

ABSTRACT
Potassium (K+) is one of the most abundant cations in the human body. Under normal conditions, the vast majority of K+ is found within cells, and the extracellular [K+] is tightly regulated to within 3.0 to 5.0 mM. However, it has recently been shown that high levels of localized necrosis can increase the extracellular concentration of K+ to above 50 mM. This raises the possibility that elevated extracellular K+ might influence a variety of biological processes that occur within regions of necrotic tissue. For example, K+ has been shown to play a central role in the replication cycles of numerous viral families, and in cases of lytic infection, localized regions containing large numbers of necrotic cells can be formed. Here, we show that the replication of the model poxvirus myxoma virus (MYXV) is delayed by elevated levels of extracellular K+. These increased K+ concentrations alter the cellular endocytic pathway, leading to increased phagocytosis but a loss of endosomal/lysosomal segregation. This slows the release of myxoma virus particles from the endosomes, resulting in delays in genome synthesis and infectious particle formation as well as reduced viral spread. Additionally, mathematical modeling predicts that the extracellular K+ concentrations required to impact myxoma virus replication can be reached in viral lesions under a variety of conditions. Taken together, these data suggest that the extracellular [K+] plays a role in determining the outcomes of myxoma infection and that this effect could be physiologically relevant during pathogenic infection.
IMPORTANCE Intracellular K+ homeostasis has been shown to play a major role in the replication of numerous viral families. However, the potential impact of altered extracellular K+ concentrations is less well understood. Our work demonstrates that increased concentrations of extracellular K+ can delay the replication cycle of the model poxvirus MYXV by inhibiting virion release from the endosomes. Additionally, mathematical modeling predicts that the levels of extracellular K+ required to impact MYXV replication can likely be reached during pathogenic infection. These results suggest that localized viral infection can alter K+ homeostasis and that these alterations might directly affect viral pathogenesis.
KEYWORDS: mathematical modeling, myxoma virus, potassium
INTRODUCTION
Potassium (K+) is the most abundant intracellular cation in the body. K+’s major role is to maintain a functional electrochemical gradient across the plasma membrane, which is critical for numerous cellular functions, including neuronal signaling (1, 2), muscle contractions (3, 4), mitochondrial function and cellular energetics (5), mRNA translation (6, 7), and many more. Because of the essential nature of the electrochemical gradient for eukaryotic life, the concentrations of intracellular and extracellular K+ are tightly regulated (8). More than 98% of the body’s K+ is contained intracellularly, where its concentration can exceed 150 mM. In contrast, the [K+] in the extracellular space is much lower, typically ranging from 3.2 to 5.0 mM (9). Prolonged changes in the extracellular [K+] are extremely hazardous, with serum concentrations beyond ~6.5 mM (hyperkalemia) causing severe medical complications, including acute heart failure and death (10).
Interestingly, while prolonged, systemic changes in extracellular K+ concentrations can be lethal, it has recently been reported that the localized concentration of this ion within the central cores of solid tumors can exceed ~50 mM (11). Critically, the authors of that work found that the driver of this increased [K+] was not a cancer-specific pathway but instead was the result of the high rate of tissue necrosis that is often found within solid tumors. This raises the possibility that other situations exhibiting high rates of localized necrosis might also display transiently increased concentrations of extracellular K+. This hypothesis, however, has never been explored.
Our laboratory is interested in the biology of the poxvirus myxoma virus (MYXV). MYXV is studied both as a model of poxviral pathogenesis and as a potential oncolytic candidate to treat solid tumors (12). Interestingly, while overall K+ homeostasis has been shown to be critical for infection by several viral families (13–18), relatively little work has been conducted on how this ion affects infection by poxviruses (19). Additionally, since most viruses encode their own K+ channels (20), most of the previous work investigating the role of K+ in viral infection has focused on how modulation of K+ channel function influences either replication or cytopathic effect. In contrast, much less attention has been paid to how overall changes in the extracellular [K+] might impact viral replication cycles (21). Since both pathogenic poxviral lesions (22) and the cores of solid tumors in which oncolytic MYXV might be used therapeutically (23) contain high levels of localized necrosis, we examined how elevated extracellular K+ concentrations might impact MYXV infection.
Here, we show that elevated concentrations of extracellular K+ directly alter the cellular endosomal/lysosomal compartment. These changes result in a delay of MYXV infection by preventing the release of viral virions from the endosomes. Additionally, mathematical modeling predicts that the concentrations of extracellular K+ needed to impact viral infection can be readily obtained in viral lesions as long as the rate of infection is not outpaced by the diffusion of K+ in the interstitial fluid. This work suggests that the localized tissue damage caused by lytic infection might transiently alter K+ homeostasis, which could influence viral infection and pathogenesis.
RESULTS
Elevated extracellular K+ inhibits MYXV infection in vitro.
K+ channel function and intracellular K+ homeostasis are known to play major roles in the replication cycles of numerous viruses (13–18, 24, 25). However, the impact of extracellular K+ concentrations on viral infection is much less well studied. Critically, it has recently been shown that high levels of localized necrosis can increase the extracellular [K+] by up to 10-fold (11). We were therefore interested in how these elevated levels of extracellular K+ might influence the replication of the model poxvirus MYXV. To address this question, we incubated a variety of cells in growth medium supplemented with excess K+ and asked how this impacted the replication of a green fluorescent protein (GFP)-expressing MYXV construct (26). We observed that cells incubated with elevated K+ and then infected with MYXV at multiplicities of infection (MOIs) ranging from 0.01 to 0.1 displayed lower rates of infection than cells incubated in normal medium after 24 h (Fig. 1A). Additionally, while this reduced infection rate could be overcome with higher MOIs (Fig. 1A), these cells still displayed a reduced intensity of virally derived GFP (vGFP) (Fig. 1B) and contained low numbers of new infectious progeny virus at 24 h postinfection compared to infection of cells grown in control medium (Fig. 1C). Titration of the extracellular [K+] indicated that the reduction in viral progeny observed at 24 h began to occur in cells grown in media containing ~25 to 50 mM K+ (Fig. 1D), which is in line with the concentrations previously observed in highly necrotic tissues (11). Critically, these changes in viral replication could not be explained by elevated extracellular K+ killing host cells since incubation in medium containing 50 mM K+ for up to 72 h did not visually alter cellular morphology or cell viability, although a slight reduction in the number of total cells was observed, likely due to a decreased division rate (Fig. 2A to D). Additionally, while the presence of elevated extracellular K+ profoundly reduced the size of MYXV foci, this effect could be rescued by placing infected cells back into normal growth medium (Fig. 2E). Taken together, these data suggest that increases in the extracellular [K+] can negatively influence MYXV replication.
