

Abstract
Within the field of lipid nanoparticles (LNPs) for RNA delivery, the focus has been mainly placed on organ level delivery, which can mask cellular level effects consequential to therapeutic applications. Here, we studied a pair of LNPs with similar physical properties and discovered how the chemistry of the ionizable amino lipid can control the endogenous LNP identity, affecting cellular uptake in the liver and altering therapeutic outcomes in a model of liver cancer. Although most LNPs accumulate in the liver after intravenous administration (suggesting that liver delivery is straightforward), we observed an unexpected behavior when comparing two similar LNP formulations (5A2-SC8 and 3A5-SC14 LNPs) that resulted in distinct RNA delivery within the organ. Despite both LNPs possessing similar physical properties, ability to silence gene expression in vitro, strong accumulation within the liver, and a shared pKa, of 6.5, only 5A2-SC8 LNPs were able to functionally deliver RNA to hepatocytes. Factor VII (FVII) activity was reduced by 87%, with 5A2-SC8 LNPs carrying FVII siRNA (siFVII), while 3A5-SC14 LNPs carrying siFVII produced baseline FVII activity levels comparable to the nontreatment control at a dosage of 0.5 mg/kg. Protein corona analysis indicated that 5A2-SC8 LNPs bind apolipoprotein E (ApoE), which can drive LDL-R receptor-mediated endocytosis in hepatocytes. In contrast, the surface of 3A5-SC14 LNPs was enriched in albumin but depleted in ApoE, which likely led to Kupffer cell delivery and detargeting of hepatocytes. In an aggressive MYC-driven liver cancer model relevant to hepatocytes, 5A2-SC8 LNPs carrying let-7g miRNA were able to significantly extend survival up to 121 days. Since disease targets exist in an organ- and cell-specific manner, the clinical development of RNA LNP therapeutics will require an improved understanding of LNP cellular tropism within organs. The results from our work illustrate the importance of understanding the cellular localization of RNA delivery and incorporating further checkpoints when choosing nanoparticles beyond biochemical and physical characterization, as small changes in the chemical composition of LNPs can have an impact on both the biofate of LNPs and therapeutic outcomes.
Keywords: lipid nanoparticles, RNA delivery, cell tropism, protein corona, cancer therapy
Graphical Abstract

INTRODUCTION
Lipid nanoparticles (LNPs) are the most clinically advanced drug delivery system for RNA medicines.1–5 In 2018, the first short interfering RNA (siRNA) LNP drug, Onpattro, was approved by the United States Food and Drug Administration (FDA) for treating the hereditary amyloidogenic transthyretin (hATTR) amyloidosis following intravenous (i.v.) infusion. More recently, similar LNPs were used to deliver messenger RNA (mRNA) encoding for the spike protein of the SARS-Cov-2 virus to vaccinate against COVID-19 following intramuscular injection.6,7
Nucleic acid therapeutics, including RNA interference (RNAi), are promising drugs for liver cancer and other diseases due to their high efficacy, selectivity, and numerous target choices. Liver cancer has limited treatment options because many current drugs have intrinsic hepatotoxicity, which can exacerbate the underlying liver disease and drastically limit a patient’s treatment options.8 As evidenced by the recent FDA approval of four siRNA drugs, it has been demonstrated that RNAi is a safe and effective therapeutic modality.9,10 However, RNA delivery still faces challenges with respect to understanding and controlling cell-specific delivery. Significant progress has been made in hepatic and extrahepatic delivery. For example, selective organ targeting (SORT) LNPs were developed that can control RNA delivery to the lungs, spleen, or liver.11–13 We showed that the chemical identity of the SORT molecule can affect biodistribution, pKa, and protein corona formation to alter organ-specific functional mRNA delivery.13 While SORT and other methods of active and endogenous targeting are beginning to open the door to organ level delivery, the specific cellular fate of LNPs within the organ remains poorly defined (including in the liver).
Understanding the cellular fate of LNPs within the organ is especially important in the context of therapeutic LNPs as delivery to an unexpected cell type could relate to adverse events. For instance, the clinical progress of nucleic acid therapy has been stymied by unexpected nanoparticle-induced stimulation of immune cells. For example, the phase I HCC trial of a miR-34a nanoparticle MRX34 was halted after severe immune-related adverse events driven by off-target nanoparticle uptake.14,15 This clinical trial failure highlights that nanoparticle cellular tropism can have negative consequences and thus is essential to understand when developing therapeutic LNP candidates.
All traditional LNPs, including those used in Onpattro and the COVID-19 vaccines, are comprised of four lipid components: an ionizable amino lipid, a phospholipid (typically DSPC), cholesterol, and a poly(ethylene glycol) (PEG) lipid.1,6,10,16–19 The ionizable amino lipid is one of the most critical components in LNPs because they bind negatively charged nucleic acids and release the encapsulated cargo of the LNP into the cytoplasm via lipid charge acquisition in acidic endosomes.19 Although many ionizable amino lipids have been studied with diverse chemical structures, applications of i.v.-administered LNPs have largely been limited to one organ (the liver) and a single cell type (hepatocytes). Analogous to very-low-density lipoprotein (V-LDL), this fate of LNPs has been shown to involve apolipoprotein E (ApoE) adsorption in the blood that subsequently mediates uptake into hepatocytes via the low-density lipoprotein receptor (LDL-R).13,18 For this study, we hypothesized that the chemical structure of the ionizable amino lipid may modulate the biofate of LNPs within the liver, which could then affect therapeutic outcomes in the treatment of liver cancer.
In our previous work, we developed a chemically diverse library of ionizable cationic lipids and identified a successful amino lipid named 5A2-SC8 that when formulated into 5A2-SC8/DSPC/Chol/PEG-DMG LNPs was able to effectively deliver siRNA, miRNA, and mRNA to the liver for therapeutic benefit.20,21 This LNP was shown to have high potency in liver hepatocytes and cancer cells while avoiding off-target toxicity to the remaining liver tissue. It is often assumed that the majority of LNPs are primarily uptaken by hepatocytes and that liver delivery is a relatively easy and solved problem in terms of targeting. However, upon re-examination of our chemical library of ionizable cationic lipids for siRNA LNP delivery, we made an unexpected observation. A structurally similar lipid, 3A5-SC14, with higher siRNA delivery potency in vitro compared to 5A2-SC8 LNPs and liver accumulation in vivo to that of 5A2-SC8 LNPs, was ineffective for functional siRNA delivery to liver hepatocytes. This observation of two related and similar LNPs, 5A2-SC8 and 3A5-SC14, that both accumulated within the liver but had distinct in vivo functional RNA delivery capabilities prompted further examination (Scheme 1).
Scheme 1. Small Changes in LNP Chemistry Can Have an Impact on Cellular Tropism within Liver and Cancer Therapeutic Efficacya.

a(A) 5A2-SC8 and 3A5-SC14 form LNPs with similar physicochemical properties. (B) Unique serum proteins recognize the surface of 5A2-SC8 and 3A5-SC14 LNPs after they are administered into the bloodstream forming different protein coronas. (C) Serum protein recognition helps direct the biofate of LNPs within the liver. (D) The cell tropism within the liver of 5A2-SC8 and 3A5-SC14 LNPs had a significant impact on liver cancer efficacy.
The phenomenon inspired us to characterize the biochemical and physical properties of 5A2-SC8 and 3A5-SC14 LNPs and to determine what factors impact the cellular location of RNA LNPs in the liver and whether cellular tropism within an organ could alter the outcomes of cancer therapy. Herein, we formulated four-component LNPs that only differed in one component—the ionizable cationic lipid. 5A2-SC8 and 3A5-SC14 LNPs each had similar physical properties such as size, charge, RNA encapsulation, and pKa. In addition, 5A2-SC8 and 3A5-SC14 LNPs were able to deliver siRNA to silence reporter luciferase expression in vitro, as well as accumulate in the liver on a gross analysis level in vivo. Despite being more potent for siRNA delivery in vitro, 3A5-SC14 LNPs were ineffective for siRNA-mediated gene silencing in hepatocytes in vivo. This phenomenon was intriguing as it challenges the dogma that the majority of LNPs targeting the liver are consumed by hepatocytes. This unique observation in differential hepatocyte delivery inspired further exploration of the two structurally similar yet functionally distinct LNPs. Through a series of in vitro studies, we found that different protein coronas help guide the biofate of LNPs in the liver. We hypothesized that the chemistry of the amino lipid plays a prominent role in which proteins bind to the surface of LNPs when administered in the bloodstream and ultimately dictates the biofate of LNPs in the liver. We further explored if this unique biofate could impact the therapeutic outcomes in a genetically engineered model of MYC-driven liver cancer and found that only 5A2-SC8 let-7g miRNA LNPS were able to extend survival in the aggressive liver cancer model.
