

Abstract
Rationale
Idiopathic pulmonary arterial hypertension (PAH) is a terminal pulmonary vascular disease characterized by increased pressure, right ventricular failure, and death. PAH exhibits a striking sex bias and is up to four times more prevalent in females. Understanding the molecular basis behind sex differences could help uncover novel therapies.
Objectives
We previously discovered that the Y chromosome is protective against hypoxia-induced experimental pulmonary hypertension (PH), which may contribute to sex differences in PAH. Here, we identify the gene responsible for Y-chromosome protection, investigate key downstream autosomal genes, and demonstrate a novel preclinical therapy.
Methods
To test the effect of Y-chromosome genes on PH development, we knocked down each Y-chromosome gene expressed in the lung by means of intratracheal instillation of siRNA in gonadectomized male mice exposed to hypoxia and monitored changes in right ventricular and pulmonary artery hemodynamics. We compared the lung transcriptome of Uty knockdown mouse lungs to those of male and female PAH patient lungs to identify common downstream pathogenic chemokines and tested the effects of these chemokines on human pulmonary artery endothelial cells. We further inhibited the activity of these chemokines in two preclinical pulmonary hypertension models to test the therapeutic efficacy.
Measurements and Main Results
Knockdown of the Y-chromosome gene Uty resulted in more severe PH measured by increased right ventricular pressure and decreased pulmonary artery acceleration time. RNA sequencing revealed an increase in proinflammatory chemokines Cxcl9 and Cxcl10 as a result of Uty knockdown. We found CXCL9 and CXCL10 significantly upregulated in human PAH lungs, with more robust upregulation in females with PAH. Treatment of human pulmonary artery endothelial cells with CXCL9 and CXCL10 triggered apoptosis. Inhibition of Cxcl9 and Cxcl10 expression in male Uty knockout mice and CXCL9 and CXCL10 activity in female rats significantly reduced PH severity.
Conclusions
Uty is protective against PH. Reduction of Uty expression results in increased expression of proinflammatory chemokines Cxcl9 and Cxcl10, which trigger endothelial cell death and PH. Inhibition of CLXC9 and CXLC10 rescues PH development in multiple experimental models.
Keywords: CXCL9, CXCL10, sex differences, endothelial cells, pulmonary arterial hypertension
At a Glance Commentary
Scientific Knowledge on the Subject
Idiopathic pulmonary arterial hypertension (PAH) is a pulmonary vascular disease that displays a marked sex bias with females being up to four times more affected. Decades of research have been spent studying the root cause of this sexual dimorphism, with an emphasis on studying the role of sex hormones. Our lab was the first to investigate the potential role of sex chromosomes in the context of PAH. We found that the male-specific Y-chromosome is protective against experimental pulmonary hypertension (PH); however, the mechanism of this protection was not known.
What This Study Adds to the Field
This study, an in-depth continuation of our previous research, identifies that the Y-chromosome gene, Uty, confers protection against the development of PAH in males by suppressing a proinflammatory pathway in the lungs. When Uty is lowly expressed in males or absent in females, increased expression of chemokines Cxcl9 and Cxcl10 result in vascular endothelial cell dysfunction and more severe PH development. We also demonstrate that targeting this downstream inflammatory pathway is sufficient to reduce PH severity in two preclinical rat models.
Pulmonary arterial hypertension (PAH) is a pulmonary vascular disease characterized by increased pressure in the pulmonary arteries leading to right ventricular (RV) hypertrophy, RV failure, and death. Pressure overload begins in the distal pulmonary arteries, which undergo vascular remodeling largely characterized by vascular endothelial cell (EC) dysfunction and aberrant smooth muscle cell proliferation (1). Inflammation plays a key role in this vascular remodeling, as it is known that PAH is concomitant with a proinflammatory lung milieu and inflammatory regulators are associated with PAH disease progression and severity (1–3).
The idiopathic form of PAH exhibits striking sex differences where females are up to four times more likely to be diagnosed than men (4). As female sex is a risk factor for developing PAH (5), the interplay between sex hormones and PAH pathogenesis has been actively investigated, with particular attention to estrogens. Estrogen signaling has been implicated in PAH pathogenesis and shown to reduce expression of bone morphogenic protein 2 (BMPR2), a gene associated with PAH due to autosomal dominant mutations (6, 7). However, animal studies have also demonstrated that circulating estrogen is protective against pulmonary hypertension (PH) severity, and estrogen treatment has been shown to prevent and rescue PH (8–10) through mediating RV compensation to pressure overload (11, 12). These powerful yet paradoxical studies indicate that other sex-biasing factors contribute to the sex differences observed in PAH.
Our group examined the influence of sex chromosomes in PH and demonstrated that, in the absence of circulating gonadal hormones, the male-specific Y chromosome (ChrY) is protective against experimental PH (13). Although this study may explain why males are more protected against developing PAH, the mechanism by which ChrY protection is achieved has yet to be discovered and may lead to a greater understanding around the pathogenesis of PAH and elucidate novel therapeutic strategies. ChrY genes are known to have widespread effects on autosomal gene regulation (14) and have been implicated in systemic diseases (15, 16).
In the present study, we investigated the role of the four ChrY genes that are expressed in lung tissue for their potential involvement in PH protection. We identified Uty (ubiquitously transcribed tetratricopeptide repeat containing, Y-linked) as the protective ChrY gene. We also identified two proinflammatory chemokines, CXCL9 and CXCL10, downstream of Uty that contribute to PH pathogenesis in a sex-specific manner and found that knockdown (KD) of these chemokines is sufficient to reduce PH severity in global Uty knockout (KO) mice. We found that inhibition of the shared CXCL9 and CXCL10 receptor significantly reduced PH development in two experimental models of PH in female rats, suggesting that targeting autosomal genes downstream of Uty is sufficient to extend ChrY protection to females with PH.
Methods
For detailed methods, see the online supplement.
Models of Experimental PH and Treatments
ChrY KD experiments
Male C57BL/6J mice underwent gonadectomy (GDX) and, 30 days later, received recurring intratracheal instillation of short interfering RNA (siRNA) targeting a ChrY gene of interest or a scrambled control. All siRNAs were formulated for in vivo use. Mice were housed in hypoxia (Hx, 10% oxygen) for 3 weeks.
Cxcl9 and Cxcl10 KD experiments
Male global Uty KO (Uty-KO) mice and wild-type (WT) controls underwent GDX. After 30 days, mice received recurring intratracheal instillation of nontargeting siRNA (WT and Uty-KO groups) or a mixture of siRNAs targeting Cxcl9 and Cxcl10 (Uty-KO Si-Cxcl group). All siRNAs were formulated for in vivo use. Mice were housed in Hx for 3 weeks.
