

Abstract
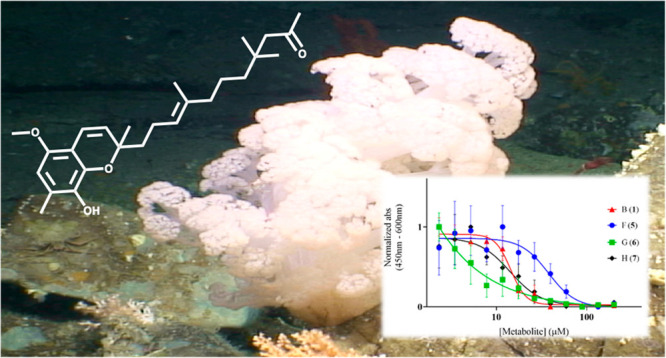
Previous chemical investigation of the Irish deep-sea soft coral Duva florida led to the identification of tuaimenal A (10), a new merosesquiterpene containing a highly substituted chromene core and modest cytotoxicity against cervical cancer. Further MS/MS and NMR-guided investigation of this octocoral has resulted in the isolation and characterization of seven additional tuaimenal analogs, B–H (1–7), as well as two known A-ring aromatized steroids (8, 9), and additional tuaimenal A (10). Tuaimenals B, F, and G (1, 5, 6), bearing an oxygen at the C5 position, as well as monocyclic tuaimenal H (7), show increased cervical cancer inhibition profiles in comparison to that of 10. Tuaimenal G further displayed potent, selective cytotoxicity with an EC50 value of 0.04 μM against the C33A cell line compared to the CaSki cell line (EC50 20 μM). These data reveal the anticancer properties of tuaimenal analogs and suggest unique antiproliferation mechanisms across these secondary metabolites.
Analysis of all the newly approved drugs spanning the past four decades has shown that roughly 60% have been inspired by nature’s secondary metabolites, with that number soaring over 80% for small-molecule anticancer drugs.1 These metabolites are chemically diverse and innately specific compounds often produced as chemical defenses against neighboring organisms to increase the survivability of a species. The ocean’s deep seas may well represent the richest remaining source of new bioactive natural products capable of inspiring the next generation of cytotoxic therapeutic agents against diseases such as cervical cancer. With three-quarters of the earth’s surface covered in oceans, of which 95% exists below 1000 m, and advances such as remotely operated vehicles (ROVs) allowing for the targeted collections of deep-sea organisms only appearing in recent decades, the biotherapeutic potential of soft-bodied corals uniquely adapted to life in extreme conditions is only beginning to be explored.2 As the mysteries of the deep continue to be probed, the perception of a barren, lifeless wasteland existing within ocean trenches has given way to the realization that biological, and potentially chemical, diversity exists comparable to that found in tropical rain forests.3 While only around 2% of all natural products emanate from the deep seas, analysis of these secondary metabolites has revealed that nearly 75% possess bioactivity, further enhancing the urgency to explore the deep.4
Human papillomavirus (HPV) is a ubiquitous virus of the Papovaviridae family linked to 99.7% of cervical squamous cell cancer cases worldwide.5 Simultaneous binding of the HPV oncoproteins E6 and E7 to the p53 tumor suppressor protein and the pRB retinoblastoma tumor suppressor protein, respectively, allows for both the inhibition of typical apoptosis and a heightened rate of cell proliferation, ultimately cascading into a deadly cancer if left untreated.6 While a tremendous effort has been undertaken to provide HPV vaccinations as a primary prevention and comprehensive screenings as secondary prevention, cervical cancer remains the fourth most common cancer among women worldwide, with approximately 570 000 new cases and 311 000 deaths in 2018 across both HPV-positive and HPV-negative incidences.7,8 These rates are exceptionally higher in low- to middle-income, resource-poor nations where education, vaccinations, and screenings are less accessible, accounting for roughly 85% of all cervical cancer deaths.6
Previous chemical investigation into a collection of four specimens
of the deep-sea Irish soft coral Duva florida afforded
tuaimenal A (10). A merosesquiterpene with a highly substituted
chromene core, 10 bears structural similarities to numerous
terrestrial and marine secondary metabolites containing a “tocopherol-esque”
backbone, perhaps most notably comparable to the two formylated tocotrienols
5-formyl-δ-tocotrienol and 7-formyl-δ-tocotrienol isolated
from the stem bark of Garcinia virgata, with all
three compounds possessing a benzaldehyde moiety.9 Justification for this unique substitution pattern about
the chromene core was posed by Avalon et al. as resulting from a divergence
from the typical tocopherol biosynthetic pathway at the homogentisate
phytyltransferase step.10 Further, tuaimenal
A showed promising potential in inhibiting the main protease of SARS-CoV-2 in silico as well as weak activity as a cytotoxic agent
against cervical cancer.10 Herein we report
further tandem mass spectrometry (MS/MS) and nuclear magnetic resonance
(NMR) guided investigation into analogous metabolites resulting in
the isolation, characterization, and cytotoxic evaluation of seven
additional tuaimenals, B–H (1–7), two known A-ring-aromatized steroids (8, 9), and tuaimenal A (10).
