

ABSTRACT
Getah virus (GETV) mainly causes disease in livestock and may pose an epidemic risk due to its expanding host range and the potential of long-distance dispersal through animal trade. Here, we used metagenomic next-generation sequencing (mNGS) to identify GETV as the pathogen responsible for reemerging swine disease in China and subsequently estimated key epidemiological parameters using phylodynamic and spatially-explicit phylogeographic approaches. The GETV isolates were able to replicate in a variety of cell lines, including human cells, and showed high pathogenicity in a mouse model, suggesting the potential for more mammal hosts. We obtained 16 complete genomes and 79 E2 gene sequences from viral strains collected in China from 2016 to 2021 through large-scale surveillance among livestock, pets, and mosquitoes. Our phylogenetic analysis revealed that three major GETV lineages are responsible for the current epidemic in livestock in China. We identified three potential positively selected sites and mutations of interest in E2, which may impact the transmissibility and pathogenicity of the virus. Phylodynamic inference of the GETV demographic dynamics identified an association between livestock meat consumption and the evolution of viral genetic diversity. Finally, phylogeographic reconstruction of GETV dispersal indicated that the sampled lineages have preferentially circulated within areas associated with relatively higher mean annual temperature and pig population density. Our results highlight the importance of continuous surveillance of GETV among livestock in southern Chinese regions associated with relatively high temperatures.
IMPORTANCE Although livestock is known to be the primary reservoir of Getah virus (GETV) in Asian countries, where identification is largely based on serology, the evolutionary history and spatial epidemiology of GETV in these regions remain largely unknown. Through our sequencing efforts, we provided robust support for lineage delineation of GETV and identified three major lineages that are responsible for the current epidemic in livestock in China. We further analyzed genomic and epidemiological data to reconstruct the recent demographic and dispersal history of GETV in domestic animals in China and to explore the impact of environmental factors on its genetic diversity and its diffusion. Notably, except for livestock meat consumption, other pig-related factors such as the evolution of live pig transport and pork production do not show a significant association with the evolution of viral genetic diversity, pointing out that further studies should investigate the potential contribution of other host species to the GETV outbreak. Our analysis of GETV demonstrates the need for wider animal species surveillance and provides a baseline for future studies of the molecular epidemiology and early warning of emerging arboviruses in China.
KEYWORDS: zoonotic pathogens, Getah virus, genomic surveillance, next-generation sequencing, phylodynamics, phylogeography
INTRODUCTION
Approximately 18% of emerging infectious diseases that affect humans originate from wild animals or livestock (1–4). In many of these host reservoir species, emerging viruses appear to be well adapted, with little or no evidence of clinical disease. However, when these viruses spillover into humans, the effects can sometimes be devastating (5, 6). Because livestock can often act as a conduit for pathogen spillover into susceptible human populations, research on emerging viral diseases is focused on livestock infections that often occur due to contact with wild animals (7). For example, the swine industry couples high-density farming with international trade, thus generating a high risk for emerging virus transmission and the potential for global spread (8). Moreover, the swine industry will increasingly represent such a risk due to its constant growth to fulfill a high demand for pork. A disease outbreak caused by a new or emerging virus may incur substantial economic burden and also endanger human health due to close human contact with pigs.
Pigs have also been shown to be a significant source of zoonotic viruses, such as Nipah virus in Malaysia (9) and influenza A virus (H1N1) that caused the “swine-origin influenza” pandemic (10). For instance, the 2009 H1N1 influenza pandemic in Mexico arose from viruses circulating in pigs for more than a decade, with a virus that originated from Eurasia owing to an expansion of influenza A virus diversity in swine resulting from long-distance live swine trade (11). The importance of pigs as a source of emerging viruses has also recently been illustrated by four cases of human acute encephalitis that were associated with a variant strain of pseudorabies virus, with all the patients having had close occupational contact with pigs (12). An efficient approach to detect both known and unexpected novel viruses in a single test is therefore crucial for emerging viral outbreak identification and management in swine worldwide.
Our ability to investigate and monitor outbreaks of potential zoonotic pathogens depends on an understanding of their ecology and evolution in reservoir hosts. Metagenomic next-generation sequencing (mNGS) technologies are particularly suitable for identifying viral etiologies. The analysis of the virome, often referred to as the assemblage of viruses in metagenomic studies, can detect known and novel viruses in environmental, human, or animal samples (13, 14). mNGS is very well suited for early diagnosis and surveillance of novel porcine viral diseases due to its high accuracy, fast response (generating large data in a short amount of time), and high sensitivity (15). By coupling the pathogen genomes assembled from mNGS with phylodynamic and phylogeographic analyses, researchers are able to achieve a comprehensive picture of the evolutionary and dispersal history of zoonotic pathogens of epidemiological importance and how it might have been shaped by external factors. In particular, recent methodological developments allow for phylodynamic and phylogeographic approaches to test epidemiological hypotheses (16–18). For instance, the skygrid coalescent model (19) has been extended to allow for testing associations between the evolution of the virus effective population size over time and time series covariates (20). Furthermore, discrete (21) or continuous (22, 23) phylogeographic reconstructions can be exploited to examine how covariates may explain the dispersal process of viral lineages (16, 24, 25). The combination of mNGS technology and state-of-the-art analytical methods allows researchers to rapidly identify emerging viruses, gain insight into the epidemiological and environmental factors that shape their evolution, transmission, and disease burden, and ultimately provide a basis for epidemic control and prevention.
Since May 2019, more than 1,500 piglets died suddenly in four intensive pig farms in the Chinese provinces of Guangxi, Henan, Hubei, and Shandong. However, the causal pathogen could not be identified by conventional diagnostic techniques. Eventually, metagenome sequencing of tissue samples from diseased piglets in our laboratory linked those cases to porcine Getah virus (GETV). GETV is an arbovirus and a member of the genus Alphavirus and can cause disease in domestic animals. For example, horses can experience fever, rashes, edema of the hindlegs, and lymph node enlargement, while infected piglets exhibit depression, tremors, hind limb paralysis, diarrhea, high mortality, and abortions (26–28). GETV has a linear, positive-sense single-stranded RNA genome of about 11.5 kb that encodes nine viral proteins (nsP1 to nsP4, E1 to E3, C, and 6K). E2 is the main glycoprotein that binds to host cell receptors when initiating cell entry, whereas the E1 glycoprotein is required for pH-triggered membrane fusion within acidified endosomes (29). Previous research has shown that GETV gradually evolved within a relative broad host range (30). Natural infections have been reported in mosquitoes, swine, cattle, horses, and blue foxes (31–34), causing reproductive disorders, fever, neurological symptoms, and death in mammals, and thus suggesting a wide distribution of susceptible animals in China. In addition, GETV neutralizing antibodies have been detected in goats, cattle, horses, pigs, and other animals (35), as well as in humans (36), suggesting a potential public health risk. In the past 50 years, numerous reemergences of alphaviruses such as chikungunya virus (CHIKV) have been documented in over 60 countries throughout Asia, Africa, Europe, and the Americas, causing over 10 million human infections (37) with irregular intervals of 2 to 20 years between outbreaks. Likewise, Venezuelan equine encephalitis, which is caused by another mosquito-borne alphavirus that commonly infects equines and humans, is endemic in at least 12 countries in South and Central America and associated with severe symptoms or death (38, 39). Therefore, a sudden outbreak of alphaviruses poses not only a threat to the breeding industry but also a potential threat to public health (37, 40).