Cxcr3 inhibition
Female gonad-intact rats received a subcutaneous injection of either monocrotaline (MCT) or Sugen 5416. In the MCT model, 2 weeks after MCT injection, animals were injected subcutaneously every 12 hours with AMG487 (1 mg/kg) or vehicle for 2 weeks. In the Sugen-Hx (SuHx) model, after the injection of Sugen 5416, animals were housed in Hx for 3 weeks and then returned to normoxia for 2 weeks, where they received a subcutaneous injection of AMG487 (1 mg/kg) or vehicle every 12 hours.
Echocardiography
Two-dimensional Doppler and B-mode echocardiography were used to quantify pulmonary artery and RV parameters.
Hemodynamics and Gross Anatomical Evaluation
RV systolic pressure (RVSP) was measured by means of direct, open-chested catherization. The heart was dissected and weighed to calculate the Fulton index, a ratio of RV weight to left ventricle (LV) plus intraventricular septum (IVS) weight: RV/(LV + IVS).
Tissue Preparation, Staining, and Imaging
Tissues were formalin fixed, embedded in optical cutting temperature compound (Tissue-Tek), and stained according to product specifications. Formalin-fixed, paraffin-embedded human lung sections were obtained from the Pulmonary Hypertension Breakthrough Initiative repository.
Real-Time qPCR
Quantitative reverse transcription PCR (RT-qPCR) was performed using iTaq Universal SYBR and reverse transcribed using the Omniscript RT Kit (Qiagen).
Bioinformatic Analysis
Total lung RNA was sequenced by the University of California, Los Angeles, Technology Center for Genomics & Bioinformatics. Differently expressed genes (DEGs) with a false discovery rate <0.1 were considered statistically significant. Public microarray data were used to define a list of inflammatory genes expressed higher in female versus male PAH patient lungs (17).
Cell Studies
Bone marrow was extracted from male Uty-KO or WT mice and incubated with colony-stimulating factor (20 ng/ml). Bone marrow–derived macrophages (BMDMs) were treated with interferon gamma (10 ng/ml) for 4 hours, and Cxcl9 and Cxcl10 expression was measured by means of RT-qPCR.
Healthy human male pulmonary artery ECs (PAECs) were spiked with CXCL9 (2 μM), CXCL10 (5 μM), or vehicle for 24 hours and stained for cleaved caspase-3.
Statistics
Mann–Whitney tests were used for comparisons of two groups. Two-way ANOVAs were used for comparisons of four groups. A significance level less than 5% (P < 0.05) was deemed significant. Values are expressed as mean ± SEM.
Results
Lung-Specific KD of Uty, but Not Other ChrY Genes Expressed in the Lung, Eliminates ChrY Protection against Hx-Induced PH
Our previously published work demonstrated that, in the absence of gonadal hormones, ChrY is protective against Hx-induced PH in mice (13). We identified only four protein-coding ChrY genes expressed in mouse lung tissue, Ddx3y (Y-linked DEAD-box helicase 3), Eif2s3y (eukaryotic translation initiation factor 2, subunit 3, structural gene Y-linked), Kdm5d (lysine demethylase 5D), and Uty (13). Three of these genes, Ddx3y, Kdm5d, and Uty, are conserved in humans, and KDM5D and UTY were both found to be downregulated in male PAH patient lungs compared with healthy patient lungs (13).
To determine which ChrY gene is responsible for PH protection, we individually knocked down each ChrY candidate genes in the lungs of GDX Hx mice and measured the effect on PH development (Figure 1A). We found no significant change in the RVSP, Fulton index, or pulmonary artery acceleration time (PAAT) of mice with KD of Kdm5d (Figure 1B; Figures E1A, E1D, and E1G in the online supplement), Ddx3Y (Figure 1C; Figures E1B, E1E, and E1H), or Eif2s3y (Figure 1D; Figures E1C, E1F, and E1I). However, KD of Uty resulted in significantly elevated RVSP compared with the Si-Scrm control group in two separate experiments (Figure 1E), which was concomitant with significantly lower PAAT and ratio of PAAT to RV ejection time (RVET) (Figure 1F; Figure E2A), although there was no significant change in RV hypertrophy (Figure 1G). As experimental validation, we confirmed that WT mice subjected to Hx exhibited significantly elevated RVSP compared with normoxic mice and that Si-Scrm treatment alone had no significant effect on RV pressure (Figure E2B).
Figure 1.
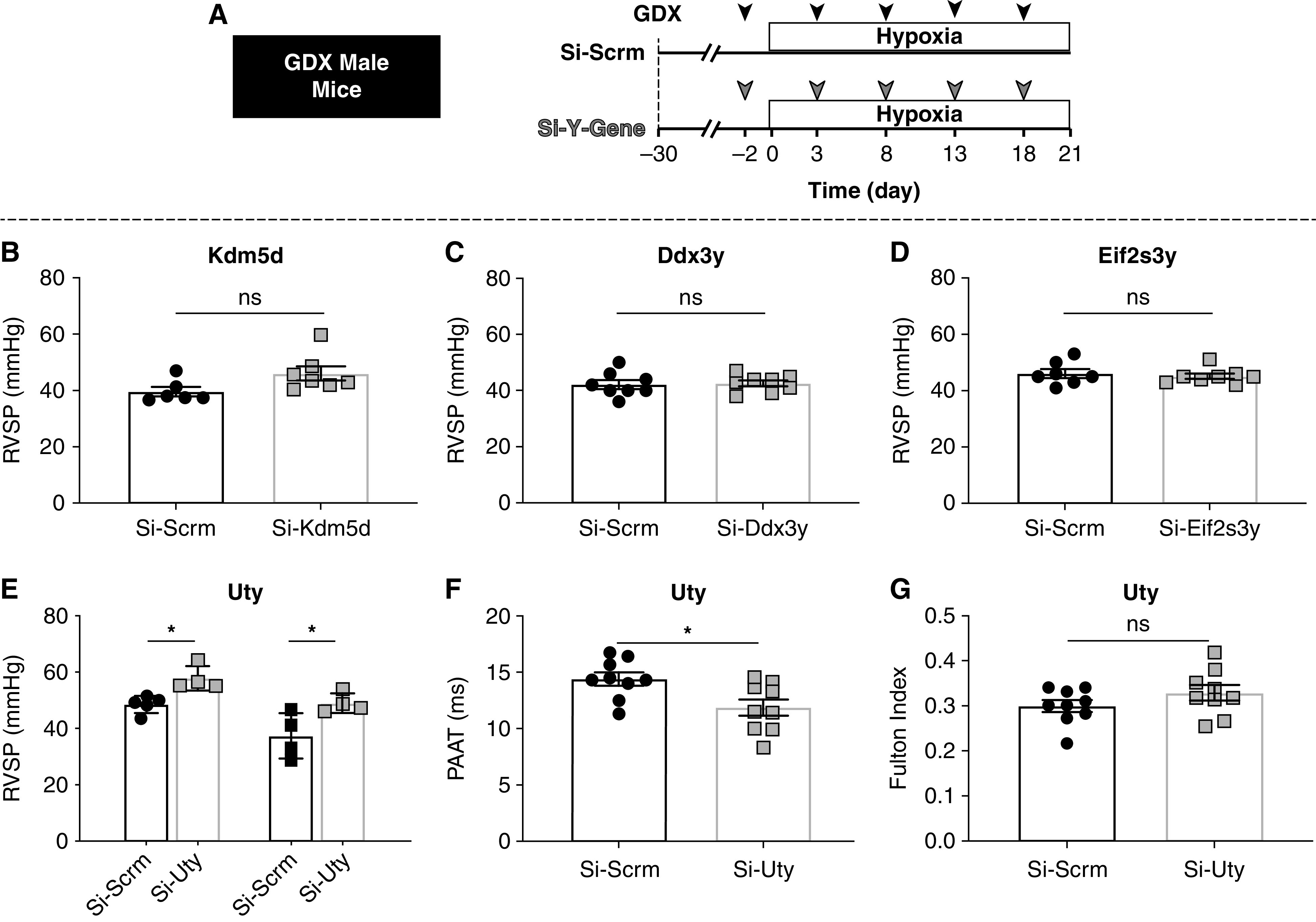
Lung-specific knockdown of Uty, but not other Y-chromosome (ChrY) genes expressed in the lung, eliminates ChrY protection against hypoxia (Hx)-induced pulmonary hypertension. (A) Experimental protocol: 30 days after gonadectomy (GDX), male mice receive scramble siRNA or siRNA targeting ChrY genes every 5 days and are placed in Hx for 3 weeks. (B–E) Right ventricular systolic pressure (RVSP) measured by direct RV catheterization for (B) Kdm5d, (C) Ddx3y, (D) Eif2s3y, and (E) Uty. (F) Pulmonary artery acceleration time (PAAT) measured by echocardiography. (G) Fulton index (RV/LV + IVS). *P < 0.05. IVS = intraventricular septum; LV = left ventricle; ns = not significant.