Tuaimenal B (1) was isolated as a pale orange oil. A molecular formula of C24H32O4 was established by HRESIMS ([M – H]−: m/z 383.2241, calcd 383.2228), corroborated by signals in the 1H and 13C NMR spectra (Table 1). Key 1H NMR signals (Figure 1) included a broad olefinic singlet H-4 (δH 6.32) demonstrating coupling in the COSY spectrum through C-3 (δC 118.0) to the benzylic methyl protons of H3-22 (δH 2.04), as well as HMBC correlations to three fully substituted carbons, C-2 (δC 147.1), C-3, and C-5 (δC 138.6), and additional four-bond correlations to C-1 (δC 137.8) and the olefinic carbon C-7 (δC 122.1). The singlet methyl protons H3-22 also displayed HMBC correlations to C-2 and C-3. The vinylic protons H-7 and H-8 (δH 6.24 and 5.57, respectively) were both observed as doublets with J-values of 9.8 Hz showing a COSY correlation to each other. The positions of H-7 and H-8 were assigned using HMBC correlations of H-7 to carbons C-5 and C-6 (δC 119.7), as well as the quaternary carbon C-9 (δC 78.6), in conjunction with HMBC correlations of H-8 to C-6, C-9, methylene carbon C-10 (δC 41.0), and methyl C-21 (δC 25.8). The singlet methyl protons on H3-21 (δH 1.33) also displayed HMBC correlations for C-9 and C-10. A triplet methylene H2-10 (δH 1.67) correlated in the HMBC spectrum to C-9, the adjacent methylene C-11 (δC 22.4) bearing two nonequivalent protons H2-11 (δH 2.07 and 2.13), as well the olefinic C-12 (δC 124.0) bearing triplet H-12 (δH 5.13). H2-11 additionally displayed correlations to C-12 as well as C-13 (δC 135.3). The olefinic proton H-12 displayed both COSY and HMBC correlations to the methyl protons H3-20 (δH 1.59) and carbon C-20 (δC 15.9) and an additional HMBC correlation to the methylene carbon C-14 (δC 39.7). The methylene triplet H2-14 (δH 1.97) correlated in the HMBC spectrum to both C-13 and methylene C-15 (δC 26.7), the latter carbon bearing a multiplet H2-15 (δH 2.06). H-16 (δH 5.09) also appeared as a triplet partially overlapping with the other side chain olefinic proton H-12; however H2-16 was placed on the basis of HMBC correlations to the two terminal methyl groups C-18 and C-19 (δC 17.7 and 25.7, respectively). Additionally, H2-16 displayed COSY correlations in both directions of the side chain to H2-15 as well as through C-17 (δC 131.3) to H3-18 (δH 1.60) and H3-19 (δH 1.68). H3-18 and H3-19 both displayed HMBC correlations to each other, as well as to C-17. H3-24 (δH 2.34) appeared as a methyl singlet with a chemical shift indicative of an acetyl group further supported by a single HMBC correlation to an ester-type carbonyl C-23 (δC 168.9). The C-12 and C-13 olefinic configuration was determined to be E by a NOESY correlation between H-12 and H2-14 linking these two protons in close proximity through space (Figure 1).
Table 1. NMR Data for Tuaimenal B (1)a.
| pos | δC,b type | δH,c (J in Hz) | HMBCd | COSYd | NOESYd |
|---|---|---|---|---|---|
| 1 | 137.8, C | ||||
| 2 | 147.1, C | ||||
| 3 | 118.0, C | ||||
| 4 | 109.8, CH | 6.32, br s | 1, 2, 3, 5, 7 | 22 | 22 |
| 5 | 138.6, C | ||||
| 6 | 119.7, C | ||||
| 7 | 122.1, CH | 6.24, d (9.8) | 4, 5, 6, 9 | 8 | 8 |
| 8 | 130.3, CH | 5.57, d (9.8) | 6, 9, 10, 21 | 21 | |
| 9 | 78.6, C | ||||
| 10 | 41.0, CH2 | 1.67, m | 9, 11, 12, 21 | ||
| 11 | 22.4, CH2 | 2.07, m | 10, 12, 13 | ||
| 2.13, m | 10, 12 | ||||
| 12 | 124.0, CH | 5.13, t (6.54) | 10, 11, 14, 20 | 20 | 14 |
| 13 | 135.3, C | ||||
| 14 | 39.7, CH2 | 1.97, t (7.63) | 12, 13, 15, 20 | ||
| 15 | 26.7, CH2 | 2.06, m | 13, 14, 16 | ||
| 16 | 124.3, CH | 5.09, t (7.08) | 15, 18, 19 | 15, 18, 19 | 19 |
| 17 | 131.3, C | ||||
| 18 | 17.7, CH3 | 1.60, s | 16, 17, 19 | ||
| 19 | 25.7, CH3 | 1.68, s | 16, 17, 18 | ||
| 20 | 15.9, CH3 | 1.59, s | 12, 13, 14 | ||
| 21 | 25.8, CH3 | 1.33, s | 8, 9, 10 | ||
| 22 | 9.3, CH3 | 2.04, s | 2, 3, 4 | ||
| 23 | 168.9, C | ||||
| 24 | 20.4, CH3 | 2.34, s | 23 |
CDCl3, ppm, type established by phase-sensitive HSQC.
150 MHz.
600 MHz.
500 MHz.
Figure 1.
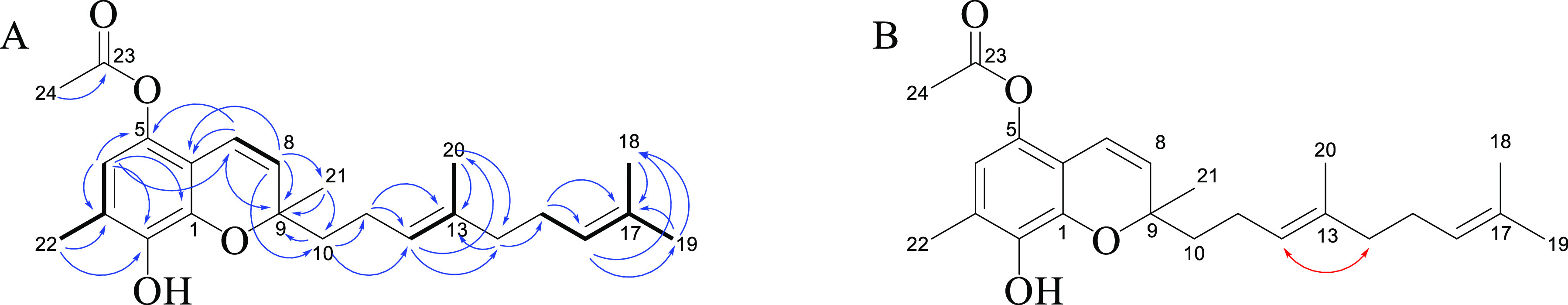
Key HMBC (→) and COSY (―) correlations (A) and key NOESY (↔) correlations (B) establishing the olefin configuration of tuaimenal B (1).
Tuaimenal C (2) was isolated as a pale yellow oil with spectral data similar to those of tuaimenal B (1). A molecular formula of C25H34O4 for 2 was established from HREIMS ([M+]: m/z 398.2452, calcd 398.2457), corroborated by 1H and 13C NMR spectra (Table 2). Tuaimenal C departed from the motif of 1 by the presence of a methoxy signal H3-23 (δH 3.84) in the 1H NMR spectrum, while retaining an acetyl group as indicated by H3-25 (δH 2.30). Assignment of the acetate on C-2 (δC 142.4) was determined through HMBC correlations of the singlet methyl H3-22 (δH 2.05) and a long-range correlation between the acetate protons H3-25 and the acetate bearing C-2. The assignment of the methoxy group on C-5 (δC 146.7) was confirmed by HMBC correlations between both H3-23 and the doublet olefinic signal H-7 (δC 6.27) to C-5. All remaining 1H and 13C NMR signals align with that of tuaimenal B including the aromatic proton H-4 (δH 6.45) on C-4 (δC 114.4) of the chromene core, as well as throughout the terpene side chain, and were confirmed through HMBC, COSY, and NOESY correlations.