FIG 1.
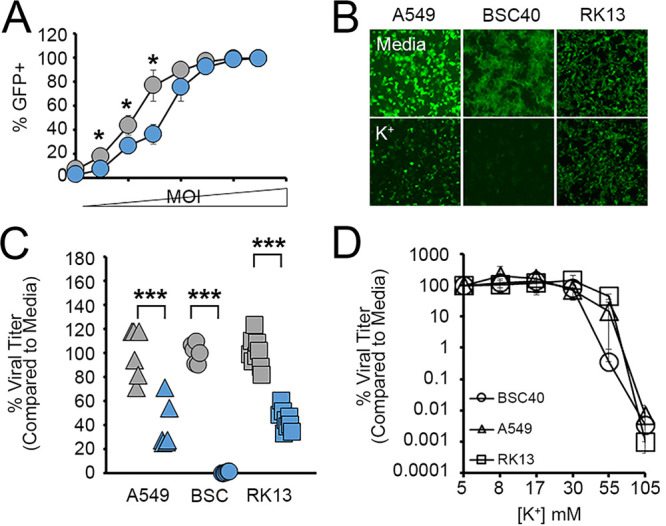
Elevated extracellular K+ inhibits MYXV infection. (A) BSC40 cells were incubated in medium containing 5 mM or 50 mM K+ as described in the text and then infected with vGFP at MOIs ranging from 0.001 to 3.0. Twenty-four hours later, rates of infection were determined by quantitating the percentage of cells expressing any level of GFP using flow cytometry. Significance was determined using Student’s t test (*, P < 0.05). (B and C) The indicated cell types were incubated in either regular medium (5 mM K+) (shown in gray) or medium containing 50 mM K+ (shown in blue) for 24 h and then infected with MYXV expressing GFP at an MOI of 10. (B) Infection was qualitatively assessed by visualizing the intensity of virally derived GFP. (C) Twenty-four hours later, the number of new infectious progeny was determined using titer assays. The data shown are normalized to the values for cells grown in medium containing 5 mM K+ and are representative of results from 1 to 3 individual experiments. Significance was determined using Student’s t test (***, P < 0.001). (D) Cells were incubated with media containing the indicated concentrations of K+ for 24 h and then infected with MYXV at an MOI of 10. At 24 h postinfection, the number of infectious progeny in each sample was determined. The data shown are normalized to the values for cells grown in medium containing 5 mM K+ and are representative of results from 2 to 3 individual experiments.
FIG 2.

Elevated extracellular K+ does not compromise cellular viability. (A to D) The indicated cell types were incubated in either regular medium (5 mM K+) (shown in gray) or medium containing 50 mM K+ (shown in blue) for 72 h. Cultures were then assayed for cellular morphology (A), cell viability using either trypan blue exclusion (B) or flow cytometry (C), or total cell numbers (D). The data shown are representative of results from at least two individual experiments for each assay. Significance was determined using Student’s t test (*, P < 0.05). (E) A549 cells were incubated in either regular medium (5 mM K+) or medium containing 50 mM K+ for 24 h and then infected with vGFP at an MOI of 0.001. The spread of individual foci was assayed using a fluorescence microscope for 48 h, after which cells initially incubated with 50 mM K+ were either transferred into normal medium (5 mM K+) or again placed into medium containing 50 mM K+. The growth of individual foci was then tracked for an additional 72 h.
Elevated extracellular K+ delays the early stages of the MYXV replication cycle.
In order to identify how increased extracellular K+ concentrations impacted MYXV infection, we next queried various stages of the viral replication cycle in the presence and absence of elevated K+. The results indicated that increasing the extracellular [K+] had no discernible impact on the binding of MYXV to the cell surface (Fig. 3A and B). Additionally, at high MOIs, where the previously observed reduced rates of infection caused by elevated K+ (Fig. 1A) could be overcome, virtually all infected cells displayed detectable GFP fluorescence by 4 h postinfection, suggesting that the virus successfully entered cells and initiated early gene expression (Fig. 3C and D). However, while amplification of the viral genome could be observed in cells grown in control medium beginning at ~4 h postinfection, this synthesis was delayed in cells incubated with elevated K+ (Fig. 3E). Consistent with this delay in genome synthesis, elevated extracellular K+ postponed the appearance of high levels of virally derived GFP (which is indicative of a switch to viral late gene expression) as well as the production of new infectious progeny by several hours (Fig. 3C to F). Taken together, these data suggest that elevated extracellular K+ impacts the MYXV replication cycle both by reducing the ability of the virus to initiate infections of cells (Fig. 1A) and by delaying genome synthesis and the production of new infectious progeny (Fig. 3E and F).
FIG 3.

Elevated extracellular K+ delays MYXV genome synthesis. (A and B) The indicated cell types were incubated in either regular medium (5 mM K+) (shown in gray) or medium containing 50 mM K+ (shown in blue) for 24 h. Cells were then incubated at 4°C with high numbers of MYXV virions containing an M094-Venus fusion protein (34). The binding of virus to the cell surface was then visualized using either fluorescence microscopy (A) or flow cytometry (B). The data shown in panels A and B are representative of results from two individual experiments for each assay. (C and D) Cells were incubated in either regular medium (5 mM K+) or medium containing 50 mM K+ for 24 h and then infected with MYXV at an MOI of 5. At the indicated times postinfection, cells were harvested, and the expression of virally derived GFP was quantitated using flow cytometry. (C) Representative histograms showing changes in GFP intensities over time. (D) Quantitation of GFP intensities. The data shown in panels C and D are representative of results from three individual experiments. Significance was determined at each time point using Student’s t test (*, P < 0.05; N.S., not significant). HPI, hours postinfection; MFI, mean fluorescence intensity. (E) RK13 cells were incubated in either regular medium (5 mM K+) or medium containing 50 mM K+ for 24 h and then infected with MYXV at an MOI of 5. At the indicated times postinfection, DNA was extracted from the cells, and the abundance of viral genomes was analyzed using rt-PCR. Significance was determined at each time point using Student’s t test (***, P < 0.001). The data shown are representative of results from two individual experiments. (F) The indicated cell types were incubated in either regular medium (5 mM K+) or medium containing 50 mM K+ for 24 h and then infected with vGFP at an MOI of 5. At the indicated times postinfection, cells were harvested, and the abundance of infectious viral progeny was quantitated using standard viral titration assays. The data shown are representative of results from at least three individual experiments for each cell line. Significance was determined at the indicated time points using Student’s t test (***, P < 0.001).