We found that Uty expression in Si-Uty–treated lungs was reduced by >50% compared with Si-Scrm lungs (Figure E3A). We further quantified cell-specific Uty knockdown and found that >50% KD was achieved in CD68+ macrophages and vascular ECs and smooth muscle cells, indicating that the siRNA successfully reduced Uty transcripts in the cell types most relevant to PH and our study (Figures E3B–E3D).
We examined Uty expression in the total lung tissue of three different rodent models of PH and found that Uty is downregulated as a result of Hx-induced PH in male mice and both the MCT and SuHx PH models in male rats, which is consistent with the downregulation of UTY found in human male PAH lungs (13) (Figure E4). We tested whether downregulation of Uty is a result of a pathogenic process in PH and PAH or a byproduct of hypoxia-inducible factor (Hif) upregulation, as Hif is upregulated in Hx and known to be associated with PH development (18). We found no change in Uty expression in response to Hif1a upregulation in alveolar macrophages (Figures E5A–E5D), indicating that Uty is not directly regulated by Hif1a. We also tested whether Hif1a is regulated by Uty expression and found no change in Hif1a expression as a result of global Uty-KO (Figures E5E–E5G). Our data suggest that Uty is downregulated as a result of the PH disease process but not because of an interaction with Hif1a alone.
In summary, these data indicate that Uty confers protection against PH. Reduction of Uty, but not the other ChrY genes expressed in the lung, is sufficient to exacerbate PH in GDX male mice, and PH and PAH pathology further downregulates Uty in a Hif1a-independent manner.
Proinflammatory Chemokines CXCL9 and CXCL10 Are Autosomal Downstream Deterrents of Uty Protection and Are Upregulated in PAH Patient Lungs in a Sex-Specific Manner
To unravel the molecular mechanism underlying Uty protection in PH, we performed RNA sequencing on Hx Si-Scrm and Si-Uty mouse lung tissue and identified 523 DEGs (Figure 2A). Pathway enrichment analysis highlighted five main pathways, with inflammation particularly enriched (Figure 2B). Because inflammation plays a major role in PAH pathogenesis and severity (2), we focused on inflammatory DEGs. We cross-referenced our inflammatory DEG set with an online microarray of PAH patient lung tissue and searched for genes that were elevated in both Si-Uty, compared with Si-Scrm, and female PAH samples, compared with male samples (17). This analysis yielded two top genes of interest: proinflammatory chemokines CXCL9 and CXCL10 (Figure 2C), both of which we validated to be upregulated in male and female PAH lungs compared with healthy lungs, with a more robust upregulation in female PAH patients compared with male patients (Figures 2D and 2F). These data are represented by immunofluorescence images of human healthy and PAH lungs (Figures 2E and 2G).
Figure 2.
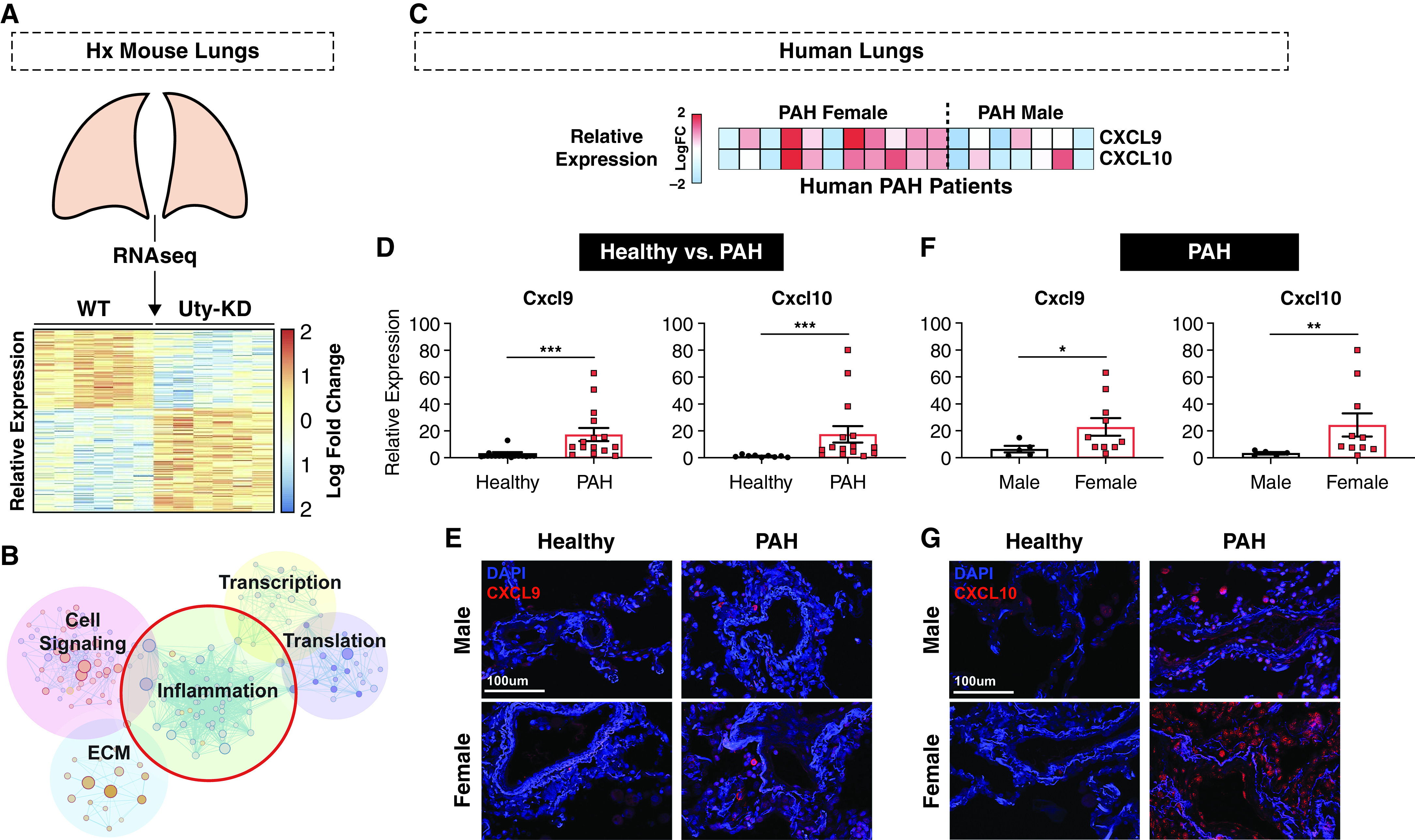
Proinflammatory chemokines CXCL9 and CXCL10 are autosomal downstream deterrents of Uty protection and are upregulated in PAH patient lungs in a sex-specific manner. (A) Comparison of RNA-sequencing (RNAseq) data from Si-Scrm and Si-Uty Hx mouse lungs revealed 523 differently expressed genes (DEGs) as represented by vertical columns on a heatmap depicting log fold change in expression. (B) Pathways enriched with DEGs include inflammation, cell signaling, extracellular matrix (ECM), transcription, and translation. (C) Integration of RNAseq data with an online microarray dataset of male and female human PAH lung samples revealed Cxcl9/CXCL9 and Cxcl10/CXCL10 as upregulated in PAH females (versus PAH males) and Si-Uty mice (versus Si-Scrm) as depicted in a heatmap representing individual patients’ relative gene expression. (D) Relative expression of CXCL9 and CXCL10 in human healthy and PAH lungs as measured by real-time quantitative PCR (qPCR). (E–G) Representative immunofluorescence staining of CXCL9 (E) and CXCL10 (G) in male and female lung tissue from healthy and diseased (PAH) patients. (F) Relative expression of CXCL9 and CXCL10 in human male and female PAH lungs as measured by real-time qPCR. Scale bars, 100 μm. *P < 0.05, **P < 0.01, and ***P < 0.001. Hx = hypoxia; KD = knockdown; WT = wild-type.