Table 2. NMR Data for Tuaimenals C, D, E, F, G, and H (2–7)a.
| tuaimenal
C |
tuaimenal
D |
tuaimenal
E |
tuaimenal
F |
tuaimenal
G |
tuaimenal
H |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pos | δC,b type | δHc (J in Hz) | δC,b type | δHc (J in Hz) | δC,b type | δHc (J in Hz) | δC,b type | δHc (J in Hz) | δC,b type | δHc (J in Hz) | δC,b type | δHc (J in Hz) |
| 1 | 143.7, C | 138.6, C | 132.2, C | 139.5, C | 147.3, C | 134.2, C | ||||||
| 2 | 142.4, C | 151.4, C | 151.4, C | 147.3, C | 139.6, C | 152.5, C | ||||||
| 3 | 124.5, C | 111.8, C | 111.8, C | 118.2, C | 118.1, C | 109.6, C | ||||||
| 4 | 114.4, CH | 6.45, s | 158.2, C | 158.2, C | 107.5, CH | 6.25, s | 107.5, CH | 6.26, s | 159.5, C | |||
| 5 | 146.7, C | 107.6, C | 107.6, C | 146.7, C | 146.7, C | 110.7, C | ||||||
| 6 | 119.9, C | 118.6, C | 118.7, C | 119.8, C | 119.8, C | 126.9, C | ||||||
| 7 | 122.5, CH | 6.27, d (9.8) | 116.5, CH | 6.87, d (10.2) | 116.5, CH | 6.86, d (10.2) | 122.6, CH | 6.24, ov d | 122.6, CH | 6.25, d, (9.8) | 23.5, CH2 | 3.60, br s |
| 8 | 129.6, CH | 5.57, d (9.8) | 132.4, CH | 5.80, d (9.8) | 132.4, CH | 5.80, d (10.2) | 129.7, CH | 5.56, d (9.8) | 129.7, CH | 5.57, d (9.8) | 121.1, CH | 5.16, br t |
| 9 | 79.0, C | 79.2, C | 79.1, C | 78.1, C | 78.1, C | 139.2, C | ||||||
| 10 | 41.6, CH2 | 1.84, m | 40.3, CH2 | 1.77, m | 40.3, CH2 | 1.78, m | 41.2, CH2 | 1.83, m | 41.2, CH2 | 1.83, m | 39.5, CH2 | 2.08, m |
| 1.67, m | 1.67, m | 1.68, m | ||||||||||
| 11 | 23.0, CH2 | 2.18, m | 22.5, CH2 | 1.43, m | 22.6, CH2 | 2.11, m | 22.8, CH2 | 2.15, m | 22.8, CH2 | 2.16, m | 26.2, CH2 | 2.11, m |
| 2.12, m | ||||||||||||
| 12 | 124.0, CH | 5.13, t (6.47) | 123.6, CH | 5.12, t (7.08) | 123.6, CH | 5.11, t (6.99) | 124.2, CH | 5.14, t (7.08) | 124.2, CH | 5.13, t (6.9) | 123.3, CH | 5.04, t (6.36) |
| 13 | 135.6, C | 135.8, C | 135.6, C | 135.2, C | 135.1, C | 135.9, C | ||||||
| 14 | 39.8, CH2 | 1.97, t | 39.9, CH2 | 1.97, t (6.18) | 39.7, CH2 | 1.96, m | 39.9, CH2 | 1.96, t (6.54) | 39.7, CH2 | 1.95, t (7.63) | 39.6, CH2 | 1.96, t (7.63) |
| 15 | 26.8, CH2 | 2.06, m | 22.6, CH2 | 2.13, m | 22.0, CH2 | 1.68, m | 22.6, CH2 | 1.44, m | 22.1, CH2 | 1.40, m | 26.6, CH2 | 2.03, m |
| 1.38, m | ||||||||||||
| 16 | 124.4, CH | 5.09, t (6.96) | 43.4, CH2 | 1.43, m | 40.2, CH2 | 1.69, m | 43.3, CH2 | 1.42, m | 40.4, CH2 | 1.67, m | 124.2, CH | 5.07, t (6.72) |
| 17 | 131.5, C | 70.1, C | 82.4, C | 71.1, C | 82.4, C | 131.4, C | ||||||
| 18 | 17.8, CH3 | 1.60, s | 29.2, CH3 | 1.22, s | 26.1, CH3 | 1.42, s | 29.2, CH3 | 1.21, s | 26.0, CH3 | 1.42, s | 17.6, CH3 | 1.60, s |
| 19 | 25.8, CH3 | 1.68, s | 29.2, CH3 | 1.22, s | 26.1, CH3 | 1.42, s | 29.2, CH3 | 1.21, s | 26.0, CH3 | 1.42, s | 25.7, CH3 | 1.67, s |
| 20 | 16.1, CH3 | 1.59, s | 15.9, CH3 | 1.58, s | 15.8, CH3 | 1.57, s | 15.8, CH3 | 1.57, s | 15.8, CH3 | 1.57, s | 16.0, CH3 | 1.59, s |
| 21 | 26.5, CH3 | 1.41, s | 25.4, CH3 | 1.43, s | 25.4, CH3 | 1.43, s | 25.9, CH3 | 1.39, s | 25.9, CH3 | 1.39, s | 16.4, CH3 | 1.82, s |
| 22 | 9.9, CH3 | 2.05, s | 7.6, CH3 | 2.14, s | 7.6, CH3 | 2.14, s | 9.0, CH3 | 2.14, s | 9.0, CH3 | 2.14, s | 7.3, CH3 | 2.12, s |
| 23 | 60.5, CH3 | 3.84, s | 191.1, CH | 10.09, s | 191.0, CH | 10.08, s | 60.4, CH3 | 3.83, s | 60.4, CH3 | 3.84, s | 192.9, CH | 9.98, s |
| 24 | 169.8, C | 170.5, C | 170.6, C | |||||||||
| 25 | 20.9, CH3 | 2.30, s | 22.5, CH3 | 1.97, s | 22.5, CH3 | 1.97, s | ||||||
CDCl3, ppm.