In this study, we used NGS to identify the causative agent of an outbreak occurring in the Chinese swine population, that is, GETV, which was not included in routine PCR-based surveillance due to its previous low prevalence. This outbreak is associated with a considerable impact on public, veterinary, and livestock health. Because the virus was only sporadically detected before 2016 in China, we considered this sudden surge in GETV cases as a reemerging infectious disease. Therefore, we subsequently performed large-scale GETV PCR-based screening on previously and recently collected samples and found a number of GETV cases starting in 2018. We then sequenced and analyzed the E2 genes of 79 strains (including 16 full genomes) collected from China since 2016 that greatly expanded the existing GETV sequence data. We detailed and demonstrated the advantages of our approach for the risk assessment of unknown disease outbreaks. Specifically, our genomic surveillance aimed at (i) analyzing the genetic diversity and amino acid mutations associated with the ongoing GETV outbreak, (ii) reconstructing the dispersal history of GETV lineages in continental China, (iii) determining which factors were related to the dynamics of GETV genetic diversity over time, and (iv) investigating the impact of environmental factors on the dispersal dynamics of GETV lineages.
RESULTS
Pathogen identification and retrospective epidemiological survey.
All sick pigs in Guangxi, Henan, Hubei, and Shandong had suffered from respiratory, digestive, and neural symptoms before dying. We performed necropsy of the dead pigs and collected the lungs, intestines, and other organs for a pathological section as well as for conventional viral pathogen PCR detection. As shown in Fig. 1A, the autopsy showed swollen mesenteric lymph nodes, and thinned intestinal walls. The samples tested negative for PCR identification of nine common viruses (porcine reproductive and respiratory syndrome virus [PRRSV], classical swine fever virus [CSFV], pseudorabies virus [PRV], porcine epidemic diarrhea virus [PEDV], porcine deltacoronavirus [PDCoV], porcine transmissible gastroenteritis virus [TGEV], porcine teschovirus [PTV], porcine kobuvirus [PKV], and porcine circovirus type 2 [PCV2]), and these pathogens were discarded as causal agents of disease. NGS and subsequent bioinformatic analyses identified GETV as the etiological agent, a pathogen not included in routine PCR-based surveillance due to its previous low prevalence (Fig. 1B). Overall, the abundance of GETV was the highest among the four infected farms compared to that of other viruses, although some GETV-positive samples were coinfected with lower concentrations of picobirnavirus or kobuvirus. In addition, we examined NGS libraries obtained in 2016 before the outbreak from the same GETV-positive swine farms in Guangxi and Shandong provinces but did not identify any GETV infection. To determine when GETV had reemerged and started to spread in China, we used Sanger sequencing and NGS to retrospectively analyze laboratory samples collected between 2016 and 2021. In addition to the identification of GETV in laboratory-preserved swine samples, it is worth noting that we also detected GETV-positive nucleic acid and sequenced the GETV E2 gene in mosquitoes, cattle, and dogs. A total of 79 samples from 17 provinces in China were detected to be positive for GETV. Among them, a total of 16 complete genomic sequences were obtained, along with an additional 63 E2 sequences. Furthermore, we also collected 104 case reports of GETV infections in a variety of animals from publications that did not release any sequence (see Fig. S2A in the supplemental material). We found that before 2015, there was only one case of swine infection in China, and that was in Henan province in 2012. Since 2015, the number of GETV cases has been increasing year by year, including infections of livestock, blue foxes and dogs, with a wide geographical range of infections (northeast, northwest, and the entire south of China). Since 2017, GETV has expanded rapidly in geographical distribution, with cases in mammals also appearing in northwest (Xinjiang) and northeast China; but eastern, central, and southern China are still the main areas of endemicity (Fig. S2A). Of note, when the cases were grouped according to seasons, GETV infection was found to occur all year round, but with a higher number of positive cases in summer and the lowest in winter (Fig. S2B). This suggested that GETV was more likely to cause disease during the warmer season, when the virus can replicate in, and be transmitted by, mosquito vectors.
FIG 1.

Isolation and characterization of GETV. (A) GETV-infected piglets showing clinical features. From left to right: cyanosis, diarrhea, thinning of the intestinal wall, and lymphadenopathy. (B) Abundance of various viruses at the genus level in GETV-positive and -negative farms. The relative abundance of each virus in each library was estimated and normalized by the number of mapped reads per million total reads (RPM). To remove contaminations, we show only RPM above 1. Guangxi-2019, Henan-2020, Shandong-2019, and Hubei-2019 are GETV-positive farms. Guangxi-2016 and Shandong-2016 correspond to the two farms that were GETV-positive in 2019. (C) GETV was successfully isolated and verified by agarose gel electrophoresis. (D and E) Vero cells were infected with GETV-GX or GETV-HN (MOI = 0.001), and PK15 cells were infected with GETV-GX or GETV-HN (MOI = 0.1). Cytopathic changes were observed at 12, 24, 36, 48, 60, and 72 hpi. (F and G) Immunofluorescence of GETV-E2 (green) detected in infected Vero cells (F) and PK15 cells (G). Nuclei are stained blue with 4′,6-diamidino-2-phenylindole (DAPI). All fluorescent images were taken at ×20 magnification. (H and I) Growth of GETV-GX or GETV-HN in Vero (H) and PK15 (I) cultures. Viral titers were determined for samples (only medium) between 12 and 72 hpi in Vero cells. Data are expressed as the mean ± standard deviation (SD) of viral titers (log10 TCID50 per 0.1 ml) derived from three infected cell cultures.
Biological characterization of reemerging GETV strains.
Two strains of GETV, named HN and GX, were isolated by inoculating Vero cells with intestinal abrasive solution after filtration and plaque purification (Fig. 1C). As shown in Fig. 1D to G, PK15 or Vero cells inoculated with purified GETV-GX or GETV-HN showed a visible cytopathic effect (CPE) in the form of shrinking, rounding and detachment of cells at 48 h postinfection (hpi) compared to that of the control. A strong signal was observed using anti-E2 antibodies in fluorescence microscopy, indicating that PK15 or Vero cells are effectively infected by GETV-GX or GETV-HN. One-step growth curves demonstrated efficient virus growth in PK15 and Vero cells, with virus titers exceeding 108 50% tissue culture infective doses (TCID50)/0.1 ml at 48 hpi (Fig. 1H and I).
