

Abstract
Background and Objective
Depleting CD20+ B cells is the primary mechanism by which ocrelizumab (OCRE) is efficient in persons with multiple sclerosis (pwMS). However, the exact role of OCRE on other immune cell subsets directly or indirectly remains elusive. The purpose of this study is to characterize the dynamics of peripheral immune cells of pwMS on OCRE.
Methods
We collected blood samples from 38 pwMS before OCRE onset (T0) and at 6 and 12 months (T6, T12) after initiation. To cover the immune cell diversity, using mass cytometry time of flight, we designed a 38-parameter panel to analyze B, T, and innate immune cell markers and CNS migratory markers. In parallel, viral-specific CD8+ T-cell responses were assessed by the quantification of interferon-γ secretion using the enzyme-linked immunospot assay on cytomegalovirus, Epstein-Barr virus, and influenza stimulations.
Results
Beside B-cell depletion, we observed a loss in memory CD8+CD20+ and central memory CD8+ T cells but not in CD4+CD20+ T cells already at T6 and T12 (p < 0.001). The loss of memory CD8+ T cells correlated with a lower CXCR3 expression (p < 0.001) and CNS-related LFA-1 integrin expression (p < 0.001) as well as a reduced antiviral cellular immune response observed at both time points (p < 0.001). Of note, we did not observe major changes in the phenotype of the other cell types studied. Seven of 38 (18.4%) patients in our cohort presented with infections while on OCRE; 4 of which were switched from dimethyl fumarate. Finally, using a mixed linear model on mass cytometry data, we demonstrated that the immunomodulation induced by previous disease-modifying therapies (DMTs) was prolonged over the period of the study.
Discussion
In addition to its well-known role on B cells, our data suggest that OCRE also acts on CD8+ T cells by depleting the memory compartment. These changes in CD8+ T cells may be an asset in the action of OCRE on MS course but might also contribute to explain the increased occurrence of infections in these patients. Finally, although more data are needed to confirm this observation, it suggests that clinicians should pay a special attention to an increased infection risk in pwMS switched from other DMTs to OCRE.
It is commonly accepted that multiple sclerosis (MS) starts in the periphery with the activation of immune cells, which then migrates to the CNS where they drive nervous system injuries. Although MS is traditionally seen as a T-cell–mediated disease, B-cell involvement is increasingly recognized.1 Indeed, the success of anti-CD20 therapies,2 which rapidly depletes B cells from the circulation but does not affect the immunoglobulin-secreting plasma cells, points to an antibody-independent effect of B-cell depletion on MS disease activity.3,4 Selective B-cell depletion successfully reduces clinical and MRI activity in active persons with MS (pwMS)2,5-7 and slows disability progression in patients with primary progressive MS.8 Ocrelizumab (OCRE), a recombinant humanized monoclonal antibody that selectively targets CD20-expressing B cells,5,7 was the first B-cell–depleting disease-modifying treatment (DMT) to demonstrate a superiority over interferon-beta-1a in phase II and III randomized trials.5,7,8 Post hoc analysis and real-world studies now show a sustained efficacy over a period of up to 7 years9 and position OCRE and other anti-CD20 treatments as high-efficacy DMTs with a similar profile as natalizumab (NTZ).10
At a mechanistic level, after binding CD20-expressing B cells, OCRE induces complement-dependent and antibody-dependent cellular cytotoxicity,11 effectively depleting CD20+ and CD19+ cells 2 weeks after treatment initiation.7,12-14 CD20 is expressed with increasing concentrations from pre-B cells to naive and memory B cells. However, it is not expressed on progenitor B cells (pro-B cells), short-lived plasma cells, and terminally differentiated antibody-producing long-lived plasma cells.15 Therefore, OCRE does not theoretically affect the capacity of B cells to reconstitute themselves.11,16
On the other hand, continuous B-cell depletion is associated with hypogammaglobulinemia and an increased risk of infection.14,17,18 This has raised many concerns during the recent SARS-CoV-2 pandemic for the care of pwMS.19,20 Anti-CD20 treatment is associated with a 2- to 3-fold increased risk of more severe COronaVIrus Disease 2019 (COVID-19) as compared to patients with MS who are not on this drug.21 Indeed, one can observe markedly attenuated humoral responses in OCRE-treated pwMS after COVID-19 infection or after vaccination by mRNA vaccine.22-26 By contrast, SARS-CoV-2–specific CD4+ and CD8+ T-cell responses do not seem to be affected,22,24-31 although selective deficits in circulating follicular helper T cells are reported.22 Nevertheless, up to now, not much is known regarding the maintenance of the virus-specific memory T-cell recall responses on OCRE, which raises the question whether this cellular immune response might be involved in the reported increased risk of infection on OCRE.17
In addition to CD20+ B cells, memory myelin-specific CD8+CD20+ T cells are affected by OCRE and have been reported to be depleted within 6 months of treatment onset.32 There is also a shift in the distribution within the T-cell compartment with enrichment of naive and depletion of effector memory CD4+ and CD8+ T cells.33,34 Whether the aforementioned changes in the T-cell compartment taking place on OCRE are accompanied by a functional deficit is not fully elucidated. Furthermore, current knowledge on how OCRE systematically affects other immune cell subsets, especially T cells, directly or indirectly is missing.
In this 1-year longitudinal observational study including patients with relapsing-remitting MS (RR-MS) and progressive MS (P-MS) treated with OCRE, using multiparameter mass cytometry time of flight, we systematically characterized the dynamics of CD20+ B- and T-cell subsets. We also assessed the changes in all other immune cell subsets in the blood of pwMS on OCRE. Finally, we examined the influence of OCRE on the function of the virus-specific memory CD8+ T-cell immune response.
Methods
Study Patients
We enrolled 38 persons with MS (pwMS) who had RR, active secondary progressive (SP), or primary progressive form. All were treated with OCRE (Table 1). The diagnosis of MS was made using the revised 2017 McDonald's criteria. Blood samples were drawn just before the first OCRE administration (T0) and then at 6 and 12 months (T6 and T12, respectively) into treatment. In parallel, we recorded any infection occurring in the first 2 years of OCRE treatment up to the day before the 24-month infusion (Table 2). Peripheral blood mononuclear cells (PBMCs) were isolated and frozen as previously described.35
Table 1.
Clinical Data of 38 Enrolled Patients With MS

Table 2.
Clinical Data of 7 Patients Developing an Infection on OCRE

Standard Protocol Approvals, Registrations, and Patient Consents
Enrolled patients were part of an ongoing open study aiming at characterizing the effects of disease-modifying therapies with every 6–12 month blood sampling of pwMS. This study was accepted by our institution's review board (2018_01622 and 2019_02197), and all patients gave their written informed consent before study initiation.
