

Abstract
Single-cell RNA sequencing (scRNA-seq) identifies cell subpopulations within tissue but does not capture their spatial distribution nor reveal local networks of intercellular communication acting in situ. A suite of recently developed techniques that localize RNA within tissue, including multiplexed in situ hybridization and in situ sequencing (here defined as high-plex RNA imaging) and spatial barcoding, can help address this issue. However, no method currently provides as complete a scope of the transcriptome as does scRNA-seq, underscoring the need for approaches to integrate single-cell and spatial data. Here, we review efforts to integrate scRNA-seq with spatial transcriptomics, including emerging integrative computational methods, and propose ways to effectively combine current methodologies.
Organ systems are composed of distinct cellular subpopulations whose spatial locations within a given tissue are deeply intertwined with their functions1. Single-cell RNA sequencing (scRNA-seq) characterizes the transcriptome of individual cells and can reveal cell subpopulations within a given organ. However, the isolation of single cells during the necessary tissue dissociation step of scRNA-seq destroys information on their spatial localization within native tissue and their proximities to each other. Given that juxtacrine and paracrine signals operate from 0 to 200 μm, such spatial information is vital to understand the intercellular communication underlying normal and diseased tissues. Interrogating intact tissue with spatial transcriptomics1 addresses this challenge by physically localizing gene sets expressed in specific cell subsets identified by scRNA-seq (FIG. 1). Current spatial transcriptomics approaches themselves cannot yet provide deep transcriptomic information on precisely localized single cells in tissue; however, they can shed light on the niches enriched for distinct gene sets. This localizes cell subpopulations to the tissue ‘neighbourhoods’ where they reside, and designates the ligands and receptors that they express to effect local intercellular communication. When used in combination, scRNA-seq and spatial transcriptomics can thus localize transcriptionally characterized single cells within their native tissue context. Integrating scRNA-seq and spatial transcriptomics data may therefore increase our understanding of the roles of specific cell subpopulations and their interactions in development, homeostasis and disease (FIG. 1).
Fig. 1 |. Adding spatial information to transcriptomes: integration of single-cell and spatial transcriptomics data.
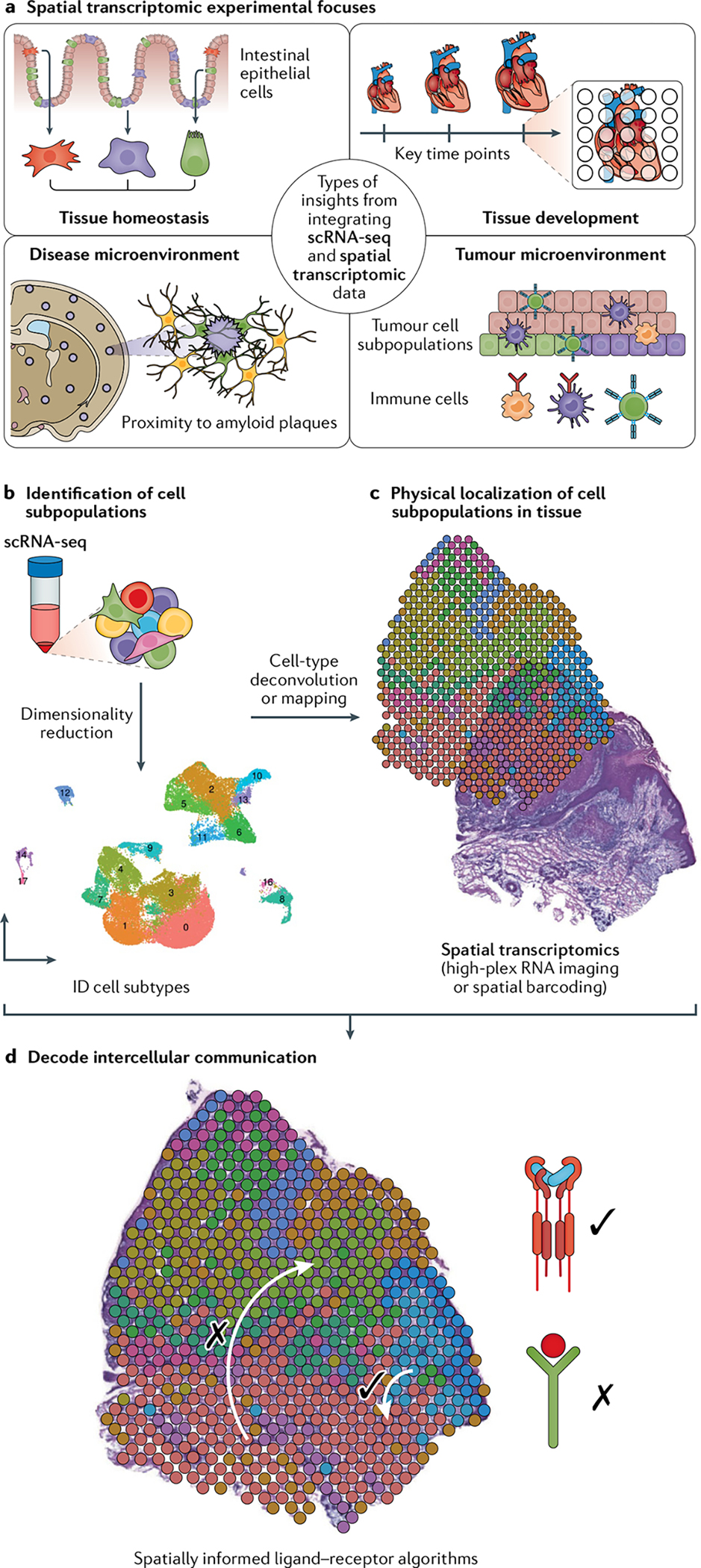
a | ‘Tissue homeostasis’ refers to elucidating spatial division of discrete cellular subtypes in a healthy tissue at a singular time point, for example, in the intestinal epithelium. ‘Tissue development’ refers to the study of how the spatial transcriptome changes in tissue at key stages in the development of a tissue. ‘Disease microenvironment’ refers to elucidating the spatial transcriptome in diseased and injured tissue niches with an eye towards proximity to relevant biological features, for example, proximity to amyloid plaques in brain tissue of patients with Alzheimer disease. ‘Tumour microenvironment’ refers to the study of spatial architecture of tumours and their interface with other cell subtypes in their environment. b | Workflows combining single-cell RNA sequencing (scRNA-seq) and spatial transcriptomic techniques begin by establishing cell subtypes typically through dimensionality reduction and clustering of scRNA-seq data. c | Deconvolution and mapping are used to localize cell subpopulations. Deconvolution is typically applied to spatial barcoding data, and mapping is typically applied to single-cell resolution spatial data (that is, high-plex RNA imaging (HPRI) data) to localize scRNA-seq subpopulations. d | Algorithms that evaluate spatial arrangement of localized subpopulations can further assess ligand–receptor interactions predicted from scRNA-seq data. Figure component in parts c and d adapted from REF.33, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
scRNA-seq has improved on bulk RNA-seq2, which alone is unable to capture cellular heterogeneity. Moreover, since its emergence in 2009 (REF.3), analytical pipelines for scRNA-seq have matured, and its limitations are increasingly understood4. For example, a key drawback of cell isolation through tissue dissociation is the potential to induce ectopic gene expression, which can lead to mischaracterization of certain cell subpopulations5. Spatial transcriptomics techniques avoid this technical artefact by assaying cells in their native tissue context. Compared with scRNA-seq, spatial transcriptomics workflows and efforts to integrate spatial transcriptomics and scRNA-seq data have emerged fairly recently and are an area of rapid evolution (TABLE 1). Building on single-molecule in situ hybridization (ISH)6, spatial methods that interrogate larger gene sets than single-molecule ISH have become more accessible7. For example, early ISH techniques8,9 have now advanced to methods that localize hundreds of genes in intact tissue through high-plex RNA imaging (HPRI) (FIG. 2A; TABLE 1), which encompasses techniques such as in situ sequencing10, multiplexed error-robust fluorescence in situ hybridization (MERFISH)11 and sequential fluorescence in situ hybridization (seqFISH)12–14. Spatial barcoding15–18 (FIG. 2B; TABLE 1) was recently developed to ‘capture’ the spatial coordinates of mRNA transcripts in an unbiased manner, which yields greater coverage. Capture spot resolution continues to improve, reaching a resolution smaller than the diameter of a typical single cell (according to two preprint publications19,20), thereby improving the likelihood that only a single cell or subtype is contributing to the RNA mixture. However, depth continues to be a limiting factor for spatial barcoding techniques. Conversely, HPRI improves on depth, but lacks transcriptome-wide coverage. Therefore, current spatial methods are still unable to create single-cell resolution spatial transcriptomics maps in which the transcriptome of each cell is captured at a depth akin to scRNA-seq (FIG. 2Ab, Bb). This limitation underscores the need to integrate current spatial transcriptomics platforms with scRNA-seq to maximize resolution in tissue.
Table 1 |.
Widely used spatial transcriptomics techniques and instruments
| Type | Technique | Characteristics | Mechanism notes | Refs |
|---|---|---|---|---|
| Hiqh-plex RNA imaging | In situ sequencing for RNA analysis in preserved tissue and cells | 25–90 reads per cell10, 69-gene panel31 | Genes of interest must be selected to design gene target-specific ‘padlock probes’ | 10 |
| STARmap | 160-gene panel, ~250 reads per cell | Uses ‘padlock probe’ mechanism and integrates hydrogel-tissue chemistry for 3D spatial resolution | 143 | |
| Multiplexed error-robust FISH (MERFISH) | 135-gene panel, ~100 reads per cell29 | Each gene has an associated binary code (wherein 1 means fluorescence for a certain part of the sequence, and 0 means no fluorescence) | 11,152 | |
| Sequential FISH (seqFISH, seqFISH+) | 249-gene panel, ~30 reads per cell13 | Each gene has an associated colour sequence code (24 colour probes per gene) with 60 different pseudo-colour options | 12–14 | |
| Spatial barcoding | Spatial transcriptomics | 100 μm diameter capture spot resolution | 100 μm from original spatial transcriptomics publication15 | 15 |
| Visium spatial gene expression | 55 μm diameter | 55 μm resolution translates to 3–30 cells per capture spot16 (original technique15is widely accessible through this 10× Genomics Visium Platform: spatialtran-scriptomics.com) | 16 | |
| High-definition spatial transcriptomics (HDST) | 2 μm diameter | Direct improvement in resolution on spatial transcriptomics15, but not as accessible | 161 | |
| Slide-seq, Slide-seq v2 | 10 μm diameter | Uses beads instead of capture spots, with the poly-T oligomers projecting radially around the bead; employs slides | 17,18 |
This table lists a widely used subset of the technologies; for more comprehensive reviews of spatial transcriptomics technologies, we refer readers to REFS162–164. Characteristics of high-plex RNA imaging (HPRI) methods are from exemplary studies that performed the method on intact tissue (rather than tissue culture). For reference, single-cell RNA sequencing (scRNA-seq) methods typically measure 104–106 reads per cell. FISH, fluorescence in situ hybridization.
Fig. 2 |. Common spatial transcriptomics techniques.