Elevated extracellular K+ alters the cellular endosomal compartment.
Our previous findings indicated that high levels of extracellular K+ negatively impact MYXV replication early after infection. However, incubation of purified MYXV virions with K+ did not detectably reduce the production of new infectious virus (Fig. 4A), suggesting that ionic imbalance was likely acting on the host cells. Based on these data, we subsequently asked what impact increasing the extracellular [K+] might have on cellular functions. To address this question, cells were incubated in either normal medium or medium containing 50 mM K+ for 24 h and then assayed for changes to their overall transcriptome using RNA sequencing (RNAseq). The results indicated that cells grown with elevated extracellular K+ displayed robust changes in gene expression corresponding to various cellular pathways (Fig. 4B). Interestingly, these pathways included endosomal trafficking (Fig. 4C), which is central to the early stages of poxviral infection (27, 28). We therefore investigated whether increased extracellular K+ might alter the cellular endosomal/lysosomal compartments. We observed that elevated extracellular K+ caused cells to take up increased amounts of fluorescein isothiocyanate (FITC)-dextran beads, suggesting increased levels of phagocytosis (Fig. 4D). Additionally, cells incubated with high levels of K+ displayed a significant mislocalization of early endosomes and a loss of endosomal/lysosomal segregation (Fig. 4E). These data suggest that elevated extracellular K+ concentrations alter the endosomal/lysosomal compartment within cells.
FIG 4.

High extracellular K+ alters the cellular endosomal/lysosomal compartment. (A) MYXV virions were incubated in either regular medium (gray) or medium containing 50 mM K+ (blue) for 1 h. Virions were then immediately used to infect BSC40 cells at an MOI of 10. At the indicated times postinfection, cells were harvested, and the amount of new infectious progeny present was quantitated using viral focus-forming assays. (B and C) ID8 cells were incubated in either regular medium (5 mM K+) (shown in gray) or medium containing 50 mM K+ (shown in blue) for 24 h. RNA was then extracted and used for transcriptomic analysis. (B) PCA comparing the overall RNA profiles of cells grown in either medium or medium with elevated K+. (C) GSEA analyzing the changes to the endosomal/lysosomal pathway induced by exposure to elevated K+. (D) A549 cells were incubated in either regular medium (5 mM K+) or medium containing 50 mM K+ for 24 h. Cells were then fed FITC-dextran beads, and the phagocytic uptake of these beads was quantitated using flow cytometry. The data shown are representative of results from more than three individual experiments. Significance was determined at the indicated time points using Student’s t test (***, P < 0.001). (E) Huh7 cells were incubated in either regular medium (5 mM K+) or medium containing 50 mM K+ for 18 h and then stained for the subcellular localization of EEA1 and LAMP1.
Elevated extracellular K+ causes delayed release of MYXV virions from endosomes.
Consistent with elevated K+ causing changes to the cellular endosomal compartment, we observed that the majority of the signal obtained from the K+-specific dye potassium-binding benzofuran isophthalate (PBFI) was found in punctate spots distributed throughout the cytoplasm (Fig. 5A). Interestingly, the intensity of this staining increased in the presence of elevated extracellular K+ (Fig. 5B), suggesting that increasing the extracellular ionic content resulted in a localized increase in K+ ions within one or more vesicular compartments. Critically, endocytosed MYXV particles could be found colocalized with these PBFI-positive (PBFI+) vesicles (Fig. 5C), with the extent of colocalization being highest ~90 min after virion binding (Fig. 5D), a time that is consistent with the kinetics of endocytosed MYXV being found in the late endosomes. In order to determine whether the delay in MYXV replication caused by the elevated extracellular [K+] might have to do with its presence in these K+-containing vesicles, we finally asked whether ionic imbalance might alter the trafficking of MYXV particles into or out of the endosomal compartment. The results indicated that 90 min after virion binding, MYXV particles could be observed still bound to the cell surface, in membrane-enclosed vesicular compartments, as well as fully released into the cytoplasm (Fig. 5E). Cells incubated with elevated extracellular K+, however, displayed fewer virions both at the cell surface and released into the cytoplasm and an increase in virions contained in membrane-bound vesicles (Fig. 5F). Taken together, these data suggest that MYXV traffics through a K+-containing vesicular compartment and that increased extracellular K+ concentrations delay the release of virions from this compartment.
FIG 5.

Elevated extracellular K+ inhibits the release of MYXV from the endosomes. (A and B) A549 cells were incubated in either regular medium (5 mM K+) (shown in gray) or medium containing 50 mM K+ (shown in blue) for 24 h. Cells were then stained with PBFI dye and analyzed for either the subcellular localization of K+ (A) or the intensity of PBFI fluorescence (B). (C and D) RK13 cells were incubated in either regular medium (5 mM K+) or medium containing 50 mM K+ for 24 h. Cells were then incubated with high numbers of MYXV virions containing an M094-Venus fusion protein (34) for the indicated times and then stained with PBFI dye. The colocalization of PBFI- and Venus-containing viral particles was then assayed using confocal microscopy. (C) Representative images of cells taken 90 min after virion adsorption. (D) Pearson colocalization scores of PBFI and Venus fluorescence obtained from images taken at the indicated time points. (E and F) Cells were incubated in either regular medium (5 mM K+) or medium containing 50 mM K+ for 24 h and then infected with MYXV at an MOI of 100. Thirty minutes after infection, cells were fixed and processed for electron microscopy. (E) Representative images showing viral virions that are bound at the cell surface (B), contained in membranous vesicles (E), or completely exposed in the cytoplasm (C). (F) Quantitation of the percentage of virions found at each subcellular location in the indicated cell types. The data shown in panel F represent data for more than 100 analyzed virions from each cell type from two independent experiments.