150 MHz, type established by phase-sensitive HSQC.
600 MHz.
Tuaimenal D (3) was isolated as a pale orange oil with spectral data similar to that of tuaimenals B and C (1, 2). A molecular formula of C23H32O5 for 3 was established from HRESIMS ([M – H]−: m/z 387.2197, calcd 387.2177), corroborated by 1H and 13C NMR spectra (Table 2). Tuaimenal D departed from the motif of 1 and 2 in four major ways observable in the 1H NMR spectrum: (1) the loss of the olefinic triplet at H-16, which now displayed as a methylene multiplet H2-16 (δH 1.43), resulting in the attached methyl groups H3-18 and H3-19 (δH 1.22) as newly equivalent, (2) a fifth oxygen noted in the chemical formula not accounted for in the 1H NMR spectrum when acquired in CDCl3, (3) a newly appearing downfield singlet H-23 (δH 10.09) indicative of the presence of an aldehyde, and (4) the loss of the aromatic proton signal H-4. The first two departures from the aforementioned motif were accounted for by the addition of a tertiary alcohol at the C-17 (δC 70.1) position on the basis of the chemical shift displaying congruence with that of a fully saturated, oxygen-bearing carbon. This placement also accounts for the loss of the C-16/C-17 olefin and would result in H3-18 and H3-19 appearing as overlapping equivalent signals in the 1H NMR spectrum, and it is not unexpected for this alcohol to be absent when acquiring NMR data with CDCl3. Additionally, HMBC correlations were observed from H3-18 to C-16 (δC 43.4), C-17, and C-19. The aldehyde moiety was determined to be at the aromatic C-5 (δC 107.6) position due to a single HMBC correlation for H-23 to C-5. The last departure from the motif of 1 and 2, being the absence of an aromatic proton at the C-4 position, was justified by the placement of a phenol on C-4 (δC 158.2). This configuration was assigned on the basis of the downfield shift of this carbon indicating an oxygen-bearing aromatic position, HMBC correlations from the new alcohol proton to C-3 (δC 111.8), C-4, and C-5, and congruence with the chemical formula proposed from the HRESIMS data.
Tuaimenal E (4) was also isolated as a pale orange oil with spectral data similar to those of tuaimenal D (3). A molecular formula of C25H34O6 for 4 was established from HRESIMS ([M – H]−: m/z 429.2296, calcd 429.2283), corroborated by 1H and 13C NMR spectra (Table 2). Tuaimenal E departed from the motif of 3 by the presence of an acetyl methyl group H3-25 (δH 1.97) and additional deshielding of the two equivalent terminal side chain methyl groups H3-18 and H3-19 (δH 1.42). The additional two carbons, two protons, and one oxygen observed in 4 thus corresponded to an acetoxy group in place of the tertiary alcohol at the C-17 (δC 82.4), as observed for 3. All 1H and 13C NMR chemical shifts, coupling, and integrations associated with the bicyclic chromene core for 4 aligned with that of 3.
Tuaimenal F (5) was isolated as a yellow oil with spectral data similar to those of tuaimenal D (3). A molecular formula of C23H34O4 for 5 was established from HRESIMS ([M – H]−: m/z 373.2399, calcd 373.2384), corroborated by 1H and 13C NMR spectra (Table 2). Tuaimenal F departed from the motif of 3 by the loss of the deshielded phenol OHa (δH 12.30) and aldehyde H-23 (δH 10.09), as well as the appearance of an aromatic singlet H-4 (δH 6.25) and methoxy singlet H3-23 (δH 3.83). The placement of H-4 and H3-23 was determined through HMBC correlations of the methyl singlet H3-22 (δH 2.14) to C-4 (δC 107.5) as well as both H3-23 and H-7 (δH 6.24) to C-5 (δC 146.7). The terminal tertiary alcohol of 3 was determined to be retained in 5 on the basis of 1H and 13C NMR shifts and HMBC correlations.
Tuaimenal G (6) was also isolated as a yellow oil with spectral data similar to those of tuaimenal F (5). A molecular formula of C25H36O5 for 6 was established from HRESIMS ([M + Na]+: m/z 439.2455, calcd 439.2455), corroborated by 1H and 13C NMR spectra (Table 2). Tuaimenal G departed from the motif of 5 by the presence of an acetyl methyl group H3-25 (δH 1.97) and additional deshielding of the two equivalent terminal side chain methyl groups H3-18 and H3-19 (δH 1.42). The additional two carbons, two protons, and one oxygen observed in 6 thus corresponded to an acetoxy group in place of the 5 tertiary alcohol at C-17 (δC 82.4). All 1H and 13C NMR chemical shifts, splitting patterns, and integrations associated with the bicyclic chromene core for 6 aligned with those of 5.
Tuaimenal H (7) was isolated as a pale orange film with spectral data similar to those of tuaimenal D (3). A molecular formula of C23H32O4 for 7 was established from HRESIMS ([M – H]−: m/z 371.2245, calcd 371.2228), corroborated by 1H and 13C NMR spectra (Table 2). Tuaimenal H departed from the motif of 3 (Figure 2) by the loss of the olefinic doublets H-7 and H-8 as well as the presence of two additional olefinic triplets resulting in three side chain double bonds H-8 (δH 5.16), H-12 (δH 5.04), and H-16 (δH 5.07). Additionally, a broad deshielded singlet H2-7 (δH 3.60) integrating to 2, indicative of the presence of a methylene group allylic to two double bonds, was observed, resulting in the determination that the ether-containing ring in 7 was absent. HMBC correlations of H3-22 (δH 2.12) to the two phenol-bearing aromatic carbons C-2 (δC 152.5) and C-4 (δH 159.5), as well as from OHa (δH 12.59) to C-3 (δC 109.6) and C-5 (δC 110.7), and the aldehyde singlet H-23 (δH 9.98) to C-4 and C-5 placed the substituents of the intact aromatic ring. H2-7 displayed COSY correlations through C-8 (δC 121.1) and C-9 (δC 139.2) to the singlet methyl H3-21 (δH 1.82) and triplet methylene H2-10 (δH 2.08), as well as HMBC correlations to the aromatic carbons C-1 (δC 134.2), C-5, and C-6 (δC 126.9) and olefinic C-8 and C-9. The olefinic singlet H-8 (δH 5.16) displayed HMBC correlations to the methyl C-21 (δC 16.4), as well as to the methylene C-10 (δC 39.5). H2-10 was shown to correlate in the HMBC to a methylene C-11 (δC 26.2), for which H-11 (δH 2.11) had a COSY correlation to the olefinic triplet H-12 (δH 5.04) and HMBC correlations to olefinic C-12 and C-13 (δC 135.9). The methyl C-20 (δC 16.0) was placed on C-13 due to correlations of H3-20 (δH 1.59) to C-13 and methylene C-14 (δC 39.6), for which the methylene triplet H2-14 (δH 1.96) displayed HMBC correlations to C-15 (δC 26.6) and C-16 (δC 124.2). The terminal end of the side chain was assigned on the basis of COSY correlations from H-16 (δH 5.07) to H2-15 (δH 2.03) and through C-17 (δC 131.4) to H3-18 and H3-19 (δH 1.60 and 1.67, respectively), as well as HMBC correlations from H3-18 to C-17 and C-19 (δC 25.7). The side chain double-bond configuration (Figure 2) was determined as 8E, 12E due to the observation of NOESY correlations of H2-7 to H3-21, H-8 to H2-10, H2-10 to H-12, and H-12 to H2-14.