Overall, this body of work focused on understanding the biochemical and biophysical factors of two similar yet unique LNPs. We learned that though two LNPs are structurally similar and share the suggested characteristics for successful liver hepatocyte targeting, these similarities do not always correlate to successful hepatocyte delivery and therapeutic efficacy. Our results illustrate the importance of understanding the suborgan cellular destination of RNA delivery and incorporating further checkpoints when choosing nanoparticles beyond biochemical and physical characterization, especially in the context of therapeutic LNPs. We anticipate that these findings can help guide others in the selection of nanoparticles for disease treatment as cellular tropism impacts cancer therapy.
EXPERIMENTAL SECTION
Materials and Reagents.
5A2-SC8 and 3A5-SC14 dendrimer amino lipids were synthesized according to our previously reported methods.20 Clodronate liposomes and PBS liposomes were purchased from Liposoma and utilized for Kupffer cell depletion studies. Additional details regarding the materials and reagents utilized can be found in the Supporting Information.
Formulation and Characterization of 5A2-SC8 and 3A5-SC14 LNPs.
5A2-SC8 or 3A5-SC14, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), cholesterol, and PEG-DMG-2000 were dissolved in ethanol (molar ratio of 50:10:38:2), and all RNAs were dissolved in citrate buffer (10 mM citrate buffer at a pH 3.9). A 3:1 ratio of the aqueous solution to the EtOH solution (by volume) was used. Under vortex mixing, the ethanol solutions (40 μL) were added to the RNA solution (120 μL). The weight ratio of 5A2-SC8 or 3A5-SC14 amino lipid to RNA was set to 25:1 for all LNP formulations. The formulated 5A2-SC8 and 3A5-SC14 LNPs were incubated for 15 min at room temperature and then diluted with 1× PBS for in vitro studies. For in vivo studies, the LNPs were purified by dialysis in sterile 1× PBS with 3.5 kD cutoff dialysis tubes for 2 h. 5A2-SC8 and 3A5-SC14 LNPs containing luciferase mRNA were prepared using the same conditions as above. 5A2-SC8 and 3A5-SC14 LNPs were diluted to a concentration of 0.1 mg/mL using 1X PBS prior to measuring size, and LNPs were diluted further to a volume of 800 μL using 1X PBS for ζ potential measurements. The LNP size and ζ potentials were measured by dynamic light scattering (DLS) using a Malvern Zetasizer Nano ZS (He–Ne laser, λ = 632 nm) prior to in vivo studies. Nucleic acid binding was determined using the Quant-iT Ribogreen assay (Fisher Scientific) following the recommended protocol, and additional details can be found in the Supporting Information. Following a reported procedure, the TNS assay was used to determine the pKa of 5A2-SC8 and 3A5-SC14 siRNA LNPs comprised of dendrimer/DMG PEG lipid/ DSPC/cholesterol (50/2/10/38 mol %) in PBS at a concentration of 100 μM total lipid.20 Briefly, the formulated LNPs were diluted to 60 μM total lipid in 100 μL volume per well in 96-well plates with a series of 10 mM HEPS/10 mM MES/10 mM ammonium acetate/130 mM NaCl buffer solution, where the pH values ranged from 2.5 to 11. The TNS buffer was diluted to 50 mM. The same volume (4 μL) of TNS stock solution was added to each well to give a final concentration of 2 μM. The plate was read with an Infinite F/M200 Pro microplate reader (Tecan) using excitation and emission wavelengths of 321 and 445 nm shaking for 200 s. Four replicates were used for each data point. The data was normalized to the values at pH 2.5, and the data was analyzed using GraphPad Prism. A nonlinear fit analysis of log (inhibitor) vs response was applied to the fluorescence data, and the pKa was measured as the pH value where the fluorescence intensity is half-maximum fluorescence (inflection point).
Evaluation of In Vitro siRNA Delivery of 5A2-SC8 and 3A5-SC14 LNPs.
HeLa-Luc cells were seeded into white 96-well plates at a density of 10,000 cells per well 24 h before transfection, and cells were incubated with DMEM medium (10% FBS, 1% PenStep). The next day, 5A2-SC8 and 3A5-SC14 LNP formulations containing antiluciferase siRNAs (siLuc) were added with a fixed 24 nM siLuc per well. After incubation of 24 h, ONE-Glo + Tox kits were used to detect luciferase expression and cytotoxicity based on Promega’s recommended protocol. Cell viability was measured using the CellTiter-Glo Luminescent Cell Viability Assay following Promega’s recommended protocol.
Evaluation of In Vivo RNA Delivery of 5A2-SC8 and 3A5-SC14 LNPs to Hepatocytes.
For in vivo small RNA delivery to hepatocytes, C57BL/6 mice received i.v. injections of PBS (negative control, n = 3), 5A2-SC8 LNPs containing anti-factor VII siRNA (siFVII) diluted in PBS (200 μL, 0.5 mg/kg of siFVII) or 3A5-SC14 LNPs containing siFVII diluted in PBS (200 μL, 0.5 mg/kg of siFVII). After 72 h, mice were anesthetized by isoflurane inhalation for blood sample collection by cheek puncture. Serum was isolated with serum separation tubes (Becton Dickinson), and factor VII protein levels were analyzed by a Biophen FVII chromogenic assay (Hyphen Biomed, Aniara Corporation) following the recommended protocol. A standard curve was constructed using samples from PBS-injected mice, and relative factor VII expression was determined by comparing treated groups to an untreated PBS control.
Analysis of siFVII-Cy5.5 Biodistribution of 5A2-SC8 and 3A5-SC14 LNPs.
C57BL/6 mice weighing approximately 20 g (6–8 weeks of age) were administered i.v. 5A2-SC8 and 3A5-SC14 LNPs containing siFVII-Cy5.5 at a dose of 0.5 mg/kg. After 6 h, mice were euthanized and major organs were removed (heart, lung, spleen, kidney, and liver) from each set of mice. Ex vivo imaging (Cy5.5 filter setting) was done 6 h post injection. Organs were analyzed using the IVIS Lumina Imaging technique (Caliper Life Sciences). Data analysis was done on IVIS Lumina software and normalized to a PBS control. Total fluorescence of the heart, lungs, liver, spleen, and kidneys was measured using Living Image Software (PerkinElmer) by drawing regions of interest around each organ.
Kupffer Cell Depletion and In Vivo RNA Delivery of 5A2-SC8 and 3A5-SC14 LNPs to Hepatocytes.
C57BL/6 mice (6–8 weeks of age) were i.v.-injected with clodronate liposomes or PBS liposome controls purchased from Liposoma at a dosage of 0.05 mg/mL (n = 3). At 24 h post injection, 5A2-SC8 or 3A5-SC14 LNPs containing siFVII were i.v.-administered to the groups of mice. The dosages used were 0.25, 0.5, and 1 mg/kg of RNA. After 72 h post LNP injection, blood was collected via cheek for FVII silencing following the Biophen FVII assay (Hyphen Biomed) above. To confirm that Kupffer cell depletion was successful, after 24 h of PBS liposome and clodronate liposome treatment, livers were removed and embedded in O.C.T to prepare frozen tissue sections. For confocal imaging, the tissue was cryosectioned (10 μm) and fixed using 4% paraformaldehyde (PFA) at room temperature for 20 min. The slides were washed three times with PBS and blocked for 30 min in PBS with 2% BSA. Sections were then incubated overnight with F4/80 primary antibody (Cell Signaling, 1:300) and Alexa 488 secondary (Thermofisher, 1:200) in 2% BSA in PBS. The next day, slides were washed three times with PBS and stained with their secondary antibody for 1 h. Afterward, slides were washed and mounted using ProLong Gold Antifade (Life Technologies). Sections were imaged using an LSM 700 point scanning confocal microscope (Zeiss) equipped with a 20× objective.
Isolation of Plasma Proteins Absorbed to 5A2-SC8 and 3A5-SC14 LNPs.
5A2-SC8 and 3A5-SC14 LNPs were prepared according to the previously described protocol and were diluted to a final lipid concentration of 1 g/L with 1× PBS.22 The plasma proteins absorbed onto the LNPs were isolated following our reported methods.13 Briefly, equal volumes of 5A2-SC8 and 3A5-SC14 LNPs (at a concentration of 1 mg/mL total lipid) were incubated in equal volumes of mouse plasma (Innovative Grade US Origin Mouse Plasma KD EDTA purchased from Fisher Scientific) at a 1:1 ratio at 37 °C for 15 min. Each sample was prepared in triplicate. A 0.7 M sucrose gradient was prepared by dissolving solid sucrose in MilliQ water. The LNP and plasma solution were added to tubes containing a 0.7 M sucrose gradient. The sucrose gradient tubes were prepared in advance by diluting 2 M sucrose in MilliQ water. Samples were centrifuged at a speed of 25,000g for 1 h at 4 °C. The supernatant was removed carefully and discarded. The samples were washed with 1× PBS 3 times at a speed of 25,000 for 5 min. After 3 washes, samples were resuspended in 50 μL of a buffer comprised of Laemli sample buffer with 50% BME. Next, excess lipids were removed using the ReadyPrep 2D Clean up kit (Bio-Rad) following the steps provided by the manufacturer. Samples were then stored at −20 °C prior to processing.
Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) Characterization of Plasma Proteins Adsorbed onto 5A2-SC8 and 3A5-SC14 LNPs.
The plasma proteins isolated from the surface of 5A2-SC8 and 3A5-SC14 LNPs were heated to 95 °C for 5 min. Ten microliters of sample in triplicate were loaded into the wells of 12% mini-PROTEAN TGX precast gels (Bio-Rad). Mouse plasma was loaded as a control (1:100 dilution). The gel was run starting at 90 V and then changed to 200 V and monitored regularly. Afterward, the gel was washed 3 times in MilliQ water for 5 min per wash. The gel was stained with SimplyBlue Safe Stain (Bio-Rad) for 1 h at room temperature, gently shaking to visualize the protein bands. The gel was destained overnight using deionized water (DI) and imaged with a Licor Scanner the following day.
Validation of ApoE and Albumin Adsorption on LNPs Using Western Blot.
The SDS-PAGE gel used for the characterization of plasma proteins adsorbed onto the surface of 5A2-SC8 and 3A5-SC14 LNPs mentioned above was rinsed in a 1× transfer buffer. After rinsing, the SDS gel was transferred using a nitrocellulose membrane via the Bio-Rad Trans-Blot transfer system according to Bio-Rad’s recommended protocol. Next, the membrane was cut into two sections based on the molecular weight of ApoE and albumin. The sample was blocked using a 5% dry milk solution (5% dry milk in 1× tris-buffered saline and 0.1% Tween 20, TBST) for 1 h. The samples were incubated overnight at 4 °C with primary antibody ApoE (Biolegend, 1:500) and albumin antibody (Genetex,1:1000) in 3% BSA in TBST overnight. After an overnight incubation, samples were washed 3× for 5 min each with TBST at room temperature while shaking. The samples were then cultured with secondary antibody goat–antirabbit IgG (H+L) (Bio-Rad, 1:3000) and secondary antibody goat antirat Ig (H+L) HRP (SantaCruz, 1:8000). The secondary antibodies were incubated for 1 h at room temperature while gently shaking. Next, the substrate of HRP (Bio-Rad) (1:1) was prepared. The protein was detected using the chemiluminescent method.
Preparation of Plasma Protein Samples for Mass Spectrometry Proteomics.
Plasma proteins isolated from the surface of 5A2-SC8 or 3A5-SC14 LNPs were loaded onto a 12% mini PROTEAM TGX Precast Protein Gel at a volume of 10 μL and run into the gel at 1 cm at 90 V. Once the samples created an even line, the gel was removed and stained with SimplyBlue Safe Stain for 1 h to fix and visualize the protein bands. After destaining for 1 h, the protein bands were excised using a sterile razor blade and sliced into 1 mm cubes. Next, the gel cubes were added to a 1.5 mL tube that had been rinsed with a 1:1 MilliQ water/ethanol solution and stored at 4 °C until being submitted to the UTSW Proteomics Core for mass spectrometry analysis. Data was analyzed via Microsoft excel and plotted using GraphPad Prism.
Luciferase mRNA Delivery Assay with and without Protein Incubation to Cell Lines.
A total of 10,000 cells per well were plated into a white-bottom 96-well plate. After 24 h, the media was replaced with 100 μL of fresh media and cells were treated with 25 ng of firefly luciferase mRNA (mFLuc) inside 5A2-SC8 and 3A5-SC14 LNPs incubated with either no serum protein, ApoE, or albumin at a ratio of 0.1 g protein/0.1 g total lipid, 0.2 g protein/0.2 g total lipid, 0.3 g protein/0.3 g total lipid, 0.4 g protein/0.4 g total lipid, 0.5 g protein/0.5 g total lipid, or 1 g protein/1 g total lipid. The next day, a One-Glo+ Tox assay was performed according to the manufacturer’s directions (Promega). All of the data was normalized to cell viability. The cell lines that were utilized in this experiment were HuH-7 cells, HepG2, and RAW 264.6 cells. Cell culture conditions can be found in the Supporting Information.
Luciferase mRNA Delivery Assay with and without Protein Incubation to Primary Cells.
Twenty-five nanograms of firefly luciferase mRNA (mFLuc) inside 5A2-SC8 and 3A5-SC14 LNPs were incubated with either no serum protein, ApoE, or albumin at a ratio of 0.1 g protein/0.1 g total lipid, 0.2 g protein/0.2 g total lipid, 0.3 g protein/0.3 g total lipid, 0.4 g protein/0.4 g total lipid, 0.5 g protein/0.5 g total lipid, or 1 g protein/1 g total lipid. The following LNPs were directly added to 96-well plates. A total of 10,000 cells per well were plated into a white-bottom 96-well plate and mixed with the LNPs. The next day, a One-Glo+ Tox assay was performed according to the manufacturer’s directions (Promega). All of the data was normalized to cell viability. The cells that were utilized in this experiment were primary Kupffer cells and primary hepatocytes. Cell culture conditions and primary cell isolation details can be found in the Supporting Information.
In Vivo Let-7g miRNA Therapeutic Study in Aggressive Liver Cancer Mouse Model Using 5A2-SC8 and 3A5-SC14 LNPs.
Twenty-one day old male transgenic c-MYC mice bearing liver tumors were randomly divided into different treatment groups of nontreatment (n = 9), 5A2-SC8 and 3A5-SC14 LNPs containing no miRNA (n = 5), or 5A2-SC8 and 3A5-SC14 LNPs containing 0.5 mg/kg of let-7g miRNA (n = 5). The groups received i.v. injections twice a week starting at day 21 and continuing until day 55. Their body weight, abdomen circumference, and survival were carefully monitored.
Immunohistochemistry and Histological Tissue Analysis.
Liver tissues from c-MYC mice were collected at 21 days, 26 days, 30 days, 45 days, and 55 days and placed in 10% formalin (Sigma-Aldrich) for 72 h for fixation. After 72 h of fixation, the formalin was replaced with PBS and the samples were sent to the UTSW tissue management shared resource core facility to perform hematoxylin and eosin (H&E) staining.
RESULTS AND DISCUSSION
5A2-SC8 and 3A5-SC14 LNPs Possess Similar Physical Properties.
During the development of LNPs, physical characterization of particle size, surface charge, and pKa are important parameters that can correlate with efficacy.23 Four-component LNPs are comprised of an ionizable cationic lipid, phospholipid, cholesterol, and PEG lipid. To examine these properties, we formulated 5A2-SC8 LNPs and 3A5-SC14 LNPs (Figure 1A), differing only in the chemistry of the ionizable amino lipid (5A-SC8 or 3A5-SC14) while keeping the other three components (PEG lipid, cholesterol, and DSPC) constant. While 5A2-SC8 and 3A5-SC14 are structurally similar dendritic amino lipids, they do have a few unique chemical differences. Both 5A2-SC8 and 3A5-SC14 have polyamine cores; they differ in the number of hydrophobic branches (5 versus 3, respectively). Further, 3A5-SC14’s alkyl tail length is comprised of 14 carbons compared to the 8 carbons on the 5A2-SC8 alkyl tail. These amine and alkyl differences could lead to different biological interactions with serum proteins and endosomal membranes. Each LNP was formulated using the same molar ratios of ionizable dendrimer (either 5A2-SC8 or 3A5-SC14), DSPC, cholesterol, and PEG2000-DMG (Figure 1B,C) (50/38/10/2, mol/mol).
Figure 1.
