

Pulmonary arterial hypertension (PAH) is a progressive disease, characterized by elevated pulmonary arterial pressure and subsequent right heart failure. These changes result from pulmonary vasculature wall thickening and remodeling. Pathogenic vascular remodeling stems in part from endothelial cell dysfunction and vascular smooth muscle cell proliferation (1). Pulmonary vascular disease progression is also driven by increased inflammatory cells and mediators, such as macrophages and cytokines, which promote further pathologic remodeling (2). Interestingly, the epidemiology of PAH reveals a fourfold greater disease prevalence in females than males, accompanied by a reciprocal increase of disease severity in males versus females (3). Insights into the sexual dimorphism observed in PAH may lead to the development of better therapeutics, as current therapies are not curative (4). But, the mechanism(s) underlying these gender differences remain poorly understood (5).
Guided by a historical focus on the role of sex hormones in PAH, a puzzling yet crucial finding has emerged: estrogen prevents disease experimentally, while clinically, females have higher disease incidence (3). Thus, sex hormone differences alone may not adequately explain the female predominance of PAH development. Accordingly, Umar and colleagues looked to chromosomal differences between the two sexes instead. Previously, they found a protective role of the Y chromosome (ChrY) in PAH, independent of gonadal hormones (6). At the same time, Yan and colleagues described pathogenic activity of the ChrY gene sry in PAH via regulation of BMPR2 gene expression in fibroblasts—findings that also suggested a role of sry in PAH gender differences (7). But, sry alone could not alone explain ChrY-dependent protection, because XY female mice lacking Sry were still protected against PAH when compared with XX females (6).
In this issue of the Journal (pp. 186–196), this group delved deeper to understand how the ChrY confers protection against the development of PAH (8). The authors concluded that the ChrY gene, uty (ubiquitously transcribed tetratricopeptide repeat containing, y-linked), is responsible for this protective role, and identified downstream inflammatory mediators as important therapeutic targets. The group first found ChrY genes that were expressed in mouse lung tissue and individually knocked down each gene in the lungs of gonadectomized hypoxic mice. Of the four genes they investigated, only knockdown of uty resulted in PAH development. RNA sequencing analysis yielded proinflammatory cytokines CXCL9 (C-X-C Motif Chemokine Ligand 9) and CXCL10 as top genes of interest. Both their in vitro and in vivo findings corroborated that decreased UTY expression in macrophages upregulated the proinflammatory cytokines CXCL9 and CXCL10. These cytokines, which are also robustly upregulated in lungs of female PAH patients, triggered endothelial dysfunction by binding the CXCR3 (C-X-C Motif Chemokine Receptor 3) receptor and promoting PAH pathophenotypes (Figure 1).