Barcoding for Mass Cytometry Analyses
PBMCs were thawed, resuspended in Roswell Park Memorial Institute medium supplemented with 10% fetal bovine serum, and left for a 4-hour resting in a cell incubator. After washing steps, to reduce sample variability, increase interassay reproducibility, increase throughput, and reduce time/reagent consumption, 2 million PBMCs were individually barcoded using a unique combination of anti-CD45–coupled Abs (see eTable 1, links.lww.com/NXI/A796, and Figure 1A for an overview of the work flow). Some of the antibodies used for mass cytometry analyses were coupled in our facility (Maxpar X8 Multimetal Labeling Kit, Fluidigm) and listed as laboratory conjugate in eTable 1 and eTable 2 (links.lww.com/NXI/A797) at room temperature (RT) for 30 minutes in cell staining medium (CSM; phosphate-buffered saline and 4% bovine serum albumin) under agitation. After washing steps, barcoded single samples from 3 donors at 3 time points were pooled together into a single reaction tube for further staining steps.
Figure 1. Immunophenotyping of the Effect of Ocrelizumab on Total PBMCs of Treated Patients With MS Using Mass Cytometry.

PBMCs from 38 patients with MS before OCRE onset (T0) and after 6 and 12 months (T6 and T12). To cover the immune cell diversity, a 38-parameter panel was designed for mass cytometry analyses (CyTOF) including B, T, NK, and innate immune cell markers as well as CNS migratory markers. (A) A schematic overview of the analysis pipeline is represented from the barcoding of PBMC individual sample to mass cytometry acquisition and analyses. The 3 longitudinal time points from 3 donors were barcoded, and all 9 samples analyzed together to minimize the batch effect. (B) FlowSOM unsupervised clustering allows to discriminate the 6 major blood cell subsets, i.e., B cells, CD4+ and CD8+ T cells, NKT cells, and monocytes/dendritic cells (see legend for marker selection [inside color pies] and metacluster definition [outside ring color]). (C) Effect of sex, age, MS type, treatment before OCRE introduction, and washout period before OCRE introduction on the expression of all 38 parameters was tested using a mixed linear model (MLM) in the total PBMC FlowSOM metaclusters. In white, pLM > 0.05 (not significant [ns]); in light gray, pLM < 0.05; in dark gray, pLM < 0.01; in black, pLM < 0.001. MS = multiple sclerosis; OCRE = ocrelizumab; PBMCs = peripheral blood mononuclear cells.
Surface Staining for Mass Cytometry Analyses
Barcoded-pooled cells were then stained in 1 mL of a combination of surface antibodies (eTable 2, links.lww.com/NXI/A797) and cell viability marker [Cell-ID Intercalator-103Rh, Fluidigm] diluted in CSM for 30 minutes at RT under agitation. To give a broad overview of the immune environment by high-dimensional analysis, a large 38-parameter panel was optimized. It included B-cell markers (CD19, CD20, CD21, CD27, CD38, Human Leukocyte Antigen-DR (HLA-DR) and IgA/G/D/M); T-cell markers (CD3, CD4, CD8, CCR7, CD45RA, CD45RO, CD127, CCR4, CCR5, CCR6, CXCR3, CXCR5, CRTH2, PD1, ICOS, CD25, and CD95); innate immune cell markers (CD16, CD56, CD7, CD14, CD1c, CD11c, CD123, and HLA-DR); and CNS migratory phenotype markers (CD49d, CD29, CD11a, and CD18). After washing steps, cells were fixed with 2.4% paraformaldehyde (10 minutes; RT). Total cells were identified by DNA intercalation (1 µM Cell-ID Intercalator, Fluidigm, South San Francisco, CA) in 1.6% PFA at 4°C overnight. Labeled samples were acquired with a Helios CyTOF System (Fluidigm). A minimum of 200,000 cells were acquired per sample.
Mass Cytometry Data Analyses
Raw mass spectrometry files were normalized to the EQ Four Element Calibration Beads using the CyTOF software (done by the CyTOF platform; Immunology and Allergy Immune Monitoring Platform, CHUV, Lausanne). For basic cytometry analysis of FCS files, including dead cell exclusion and debarcoding, raw files were loaded into Cytobank data analysis software. Once debarcoded, each individual FCS file was analyzed using different softwares including FlowJo (TreeStar, v10.7.1) for cell subset analyses and R studio (R version 4.0.2) using the flowWorkspace framework.36-39 Marker intensity values were arcsinh (hyperbolic inverse sine) with cofactor 5 transformed and centered using the formula 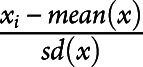 , where x is an individual marker. Unsupervised clustering was run using self-organizing map in combination with consensus clustering (FlowSOM R package). All FlowSOM-based clustering was performed on the combined dataset to enable the identification of populations of interest and manually annotated after automated unsupervised clustering (see figure legend for the detailed list of specific markers used). To take into account the depletion of CD20+ cell subsets on OCRE, nodes or clusters' individual frequencies were subsequently corrected for total CD20-negative cell counts.
, where x is an individual marker. Unsupervised clustering was run using self-organizing map in combination with consensus clustering (FlowSOM R package). All FlowSOM-based clustering was performed on the combined dataset to enable the identification of populations of interest and manually annotated after automated unsupervised clustering (see figure legend for the detailed list of specific markers used). To take into account the depletion of CD20+ cell subsets on OCRE, nodes or clusters' individual frequencies were subsequently corrected for total CD20-negative cell counts.
CD8+ T-Cell Isolation and Coculture
CD8+ T cells were sorted from thawed PBMCs by magnetic-activated cell sorting (MACS) using anti-CD8 MicroBeads (Miltenyi Biotec, North Rhine-Westphalia, Germany) with an autoMACS Pro Separator (Miltenyi Biotec) according to the manufacturer's instructions. The CD8-negative fraction from the baseline time point (T0; CD8−0) was kept for coculture purposes, with CD8+ fractions at all time points (T0, T6, and T12; CD8+0, CD8+6, and CD8+12). The purity of sorted cells was checked by flow cytometry (see section Flow Cytometry). In vitro coculture was performed only if the purity of CD3+CD8+ sorted fractions reached at least 90%, and if CD8-negative populations at T0 contained a maximum of 5% remaining CD3+CD8+ T cells. A total of 20,000 CD8+ T cells isolated at T0, T6, or T12 were cocultured with autologous cells from the CD8-negative fraction from the baseline time point (T0) at a ratio of 1:10 cells (CD8+:CD8−). The evolution of antiviral T-cell response was assessed by enzyme-linked immunospot (ELISPOT) assay (see section ELISPOT Assay).