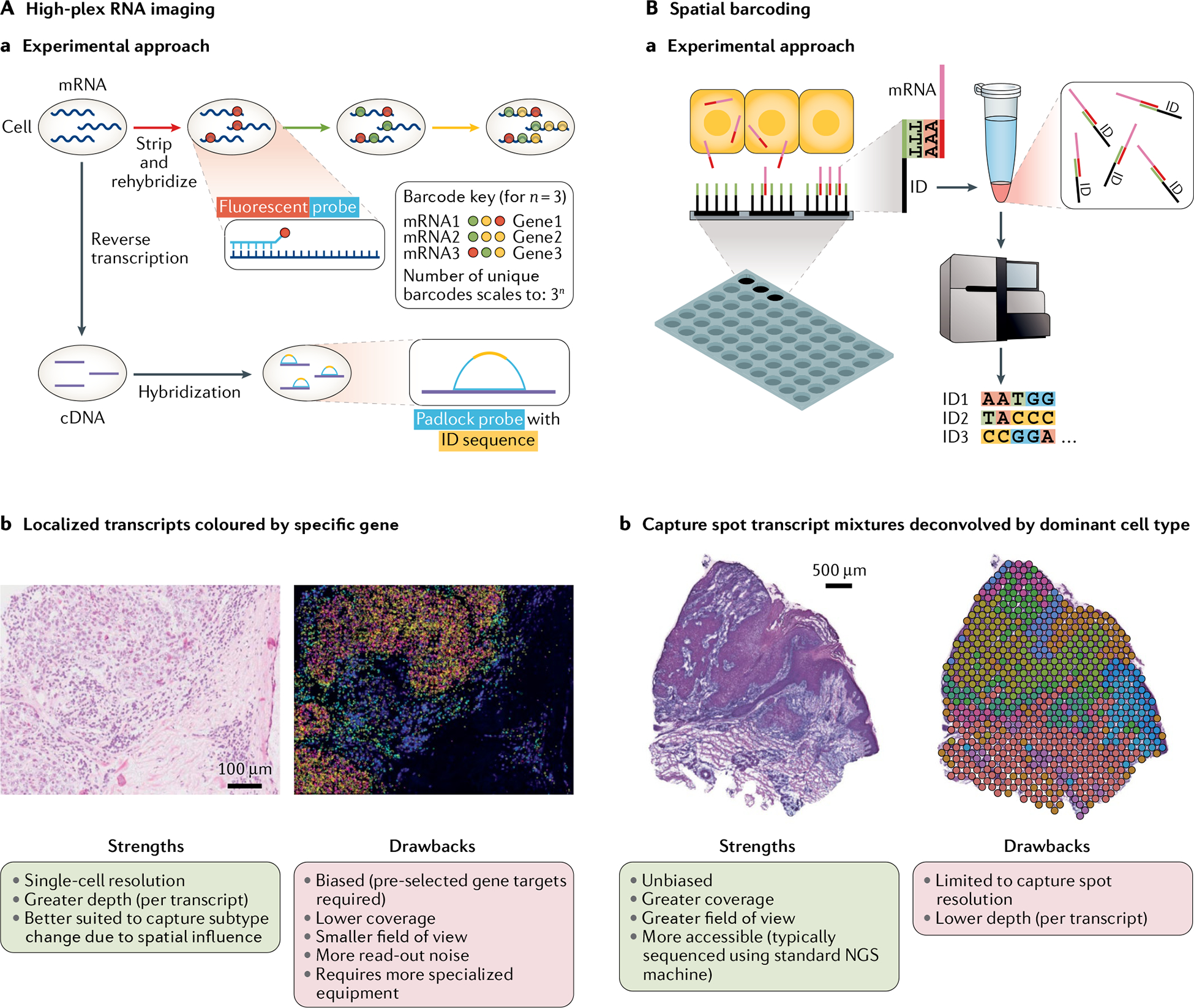
Aa | High-plex RNA imaging (HPRI) methods localize mRNA transcripts through probes that target specific genes. Fluorescent probe methods typically employ an encoding scheme whereby each gene can be identified through a unique sequence of fluorescent probe signals obtained through multiple rounds of hybridization. Padlock probe methods typically use probes that target the complementary DNA (cDNA) of target genes. Each probe has an identification (ID) sequence of nucleotides specific to each gene. The strategy for fluororescent sequencing of this ID varies by method. Ab | HPRI map of human breast cancer tissue cross-section with mRNA transcripts decoded based on gene fluorescent signals (not yet annotated by cell type, which can be done through mapping). Left: associated haematoxylin and eosin (H&E) stain (scale bar = 100 μm). Strengths and drawbacks of HPRI methods are listed. Pre-selected gene target panels typically range from 100 to 200 genes for intact tissue sections (proof-of-concept literature indicates an ~10,000 gene limit in tissue culture that is not easily scaled to intact tissue), and for some well-established methods only long RNA species (greater than 1,000 nucleotides) can be included. More specialized equipment typically makes for a more labour-intensive workflow. Ba | Spatial barcoding uses spatially barcoded (ID = location barcode) poly-T oligonucleotide capture of mRNA transcripts across tissue cross-sections followed by detachment and deep sequencing. Following sequencing, each transcript is de-multiplexed for assignment to its capture spot of origin based on its ID. Bb | Spatial barcoding map of human squamous cell carcinoma cross-section with capture spot mRNA mixtures deconvolved by cell type. Left: associated H&E stain (scale bar = 500 μm). Strengths and drawbacks of spatial barcoding methods are listed. Unbiased refers to the fact that the method does not involve selection of target genes. Greater accessibility also comes from the commercialization of spatial barcoding methods. NGS, next-generation sequencing. Part Ab reprinted from REF.10, Springer Nature Limited.
This Review provides an overview of the types of biological insights gained from integrating spatial transcriptomics and scRNA-seq data, and describes approaches to help optimize spatial analyses to deeply characterize single cells in tissue. This includes how spatial barcoding capture spots can be deconvolved to establish cell-type proportions and how scRNA-seq cell-type data can be mapped onto HPRI data. Additionally, the enhancement of ligand–receptor intercellular communication analysis with spatial resolution will be addressed. Finally, we discuss newly emerging experimental modalities and algorithms for interrogating the spatial transcriptome.
Biological insight from integrated data
Spatial transcriptomics has accelerated the capacity to elucidate the inner workings of discrete cell subpopulations within various contexts and organ systems (FIG. 1a). These approaches have been applied to assess the homeostasis and development of healthy tissue in the liver21–24, intestine25,26, bone marrow27, mouse embryo28, brain13,29, reproductive system30 and heart31,32. The widest application of spatial transcriptomics to studying disease has been the tumour microenvironment of cancers33–35. Spatial transcriptomics has also been applied to other diseased and injured microenvironments, such as the brain36,37 and spine38 in neurodegenerative diseases, the heart after myocardial infarction39 or other injury40 and the lungs during respiratory infection41. Recent work has begun to integrate spatial transcriptomics data with additional modalities, namely scRNA-seq, to provide new insights into tissue composition and function (TABLE 2).
Table 2 |.
Studies resolving scRNA-seq and spatial transcriptomics data
| Tissue sample | Input data | Analyses | Highlights | Ref. |
|---|---|---|---|---|
| Tissue homeostasis | ||||
| Liver (mouse) | scRNA-seq, smFISH | DR (t-SNE), single cell assigned to smFISH zones | Discovery of novel class of intermediate lobule cells along the porto-central axis | 21 |
| Liver (mouse) | FACS + bulk RNA-seq, immunofluorescence | GRN, cell assigned to zones | Wnt signalling pathway has spatially zonated processing machinery key for metabolic zonation and regeneration | 22 |
| Liver (mouse) | LCM + bulk RNA-seq | DR-PCA | Spatial resolution of epigenomic data from other studies to characterize Wnt and Notch signalling dynamics | 23 |
| Intestinal epithelium (mouse) | scRNA-seq, LCM + bulk RNA-seq, smFISH, lineage tracing | DR (t-SNE), pseudo-time | Characterized previously unknown spatial zonation of enterocyte function along villus axis | 25 |
| Bone marrow (mouse) | scRNA-seq, LCM + bulk RNA-seq, immunofluorescence | Bulk deconvolution (CIBERSORT) + DR (t-SNE) LRI (RNA-Magnet) | Localization of previously unknown bone barrow cell populations to distinct niches | 27 |
| Brain — hypothalamic preoptic region (mouse) | scRNA-seq, HPRI (MERFISH), smFISH | DR (t-SNE), CTA (through separate clustering of HPRI data) | Mapping specific cell subtypes and identifying behaviours associated with these cell types via gene activity | 29 |
| Spermatogenesis | SB, smFISH | DR (PCA), pseudo-time, SB deconvolution (NMFReg17 method) | Comparison with diabetic mouse testis reveals spatial dysregulation of seminiferous tubes | 30 |
| Tissue development | ||||
| Embryonic intestine (human) | scRNA-seq, SB | DR (UMAP), SB deconvolution (but used Seurat Integration), LRI (SingleCellSingalR) | Charting of spatio-temporal dynamics of small intestine morphogenesis across key time points, with an eye to cell types involved in intestinal defects | 26 |
| Embryonic heart (human) | scRNA-seq, SB, HPRI, smFISH | DR (t-SNE), SB-CTA, HPRI-CTM (pciSeq) | 3D charting of cardiac morphogenesis transcriptomic patterns in distinct anatomical regions across key time points | 31 |
| Spinal cord (mouse) | SB, immunofluorescence | SB-CTA (labelled as gene modules) | Spatial mapping of 31 distinct, disease-relevant gene modules for amyotrophic lateral sclerosis mice across key time points | 38 |
| Tumour microenvironment | ||||
| Squamous cell carcinoma (human) | scRNA-seq, SB, MIBI | DR (UMAP), SB-CTA (Seurat enrichment scoring), LRI (NicheNet) | Characterization of human squamous cell carcinoma through comparison with matched normal skin discovers tumour-specific keratinocyte | 33 |
| Pancreatic ductal adenocarcinoma (human) | scRNA-seq, SB, immunofluorescence | DR (t-SNE), SB-CTA (enrichment scoring) | Characterization of human pancreatic ductal adenocarcinoma discovers co-localization of stress-response cancer cell state and inflammatory fibroblasts | 34 |
| Cutaneous malignant melanoma | SB | DR (PCA) | Characterized melanoma lymph node biopsies of patients with different survival outcomes | 35 |
| Other disease and injury microenvironments | ||||
| Brain (Alzheimer disease mouse model and human) | SB, HPRI, immunofluorescence | DR (t-SNE), gene module enrichment by brain regions | Identified two gene networks expressed by cells in proximity to amyloid plaques across different disease progression time points | 37 |
| Brain (Alzheimer disease mouse model) | scRNA-seq, smFISH, immunofluorescence | DR (t-SNE), pseudo-time | Identified a novel disease-associated microglia cell that localizes near amyloid plaques | 36 |
| Heart post-myocardial infarction (human) | snRNA-seq, scATAC-seq, SB, ISH | DR (UMAP), SB-CTA (but used Seurat Integration) pseudo-time | Integrated snATAC-seq data to localize spatially distinct enhancer accessibility or transcription factor binding relative to the infarcted area | 39 |
| Heart (zebrafish) | Cryosection + bulk RNA-seq, smFISH, immunofluorescence | CTA | Spatial division of cardiomyocyte regeneration signalling in wound border zones of cryo-injured hearts | 40 |
Input data: single-cell RNA sequencing (scRNA-seq), single-molecule fluorescent in situ hybridization (smFISH), spatial barcoding (SB), high-plex RNA imaging (HRPI), laser-capture microdissection (LCM), bulk RNA-seq, fluorescent-activated cell sorting (FACS), cryo-sectioning, multiplexed ion beam imaging (MIBI), multiplexed error-robust fluorescence in situ hybridization (MERFISH) and single-cell sequencing assay for transposase-accessible chromatin using sequencing (scATAC-seq). Analyses performed: dimensionality reduction (DR) to establish cell type (uniform manifold approximation and projection (UMAP), t-distributed stochastic neighbour embedding (t-SNE), principal component analysis (PCA)), other (clustering techniques (k-means, hierarchical), RNA-seq-based cell-type annotation based on cell type-specific gene modules (SB-CTA), mapped (cell-type mapping (CTM)) onto HPRI-based data (HPRI-CTM)) and single-cell analyses (ligand-receptor interactions (LRI), pseudo-time development or cell fate trajectories (pseudo-time), gene regulatory network analysis (GRN)).
Normal tissue homeostasis and development.
A major goal of spatial transcriptomics is to provide an increased understanding of the roles of distinct cell types in steady-state tissue homeostasis. For example, each liver lobule unit along a radial axis surrounding a blood vessel contains a distinct gradient of oxygen, nutrients and hormones42. As the liver is classically divided into two periportal and pericentral hepatocyte zones, one study used a probabilistic inference model to assign each cell from scRNA-seq-based single-molecule fluorescence in situ hybridization into nine discrete zones21, and thereby revealed the roles for a novel class of intermediate lobule cells. Other studies used fluorescence-activated cell sorting (FACS) with zonated liver cell surface markers22 and laser-capture microdissection (LCM)23 to elucidate the spatial dynamics of signalling pathways regulating metabolic zonation and regeneration. Although it was known that intestinal epithelial cells migrate up from crypts along the villus axis, a recent analysis defined three functionally distinct regions along the villus axis in the absence of any pre-existing knowledge of landmark genes unique to each section25, providing new insight into cell subpopulations that mediate tissue homeostasis.
In cases in which homeostatic cellular division of labour is less understood, physical partitioning-based spatial transcriptomics may provide meaningful insight. One such approach mapped bone marrow-resident cell types to previously unrecognized, spatially distinct niches and defined cellular origins of pro-haematopoietic factors27. The work first used scRNA-seq to establish the transcriptomes of discrete cellular subtypes found in bone marrow. Then, on separate samples, classic immunostaining guided LCM of bone marrow cross-sections, followed by bulk RNA-seq for each LCM niche. mRNA mixtures from LCM niches were deconvolved to localize scRNA-seq-based bone marrow subtypes. Immunofluorescence based on gene markers specific to the cell types validated deconvolution findings, thereby providing insight into resident cell subpopulations active in normal bone marrow. LCM on single mid-gastrulation mouse embryos followed by RNA-seq was performed to render a 3D understanding of the regionalization of cell fates in the embryo28.
In the absence of such physical manipulation, spatial transcriptomics can help interrogate tissues without having an idea of where cell types localize. For example, an analysis of the functional organization of neurons in the preoptic hypothalamus combined scRNA-seq and HPRI to discover novel cell subtypes, their spatial localization and their activation during certain behaviours29. A preliminary study investigated the dynamics of spermatogenesis using spatial barcoding to discover two spatially segregated populations, each comprising stem cells at unique transcriptional states30. Moreover, a map of development stages was constructed through pseudo-time analysis43 deconvolved from the gene signature at each capture spot.
Excitingly, spatial transcriptomics is also amenable to discovering the dynamics of tissue development across multiple key time points during development. One effort used scRNA-seq to define the transcriptomes of cardiomyocyte subtypes, and then applied spatial barcoding to study subtype spatial localization during human embryonic heart development at three key development time points31. Transcriptomic data from both spatial barcoding and scRNA-seq were then synthesized to select probes for HPRI to ultimately produce a 3D map of the developing heart at single-cell resolution. To discover the spatial coordination of intestine development, another group integrated spatial barcoding with scRNA-seq across three critical developmental time points26. Notably, the authors analysed how morphogen gradients direct the dynamic development of cell types linked to developmental intestinal disorders, which are challenging to study in utero. Algorithms for aligning spatial barcoding data across spatio-temporal development are still in their nascency44,45.