Figure 2.
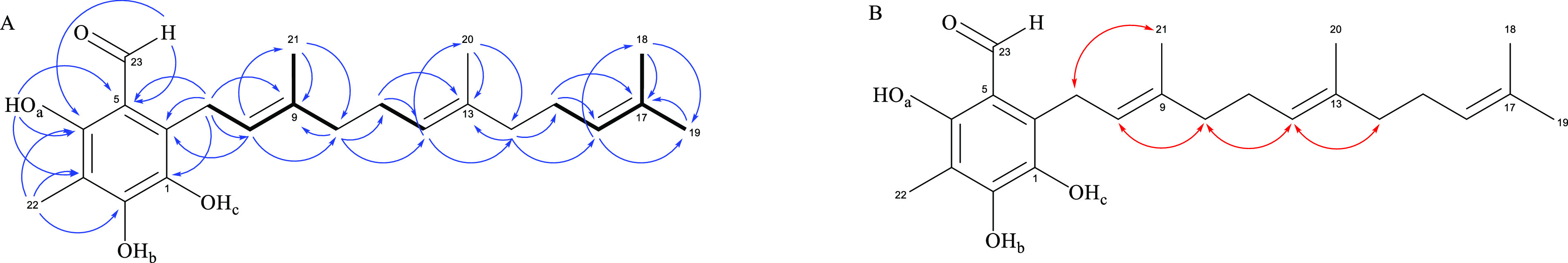
Key HMBC (→) and COSY (―) correlations (A) and key NOESY (↔) correlations (B) establishing the olefinic configurations of tuaimenal H (7).
Oxidized steroid 8 was isolated as a white, crystalline solid. A molecular formula of C23H32O3 was established from high-resolution electrospray ionization mass spectrometry (HRESIMS) ([M – H]−: m/z 355.2298, calcd 355.2279). Comparison of the HSQC using SMARTNMR, an online artificial intelligence-based database of natural products made available by the University of California San Diego, revealed a strong correlation (cos > 0.992) to that of 3-hydroxy-19-norchola-1,3,510-trien-24-oic acid, a ring-A-aromatized bile acid originally reported from the marine sponge Sollasella moretonensis.11,12 Further comparison of the literature 13C NMR chemical shifts (Table S2) confirmed the structure of 8. Absolute stereochemical determination on the gonane backbone of 8 was determined through X-ray crystallography (Figure S74).
Oxidized steroid 9 was isolated as a white, crystalline solid with spectral data similar to those of 8. A molecular formula of C24H34O3 (9) was established from HRESIMS ([M + H]+: m/z 371.2589, calcd 375.2581), corroborated by the 13C NMR spectrum (Table S2). The presence of a methyl ester H3-25 (δH 3.69), correlating in the HMBC spectrum to the side chain carbonyl C-24 (δC 175.0), was the major departure of 9 from 8.
Additional mass of tuaimenal A (10) was isolated from previously uninvestigated fractions of D. florida extract as a yellow oil matching the previously reported configuration of this metabolite on the basis of MS and NMR spectral data. A molecular formula of C23H30O4 for 10 was confirmed from HRESIMS ([M – H]−: m/z 369.2089, calcd 369.2071), corroborated by 1H and 13C NMR spectra. Interestingly, 10 was previously assigned by Avalon et al. (2022) as existing in the R configuration about the C-9 stereocenter on the basis of comparisons of experimental vibrational circular dichroism (VCD) data to computationally predicted values for each enantiomer.10 While significant overlap between experimental data and in silico predictions did exist indicating this assignment of absolute configuration, the observed VCD and optical rotation (OR) signals were both inconsistently low, incongruent with an enantiomerically pure metabolite. To determine if this trait was maintained for compounds 1–7, optical rotation data were recorded at the maximal concentrations for measurement for each compound using a 50 μL cell, with the results displayed in Table 3.
Table 3. Measured Optical Rotations in CHCl3 of All Tuaimenals Bearing a Stereocenter at the C-9 Position.
| compound | concentration (g/mL) | optical rotation ([α]25D) |
|---|---|---|
| tuaimenal B (1) | 0.058 | 0.0 |
| tuaimenal C (2) | 0.008 | 0.0 |
| tuaimenal D (3) | 0.018 | 1.1 |
| tuaimenal E (4) | 0.034 | 2.6 |
| tuaimenal F (5) | 0.059 | 1.5 |
| tuaimenal G (6) | 0.065 | 1.1 |
| tuaimenal A (10) | 0.065 | 0.5 |
The lack of significant optical rotation observed for all of the tuaimenals bearing the C-9 stereocenter indicated that these metabolites may in fact be present as racemic mixtures at the time of analysis, or at least with exceedingly low values for enantiomeric excess (ee). To confirm this finding, tuaimenal E (4), which showed, while still incredibly low, the highest value for optical rotation at [α]25D 2.6, was selected to be analyzed with a chiral-phase HPLC column with integrations calculated for the area under the curve of the signal from each enantiomer.