Mathematical modeling predicts that localized viral infection will alter extracellular K+ homeostasis.
It has been previously shown that the high rates of localized necrosis found within solid tumors can increase the levels of extracellular K+ within a confined space (11). However, the potential impact of localized necrosis on extracellular K+ in noncancerous settings has never been explored. We hypothesized that the localized cell death caused by a lytic viral infection might also increase the concentration of extracellular K+ within a confined space. Unfortunately, attempts to directly measure the extracellular [K+] inside a MYXV lesion in rabbits were technically unsuccessful (our unpublished observations). Therefore, to test the plausibility of our hypothesis, we created a mathematical model based on the growth of primary MYXV lesions in rabbits (see Table S1 in the supplemental material). To validate this model, five susceptible Oryctolagus rabbits were inoculated intradermally with 1,000 focus-forming units (FFU) of MYXV and then euthanized at 7 days postinfection. Primary lesions were excised, sectioned, and histologically stained for virally expressed GFP. The diameter of the GFP+ region was then measured with calipers (Fig. 6A, top) and compared to the size of the infected region predicted by our in silico model (Fig. 6A, bottom). Consistent with our previously observed experimental results (22), our model predicted that the diameter of the infected region within a myxomatosis lesion would expand over time (Fig. 6A). Critically, the predicted size of the infected region correlated extremely well with the empirically observed size of the GFP+ region within actual myxomatosis lesions 7 days after infection. In addition to the lesion increasing in size, the model also predicted that the majority of the infection in this lesion would be found throughout the central core (Fig. 6B), a phenotype that appeared consistent with the observed localization of MYXV-derived GFP (Fig. 6A, top). Critically, in addition to accurately modeling the expansion of a primary MYXV lesion, our model also predicted that the extracellular [K+] within this lesion would increase over time, exceeding 50 mM (the point at which extracellular K+ began to inhibit MYXV replication in vitro) at between 60 and 120 h postinfection (Fig. 6C).
FIG 6.
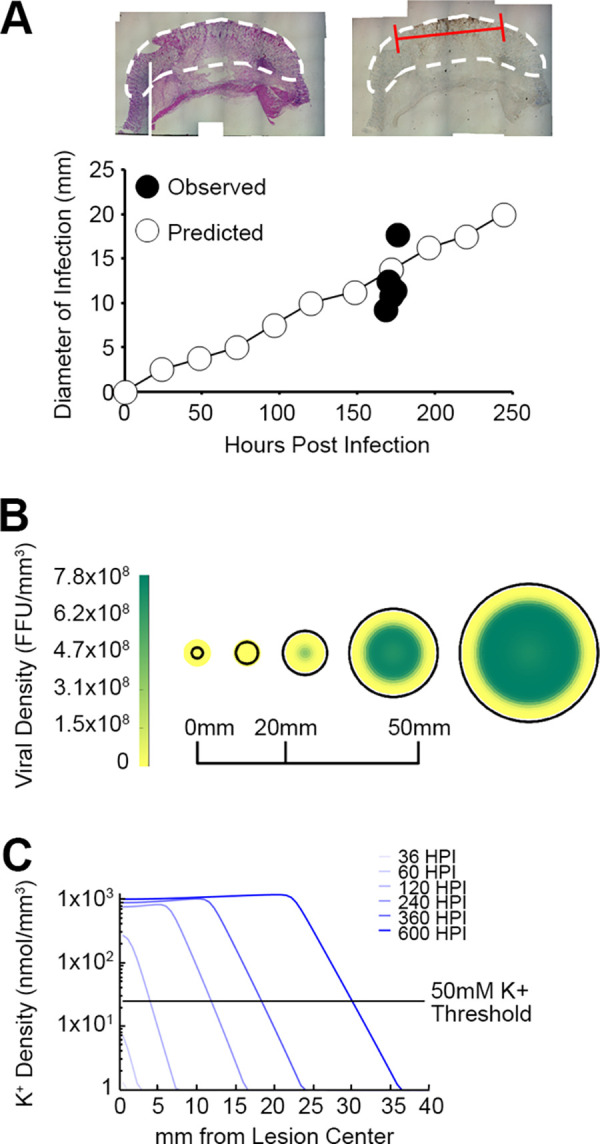
Mathematical modeling predicts that localized MYXV infection will result in an increased extracellular [K+]. (A) Comparison of the diameter of the region of MYXV infection predicted by our mathematical model to that which has been previously experimentally observed. Predicted results were calculated by computationally predicting the diameter at which 1% of the total cells were infected with virus every 24 h. The observed results were calculated by infecting susceptible Oryctolagus rabbits intradermally with vGFP for 7 days, harvesting the resulting primary lesions, histologically staining these lesions for the presence of virally derived GFP, and then measuring the diameter of the GFP+ region with calipers. The size of the phenotypically inflamed lesion is marked with a white line, while the area of GFP+ infection is marked with a red line. The data shown represent the results of a reanalysis of existing samples from a previous study (22). (B) Predicted localization of free MYXV virions within a viral lesion. (C) Predicted extracellular [K+] within a viral lesion. The extracellular [K+] throughout the viral lesion was predicted at the indicated time points. The experimentally determined threshold at which extracellular K+ begins to inhibit MYXV infection (50 mM) is indicated for reference.
Since our previous modeling predicted that extracellular K+ would increase within a viral lesion, we finally wished to determine whether this increased K+ could potentially impact MYXV pathogenesis. To address this, we modified our previous in silico model such that elevated extracellular K+ would both inhibit the initiation of infection and delay the production of new infectious progeny and the death of infected cells. Interestingly, despite elevated extracellular K+ being present within the lesion and multiple stages of the viral replication cycle being negatively impacted by this K+, the results from this new model indicated that the overall lesion size remained relatively unchanged (Fig. 7A). However, the inhibitory effects of extracellular K+ significantly reduced both the density of infectious virions and the total number of these viruses found in the virion. Additionally, elevated K+ restricted the spatial localization of these infectious particles more tightly into the middle of the viral lesion (Fig. 7B). Taken together, these predictive results suggest that lytic viral lesions can increase the local concentration of extracellular K+ and that this can modify, but not prevent, pathogenesis.