Taken together, we found that downregulation of Uty is concomitant with upregulation of Cxcl9 and Cxcl10 in Uty-KD mouse lungs (Figures E6A and E6B) and that CXCL9 and CXCL10 expression levels are upregulated in human PAH lungs in a sex-specific manner.
Global Uty-KO Promotes More Severe PH that Is Prevented by KD of Cxcl9 and Cxcl10
Because lung-specific Uty-KD resulted in increased PH severity and elevated Cxcl9 and Cxcl10 expression, we tested 1) whether global Uty-KO mice also developed more severe PH and 2) whether knocking down Cxcl9 and Cxcl10 in Uty-KO mice would be sufficient to reduce PH severity (Figure 3A). We found that GDX UTY-KO male mice exposed to chronic Hx exhibited elevated RV pressures and a reduced PAAT and PAAT/RVET ratio compared with GDX WT males (Figures 3B and 3C; Figure E7). More severe PH was also observed in gonad-intact Hx Uty-KO animals compared with intact Hx WT males, although there was no significant interaction between gonadal hormones and PH severity (Figures E8A–E8E). Combined KD of Cxcl9 and Cxcl10 in the lungs of Hx GDX Uty-KO mice resulted in a significant reduction in RV pressures and RV hypertrophy, coupled with a significant increase in PAAT and PAAT/RVET ratio compared with that in Uty-KO controls (Figures 3B–3D; Figure E7). We validated that Cxcl9 and Cxcl10 expressions were significantly reduced in the lungs of the Uty-KO mice that received targeting siRNA compared with Uty-KO mice that received nontargeting Si-Scrm (Figure 3E).
Figure 3.

Global Uty-KO promotes more severe PH, which is prevented by KD of Cxcl9 and Cxcl10. (A) Experimental protocol: 30 days after GDX, age-matched global Uty-KO or WT male mice receive scramble siRNA or siRNA targeting Cxcl9 and Cxcl10 genes every 3–5 days and are placed in Hx for 3 weeks. (B) RVSP measured by direct RV catheterization. (C) PAAT measured by echocardiography. (D) Fulton index (RV/LV + IVS). (E) Quantification of Cxcl9 and Cxcl10 transcripts in whole lung tissue by means of real-time qPCR (normalized to Si-Scrm). *P < 0.05 and ***P < 0.001. GDX = gonadectomy; HX = hypoxia; IVS = intraventricular septum; KD = knockdown; KO = knockout; LV = left ventricle; ns = not significant; PAAT = pulmonary artery acceleration time; PH = pulmonary hypertension; RV = right ventricle; RVSP = right ventricular systolic pressure; WT = wild type.
Overall, Uty-KO male mice exhibited worse PH severity compared with WT mice when exposed to Hx. Lung-specific KD of Cxcl9 and Cxcl10 is sufficient to attenuate PH development in GDX Uty-KO mice.
Uty, Cxcl9, and Cxcl10 Are Colocalized in Macrophages, and Uty Expression Is Inversely Related to Cxcl9 and Cxcl10 Expression
Given the increasing importance of macrophages in PH pathobiology (19–22) and macrophage expression of both CXCL9 and CXCL10 (23), we tested for Uty expression in lung macrophages. Stained tissue sections from mouse and human lungs confirmed Uty expression within Cd68+ macrophages in male tissue but not in female tissue (Figures E9A and E9B). Although we found Uty inversely related to chemokines Cxcl9 and Cxcl10, we examined lung tissue for colocalization within macrophages and found Uty transcript colocalized with Cxcl9 (Figures 4A and 4C) and Cxcl10 (Figures 4B and 4D).
Figure 4.