The results of this analysis showed relative areas of 49.8% and 51.2% for the two enantiomers present in the sample, leading to the conclusion that the tuaimenals, including tuaimenal A (10), which was reisolated, exist with sufficiently low ee that any further attempts at deciphering the predominant configuration would yield inconclusive results, and thus should be considered racemic mixtures. Further support of this theory is drawn from the recent review of naturally occurring 6-hydroxy-chromanols and 6-hydroxy-chromenols, which cited 73% of marine-derived chromanols as having been isolated in the R configuration, while only 26% of chromenols displayed the same stereochemistry, indicating that the chromenols as described herein may originate via a cyclization reaction that is not biosynthetically enzymatically catalyzed.13
Compounds isolated with sufficient mass for biological evaluation were tested for antiproliferative effects in vitro against two cervical cancer cell lines (Figure 3) at metabolite concentrations ranging from 2 to 200 μM. While none of the tested compounds revealed <10 μM effects on the HPV-positive CaSki cell line, all new metabolites showed increased activity in comparison to tuaimenal A (10) for both cell lines. Tuaimenal G (6) displayed potent activity in the C33A cell line with an EC50 value of 0.04 μM (Table 4). In contrast to the other evaluated metabolites, tuaimenal G additionally showed high specificity toward the HPV-negative cell line with a 500-fold difference in EC50 values between the C33A and CaSki cell lines. The 2018 review by Birringer et al. of naturally occurring chromanols and chromenols analyzed the structural motifs of such meroterpenoids in relation to anticancer activity and drew several conclusions of importance to the current study: (1) the chromanol/chromenol core is indespensible for activity, (2) the degree of methylation about this core is inversely related to activity, and (3) side chain olefins at C-12 and C-16 have little influence on activity.13 To these points, we have found (1) the tuaimenal core is able to retain comparable levels of cytotoxicity against the cell line C33A despite decylcization of the dihydropyran ring, (2) there exists a great deal of variability in activity among metabolites with a singular methyl group on the aromatic ring, and (3) it is correct in assessing that the side chain olefins play little role in activity. Additionally, on the basis of a nearly 1000-fold increase in activity from tuaimenal G (6) to tuaimenal F (5), it may be concluded that functionalization at C-17 as well as C-4 plays a pivotal role in activity against this cancerous cell line. Positive controls using 50 μM etoposide and negative controls in the absence of metabolite were conducted simultaneously for both cell lines to ensure viability of the assays (Figure 3). This observed specificity should be further probed using a larger panel of cancerous cell lines. Additional testing revealed no reportable antifungal activity against seven strains of Candida, antibiotic activity against the ESKAPE pathogens and Mycobacterium tuberculosis, or antiviral activity against human respiratory syncytial virus, indicating selective cytotoxicity.
Figure 3.

Antiproliferative effect of 1 and 5–7 on CaSki (A) and C33A cells (B) and 10 in CaSki and C33A cells (C), summary of antiproliferative effects of all tested tuaimenals (D), and DMSO (negative) and etoposide (positive) control data (E). All experiments were seeded at 1000–3000 cells per well in a 96-well plate and treated with varying concentrations of each metabolite (N = 3). At 48 h post-treatment proliferation was observed after a 2 h incubation with WST reagent (10 μL in 190 μL media/well). Nonlinear regression curves were fitted with GraphPad Prism software.
Table 4. Calculated EC50 Values for Antiproliferative Effect of Tuaimenals B, F, G, H, and A (1, 5–7, 10) against the Cervical Cancer CaSki and C33A Cell Lines.
| compound | CaSki EC50 (μM) | C33A EC50 (μM) |
|---|---|---|
| tuaimenal B (1) | 25 | 14 |
| tuaimenal F (5) | 41 | 38 |
| tuaimenal G (6) | 20 | 0.04 |
| tuaimenal H (7) | 23 | 14 |
| tuaimenal A (10) | 46 | 58 |
This study represents the second chemical investigation into the secondary metabolome of the Irish deep-sea coral D. florida for which only tuaimenal A (10) was previously reported. We report the isolation, structural elucidation, and biological evaluation of seven new analogous tuaimenals, B–H (1–7), all of which display increased levels of antiproliferative effects against both HPV-positive and HPV-negative cervical cancer cell lines, with tuaimenal G possessing potent in vitro cytotoxicity and selectivity against HPV-negative cervical cancer cell lines.
Experimental Section
General Experimental Procedures
Solvents were obtained from Fisher Scientific Co. and were HPLC grade (>99% purity) unless otherwise stated. UV absorptions were measured with an Agilent Cary 60 UV–vis spectrophotometer with a 10 mm path length fiber optic probe in acetonitrile. IR spectra were recorded as a thin film with an Agilent Cary 630 FTIR. NMR spectra were acquired in CDCl3 with residual solvent referenced as the internal standard (δH 7.27; δC 77.0) for 1H and 13C NMR spectra, respectively. The 1H and 13C spectra were recorded on a Bruker 600 MHz broadband instrument with the 13C spectra recorded at 150 MHz. 2D NMR experiments were recorded on a Varian 500 MHz broadband instrument. GC/MS analysis was performed on an Agilent 7890A GC using a Zebron ZB-5HT Inferno (30 m × 0.25 mm, 0.25 μm film thickness) column coupled to an Agilent 7200 accurate-mass QToF with electron impact ionization. LC/MS analysis was performed on an Agilent 1260 Infinity LC coupled to an Agilent 6540 UHD accurate-mass QToF with electrospray ionization. MPLC fractionation and analysis was performed on a Teledyne-Isco CombiFlash Rf system equipped with built-in UV detection at 254 and 280 nm. HPLC fractionation and analysis were performed on a Shimadzu LC-20AR system equipped with a Shimadzu SPD-20A UV/vis detector using preparative silica or semipreparative C18 ((250 × 21.2 mm, 5 μm) or (250 × 10.0 mm, 5 μm)) conditions.
Biological Materials
Four specimens of Duva florida (Cnidaria, Alcyonacea, Alcyoniina, Neptheidae) were collected at a depth of 823 m along the Irish continental margin (54.26007932 N, 11.58046619 W) during a 2018 cruise using the ROV Holland I deployed from the Irish national research vessel R/V Celtic Explorer. Specimens were stored in bioboxes on the ROV and immediately pooled, logged, labeled, and frozen at −80 °C when the ROV was recovered to the vessel. Specimens were freeze-dried on return to land and then stored until analysis at −20 °C.