FIG 7.
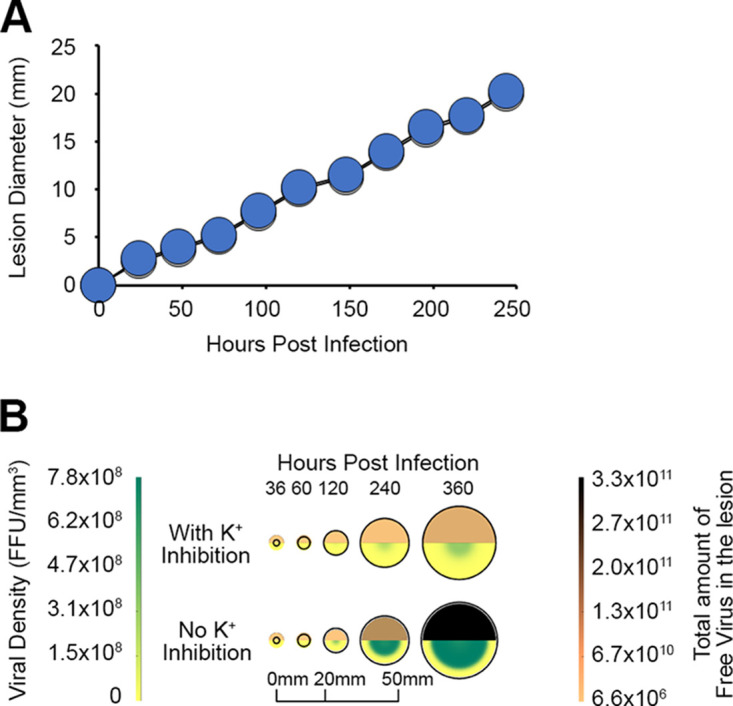
Modeling predicts that elevated extracellular K+ will alter the abundance of MYXV particles within lesions. (A) Comparison of predicted viral lesion diameters with (gray) and without (blue) inhibition by K+. (B) Impact of K+ inhibition on viral distribution within a lesion with and without inhibition by K+. Both the viral density (bottom of each circle) and the total amount of infectious virus contained within the entire lesion (top of each circle) were calculated throughout the infected lesion with and without inhibition by elevated extracellular K+.
DISCUSSION
Numerous studies have examined the potential roles of K+ channels and K+ homeostasis in viral infection (13–18). Very few of these studies, however, have examined how changes in the extracellular [K+] might impact infection (21). Interestingly, this limited body of literature suggested that elevated extracellular K+ concentrations can enhance the infectivity of bunyavirus by directly acting on the viral spike protein to enhance endosomal fusion (21). In contrast, our data suggest that elevated extracellular K+ concentrations negatively impact infection with MYXV in multiple cell lines (Fig. 1). In vitro, this inhibition occurs only at relatively high concentrations of extracellular K+ (above 25 mM). Critically, these concentrations are unlikely to be found for any prolonged period of time in any systemic location since elevating serum K+ above 6.0 to 7.0 mM is lethal in most eukaryotes (10). However, the recently reported observation that extracellular K+ concentrations in the necrotic cores of solid tumors can exceed 50 mM (11) suggests that these ionic levels can transiently exist within confined spaces. It is therefore critical to note that our mathematical modeling predicts that localized lytic infection can rapidly increase the extracellular [K+] above 50 mM (Fig. 6). It is also noteworthy that our model does not contain any variables that are unique to MYXV. These modeling results therefore suggest that a variety of lytic viruses, or even pathogenic bacteria, might increase the local extracellular [K+] during infection. Interestingly, despite directly delaying the time that it takes to produce new infectious progeny and initiate a viral burst, our model predicted that elevated extracellular K+ will not significantly reduce the overall size of a viral lesion (Fig. 7). Instead, the effect is largely limited to altering the abundance and distribution of virions within the lesion. While the impact of this on actual pathogenesis is unclear, it is easy to envision it playing a major role in viral transmission for pathogens such as MYXV whose spread by insect vectors is dependent on intralesional viral titers. Unfortunately, attempts to directly confirm these predictions in vivo by sampling the [K+] from the interstitial fluid of MYXV lesions were unsuccessful due to the lack of established sampling methods in rabbits (our unpublished observations). It will therefore be critical for future experiments to validate that lytic infections truly increase the local K+ concentrations within confined lesions.
In the case of MYXV, our data show that incubation of viral virions with elevated K+ has no effect on subsequent infection, while incubation of noninfected cells altered both phagocytosis and the stability of the endosomal/lysosomal pathway (Fig. 4 and 5). These data suggest a model in which elevated extracellular K+ delays MYXV infection through an indirect mechanism involving alterations to the cellular vesicular compartment, which slows the release of viral particles. Critically, this effect on treated cells is not simply due to reductions in cell viability since cells incubated with high K+ concentrations remain viable, and the removal of the elevated K+ ions rescues the infectivity of already adsorbed virus (Fig. 2). Unfortunately, our data do not identify any specific molecular changes within the endosomes that might directly inhibit the release of MYXV particles. It is attractive to hypothesize that changes in the extracellular [K+] might affect endosomal/lysosomal pH since K+ has been shown to play an essential role in ionic balancing during endosomal/lysosomal acidification (29, 30). However, our experiments failed to detect any changes in endosomal/lysosomal pH following incubation of cells in medium containing elevated extracellular K+, suggesting that this is not the mechanism through which viral release is inhibited (see Fig. S1 in the supplemental material). We do clearly observe a loss of endosomal/lysosomal stability; however, additional experiments will be needed to elucidate the mechanisms mediating this loss and whether it is responsible for the observed inhibition of MYXV replication. Interestingly, since cellular endocytosis plays a major role in the infection cycles of numerous viruses, it is easy to hypothesize that elevated extracellular K+ would inhibit all of these viral families. This hypothesis, however, is contradicted by previously reported results with bunyavirus (21), in which K+ plays a key role in the function of the viral spike protein. More work will therefore be needed to elucidate the role of elevated extracellular K+ in additional viral families.