Of note, GETV can replicate in a variety of animal and primate cell lines, including human cell lines such as 293T and U251 (Fig. S3A and B), which suggests a potential to infect humans. In addition, GETV-GX was shown to be pathogenic in mouse models, which serve as effective models to explore the pathogenic molecular mechanism and tissue tropism of GETV (41, 42). GETV-GX was intracranially inoculated into 3-day-old ICR suckling mice with 25 μl of a 106.5 TCID50/0.1 ml GETV viral solution. In the infected group, the weight of the suckling mice ceased to increase after 24 h, and after 48 h, some suckling mouse began to die with hunched back, tremor, and difficulty in eating. All the suckling mice in the infected group died 80 h after inoculation (Fig. S3C to E).
Sequence, mutation, and selection analyses.
Analysis of all GETV genomes and E2 genes revealed no recombinant signal. More than 50 amino acid substitutions were observed between the recently obtained GETV viruses. Here, we explored the effect of seven amino acid substitutions in E1 and 20 substitutions in E2 relative to a prototype strain and found four interesting substitutions in the E2 gene, three of which are potential sites under positive selection (Table S1). Overall, selection pressure analysis revealed that the GETV E2 gene was under purifying selection, and only two amino acid sites, E2-86 and E2-323, were found to show evidence for positive selection across all three methods used (fixed-effects likelihood [FEL], fast unconstrained Bayesian approximation [FUBAR], and mixed effects model of evolution [MEME]). In addition, site E2-253 was also found to be subject to positive selection according to the FUBAR analysis (posterior probability = 0.986), and we found no evidence for positive selection on any individual lineage in the GETV phylogeny. The important mutations and potentially positively selected sites in the ectodomain are highlighted in the crystal structure of the E1/E2 dimer of the closely related CHIKV (43) (Fig. 2A). The four interesting mutations in E2 are also depicted in the trimeric E1/E2 spike, which is shown as a top view in Fig. 2B. Residue 323, which is characterized by a conservative Asp-to-Glu substitution, is exposed at the surface of the molecule near the membrane. It is thus unlikely to be involved in receptor binding or to act as an antibody epitope; its side chain does not form contacts with other amino acids. The His86Tyr substitution is located in the central cavity between E1/E2 dimers, which contains heparan sulfate-binding sites in many alphaviruses (44). Site 207 (Asn207His) is located in a loop at the edge of the spike and exposed at the cell surface (Fig. 2C). This region contains epitopes for cross-reactive neutralizing antibodies that compete with binding to the Mxra8 receptor in other alphaviruses (45, 46). Residue 253 is located at the base of the viral spike near E3; the side chain of Lys interacts with Tyr47 of E3 (Fig. 2D). Furthermore, in close vicinity of residue 253 are two other basic amino acids, Arg250 and Lys251, and the former forms an electrostatic interaction with Asp40 in E3. These amino acids are conserved in other alphaviruses, but Lys is replaced by Arg in some of GETV variants.
FIG 2.
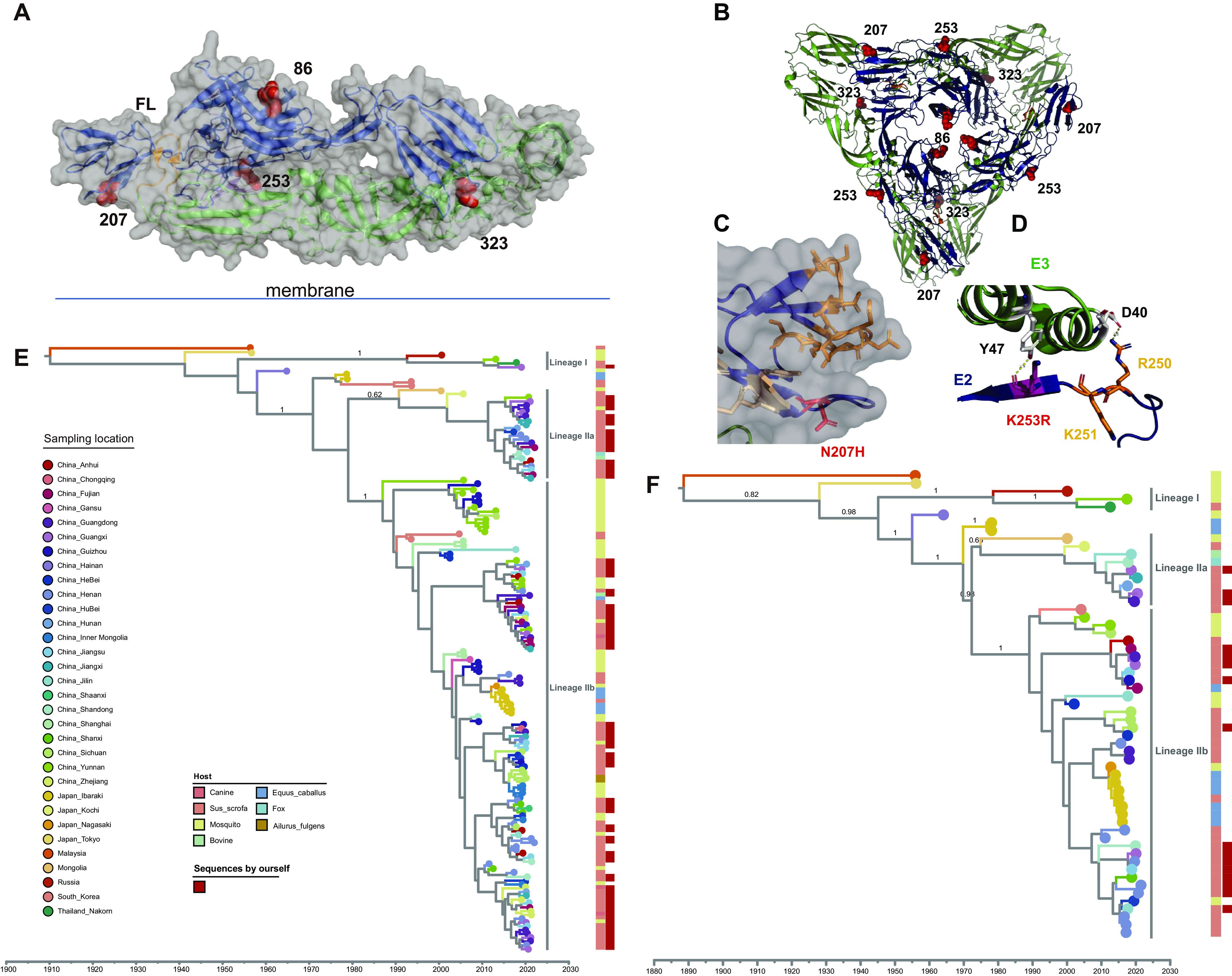
Mapping of amino acid substitutions and selected sites in E2 and evolution of GETV. (A) Structure of a heterodimer containing the E1 (green cartoon) and E2 (blue cartoon) subunit. The small E3 subunit (magenta cartoon) is still associated with E2. Amino acid exchanges are highlighted by red spheres. The horizontal line symbolizes the viral membrane, in which both proteins are anchored by a transmembrane region. FL, fusion loop in E1. (B) Top view of a hexameric spike composed of three E1 (green cartoon) and three E2 (blue) subunits. Positively selected and other interesting sites in E2 are highlighted by red spheres. (C) Detail of the E2 structure in a semitransparent surface projection showing the location of residue 207 as a red stick. Epitopes for antibodies that prevent binding of alphaviruses to the Mxra8 receptor are shown as orange sticks (46), and those for other broadly neutralizing antibodies are shown as beige sticks (45). (D) Detail of the interface of E2 (blue) with E3 (green) showing the location of the selected site 253 as a stick. Lys253 interacts with Tyr47 in the E3 subunit. Shown as sticks are also two other basic amino acids in the vicinity; one of them (Arg250) forms an ionic interaction with Asp40 in E3. After removal of E3 during virus entry, the three basic amino acids might form a heparan sulfate (HS)-binding site. The Lys253Arg conservative exchange might affect HS binding or removal of E3. The figures were created with PyMol from PDB files 3N40 (A, C, and D) and 2XFB (B). (E) Maximum clade credibility (MCC) tree based on the phylogenetic analysis of GETV E2 gene sequences. (F) MCC tree based on the phylogenetic analysis of whole GETV genome sequences. Red squares represent new sequences obtained in this study.