The tumour microenvironment.
Although spatial transcriptomics analyses of several disease microenvironments exist, tumour microenvironments are presently the most extensively studied46. Tumour microenvironments are particularly heterogeneous and have complex, spatially restricted interactions with the immune system47,48. Until recently, the ability to interrogate the inner workings and heterogeneity of the tumour microenvironment has relied largely on bulk RNA-seq49. scRNA-seq has advanced our understanding by identifying cancer subpopulations that can drive drug resistance, predict metastatic risk and provide prognostic value50,51.
A recent study combined spatial barcoding and scRNA-seq to localize an immunosuppressive tumour-specific keratinocyte subpopulation to a fibrovascular niche at the tumour borders (as defined by aligning haematoxylin and eosin images) in human squamous cell carcinoma33. This population expressed genes associated with immunotherapy resistance and numerous ligands inferred to modulate cancer-associated fibroblasts, suggesting ways tumour subpopulations may promote local immunosuppression. In pancreatic ductal adenocarcinoma, another study intersected scRNA-seq and spatial barcoding with annotated haematoxylin and eosin images to reveal that inflammatory fibroblasts play a significant role in cancer stress responses34. In addition to providing insights into tumour resistance and stress responses, such integrated data may offer insight into clinical prognosis. For example, one study observed that greater heterogeneity of a transition area within cross-sections of melanoma metastases was associated with poorer patient survival35. Furthermore, spatial data enabled the identification of the most abundantly expressed genes in the transition area and could guide microdissection of this area to provide more a detailed characterization in the future. Ultimately, spatial data enabled a more detailed analysis of clinically relevant characteristics of the tumour microenvironment, thereby yielding greater prognostic power and potential therapeutic targets.
Other diseased and injured microenvironments.
Integrating spatial data with scRNA-seq can elucidate the molecular pathogenesis of localized gene expression programmes proximal to disease biomarkers, such as amyloid plaques in Alzheimer disease52,53. One study discovered an Alzheimer disease-associated subpopulation of microglia in mouse brains using single-molecule fluorescent ISH (in which probes were selected based on the most highly expressed genes in the disease-associated subpopulations of their scRNA-seq data); annotating the spatial cross-sections from diseased brain tissues revealed that this disease-associated population localized to amyloid plaques36. Similarly, alignment of spatial barcoding data with immunostained adult mouse brain cross-sections identified gene networks common to cells in areas with higher densities of amyloid plaques37. HPRI was then used to develop a single-cell resolution map of these gene networks and to capture their spatial dynamics at different disease stages. Elucidating gene networks that are upregulated adjacent to amyloid plaques provided candidate therapeutic targets of greater interest compared with those from gene networks obtained from scRNA-seq alone in which proximity to the plaque biological feature was lost37.
Spatial analysis around disease-relevant biological features has also been performed outside the neurological context. A spatial multiomics analysis integrating single-nucleus RNA-seq (snRNA-seq), spatial barcoding and, uniquely, single-cell sequencing assay for transposase-accessible chromatin using sequencing (scATAC-seq) spatially mapped the gene regulatory networks controlling the fibroblast to myofibroblast differentiation that drives cardiac scar formation in the myocardial border zone area39 (which is the leading cause of death in patients with myocardial infarction54). To spatially map the gene regulatory networks, snRNA-seq and scATAC-seq were integrated first, and then snRNA-seq cell-type annotations were used to score each capture spot. One study characterized zebrafish cardiomyocyte regeneration following cardiac injury40. Analysis of cryo-sections parallel to the wound border revealed three distinct spatially restricted zones with unique signalling patterns.
Spatial transcriptomics can also be leveraged to map disease pathogenesis across key time points. For example, given that a lack of spatial resolution has hampered efforts to understand amyotrophic lateral sclerosis, spatial barcoding was applied to profile the spatial transcriptome of spinal cord cross-sections during three key disease states38. The resulting co-expression analysis identified 31 major disease-relevant co-expression modules and charted their dynamic expression levels across time points of disease progression, ultimately nominating potential therapeutic targets. Ultimately, spatial transcriptomics data can validate what scRNA-seq can merely impute: the spatial coordinates of the discrete cellular subpopulations, dynamic changes in spatial arrangement during development and pathogenic progression, and proximity to disease-relevant bio-features.
A model workflow for integration
The lack of spatial transcriptomics methods yielding depth and coverage comparable with scRNA-seq underscores the need to integrate data from both techniques (FIG. 3).
Fig. 3 |. Model workflow integrating scRNA-seq and spatial transcriptomics: the four As.
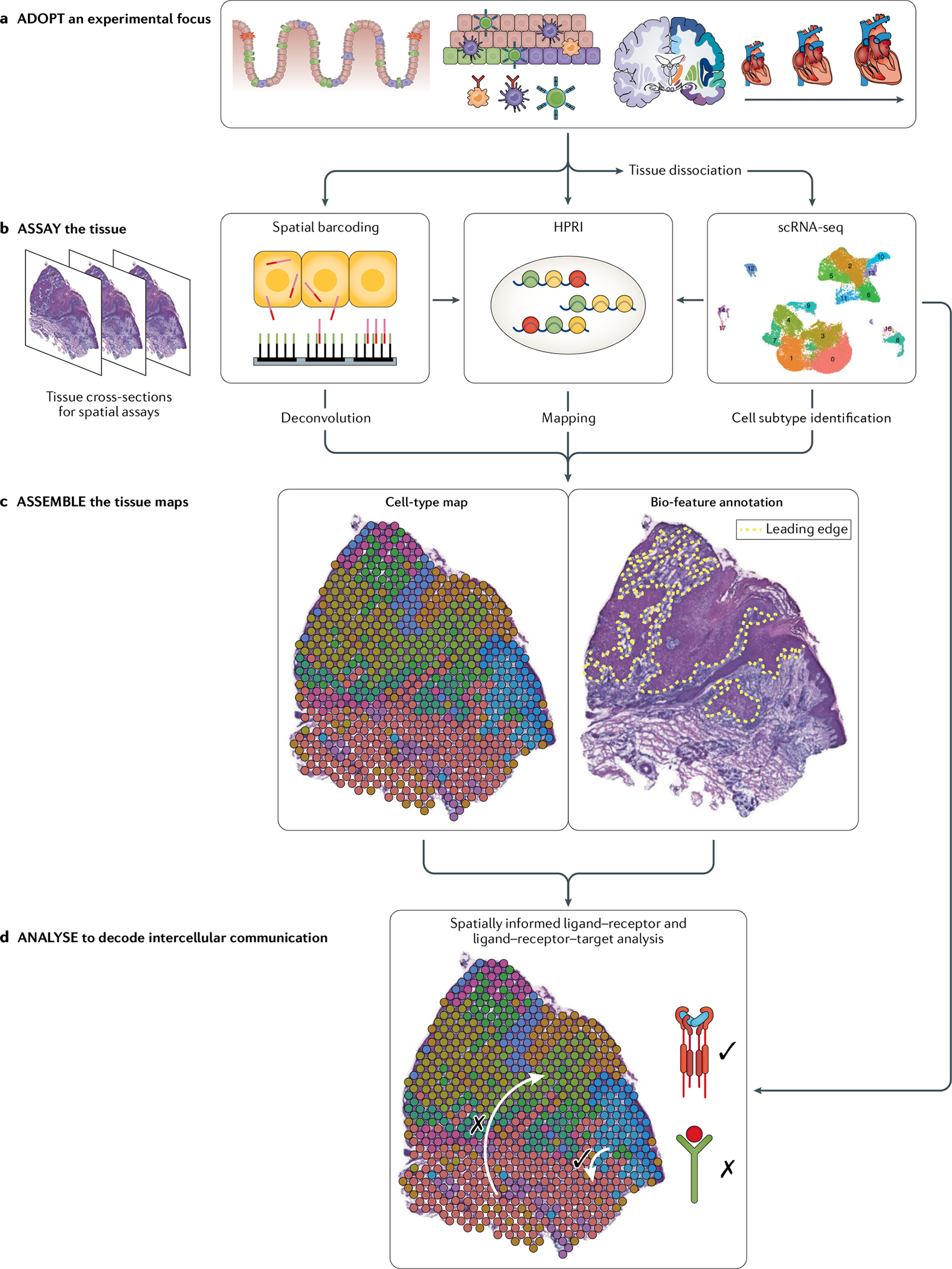
a | Adopt a rationale for assessing the spatial transcriptome in a particular tissue type. Common applications are depicted in FIG. 1a. b | Assay using single-cell RNA sequencing (scRNA-seq) to identify discrete cell subpopulations in a given tissue, and then with spatial barcoding to ascertain their physical locations in situ. Given their unbiased nature, scRNA-seq and spatial barcoding can help identify optimal gene panels for high-plex RNA imaging (HPRI) studies. Additional genes of interest for HPRI studies can be obtained by algorithms that identify spatially differentially expressed genes165,166 and through literature research. c | Assemble maps that localize specific cell subtypes spatially within the tissue. For spatial barcoding data, deconvolution can be used to localize cell types. For HPRI data, mapping can be used to localize cell types. Matched histology images can be annotated for landmarks of interest such as the tumour leading edge to further inform spatial analysis. d | Analyse assembled cell-type maps to nominate the cell types, tissue niches and ligand–receptor interactions involved in intercellular communication that drive the tissue phenotype. Figure component used in parts b and c adapted from REF.33, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
Establishing discrete cell subtypes through scRNA-seq.
scRNA-seq typically detects a significantly greater number of unique genes per cell compared with any spatial transcriptomics method55,56, thereby revealing novel cell subtypes as well as more detail on transcriptomes of each subpopulation. Furthermore, many scRNA-seq analytic pipelines plot cellular differentiation trajectories and gene regulatory networks17,57–59. Classifying cell types leverages several clustering methods60–65. Seurat66, an unsupervised clustering method using the Louvain method, has emerged as one of the most commonly employed, with SCANPY58 and SC3 (REF.63) being two other popular scRNA-seq subtype clustering tools. In silico sub-clustering can identify subpopulations within cell types of interest, which can then be validated by additional scRNA-seq experiments67. Because of its depth and single-cell precision, scRNA-seq is thus the current optimal platform for defining cell subpopulations in a given tissue.
Investigating tissue niches of interest from spatial data.
HPRI and spatial barcoding both benefit from inference of cell types based on scRNA-seq data (FIG. 3b,c). To more clearly define and validate cell types in niches of interest from spatial data, cells can be labelled based on their niche in situ and then sequenced. One approach to niche-labelling uses photoactivatable reporters, which are activated by a particular wavelength of light to label a given tissue niche. For example, GeoMx Digital Spatial Profiling allows the user to select small regions of interest to profile while viewing the tissue slide stained with desired imaging reagents68. Indexing oligonucleotides are attached to targets of interest (primary antibodies or to mRNA hybridization probes); upon exposure to UV light, these probes are cleaved and then quantified. Similarly, ZipSeq69 anneals a particular ‘zip code’ DNA barcode to cells targeted with UV light. The annealed barcode can then be read out during typical scRNA-seq workflows to localize the cell’s signature at the niche associated with the barcode. Transcriptome in vivo analysis poly-U oligo tags with photocleavable linkers70 can be loaded into tissues and photo-activated to capture mRNA in the niche of interest. In NICHE-seq71, a cell-expressed photoactivatable green fluorescent protein is activated by two-photon irradiation. Following activation, FACS-based sorting captures labelled cells for scRNA-seq. An alternative FACS-based approach uses a labelling system in which metastatic cancer cells engineered to secrete the sLP-mCherry protein are injected into metastatic niches of interest72. Unlabelled cells in the niche take up the sLP-mCherry protein and are then sorted by FACS for subsequent scRNA-seq analysis to profile metastatic niche cells involved in colonization. Each of these methods may augment conventional spatial transcriptomics approaches by enabling further characterization of tissue regions of interest.
Corroborating cell-type classifications made from mapping.
Given that mRNA is a proxy for protein expression, a valuable means of corroborating scRNA-seq-based cell-type assignments for HPRI data is multiplexed epitope-based tissue imaging73–76 using immunofluorescence microscopy77–80 or imaging mass cytometry81–86. Whereas immunofluorescence microscopy reveals more detailed organelle architecture, imaging mass cytometry commonly maps more protein targets (up to 100)80; both approaches require validated antibody reagents. Emerging integrative approaches include an imaging mass cytometry method that simultaneously measures mRNA transcripts and 16 different proteins87. In this regard, algorithms such as CellProfiler are helpful in quantitatively characterizing cellular phenotypes of single-cell resolution images from various assays88. These approaches provide valuable protein-based corroboration of spatial transcriptomics data in tissue.
Integrating scRNA-seq and spatial data
Given that spatial transcriptomics methods do not yet generate deep single-cell resolution transcriptomic maps in tissue, analyses that can successfully integrate both single-cell and spatial transcriptomics data will be helpful in understanding the architecture of the cell-type distribution (FIG. 3c) and the putative mechanisms of intercellular communication that underlie this architecture (FIG. 3d). There are two primary approaches for integrating scRNA-seq and spatial data: deconvolution and mapping (TABLE 3). Deconvolution seeks to disentangle discrete cellular subpopulations from mixtures of mRNA transcripts from each capture spot based on single-cell data (FIG. 4a). Mapping has two facets: mapping of assigned scRNA-based cell subtypes to each cell on HPRI maps (FIG. 4b) and mapping each scRNA-seq cell to a specific niche or region of a tissue. Such analyses can provide spatial context for putative ligand–receptor interactions obtained from analysis of scRNA-seq data.