Additionally, while our data clearly demonstrate that an elevated extracellular [K+] inhibits the release of the MYXV particle from the endosomes, it is unclear whether it might also impact additional steps in the viral replication cycle downstream of endosomal release. In this context, it is noteworthy that the functions of both the RNA and DNA polymerases from vaccinia virus have been shown to be impacted by the [K+] in vitro (31, 32). It is therefore entirely possible that increasing the extracellular [K+] can inhibit or delay the MYXV replication cycle through multiple distinct mechanisms. Unfortunately, attempts to bypass the block in endosomal release through pH-mediated fusion were unsuccessful as MYXV does not appear to possess this entry mechanism (33).
MATERIALS AND METHODS
Cell lines and reagents.
A549 (ATCC CCL-185), BSC40 (ATCC BRL-2761), and RK13 (ATCC CCL-37) cells were obtained directly from the American Type Culture Collection (Manassas, VA). Cells were grown in standard Dulbecco’s modified Eagle’s medium (DMEM) (Mediatech, Manassas, VA) supplemented with 10% fetal bovine serum (VWR, Radnor, PA) and 1× penicillin-streptomycin-glutamine (Corning, Corning, NY). Cells were passaged for <6 months prior to use and checked quarterly for mycoplasma contamination by PCR. High-K+ DMEM was made by dissolving KCl in medium at the desired concentration. The cell-permeant dye potassium-binding benzofuran isophthalate (PBFI) was obtained from Thermo Fisher (Waltham, MA) and used according to the manufacturer’s recommendations. Cellular viability was determined using the Cell-Titer 96 nonradioactive cell proliferation assay [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT)] (Promega, Madison, WI) and Live/Dead fixable dyes (Thermo Fisher, Waltham, MA). The following antibodies were used in these studies: EEA1 (catalog number 3288; Cell Signaling Technologies) and LAMP1 (catalog number 9091; Cell Signaling Technologies) antibodies.
Virus and viral infections.
All MYXV constructs used are based on the Lausanne strain. Viral constructs expressing GFP (26) as well as an M093/Venus fusion protein (vM093/Venus [34]) were described previously. All viruses were amplified in BSC40 cells and purified using gradient centrifugation as previously described (35). Unless otherwise noted, infections were carried out by adsorbing virus to cells for 30 min at a multiplicity of infection (MOI) of 10, removing unbound virus, and then replacing the viral inoculum with fresh medium. Cells treated with high-K+ medium were typically incubated in this medium for 24 h prior to infection. The viral titer was determined by releasing intracellular virions using mechanical disruption, serially diluting the resulting supernatants onto confluent BSC40 cells, and counting the resulting viral foci after 48 h. Viral binding was assayed by incubating cells at 4°C with high MOIs of vM093/Venus and then measuring the intensity of Venus fluorescence using flow cytometry.
Real-time PCR.
Real-time PCR (rt-PCR) analysis was conducted as previously described (36). In short, viral genomic DNA was extracted from cells using the RNeasy kit (Qiagen, Hilden, Germany). The abundance of genomic DNA was then quantitated using a CFX96 real-time system (Bio-Rad, Hercules, CA). Data were analyzed with the accompanying Bio-Rad CFX Manager software. Amplified products were analyzed by gel electrophoresis to ensure specificity. The PCR primers used in this study are shown in Table 1.
TABLE 1.
Primer sequences used in this study
| Target gene | Orientationa | Sequence |
|---|---|---|
| M053 | FWD | ATCGTAAGCGTCGACTGCAA |
| REV | ATCCAGATTTCCCGGCGTTT | |
| M084 | FWD | CGGGTCGATTCGATTCCTCA |
| REV | GACGATAGGCGGGTTCATGT | |
| M105 | FWD | AAGCGTACCGTTTCCAACA |
| REV | GTGGGATTCGATAACACTACGG | |
| M140 | FWD | ACGCCCGACAGTTCTTTAG |
| REV | GAGGAGGACACTCTTTCCC |
FWD, forward; REV, reverse.
Immunofluorescence.
Cells were seeded onto coverslips coated with 5 μg/mL fibronectin (Sigma-Aldrich, St. Louis, MO) at 40,000 cells per well. After treatment, the cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA), permeabilized with 0.2% Triton X-100, and blocked in 0.2% bovine serum albumin (Sigma-Aldrich, St. Louis, MO). Cells were then sequentially incubated with primary and secondary antibodies and mounted onto glass slides. Cells were imaged using a Leica SP8 confocal microscope (Carl Zeiss AG, Oberkochen, Germany). The final composite images were created using ImageJ (National Institutes of Health, Bethesda, MD).
Electron microscopy.
Electron microscopy analysis of MYXV-infected cells was conducted as previously described (22). In short, cells were fixed in 3% formaldehyde plus 2% glutaraldehyde, rinsed in 0.1 M cacodylate buffer, and then fixed in 2% aqueous osmium tetroxide. After rinsing in distilled water, the pellets were dehydrated through a series of graded ethyl alcohol washes and then embedded in a mixture of propylene oxide and EMbed 812 (Electron Microscopy Sciences, Hatfield, PA). Pellets were then thin sectioned, double stained with uranyl acetate in methanol and Reynolds lead citrate, and viewed using a JEOL 1010 transmission electron microscope.
RNA sequencing and bioinformatics.
RNAseq analysis was conducted using ID8 cells. Cells were incubated in normal DMEM or DMEM supplemented with 50 mM K+ for 24 h. Following incubation, cells were harvested, and total RNA was extracted using an RNeasy kit (Qiagen, Hilden, Germany). RNAseq was then performed by Novogene (Cambridge, UK) using an Illumina sequencer. Raw RNAseq data were downloaded in compressed FASTQ format, mapped against the Mus musculus reference cDNA sequence (GRCm38.p5; ENSEMBL release v98), quantified at the transcript level with kallisto (v0.46.0), and summarized at the gene level using BioMART’s gene-to-transcript mapping (accessed on 22 January 2019) via tximport (v1.15.6). Principal-component analysis (PCA) was conducted using finalized transcript counts. Gene set enrichment analysis (GSEA) was conducted by identifying differentially expressed genes having a log2 fold change (Log2FC) of ≥2 and a P value of <0.05 and subsequently analyzing the enrichment of these genes in the KEGG process for endosomal transport.