Flow Cytometry Analyses
Sorted CD8-positive and CD8-negative fractions were stained as described previously40 using the following antibodies: anti-CD8 Pacific Blue (clone RPA-T8, BD), anti-CCR7 FITC (clone REA546, Miltenyi), anti-CD4 ECD (clone T4, Beckman Coulter, Brea, CA), anti-CD45RA PerCP-Cy5.5 (clone HI100, BD), anti-CD20 AF700 (clone 2H7, BioLegend, San Diego, CA), and anti-CD3 APC-H7 (clone SK7, BD). Dead cells were excluded using the Aqua LIVE/DEAD stain kit (Invitrogen AG, Basel, Switzerland). Data were acquired on a LSRII flow cytometer (Becton Dickinson, Franklin Lakes, NJ). A blinded investigator acquired the data on a LSRII flow cytometer (Becton Dickinson) and analyzed using FlowJo software (TreeStar Inc., version 10.7.1). All samples from a given study participant were analyzed on the same day; each set of experiments included pwMS from different baseline, untreated, or previously treated so that potential sources of variation and technical side effects were minimized.
ELISPOT Assay
To detect interferon-γ–secreting activated T cells specific to CMV, EBV, and FLU (CEF), we used an ELISPOT assay as described previously.35 Briefly, 200,000 thawed PBMCs or 200,000 CD8−0 alone or 200,000 CD8−0 cocultured with 20,000 autologous CD8+0, CD8+6, or CD8+12 cells were stimulated overnight. Conditions used were medium only (negative control), phytohemagglutinin lectin (positive control, 5 µg/mL, used as a saturating condition), and CEF pool of different HLA Class I-restricted 9mer peptide immunodominant CD8+ T-cell epitopes (CEF pool, 1 µg/mL).41 Spot counts and mean responses were generated as previously described.35 All conditions were performed in triplicates, and a mean of the results was calculated for each condition. Responses were expressed as net spot-forming cells (SFCs) per 106 cells. The assay was considered valid if, in the absence of peptide (i.e., negative control), there were fewer than 40 SFCs per 106 cells. A CEF-specific response was considered as positive if it was >4 times higher than the negative control. Validated background values were subtracted from the peptide-stimulated data before analysis. Notably, to take the lymphopenic effect of OCRE into account, all SFCs were normalized to 106 CD3+CD8+ T cells, whose frequency was determined by unsupervised clustering of CD8+ T cells of mass cytometry acquisitions (Figure 1B, cluster 5), such as described previously.42 To dismiss baseline CD8−0 residual response, spots from the CD8−0 alone CEF control conditions were removed for all CEF counts in the coculture conditions with autologous CD8+0, CD8+6, or CD8+12 cells. For CD8 coculture, all SFCs were normalized to 106 memory CD8+ T cells, whose frequency was determined by unsupervised clustering of CD8+ T cells of mass cytometry acquisitions (Figure 4A, clusters 2 and 3).42
Figure 4. Loss of Functional Activated Central Memory CD8+ T Cells on OCRE Treatment.

(A) FlowSOM unsupervised subclustering of PBMC CD8+ metacluster 5 discriminates the circulating naive, central memory (CM), effector memory (EM), and effector (Eff) CD8+ T cells (see legend for marker selection and submetacluster definition). (B) Modification in the frequency of FlowSOM unsupervised naive and memory CD8+ T-cell subsets overtime (T0, T6, and T12) is represented. The asterisks (*) represent significant differences for the effect of time for a given treatment on a given metacluster: **p < 0.01; ***p < 0.001 using a nonparametric paired post hoc Nemenyi test. (C) Statistical heatmap analyses of markers of phenotype, function/activation, and CNS-migration properties of CD8+ cell subsets overtime (T0, T6, and T12). Effect of the treatment overtime and over CD8+submetaclusters was tested using a nonparametric paired Friedman test as compared to baseline (T0, squares). If significant, a nonparametric paired post hoc Nemenyi test was run to compare baseline values with subsequent time points (T0 vs T6 and T0 vs T12). Post hoc results are depicted as circles (the smallest being not significant; the lower intermediate, p < 0.05; the upper intermediate, p < 0.01; and the largest, p < 0.001, see the figure for details). (D) IFN-γ–mediated cellular immune responses against a pool of immunodominant peptides of CMV, EBV, and influenza (CEF) were tested using an ELISPOT assay at 3 time points of OCRE administration (T0, T6, and T12) in n = 22 of 38 patients tested with valid ELISPOT at T0 and more than 4 times the unstimulated condition. Spot-forming units (SFUs) were normalized to total CD8+ T-cell counts (frequencies from cluster 5, Figure 1, B and C). (E) In a subset of patients tested in panel D (11/22), CD8+ T-cell fractions were MACS sorted at 3 time points of OCRE administration (T0, T6, and T12) and cocultured with autologous CD8-depleted PBMCs (CD80−) that were collected only at T0, at a ratio of 1:10 (CD8+:CD80−). SFUs were normalized to total memory CD8+ T-cell counts (frequencies from clusters 2 and 3, panel A). (D-E) The asterisks (*) represent significant differences for the effect of time: ***p < 0.001 using a nonparametric paired Friedman test followed by a post hoc Dunn test (pD). Colored dots represent patients who developed infections on OCRE treatment over the period of the study. In green, patients previously under IFN; in red, previously under DMF; and in blue, previously under GA. ELISPOT = enzyme-linked immunospot; MACS = magnetic-activated cell sorting; OCRE = ocrelizumab; PBMCs = peripheral blood mononuclear cells.
Statistical Analyses
All plots and statistical analyses were performed using the R software (ggplot, rstatix, PMCMR packages; version 4.0.2) and GraphPad Prism (version 9.1.0 [221]). The effect of sex, age, MS type, DMT used before OCRE introduction, and washout period between previous DMT and OCRE onset was tested using a mixed linear model (R “lm” function in R software). Because age and previous DMT were found to be the most significant parameters, all data were adjusted accordingly for statistical analyses. p Values obtained from these linear models are mentioned in the text as pLM. A nonparametric paired Friedman analysis of variance (ANOVA) was run to test the effect of OCRE treatment, all along the treatment duration (T0 vs T6 vs T12). p Values obtained from these ANOVAs are mentioned in the text as pF. Comparisons include frequencies of immune populations and modulation of marker expressions. If the effect of the treatment reached significance, the differences between immune responses measured at T0 with the various subsequent time points were analyzed with the Nemenyi or Dunn-corrected post hoc test (nonparametric paired rank test corrected for multiple comparisons; pN and pD, respectively, in the text). p < 0.05 was considered as significant for adjusted p values.