Figure 1.
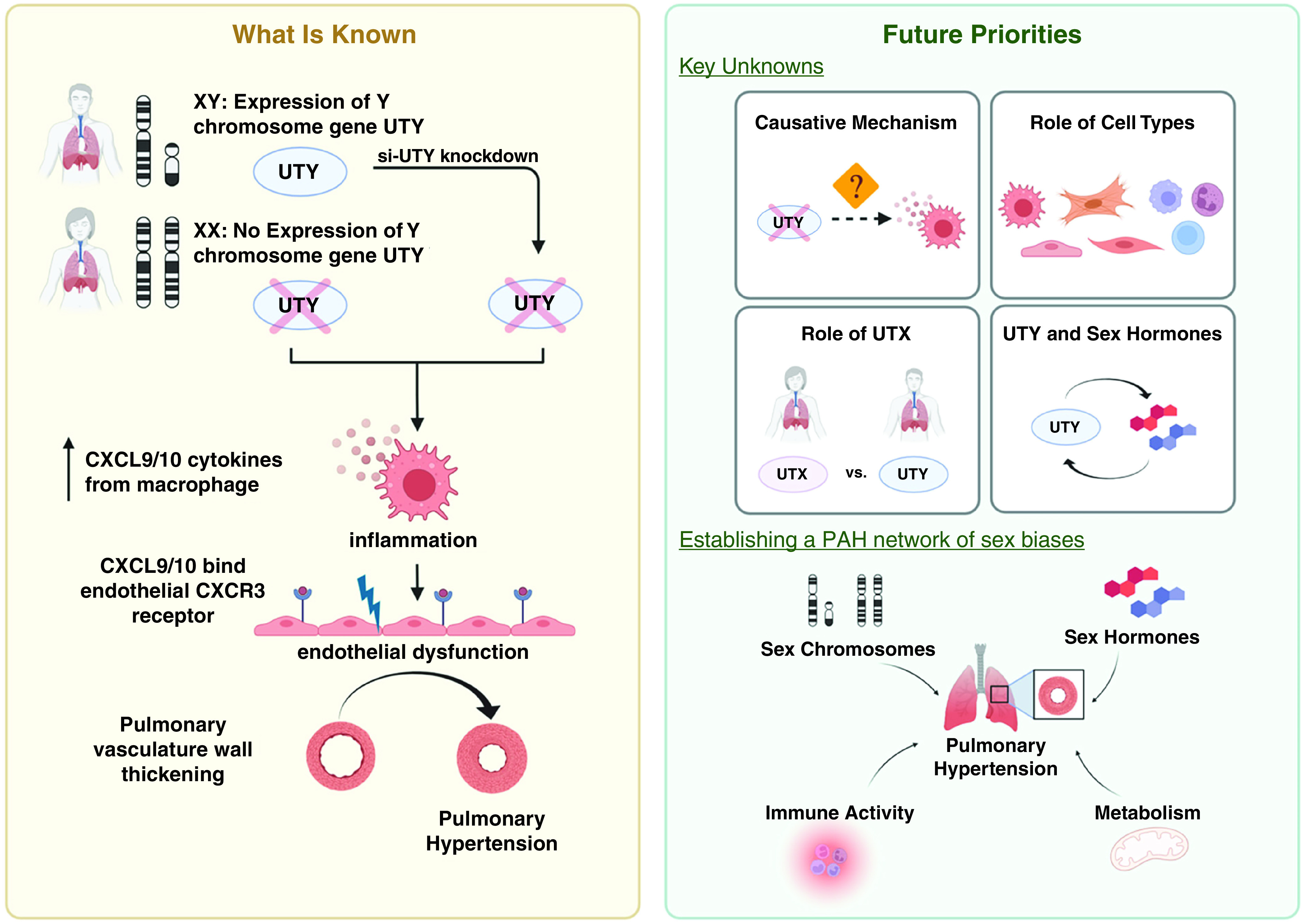
Decreased UTY (ubiquitously transcribed tetratricopeptide repeat containing, y-linked) causes increased inflammation via upregulation of proinflammatory cytokines CXCL9/10 (C-X-C motif chemokine ligand 9/10) release from macrophages. Binding of these cytokines to CXCR3 (C-X-C motif chemokine receptor 3) results in endothelial dysfunction and subsequent development of pulmonary arterial hypertension (left panel). Further understanding of sex biases in physiologic and pathologic processes may provide insight necessary for the successful development of gender-based targeted therapeutics (right panel). Created with BioRender.com. PAH = pulmonary arterial hypertension; UTX = female paralog of UTY.
Overall, this work describes a potentially important discovery of a protective role of the UTY-CXCL9/10 axis in PAH, independent of sex hormone alterations. Considering expression patterns of UTY documented in both lung tissue and macrophages, these findings offer molecular insight into how ChrY can directly regulate gene expression in somatic cells and invoke a greater complexity in PAH sex differences than previously appreciated (9, 10). Additionally, blocking CXCL9/10 activity prevented PAH, representing a promising step forward for the development of targeted and gender-specific therapeutics.
However, these key findings raise several new questions to be addressed by future studies. First, the causative mechanism linking reduced UTY expression and upregulation of proinflammatory cytokines CXCL9 and CXCL10 remains unclear. Although UTY is a known member of the Jumonji family of histone 3 lysine 27 (H3K27) demethylases, Cunningham and colleagues found that UTY did not influence methylation levels in the lung, consistent with prior studies showing that UTY retains low levels of catalytic activity (11). Thus, future work is needed to define how UTY controls this downstream chemokine axis. Second, while evidence is suggestive of both macrophage and endothelial cell involvement, more precise proof is needed to show that these cell types and potentially others are essential for the protective effects of the UTY-CXCL9/10 axis. Given the pleiotropic roles of the CXCL9/10 chemokines even beyond inflammation, it is possible that other genetic, environmental, and even metabolic perturbations may be relevant to this disease pathway (12). Third, it is also known that UTY carries a high degree of homology with UTX, the female paralog of UTY (10). Thus, assessing the role of UTX in PAH may lead to further insight into gender differences in this disease. Finally, while Cunningham and colleagues present evidence that UTY can act independently of sex hormones, a more nuanced interplay between UTY and sex hormones is possible and even likely in PAH. Future work to define that complex mechanism may help to unravel the still unsolved estrogen paradox as well as the clinical observation of greater disease severity in males (3).
