021;90:887–900. doi: 10.1002/ana.26245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaser A, Blumberg RS. Endoplasmic reticulum stress in the intestinal epithelium and inflammatory bowel disease. Semin Immunol. 2009;21:156–63. doi: 10.1016/j.smim.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cosin-Roger J, Ortiz-Masia D, Calatayud S, Hernandez C, Esplugues JV, Barrachina MD. The activation of Wnt signaling by a STAT6-dependent macrophage phenotype promotes mucosal repair in murine IBD. Mucosal Immunol. 2016;9:986–98. doi: 10.1038/mi.2015.123. [DOI] [PubMed] [Google Scholar]
- 25.Smith AM, Rahman FZ, Hayee B, Graham SJ, Marks DJ, Sewell GW, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J Exp Med. 2009;206:1883–97. doi: 10.1084/jem.20091233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen Y, Kapfhamer D, Minnella AM, Kim JE, Won SJ, Chen Y, et al. Bioenergetic state regulates innate inflammatory responses through the transcriptional co-repressor CtBP. Nat Commun. 2017;8:624. doi: 10.1038/s41467-017-00707-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moreno-Fernandez ME, Giles DA, Oates JR, Chan CC, Damen M, Doll JR, et al. PKM2-dependent metabolic skewing of hepatic Th17 cells regulates pathogenesis of non-alcoholic fatty liver disease. Cell Metab. 2021;33:1187–204.e189. doi: 10.1016/j.cmet.2021.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Torretta S, Scagliola A, Ricci L, Mainini F, Di Marco S, Cuccovillo I, et al. D-mannose suppresses macrophage IL-1beta production. Nat Commun. 2020;11:6343. doi: 10.1038/s41467-020-20164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hotta N, Sakamoto N, Shigeta Y, Kikkawa R, Goto Y. Clinical investigation of epalrestat, an aldose reductase inhibitor, on diabetic neuropathy in Japan: multicenter study. Diabetic Neuropathy Study Group in Japan. J Diabetes Complications. 1996;10:168–72. doi: 10.1016/1056-8727(96)00113-4. [DOI] [PubMed] [Google Scholar]
- 30.Grabinger T, Luks L, Kostadinova F, Zimberlin C, Medema JP, Leist M, et al. Ex vivo culture of intestinal crypt organoids as a model system for assessing cell death induction in intestinal epithelial cells and enteropathy. Cell Death Dis. 2014;5:e1228. doi: 10.1038/cddis.2014.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table 1 - Primer sequences
Data Availability Statement
All data associated with this study are presented in the paper or Supplementary Materials and are available from the corresponding author upon reasonable request.