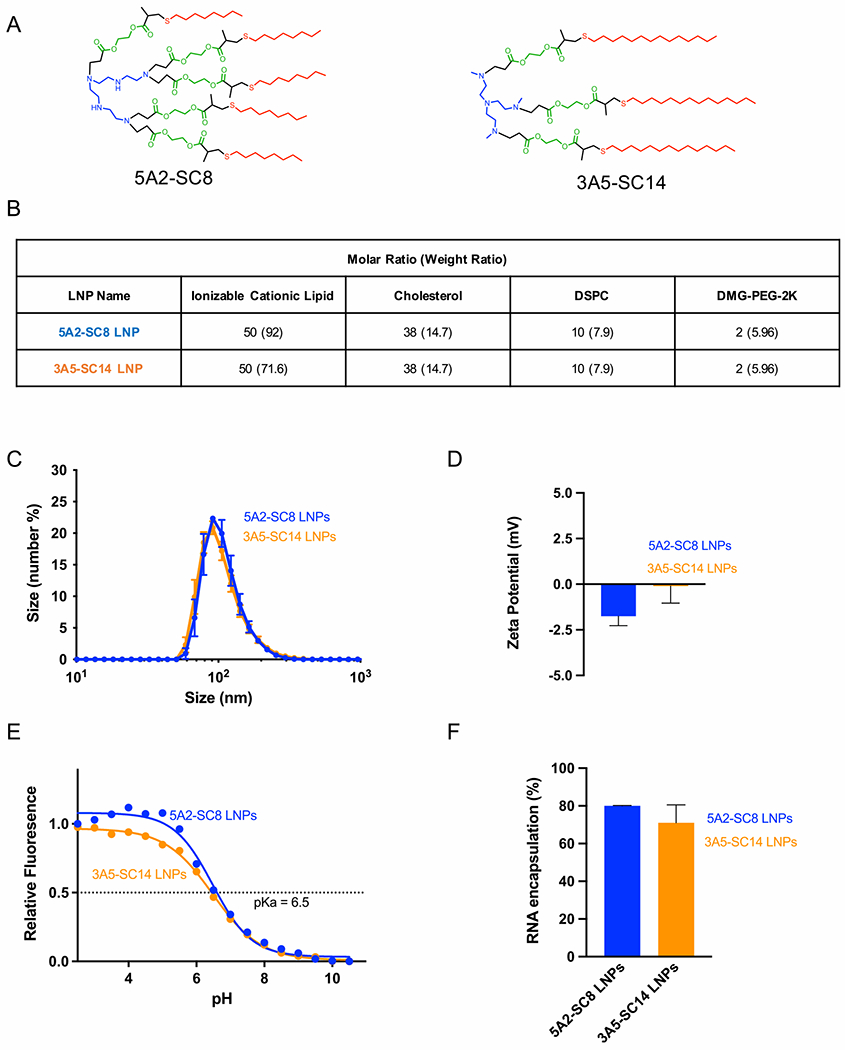
5A2-SC8 and 3A5-SC14 LNPs containing siRNA from LNPs with similar physical properties. (A) Chemical structures of selected ionizable cationic amino lipids, 5A2-SC8 and 3A5-SC14. (B) Formulation composition of 5A2-SC8 and 3A5-SC14 LNPs. (C) Size of 5A2-SC8 siFVII LNPs and 3A5-SC14 siFVII LNPs. (D) ζ-Potential surface charge of 5A2-SC8 siFVII LNPs and 3A5-SC14 siFVII LNPs. (E) pKa of 5A2-SC8 LNPs and 3A5-SC14 LNPs containing a negative control siRNA. (F) RNA encapsulation of 5A2-SC8 siFVII LNPs and 3A5-SC14 siFVII LNPs.
We then measured the physical properties including size, surface charge, and RNA encapsulation (Figure 1D–F). Both 5A2-SC8 and 3A5-SC14 LNPs shared similar physical properties. 5A2-SC8 and 3A5-SC14 LNP diameters were each around 80 nm with similar surface charge and similar encapsulation of siRNA. It has been shown in the literature that efficacious liver targeting LNPs possess a pKa value ranging from 6.2 to 6.5.23 We measured the apparent pKa for 5A2-SC8 and 3A5-SC14 LNPs using the 6-(p-toluidino)-2-naphthalenesulfonic acid (TNS) assay. Both 5A2-SC8 and 3A5-SC14 LNPs have an identical pKa value of 6.5, which falls within the preferable range for hepatocyte delivery (Figure 1E). Further, neither 5A2-SC8 or 3A5-SC14 LNPs elevated liver (AST and ALT) and kidney function enzymes (CREA and BUN) when delivering siRNA (Figure S1), indicating that observed differences would also not be due to toxicity. Taken together, we found that both 5A2-SC8 and 3A5-SC14 LNPs exhibit similar physical properties. These physical attributes, therefore, could not sufficiently explain differences in their activities within the liver. We wanted to explore if this potential factor attributing to the different activities within the liver was a mechanistic difference between the two LNPs or a difference in overcoming the delivery barrier to reach specific liver cells.
5A2-SC8 and 3A5-SC14 LNPs Exhibit Differential siRNA Delivery Capabilities to Hepatocytes.
To examine first the barriers of cellular uptake and endosomal escape of siRNA, we compared the activity of 5A2-SC8 and 3A5-SC14 LNPs in vitro. 5A2-SC8 and 3A5-SC14 LNPs containing antiluciferase siRNA (siLuc) were delivered into HeLa cells, which were stably expressing the luciferase gene (HeLa-Luc). After 24 h, cytotoxicity and luciferase activity were quantified. Both 5A2-SC8 and 3A5-SC14 were not toxic to cells (Figure 2A). While both 5A2-SC8 siLuc LNPs and 3A5-SC14 siLuc LNPs were able to silence luciferase in HeLa-Luc cells, 3A5-SC14 LNPs were more potent (Figure 2B). These results indicated that both LNPs can overcome intracellular barriers such as cellular uptake and endosomal escape to deliver siRNA effectively in vitro. To examine the accumulation of 5A2-SC8 and 3A5-SC14 LNPs in vivo, we i.v.-administered Cy5.5 dye-labeled siRNA encapsulated inside 5A2-SC8 and 3A5-SC14 LNPs and quantified the explanted tissue fluorescence using in vivo imaging. 5A2-SC8 and 3A5-SC14 LNPs were each sequestered in the liver as the major organ of accumulation, with 3A5-SC14 LNPs accumulating more (Figures 2C and S2). To test the functional RNA delivery to the liver in vivo, we prepared both LNPs encapsulating mFluc and administered i.v. at a time point of 6 h and a dose of 0.1 mg/kg. The results indicated that both 5A2-SC8 and 3A5-SC14 LNPs delivered functional mRNA to the liver (Figure S3B). We next examined in vivo siRNA-mediated gene silencing in the liver. We choose factor VII (FVII) siRNA (siFVII) as the in vivo RNA delivery efficacy test because the assay is well established and commonly used to access functional delivery to the hepatocytes in the liver.24 FVII, a blood clotting protein, is specifically produced by the hepatocytes and secreted into the blood, where it can be readily measured in serum. We administered 5A2-SC8 and 3A5-SC14 LNPs containing siFVII to mice through i.v. injection. After 72 h, the FVII levels in the serum were quantified utilizing chromogenic FVII kit. Interestingly, only 5A2-SC8 LNPs could enable FVII silencing in hepatocytes. 3A5-SC14 LNPs were unable to silence FVII at doses of 0.25 and 0.5 mg/kg despite having such similar physical properties and being more potent in vitro compared to 5A2-SC8 LNPs. The FVII activity for 5A2-SC8 LNP-treated groups was reduced by 87% compared with that of the nontreated groups. However, the FVII activity in the groups treated with 3A5-SC14 LNPs was not reduced and remained comparable to the nontreated groups (Figure 2D). Hepatocytes make up 80% of the liver cell mass, and LNPs have to first pass through the endothelial and Kupffer cell barriers to reach hepatocytes.25 However, the distinct FVII activity reduced by 5A2-SC8 and 3A5-SC14 LNPs illustrates the unique cell tropism of these LNPs in vivo.
Figure 2.

5A2-SC8 and 3A5-SC14 LNPs containing siRNA have distinct in vivo activity in regard to hepatocyte delivery. (A) Cell viability after in vitro siLuc delivery to HeLa-Luc cells using 5A2-SC8 and 3A5-SC14 LNPs containing siLuc at a dose of 25 nM for 24 h. Control LNPs were LNPs containing no siRNA. (B) In vitro siLuc delivery to HeLa-Luc cells using 5A2-SC8 and 3A5-SC14 LNPs containing siLuc at a dose of 25 nM for 24 h. Control LNPs were LNPs containing no siRNA. (C) Quantification of organ biodistribution of 5A2-SC8 and 3A5-SC14 LNPs containing siFVII-Cy5.5 (0.5 mg/kg) (6 h). (D) Quantification of in vivo FVII gene silencing activity of 5A2-SC8 and 3A5-SC14 LNPs containing siFVII at 0.5 mg/kg (72 h). Data is shown as mean +/− SEM. Statistical significance was determined using a one-way ANOVA with multiple comparison test (*p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.0001).
Kupffer Cells Impact 3A5-SC14 LNP RNA Delivery to Hepatocytes and Do Not Affect 5A2-SC8 LNPs.
Since 5A2-SC8 LNPs silenced FVII in liver hepatocytes but 3A5-SC14 LNPs did not, we hypothesized that the cellular distribution of these LNPs to cells within the liver is different. The liver structure is complex and is comprised of multiple cell types, where 60–80% of parenchymal cells are hepatocytes and the remaining 20% are nonparenchymal cells such as Kupffer cells, endothelial cells, and hepatic stellate cells.26 Hepatocytes are involved in functions such as protein synthesis, protein storage, detoxification, and metabolism.27 When LNPs are administered i.v. and enter the liver, one of the first cell types they are exposed to is the Kupffer cell.26 Kupffer cells are an important first line of defense against foreign materials as these cells are tissue-resident macrophages that will phagocytose and destroy pathogens and other foreign materials within the blood.26 They are responsible for the majority of phagocytic activity within the liver and make up 80–90% of the total macrophage population within the body.26,28 We aimed to investigate whether the Kupffer cells were playing a role in the difference in RNA delivery of 5A2-SC8 and 3A5-SC14 LNPs, theorizing that Kupffer cells could be acting as a barrier for delivery to hepatocytes.