Data Availability
Anonymized data not published within this article will be made available by request from any qualified investigator.
Results
To better characterize the overall effect of OCRE on circulating immune cells, a 38-parameter panel was designed for mass cytometry analyses (CyTOF) to cover the immune cell diversity including B, T, NK, and other innate immune cell markers as well as CNS migratory markers. Because of the high number of parameters to be acquired and to circumvent any experimentator-dependent gating strategy, we followed an unsupervised clustering approach (Figure 1, A and B) using FlowSOM. We were able to define all 6 major leukocyte subsets: B, CD4 and CD8 T cells, NK and NKT cells, and myeloid cells. On all time points and parameters tested, we found that age, MS type, and previous DMT used before OCRE onset were the 3 determinants that mostly influenced the immune parameters tested (Figure 1C). Therefore, for statistical analyses, the data were corrected for these parameters. Nevertheless, because MS type and age are highly correlated (patients with P-MS are older than patients with RR-MS), we corrected only for age and prior DMT use, all along the manuscript. Indeed, age is a continuous variable, thus leading to finer and more robust correction as compared to MS type, which is a categorical variable (with only 3 variables, RR vs SP vs PP). This observation suggests that DMTs given before OCRE have a long-standing effect on the immune repertoire that persists even on OCRE. Of interest, this effect seems to be independent of the length of the washout, i.e., the period between the end of the previous DMT and the start of OCRE. Of note, 7 of 38 patients (18%) developed infections on OCRE during the study: 1 was previously on interferon (IFN)-β, 1 on glatiramer acetate (GA), 4 on dimethyl fumarate (DMF), and 1 on NTZ (Table 2). None of them had severe lymphopenia or significant hypogammaglobulinemia at the time of infection.
We then studied the overall expression of the CD20 marker across all immune cell subsets. We were able to identify 8 CD20+ nodes: 3 among the B-cell cluster (at T0; 2 naive CD27−IgD+ [7, 8] and 1 memory IgG+ [16]), 3 among the CD4+ T-cell cluster (2 naive CCR7+CD45RA+ [47, 56] and 1 central memory CCR7+CD45RA− [61] [CM]), and 2 among the CD8+ T-cell cluster (naive CCR7+CD45RA+ [24] and effector memory CCR7−CD45RA− [36] [EM]) (Figure 2A). Regarding the first CD20+ B-cell compartment, we confirmed the rapid depletion of all CD20+ B cells (Figure 2B, pF < 0.001) already at T6 (pN < 0.001) and T12 (pN < 0.001).
Figure 2. Depletion and Phenotypical Changes of CD20+ B-Cell Subsets on OCRE.

PBMCs were analyzed as in Figure 1. (A) CD20 expression across FlowSOM unsupervised clustering allows to discriminate the major CD20-expressing nodes including B cells (nodes 7, 8, and 16), CD4+ T cells (nodes 47, 56, and 61), and CD8+ T cells (nodes 24 and 36). (B) Changes in the frequency of FlowSOM unsupervised naive and memory CD20+ B-cell subsets overtime (T0, T6, and T12). The asterisks (*) represent significant differences for the effect of time for a given treatment on a given metacluster: ***p < 0.001 using a nonparametric paired post hoc Nemenyi test. (C) Statistical heatmap expression analyses of markers of function/activation of CD20+ B-cell subsets overtime (T0, T6, and T12). Effect of the treatment overtime and over CD20+ nodes was tested using a nonparametric paired Friedman test as compared to baseline (T0, squares). If significant, a nonparametric paired post hoc Nemenyi test was run to compare baseline values with subsequent time points (T0 vs T6 and T0 vs T12). Post hoc results are depicted as circles (the smallest being not significant and the largest, p < 0.001, see the figure for details). OCRE = ocrelizumab; PBMCs = peripheral blood mononuclear cells.
To go a step further, we analyzed the phenotypic and functional evolution of the CD20+ B-cell nodes (Figure 2C). On OCRE, there was a significant decrease in the level of expression of IgD (pF < 0.001), HLA-DR (nodes 7, 16: pF < 0.001), CD21 (pF < 0.001), and CD19 (pF < 0.001). By contrast, there was an increase in the level of expression of CD38 (pF < 0.001), CD27 (node 8: pF < 0.001), IgA (nodes 7, 8: pF < 0.001), and IgG (node 8: pF < 0.001) (Figure 2C). With our unsupervised systematic approach, we were thus able to confirm the rapid occurrence of naive CD27-IgD+CD21+ and nonswitched memory [CD27+IgD+] B-cell depletion. Our data further suggest that remaining B cells are rapidly switched to memory CD27+IgD− and plasma cells IgG/IgA CD27+CD38+ rapidly after OCRE introduction, such as seen already at T6 and T12. These findings, in line with previous results on OCRE effect on B cells, validate our unsupervised clustering approach and pave the way for the analyses of other immune compartments.43
We also wanted to study the effect of OCRE on CD20+ cells outside CD20+ B cells, i.e., first on CD20+ T cells. Of interest, we found that as expected memory CD8+ CD20+ T cells were significantly depleted at both time points (Figure 3A, node 36, pF < 0.001), but strikingly not naive CD8+ CD20+ T cells (Figure 3A; node 24) nor naive and memory CD4+ T cells (Figure 3, A and B, nodes 47, 56, and 61).
Figure 3. Depletion of Memory CD20+ CD8+ T Cells and Phenotype Changes of CD20+ T-Cell Subsets on OCRE.

PBMCs were analyzed as in Figure 1. CD8+CD20+ and CD4+CD20+-expressing nodes were selected as depicted in Figure 2A. CD20 expression across FlowSOM unsupervised clustering allows to discriminate the major CD20-expressing T-cell nodes including CD8+ T cells (nodes 24 and 36) and CD4+ T cells (nodes 47, 56, and 61) as identified in Figure 2. Changes in the frequency of FlowSOM unsupervised naive and memory CD8+CD20+ (A) and CD4+CD20+ (B) T-cell subsets overtime (T0, T6, and T12). The asterisks (*) represent significant differences for the effect of time for a given treatment on a given metacluster: ***p < 0.001 using a nonparametric paired post hoc Nemenyi test. Statistical heatmap expression analyses of markers of function/activation and CNS-migration properties of CD8+CD20+ (C) and CD4+CD20+ (D) T-cell subsets overtime (T0, T6, and T12). Effect of the treatment overtime and over CD20+ nodes was tested using a nonparametric paired Friedman test as compared to baseline (T0, squares). If significant, a nonparametric paired post hoc Nemenyi test was run to compare baseline values with subsequent time points (T0 vs T6 and T0 vs T12). Post hoc results are depicted as circles (the smallest being not significant and the largest, p < 0.001, see the figure for details). OCRE = ocrelizumab; PBMCs = peripheral blood mononuclear cells.