Phylogenetic, phylodynamic, and phylogeographic analyses.
By regressing root-to-tip divergences against sampling times, we confirmed the presence of a temporal signal for both the whole-genome and E2 gene maximum likelihood (ML) tree using the program TempEst, with a coefficient of determination (R2) equal to 0.68 and 0.31, respectively (Fig. S4). In the absence of clear criteria for genotyping of GETV, we refrain from providing a formal genotype classification. We used the program ClusterPicker v1.2.5 (47) to delineate lineages within both the ML trees based on E2 gene and genomic sequences, considering a bootstrap support of >90 at its most ancestral node and a genetic distance of <0.04 within it. Using ClusterPicker, we identified three lineages englobing all viruses sampled after 2000, which we refer to as lineages I, IIa, and IIb, and show them in the maximum clade credibility (MCC) trees (Fig. 2E and F). While lineage I has few representatives and contains two mosquito-borne GETV sequences and two swine-borne GETV sequences, lineages IIa and IIb are responsible for the major epidemic strains from pigs in China and for GETV from mosquitoes, cattle, blue foxes, horses, lesser panda, and canines. To infer the time of emergence of GETV, the time of the most recent common ancestor (TMRCA) of GETV and of each lineage was estimated based on whole-genome and E2 sequences. The TMRCA was estimated around 1880 (95% highest posterior density [HPD] = 1799 to 1943) for the complete genome data set, and around 1904 (1846 to 1947) for the E2 gene. Based on the analysis of the E2 gene, the estimated TMRCAs for the three lineages were 1990 (1972 to 2000), 1989 (1976 to 2000), and 1986 (1979 to 1991), respectively. The estimated divergence times for each lineage based on whole-genome sequences were similar to the E2 gene estimates. The mean nucleotide substitution rate (substitutions/site/year) estimated using the whole-genome data set of GETV was 3.19 (2.23, 4.18) ×10−4 substitutions/site/year and 6.26 (4.75, 7.76) ×10−4 substitutions/site/year using the E2 gene.
We also performed skygrid generalized linear model (skygrid-GLM) analyses to investigate which factors may be associated with the dynamics of GETV genetic diversity over time. In the case of the per capita meat consumption covariate, we inferred a mean effect size of 0.16 with a 95% HPD interval of (0.01, 0.30). Because the 95% HPD interval excludes zero, there is strong support of a positive relationship between the viral effective population size and per capita meat consumption, meaning that an increase in meat consumption was associated with an increase in the effective size of the viral population. On the other hand, the skygrid-GLM effect size coefficients for all other covariates have 95% HPD intervals that include zero, suggesting a lack of evidence of a relationship between the viral effective population size and each of the four remaining covariates (Fig. S5). In particular, we inferred a mean effect size of 0.8 (–0.78, 2.35) for the temperature, a mean effect size of 0.06 (–0.12, 0.22) for the precipitation, a mean effect size of 0.06 (–0.03, 0.14) for the annual forest area, and a mean effect size of 0.0004 (–0.0009, 0.0016) for the pork production, which thus indicated that those covariates were not associated with inferred variations in the effective population size of the virus.
The demographic reconstruction resulting from the skygrid-GLM analysis with the per capita meat consumption covariate is shown in Fig. 3: the white trajectory represents the mean log-transformed viral effective population size, and its corresponding 95% HPD interval region is shown in light blue. This figure also includes per capita meat consumption (shown as a red line) as well as the standard skygrid reconstruction that is based only on genetic sequence data (shown as an orange curve corresponding to the mean log-transformed effective population size, with the associated 95% HPD interval region in light orange). Notably, the mean demographic trajectory inferred by the skygrid-GLM analysis follows the trajectory of the covariate more closely. Further, the 95% HPD interval region inferred using the skygrid-GLM is narrower than, and almost entirely contained within, the 95% HPD interval region inferred with the standard skygrid approach, yielding a more precise demographic reconstruction and revealing a higher level of viral genetic diversity from 2015 onward.
FIG 3.

Skygrid reconstructions. The dark orange line and shaded light orange region represent the mean log-transformed viral effective population size and its 95% highest posterior density (HPD) interval region, respectively, inferred using a standard skygrid approach based solely on the analysis of sequence data. The white line and shaded light blue region represent the mean log-transformed viral effective population size and its 95% HPD interval region, respectively, inferred using a skygrid-GLM approach based on the analysis of both sequence data and per capita meat (pork, beef, mutton) consumption covariate data. The per capita meat consumption is represented by a red line. The skygrid-GLM analysis yields an effect size coefficient with a mean of 0.16 and a 95% HPD interval (0.01, 0.30), supporting a positive association between the viral effective population size and per capita meat consumption.
The discrete phylogeographic reconstruction coupled with a GLM analysis does not identify support for particular predictors of the dispersal frequency of GETV lineages among Chinese provinces, including the live swine trade. Indeed, only the sampling sizes at the province of origin and at the province of destination are associated with Bayes factor values >20, which correspond to strong statistical supports (48). Nevertheless, we found that the Henan province in central China and the eastern region in China should be the one of hubs for GETV spread (Fig. S6).