We designed a series of experiments to determine the impact of liver Kupffer cells on the siRNA delivery of LNPs to hepatocytes (Figure 3A). First, we depleted the Kupffer cells in the liver using dichloromethylenediphosphonic acid (clodronate) liposomes.29 Immunohistochemistry (IHC) was used to demonstrate that Kupffer cells in liver were successfully removed after 24 h of treatment of clodronate liposomes (Figure 3A). When Kupffer cells were depleted, 5A2-SC8 and 3A5-SC14 LNPs still accumulated strongly within the liver following i.v. administration of 5A2-SC8 and 3A5-SC14 LNPs containing Cy5.5 dye-labeled siFVII (Figure 3B). Next, 5A2-SC8 and 3A5-SC14 LNPs containing FVII siRNA were administrated into mice via i.v. injection at three different doses (0.25, 0.5, and 1 mg/kg siRNA) 24 h after the depletion of Kupffer cells. Mouse serum was collected 72 h later for quantification of FVII levels. After the administration of 5A2-SC8 siFVII LNPs, FVII levels were distinctly reduced in wild-type (WT) mice and Kupffer cell-depleted mice (Figure 3C). The presence or absence of Kupffer cells did not significantly affect siRNA delivery to hepatocytes using 5A2-SC8 siFVII LNPs at all doses tested (0.25, 0.5, and 1 mg/kg). In contrast, Kupffer cell depletion was required for 3A5-SC14 siFVII LNP-mediated siRNA gene silencing in hepatocytes. 3A5-SC14 siFVII LNPs were only able to modestly affect FVII levels in WT mice at the highest dose of 1 mg/kg. However, FVII levels were significantly reduced after Kupffer cells were depleted including at the lowest dose tested (0.25 mg/kg) (Figure 3C). This data suggests that 5A2-SC8 siFVII LNPs pass through the first barrier of Kupffer cells in the liver and internalize into hepatocytes, whereas 3A5-SC14 siFVII LNPs may end up trapped within Kupffer cells, unable to pass through this barrier to reach hepatocytes. We next aimed to understand, mechanistically, whether the endogenous identity of LNPs formed when the LNPs come in contact with the plasma could explain their transport to Kupffer cells and hepatocytes.
Figure 3.

Depletion of Kupffer cells enables the RNA delivery of 3A5-SC14 LNPs to hepatocytes but does not impact the RNA delivery of 5A2-SC8 LNPs. (A) Experimental design scheme for evaluating siFVII LNPs after the depletion of Kupffer cells using clodronate liposomes (5 mg/kg). PBS liposomes (PBS-L) were used as a control, and the treatment groups consist of clodronate liposomes (clodronate-L) i.v.-administered 24 h prior to i.v. administration of 5A2-SC8 siFVII LNPs or 3A5-SC14 siFVII LNPs. F4/80 antibody staining was used to validate Kupffer cell depletion. (B) Quantification of organ biodistribution for 5A2-SC8 siFVII-Cy5.5 LNPs and 3A5-SC14 siFVII-Cy5.5 LNPs (0.5 mg/kg) (6 h), and the nontreatment (NT) group was untreated C57BL/6 mice. Data is shown as mean +/− SEM. Statistical significance was determined using one-way ANOVA with multiple comparisons (*p < 0.05). (C) The evaluation of siFVII delivery in vivo using 5A2-SC8 and 3A5-SC14 LNPs at 0.25, 0.5, and 1 mg/kg siFVII (n = 3). Data is shown as mean +/− SEM. Statistical significance was determined using one-way ANOVA with multiple comparisons. Each data set group was compared to the nontreatment (NT) group (****p < 0.0001, ***p < 0.005, p < 0.01).
Protein Corona Formed on 5A2-SC8 and 3A5-SC14 LNPs Aids or Restricts Delivery to Hepatocytes.
To further explore the mechanism of the in vivo biofate of 5A2-SC8 and 3A5-SC14 LNPs (Figure 2D), we examined the interaction of LNPs with serum proteins. Once LNPs are administered into the bloodstream, the nanoparticle’s surface becomes modified by a layer of proteins from the biological fluid, known as the protein corona, which creates an endogenous identity with biological impact.30,31 For example, individual proteins associated with the nanoparticle surface can serve as ligands guiding nanoparticles to specific cell surface receptors.32 For LNP delivery to the liver, the adsorbed ApoE enables by hepatocyte uptake via binding to the LDL-R on the surface of hepatocytes and undergoing receptor-mediated endocytosis.32,33 Furthermore, in our previous work, it was shown that LNPs undergo a three-step endogenous targeting mechanism to reach specific organs (such as the lungs, liver, or spleen).13 proteinsEG lipid displacement first exposes the underlying molecules on the LNP, in this case, 5A2-SC8 or 3A5-SC14, to serum proteins in the blood. Next, distinct proteins bind to the surface of the LNP. Third, interactions between specific surface-bound proteins and cognate receptors can drive endogenous targeting.13
Thus, it is reasonable to expect that the chemistry of surface-exposed amino lipids (5A2-SC8 or 3A5-SC14) could control protein adsorption that would affect cellular uptake within the liver. LNPs were incubated with mouse plasma, and the plasma proteins, which bind to the surface of 5A2-SC8 and 3A5-SC14 LNPs, were isolated using differential centrifugation.34 Once the LNP protein coronas were isolated, we utilized an SDS-PAGE gel to separate the different proteins present on the protein coronas of 5A2-SC8 and 3A5-SC14 LNPs. While the set of plasma proteins on the surface of 5A2-SC8 and 3A5-SC14 LNPs share similarities, there was a striking difference at a molecular weight of around 35 kDa (Figure 4A), which is close to the molecular weight of ApoE. Western blotting (WB) using an ApoE antibody demonstrated that 5A2-SC8 LNPs were enriched in ApoE, while there was little to no ApoE present on 3A5-SC14 LNPs. In addition, we noticed a pronounced band around 69 kDa, particularly for 3A5-SC14 LNPs. The WB result validated that this protein was albumin. Therefore, these results indicate that 5A2-SC8 LNPs are enriched in ApoE, whereas 3A5-SC14 LNPs are enriched in albumin (Figure 4B). Next, we quantified this result using mass spectrometry proteomics, which confirmed that 5A2-SC8 LNP corona had a higher abundance of ApoE and 3A5-SC14 LNP corona had a slightly higher abundance of albumin (Figure 4C,D).
Figure 4.

3A5-SC14 and 5A2-SC8 LNPs have unique protein coronas. (A) SDS-PAGE of protein coronas isolated from 5A2-SC8 and 3A5-SC14 LNPs. (B) ApoE and albumin on the surface of 3A5-SC14 LNPs were validated by Western blot. (C) Quantification of ApoE enrichment in the protein coronas of 5A2-SC8 LNPs using mass spectrometry proteomics (n = 6). (D) Quantification of albumin enrichment in the protein corona of 3A5-SC14 LNPs using mass spectrometry proteomics (n = 6). Data is shown as mean +/− SEM. Statistical significance was determined using Student t-test. (E, F) Activity of functional luciferase protein translated from uncoated or protein-coated 5A2-SC8 and 3A5-SC14 LNPs in HuH-7 cells (25 ng mRNA, 24 h, n = 4). (G, H) Activity of functional luciferase protein translated from uncoated or protein-coated 5A2-SC8 and 3A5-SC14 LNPs in primary Kupffer cells (25 ng mRNA, 24 h, n = 4). Data is shown as mean +/− SEM. Statistical significance was determined using one-way ANOVA with multiple comparisons. Each data set group was compared to the uncoated group (****p < 0.0001, *** p < 0.0009, **p < 0.005, *p < 0.05).
To further investigate whether the proteins on the surface of 5A2-SC8 and 3A5-SC14 LNPs aid cell-specific uptake, functional mRNA delivery experiments using 5A2-SC8 and 3A5-SC14 LNPs were performed in vitro using relevant cell lines. First, LDL-R expressing HepG2 and HuH-7 cells was used to examine ApoE-mediated uptake. 5A2-SC8 and 3A5-SC14 LNPs encapsulating mFLuc were preincubated with recombinant ApoE or albumin prior to the treatment of cultured cells. Next, luciferase activity was quantified after 24 h of incubation as a readout for successful cellular uptake, endosomal escape, and mRNA translation to a functional protein. The results indicated that when 5A2-SC8 LNPs were preincubated with ApoE, there was an improvement in mRNA delivery to HuH-7 (Figure 4E), HepG2 (Figure S4A,E), and primary hepatocytes (Figure S5C). Preincubation of 5A2-SC8 with albumin eliminated efficacy in both HuH-7 (Figure 4E) and HepG2 cells (Figure S4A,E), suggesting that 5A2-SC8 LNPs may utilize ApoE LDL-R uptake pathways, as previously identified.13 In contrast, preincubation of 3A5-SC14 LNPs with ApoE did not improve mRNA delivery to HuH-7 (Figure 4F) or HepG2 cells (Figure S4B). It is speculated that the inability of 3A5-SC14 LNPs to bind ApoE led to free ApoE in the media that blocked LDL-R (Figure S6B). Preincubation of 3A5-SC14 LNPs with albumin led to a slight increase in mRNA delivery to HuH-7 (Figure 4F) and HepG2 cells (Figure S4B,F), suggesting possible ApoE-independent mechanisms although this will have to be further studied in the future.35,36 To model the delivery of LNPs to Kupffer cells, primary mouse Kupffer cells and RAW 264.7 murine peritoneal macrophage cell lines were employed. 5A2-SC8 LNPs were unable to effectively deliver mRNA to primary Kupffer cells (Figure 4G) or RAW 264.7 (Figure S4C,G) with or without ApoE or albumin protein preincubation. In comparison, preincubation of 3A5-SC14 LNPs with albumin did maintain efficacy in primary Kupffer cells (Figure 4H) and RAW 264.7 cells incubated with FBS media (Figure S4D). Together, these results confirm the mechanism of ApoE adsorption on 5A2-SC8 LNPs for delivery to LDL-R expressing cells while also revealing possible ApoE-independent mechanisms. Multiple studies have reported that LNP delivery to liver hepatocytes is ApoE-dependent,35,36 evidenced by the loss of efficacy in ApoE knockout mice.33 Although albumin is an abundant protein, reports have indicated that it can become structurally altered once it binds to the surface of a nanocarrier, and this altered form of albumin can become a ligand for cell membrane receptors such as glycoprotein receptors and macrophage class A1 scavenger receptors (SR-A1).37 It has also been suggested that albumin may aid in preferential uptake into macrophage populations and impact the mechanisms of endocytosis due to gp18 and gp30 receptor binding.36–39 Albumin has been identified on the protein corona of Syn-3 mRNA LNPs, wherein it aided liver delivery.36 Together, these results show that protein corona adsorption can be linked to lipid chemistry and guide the biofate of LNPs within the liver.