Mathematical modeling of MYXV infection.
We modeled the virus infection process using a reaction-diffusion system that incorporates the various effects of elevated extracellular K+ observed in our in vitro experiments. In short, the model predicts that myxoma virus infects cells and that, after some delay (37), these infected cells die, releasing both new infectious viruses as well as K+ into the extracellular matrix. The newly produced infectious viruses infect neighboring cells according to the law of mass action as they diffuse through the matrix (37, 38). K+ also diffuses through the extracellular matrix. We use an approximation of the Heaviside function to incorporate a threshold value of K+ beyond which virus production, viral infectivity, and infected cell death are impacted. Since the model involves both intracellular and extracellular values for both potassium and virus concentrations, we incorporated a conversion factor between the intracellular and extracellular spaces and developed a computer algorithm for solving the model system. Parameter values were estimated from both our experimental results as well as the existing literature (8, 22, 37, 39–41). Although our mathematical model is based on well-established infectious disease models (38, 42–45), it is the first to incorporate the potential inhibitory effects of elevated K+. It should be considered a first-order approximation of the in vivo processes since the dynamics and effects of K+ on viral infection are complex, and numerous additional factors that might influence viral replication and spread (such as the presence of inflammatory and antiviral immune responses) are not modeled. The specific details of the mathematical model and computational methods are included in the supplemental material.
Footnotes
Supplemental material is available online only.
Contributor Information
Eric Bartee, Email: ebartee@salud.unm.edu.
Derek Walsh, Northwestern University Feinberg School of Medicine.
REFERENCES
- 1.Bennett MV. 1964. Nervous function at the cellular level. Annu Rev Physiol 26:289–340. 10.1146/annurev.ph.26.030164.001445. [DOI] [PubMed] [Google Scholar]
- 2.Pongs O. 2008. Regulation of excitability by potassium channels. Results Probl Cell Differ 44:145–161. 10.1007/400_2007_032. [DOI] [PubMed] [Google Scholar]
- 3.Conway EJ. 1957. Nature and significance of concentration relations of potassium and sodium ions in skeletal muscle. Physiol Rev 37:84–132. 10.1152/physrev.1957.37.1.84. [DOI] [PubMed] [Google Scholar]
- 4.Cox RH. 2005. Molecular determinants of voltage-gated potassium currents in vascular smooth muscle. Cell Biochem Biophys 42:167–195. 10.1385/CBB:42:2:167. [DOI] [PubMed] [Google Scholar]
- 5.Laskowski M, Augustynek B, Kulawiak B, Koprowski P, Bednarczyk P, Jarmuszkiewicz W, Szewczyk A. 2016. What do we not know about mitochondrial potassium channels? Biochim Biophys Acta 1857:1247–1257. 10.1016/j.bbabio.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 6.Rozov A, Khusainov I, El Omari K, Duman R, Mykhaylyk V, Yusupov M, Westhof E, Wagner A, Yusupova G. 2019. Importance of potassium ions for ribosome structure and function revealed by long-wavelength X-ray diffraction. Nat Commun 10:2519. 10.1038/s41467-019-10409-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber LA, Hickey ED, Nuss DL, Baglioni C. 1977. 5′-terminal 7-methylguanosine and mRNA function: influence of potassium concentration on translation in vitro. Proc Natl Acad Sci USA 74:3254–3258. 10.1073/pnas.74.8.3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmer BF. 2015. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 10:1050–1060. 10.2215/CJN.08580813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee-Lewandrowski E, Burnett R, Lewandrowski K. 2002. Electrolytes and acid-base balance, p 347–365. In McClatchey KD (ed), Clinical laboratory medicine, 2nd ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 10.Palmer BF, Clegg DJ. 2016. Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ 40:480–490. 10.1152/advan.00121.2016. [DOI] [PubMed] [Google Scholar]
- 11.Eil R, Vodnala SK, Clever D, Klebanoff CA, Sukumar M, Pan JH, Palmer DC, Gros A, Yamamoto TN, Patel SJ, Guittard GC, Yu Z, Carbonaro V, Okkenhaug K, Schrump DS, Linehan WM, Roychoudhuri R, Restifo NP. 2016. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature 537:539–543. 10.1038/nature19364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stanford MM, McFadden G. 2007. Myxoma virus and oncolytic virotherapy: a new biologic weapon in the war against cancer. Expert Opin Biol Ther 7:1415–1425. 10.1517/14712598.7.9.1415. [DOI] [PubMed] [Google Scholar]
- 13.Ashbrook AW, Lentscher AJ, Zamora PF, Silva LA, May NA, Bauer JA, Morrison TE, Dermody TS. 2016. Antagonism of the sodium-potassium ATPase impairs chikungunya virus infection. mBio 7:e00693-16. 10.1128/mBio.00693-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi B, Gatti PJ, Haislip AM, Fermin CD, Garry RF. 1998. Role of potassium in human immunodeficiency virus production and cytopathic effects. Virology 247:189–199. 10.1006/viro.1998.9251. [DOI] [PubMed] [Google Scholar]
- 15.Hackstadt T, Mallavia LP. 1982. Sodium and potassium transport in herpes simplex virus-infected cells. J Gen Virol 60:199–207. 10.1099/0022-1317-60-2-199. [DOI] [PubMed] [Google Scholar]
- 16.Hover S, King B, Hall B, Loundras EA, Taqi H, Daly J, Dallas M, Peers C, Schnettler E, McKimmie C, Kohl A, Barr JN, Mankouri J. 2016. Modulation of potassium channels inhibits bunyavirus infection. J Biol Chem 291:3411–3422. 10.1074/jbc.M115.692673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levine AS, Bond PH, Scala AR, Eaton MD. 1956. Studies on the relationship of potassium to influenza virus multiplication. J Immunol 76:386–392. [PubMed] [Google Scholar]