Table 3 |.
Single-cell and spatial integration strategies
| Algorithm | Strategy type | Recommended data | Output | Refs |
|---|---|---|---|---|
| Deconvolution | ||||
| SPOTlight | Non-negative least squares regression | scRNA-seq and spatial barcoding | Estimated cell-type proportions for each capture spot | 91 |
| SpatialDWLS | Dampened weighted least squares regression | scRNA-seq and spatial barcoding | Estimated cell-type proportions for each capture spot | 102 |
| stereoscope | Probabilistic modelling: negative binomial distribution | scRNA-seq and spatial barcoding | Estimated cell-type proportions for each capture spot based on MAP estimation | 92 |
| Robust cell-type decomposition (RCTD) | Probabilistic modelling: Poisson distribution | scRNA-seq and spatial barcoding | Estimated cell-type proportions for each capture spot based on MAP estimation | 90 |
| cell2location | Probabilistic modelling: negative binomial distribution | scRNA-seq and spatial barcoding | Estimated cell-type proportions for each capture spot on MAP estimation | 103 |
| Multimodal intersection analysis (MIA) | Enrichment analysis | Cell type-specific genes from scRNA-seq and region-specific genes from spatial barcoding | Enrichment or depletion values for certain scRNA-seq cell types in tissue regions annotated from H&E of spatial barcoding data | 34 |
| Relative expression scoring | Deconvolution and mapping (enrichment analysis) | scRNA-seq (for cell subtypes) applicable to spatial barcoding and HPRI | Relative cell-type expression scores for each capture spot or single cell | 62,66 |
| Mapping | ||||
| pciSeq | Probabilistic model: variation Bayesian mean-field approximation | scRNA-seq and HPRI | Pie chart of cell-type assignment probabilities for each cell | 89 |
| Harmony | Principal component analysis followed by k-means clustering | scRNA-seq and HPRI | Each cell in the HPRI data is assigned a cell type derived from scRNA-seq based on the shared clusters | 115 |
| LIGER | Integrative non-negative matrix factorization followed by shared factor neighbourhood graph clustering | scRNA-seq and HPRI | Each cell in the HPRI data is assigned a cell type derived from scRNA-seq based on the shared clusters; gene expression of spatially resolved single cells is imputed through average of nearest scRNA-seq neighbours in aligned factor space | 113 |
| Seurat Integration | Canonical correlation analysis followed by mutual nearest neighbour clustering | scRNA-seq and HPRI | Cell type for HPRI cell is assigned based on anchor’s cell type; gene expression of each HPRI cell is imputed through its anchored scRNA-seq pair | 114 |
| SpaGE | Principal component analysis followed by k-nearest-neighbour clustering | scRNA-seq and HPRI | Gene expression of each HPRI cell is imputed through k-nearest-neighbour regression | 117 |
| Spatially informed ligand-receptor analysis | ||||
| Fawkner-Corbett et al. (2021) | Fit model to test whether receptor expression is dependent on ligand expression | scRNA-seq and spatial barcoding | p value and coefficient used to determine whether the pair spatially co-localizes; output can be generated for within individual capture spots or between adjacent capture spots | 26 |
| Giotto | Proximity of ligand-receptor co-expression | HPRI or spatial barcoding | Cell-cell communication score for every pair of cell types | 128 |
| SpaOTsc | Optimal transport problem and downstream signalling targets | scRNA-seq and HPRI | Cell-cell signalling map in 2D or 3D | 129 |
| SVCA (spatial variance component analysis) | Gaussian mixture model that considers three factors: intrinsic, environmental and cell-cell interaction | HPRI | Quantifies how much variance for each gene can be explained by cell-cell interactions (among other factors) | 133 |
Listed on the deconvolution to mapping spectrum (FIG. 4). Also includes the main spatially informed intercellular communication algorithms. H&E, haematoxylin & eosin; HPRI, high-plex RNA imaging; MAP, maximum a posteriori; scRNA-seq, single-cell RNA sequencing.
Fig. 4 |. Deconvolution and mapping methods.
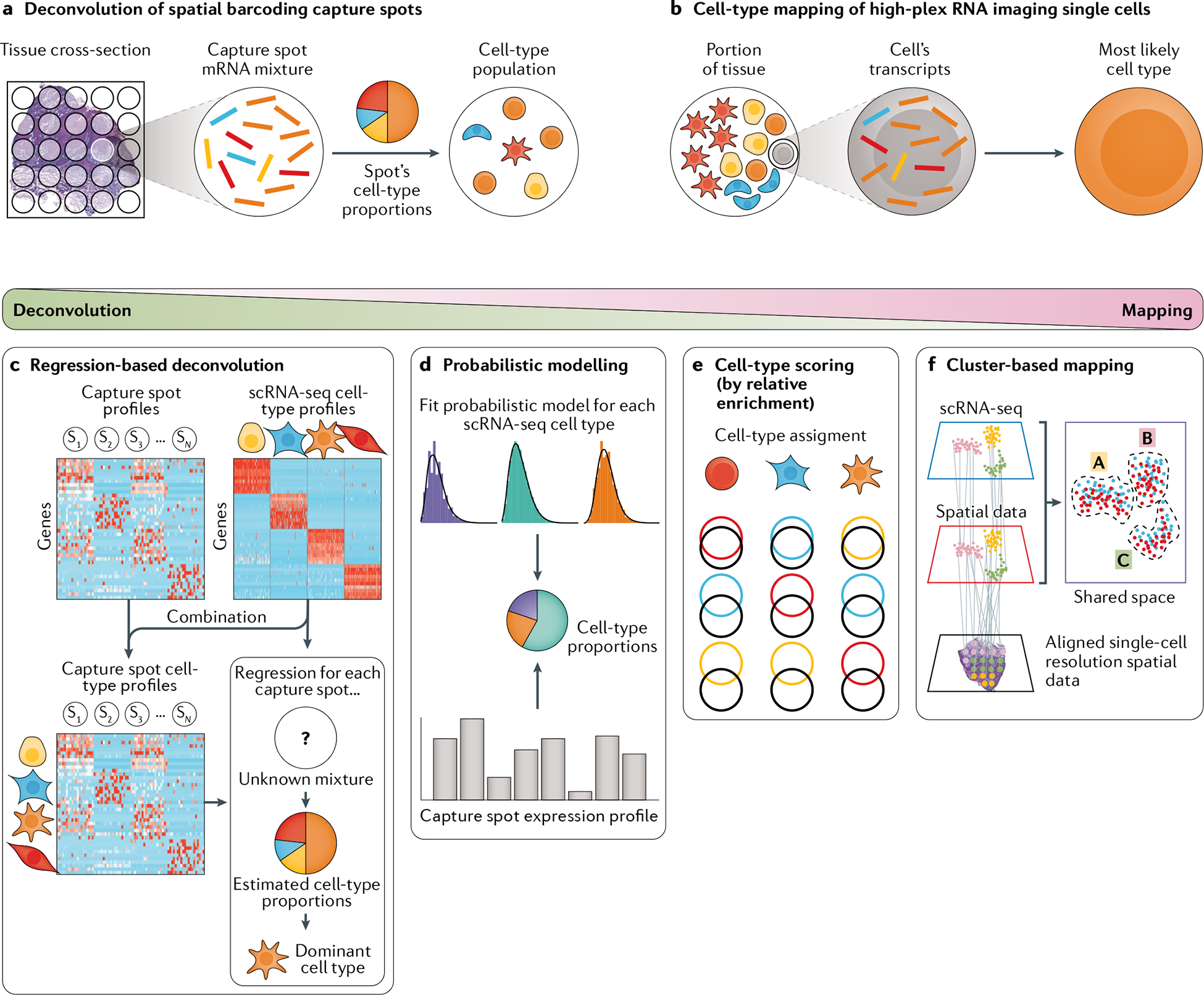
Computational approaches that localize cellular subtypes through integration of single-cell and spatial transcriptomics data. As spatial transcriptomics can only measure a fraction of the genes compared with single-cell RNA sequencing (scRNA-seq), deconvolving and mapping scRNA-seq-based cell types enriches spatial transcriptomics cell-type tissue maps. a | For spatial barcoding data, cell subpopulations can be localized by deconvolving the mixture of mRNA transcripts from each capture spot to predict the proportions of each cell type from the mixture of cells at each spot. b | For high-plex RNA imaging (HPRI) data, cell subpopulations can be localized by mapping scRNA-seq-based cell types onto each spatially resolved cell. Deconvolution and mapping methods to characterize spatial transcriptomics data using scRNA-seq cell subtypes exist on a weighted spectrum as each type of method can theoretically be applied to elucidate the subtype composition for both capture spots and single-cell transcript mixtures. Statistical regression (left) is most commonly applied to capture spot deconvolution and cluster-based mapping methods mostly towards HPRI cell-type mapping. c | Regression-based deconvolution combines scRNA-seq data clustered by cell type with capture spot data to yield a matrix containing capture spot profiles with scRNA-seq cell subtypes overlaid. Regression is used to find the scRNA-seq subtype profiles that best explain each capture spot mixture. d | Probabilistic distribution models are fitted to scRNA-seq transcript distributions for each cell type, with each capture spot characterized by determining the degree to which each cell type fits that spot’s transcript distribution. e | Cell-type scoring assigns cell types to single cells or capture spots based on overlap of spatial gene expression (black circles) with the marker genes for each cell type (coloured circles). f | Cluster-based mapping involves integrating scRNA-seq data (blue plot) and single-cell resolution spatial data (red plot) into a shared low-dimensional space (purple plot) whose clusters represent scRNA-seq cell types correspondening to spatial assay cell types. Tissue cross-section in part a adapted from REF.33, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0). Parameter estimation distribution graphs in part d adapted from REF.92, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). Part f adapted from REF.33, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
Deconvolution: disentangling discrete cellular subtypes from a single capture spot.
There are two main ways to approach deconvolution: inferring the proportions of cellular subtypes for a given spot89–92 (FIG. 4c,d) and scoring a given spatial transcriptomics spot for how strongly it corresponds to a single cellular subtype29,31,33,34 (FIG. 4e). There are many models that can deconvolve cell types from an mRNA mixture of an entire tissue sample, as from bulk RNA-seq17,93–101; however, given that spatial barcoding methods have only recently emerged as an accessible technique, a limited number of models are specifically tailored towards deconvolving mRNA mixtures from capture spots90–92,101,102. Regardless of which approach is taken, the first step in deconvolution is establishing which cellular subtypes exist in a given tissue sample (as described in the section ‘Establishing discrete cell subtypes through scRNA-seq’).
Inference-based deconvolution techniques involve estimating proportions of each cell type for a given capture spot. One approach to this form of deconvolution employs models based on statistical regression, which use a matrix of scRNA-seq data containing cell-type classifications for each cell to deconvolve the mRNA mixture for each capture spot (FIG. 4c). Various linear regression models have been applied to deconvolving bulk RNA-seq mixtures17,93–101. Non-negative least squares91 and dampened weighted least squares102 linear regression have been specifically applied to the deconvolution of capture spot mixtures.
A complementary approach to estimating exact proportions of each cell type in a given capture spot is through a Bayesian statistical framework that fits a probability distribution, often a negative binomial distribution92,103 (alternatively a Poisson distribution90), to the distribution of gene counts from scRNA-seq data (FIG. 4d). Once this probabilistic model is fit to the scRNA-seq data for each cell type and gene, empirical data for each capture spot are input as a prior to the model to yield a maximum a posteriori estimation of a cell-type distribution given gene distributions at that particular capture spot. Additional approaches that are specifically tailored towards addressing the spatial barcoding deconvolution problem are likely to emerge. Given the recent and rapid development of these models, a standardized approach to benchmark model performance has yet to be developed. In cases where two samples of matched cell-type composition can be obtained, the efficacy between one processed by scRNA-seq and the other through bulk RNA-seq followed by deconvolution can be compared96,99,100. However, a more convenient strategy for assessing efficacy is to create mixtures in silico by combining the transcript measurements from multiple well-characterized scRNA-seq cells90–92,95,96,100 or bulk RNA-seq mixtures of known cell-type composition97,99. The model’s deconvolution output can be compared with the ground-truth cell subtype proportion values to assess efficacy. SPOTlight’s benchmarking strategy is among the most thorough: assessing the accuracy, sensitivity and specificity of cell-type detection and overall correlation with the ground truth91. Furthermore, physical validation of subtype spatial localization at a higher resolution can be obtained through HPRI91,92,102. However, until HPRI improves the transcriptome coverage and applicability of untargeted methods to intact tissue, spatial barcoding may remain advantageous, especially for obtaining an unbiased characterization of the spatial transcriptome (FIG. 2Ab,Bb). As more deconvolution methods develop, more systematic unbiased benchmarking studies will be necessary.