The continuous phylogeographic reconstruction does not allow us to trace the precise origin of the spread of GETV lineages, because the uncertainty associated with the location inferred for the root of the tree is relatively pronounced (Fig. 4). However, the reconstructed dispersal history of GETV lineages clearly highlights that some southern and eastern Chinese provinces (Guangxi, Guangdong, Jiangxi, Fujian, and Zhejiang) were more recently colonized (later than 2015) (Fig. 4). Taking advantage of the continuous phylogeographic reconstruction, we have estimated the weighted dispersal velocity of GETV lineages as follows: 151.0 (110.7 to 203.2) km/year when considering the entire phylogenetic tree, 139.4 (99.3 to 192.3) km/year when considering only phylogenetic branches occurring before 2015 (<2015), and 157.8 (112.2 to 216.3) km/year when considering only phylogenetic branches occurring after the beginning of 2015 (>2015). While the median value estimated for <2015 is slightly lower than the median value for >2015, their 95% HPD intervals largely overlap. Similarly, we did not identify that more recent (>2015) long-distance lineage dispersal events tended to be associated with relatively higher dispersal velocity (i.e., smaller MCC phylogenetic branch durations for similar geographic distances travelled by those branches) (Fig. S6).
FIG 4.
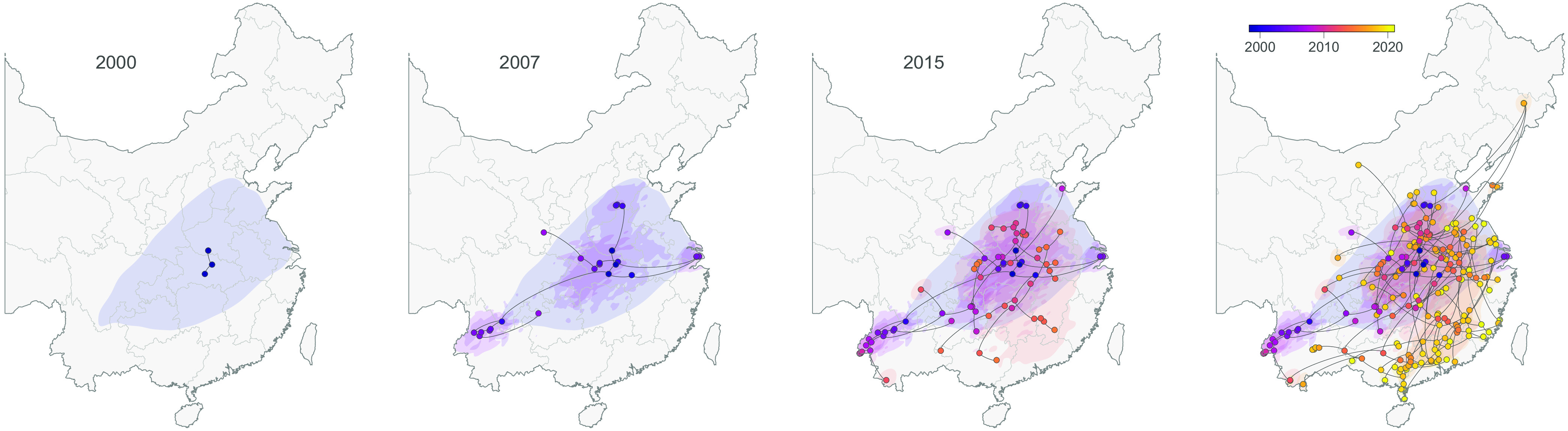
Dispersal history of GETV lineages in China as inferred by a continuous phylogeographic analysis: maximum clade credibility (MCC) tree and 80% highest posterior density (HPD) regions reflecting the uncertainty related to the phylogeographic inference and based on 1,000 trees subsampled from the posterior distribution. MCC tree nodes are colored according to their time of occurrence, and 80% HPD regions were computed for successive time layers and then superimposed using the same color scale reflecting time. In addition to the overall continuous phylogeographic reconstruction, we also mapped the dispersal history of GETV inferred until three years in the past, i.e., 2000, 2007, and 2015, which allows visualization of the progression of the virus spread.
We further tested whether lineage dispersal locations tended to be associated with specific environmental conditions. In particular, we started by computing the E statistic, which measures the mean environmental values at tree node positions. These values were extracted from raster (geo-referenced grids) that summarized the different environmental factors to be tested (Fig. S1). The analyses of the impact of environmental factors on the dispersal location of viral lineages reveal that sampled GETV lineages have tended to avoid circulating in areas with higher altitude and to preferentially circulate within areas associated with relatively higher mean annual temperature and pig population density (Fig. S1 and Table S2).
The analyses of the impact of environmental factors on the dispersal velocity of viral lineages indicate that none of the environmental variables appears to significantly impact the dispersal velocity of GETV lineages: when treated either as conductance or resistance factors and with both path models considered, none of the tested environmental factor is associated with both a positive Q distribution and a Bayes factor support >20. This overall result thus indicates that none of these environmental variables improve the correlation between branch durations and spatial distances (here approximated by the environmental distance computed on a uniform “null” raster), this correlation being already relatively high: R2 = 0.21 (0.08 to 0.39) when spatial distances are computed with the least-cost path model, and R2 = 0.13 (0.04 to 0.27) when spatial distances are computed with the Circuitscape path model. In other words, our results reveal that among the environmental factors that we tested, the spatial distance remains the best predictor of the duration associated with GETV lineage dispersal events.
DISCUSSION
Most alphaviruses circulate between specific hematophagous mosquito vectors and susceptible vertebrate hosts, some of which are major public health threats and result in disasters to humans upon spillover (49). GETV is a member of the Alphavirus genus, and while some serologic data and cell culture experiments suggest that humans are a potential host for GETV (36, 50), clear evidence for this possibility is still lacking. Up to now, epidemiological surveillance studies and available GETV sequences from swine have been rare (28, 51, 52). In this study, we performed a state-of-the-art genomic surveillance using mNGS coupled with phylodynamic and phylogeographic analyses. We found a large amount of GETV in dead pig samples and identified its link to an outbreak among swine herds in China. We also showed that GETV has a broader host range than previously anticipated, which complicates prevention and control because of its diverse reservoir and multiple hosts. We analyzed the genetic diversity, dispersal history, and the external factors that may have impacted the spatial spread of the virus in the early stage of an outbreak/reemergence in a Chinese pig herd.