Going one step further into the detailed characterization of the effect of OCRE on the CD20-expressing T cells, we ran a functional phenotype analysis on both CD8+CD20+ and CD4+CD20+ T-cell nodes. To this end, we used a combination of 17 function/activation markers and CNS-related integrins (Figure 3, C and D). We found that both the CD8+CD20+ and the CD4+CD20+ exhibited changes in their phenotypes as compared to T0, but these changes involved only the memory population (node 36 for CD8+CD20+ T cells and node 61 for CD4+CD20+ T cells; Figure 3, C and D). However, the modified markers were not always the same between CD4+CD20+ and CD8+CD20+ T cells. Indeed, although CD45RO, CXCR3, or PD1 was decreased in both populations of T cells, there was an additional effect of OCRE on the CD8+CD20+ T-cell population because it seemed to decrease the expression of lymphocyte function-associated antigen 1 (LFA-1) subunits (CD18 [pF < 0.001; −20%] and CD11a [pF < 0.001; −20%]), LFA-1 being an important CNS-related integrin (Figure 3D). Altogether, OCRE, in addition to significantly reducing the number of CD8+CD20+ T cells, seems to render them less active and less prone to migrate to the brain, thus markedly impairing this population.
Whereas NK, NKT, and CD4+ T cells were not significantly modulated by OCRE at the total cell level, there was an overall decrease in CD8+ T cells (−13.2%; Figure 1B, cluster 5) at T6 (pN = 0.005) and T12 (pN = 0.005). Thus, to better characterize the effect of OCRE on the overall CD8+ T-cell subsets, we performed a submetaclustering on the CD8+ T-cell cluster 5 (Figures 1B and 4). This unsupervised approach allowed to discriminate CCR7+CD45RO−CD45RA+ naive, CCR7+CD45RO+CD45RA− CM, CCR7−CD45RO−CD45RA− EM, and CCR7−CD45ROintCD45RA+ effector CD8+ T cells (Figure 4A). Of interest, as compared to T0, there was a loss of CM CD8+ T cells at T6 (−14.4%) and T12 (−15.9%, Figure 4B; cluster 2, pF < 0.001) along with an expansion of naive CD8+ T-cell subsets (at T6 [+8.8%] and T12 [+11.8%; Figure 4B; cluster 3, pF < 0.001]). Using a combination of activation markers listed in Figure 4C, we could show that the phenotype of the remaining CM CD8+ T cells was also altered and displayed a decreased expression of CD45RO (−5%) and CXCR3 (÷5; pF < 0.001) as well as a gain in the expression of ICOS (pF < 0.001; ×2.3) (Figure 4C). Of interest, CXCR3 expression was also downregulated on both EM (÷4,8) and effector (÷3; Figure 4C; pF < 0.001) CD8+ T cells.
Then, we wanted to investigate whether the phenotypical changes observed by mass cytometry in CD8+ T cells were associated with an actual loss of function. To this end, using an ELISPOT assay, we measured the IFN-γ–mediated cellular immune responses against a pool of immunodominant peptides of CMV, EBV, and influenza (CEF) at all 3 OCRE time points (Figure 4D). Of note, to take into account the overall diminution in CD8+ T cells, spot-forming units (SFUs) were normalized to total CD8+ T-cell counts as determined by unsupervised FlowSOM clustering (Figure 1B, cluster 5). Among the 38 patients tested by mass cytometry, 33 had a valid ELISPOT at T0. Of those, 22 exhibited positive specific-CEF memory responses at T0 (see Methods section for details). In these 22 patients, there was a sharply reduced antiviral immune response at both time points (pD < 0.001; T6: ÷2.2; T12: ÷2.4) (Figure 4D). Furthermore, 22.7% (n = 5/22) showed no reactivity at T6 and T12, and half of the study patients who had a response at T0 experienced a minimal 50% reduction in the CEF-specific CD8+ T-cell responses at T6 and T12.
Because B cells can present antigens to T cells, we wanted to rule out that the reduced CD8 T-cell antiviral response was due to the lack of B cells in the in vitro ELISPOT assay. Therefore, we performed cocultures between (1) CD8+ T cells isolated at all 3 time points and (2) antigen-presenting cells consisting in autologous PBMCs depleted in CD8+ T cells sampled before OCRE treatment (i.e., T0). These highly controlled experiments were performed in a subset of 11 of 22 CEF-responsive patients including 5 pwMS untreated at T0; 2 previously on DMF and 4 infectious patients (1 previously on IFN, 2 previously on DMF, and 1 previously on GA) (Figure 4E). To take into account the overall reduction in memory CD8+ T-cell frequency among CD8+ T cells, SFUs were normalized to total memory CD8+ T-cell counts as determined by unsupervised FlowSOM clustering (Figure 4A, clusters 2 and 3). In these 11 patients, CEF-specific immune response was already diminished at T6 (−80.5%, pD = 0.057) and significantly leveled off at T12 (−87.4%, pD < 0.001), showing that there is a specific loss of the remaining function of memory CD8+ T cells on OCRE.
We then examined the viral-specific immune response of the 7 tested study patients who developed an infection during the time of this study, thus while on OCRE. They did not present with lymphopenia nor with hypogammaglobulinemia G, and their ELISPOT response was not significantly lower than the one of the other study patients (Figure 4, colored spots; 4/7 being CEF positive at T0). However, and interestingly, 4 of them were treated with DMF before receiving OCRE. Of note, all patients on DMF before receiving OCRE had as expected a lower total CD8+ T-cell count as compared to the other study patients (data not shown).
Discussion
OCRE has a major and predominant effect consisting in depleting CD20+ B cells. Nevertheless, how OCRE may affect the phenotype and functions of other immune cell subsets is less clear. Using the unique combination of an unbiased comprehensive mass cytometry approach and a functional T-cell assay in OCRE-treated pwMS followed over 1 year, we show that OCRE also significantly affects the CD8+ T-cell population.