There are many enrichment score-based techniques to deconvolution (FIG. 4e). Seurat 3.0 (REF.66) calculates relative expression scores of a cell type for a given capture spot by subtracting the average relative expression of the control gene set from the average relative expression of the gene set for that cell type62. Alternatively, multimodal intersection analysis34 computes the overlap between cell-type gene programmes established from scRNA-seq and tissue-type gene programmes established from spatial barcoding data to elucidate to what degree certain cell types are enriched or depleted in each region. Importantly, scRNA-seq-based scoring of a spot does not have to be limited to scoring for a cell type — one can score for characteristics such as cell cycle phase, tissue type, cancerous versus non-cancerous and specific gene expression programmes62. The limitation to this scoring approach is finding truly unique genes expressed by cell types of interest because overlapping genes may confound scoring.
Principles of data set mismatch and deconvolution method strategies to address it.
Mismatch between cell subtypes present in scRNA-seq data and those in spatial data (spatial barcoding and HPRI) can complicate deconvolution and mapping. Broadly, mismatch can result from errors in the pre-sequencing steps and/or in the post-sequencing analysis. Furthermore, mismatch can be exacerbated when using independent scRNA-seq atlases as references whose tissue conditions do not align well to newly generated spatial data.
Pre-sequencing mismatch causes are similar for both spatial barcoding and HPRI. More subtypes in scRNA-seq data than in spatial data could reflect pre-sequencing issues, such as the artificial creation of subtypes from stress response to scRNA-seq tissue dissociation5, the relatively low transcript capture depth per cell unit area in spatial barcoding and the relatively low transcript coverage with HPRI or an inability of HPRI or spatial barcoding to detect transcripts coming from a certain cell subtype due to tissue structure. Although probably rare, it is also possible that tissue dissociation may cause loss of subtypes whose phenotype can be acutely altered by the disruption of in situ spatial dynamics (a systemic analysis on this has yet to be performed); however, spatial barcoding is unlikely to be sensitive enough to capture changes in gene expression responsible for such subtle differences in phenotype. But if such genes can be pinpointed, HPRI could leverage its greater depth to capture these subtypes (FIG. 2Ab). Additional or missing subtypes detected from either assay can stem from sampling bias (of the tissue section used for scRNA-seq or spatial transcriptomics).
Post-sequencing mismatch can result from poor clustering choices (that is, over-annotating or under-annotating subtypes). A common strategy to assess the validity of scRNA-seq clusters is to see whether marker genes104 are observed in the proper cell-type clusters105. As capture spot deconvolution models typically treat scRNA-seq as a gold standard for establishing cellular subtypes, a subtype only present in spatial barcoding data is treated as noise by inference-based models90,92,103 whose presence hinders optimal fit. However, as we and others have found previously29,31,33,39,106, certain cell types either are difficult to dissociate or do not survive the scRNA-seq workflow, and therefore are not detected in scRNA-seq despite their verified presence in spatial data. To assess whether scRNA-seq has missed cell types, clustering capture spots29,31,39 (as one would cluster scRNA-seq data to obtain cell subtypes) may reveal subtypes only captured by spatial barcoding. Clustered captured spots may serve as a surrogate reference in deconvolution when no suitable scRNA-seq data set is available35,107.
Although inadequate depth and subtype detection cannot easily be computationally remedied, creation of cellular subtypes during dissociation is not likely to confound cell-type modelling, because these artificial cell types usually occur at low frequencies and do not pass confidence thresholding. In fact, SPOTlight91 has a minimum frequency threshold. Furthermore, SpatialDWLS102 performs an additional round of capture spot deconvolution, with subtypes below the minimum frequency removed. Under the premise that capture spots with three or more cell types are rare, robust cell-type decomposition90 wields the ‘doublet mode’ to mitigate overfitting by determining whether a model fit to one cell type or two cell types better explains the capture spot’s transcript mixture. The doublet mode can be extended to triplets and beyond. As higher resolution spatial barcoding assay techniques emerge (thereby decreasing the typical number of cell types per capture spot), the doublet mode will serve as a model deconvolution strategy. The aforementioned strategies reflect the ways in which the spatial barcoding capture spot deconvolution problem is distinct from the bulk RNA-seq deconvolution problem. As additional matched scRNA-seq and spatial barcoding data sets are generated, we anticipate the refinement of current deconvolution strategies and development of novel algorithms to maximize deconvolution accuracy.
Mapping: creating spatially resolved cell-type maps at single-cell resolution.
Just as with deconvolution, the first step to mapping is establishing cellular subtypes based on scRNA-seq data. Then, the primary challenge of mapping is to assign scRNA-seq-based cell types onto each cell from HPRI data (FIG. 4b). Intuitively, scRNA-seq-based cell-type scoring can be applied to each individual cell from HPRI similarly to how it is applied to scoring capture spots (FIG. 4e). Just as with deconvolution, there are also more computationally sophisticated approaches to addressing the mapping problem. Early efforts to spatially map scRNA-seq data relied on ISH of a small number of landmark genes to probabilistically map scRNA-seq cells to user-defined regions21,108,109 or single-cell coordinates110 in 2D or 3D space. However, these methods rely on the tissue having a prototypical structure. Furthermore, given rapid advances in throughput of HPRI techniques, most mapping algorithms have not yet caught up to specifically address integration of scRNA-seq with HPRI. Instead, they have focused on batch-correcting two different scRNA-seq experiments, but nevertheless have demonstrated successful application to single-cell resolution spatial data111.
A systematic evaluation of 14 published algorithms that implement batch correction strategies for mapping through cluster-based analysis112 identified three algorithms that most effectively integrate scRNA-seq data with single-cell resolution spatial data: LIGER113, Seurat Integration114 (from Seurat 3.0) and Harmony115 (FIG. 4f). All three algorithms ultimately obtain cell types from community detection of clusters that have been integrated into a low-dimensional space through different methods. Harmony projects both mapping data sets into low-dimensional space using principal component analysis and then iteratively removes batch effects by favouring the clustering of cells with a greater diversity of batches, while simultaneously maintaining cell-type similarity in the k-means clusters. Intuitively, mismatch would increase the odds of spuriously assigning data set-unique subtypes to the most similar cell type of the other data set; however, Harmony incorporates other penalties to successfully address mismatch. Seurat Integration begins by projecting scRNA-seq and HPRI mapping data sets into low-dimensional space using canonical correlation analysis, and then finds anchor cell pairs encoding cellular relationships across the two data sets through mutual nearest neighbour clustering116. Anchors can then be evaluated: higher-scoring anchors are those wherein many similarly clustered cells in one data set are predicted to correspond to that same group of similar cells in the other data set. Mutual nearest neighbour clustering has proved to be robust to mismatch as cells in non-overlapping populations displayed lower median anchor scores114; therefore, they were given less weight when transforming the data sets into a shared space.
As Harmony and Seurat Integration both attempt to ameliorate batch effects by mapping data sets into a completely shared latent space, they both implicitly assume that differences between the data sets are entirely due to technical variations. LIGER does not make the same assumption. LIGER addresses this during the dimensionality reduction step through employing integrative non-negative matrix factorization, wherein each cell is defined by a set of data-set-specific genes and a separate set of shared genes. Using the values for these genes, each cell is assigned a type based on whichever cell type corresponds to its maximum factor loading. Then, shared factor neighbourhood graph clustering is performed on both data sets to connect cells with similar factor loading neighbourhoods, thereby linking scRNA-seq and HPRI data. Given that the factor loadings often successfully characterized biological sources of variation, LIGER has proved to be robust to mismatch during mapping from scRNA-seq to HPRI cell types.
In addition to ascertaining community clusters of cell types, Seurat Integration and LIGER can impute expression of genes not measured for the spatially resolved single cells. Seurat Integration readily imputes by using the scRNA-seq gene expression profile of the spatial cell’s highest scoring anchor. Higher-scoring pairs have more shared nearest neighbours in low-dimensional space. By contrast, LIGER and SpaGE117, a newly published mapping algorithm tailored towards integration of scRNA-seq and single-cell spatial data, impute by obtaining the average expression of a certain number of corresponding scRNA-seq nearest neighbours in low-dimensional space. Imputation of spatial gene expression can be validated by comparing actual HPRI data with the predicted values from mapping for each HPRI gene assayed114,115.
Instead of a clustering approach, pciSeq89 uses a mean-field approximation118 Bayesian algorithm that leverages scRNA-seq data to simultaneously estimate the probability of assigning each read to each cell from the HPRI data. For each gene, pciSeq assesses the probability distribution for numerous gene spots for a given gene and their locations, known as a spatial point process. To obtain cell-type classification probabilities, the model calculates the expected number of copies of each gene assigned to the cell relative to the number of gene assignments predicted from scRNA-seq counts, cell area and estimated efficiency.
As additional matched scRNA-seq and spatial data sets are generated, algorithms specifically suited to address the challenges of both mapping and deconvolution will likely evolve. Eventually, improvement of spatial barcoding towards single-cell resolution could obviate the need for deconvolution and convert it into a mapping problem. In fact, given that the spatial barcoding deconvolution algorithms (both linear regression and Bayesian models) generally have lower accuracy on sparse data103, that is, many fewer transcripts per capture spot, mapping algorithms may begin to yield more accurate cell-type predictions for single-cell resolution spatial barcoding data. This may be especially true in tissues where there is a low degree of cell-type intermixing26.
Incorporating spatial data into analysis of intercellular communication.
Interactions between cell subpopulations mediate tissue homeostasis, development and disease. Given that much cellular cross-talk, notably juxtracrine and paracrine communication, is spatially restricted109,119, spatial transcriptomics data are well suited to evaluate the reliability of the ligand–receptor interactions computed from scRNA-seq (FIG. 5). Standard algorithms for predicting the ligand–receptor interaction pairs involved in intercellular communication primarily incorporate scRNA-seq data and a database of known ligand–receptor interactions120–127. When a landmark of interest is known, such as the tumour leading edge33, the co-expression of ligands and receptors at proximal spots in the region can be evaluated for statistical significance above the background to extend insight beyond the cell subtypes present to the level of the potential proteins they use to communicate with each other to drive local phenotypic features.
Fig. 5 |. Principles used to decode mechanisms of intercellular communication via expression of ligands and receptors in physically proximal cell subpopulations.
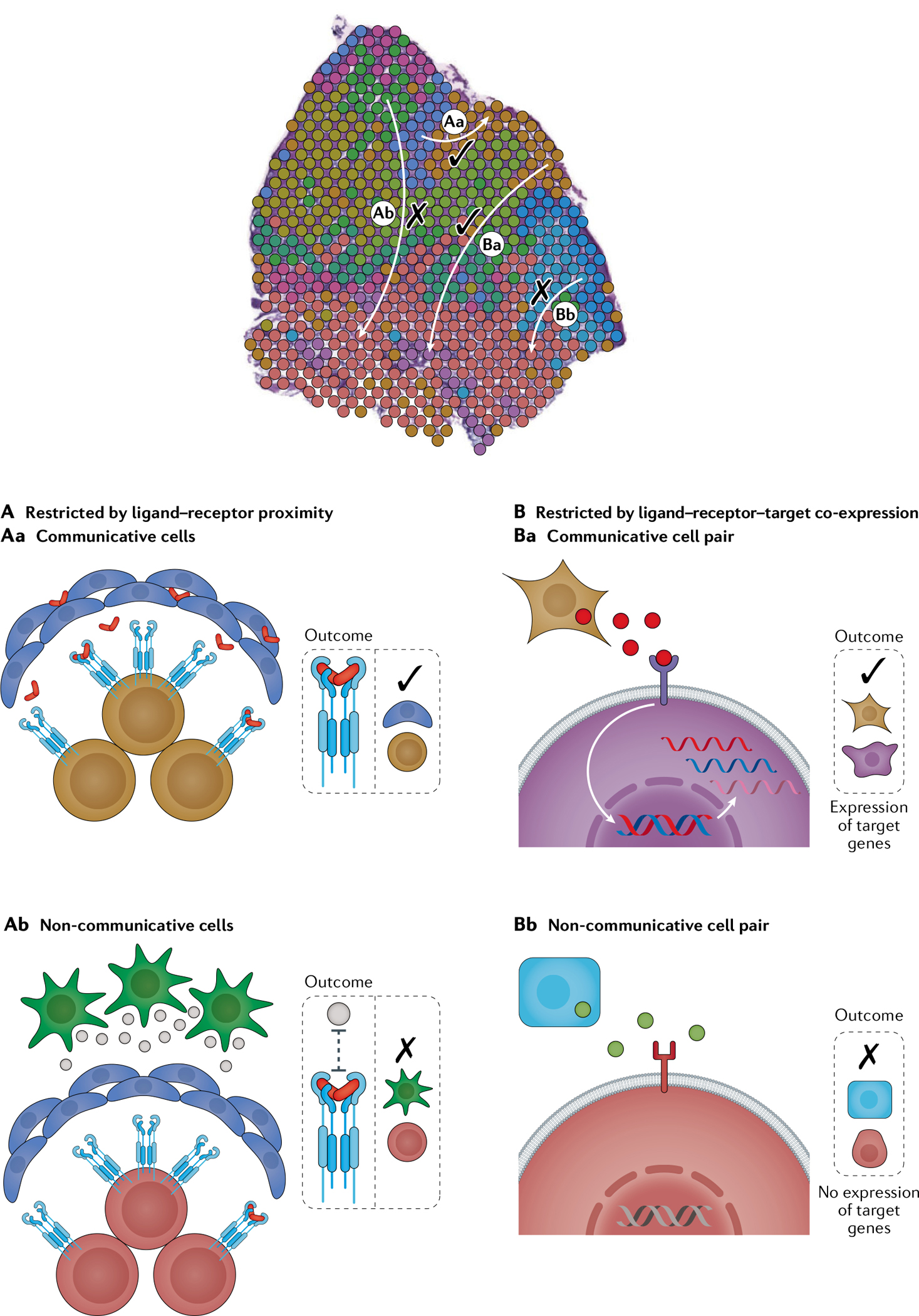
In a given tissue niche, cell subpopulations are more likely to be in communication if they are spatially proximal to each other (part A) and exhibit appropriate target gene signatures in signal-receiving cells (part B). A | Communicative cell types are established by evaluating co-localization to a given capture spot and/or expression of cell types in adjacent capture spots or cells. B | To account for longer-range communicative cell types that might be missed by the aforementioned strategies, the SpaOTsc algorithm predicts the maximum communication range for a given ligand–receptor pair through ligand–receptor target gene co-expression. Whereas ligand–receptor and ligand–receptor–target co-expression restriction can be used to establish intercellular communications from single-cell RNA sequencing (scRNA-seq) data alone, the spatial context can enhance this analysis by predicting maximum communication ranges and may disprove communications if the distance between the pairs is much farther than expected based on typical ranges recorded in the literature. Tissue cross-section adapted from REF.33, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
Numerous approaches exist to decode such mechanisms of intercellular communication. For example, one group fit a generalized linear model to test whether ligand–receptor pairs co-localize within a capture spot or among adjacent capture spots26. Similarly, for each pair of interacting cells from two different cell types, the Giotto workflow128 evaluates the likelihood that a given ligand–receptor interaction is used more or less based on the proximity of all of the co-expressing cells (FIG. 5A). The SpaOTsc algorithm129 combines scRNA-seq and spatial data to create a 3D single-cell resolution map with intercellular communication networks charted. It treats mapping cells and their ligand–receptor interactions as an optimal transport problem130 in which the cell–cell distance, obtained from mapping scRNA-seq cells to spatial positions from HPRI data, is used as a transport cost to spatially constrain the ligand–receptor signalling network. The yielded optimal transport plan represents likelihoods of cell–cell communication events between each pair of cells. An additional constraint is the maximum spatial range for the signalling, which is calculated by evaluating expression levels of a ligand’s downstream signalling targets (FIG. 5B), and the distance between the two cells with the strongest downstream correspondence is the maximum range.