We highlight that the current reemergence of GETV can be divided into three main lineages that primarily evolved and spread in livestock, and which are geographically widespread. Interestingly, the relatively strong geographical clustering observed in some early mosquito sequences may be related to the limited long-distance travel of mosquitoes or caused by a lack of early sequence samples in livestock, as we found only two recent lineage I sequences in swine. The results of the selection analysis showed that the E2 gene was mostly subject to purifying selection. This is consistent with the widely supported “trade-off” hypothesis for mosquito-borne alphaviruses, i.e., alternate replication in two distinct hosts (vertebrate and invertebrate) limits the evolution of arboviruses, as enhanced fitness in one host may be detrimental to replication in the other host (53). In addition, the estimated nucleotide substitution rate of GETV is similar to that of other alphaviruses, such as Ross River virus (RRV), which is most similar to GETV genetically (54). Of note, we found some evidence for potential adaptive evolution or important amino acid mutations such as His86Tyr, Lys253Arg, and Asn207His in the GETV currently circulating in China. Mapping mutations onto structural models revealed that two sites might affect binding of GETV to negatively charged heparan sulfate (HS). Different HS-binding sites, basic amino acids, have been identified for equine encephalitis virus (EEV), peripheral sites at the base and axial sites in the central cavity of the viral spike (55). The selected site His86Tyr in E2 of GETV was also exposed to the central cavity of the viral spike, but the exchange of the weakly basic His for the uncharged Tyr rather decreases HS binding. HS-mediated attachment usually increases virus replication in cell culture but, depending on the virus, either increases or decreases virulence in vivo (44, 56). The location of the Lys253Arg site corresponds to a peripheral HS-binding site in EEV (55). Lysine 253 as well as other basic residues in the vicinity interacts with amino acids in the E3 subunit. After removal of E3, which detaches from E2 upon virus entry, these basic residues might bind to HS. The Lys253Arg substitution, although conservative, might directly affect the HS interaction. Alternatively, it could facilitate or hinder the detachment of E3, which is, in other alphaviruses, a prerequisite for binding to the cellular receptor and hence for viral infectivity (45, 46, 57). The other important mutation, Asn207His, is located at the surface of E2. Epitopes for broadly neutralizing antibodies, which prevent virus attachment to the Mxra8 receptor, are located in the same region (45, 46). Mxra8 has been identified as an entry receptor for a variety of alphaviruses, but whether this is also the case for GETV has not been demonstrated so far (44, 58). Nevertheless, the Asn207His substitution may affect cell attachment of GETV. Importantly, previous examples of epidemic-enhancing mutations in alphaviruses include CHIKV adaptation to Aedes albopictus and Venezuelan equine encephalitis virus (VEEV) adaptive mutations that increase replication in horses (59). Hence, it is possible that GETV may have undergone a similar adaptive evolution in Chinese mammals that may lead to a public health risk. Therefore, in the wake of the recently sudden outbreak of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in the human population from an unknown animal origin, our results highlight the importance of genome surveillance and early warning of emerging infectious diseases in animals.
Moreover, by expanding the sample size of available GETV sequences, our study has enabled a more robust analysis of GETV evolutionary history (30). We estimate that the overall genetic diversity of GETV started to increase in ~2015, which corresponds to the time when the number of publications related to GETV started to increase. GETV has a wide geographical distribution in China, especially in the southern region as well as in areas used for livestock (see Fig. S1 in the supplemental material), which might be related to the distribution of its mosquito vector. Therefore, we examined the trajectory of the GETV effective population size over time and compared it to a number of different factors that have been hypothesized to be related to GETV population dynamics. Of note, we found evidence of a positive association between the viral effective population size and the per capita livestock meat consumption, but there is a lack of evidence of an association between the viral effective population size and pork production. While meat consumption is certainly not a direct cause of GETV population dynamics, the consumption of meat, such as cattle, sheep, and pigs, may greatly impact the frequency and volume of livestock transportation and distribution. This may, for instance, lead to potential contamination of transport vehicles. This result suggests that it may not simply be the breeding and trade of swine themselves that caused the GETV outbreak but rather that GETV may have been prevalent in other livestock for some time and may have been partly responsible for causing the outbreak in pigs via mosquitos. In addition, vaccine contamination is a potential cause of GETV transmission, as one study isolated live GETV virus in a commercial modified live vaccine against PRRSV (60).
Finally, by testing the association between environmental factors and the locations of lineage dispersal, we demonstrated that, overall, sampled GETV lineages have preferentially circulated under specific environmental conditions (higher temperature) and in regions with higher swine population density. In this respect, it is important to note that the highest mosquito density in China occurs near livestock farms (61) and that GETV incidence in mammals is significantly higher in summer than in winter. Furthermore, our landscape phylogeographic investigations of the association between viral lineage locations and local environmental conditions are known to be sensitive to sampling vagaries (62). Indeed, half of the environmental values used in the analysis are extracted at tip node locations, which are directly determined by the sampling effort. Therefore, this particular landscape phylogeographic analysis serves more as a description of the environmental conditions related to the dispersal locations of inferred viral lineages than an actual test of the impact of those conditions on their dispersal location (62). Nonetheless, our results provide insight into the evolution and diffusion of GETV that may help to prevent and control GETV infections in livestock. We recommend increased sample collection from and surveillance of a wider range of species and geographic regions, as uncovering the transmission routes and major sources of GETV in animals will help prevent future outbreaks of GETV disease among livestock and emergence in humans.
Our findings, as alluded to above, should be interpreted in the light of particular limitations. Uneven sampling may affect our results, and although our pig sampling covers well the sites we surveil, we may lack a large number of samples from other sites as well as from other livestock and wildlife animals. In addition, as detailed above, our phylogeographic analyses will be impacted by the sampling effort.
Our research is the first to integrate transcriptomic, genomic, phylogenetic, and landscape phylogeographic analyses of GETV, a virus that our analyses confirm as the cause of pig herd deaths in China. Although GETV cases have recently increased and this virus has become a target pathogen for surveillance, there is still no commercial vaccine for prevention and control. Early detection and monitoring of future pandemics will also require intensified viral surveillance and an understanding of how economic forces and livestock trade policies affect changes in animal movements and production practices that drive viral emergence. In the present study, we further demonstrate that modern technological platforms, such as NGS and phylodynamic analyses, allow more rapid identification of virus outbreaks than traditional methods, such as PCR and virus isolation. Furthermore, our study suggests that GETV could have the potential to cause further epidemics in the animal population, especially in areas with high livestock production in China.
MATERIALS AND METHODS
Collection and processing of veterinary samples.
(i) Sample collection from dead piglets of unknown etiology. From May 2019 to September 2020, more than 1,500 piglet deaths of unknown causes occurred at several pig farms in the Chinese provinces of Henan, Guangxi, Hubei, and Shandong. Before the piglets died, they showed clinical symptoms such as diarrhea, wasting, panting, skin rash, and some neurological symptoms. To investigate the cause of the piglet deaths, we collected swabs, feces, and tissue samples of dead piglets from these farms. During the transportation of samples, sufficient cryogenic ice packs were added to the carrying case to maintain a low-temperature environment.