First, we find that OCRE not only depletes the memory CD8+CD20+ T cells but also impairs their phenotype, especially by reducing CXCR3 and CNS-related LFA-1 integrin expression. Of interest, memory MOG-specific double-positive CD8+CD20+ T cells, a subset that is increased in pwMS in comparison with HD, are rapidly depleted on anti-CD20 treatment.32 Thus, it is tempting to postulate, as already suggested by others, that the disappearance of this subset of CD8+ T cells and the loss of migratory functions could contribute to the clinical efficacy observed in pwMS on OCRE and on other anti-CD20 therapy.32,44,45
Second, we observe that, within the total CD8+ T-cell population, OCRE induces a shift in the distribution of cells with an enrichment in naive and a depletion in CM CD8+ T cells, such an effect on the T-cell compartment being reported by others.33,34 Mirroring what was observed for memory CD8+CD20+ T cells, the expression of CXCR3, a key chemokine receptor for memory T-cell recruitment in inflamed tissues including the brain, was fully repressed on both CM and EM CD8+ T cells.46 Altogether, these results suggest that OCRE, by reducing the ability of potential pathogenic memory CD8+ T cells to migrate into the brain, may exert its therapeutical effect not only through the B-cell depletion but also through T cells, particularly CD8+ T cells.
Nevertheless, as the other side of the medal, we also demonstrate that the virus-specific memory CD8+ T-cell response is significantly reduced already after 6 months on OCRE, even after normalization of the data for the total number of CD8+ T cells per individual counts (Figure 4D). Furthermore, we demonstrate that this loss of function of memory CD8 T cells does not seem to be due to the absence of B cells in the assay but well to a broader effect of OCRE in vivo including on circulating memory CD8+ T cells (Figure 4E).
Our results contrast with what has been reported to date regarding the SARS-CoV-2–specific T-cell response. These data show that OCRE does not impair a de novo induction of new virus-specific effector cellular immune response such as extensively reported in the context of vaccination against22,24-31 or infection by SARS-CoV-2.47-49 Here, we do not claim that a patient on OCRE is not able to mount a virus-specific immune response; however, our longitudinal systematic study suggests that the persistence of a classical CM immune response such as a CEF-specific CD8+ T-cell response is impaired.
Precisely, 7 of 38 (18.4%) patients in our cohort presented with infections while on OCRE (Table 2). We did not identify any specific hallmark of immune suppression either general (lymphopenia and hypogammaglobulinemia) or issued from our study (CEF viral-specific immune responses in infectious patients were similar to the ones of the noninfectious patients) that would support the occurrence of these infections. Nevertheless, we also show that the pre-OCRE DMTs do have an enduring influence on the phenotype of the cells, which is independent on the length of the washout period (Figure 1C). Of interest, other authors have recently reported that a FTY-caused T-cell lymphopenia persisted in patients once they were on OCRE, suggesting that immune dysfunction could be carried over from the previous treatment to OCRE.50 Here, we demonstrate that the overall immune landscape is imprinted by the DMT used before OCRE. Of these 7 patients reporting benign to severe infections on OCRE, 4 were previously on DMF, representing two-thirds of the total number of patients switching from DMF (4/6). Knowing that DMF affects particularly the CD8+ T-cell population including the CD8+CD20+ T cells,45 it is tempting to hypothesize that there is a legacy effect of DMF on the CD8+ T cells, a population of cells that was further affected by OCRE.51 Nevertheless, such as mentioned above, we did not detect significant differences between the CD8+ T cells of OCRE-infected vs OCRE-noninfected study patients. This absence of difference could be explained by the low number of study patients and/or by markers that we did not identify. Our results may open new axes of research to stratify the risk of developing opportunistic infection in patients on anti-CD20 treatments. Whatever the cause, it might be important for the clinician to pay a particular attention to patients who are switched from DMF to OCRE.
To conclude, our study contributes to a better understanding of the mechanisms of actions of OCRE in pwMS, especially regarding its impact on memory CD8+ T cells suspected to be major players in MS pathogenesis. This article also suggests that a special attention should be paid to patients switching from DMF to OCRE.
Acknowledgment
The authors thank G. Le Goff for helping in enrolling patients and obtaining blood samples.
Glossary
- ANOVA
analysis of variance
- CSM
cell staining medium
- DMF
dimethyl fumarate
- ELISPOT
enzyme-linked immunospot
- GA
glatiramer acetate
- HLA-DR
Human Leukocyte Antigen-DR
- IFN
interferon
- LFA-1
lymphocyte function-associated antigen 1
- MACS
magnetic-activated cell sorting
- MS
multiple sclerosis
- NTZ
natalizumab
- OCRE
ocrelizumab
- P-MS
progressive MS
- PBMCs
peripheral blood mononuclear cells
- pwMS
persons with MS
- RR-MS
relapsing-remitting MS
- RT
room temperature
- SFCs
spot-forming cells
- SFU
spot-forming unit
Appendix. Authors
Study Funding
This work was supported by a grant from the Swiss National Science Foundation (320030-179531).
Disclosure
A. Mathias, S. Perriot, M. Canales, S. Jones, L. Oberholster, R. Bernard-Valnet, M. Moulin, and C. Fenwick report no disclosures relevant to the manuscript. V. Pantazou: received funding for travel from Biogen Idec and Merck, none related to this work. M. Theaudin: has served as an expert in advisory boards for Biogen, Genzyme-Sanofi, Merck, Novartis, and Roche; received travel grants from Biogen, Genzyme-Sanofi, Merck, Novartis, and Roche; and received speaker honoraria from Biogen, Novartis, and Merck. None were related to this work. C. Pot: has served as an expert in advisory boards for Biogen, Genzyme-Sanofi, Merck, Novartis, and Roche, none related to this work. R. Du Pasquier: has served on scientific advisory boards for Biogen, Novartis, Roche, and Sanofi-Genzyme and has received funding for travel or speaker honoraria from Biogen Idec, Celgene, Merck, Roche, and Sanofi-Genzyme. Go to Neurology.org/NN for full disclosure.