Uty, Cxcl9, and Cxcl10 are colocalized in macrophages, and Uty expression is inversely related to Cxcl9 and Cxcl10 expression. (A–D) Representative images depicting colocalization of in situ probes labeling Cd68/CD68 (red), Uty/UTY (green), and Cxcl9/CXCL9 (white) in mouse (A) and human lung sections (C). Representative images depicting colocalization of in situ probes labeling Cd68/CD68 (red), Uty/UTY (green), and Cxcl10/CXCL10 (white) in mouse (B) and human lung sections (D). (E) Schematic of bone marrow–derived macrophage (BMDM) in vitro experiments. (F and G) Relative expression of Cxcl9 (F) and Cxcl10 (G), as measured by real-time qPCR in BMDMs isolated from WT and Uty-KO mice. *P < 0.05 and **P < 0.01. Scale bars, 50 μm. KO = knockout; WT = wild type.
To identify whether Uty expression in macrophages directly influences Cxcl9 and Cxcl10 production, we measured Cxcl9 and Cxcl10 expression in BMDMs from male WT and Uty-KO mice (Figure 4E). BMDMs from Uty-KO mice expressed significantly more Cxcl9 and Cxcl10 compared with WT mice (Figures 4F and 4G).
UTY is a member of the Jumonji family of histone demethylases, but its role as an H3K27 demethylase is highly contested in literature (24–27). We examined whether the effects of UTY in the lung were mediated through demethylation and found that UTY expression in whole lung and isolated lung macrophages did not influence levels of H3K27 trimethylated protein as measured by Western blot (Figure E10).
In summary, we found that Uty, which colocalizes with Cxcl9 and Cxcl10 in lung macrophages, regulates Cxcl9 and Cxcl10 expression but has no apparent effect on H3K27 demethylation.
CXCL9 and CXCL10 Expression Triggers Pulmonary Artery EC Dysfunction
To identify the mechanism by which Uty downstream autosomal genes Cxcl9 and Cxcl10 promote PH severity, we performed in vitro cell studies using human PAECs. PAECs express the shared CXCL9 and CXCL10 receptor, CXCR3; and PAEC dysfunction, a hallmark of PAH pathogenies, is known to increase PH severity through dysregulation of angiogenesis, altered secretion of vasoactive agents, and increasing vascular permeability (28). We incubated human PAECs with recombinant human CXCL9 and CXCL10 protein and measured the effect on EC dysfunction (Figure 5A). Treatment of PAECs with either CXCL9 or CXCL10 resulted in increased PAEC apoptosis, as measured by quantification of cleaved caspase-3 staining (Figures 5B–5D).
Figure 5.

CXCL9 and CXCL10 recombinant protein triggers pulmonary artery endothelial cell (PAEC) dysfunction. (A) Schematic of human PAEC (hPAEC) experiments. (B and C) Quantification of percent apoptosis measured in PAECs treated with CXCL9 (B) or CXCL0 (C), as measured by cleaved caspase-3 immunofluorescence. (D) Representative images of cleaved caspase-3 (CC-3; red) immunofluorescence in vehicle, CXCL9-treated, and CXCL10-treated cells. *P < 0.05. Scale bar, 50 μm.
These data demonstrate that proinflammatory chemokines Cxcl9 and Cxcl10, which are upregulated in the absence of Uty, induce PAEC apoptosis in vitro.
Blocking the Activity of CXCL9 and CXCL10 in Females Is Sufficient to Rescue PH in Two Preclinical Rat Models
Our data suggest that Uty confers PH protection through downregulating Cxcl9 and Cxcl10 expression (Figure 3) and that CXCL9 and CXCL10 promote PAEC dysfunction (Figure 5). Because we found these proinflammatory chemokines elevated in human PAH patient lungs and particularly high in female PAH patients (Figure 2), we tested whether we could extend the mechanism of Uty protection to females with PH by blocking the activity of CXCL9 and CXCL10 using a small-molecular inhibitor (AMG487) targeting CXCR3 (Figures 6A and 6B).
Figure 6.

Blocking the activity of CXCL9 and CXCL10 is sufficient to rescue PH in two preclinical rat models. Schematic of in vivo experiments in female rats with (A) monocrotaline (MCT)-induced PH and (B) Sugen 5416-Hx (SuHx)–induced PH. All MCT model measurements are listed in the left column (A, C, E, G, I, and K), and SuHx measurements are listed in the right column (B, D, F, H, J, and L). (C and D) RVSP, (E and F) PAAT, and (G and H) Fulton index (RV/LV + IVS) measured in AMG487 (AMG)-treated PH rats compared with vehicle-treated PH controls. (I and J) Quantification and (K and L) representative images of apoptotic EC cells in CD31 (yellow) and cleaved caspase-3 (CC-3; pink) labeled sections from vehicle (VEH) and AMG-treated lungs. *P < 0.05 and **P < 0.01. Scale bars, 50 μm. CTRL = control; EC = endothelial control; INH = inhibition; IVS = intraventricular septum; LV = left ventricle; ns = not significant; PAAT = pulmonary artery acceleration time; RV = right ventricle; RVSP = right ventricular systolic pressure.
To better examine for protective effects, we used two preclinical models of PH in rats, the MCT and SuHx models, which are more severe than Hx in mice. We tested our preclinical treatment strategy on gonad-intact females to determine whether targeting CXCL9 and CXCL10 would be a viable treatment strategy for PAH. We found that AMG487 sufficiently rescued PH development in both the MCT and SuHx models, as rats treated with AMG487 2 or 3 weeks after PH stimulus exhibited significantly lower RV pressures concomitant with increased PAAT, PAAT/RVET ratios, and RV fractional area change (Figures 6C–6F; Figures E11A–E11D). In addition, RV hypertrophy was significantly reduced in AMG487-treated rats in the MCT model, and there was a trend toward reduced RV hypertrophy in the SuHx model (Figures 6G–6H). We validated that the RV pressures were elevated in both preclinical models compared with that in healthy control rats (Figure E12).
Because CXCL9 and CXCL10 induce PAEC apoptosis in vitro (Figure 5), we aimed to delineate whether this cellular mechanism contributes to the PH protection conferred by AMG487 treatment in rats. We found reduced EC apoptosis in the lungs of treated MCT and SuHx rats, which indicates that blocking the actions of CXCL9 and CXCL10 reduced pulmonary vascular EC dysfunction (Figures 6I–6L).
Taken together, our findings demonstrate that inhibiting the shared CXCL9 and CXCL10 receptor in gonad-intact female rats is a viable preclinical treatment strategy, as it reduced PH severity and lung vascular EC dysfunction in two experimental models of PH.