(ii) Sample processing. Further processing of samples was carried out in a biosafety cabinet. Tissue samples from the lesion area of tissues were cut into small pieces using scissors for surgery and put in 1.5-ml autoclaved tubes. Sterile phosphate-buffered saline buffer solution was added after the fecal samples, and the mixture was divided into equal parts. Swab samples could be directly divided into 200 μl for each sterilized tube and stored. All samples were stored in the laboratory at −80°C before use.
Pathogen identification and retrospective epidemiological survey.
(i) Etiology investigation. According to the clinical symptoms of the dead piglets, several routine PCR-based pathogen detections were performed on the collected samples and the pathogens detected included porcine reproductive and respiratory syndrome virus (PRRSV), classical swine fever virus (CSFV), pseudorabies virus (PRV), porcine epidemic diarrhea virus (PEDV), porcine deltacoronavirus (PDCoV), porcine transmissible gastroenteritis virus (TGEV), porcine teschovirus (PTV), porcine kobuvirus (PKV), and porcine circovirus type 2 (PCV2). We also performed dissections on the dead piglets to further characterize the pathological changes of tissues and organs.
(ii) Next-generation sequencing and genome assembly. Total RNA was extracted using an RNA Clean & Concentrator kit (Zymokit) in accordance with the manufacturer’s instructions. An RNA library was built using TruSeq stranded total RNA sample preparation kits from Illumina (San Diego, CA) per their protocol. Ribo-Zero rRNA removal kits from Illumina (San Diego, CA) were used to remove rRNA. After fragmenting the RNA sample, cDNA synthesis, end repair, A-base addition, and ligation of Illumina-indexed adaptors, paired-end (150-bp reads) sequencing of the RNA library was performed on the NovaSeq platform (Illumina). For the genome assembly workflow, we refer to our previous research (63). All the GETV genomes from single samples were confirmed by Sanger sequencing.
(iii) RNA extraction, pathogen screening, and retrospective epidemiological survey of GETV in China. After further determination of pathogenicity by virus isolation, a retrospective investigation was conducted on some randomly selected samples from pigs with similar clinical symptoms collected between 2016 and 2021. Meanwhile, to explore GETV host diversity, we also monitored lab samples from mosquitoes, pet dogs and cattle collected over this period. All clinical samples were subsequently screened using primers designed according to the GETV sequences available in GenBank (https://www.ncbi.nlm.nih.gov). To obtain GETV genome and E2 glycoprotein sequences, the sample RNA was extracted using the Omega viral RNA isolation kit (Omega, USA), by strictly following the manufacturer's instructions. A HiScript II first-strand cDNA synthesis kit (Vazyme, China) was used for cDNA synthesis. Then, PCR was performed with the GETV detection primers. Samples identified as positive were selected and further subjected to an amplification reaction with Phanta max super-fidelity DNA polymerase (Vazyme, China) and a set of primers for GETV genome amplification that were designed based on reference genomes. Subsequently, purified PCR amplification products were sequenced by the Sanger dideoxy chain termination method.
Biological characterization of reemerging GETV strains.
Two GETV isolations were performed on samples originating from Guangxi and Henan provinces. The samples were ground with steel balls under aseptic clean conditions to homogenize them. The homogenized tissues were centrifuged at 16,500 × g for 10 min at 4°C. The supernatant was filtered through a 0.22-μm filter (Millipore, USA), diluted 1:50 with Dulbecco's modified Eagle medium (DMEM; Gibco, USA), and then inoculated onto Vero cells cultured in a monolayer. After incubation at 37°C for 1 h with 5% CO2, the inoculum was discarded and maintained in fresh DMEM containing 2% (vol/vol) fetal bovine serum (FBS; Biological Industries, Israel) and 1% (vol/vol) penicillin-streptomycin for 48 h. Continuous passage like this, when the HN isolate was passaged to the fifth generation and the GX isolate was passaged to the eighth generation, a plaque purification assay was performed to purify the virus. Next, virus isolates were confirmed by reverse transcription (RT)-PCR and an indirect immunofluorescence assay. Virus titration, one-step growth curve determination, immunofluorescence assay (IFA), and the mouse infection test of GETV are detailed in the supplemental material. All animal sample collections and animal experiments were approved by the Institutional Animal Care and Use Committee of Nanjing Agricultural University, Nanjing, China (no. SYXK2017-0007; February 2017).
Bioinformatic analyses.
(i) Analysis of genomic sequences. All available GETV genomic and E2 gene sequences were collected from the NCBI GenBank database (https://www.ncbi.nlm.nih.gov) up to December 2, 2021. Some sequences were removed because they (i) presented duplicate strain names for the same strain or (ii) corresponded to cell passages of the same original isolate. A total of 159 E2 genes and 59 genomic sequences were used for analysis, including 16 newly obtained genomic sequences and 63 additional newly obtained E2 genes (GenBank accession numbers MZ736724 to MZ736801, and Project ID: CNP0003775). E2 is the main protein that mediates virus entry into the host cell during infection and the key antigen eliciting virus-specific antibodies that affect the ability to spread infection (30, 64). Therefore, we used E2 as a molecular marker and preferentially sequenced E2 genes to understand the evolution and transmission of GETV during the outbreak. We performed multiple sequence alignments using the program MAFFT (version 7.475) (65) and manually edited the resulting alignment in MEGA (version 7) (66). We then performed statistical analyses of the variable amino acid sites to calculate the frequency of substitutions based on the structural and nonstructural protein regions of the GETV genome.
(ii) Analysis of recombination. We used the program RDP4 4.97 to perform recombination analysis of the genomic and E2 gene sequences (67). Seven methods, i.e., LARD (68), 3Seq (69), GENECONV (70), SiScan (71), Chimaera (71), MaxChi (72), and RDP (73), were used to detect recombinant events. We considered that p-values had to be below the 0.05 threshold for at least three of these seven methods for detection of an actual recombinant event (74).
(iii) Characterization of selective pressure. We used the program HyPhy (75) to estimate the nonsynonymous to synonymous substitution ratios (dN/dS). Specifically, the mixed effects model of evolution (MEME) (76), the single likelihood ancestor counting (SLAC) (77), the fast unconstrained Bayesian approximation (FUBAR) (78), and the fixed effects likelihood (FEL) (77) methods were used to estimate the positively selected amino acid sites during evolution. We also used the adaptive branch site random effects likelihood (aBSREL) method to identify specific branches under positive selection during the evolutionary process (79). A specific site was considered to have undergone positive selection when a posterior probability was >0.95 or a p-value was <0.05 for more than two methods. E2 and E1 protein cartoon structures were created with PyMol version 2.1.1 from PDB file 3N40 (Fig. 2A and C), 2XFB (Fig. 2B), or 3N41 (Fig. 2D) (43), which all contain the structure of an E1/p62 dimer of CHIKV that shows relatively high amino acid identity (E1, 60%; E2, 54%) and similarity (E1, 77%; E2, 68%) to E1 and E2 of GETV.