References
- 1.Greenfield AL, Hauser SL. B-cell therapy for multiple sclerosis: entering an era. Ann Neurol. 2018;83(1):13-26. doi: 10.1002/ana.25119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hauser SL, Waubant E, Arnold DL, et al. . B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358(7):676-688. doi: 10.1056/NEJMoa0706383 [DOI] [PubMed] [Google Scholar]
- 3.Krumbholz M, Derfuss T, Hohlfeld R, Meinl E. B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nat Rev Neurol. 2012;8(11):613-623. doi: 10.1038/nrneurol.2012.203 [DOI] [PubMed] [Google Scholar]
- 4.Meier UC, Giovannoni G, Tzartos JS, Khan G. Translational mini-review series on B cell subsets in disease. B cells in multiple sclerosis: drivers of disease pathogenesis and Trojan horse for Epstein–Barr virus entry to the central nervous system? Clin Exp Immunol. 2011;167(1):1-6. doi: 10.1111/j.1365-2249.2011.04446.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kappos L, Li D, Calabresi PA, et al. . Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011;378(9805):1779-1787. doi: 10.1016/S0140-6736(11)61649-8 [DOI] [PubMed] [Google Scholar]
- 6.Barr TA, Shen P, Brown S, et al. . B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J Exp Med. 2012;209(5):1001-1010. doi: 10.1084/jem.20111675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hauser SL, Bar-Or A, Comi G, et al. . Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. 2017;376(3):221-234. doi: 10.1056/NEJMoa1601277 [DOI] [PubMed] [Google Scholar]
- 8.Montalban X, Hauser SL, Kappos L, et al. . Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376(3):209-220. doi: 10.1056/NEJMoa1606468 [DOI] [PubMed] [Google Scholar]
- 9.Wolinsky JS, Arnold DL, Brochet B, et al. . Long-term follow-up from the ORATORIO trial of ocrelizumab for primary progressive multiple sclerosis: a post-hoc analysis from the ongoing open- label extension of the randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2020;19(12):998-1009. doi: 10.1016/S1474-4422(20)30342-2 [DOI] [PubMed] [Google Scholar]
- 10.Li C, Zhang L, Qiao L, et al. . Combination therapy with anti-CD20 mAb and IL-10 gene to reverse type 1 diabetes by attenuating pancreatitis and inhibiting apoptosis in NOD mice. Life Sci. 2020;256:117985. doi: 10.1016/j.lfs.2020.117985 [DOI] [PubMed] [Google Scholar]
- 11.Klein C, Lammens A, Schafer W, et al. . Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. MAbs. 2013;5(1):22-33. doi: 10.4161/mabs.22771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palanichamy A, Jahn S, Nickles D, et al. . Rituximab efficiently depletes increased CD20- expressing T cells in multiple sclerosis patients. J Immunol. 2014;193(2):580-586. doi: 10.4049/jimmunol.1400118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barkhof F, Kappos L, Wolinsky JS, et al. . Onset of clinical and MRI efficacy of ocrelizumab in relapsing multiple sclerosis. Neurology. 2019;93(19):e1778–e1786. doi: 10.1212/WNL.0000000000008189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baker D, Pryce G, James LK, Marta M, Schmierer K. The ocrelizumab phase II extension trial suggests the potential to improve the risk: benefit balance in multiple sclerosis. Mult Scler Relat Disord. 2020;44:102279. doi: 10.1016/j.msard.2020.102279 [DOI] [PubMed] [Google Scholar]
- 15.Roach CA, Cross AH. Anti-CD20 B cell treatment for relapsing multiple sclerosis. Front Neurol. 2020;11:595547. doi: 10.3389/fneur.2020.595547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DiLillo DJ, Hamaguchi Y, Ueda Y, et al. . Maintenance of long-lived plasma cells and serological memory despite mature and memory B cell depletion during CD20 immunotherapy in mice. J Immunol. 2008;180(1):361-371. doi: 10.4049/jimmunol.180.1.361 [DOI] [PubMed] [Google Scholar]
- 17.Hauser SL, Kappos L, Montalban X, et al. . Safety of ocrelizumab in patients with relapsing and primary progressive multiple sclerosis. Neurology. 2021;97(16):e1546-e1559. doi: 10.1212/wnl.0000000000012700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perriguey M, Maarouf A, Stellmann JP, et al. . Hypogammaglobulinemia and infections in patients with multiple sclerosis treated with rituximab. Neurol Neuroimmunol Neuroinflamm. 2022;9(1):e1115. doi: 10.1212/nxi.0000000000001115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sormani MP, Salvetti M, Labauge P, et al. . DMTs and Covid-19 severity in MS: a pooled analysis from Italy and France. Ann Clin Transl Neurol. 2021;8(8):1738-1744. doi: 10.1002/acn3.51408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng C, Kar I, Chen CK, et al. . Multiple sclerosis disease-modifying therapy and the COVID-19 pandemic: implications on the risk of infection and future vaccination. CNS Drugs. 2020;34(9):879-896. doi: 10.1007/s40263-020-00756-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hada M, Mosholder AD, Leishear K, Perez-Vilar S. Systematic review of risk of SARS-CoV-2 infection and severity of COVID-19 with therapies approved to treat multiple sclerosis. Neurol Sci. 2022;43(3):1557-1567. doi: 10.1007/s10072-021-05846-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Apostolidis SA, Kakara M, Painter MM, et al. . Cellular and humoral immune responses following SARS-CoV-2 mRNA vaccination in patients with multiple sclerosis on anti-CD20 therapy. Nat Med. 2021;27(11):1990-2001. doi: 10.1038/s41591-021-01507-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sormani MP, Inglese M, Schiavetti I, et al. . Effect of SARS-CoV-2 mRNA vaccination in MS patients treated with disease modifying therapies. eBioMedicine. 2021;72:103581. doi: 10.1016/j.ebiom.2021.103581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tallantyre EC, Vickaryous N, Anderson V, et al. . COVID-19 vaccine response in people with multiple sclerosis. Ann Neurol. 2022;91(1):89-100. doi: 10.1002/ana.26251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tortorella C, Aiello A, Gasperini C, et al. . Humoral- and T-cell–specific immune responses to SARS-CoV-2 mRNA vaccination in patients with MS using different disease-modifying therapies. Neurology. 2022;98(5):e541–e554. doi: 10.1212/WNL.0000000000013108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brill L, Raposo C, Rechtman A, et al. . Severe acute respiratory syndrome coronavirus 2 third vaccine immune response in multiple sclerosis patients treated with ocrelizumab. Ann Neurol. 2022;91(6):796-800. doi: 10.1002/ana.26343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brill L, Rechtman A, Zveik O, et al. . Humoral and T-cell response to SARS-CoV-2 vaccination in patients with multiple sclerosis treated with ocrelizumab. JAMA Neurol. 