Discussion
We previously demonstrated that ChrY is protective against experimental PH (13), and this study is the first in the field to investigate the protective role of each ChrY gene expressed in the lung. We found that lung-specific KD of Uty—but not ChrY genes Eif2s3y, Ddx3y, or Kdm5d—in GDX male mice exposed to Hx increased PH severity, thereby eliminating ChrY protection (Figure 1). In agreement with our Uty-KD studies, our data also show global Uty-KO mice develop more severe PH than WT controls (Figure 3). We found that loss of Uty expression in PH lungs and cultured macrophages resulted in an upregulation of proinflammatory chemokines Cxcl9 and Cxcl10 (Figure 4; Figure E6). Interestingly, CXCL9 and CXCL10 are upregulated in PAH patient lungs with a more robust upregulation in female patients compared with males (Figure 2). We found that knocking down Cxcl9 and Cxcl10 was sufficient to prevent more severe PH in Uty-KO mice, indicating that Cxcl9 and Cxcl10 are downstream of Uty protection (Figure 3). Stimulating human PAECs with exogenous CXCL9 or CXCL10 was sufficient to trigger PAEC dysfunction, a hallmark of PAH pathogenesis (Figure 5). Furthermore, we found that blocking CXCL9 and CXCL10 activity is a novel PH therapeutic strategy, as it reduced PH severity and EC apoptosis in two preclinical models (Figure 6). Figure 7 summarizes our findings.
Figure 7.

Proposed mechanism of Uty/Cxcl9/Cxcl10 axis in PH pathogenesis. Uty absence (in females) or reduced expression (in males with PAH or Uty-KD or in KO mice) results in an upregulation of proinflammatory chemokines Cxcl9 and Cxcl10 in the lung. CXCL9 and CXCL10 trigger vascular EC dysfunction, resulting in increased PH severity. Blocking CXCL9 and CXCL10 activity by pharmacologically inhibiting their shared receptor, CXCR3 is a novel treatment strategy to rescue PH development. EC = endothelial control; KD = knockdown; KO = knockout; PAH = pulmonary arterial hypertension; PH = pulmonary hypertension.
We believe that our study is the first to directly explain ChrY protection against PH. A recent study from Yan and colleagues noted a connection between the ChrY testis-determining gene Sry and the PAH-related gene Bmpr2 in cultured dermal fibroblasts (29). Unlike Uty, Sry expression is not detected in mouse or human lung tissue. Additionally, our previous study demonstrating ChrY protection utilized the well-published four core genotypes mouse model, which produces gonadal male and female mice with both XX and XY chromosomes (30). XY females, which were protected against PH compared with XX females, lack the testis-determining Sry gene (13). This indicates that Sry, a gene absent from lung tissue and the XY female mouse model, is not responsible for ChrY protection against PH.
Our study found that Uty regulates the expression of the proinflammatory chemokines CXCL9 and CXCL10. Dysregulation of these small proteins and their receptor (23), which are known to promote chemotaxis and immune cell differentiation, is associated with EC apoptosis (31–33) and a myriad of systemic inflammatory diseases (34–38). Before this study, CXCL9 and CXCL10 were already associated with PAH severity, and plasma levels of CXCL10 were found to be upregulated in PAH patients compared with healthy controls (36, 37). However, sex differences in CXCL9 and CXCL10 expression in PAH patients and the mechanism by which they promote PAH development were not known. Our study identifies a sexually dimorphic CXCL9 and CXCL10 upregulation in human PAH lung tissues (Figure 2), mechanistically links these chemokines to Uty expression and PH pathogenesis through PAEC dysfunction, and shows that targeting their activity is a novel PH therapy.
UTY is a member of the Jumonji family of H3K27 histone demethylases; however, it has undergone biochemical changes, and the demethylase activity of UTY is contested (24, 26, 27). Although Walport and colleagues demonstrated some residual demethylase activity in the human UTY enzyme (25), our study is in agreement with Lan and colleagues, Hong and colleagues, and Shpargel and colleagues, who found no UTY enzymatic activity (24, 26, 27). Regardless, Uty expression has a widespread effect on autosomal gene expression (14), and dysregulation of Uty is associated with disease (15, 39). We found Uty expression is downregulated in male PAH patient lung tissue (13) as well as in the lung tissue of three different experimental models of PH (Figure E4). Reduced UTY expression, specifically in macrophages, is associated with increased atherosclerosis risk in men (15, 40, 41), and macrophage-specific Uty-KD impairs immune activation (41). We confirmed colocalization of Uty with Cxcl9 and Cxcl10 in lung macrophages using RNAscope in situ hybridization and found that macrophages derived from Uty-KO BM have higher expression of Cxcl9 and Cxcl10, indicating that Uty expression in macrophages regulates Cxcl9 and Cxcl10 production (Figure 4). Although Hif1a signaling is associated with Hx and PH development through chemokine signaling (18), we did not see evidence of a Hif1a/Uty interaction (Figure E5). RNAscope in situ probes were used for imaging after we determined that UTY antibodies likely detected ChrX paralog UTX, as they revealed a signal in females. We believe that our RNAscope images are the first to depict specific Uty expression in mouse and human lung tissue (Figure E9B).
Our study provides mechanistic insight into ChrY protection in PH and elucidates a novel therapeutic strategy targeting the activity of Uty downstream mediators upregulated in male and female PAH patients (Figure 7). Although these results help explain why females who lack UTY have a higher incidence of PAH, they do not explain why PAH males have worse prognoses and clinical outcomes (42). We hypothesize that reduced UTY expression in some males, potentially because of a paternal haplotype that is known to influence UTY expression (43), could prime them for PAH development and elevated Cxcl9 and Cxcl10 expression compared with males with higher UTY expression. Additionally, there are known sex differences in the immune system (44, 45), and further reduction of UTY expression in male lungs as a result of PAH pathology may trigger a harmful immune cascade that contributes to more severe PAH. There is also a known relationship between RV remodeling and PAH prognosis, which is linked to sex hormones (46, 47). In particular, testosterone was shown to contribute to more severe RV remodeling in PH, which could account for male PAH patients having worse prognoses (48). Although Uty expression may initially protect males from developing PAH compared with females, increased testosterone in males may lead to worse outcomes. Our studies did not find a significant interaction of gonadal hormones and PH severity in global Uty-KO mice (Figure E8E); however, more investigation into the interaction between Uty and sex-biasing factors could provide additional insight into the complex and paradoxical sex differences in PAH.
Acknowledgments
Acknowledgment
The authors thank Mylène Vaillancourt, Crystal Eshraghi, and Xuqi Chen for their help with experimental procedures; Tristin Grogan for help with statistics; and Karl Shpargel for helpful discussions. We would also like to acknowledge the Translational Pathology Core Laboratory, the Technology Center for Genomics & Bioinformatics, and the Flow Cytometry Core Facility at the University of California, Los Angeles, for their assistance with tissue preparation, RNA sequencing, and cell sorting, respectively. We thank the Pulmonary Hypertension Breakthrough Initiative for their biobank of human PAH tissues. Global Uty-KO mice were obtained from the Mutant Mouse Resource & Research Centers.