Ultimately, the fundamental contributions of this study could pave the way for a more precise understanding of the complex PAH network of sex biases across genetic, environmental, immunologic, and metabolic factors (Figure 1; 1, 6, 13, 14). If successful, such endeavors bring us closer to precision medicine in PAH and long-awaited, gender-based clinical treatment strategies in this challenging disease.
Footnotes
Supported by NIH grants R01 HL124021 and HL 122596 and the American Heart Association grant 18EIA33900027 (S.Y.C.).
Originally Published in Press as DOI: 10.1164/rccm.202204-0653ED on May 11, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Rafikova O, Al Ghouleh I, Rafikov R. Focus on early events: pathogenesis of pulmonary arterial hypertension development. Antioxid Redox Signal . 2019;31:933–953. doi: 10.1089/ars.2018.7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Price LC, Wort SJ, Perros F, Dorfmüller P, Huertas A, Montani D, et al. Inflammation in pulmonary arterial hypertension. Chest . 2012;141:210–221. doi: 10.1378/chest.11-0793. [DOI] [PubMed] [Google Scholar]
- 3. Lahm T, Tuder RM, Petrache I. Progress in solving the sex hormone paradox in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol . 2014;307:L7–L26. doi: 10.1152/ajplung.00337.2013. [DOI] [PubMed] [Google Scholar]
- 4. Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT. Pulmonary arterial hypertension: the clinical syndrome. Circ Res . 2014;115:115–130. doi: 10.1161/CIRCRESAHA.115.301146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Batton KA, Austin CO, Bruno KA, Burger CD, Shapiro BP, Fairweather D. Sex differences in pulmonary arterial hypertension: role of infection and autoimmunity in the pathogenesis of disease. Biol Sex Differ . 2018;9:15. doi: 10.1186/s13293-018-0176-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Umar S, Cunningham CM, Itoh Y, Moazeni S, Vaillancourt M, Sarji S, et al. The Y chromosome plays a protective role in experimental hypoxic pulmonary hypertension. Am J Respir Crit Care Med . 2018;197:952–955. doi: 10.1164/rccm.201707-1345LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yan L, Cogan JD, Hedges LK, Nunley B, Hamid R, Austin ED. The Y chromosome regulates BMPR2 expression via SRY: a possible reason “why” fewer males develop pulmonary arterial hypertension. Am J Respir Crit Care Med . 2018;198:1581–1583. doi: 10.1164/rccm.201802-0308LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cunningham CM, Li M, Ruffenach G, Doshi M, Aryan L, Hong J, et al. Y-chromosome gene, Uty, protects against pulmonary hypertension by reducing proinflammatory cytokines. Am J Respir Crit Care Med . 2022;206:186–196. doi: 10.1164/rccm.202110-2309OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wijchers PJ, Festenstein RJ. Epigenetic regulation of autosomal gene expression by sex chromosomes. Trends Genet . 2011;27:132–140. doi: 10.1016/j.tig.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 10. Deschepper CF. Regulatory effects of the Uty/Ddx3y locus on neighboring chromosome Y genes and autosomal mRNA transcripts in adult mouse non-reproductive cells. Sci Rep . 2020;10:14900. doi: 10.1038/s41598-020-71447-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gažová I, Lengeling A, Summers KM. Lysine demethylases KDM6A and UTY: the X and Y of histone demethylation. Mol Genet Metab . 2019;127:31–44. doi: 10.1016/j.ymgme.2019.04.012. [DOI] [PubMed] [Google Scholar]
- 12. Van Raemdonck K, Van den Steen PE, Liekens S, Van Damme J, Struyf S. CXCR3 ligands in disease and therapy. Cytokine Growth Factor Rev . 2015;26:311–327. doi: 10.1016/j.cytogfr.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 13. Meester I, Manilla-Muñoz E, León-Cachón RBR, Paniagua-Frausto GA, Carrión-Alvarez D, Ruiz-Rodríguez CO, et al. SeXY chromosomes and the immune system: reflections after a comparative study. Biol Sex Differ . 2020;11:3. doi: 10.1186/s13293-019-0278-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Culley MK, Perk D, Chan SY. NFU1, iron-sulfur biogenesis, and pulmonary arterial hypertension: a (metabolic) shift in our thinking. Am J Respir Cell Mol Biol . 2020;62:136–138. doi: 10.1165/rcmb.2019-0309ED. [DOI] [PMC free article] [PubMed] [Google Scholar]