Additionally, spatial data can be used to evaluate scRNA-seq map reconstructions and imputed ligand–receptor interactions. The novoSpaRc algorithm131 does this by reconstructing scRNA-seq single cells into a virtual tissue map through the assumption that physically close cells have similar transcription profiles. CSOmap132 combines scRNA-seq gene expression profiles and a database of ligand–receptor interactions to reconstruct single-cell resolution maps through dimensionality reduction. Once the cells are mapped, the contribution of each ligand–receptor pair to interactions between each cell-type cluster can be calculated. These approaches involving scRNA-seq integrated with spatial information can be used to nominate the receptors and ligands that mediate communication between proximal cell subpopulations. Going one step beyond, models to quantify the influence of cell–cell interactions on specific gene expression markers in the cell’s neighbourhood133 or on tissue-wide expression134 are beginning to emerge.
Several methods to further validate and characterize intercellular communication have been recently developed. For example, PIC-seq135 captures physically interacting pairs of cells (doublets) and performs scRNA-seq on them. Another means of measuring ligand–receptor interactions between cells in tissue culture is through an enzyme-based labelling technique. The ID-PRIME method136 labels static interactions between cells. To track more transient interactions, LIPSTIC137 has been developed and applied to the immune system. Such assays complement recent approaches designed to integrate scRNA-seq with spatial transcriptomics to nominate communicating cell subtypes and the receptors and ligands they use within specific spatial niches in tissue.
Future directions
Integrating single-cell expression data with existing spatial transcriptomics techniques will sharpen the resolution of physically proximal cell subpopulations within healthy and diseased tissue. The insights from such integrative efforts may help uncover previously opaque mechanisms of intercellular communication and biologic function operating below the signal to noise ratio in diverse tissues. In doing this, such advances may help define disease subtypes, guide prognosis and enable targeting of cell populations, along with the ligands and receptors critical for their actions, for precision therapy. Accomplishing such goals for human health will benefit from additional innovations that enhance spatial transcriptomics efficiency and resolution, incorporate more information about tissue architecture and apply new learning algorithms to discern patterns of cell subpopulations linked to disease behaviour.
Integration of additional modalities.
Currently, spatial transcriptomics techniques are largely focused on measuring mRNA transcripts through next-generation sequencing (spatial barcoding) or fluorescent markers (HPRI). However, spatial transcriptomics experiments generate a rich set of data that are often left untapped, namely, histologic images of tissue sections. Based on the premise that a significant amount of spatial variance manifests visually at the level of tissue architecture, a group developed ST-Net138, a deep learning algorithm that predicts the spatial variation in expression of 102 genes for each spatial barcoding capture spot superimposed onto tissue histology. Furthermore, XFuse incorporates spatial barcoding and histology sections to predict expression at single-cell resolution139. Saliency maps of these deep learning models are essential to extract novel spatial features linked to the expression of individual genes in the transcriptome. Such deep learning approaches, if successfully applied on large series of well-characterized data sets, may complement integrated scRNA-seq and spatial transcriptomics data sets by capturing information on cell types and gene expression via conventional histology. In addition to improving deconvolution and mapping algorithms, one needed focus centres on developing additional deep learning models138–141 to help disentangle which features of a given spatial transcriptome are most biologically relevant.
Defining the 3D spatial transcriptome and real-time cell tracking offer additional frontiers for future progress. Currently, most studies of the 3D spatial transcriptome take high-density cross-sections and computationally reconstruct them31,45,142 or infer scRNA-seq cell locations through sparse 3D single-molecule fluorescence in situ hybridization data108,110. However, STARmap143 and ExSeq144 are newly developed methods that pair HPRI with transformation of intact tissue into a hydrogel to preserve the 3D arrangement of the amplicons. Furthermore, ExSeq boasts an untargeted HPRI method that successfully leverages expansion microscopy to better resolve the normally densely packed RNA transcripts associated with untargeted HPRI methods.
Although it is true that the spatial transcriptome across a time course of development or tissue pathogenesis can be charted, spatial transcriptomics techniques do not monitor the physical dynamics of cell subtypes in real time. Real-time tissue tracking of cellular subtypes is especially important to understanding the inner workings of the tumour microenvironment interface with immune cells. In this regard, optical coherence tomography has been used to track the migration of tumour-associated myeloid cells145, and CellGPS has been used with positron emission tomography to track human breast cancer cells loaded with radioisotope146. When paired with spatial transcriptomics, both of these live tracking techniques could be applied to cell types of interest from spatial data to elucidate cell kinetics in settings, such as metastatic progression and immune cell dynamics during cancer immunotherapy. RNA timestamping represents an alternative means to elucidate the temporal dynamics of gene expression programmes in which the age of transcripts can be recorded on a 1-h timescale147. Recording transcriptional dynamics on this timescale is crucial to chart responses to perturbations. The application of such newly emerging efforts in 3D mapping and real-time analysis promises to extend current opportunities in understanding spatial tissue dynamics at the resolution of single cells.
An even deeper understanding of tissue function can be developed by going beyond resolution of the spatio-temporal transcriptome by spatially resolving other biomolecules integral to the central dogma of molecular biology148. DBiT-seq can spatially resolve proteins and mRNA transcripts on the same tissue by applying a mixture of antibody-derived DNA tags to localize proteins of interest prior to ligating two barcode sets through crossflow of two perpendicular microfluidic channels149. Remarkably, given the high barcoding resolution (10 × 10 μm2 squares), DBiT-seq used Seurat Integration (designed for integration of two single-cell data sets)114 to map scRNA-seq cell types to spatially resolved cells. In a recent preprint, the creators of DBiT-seq employed a similar barcoding strategy to localize the spatial epigenome on a tissue-wide scale and were able to successfully integrate these data with ISH at single-cell resolution and overlay scRNA-seq cell-type annotations150. 3D imaging of the sequenced genome in situ151, subcellular resolution of RNA152 and simultaneous imaging of 3D chromatin organization within the context of nucleoli and RNA153 all exist now at single-cell scale. They hold promise to advance to application on intact tissue and to revolutionize our understanding of how the machinery of the central dogma functions in the 3D context of a cell153 to reveal the inner workings of developmental trajectories151,152 and disease, namely cancer151.
Clinical relevance.
Spatial transcriptomics studies that perform comparative analysis between diseased and healthy tissue have begun to elucidate prognosis, optimal therapeutic treatment and potential therapeutic targets. Such efforts, however, have been limited in sample size and have thus far been exploratory. Aggregating the spatially resolved transcriptome of diseased and healthy cells across many patients may be important to enhance the clinical relevance and predictive prognostic power of such data. To increase the pace of data generation, analyses can focus on characterizing a much smaller number of regions of interest that drive disease-relevant phenotypes. In addition to describing trends in patient outcomes, interrogating how existing drugs, especially repurposed ones, affect spatio-temporal gene expression patterns in disease-driving cell types may lend insight into potential therapeutic agents. In this regard, monitoring mRNA transcripts in response to stimuli through methods such as NASC-seq154 may be useful in better understanding how drug perturbations affect the spatial transcriptome of diseased cells. Once these patient tissue data are aggregated, deep learning models could help identify the most relevant spatial expression patterns associated with survival outcomes or therapeutic response, potentially highlighting favourable landmarks to recapitulate or additional nodes of intervention during treatment. Cancer serves as a prototype for the promise of such strategies. Although therapeutic resistance in cancer remains a daunting challenge, the presence of tertiary lymphoid structures155 or densely fibrous stroma156 have served as positive and negative prognosticators of response to immune checkpoint blockade, a reminder that additional useful features may be waiting to be uncovered through spatial transcriptomics.
As more spatial transcriptomics analyses are performed, it will be increasingly challenging to disentangle definitive, disease-relevant cell types and their gene modules. As more cell types are identified and mapped in tissue, tools such as Seurat Integration114, Harmony115 and LIGER113 may evolve to integrate data across different experimental assays to determine whether specific cell types are consistently observed in each tissue. Furthermore, efforts to centralize spatial transcriptomics data for each organ system and disease will be valuable157, as the Allen Brain Atlas158 has done for the mouse brain, the National Institutes of Health (NIH) Blueprint Non-Human Primate159 has done for the rhesus macaque monkey brain and SpatialDB160 has done for multiple species and spatial transcriptomics techniques. Ultimately, better-defined spatial transcriptomes for disease-driving cell types, especially for cases in which cellular function is particularly dependent on an in situ context and adjacent cell populations, may yield more fruitful biological mechanisms for therapeutic targeting.
Conclusions
Integration of scRNA-seq with spatial transcriptomics can produce high-resolution maps of cellular subpopulations in tissue. Techniques for examining the spatial transcriptome are rapidly evolving, and therefore no singular spatial transcriptomics technique is best for all applications. Depending on the biological questions being asked, the experimental methodology can be designed to integrate any spatial transcriptomics approach with scRNA-seq. In addition to developing enhanced methods, carefully selecting algorithms for integrating such data is paramount, because spatial transcriptomics methods that spatially resolve tissue at single-cell resolution with scRNA-seq depth and whole-transcriptome coverage do not yet exist. Such integrative approaches are beginning to spatially map specific cell subpopulations in development and disease, and to shed light on the mechanisms whereby such populations collaboratively shape tissue phenotypes.
Acknowledgements
The authors thank members of the Khavari, Nolan, Altman, Zou and Lundeberg laboratories for helpful discussions. This work was supported by the US Veterans Affairs Office of Research and Development I01BX00140908, National Institutes of Health, National Cancer Institute (NIH/NCI) CA142635, NIH, National Institute for Arthritis and Musculoskeletal and Skin Diseases (NIH/NIAMS) AR43799 and AR49737 (P.A.K.), and a Physician-Scientist Training Award from the Damon Runyon Cancer Research Foundation (A.L.J.).