Cellular Tropism within the Liver Affects Therapeutic Outcomes in an Aggressive Liver Cancer Mouse Model.
Understanding the biofate of LNPs can open up new avenues for therapeutic development as disease targets exist not only in specific organs but also in specific cell types. To move RNA LNPs into clinical development, an improved understanding of LNP cellular tropism within the liver and its impact on therapeutic efficacy will be valuable. We have seen in the context of failed clinical trials that it is crucial to understand cellular tropism.
We hypothesized that 5A2-SC8 LNPs would provide a therapeutic benefit when delivering a therapeutic RNA as this LNP was able to deliver RNA more effectively to hepatocytes. Liver cancer is one of the most common malignancies in the world with few promising effective treatments available, and current treatments only extend life by 3 months.40,41 One of the most frequently activated oncogenes associated with the pathogenesis of liver tumors in liver cancer is the MYC oncogene.42 Since MYC is an oncogene deregulated in hepatocellular carcinoma (HCC), an aggressive form of liver cancer, we chose to carry out our therapeutic study with a genetically engineered mouse model of MYC-driven cancer where cancer develops in the liver from hepatocytes and disease progresses quickly.20
In this transgenic mouse model, the overexpression of human c-MYC is controlled by a liver-specific promoter wherein doxycycline (dox) is used to turn on or off MYC in hepatocytes.42,43 Upon the removal of dox, rapid tumor growth leads to death within 60 days without treatment. Importantly, hepatocytes differentiate into tumor cells in this model, which further allowed us to test cellular tropism and consequences in regard to liver cancer treatment. Without any treatment, liver tumors become visible around days 20–26 and tumors take over the entire liver by days 45–55 (Figure 5A).
Figure 5.

Therapeutic efficacy of 5A2-SC8 let-7g miRNA LNPs and 3A5-SC14 let-7g miRNA LNPs (0.5 mg/kg) were evaluated in MYC-driven liver cancer model. (A) Tumors grow aggressively after initiation in the human c-MYC genetically engineered mouse model. Representative H&E liver sections are shown at 10× magnification. (B) 5A2-SC8 let-7g miRNA and 3A5-SC14 let-7g miRNA LNPs (0.5 mg/kg) were administered i.v. to treatment groups twice a week starting at day 21 until day 55 (n = 5). (C) Representative abdominal images of the various treatment groups on day 21 and day 60. (D) Abdominal circumference measurements spanning day 21 to day 87. (E) Kaplan–Meier survival curve of the various treatment groups. Treated mice were i.v.-administered twice a week starting at day 21 until day 55 with 0.5 mg/kg of 5A2-SC8 let-7g miRNA LNPs (n = 5) and 3A5-SC14 let-7g miRNA LNPs (n = 5). A nontreatment group (n = 9) and empty 5A2-SC8 and 3A5-SC14 LNP group (n = 5) were used as control groups. 5A2-SC8 let-7g miRNA LNP-treated groups had improved survival.
We followed a treatment regimen (Figure 5B) to test the therapeutic outcome of 5A2-SC8 and 3A5-SC14 LNPs. MYC is a challenging target and is an oncogene deregulated in many types of cancers.44,45 Tumors with elevated MYC expression are a challenge to treat due to the highly proliferative and aggressive phenotypes.46 When MYC is turned on, it drives cellular growth and proliferation.46 microRNAs have emerged as key posttranscriptional regulators of gene expression, offering a promising approach for cancer therapy.47 Let-7g is part of the let-7 family and is known to be downregulated in many tumor types, especially in HCC.48 We hypothesized that 5A2-SC8 LNPs would bind ApoE to aid delivery to LDL-R expressing hepatocytes and LDL-R expressing cancer cells, while 3A5-SC14 LNPs would not. Instead, albumin recognition on 3A5-SC14 LNPs is likely engulfed by the Kupffer cells that are unable to deliver therapeutic miRNA to hepatocytes or cancer cells.
We administered 5A2-SC8 LNPs and 3A5-SC14 LNPs encapsulating let-7g miRNA to the cancer mouse model. Let-7g was chosen as the therapeutic small RNA because it is an important tumor suppressor that is downregulated in liver cancer49,50 and can lead to the silencing of multiple oncogenes beneficial for cancer therapy.51 The use of a validated therapeutic in the MYC model also allowed a direct comparison of 5A2-SC8 and 3A5-SC14 LNP efficacies. MYC was induced at birth by the removal of Dox water, and male pups were randomly divided into five groups of nontreatment, empty 5A2-SC8 LNPs, empty 3A5-SC14 LNPs, 5A2-SC8 let-7g miRNA LNPs, and 3A5-SC14 let-7g miRNA LNPs. Let-7g miRNA was delivered twice a week, with 5A2-SC8 and 3A5-SC14 let-7g miRNA LNPs at a dose of 0.5 mg/kg starting at day 21 and continuing until day 55 (Figure 5A). At specific time points, the liver tissue was collected for H&E staining to assess tumor burden (21 days, 26 days, 30 days, 45 days, and 55 days). By day 55, the liver was entirely full of tumors. (Figure 5A) The various treatment groups were monitored weekly for their abdomen size and body weight. The results showed that empty LNPs had comparable results to the nontreatment in terms of histology, abdomen size, and overall survival. The abdominal growth of groups treated with controls and 3A5-SC14 LNPs had a steady increase in the abdominal size, consistent with the livers becoming overcome by tumor growth (Figure 5C,D). In comparison, 5A2-SC8 let-7g miRNA LNP treatment led to a slower increase in abdominal size that flattened out, consistent with our hypothesis of anticancer benefit and overall survival (Figure 5C,D).
Further, our results showed that 5A2-SC8 let-7g miRNA LNPs extended survival in the MYC mouse model to day 121, while 3A5-SC14 let-7g miRNA LNPs were not able to extend survival past day 67 (Figure 5E). Delivery of let-7g miRNA or a therapeutic miRNA to hepatocytes and cancer cells is beneficial for treatment in this MYC-driven liver cancer model. 3A5-SC14 LNPs are unable to release let-7g miRNA into hepatocytes and thus are not able to provide a therapeutic benefit, likely due to Kupffer cell engulfment and potential degradation. This result emphasizes the importance of understanding the cell type delivery of LNPs as two similar LNPs with promising liver targeting characteristics do not provide a similar therapeutic benefit. We anticipate that understanding the suborgan cellular destination of RNA delivery can help guide others in the selection of nanoparticles for specific diseases and cancers.