Extraction and Isolation
Following a Soxhlet extraction in CHCl2 of 88 g of four lyophilized organisms, 14.3 g of crude extract was obtained and underwent a liquid/liquid partition with CH2Cl2/H2O, resulting in 13.7 g of extract in the organic layer. A crude separation was achieved utilizing NP MPLC with a gradient from 1% to 100% EtOAc in hexanes over 30 min followed by a 20% MeOH in EtOAc wash following a period of 100% EtOAc for 8 min on a silica column. All fractions were dried under either passive air or nitrogen, and fractions 5–9, eluting from roughly 20–50% EtOAc, were prioritized for further purification based on comparisons of the 1H NMR spectra to that of the known tuaimenal A (10). Crude MPLC fractions of interest underwent iterative rounds of normal and reversed-phase HPLC until pure compounds were achieved as determined from 1H and 13C NMR spectra, as well as MS, analyses.
Stereochemical Evaluation
Optical rotation data were acquired in MeOH or CHCl3 using an AutoPol IV polarimeter at a wavelength of 589 nm with a 10 mm path length cell. Chiral-phase chromatography was performed on a semipreparative Phenomenex Lux Amylose-3 column utilizing a gradient from 50% to 100% MeOH in H2O over 30 min with results recorded at a wavelength of 254 nm and integrations performed using Shimadzu LabSolutions software.
X-ray Crystallography
Crystals of 8 were obtained from a concentrated solution of MeOH in a refrigerator with limited oxygen exchange into or out of the sample vial to slow the progression of crystal growth for a period of 5 days. A Bruker D8 Venture PHOTON II CPAD diffractometer with a Cu Kα INOCOATEC ImuS microfocus source was used to measure C-ray diffraction. The Difference Vectors method APEX3 was utilized for indexing with SaintPlus employed for data integration and reduction. Multiscan methods in SADABS were employed for absorption correction, and XPREP in APEX3 was used for space group determination. SHELXT was used to solve the structure with refinements from SHELXL-2018 in an OLEX2 interface program. A riding model with isotropic thermal parameters was utilized for the placement of hydrogen atoms prior to refinement in geometrically calculated positions.
Crystallographic data for the structure reported in this article (8) were deposited at the Cambridge Crystallographic Data Centre under the deposition number CCDC 2207359. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif.
Cell Culture
The cervical cancer cell lines CaSki and C33A were cultured in Rosewell Park Memorial Institute (RPMI) 1640 (1×) complete media and Dulbecco’s Modified Eagle Medium (DMEM), respectively, supplemented with 10% fetal bovine serum (FBS), 100 units/mL of penicillin, 100 mg/mL of streptomycin, 2 mM l-glutamines, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids (NEAA), 0.05 mM 2-mercaptoethanol, 0.5 mg/mL amphotericin B, and 0.5 mg/mL gentamycin. CaSki (ATCC, CRM-CRL-1550) and C33A (ATCC, HTB-31) cell cultures were kept at passages less than 10 and maintained in an incubator at 37 °C and 5% CO2 atmosphere.
Analysis of Cellular Proliferation
Cells were seeded in a 96-well plate at 1000–3000 cells per well with 200 μL of media to achieve 50% confluency. Cells were treated with dimethyl sulfoxide (DMSO negative control, N = 3), 50 μM etoposide (positive control, N = 3), and serial dilutions of tuaimenal A (10) and its derivatives B, F, G, and H (1, 5–7) (200, 133, 89, 59, 40, 26, 18, 12, 8, 5, 4, and 2 μM, N = 3) diluted in DMSO. After 48 h of treatment, the cell proliferation was analyzed using water-soluble tetrazolium (WST-1, Roche, 5015944001) at wavelengths of 450 and 600 nm. The plates were read on a BioTek SynergyHT microplate reader. End point absorbances (450–600 nm) were normalized, and nonlinear regression curves were plotted and fitted using GraphPad Prism software.
Tuaimenal B (1):
pale orange oil, [α]25D 0.0 (c 0.021, CHCl3); UV (C2H3N) λmax (log ε) 222 nm (6.45); IR ν (thin film) 3426, 2972, 2928, 2861, 1774, 1744, 1588, 1461, 1379, 1215, 1089, 1029 cm–1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 383.2241 [M – H]− (calcd for C24H32O4, 383.2228).
Tuaimenal C (2):
pale yellow oil, [α]25D 0.0 (c 0.008, CHCl3); UV (C2H3N) λmax (log ε) 227 nm (6.35); IR ν (thin film) 3508, 2928, 2861, 1766, 1580, 1461, 1379, 1215, 1096, 1029 cm–1; 1H and 13C NMR data, see Table 2; 70 eV HREIMS m/z 398.2452 [M]+ (calcd for C25H34O4, 398.2457).
Tuaimenal D (3):
pale orange oil, [α]25D 1.1 (c 0.018, CHCl3); UV (C2H3N) λmax (log ε) 250 nm (6.38); IR ν (thin film) 3397, 2972, 2935, 1729, 1617, 1429, 1379, 1342, 1290, 1171, 1126, 1059 cm–1; 1H and 13C NMR data, see Table 2; HRESIMS m/z 387.2197 [M – H]− (calcd for C23H32O5, 387.2177).
Tuaimenal E (4):
pale orange oil, [α]25D 2.6 (c 0.034, CHCl3); UV (C2H3N) λmax (log ε) 250 nm (6.55); IR ν (thin film) 3419, 2980, 2935, 2876, 1729, 1640, 1491, 1439, 1379, 1342, 1282, 1171, 1134, 1059 cm–1; 1H and 13C NMR data, see Table 2; HRESIMS m/z 429.2296 [M – H]− (calcd for C25H34O6, 429.2283).
Tuaimenal F (5):
yellow oil, [α]25D 1.5 (c 0.059, CHCl3); UV (C2H3N) λmax (log ε) 228 nm (6.53); IR ν (thin film) 3374, 2972, 2943, 1647, 1588, 1461, 1431, 1394, 1208, 1104, 1029 cm–1; 1H and 13C NMR data, see Table 2; HRESIMS m/z 373.2399 [M – H]− (calcd for C23H34O4, 373.2384).
Tuaimenal G (6):
yellow oil, [α]25D 1.1 (c 0.065, CHCl3); UV (C2H3N) λmax (log ε) 230 nm (6.58); IR ν (thin film) 3426, 2972, 2935, 1737, 1707, 1588, 1461, 1431, 1372, 1260, 1208, 1104, 1029 cm–1; 1H and 13C NMR data, see Table 2; HRESIMS m/z 439.2455 [M – H]− (calcd for C25H36O5, 439.2455).