Footnotes
Supported by American Heart Association grants 17PRE33420159 (C.M.C.), 20POST35210727, 17POST33670424, and 20CDA353550059 (G.R.); the Iris Cantor ‐ UCLA Women’s Health Center Executive Advisory Board NCATS UCLA CTSI grant UL1TR001881 (C.M.C.); and National Institutes of Health grants R01HL129051, R01HL147586, R01HL159865 (M.E.), and R01HL131182 (M.E. and A.P.A.).
Author Contributions: C.M.C., G.R., S.U., J.H., J.P., L.M., A.P.A., and M.E. provided intellectual input. C.M.C., G.R., S.U., M.L., M.D., and L.A. collected data. J.H. and G.R. analyzed bioinformatic data. H.H. performed gonadectomies and animal breeding. C.M.C., A.P.A., S.U., and M.E. interpreted data. C.M.C. analyzed data and wrote the manuscript. M.E. supervised the study.
This article has a data supplement, which is accessible from this issue’s table of contents online at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202110-2309OC on May 2, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest . 2012;122:4306–4313. doi: 10.1172/JCI60658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res . 2014;115:165–175. doi: 10.1161/CIRCRESAHA.113.301141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guignabert C, Tu L, Le Hiress M, Ricard N, Sattler C, Seferian A, et al. Pathogenesis of pulmonary arterial hypertension: lessons from cancer. Eur Respir Rev . 2013;22:543–551. doi: 10.1183/09059180.00007513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Walker AM, Langleben D, Korelitz JJ, Rich S, Rubin LJ, Strom BL, et al. Temporal trends and drug exposures in pulmonary hypertension: an American experience. Am Heart J . 2006;152:521–526. doi: 10.1016/j.ahj.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 5. Foderaro A, Ventetuolo CE. Pulmonary arterial hypertension and the sex hormone paradox. Curr Hypertens Rep . 2016;18:84. doi: 10.1007/s11906-016-0689-7. [DOI] [PubMed] [Google Scholar]
- 6. Fessel JP, Chen X, Frump A, Gladson S, Blackwell T, Kang C, et al. Interaction between bone morphogenetic protein receptor type 2 and estrogenic compounds in pulmonary arterial hypertension. Pulm Circ . 2013;3:564–577. doi: 10.1086/674312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hamada H, Kim MK, Iwakura A, Ii M, Thorne T, Qin G, et al. Estrogen receptors alpha and beta mediate contribution of bone marrow-derived endothelial progenitor cells to functional recovery after myocardial infarction. Circulation . 2006;114:2261–2270. doi: 10.1161/CIRCULATIONAHA.106.631465. [DOI] [PubMed] [Google Scholar]
- 8. Umar S, Iorga A, Matori H, Nadadur RD, Li J, Maltese F, et al. Estrogen rescues preexisting severe pulmonary hypertension in rats. Am J Respir Crit Care Med . 2011;184:715–723. doi: 10.1164/rccm.201101-0078OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rabinovitch M, Gamble WJ, Miettinen OS, Reid L. Age and sex influence on pulmonary hypertension of chronic hypoxia and on recovery. Am J Physiol . 1981;240:H62–H72. doi: 10.1152/ajpheart.1981.240.1.H62. [DOI] [PubMed] [Google Scholar]
- 10. Farhat MY, Chen MF, Bhatti T, Iqbal A, Cathapermal S, Ramwell PW. Protection by oestradiol against the development of cardiovascular changes associated with monocrotaline pulmonary hypertension in rats. Br J Pharmacol . 1993;110:719–723. doi: 10.1111/j.1476-5381.1993.tb13871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frump AL, Goss KN, Vayl A, Albrecht M, Fisher A, Tursunova R, et al. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: effects of endogenous and exogenous sex hormones. Am J Physiol Lung Cell Mol Physiol . 2015;308:L873–L890. doi: 10.1152/ajplung.00006.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nadadur RD, Umar S, Wong G, Eghbali M, Iorga A, Matori H, et al. Reverse right ventricular structural and extracellular matrix remodeling by estrogen in severe pulmonary hypertension. J Appl Physiol . 2012;113:149–158. doi: 10.1152/japplphysiol.01349.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Umar S, Cunningham CM, Itoh Y, Moazeni S, Vaillancourt M, Sarji S, et al. The Y chromosome plays a protective role in experimental hypoxic pulmonary hypertension. Am J Respir Crit Care Med . 2018;197:952–955. doi: 10.1164/rccm.201707-1345LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deschepper CF. Regulatory effects of the Uty/Ddx3y locus on neighboring chromosome Y genes and autosomal mRNA transcripts in adult mouse non-reproductive cells. Sci Rep . 2020;10:14900. doi: 10.1038/s41598-020-71447-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maan AA, Eales J, Akbarov A, Rowland J, Xu X, Jobling MA, et al. The Y chromosome: a blueprint for men’s health? Eur J Hum Genet . 2017;25:1181–1188. doi: 10.1038/ejhg.2017.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Case LK, Teuscher C. Y genetic variation and phenotypic diversity in health and disease. Biol Sex Differ . 2015;6:6. doi: 10.1186/s13293-015-0024-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miao R, Leng D, Wang Y, Li JF, Gong JN, Liang Y, et al. [Differentially expressed genes analysis in expression profile data of pulmonary fibrosis with pulmonary hypertension] Zhonghua Yi Xue Za Zhi . 2017;97:1240–1243. doi: 10.3760/cma.j.issn.0376-2491.2017.16.012. [DOI] [PubMed] [Google Scholar]
- 18. Pullamsetti SS, Mamazhakypov A, Weissmann N, Seeger W, Savai R. Hypoxia-inducible factor signaling in pulmonary hypertension. J Clin Invest . 2020;130:5638–5651. doi: 10.1172/JCI137558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li M, Riddle S, Kumar S, Poczobutt J, McKeon BA, Frid MG, et al. Microenvironmental regulation of macrophage transcriptomic and metabolomic profiles in pulmonary hypertension. Front Immunol . 2021;12:640718. doi: 10.3389/fimmu.2021.640718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pugliese SC, Kumar S, Janssen WJ, Graham BB, Frid MG, Riddle SR, et al. A time- and compartment-specific activation of lung macrophages in hypoxic pulmonary hypertension. J Immunol . 2017;198:4802–4812. doi: 10.4049/jimmunol.1601692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qiu H, Zhang Y, Li Z, Jiang P, Guo S, He Y, et al. Donepezil ameliorates pulmonary arterial hypertension by inhibiting M2-macrophage activation. Front Cardiovasc Med . 2021;8:639541. doi: 10.3389/fcvm.2021.639541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, Fink L, et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med . 2012;186:897–908. doi: 10.1164/rccm.201202-0335OC. [DOI] [PubMed] [Google Scholar]