CONCLUSIONS
Understanding the cellular fate of LNPs within the liver is important in the context of cancer therapy because delivery to an unexpected cell type could result in a lack of efficacy and even in adverse events. Here, we studied two LNPs, 5A2-SC8 and 3A5-SC14, that accumulate in the liver and share similar physical properties. We found that altering the chemistry of the amino lipid can affect protein corona formation, which then likely drives the uptake into hepatocytes or Kupffer cells. We identified ApoE and albumin as key mediators of this difference between 5A2-SC8 and 3A5-SC14 LNPs. 3A5-SC14 LNPs were only able to silence gene expression in hepatocytes after Kupffer cells were depleted. Consequently, in a liver cancer model dependent on hepatocyte and cancer cell delivery, only 5A2-SC8 LNPs carrying let-7g miRNA provided a therapeutic benefit. Overall, these results illustrate the importance of understanding the suborgan cellular destination of RNA delivery and incorporating further checkpoints when choosing nanoparticles beyond biochemical and physical characterization. We hope that our research can help guide others in the future selection of nanoparticles for specific diseases.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Prof. Hao Zhu for helpful advice. D.J.S. acknowledges support from the Cancer Prevention and Research Institute of Texas (CPRIT) (RP190251), the National Institutes of Health (NIH) (R01 EB025192-01A1, R01 CA269787-01), American Cancer Society (ACS) (RSG-17-012-01), the Cystic Fibrosis Foundation (CFF) (SIEG-WA18XX0), and the Welch Foundation (I-2123-20220331). L.T.J. acknowledges support from the PhRMA Foundation. S.M.L. acknowledges support from the NIH Chemical Biology Training Grant (T32GM127216). S.A.D. acknowledges financial support from the NIH Pharmacological Sciences Training Grant (GM007062) and the NIH Molecular Medicine Training Grant (GM109776). The authors acknowledge the UTSW Small Animal Imaging Shared Resource, the UTSW Proteomics Core, the UTSW Metabolic Phenotyping Core, and the UTSW Tissue Resource, which are supported, in part, by the NIH, National Cancer Institute Support Grant (P30 CA142543). Scheme images were created using Biorender.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.2c00442.
The Supporting Information contains supporting figures and experimental details (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.molpharmaceut.2c00442
Contributor Information
Lindsay T. Johnson, Department of Biochemistry, Simmons Comprehensive Cancer Center, The University of Texas Southwestern Medical Center, Dallas 75390 Texas, United States
Di Zhang, Department of Biochemistry, Simmons Comprehensive Cancer Center, The University of Texas Southwestern Medical Center, Dallas 75390 Texas, United States.
Kejin Zhou, Department of Biochemistry, Simmons Comprehensive Cancer Center, The University of Texas Southwestern Medical Center, Dallas 75390 Texas, United States.
Sang M. Lee, Department of Biochemistry, Simmons Comprehensive Cancer Center, The University of Texas Southwestern Medical Center, Dallas 75390 Texas, United States
Shuai Liu, Department of Biochemistry, Simmons Comprehensive Cancer Center, The University of Texas Southwestern Medical Center, Dallas 75390 Texas, United States.
Sean A. Dilliard, Department of Biochemistry, Simmons Comprehensive Cancer Center, The University of Texas Southwestern Medical Center, Dallas 75390 Texas, United States
Lukas Farbiak, Department of Biochemistry, Simmons Comprehensive Cancer Center, The University of Texas Southwestern Medical Center, Dallas 75390 Texas, United States.
Sumanta Chatterjee, Department of Biochemistry, Simmons Comprehensive Cancer Center, The University of Texas Southwestern Medical Center, Dallas 75390 Texas, United States.
Yu-Hsuan Lin, Children’s Research Institute, Departments of Pediatrics and Internal Medicine, Center for Regenerative Science and Medicine, Children’s Research Institute Mouse Genome Engineering Core, The University of Texas Southwestern Medical Center, Dallas 75390 Texas, United States.
Daniel J. Siegwart, Department of Biochemistry, Simmons Comprehensive Cancer Center, The University of Texas Southwestern Medical Center, Dallas 75390 Texas, United States
REFERENCES
- (1).Xiao YL; Shi JJ, Lipids and the Emerging RNA Medicines. Chem. Rev 2021, 121, 12109–12111. [DOI] [PubMed] [Google Scholar]
- (2).Whitehead KA; Langer R; Anderson DG, Knocking Down Barriers: Advances in siRNA Delivery. Nat. Rev. Drug Discovery 2009, 8, 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hajj KA; Whitehead KA Tools for Translation: Non-Viral Materials for Therapeutic mRNA Delivery. Nat. Rev. Mater 2017, 2, No. 17056. [Google Scholar]
- (4).Wei T; Cheng Q; Farbiak L; Anderson DG; Langer R; Siegwart DJ Delivery of Tissue-Targeted Scalpels: Opportunities and Challenges for In Vivo CRISPR/Cas-Based Genome Editing. ACS Nano 2020, 14, 9243–9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Zhang G; Wang Q; Xu R, Therapeutics Based on MicroRNA: A New Approach for Liver Cancer. Curr. Genomics 2010, 11, 311–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Baden LR; El Sahly HM; Essink B; Kotloff K; Frey S; Novak R; Diemert D; Spector SA; Rouphael N; Creech CB; McGettigan J; Khetan S; Segall N; Solis J; Brosz A; Fierro C; Schwartz H; Neuzil K; Corey L; Gilbert P; Janes H; Follmann D; Marovich M; Mascola J; Polakowski L; Ledgerwood J; Graham BS; Bennett H; Pajon R; Knightly C; Leav B; Deng WP; Zhou HH; Han S; Ivarsson M; Miller J; Zaks T; Grp CS, Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med 2021, 384, 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chaudhary N; Weissman D; Whitehead KA, mRNA Vaccines for Infectious Diseases: Principles, Delivery and Clinical Translation. Nat. Rev. Drug Discovery 2021, 20, 817–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhu J; Yin TL; Xu Y; Lu XJ, Therapeutics for advanced hepatocellular carcinoma: Recent advances, current dilemma, and future directions. J. Cell. Physiol 2019, 234, 12122–12132. [DOI] [PubMed] [Google Scholar]
- (9).Zhang MM; Bahal R; Rasmussen TP; Manautou JE; Zhong XB, The Growth of siRNA-Based Therapeutics: Updated Clinical Studies. Biochem. Pharmacol 2021, 189, No. 114432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Mendes BB; Conniot J; Avital A; Yao D; Jiang X; Zhou X; Sharf-Pauker N; Xiao Y; Adir O; Liang H; Shi J; Schroeder A; Conde J, Nanodelivery of nucleic acids. Nat. Rev. Methods Primers 2022, 2, No. 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Liu S; Cheng Q; Wei T; Yu X; Johnson LT; Farbiak L; Siegwart DJ Membrane-Destabilizing Ionizable Phospholipids for Organ-Selective mRNA Delivery and CRISPR-Cas Gene Editing. Nat. Mater 2021, 20, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Cheng Q; Wei T; Farbiak L; Johnson LT; Dilliard SA; Siegwart DJ Selective Organ Targeting (SORT) Nanoparticles for Tissue-Specific mRNA Delivery and CRISPR-Cas Gene Editing. Nat. Nanotechnol 2020, 15, 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dilliard SA; Cheng Q; Siegwart DJ, On the Mechanism of Tissue-Specific mRNA Delivery by Selective Organ Targeting Nanoparticles. Proc. Natl. Acad. Sci. U.S.A 2021, 118, No. e2109256118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hanna J; Hossein GS; Kocerha J, The Potential for microRNA Therapeutics and Clinical Research. Front. Genet 2019, 10, No. 478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chakraborty C; Sharma AR; Sharma G; Doss CGP; Lee SS, Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther. Nucleic Acids 2017, 8, 132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Cai X; Li JJ; Liu T; Brian O; Li J, Infectious disease mRNA vaccines and a review on epitope prediction for vaccine design. Briefings Funct. Genomics 2021, 20, 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Polack FP; Thomas SJ; Kitchin N; Absalon J; Gurtman A; Lockhart S; Perez JL; Perez Marc G; Moreira ED; Zerbini C; Bailey R; Swanson KA; Roychoudhury S; Koury K; Li P; Kalina WV; Cooper D; Frenck RW Jr.; Hammitt LL; Tureci O; Nell H; Schaefer A; Unal S; Tresnan DB; Mather S; Dormitzer PR; Sahin U; Jansen KU; Gruber WC; Group CCT. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med 2020, 383, 2603–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Akinc A; Maier MA; Manoharan M; Fitzgerald K; Jayaraman M; Barros S; Ansell S; Du XY; Hope MJ; Madden TD; Mui BL; Semple SC; Tam YK; Ciufolini M; Witzigmann D; Kulkarni JA; van der Meel R; Cullis PR, The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol 2019, 14, 1084–1087. [DOI] [PubMed] [Google Scholar]