- 18.Punch EK, Hover S, Blest HTW, Fuller J, Hewson R, Fontana J, Mankouri J, Barr JN. 2018. Potassium is a trigger for conformational change in the fusion spike of an enveloped RNA virus. J Biol Chem 293:9937–9944. 10.1074/jbc.RA118.002494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Norrie DH, Wolstenholme J, Howcroft H, Stephen J. 1982. Vaccinia virus-induced changes in [Na+] and [K+] in HeLa cells. J Gen Virol 62(Part 1):127–136. 10.1099/0022-1317-62-1-127. [DOI] [PubMed] [Google Scholar]
- 20.Thiel G, Moroni A, Blanc G, Van Etten JL. 2013. Potassium ion channels: could they have evolved from viruses? Plant Physiol 162:1215–1224. 10.1104/pp.113.219360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hover S, Foster B, Fontana J, Kohl A, Goldstein SAN, Barr JN, Mankouri J. 2018. Bunyavirus requirement for endosomal K+ reveals new roles of cellular ion channels during infection. PLoS Pathog 14:e1006845. 10.1371/journal.ppat.1006845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolfe AM, Dunlap KM, Smith AC, Bartee MY, Bartee E. 2018. Myxoma virus M083 is a virulence factor which mediates systemic dissemination. J Virol 92:e02186-17. 10.1128/JVI.02186-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richards CH, Mohammed Z, Qayyum T, Horgan PG, McMillan DC. 2011. The prognostic value of histological tumor necrosis in solid organ malignant disease: a systematic review. Future Oncol 7:1223–1235. 10.2217/fon.11.99. [DOI] [PubMed] [Google Scholar]
- 24.McClenaghan C, Hanson A, Lee SJ, Nichols CG. 2020. Coronavirus proteins as ion channels: current and potential research. Front Immunol 11:573339. 10.3389/fimmu.2020.573339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Voss TG, Fermin CD, Levy JA, Vigh S, Choi B, Garry RF. 1996. Alteration of intracellular potassium and sodium concentrations correlates with induction of cytopathic effects by human immunodeficiency virus. J Virol 70:5447–5454. 10.1128/JVI.70.8.5447-5454.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bartee MY, Dunlap KM, Bartee E. 2017. Tumor-localized secretion of soluble PD1 enhances oncolytic virotherapy. Cancer Res 77:2952–2963. 10.1158/0008-5472.CAN-16-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moss B. 2006. Poxvirus entry and membrane fusion. Virology 344:48–54. 10.1016/j.virol.2005.09.037. [DOI] [PubMed] [Google Scholar]
- 28.Moss B. 2016. Membrane fusion during poxvirus entry. Semin Cell Dev Biol 60:89–96. 10.1016/j.semcdb.2016.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishida Y, Nayak S, Mindell JA, Grabe M. 2013. A model of lysosomal pH regulation. J Gen Physiol 141:705–720. 10.1085/jgp.201210930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Courville P, Mindell JA. 2014. Roles of cytoplasmic ions in lysosomal acidification. Biophys J 106:147A. 10.1016/j.bpj.2013.11.847. [DOI] [Google Scholar]
- 31.Munyon W, Paoletti E, Grace JT, Jr.. 1967. RNA polymerase activity in purified infectious vaccinia virus. Proc Natl Acad Sci USA 58:2280–2287. 10.1073/pnas.58.6.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lazier C, Helleiner CW. 1970. The DNA polymerases of vaccinia virus-infected animal cells. J Gen Virol 8:149–152. 10.1099/0022-1317-8-2-149. [DOI] [PubMed] [Google Scholar]
- 33.Villa NY, Bartee E, Mohamed MR, Rahman MM, Barrett JW, McFadden G. 2010. Myxoma and vaccinia viruses exploit different mechanisms to enter and infect human cancer cells. Virology 401:266–279. 10.1016/j.virol.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan WM, Bartee EC, Moreb JS, Dower K, Connor JH, McFadden G. 2013. Myxoma and vaccinia viruses bind differentially to human leukocytes. J Virol 87:4445–4460. 10.1128/JVI.03488-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smallwood SE, Rahman MM, Smith DW, McFadden G. 2010. Myxoma virus: propagation, purification, quantification, and storage. Curr Protoc Microbiol Chapter 14:Unit 14A.1. 10.1002/9780471729259.mc14a01s17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flores EB, Bartee MY, Bartee E. 2020. Reduced cellular binding affinity has profoundly different impacts on the spread of distinct poxviruses. PLoS One 15:e0231977. 10.1371/journal.pone.0231977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friedman A, Tian JP, Fulci G, Chiocca EA, Wang J. 2006. Glioma virotherapy: effects of innate immune suppression and increased viral replication capacity. Cancer Res 66:2314–2319. 10.1158/0008-5472.CAN-05-2661. [DOI] [PubMed] [Google Scholar]
- 38.Murray JD. 2002. Interdisciplinary applied mathematics. Mathematical biology II: spatial models and biomedical applications, vol 18, 3rd ed. Springer, New York, NY. [Google Scholar]
- 39.Harned H, Nuttall R. 1949. The differential diffusion coefficient of potassium chloride in aqueous solutions. J Am Chem Soc 71:1460–1463. 10.1021/ja01172a090. [DOI] [PubMed] [Google Scholar]
- 40.Chizhov AV, Sanin AE. 2020. A simple model of epileptic seizure propagation: potassium diffusion versus axo-dendritic spread. PLoS One 15:e0230787. 10.1371/journal.pone.0230787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luby-Phelps K. 2000. Cytoarchitecture and physical properties of cytoplasm: volume, viscosity, diffusion, intracellular surface area. Int Rev Cytol 192:189–221. 10.1016/s0074-7696(08)60527-6. [DOI] [PubMed] [Google Scholar]
- 42.Brauer F. 2017. Mathematical epidemiology: past, present, and future. Infect Dis Model 2:113–127. 10.1016/j.idm.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Daley DJ, Gani J. 2005. Epidemic modelling: an introduction. Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 44.Hethcote H. 2000. The mathematics of infectious diseases. SIAM Rev 42:599–653. 10.1137/S0036144500371907. [DOI] [Google Scholar]
- 45.Nowak MA, May RM. 2001. Virus dynamics: mathematical principles of immunology and virology. Oxford University Press, Oxford, United Kingdom. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1, Tables S1 and S2, and supplemental text. Download jvi.01294-22-s0001.pdf, PDF file, 0.4 MB (459KB, pdf)