- 23. Comerford I, McColl SR. Mini-review series: focus on chemokines. Immunol Cell Biol . 2011;89:183–184. doi: 10.1038/icb.2010.164. [DOI] [PubMed] [Google Scholar]
- 24. Shpargel KB, Sengoku T, Yokoyama S, Magnuson T. UTX and UTY demonstrate histone demethylase-independent function in mouse embryonic development. PLoS Genet . 2012;8:e1002964. doi: 10.1371/journal.pgen.1002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Walport LJ, Hopkinson RJ, Vollmar M, Madden SK, Gileadi C, Oppermann U, et al. Human UTY(KDM6C) is a male-specific Nϵ-methyl lysyl demethylase. J Biol Chem . 2014;289:18302–18313. doi: 10.1074/jbc.M114.555052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lan F, Bayliss PE, Rinn JL, Whetstine JR, Wang JK, Chen S, et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature . 2007;449:689–694. doi: 10.1038/nature06192. [DOI] [PubMed] [Google Scholar]
- 27. Hong S, Cho Y-W, Yu L-R, Yu H, Veenstra TD, Ge K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci USA . 2007;104:18439–18444. doi: 10.1073/pnas.0707292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation . 2004;109:159–165. doi: 10.1161/01.CIR.0000102381.57477.50. [DOI] [PubMed] [Google Scholar]
- 29. Yan L, Cogan JD, Hedges LK, Nunley B, Hamid R, Austin ED. The Y chromosome regulates BMPR2 expression via SRY: a possible reason ‘why’ fewer males develop PAH. Am J Respir Crit Care Med . 2018;198:1581–1583. doi: 10.1164/rccm.201802-0308LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burgoyne PS, Arnold AP. A primer on the use of mouse models for identifying direct sex chromosome effects that cause sex differences in non-gonadal tissues. Biol Sex Differ . 2016;7:68. doi: 10.1186/s13293-016-0115-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Darakhshan S, Hassanshahi G, Mofidifar Z, Soltani B, Karimabad MN. CXCL9/CXCL10 angiostasis CXC-chemokines in parallel with the CXCL12 as an angiogenesis CXC-chemokine are variously expressed in pre-eclamptic-women and their neonates. Pregnancy Hypertens . 2019;17:36–42. doi: 10.1016/j.preghy.2019.05.001. [DOI] [PubMed] [Google Scholar]
- 32. Green LA, Petrusca D, Rajashekhar G, Gianaris T, Schweitzer KS, Wang L, et al. Cigarette smoke-induced CXCR3 receptor up-regulation mediates endothelial apoptosis. Am J Respir Cell Mol Biol . 2012;47:807–814. doi: 10.1165/rcmb.2012-0132OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang X, Zhao Z, Zhu K, Bao R, Meng Y, Bian J, et al. Effects of CXCL4/CXCR3 on the lipopolysaccharide-induced injury in human umbilical vein endothelial cells. J Cell Physiol . 2019;234:22378–22385. doi: 10.1002/jcp.28803. [DOI] [PubMed] [Google Scholar]
- 34. Rabquer BJ, Tsou P-S, Hou Y, Thirunavukkarasu E, Haines GK, III, Impens AJ, et al. Dysregulated expression of MIG/CXCL9, IP-10/CXCL10 and CXCL16 and their receptors in systemic sclerosis. Arthritis Res Ther . 2011;13:R18. doi: 10.1186/ar3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - a target for novel cancer therapy. Cancer Treat Rev . 2018;63:40–47. doi: 10.1016/j.ctrv.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ross DJ, Strieter RM, Fishbein MC, Ardehali A, Belperio JA. Type I immune response cytokine-chemokine cascade is associated with pulmonary arterial hypertension. J Heart Lung Transplant . 2012;31:865–873. doi: 10.1016/j.healun.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 37. Mamazhakypov A, Viswanathan G, Lawrie A, Schermuly RT, Rajagopal S. The role of chemokines and chemokine receptors in pulmonary arterial hypertension. Br J Pharmacol . 2021;178:72–89. doi: 10.1111/bph.14826. [DOI] [PubMed] [Google Scholar]
- 38. Keane MP. The role of chemokines and cytokines in lung fibrosis. Eur Respir Rev . 2008;17:151–156. [Google Scholar]
- 39. Ahn J, Kim KH, Park S, Ahn Y-H, Kim HY, Yoon H, et al. Target sequencing and CRISPR/Cas editing reveal simultaneous loss of UTX and UTY in urothelial bladder cancer. Oncotarget . 2016;7:63252–63260. doi: 10.18632/oncotarget.11207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Charchar FJ, Tomaszewski M, Padmanabhan S, Lacka B, Upton MN, Inglis GC, et al. The Y chromosome effect on blood pressure in two European populations. Hypertension . 2002;39:353–356. doi: 10.1161/hy0202.103413. [DOI] [PubMed] [Google Scholar]
- 41. Eales JM, Maan AA, Xu X, Michoel T, Hallast P, Batini C, et al. Human Y chromosome exerts pleiotropic effects on susceptibility to atherosclerosis. Arterioscler Thromb Vasc Biol . 2019;39:2386–2401. doi: 10.1161/ATVBAHA.119.312405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marra AM, Benjamin N, Eichstaedt C, Salzano A, Arcopinto M, Gargani L, et al. Gender-related differences in pulmonary arterial hypertension targeted drugs administration. Pharmacol Res . 2016;114:103–109. doi: 10.1016/j.phrs.2016.10.018. [DOI] [PubMed] [Google Scholar]
- 43. O’Keeffe LM, Howe LD, Fraser A, Hughes AD, Wade KH, Anderson EL, et al. Associations of Y chromosomal haplogroups with cardiometabolic risk factors and subclinical vascular measures in males during childhood and adolescence. Atherosclerosis . 2018;274:94–103. doi: 10.1016/j.atherosclerosis.2018.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol . 2016;16:626–638. doi: 10.1038/nri.2016.90. [DOI] [PubMed] [Google Scholar]
- 45. Takahashi T, Ellingson MK, Wong P, Israelow B, Lucas C, Klein J, et al. Yale IMPACT Research Team Sex differences in immune responses that underlie COVID-19 disease outcomes. Nature . 2020;588:315–320. doi: 10.1038/s41586-020-2700-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hester J, Ventetuolo C, Lahm T. Sex, gender, and sex hormones in pulmonary hypertension and right ventricular failure. Compr Physiol . 2019;10:125–170. doi: 10.1002/cphy.c190011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. D’Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med . 1991;115:343–349. doi: 10.7326/0003-4819-115-5-343. [DOI] [PubMed] [Google Scholar]
- 48. Hemnes AR, Maynard KB, Champion HC, Gleaves L, Penner N, West J, et al. Testosterone negatively regulates right ventricular load stress responses in mice. Pulm Circ . 2012;2:352–358. doi: 10.4103/2045-8932.101647. [DOI] [PMC free article] [PubMed] [Google Scholar]