- (19).Han X; Zhang H; Butowska K; Swingle KL; Alameh MG; Weissman D; Mitchell MJ, An ionizable lipid toolbox for RNA delivery. Nat. Commun 2021, 12, No. 7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhou K; Nguyen LH; Miller JB; Yan Y; Kos P; Xiong H; Li L; Hao J; Minnig JT; Zhu H; Siegwart DJ Modular Degradable Dendrimers Enable Small RNAs to Extend Survival in an Aggressive Liver Cancer Model. Proc. Natl. Acad. Sci. U.S.A 2016, 113, 520–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Cheng Q; Wei T; Jia Y; Farbiak L; Zhou K; Zhang S; Wei Y; Zhu H; Siegwart DJ Dendrimer-Based Lipid Nanoparticles Deliver Therapeutic FAH mRNA to Normalize Liver Function and Extend Survival in a Mouse Model of Hepatorenal Tyrosinemia Type I. Adv. Mater 2018, 30, No. e1805308. [DOI] [PubMed] [Google Scholar]
- (22).Zhou KJ; Nguyen LH; Miller JB; Yan YF; Kos P; Xiong H; Li L; Hao J; Minnig JT; Zhu H; Siegwart DJ, Modular Degradable Dendrimers Enable Small RNAs to Extend Survival in an Aggressive Liver Cancer Model. Proc. Natl. Acad. Sci. U.S.A 2016, 113, 520–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Jayaraman M; Ansell SM; Mui BL; Tam YK; Chen J; Du X; Butler D; Eltepu L; Matsuda S; Narayanannair JK; Rajeev KG; Hafez IM; Akinc A; Maier MA; Tracy MA; Cullis PR; Madden TD; Manoharan M; Hope MJ, Maximizing the Potency of siRNA Lipid Nanoparticles for Hepatic Gene Silencing in vivo. Angew. Chem., Int. Ed 2012, 51, 8529–8533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chen S; Tam YY; Lin PJ; Leung AK; Tam YK; Cullis PR, Development of Lipid Nanoparticle Formulations of siRNA for Hepatocyte Gene Silencing Following Subcutaneous Administration. J. Control. Release 2014, 196, 106–112. [DOI] [PubMed] [Google Scholar]
- (25).Poon W; Zhang YN; Ouyang B; Kingston BR; Wu JLY; Wilhelm S; Chan WCW Elimination Pathways of Nanoparticles. ACS Nano 2019, 13, 5785–5798. [DOI] [PubMed] [Google Scholar]
- (26).Zhang YN; Poon W; Tavares AJ; McGilvray ID; Chan WCW, Nanoparticle-Liver Interactions: Cellular Uptake and Hepatobiliary Elimination. J. Control. Release 2016, 240, 332–348. [DOI] [PubMed] [Google Scholar]
- (27).Zhou Z; Xu MJ; Gao B, Hepatocytes: A Key Cell Type for Innate Immunity. Cell Mol. Immunol 2016, 13, 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wang H; Thorling CA; Liang X; Bridle KR; Grice JE; Zhu Y; Crawford DHG; Xu ZP; Liu X; Roberts MS Diagnostic Imaging and Therapeutic Application of Nanoparticles Targeting the Liver. J. Mater. Chem. B 2015, 3, 939–958. [DOI] [PubMed] [Google Scholar]
- (29).Tavares AJ; Poon W; Zhang YN; Dai Q; Besla R; Ding D; Ouyang B; Li A; Chen J; Zheng G; Robbins C; Chan WCW Effect of Removing Kupffer Cells on Nanoparticle Tumor Delivery. Proc. Natl. Acad. Sci. U.S.A 2017, 114, E10871–E10880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Monopoli MP; Aberg C; Salvati A; Dawson KA Biomolecular Coronas Provide the Biological Identity of Nanosized Materials. Nat. Nanotechnol 2012, 7, 779–786. [DOI] [PubMed] [Google Scholar]
- (31).Ritz S; Schottler S; Kotman N; Baier G; Musyanovych A; Kuharev J; Landfester K; Schild H; Jahn O; Tenzer S; Mailander V, Protein Corona of Nanoparticles: Distinct Proteins Regulate the Cellular Uptake. Biomacromolecules 2015, 16, 1311–1321. [DOI] [PubMed] [Google Scholar]
- (32).Yan XD; Kuipers F; Havekes LM; Havinga R; Dontje B; Poelstra K; Scherphof GL; Kamps JAAM The Role of Apolipoprotein E in the Elimination of Liposomes from Blood by Hepatocytes in the Mouse. Biochem. Biophys. Res. Commun 2005, 328, 57–62. [DOI] [PubMed] [Google Scholar]
- (33).Akinc A; Querbes W; De SM; Qin J; Frank-Kamenetsky M; Jayaprakash KN; Jayaraman M; Rajeev KG; Cantley WL; Dorkin JR; Butler JS; Qin LL; Racie T; Sprague A; Fava E; Zeigerer A; Hope MJ; Zerial M; Sah DWY; Fitzgerald K; Tracy MA; Manoharan M; Koteliansky V; de Fougerolles A; Maier MA Targeted Delivery of RNAi Therapeutics With Endogenous and Exogenous Ligand-Based Mechanisms. Mol. Ther 2010, 18, 1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Docter D; Distler U; Storck W; Kuharev J; Wunsch D; Hahlbrock A; Knauer SK; Tenzer S, Stauber RH, Quantitative Profiling of the Protein Coronas that Form Around Nanoparticles. Nat. Protoc 2014, 9, 2030–2044. [DOI] [PubMed] [Google Scholar]
- (35).Da Silva Sanchez AJ; Dobrowolski C; Cristian A; Echeverri ES; Zhao K; Hatit MZC; Loughrey D; Paunovska K; Dahlman JE Universal Barcoding Predicts In Vivo ApoE-Independent Lipid Nanoparticle Delivery. Nano Lett. 2022, 22, 4822–4830. [DOI] [PubMed] [Google Scholar]
- (36).Miao L; Lin JQ; Huang YX; Li LX; Delcassian D; Ge YF; Shi YH; Anderson DG, Synergistic Lipid Compositions for Albumin Receptor Mediated Delivery of mRNA to the Liver. Nat. Commun 2020, 11, No. 2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Vincent MP; Bobbala S; Karabin NB; Frey M; Liu Y; Navidzadeh JO; Stack T; Scott EA Surface Chemistry-Mediated Modulation of Adsorbed Albumin Folding State Specifies Nanocarrier Clearance by Distinct Macrophage Subsets. Nat. Commun 2021, 12, No. 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Schnitzer JE; Sung A; Horvat R; Bravo J, Preferential Interaction of Albumin-Binding Proteins, Gp30 and Gp18, with Conformationally Modified Albumins - Presence in Many Cells and Tissues with a Possible Role in Catabolism. J. Biol. Chem 1992, 267, 24544–24553. [PubMed] [Google Scholar]
- (39).Schnitzer JE; Bravo J, High Affinity Binding, Endocytosis, and Degradation of Conformationally Modified Albumins. Potential Role of gp30 and gp18 as Novel Scavenger Receptors. J. Biol. Chem 1993, 268, 7562–7570. [PubMed] [Google Scholar]
- (40).Kobayashi M; Kudo M; Izumi N; Kaneko S; Azuma M; Copher R; Meier G; Pan J; Ishii M; Ikeda S, Cost-effectiveness Analysis of Lenvatinib Treatment for Patients with Unresectable Hepatocellular Carcinoma (uHCC) Compared with Sorafenib in Japan. J. Gastroenterol 2019, 54, 558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Roberts LR, Sorafenib in Liver Cancer - Just the Beginning. N. Engl. J. Med 2008, 359, 420–422. [DOI] [PubMed] [Google Scholar]
- (42).Shachaf CM; Kopelman AM; Arvanitis C; Karlsson A; Beer S; Mandl S; Bachmann MH; Borowsky AD; Ruebner B; Cardiff RD; Yang QW; Bishop JM; Contag CH; Felsher DW MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117. [DOI] [PubMed] [Google Scholar]
- (43).Kistner A; Gossen M; Zimmermann F; Jerecic J; Ullmer C; Lubbert H; Bujard H, Doxycycline-Mediated Quantitative and Tissue-Specific Control of Gene Expression in Transgenic Mice. Proc. Natl. Acad. Sci. U.S.A 1996, 93, 10933–10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Whitfield JR; Beaulieu ME; Soucek L, Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol 2017, 5, No. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Dang CV c-Myc Target Genes Involved in Cell Growth, Apoptosis, and Metabolism. Mol. Cell Biol 1999, 19, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Dang CV, MYC, Metabolism, Cell Growth, and Tumorigenesis. Cold Spring Harh. Perspect. Med 2013, 3, No. a014217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Sun J; Lu H; Wang X; Jin H, MicroRNAs in Hepatocellular Carcinoma: Regulation, Function, and Clinical Implications. Sci. World J 2013, 2013, No. 924206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Nguyen LH; Robinton DA; Seligson MT; Wu LW; Li L; Rakheja D; Comerford SA; Ramezani S; Sun XK; Parikh MS; Yang EH; Powers JT; Shinoda G; Shah SP; Hammer RE; Daley GQ; Zhu H Lin28b Is Sufficient to Drive Liver Cancer and Necessary for Its Maintenance in Murine Models. Cancer Cell 2014, 26, 248–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Roush S; Slack FJ, The let-7 family of microRNAs. Trends Cell Biol. 2008, 18, 505–516. [DOI] [PubMed] [Google Scholar]
- (50).Wu L; Nguyen LH; Zhou K; de Soysa TY; Li L; Miller JB; Tian J; Locker J; Zhang S; Shinoda G; Seligson MT; Zeitels LR; Acharya A; Wang SC; Mendell JT; He X; Nishino J; Morrison SJ; Siegwart DJ; Daley GQ; Shyh-Chang N; Zhu H, Precise let-7 expression levels balance organ regeneration against tumor suppression. eLife 2015, 4, No. e09431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Lam JKW; Chow MY; Zhang Y; Leung SW, siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol. Ther. Nucleic Acids 2015, 4, No. e252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.