Tuaimenal H (7):
pale orange film, UV (C2H3N) λmax (log ε) 299 nm (6.44); IR ν (thin film) 3374, 2972, 2920, 2861, 1617, 1439, 1335, 1268, 1178, 1052 cm–1; 1H and 13C NMR data, see Table 2; HRESIMS m/z 371.2245 [M – H]− (calcd for C23H32O4, 371.2228).
3-Hydroxy-19-norchola-1,3,510-trien-24-oic acid (8):
white crystalline solid, [α]25D 44.4 (c 0.024, CHCl3); UV (C2H3N) λmax (log ε) 281 nm (6.00); IR ν (thin film) 3255, 2935, 2868, 1707, 1617, 1506, 1454, 1387, 1290, 1245, 1186, 1104, 1029 cm–1; 13C NMR data, see Table S2; HRESIMS m/z 355.2298 [M – H]− (calcd for C23H32O3, 355.2279).
Methyl 3-hydroxy-19-norchola-1,3,510-trien-24-oic acid (9):
white crystalline solid, [α]25D 45.1 (c 0.065, CHCl3); UV (C2H3N) λmax (log ε) 280 nm (6.02); IR ν (thin film) 3426, 2943, 2868, 1744, 1617, 1506, 1446, 1387, 1290, 1253, 1186 cm–1; 13C NMR data, see Table S2; HRESIMS m/z 371.2589 [M + H]+ (calcd for C24H34O3, 371.2581).
Tuaimenal A (10):
yellow oil, [α]25D 0.5 (c 0.058, CHCl3); UV (C2H3N) λmax (log ε) 253 nm (6.37); IR ν (thin film) 3412, 2972, 2928, 1610, 1491, 1439, 1335, 1282, 1171, 1119, 1052 cm–1; 1H and 13C NMR data, see Table S1; HRESIMS m/z 369.2089 [M – H]− (calcd for C23H30O4, 369.2071).
Acknowledgments
The authors wish to thank the crew and scientists of research expedition CE18012 aboard R/V Celtic Explorer. We also thank the USF Chemical Purification, Analysis, and Screening core facility and its director Dr. Laurent Calcul, as well as the USF NMR core facility. This work was supported by Science Foundation Ireland (SFI) and the Marine Institute under the Investigators Programme Grant No. SFI/15/1A/3100, cofunded under the European Regional Development Fund 2014-2020, to A.L.A. along with the project NMBLI Grant-Aid Agreement PBA/MB/16/01, and the U.S. National Institutes of Health grant R35 GM143004 to J.M.B. and R56 AI154922 to B.J.B. Subsea photographs taken by NUI Galway, copyright Marine Institute, during cruise CE18012 funded under SFI/15/1A/3100.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jnatprod.2c00898.
NMR spectra of tuaimenals B–H (1–7), 3-hydroxy-19-norchola-1,3,510-trien-24-oic acid (8), methyl 3-hydroxy-19-norchola-1,3,510-trien-24-oic acid (9), and tuaimenal A (10); HRESIMS of 1, 3–10; HREIMS of 2; UV λmax of 1–10; IR spectra of 1–10; X-ray crystallographic data for 8; comparisons to literature 1H and 13C spectra of 8–10, chiral-phase HPLC chromatogram of 4 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Newman D. J.; Cragg G. M. J. Nat. Prod. 2020, 83, 770–803. 10.1021/acs.jnatprod.9b01285. [DOI] [PubMed] [Google Scholar]
- Skropeta D. Nat. Prod. Rep. 2008, 25, 1131–166. 10.1039/b808743a. [DOI] [PubMed] [Google Scholar]
- Travis J. Science 1993, 259, 1123–1124. 10.1126/science.259.5098.1123. [DOI] [PubMed] [Google Scholar]
- Skropeta D.; Wei L. Nat. Prod. Rep. 2014, 31, 1–27. 10.1039/C3NP70118B. [DOI] [PubMed] [Google Scholar]
- Burd E. M. Clin. Microbiol. Rev. 2003, 16, 1–17. 10.1128/CMR.16.1.1-17.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedell A. L; Goldstein L. S; Goldstein A. R; Goldstein A. T. Med. Rev. 2020, 8, 28–37. 10.1016/j.sxmr.2019.09.005. [DOI] [PubMed] [Google Scholar]
- Canfell K. Papillomavirus Res. 2019, 8, 1–3. 10.1016/j.pvr.2019.100170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbyn M.; Weiderpass E.; Bruni L.; Sanjose S. D; Saraiya M.; Ferlay J.; Bray F. Lancet Glob. Health. 2020, 8, 191–203. 10.1016/S2214-109X(19)30482-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merza J.; Aumond M. C; Rondeau D.; Dumontet V.; Le Ray A. M; Seraphin D.; Richomme P. Phytochemistry 2004, 65, 2915–2920. 10.1016/j.phytochem.2004.06.037. [DOI] [PubMed] [Google Scholar]
- Avalon N. E; Nafie J.; Verissimo C. D. M.; Warrensford L. C.; Dietrick S. G.; Pittman A. R.; Young R. M.; Kearns F. L.; Smalley T.; Binning J. M.; Dalton J. P.; Johnson M. P.; Woodcock H. L; Allcock A. L.; Baker B. J. J. Nat. Prod. 2022, 85, 1315–1323. 10.1021/acs.jnatprod.2c00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reher R.; Kim H. W.; Zhang C.; Mao H. H.; Wang M.; Nothias L.-F.; Caraballo-Rodriguez A. M.; Glukhov E.; Teke B.; Leao T.; Alexander K. L.; Duggan B. M.; Van Everbroeck E. L.; Dorrestein P. C.; Cottrell G. W.; Gerwick W. H. J. Am. Chem. Soc. 2020, 142, 4114–4120. 10.1021/jacs.9b13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z.; Van Wagoner R. M.; Harper M. K.; Hooper J. N. A.; Ireland C. M. Nat. Prod. Commun. 2010, 5, 1571–1574. 10.1177/1934578X1000501011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birringer M.; Siems K.; Maxones A.; Frank J.; Lorkowski S. RSC Adv. 2018, 8, 4803–4841. 10.1039/C7RA11819H. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.