Glossary
- Single-cell RNA sequencing
(scRNA-seq). A method that sequences RNA transcripts (primarily mRNA) isolated from each cell of a tissue, thereby characterizing individual cells’ transcriptomes; aggregated data characterize the gene expression distribution of tissue cell subpopulations
- Intercellular communication
Communication between cells, typically through ligand–receptor interaction. Juxtracrine denotes direct cellular contact for signalling molecules to be passed between cells, whereas paracrine refers to diffusion of signalling molecules from a sender to a receiver cell
- Spatial transcriptomics
A method that localizes mRNA transcripts to precise spatial locations (single cells or capture spots, formerly regions) in a tissue. In this Review, the term refers broadly to all spatial transcriptomics methods, not exclusively spatial barcoding
- Bulk RNA-seq
A method that sequences a mixture of RNA transcripts (primarily mRNA) from the whole tissue to generate an average expression level for each gene across all cells sequenced
- High-plex RNA imaging (HPRI)
A targeted spatial transcriptomics method that localizes and quantifies RNA transcripts through multiplexed fluorescent microscopy imaging. Can typically target up to ~100–200 genes simultaneously in intact tissue sections
- Spatial barcoding
Spatial transcriptomic methods that use a microarray of poly-T oligonucleotides to ‘capture’ mRNA transcripts of tissue cross-sections, typically followed by next-generation sequencing
- Coverage
In the context of single-cell or spatial transcrtipomics assays, the number of distinct genes that are represented from captured RNA molecules
- Capture spots
Individual coordinates or capture locations on the microarray used to ‘capture’ mRNA transcripts for spatial barcoding and identified by a DNA barcode; each capture spot generally captures mRNA from multiple cells
- Resolution
In spatial data, the distance between spatial coordinates that identify the source of molecules. For spatial barcoding, higher resolution refers to a smaller distance between capture spot coordinates with smaller capture spot diameter
- Depth
In the context of single-cell or spatial transcriptomics assays, the number of unique RNA molecules captured for a particular gene
- Fluorescent-activated cell sorting
(FACS). A cell sorting technique based on flow cytometry for isolating and identifying different cell types using fluorescent antibodies targeting known cell type-specific cell surface proteins
- Microdissection
A method, such as laser-capture microdissection (LCM), that finely isolates different tissue parts from cryo-sections. LCM-seq is a spatial transcriptomics method that couples LCM with RNA sequencing
- Deconvolution
The process of predicting cell-type proportions at a given spatial barcoding capture spot based on its mRNA mixture. Cell types are typically derived from single-cell RNA sequencing profiles of the tissue
- Pseudo-time analysis
Computational ordering of single cells, namely of single-cell RNA sequencing, based on a gradual evolution of their transcriptomes to measure progress through a given biological process (for example, differentiation or proliferation
- Mapping
The process of assigning a single-cell RNA sequencing (scRNA-seq)-based cell type to each cell spatially resolved by high-plex RNA imaging assays; secondarily, predicting the spatial location of each scRNA-seq cell based on its transcriptome
- Statistical regression
In the context of deconvolution, linear regression models are fit to determine to what extent each cell type explains the gene expression values for each capture spot
- Bayesian statistical framework
In the deconvolution context, statistical models that rely on inferences about the distribution of transcripts for each cell type to yield a probability that a mixture of transcripts can be explained by a specific single-cell RNA sequencing cell type
- Maximum a posteriori
A Bayesian estimate of an unknown conditional. In the context of spatial transcriptomics integration methods, the maximum a posteriori estimate most often refers to the cell-type distribution given the gene distribution at a capture spot
- Enrichment
Refers to a particular class of genes that is over-represented in a large set of genes. In the deconvolution and mapping contexts, this class is often the cell type
- Mismatch
Incongruity between cell types detected as present in single-cell RNA sequencing and spatial transcriptomics data that can complicate spatial barcoding deconvolution, but less so high-plex RNA imaging mapping
- Mutual nearest neighbor
A robust algorithm for clustering single cells between two different single-cell transcriptomic data sets (that is, single-cell RNA sequencing with high-plex RNA imaging) based on the cell subtype
- Dimensionality reduction
A mathematical technique to represent high-dimensional data in lower dimensions. Mostly used in transcriptomic analysis for visualization of cell subtype clustering in 2D, sometimes 3D, space to establish subtypes
- Non-negative matrix factorization
A method commonly used in bioinformatics for dimensionality reduction of gene expression data as the non-negativity constraint reflects that genes are either expressed or not and cannot be negatively expressed
- Factor loading
In the context of transcriptomics, correlation coefficients between known gene transcript levels and latent cell subtype information. Can be plotted in low-dimensional space prior to joint clustering of single-cell RNA sequencing and high-plex RNA imaging data
- Impute
In the context of spatial transcriptomics, the computational process of determining unknown gene expression values of spatially resolved single cells (high-plex RNA imaging) with corresponding values from mapped single-cell RNA sequencing data
Footnotes
Competing interests
The authors declare no competing interests.
Peer review information
Nature Reviews Genetics thanks O. Bayraktar and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
References
- 1.[No authors listed] Method of the Year 2020: spatially resolved transcriptomics. Nat. Methods 18, 1 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Stegle O, Teichmann S & Marioni J Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet. 16, 133–145 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Tang F et al. mRNA-seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Chen G, Ning B & Shi T Single-cell RNA-seq technologies and related computational data analysis. Front. Genet. 10, 317 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van den Brink SC et al. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 14, 935–936 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Femino AM, Fay FS, Fogarty K & Singer RH Visualization of single RNA transcripts in situ. Science 280, 585–590 (1998). [DOI] [PubMed] [Google Scholar]
- 7.Liao J, Lu X, Shao X, Zhu L & Fan X Uncovering an organ’s molecular architecture at single-cell resolution by spatially resolved transcriptomics. Trends Biotechnol. 39, 43–58 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Levsky JM, Shenoy SM, Pezo RC & Singer RH Single-cell gene expression profiling. Science 297, 836–840 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Lyubimova A et al. Single-molecule mRNA detection and counting in mammalian tissue. Nat. Protoc. 8, 1743–1758 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Ke R et al. In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods 10, 857–860 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Chen KH, Boettiger AN, Moffitt JR, Wang S & Zhuang X Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, 1360–1363 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M & Cai L Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 11, 360–361 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shah S, Lubeck E, Zhou W & Cai L In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron 92, 342–357 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eng CL et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 568, 235–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ståhl PLPL et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016). [DOI] [PubMed] [Google Scholar]
- 16.10× Genomics. Inside Visium spatial capture technology https://pages.10xgenomics.com/rs/446-PBO-704/images/10x_BR060_Inside_Visium_Spatial_Technology.pdf (2019) [Google Scholar]
- 17.Rodriques SG et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stickels RR et al. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat. Biotechnol. 10.1038/s41587-020-0739-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho C-S et al. Seq-Scope: submicrometer-resolution spatial transcriptomics for single cell and subcellular studies. bioRxiv 10.1101/2021.01.25.427807 (2021). [DOI] [Google Scholar]
- 20.Chen A et al. Large field of view-spatially resolved transcriptomics at nanoscale resolution. bioRxiv 10.1101/2021.01.17.427004 (2021). [DOI] [Google Scholar]
- 21.Halpern KB et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542, 353–356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ben-Moshe S et al. Spatial sorting enables comprehensive characterization of liver zonation. Nat. Metab. 1, 899–907 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brosch M et al. Epigenomic map of human liver reveals principles of zonated morphogenic and metabolic control. Nat. Commun. 9, 4150 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saviano A, Henderson NC & Baumert TF Review single-cell genomics and spatial transcriptomics: discovery of novel cell states and cellular interactions in liver physiology and disease biology. J. Hepatol. 73, 1219–1230 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moor AE et al. Spatial reconstruction of single enterocytes uncovers broad zonation along the intestinal villus axis. Cell 175, 1156–1167.e15 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Fawkner-Corbett D et al. Spatiotemporal analysis of human intestinal development at single-cell resolution. Cell 184, 810–826.e23 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baccin C et al. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat. Cell Biol.22, 38–48 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng G et al. Spatial transcriptome for the molecular annotation of lineage fates and cell identity in mid-gastrula mouse embryo. Dev. Cell 36, 681–697 (2016). [DOI] [PubMed] [Google Scholar]
- 29. Moffitt JR et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, eaau5324 (2018). This article is an exemplary application of integrating multiplexed ISH with scRNA-seq to reveal the spatial organization and circuitry of neuronal subpopulations pertinent to social behaviours at a single-cell resolution.
- 30.Chen H et al. Dissecting mammalian spermatogenesis using spatial transcriptomics. bioRxiv 10.1101/2020.10.17.343335 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Asp M et al. A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell 179, 1647–1660 (2019). This analysis generates one of the first organ-wide, human developmental transcriptional atlases with single-cell spatial resolution by integrating scRNA-seq and spatial barcoding to yield optimized in situ sequencing.
- 32.Burkhard SB & Bakkers J Spatially resolved RNA-sequencing of the embryonic heart identifies a role for Wnt/β-catenin signaling in autonomic control of heart rate. eLife 7, e31515 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ji AL et al. Multimodal analysis of composition and spatial architecture in human squamous cell carcinoma. Cell 182, 497–514 (2020). This exemplary application of multimodal spatial analysis integrates scRNA-seq with spatial barcoding and multiplexed ion beam imaging (akin to spatial proteomics) to inform an in vivo CRISPR screen that identifies gene networks essential to the function of tumorigenic subpopulations.
- 34.Moncada R et al. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol. 38, 333–342 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Thrane K, Eriksson H, Maaskola J, Hansson J & Lundeberg J Spatially resolved transcriptomics enables dissection of genetic heterogeneity in stage III cutaneous malignant melanoma. Cancer Res. 78, 5970–5979 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Keren-Shaul H et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17 (2017). [DOI] [PubMed] [Google Scholar]
- 37. Chen W et al. Spatial transcriptomics and in situ sequencing to study Alzheimer’s disease. Cell 182, 976–991 (2020). This exemplary study characterizes Alzheimer disease-revelant cell types based on biofeature proximity by integrating spatial barcoding and in situ sequencing with histological stainings of disease mouse brain tissue cross-sections, effectively demonstrating how these integrated approaches can help map the spatio-temporal transcriptome at key disease stages.
- 38.Maniatis S et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science 364, 89–93 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Kuppe C et al. Spatial multi-omic map of human myocardial infarction. bioRxiv 10.1101/2020.12.08.411686 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu C-C et al. Spatially resolved genome-wide transcriptional profiling identifies BMP signaling as essential regulator of zebrafish cardiomyocyte regeneration. Dev. Cell 36, 36–49 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Boyd DF et al. Exuberant fibroblast activity compromises lung function via ADAMTS4. Nature 587, 466–471 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moor AE & Itzkovitz S Spatial transcriptomics: paving the way for tissue-level systems biology. Curr. Opin. Biotechnol. 46, 126–133 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Trapnell C et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tarmo Ä et al. Splotch: robust estimation of aligned spatial temporal gene expression data. bioRxiv 10.1101/757096 (2019). [DOI] [Google Scholar]
- 45.Bergenstråhle J, Larsson L & Lundeberg J Seamless integration of image and molecular analysis for spatial transcriptomics workflows. BMC Genomics 21, 482 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maniatis S, Petrescu J & Phatnani H Spatially resolved transcriptomics and its applications in cancer. Curr. Opin. Genet. Dev. 66, 70–77 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Balkwill FR, Capasso M & Hagemann T The tumor microenvironment at a glance. J. Cell Sci.125, 5591–5596 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Whiteside TL The tumor microenvironment and its role in promoting tumor growth. Oncogene 27, 5904–5912 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fan J, Slowikowski K & Zhang F Single-cell transcriptomics in cancer: computational challenges and opportunities. Exp. Mol. Med. 52, 1452–1465 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Puram SV et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171, 1611–1624.e24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patel AP et al. Single-cell RNA-seq highlights intratumoral hetereogeneity in primary glioblastoma. Science 344, 1396–1402 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu L, He D & Bai Y Microglia-mediated inflammation and neurodegenerative disease. Mol. Neurobiol. 53, 6709–6715 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Lein E, Borm LE & Linnarsson S The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science 358, 64–69 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Gulati A et al. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA 309, 896–908 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Papalexi E & Satija R Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 18, 35–45 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Haque A, Engel J, Teichmann SA & Lönnberg T A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 9, 75 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hwang B, Lee JH & Bang D Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 50, 96 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wolf FA, Angerer P & Theis FJ SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luecken MD & Theis FJ Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol.15, e8746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kiselev VY, Andrews TS & Hemberg M Challenges in unsupervised clustering of single-cell RNA-seq data. Nat. Rev. Genet. 20, 273–282 (2019). [DOI] [PubMed] [Google Scholar]
- 61.Stegle O, Teichmann SA & Marioni JC Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet. 16, 133–145 (2015). [DOI] [PubMed] [Google Scholar]
- 62. Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). This study exemplifies how scRNA-seq can be wielded to characterize single cells beyond the cell subtype.