2021;78(12):1510-1514. doi: 10.1001/jamaneurol.2021.3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palomares Cabeza V, Kummer LYL, Wieske L, et al. . Longitudinal T-cell responses after a third SARS-CoV-2 vaccination in patients with multiple sclerosis on ocrelizumab or fingolimod. Neurol Neuroimmunol Neuroinflamm. 2022;9(4):e1178. doi: 10.1212/nxi.0000000000001178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwarz T, Otto C, Jones TC, et al. . Preserved T cell responses to SARS-CoV-2 in anti-CD20 treated multiple sclerosis. Mult Scler. 2022;28(7):1041-1050. doi: 10.1177/13524585221094478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Madelon N, Heikkilä N, Sabater Royo I, et al. . Omicron-specific cytotoxic T-cell responses after a third dose of mRNA COVID-19 vaccine among patients with multiple sclerosis treated with ocrelizumab. JAMA Neurol. 2022;79(4):399-404. doi: 10.1001/jamaneurol.2022.0245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guerrera G, Mandelli A, Finardi A, et al. . Anti-SARS-CoV-2 T-stem cell memory persists in ocrelizumab-treated MS patients. Mult Scler J. 2022;28(12):1937-1943. doi: 10.1177/13524585221102158 [DOI] [PubMed] [Google Scholar]
- 32.Sabatino JJ, Wilson MR, Calabresi PA, Hauser SL, Schneck JP, Zamvil SS. Anti-CD20 therapy depletes activated myelin-specific CD8+ T cells in multiple sclerosis. Proc Natl Acad Sci USA. 2019;116(51):25800-25807. doi: 10.1073/pnas.1915309116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Capasso N, Nozzolillo A, Scalia G, et al. . Ocrelizumab depletes T-lymphocytes more than rituximab in multiple sclerosis. Mult Scler Relat Disord. 2021;49:102802. doi: 10.1016/j.msard.2021.102802. [DOI] [PubMed] [Google Scholar]
- 34.Fernández-Velasco JI, Kuhle J, Monreal E, et al. . Effect of ocrelizumab in blood leukocytes of patients with primary progressive MS. Neurol Neuroimmunol Neuroinflamm. 2021;8(2):e940. doi: 10.1212/NXI.0000000000000940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jilek S, Jaquiery E, Hirsch HH, et al. . Immune responses to JC virus in patients with multiple sclerosis treated with natalizumab: a cross-sectional and longitudinal study. Lancet Neurol. 2010;9(3):264-272. doi: 10.1016/S1474-4422(10)70006-5 [DOI] [PubMed] [Google Scholar]
- 36.Noto A, Procopio FA, Banga R, et al. . CD32+ and PD-1+ lymph node CD4 T cells support persistent HIV-1 transcription in treated aviremic individuals. J Virol. 2018;92(20):e00901-e00918. doi: 10.1128/jvi.00901-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leipold MD, Obermoser G, Fenwick C, et al. . Comparison of CyTOF assays across sites: results of a six-center pilot study. J Immunol Methods. 2018;453:37-43. doi: 10.1016/j.jim.2017.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mrdjen D, Pavlovic A, Hartmann FJ, et al. . High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity. 2018;48(2):380-395.e6. doi: 10.1016/j.immuni.2018.01.011 [DOI] [PubMed] [Google Scholar]
- 39.Galli E, Hartmann FJ, Schreiner B, et al. . GM-CSF and CXCR4 define a T helper cell signature in multiple sclerosis. Nat Med. 2019;25(8):1290-1300. doi: 10.1038/s41591-019-0521-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mathias A, Perriot S, Canales M, et al. . Impaired T-cell migration to the CNS under fingolimod and dimethyl fumarate. Neurol Neuroimmunol Neuroinflamm. 2017;4(6):e401. doi: 10.1212/nxi.0000000000000401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harari A, Bart PA, Stohr W, et al. . An HIV-1 clade C DNA prime, NYVAC boost vaccine regimen induces reliable, polyfunctional, and long-lasting T cell responses. J Exp Med 2008;205(1):63-77. doi: 10.1084/jem.20071331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mathias A, Perriard G, Canales M, et al. . The VZV/IE63-specific T cell response prevents herpes zoster in fingolimod-treated patients. Neurol Neuroimmunol Neuroinflamm. 2016;3(2):e209. doi: 10.1212/NXI.0000000000000209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nissimov N, Hajiyeva Z, Torke S, et al. . B cells reappear less mature and more activated after their anti-CD20 – mediated depletion in multiple sclerosis. Proc Natl Acad Sci USA. 2020;117(41):25690-25699. doi: 10.1073/pnas.2012249117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.von Essen MR, Hansen RH, Højgaard C, Ammitzbøll C, Wiendl H, Sellebjerg F. Ofatumumab modulates inflammatory T cell responses and migratory potential in patients with multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. 2022;9(4):e200004. doi: 10.1212/NXI.0000000000200004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quendt C, Ochs J, Häusser-Kinzel S, Häusler D, Weber MS. Proinflammatory CD20+ T cells are differentially affected by multiple sclerosis therapeutics. Ann Neurol. 2021;90(5):834-839. doi: 10.1002/ana.26216 [DOI] [PubMed] [Google Scholar]
- 46.Smolders J, van Luijn MM, Hsiao C-C, Hamann J. T-cell surveillance of the human brain in health and multiple sclerosis. Semin Immunopathol. 2022;44(6):855-867. doi: 10.1007/s00281-022-00926-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iannetta M, Landi D, Cola G, et al. . T-cell responses to SARS-CoV-2 in multiple sclerosis patients treated with ocrelizumab healed from COVID-19 with absent or low anti-spike antibody titers. Mult Scler Relat Disord. 2021;55:103157. doi: 10.1016/j.msard.2021.103157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zabalza A, Arrambide G, Tagliani P, et al. . Humoral and cellular responses to SARS-CoV-2 in convalescent COVID-19 patients with multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. 2022;9(2):e1143. doi: 10.1212/NXI.0000000000001143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kister I, Patskovsky Y, Curtin R, et al. . Cellular and humoral immunity to SARS-CoV-2 infection in multiple sclerosis patients on ocrelizumab and other disease-modifying therapies: a multi-ethnic observational study. Ann Neurol. 2022;91(6):782-795. doi: 10.1002/ana.26346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Landi D, Grimaldi A, Bovis F, et al. . Influence of previous disease-modifying drug exposure on T-lymphocyte dynamic in patients with multiple sclerosis treated with ocrelizumab. Neurol Neuroimmunol Neuroinflamm. 2022;9(3):e1157. doi: 10.1212/NXI.0000000000001157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Borrelli S, Mathias A, Goff GL, Pasquier RD, Théaudin M, Pot C. Delayed and recurrent dimethyl fumarate induced-lymphopenia in patients with multiple sclerosis. Mult Scler Relat Disord. 2022;63:103887. doi: 10.1016/j.msard.2022.103887 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data not published within this article will be made available by request from any qualified investigator.