- 63.Kiselev VY et al. SC3: consensus clustering of single-cell RNA-seq data. Nat. Methods 14, 483–486 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Andrews TS & Hemberg M Identifying cell populations with scRNASeq. Mol. Asp. Med. 59, 114–122 (2018). [DOI] [PubMed] [Google Scholar]
- 65.Duò A, Robinson MD & Soneson C A systematic performance evaluation of clustering methods for single-cell RNA-seq data. F1000Res. 7, 1141 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Butler A et al. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Villani AC et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, eaah4573 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Merritt CR et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol.38, 586–599 (2020). [DOI] [PubMed] [Google Scholar]
- 69.Hu KH et al. ZipSeq: barcoding for real-time mapping of single cell transcriptomes. Nat. Methods 17, 833–843 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lovatt D et al. Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat. Methods 11, 190–196 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Medaglia C et al. Spatial reconstruction of immune niches by combining photoactivatable reporters and scRNA-seq. Science 1626, 1622–1626 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ombrato L et al. Metastatic-niche labelling reveals parenchymal cells with stem features. Nature 572, 603–608 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bodenmiller B Multiplexed epitope-based tissue imaging for discovery and healthcare applications. Cell Syst. 2, 225–238 (2016). [DOI] [PubMed] [Google Scholar]
- 74.Lundberg E & Borner GHH Spatial proteomics: a powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 20, 285–302 (2019). [DOI] [PubMed] [Google Scholar]
- 75.Rimm DL Next-gen immunohistochemistry. Nat. Methods 20, 436–442 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Levenson RM, Borowsky AD & Angelo M Immunohistochemistry and mass spectrometry for highly multiplexed cellular molecular imaging. Lab. Investig. 95, 397–405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gut G, Herrmann MD & Pelkmans L Multiplexed protein maps link subcellular organization to cellular states. Science 361, eaar7042 (2018). [DOI] [PubMed] [Google Scholar]
- 78.Gerdes MJ et al. Highly multiplexed single-cell analysis of formalinfixed, paraffin-embedded cancer tissue. Proc. Natl Acad. Sci. USA 110, 11982–11987 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zrazhevskiy P & Gao X Quantum dot imaging platform for single-cell molecular profiling. Nat. Commun. 4, 1619 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lin JR, Fallahi-Sichani M & Sorger PK Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat. Commun. 6, 8390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Angelo M et al. Multiplexed ion beam imaging of human breast tumors. Nat. Med. 20, 436–442 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Goltsev Y et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 174, 968–981.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Damond N et al. A map of human type 1 diabetes progression by imaging mass cytometry. Cell Metab. 29, 755–768 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ali HR et al. Imaging mass cytometry and multiplatform genomics define the phenogenomic landscape of breast cancer. Nat. Cancer 1, 163–175 (2020). [DOI] [PubMed] [Google Scholar]
- 85.Giesen C et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422 (2014). [DOI] [PubMed] [Google Scholar]
- 86.Bendall SC et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332, 687–695 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schulz D et al. Simultaneous multiplexed imaging of mRNA and proteins with subcellular resolution in breast cancer tissue samples by mass cytometry. Cell Syst. 6, 25–36 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Carpenter AE et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Qian X et al. Probabilistic cell typing enables fine mapping of closely related cell types in situ. Nat. Methods 17, 101–106 (2020). This report presents pciSeq for mapping scRNA-seq cell types to multiplexed ISH and in situ sequencing data through probablistic modelling that implements the spatial point process. This is one of very few mapping algorithms specifically tailored towards capture spot deconvolution.
- 90.Cable DM et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. 10.1038/s41587-021-00830-w (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Elosua M et al. SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res. 49, e50 (2021). This article presents SPOTlight, to date the only deconvolution technique published in a peer-reviewed journal that is tailored towards deconvolving spatial barcoding capture spots through regression. The article also presents a comprehensive strategy for benchmarking emerging deconvolution methods.
- 92. Andersson A et al. Single-cell and spatial transcriptomics enables probabilistic inference of cell type topography. Commun. Biol. 3, 565 (2020). This report presents stereoscope, an effective approach for deconvolving spatial barcoding capture spots through probabilistic modelling and one of the few spatial barcoding deconvolution strategies published in a peer-reviewed journal to date.
- 93.Avila Cobos F, Vandesompele J, Mestdagh P & De Preter K Computational deconvolution of transcriptomics data from mixed cell populations. Bioinformatics 34, 1969–1979 (2018). [DOI] [PubMed] [Google Scholar]
- 94.Baron M et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. 3, 346–360 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang X, Park J, Susztak K, Zhang NR & Li M Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat. Commun. 10, 380 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dong M et al. SCDC: bulk gene expression deconvolution by multiple single-cell RNA sequencing references. Brief. Bioinform. 22, 416–427 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gong T & Szustakowski JD DeconRNASeq: a statistical framework for deconvolution of heterogeneous tissue samples based on mRNA-seq data. Bioinformatics 29, 1083–1085 (2013). [DOI] [PubMed] [Google Scholar]
- 98.Du R, Carey V & Weiss ST DeconvSeq: deconvolution of cell mixture distribution in sequencing data. Bioinformatics 35, 5095–5102 (2019). [DOI] [PubMed] [Google Scholar]
- 99.Newman AM et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Newman AM et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37, 773–782 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Aliee H & Theis F AutoGeneS: automatic gene selection using multi-objective optimization for RNA-seq deconvolution. bioRxiv 10.1101/2020.02.21.940650 (2020). [DOI] [PubMed] [Google Scholar]
- 102.Dong R, Yuan GC. SpatialDWLS: accurate deconvolution of spatial transcriptomic data. Genome Biol. 22, 145 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kleshchevnikov V et al. Comprehensive mapping of tissue cell architecture via integrated single cell and spatial transcriptomics. bioRxiv 10.1101/2020.11.15.378125 (2020). [DOI] [Google Scholar]
- 104.Zhang X et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 47, D721–D728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cao Y, Wang X & Peng G SCSA: a cell type annotation tool for single-cell RNA-seq data. Front. Genet. 11, 490 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McGinnis CS et al. MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nat. Methods 16, 619–626 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Andersson A et al. Spatial deconvolution of HER2-positive breast tumors reveals novel intercellular relationships. bioRxiv 10.1101/2020.07.14.200600 (2020). [DOI] [Google Scholar]
- 108.Satija R, Farrell JA, Gennert D, Schier AF & Regev A Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Karaiskos N et al. The Drosophila embryo at single-cell transcriptome resolution. Science 358, 194–199 (2017). [DOI] [PubMed] [Google Scholar]
- 110.Achim K et al. High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nat. Biotechnol. 33, 503–509 (2015). [DOI] [PubMed] [Google Scholar]
- 111.Forcato M, Romano O & Bicciato S Computational methods for the integrative analysis of single-cell data. Brief. Bioinform. 22, 20–29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Thi H et al. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 21, 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Welch JD et al. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell 177, 1873–1887.e17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Stuart T et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). This report details the inner workings of the Seurat Integration method, which is an exemplary method for mapping scRNA-seq cell types onto single-cell resolution spatial data. Seurat Integation is one part of a widely used R toolkit for analysing single-cell genomics data.
- 115.Korsunsky I et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1295 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Haghverdi L, Lun ATL, Morgan MD & Marioni JC Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421–427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Abdelaal T, Mourragui S, Mahfouz A & Reinders MJT SpaGE: Spatial Gene Enhancement using scRNA-seq. Nucleic Acids Res. 48, E107–E107 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bishop CM Pattern Recognition and Machine Learning. Oxidation Communications Vol. 27 (Springer, 2004). [Google Scholar]
- 119.Armingol E, Officer A, Harismendy O & Lewis NE Deciphering cell–cell interactions and communication from gene expression. Nat. Rev. Genet. 22, 71–88 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Browaeys R, Saelens W & Saeys Y NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020). [DOI] [PubMed] [Google Scholar]
- 121.Efremova M, Vento-Tormo M, Teichmann SA & Vento-Tormo R CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 15, 1484–1506 (2020). [DOI] [PubMed] [Google Scholar]
- 122.Noël F et al. Dissection of intercellular communication using the transcriptome-based framework ICELLNET Floriane. Nat. Commun. 12, 1089 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Choi H et al. Transcriptome analysis of individual stromal cell populations identifies stroma–tumor crosstalk in mouse lung cancer model. Cell Rep. 10, 1187–1201 (2015). [DOI] [PubMed] [Google Scholar]
- 124.Wang S, Karikomi M, Maclean AL & Nie Q Cell lineage and communication network inference via optimization for single-cell transcriptomics. Nucleic Acids Res. 47, e66 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cillo AR et al. Immune landscape of viral- and carcinogen-driven head and neck cancer. Immunity 52, 183–199.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cabello-Aguilar S et al. SingleCellSignalR: inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Res. 48, e55 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jin S et al. Inference and analysis of cell–cell communication using CellChat. Nat. Commun. 12, 1088 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dries R et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 22, 78 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Cang Z & Nie Q Inferring spatial and signaling relationships between cells from single cell transcriptomic data. Nat. Commun. 11, 2084 (2020). This report presents SpaOTsc, one of the few peer-reviewed methods that formally integrate spatial and scRNA-seq data into an algorithm to decode intercellular communication in tissues. SpaOTsc predicts the maximum signalling range for ligand–receptor pairs through a spatial transcriptomic analysis of each ligand–receptor pair’s target genes.
- 130.Villani C Optimal Transport, Old and New Vol. 338 (Springer, 2009). [Google Scholar]
- 131.Nitzan M, Karaiskos N, Friedman N & Rajewsky N Gene expression cartography. Nature 576, 132–137 (2019). [DOI] [PubMed] [Google Scholar]
- 132.Ren X et al. Reconstruction of cell spatial organization from single-cell RNA sequencing data based on ligand–receptor mediated self-assembly. Cell Res. 30, 763–778 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Arnol D, Schapiro D, Bodenmiller B, Saez-Rodriguez J & Stegle O Modeling cell–cell interactions from spatial molecular data with spatial variance component analysis. Cell Rep. 29, 202–211 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tanevski J, Gabor A, Flores ROR, Schapiro D & Saez-Rodriguez J Explainable multi-view framework for dissecting inter-cellular signaling from highly multiplexed spatial data. bioRxiv 10.1101/2020.05.08.084145 (2020). [DOI] [Google Scholar]
- 135.Giladi A et al. Dissecting cellular crosstalk by sequencing physically interacting cells. Nat. Biotechnol.38, 629–637 (2020). [DOI] [PubMed] [Google Scholar]
- 136.Liu DS, Loh KH, Lam SS, White KA & Ting AY Imaging trans-cellular neurexin–neuroligin interactions by enzymatic probe ligation. PLoS ONE 8, e52823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pasqual G et al. Monitoring T cell-dendritic cell interactions in vivo by intercellular enzymatic labelling. Nature 553, 496–500 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. He B et al. Integrating spatial gene expression and breast tumour morphology via deep learning. Nat. Biomed. Eng. 4, 827–834 (2020). This pioneering work presents ST-Net, one of the first applications of deep learning models for analysing spatial transcriptomic data, and one of the first methods to formally automate analysis of histological images for characterizing the spatial transcriptome.
- 139.Bergenstråhle L et al. Super-resolved spatial transcriptomics by deep data fusion. bioRxiv 10.1101/2020.02.28.963413 (2020). [DOI] [PubMed] [Google Scholar]
- 140.Yuan Y & Bar-Joseph Z GCNG: graph convolutional networks for inferring gene interaction from spatial transcriptomics data. Genome Biol. 21, 300 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Xu Y & McCord RP CoSTA: unsupervised convolutional neural network learning for spatial transcriptomics analysis. bioRxiv 10.1101/2021.01.12.426400 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Junker JP et al. Genome-wide RNA tomography in the zebrafish embryo. Cell 159, 662–675 (2014). [DOI] [PubMed] [Google Scholar]
- 143.Wang X et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361, eaat5691 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Alon S et al. Expansion sequencing: spatially precise in situ transcriptomics in intact biological systems. Science 371, eaax2656 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.SoRelle ED et al. Spatiotemporal tracking of brain-tumor-associated myeloid cells in vivo through optical coherence tomography with plasmonic labeling and speckle modulation. ACS Nano 13, 7985–7995 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jung KO et al. Whole-body tracking of single cells via positron emission tomography. Nat. Biomed. Eng.4, 835–844 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rodriques SG et al. RNA timestamps identify the age of single molecules in RNA sequencing. Nat. Biotechnol. 39, 320–325 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Crick F Central dogma. Nature 227, 561–563 (2008). [DOI] [PubMed] [Google Scholar]
- 149.Liu Y et al. High-spatial-resolution multi-omics sequencing via deterministic barcoding in tissue. Cell 183, 1665–1681.e18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Deng Y et al. Spatial epigenome sequencing at tissue scale and cellular level. bioRxiv 10.1101/2021.03.11.434985 (2021). [DOI] [Google Scholar]
- 151.Payne AC et al. In situ genome sequencing resolves DNA sequence and structure in intact biological samples. Science 371, eaay3446 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Xia C, Fan J, Emanuel G, Hao J & Zhuang X Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc. Natl Acad. Sci. USA 116, 19490–19499 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Su JH, Zheng P, Kinrot SS, Bintu B & Zhuang X Genome-scale imaging of the 3D organization and transcriptional activity of chromatin. Cell 182, 1641–1659.e26 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hendriks G-J et al. NASC-seq monitors RNA synthesis in single cells. Nat. Commun. 10, 3138 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Helmink B et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mariathasan S et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Regev A et al. Science forum: the Human Cell Atlas. eLife 6, e27041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lein ES et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 (2007). [DOI] [PubMed] [Google Scholar]
- 159.Bakken TE et al. A comprehensive transcriptional map of primate brain development. Nature 535, 367–375 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Fan Z, Chen R & Chen X SpatialDB: a database for spatially resolved transcriptomes. Nucleic Acids Res. 48, D233–D237 (2020). This article is one of the first publications describing a database directly geared towards aggregating spatial transcriptomic data.
- 161.Vickovic S et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 16, 987–990 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Asp M, Bergenstråhle J & Lundeberg J Spatially resolved transcriptomes — next generation tools for tissue exploration. BioEssays 42, 1–16 (2020). [DOI] [PubMed] [Google Scholar]
- 163.Zhuang X Spatially resolved single-cell genomics and transcriptomics by imaging. Nat. Methods 18, 18–22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Waylen LN, Nim HT, Martelotto LG & Ramialison M From whole-mount to single-cell spatial assessment of gene expression in 3D. Commun. Biol. 3, 602 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Svensson V, Teichmann SA & Stegle O SpatialDE: identification of spatially variable genes. Nat. Methods 15, 343–346 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sun S, Zhu J & Zhou X Statistical analysis of spatial expression patterns for spatially resolved transcriptomic studies. Nat. Methods 17, 193–200 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]