Phylogenetic and phylodynamic analyses.
(i) Phylogenetic and molecular clock analysis of the genomes and E2 gene sequences. A first phylogenetic analysis of both the genome and E2 gene sequences was performed with the maximum likelihood (ML) method implemented in RAxML (version 8.2.12) (80) using a general time-reversible (81) nucleotide substitution model with a discretized gamma distribution (82) to model rate heterogeneity among sites (GTR + Γ) and 1,000 bootstrap replicates to assess branch support. The temporal signal in our data set was visually assessed using TempEst (1.5.1) (83). Time-scaled phylogenetic inference was performed using BEAST 1.10.4 software (84) coupled with the high-performance computing library BEAGLE 3 (85). We assessed the best-fitting molecular clock model through marginal likelihood estimation (MLE) using path-sampling and stepping-stone estimation approaches. With these approaches, we identified the relaxed molecular clock with an underlying log-normal distribution as the best-fitting clock model. We specified a GTR+Γ nucleotide substitution model with three partitions for codon positions, an uncorrelated log-normal relaxed molecular clock, and a coalescent-based non-parametric skygrid prior for the phylogenetic tree, which enables inference of the effective population size over time (19). Two independent simulated Markov chains of 1 × 108 iterations converged to indistinguishable posterior distributions. Convergence and mixing were examined using the program Tracer 1.7 (86) with consideration of a burn-in of 10% of the total chain length. All parameter estimates were based on effective sample sizes over 200. A maximum clade credibility (MCC) tree summary was generated by TreeAnnotator (1.10.4) and visualized using Figtree 1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/). We used the program ClusterPicker v1.2.5 (47) for cluster analysis to delineate lineages.
(ii) Testing the impact of environmental factors on the viral diversity over time based on the E2 gene. We employed a coalescent-based skygrid generalized linear model (skygrid-GLM) framework (20) to examine the relationship over time between the viral effective population size and several time-varying covariates. The skygrid-GLM posits a log linear relationship between the effective population size and a given covariate and enables the inference of an effect size coefficient that quantifies this relationship. Importantly, under the skygrid-GLM, the effective population size and the effect size coefficient that relate it to a covariate are inferred jointly. This ensures that the effect size coefficient takes the uncertainty in the effective population size reconstruction into account. Finally, in the case of a clear relationship between the effective population size and a covariate, the skygrid-GLM model provides a demographic reconstruction that is based on genetic sequence data as well as the covariate data (in contrast to standard coalescent-based approaches that reconstruct the demographic history exclusively from genetic sequence data). We performed a separate analysis for each of the following five covariates: annual mean temperature, annual precipitation, annual forest area, pork production, and per capita meat (pork, beef, mutton) consumption. Annual mean temperature and annual precipitation data were retrieved from the WorldClim 2 database (https://worldclim.org/), annual forest area data were taken from the Food and Agriculture Organization of the United Nations (http://www.fao.org/home/en/), and pork production and per capita meat consumption data were collected from the National Bureau of Statistics of China (http://www.stats.gov.cn/).
(iii) Phylogeographic analyses based on the E2 gene. We performed both discrete (21) and continuous (22) phylogeographic reconstructions based on the alignment of E2 gene sequences, using the discrete trait and relaxed random walk diffusion models implemented in BEAST 1.10.4 (84), respectively, with the BEAGLE 3 library (85) to improve computational performance. For both kinds of phylogeographic inference, a distinct reconstruction was performed for each of the two major GETV clades (see below) but within the same BEAST analysis in order to share the estimation of substitution, molecular clock, coalescent, and diffusion model parameters. Specifically, we specified a flexible substitution model with a GTR+Γ parametrization, a relaxed clock model with rates drawn from an underlying log-normal distribution (87), and a flexible non-parametric skygrid coalescent model as the phylogenetic tree prior (19).
For the discrete phylogeographic analysis, we used the GLM extension of the discrete diffusion model (16) to jointly infer the dispersal history of lineages among discrete locations as well as the potential relationship of external predictors with the transition rates between pairs of locations. Notably, such a procedure allows investigation of the potential impact of external factors on the dispersal frequency of GETV lineages among discrete locations. For each tested predictor, the association is characterized by the GLM coefficients, and the related statistical support is estimated through a Bayes factor. In the context of this study, we treated the provinces of origin of each sample as discrete locations, and we tested the following predictors using the GLM approach: geographic distance among provinces (great-circle distance between province centroid points; in kilometers), pig trade among provinces (10,000 heads/km2) computed for three different time periods (2017 to 2018, 2017 to 2019, and 2020), the number of pigs slaughtered in the province of origin and the province of destination during two different time periods (2017 to 2018 and 2017 to 2019), the number of pigs raised in the province of origin and the province of destination during two different time periods (2017 to 2018 and 2017 to 2019), as well as the breeding density of pigs (1,000 heads/km2) in the province of origin and the province of destination during two different time periods (2017 to 2018 and 2017 to 2019). In addition, we also included as predictors the numbers of sequences sampled at the province of origin and at the province of destination, which allows assessment of the impact of sampling efforts on predictor support (16). For this analysis, a Markov chain Monte Carlo (MCMC) simulation was run for 1 × 109 iterations while sampling every 5 × 104 iterations and discarding the first 10% of trees sampled from the posterior distribution as burn-in. Convergence and mixing were examined using the program Tracer 1.7 (86), and each parameter estimate was based on an effective sample size (ESS) greater than 200.
For the continuous phylogeographic analyses, we used a gamma distribution to model the among-branch heterogeneity in diffusion velocity. The MCMC simulation was run for 2 × 109 iterations while sampling was done every 1 × 105 iterations and the first 10% of samples was discarded from the posterior distribution as burn-in. Convergence and mixing were again examined using Tracer, and all parameter estimates were associated with an ESS greater than 200. MCC trees were summarized using TreeAnnotator 1.10 (84) based on 1,000 trees regularly sampled from the posterior distribution of trees obtained for each of the two major GETV clades considered here. We used R functions available in the package “seraphim” (88, 89) to extract the spatiotemporal information embedded within posterior trees and to visualize the dispersal history of GETV lineages and to estimate the weighted lineage dispersal velocity. We further used “seraphim” to perform post hoc analyses of the potential impact of the following continuous environmental factors on the dispersal location (62) and velocity (90) of viral lineages (Fig. S1): annual mean temperature and annual precipitation data retrieved from the WorldClim 2 database (https://worldclim.org/), the pig population density data obtained from the Food and Agriculture Organization (FAO) database (http://www.fao.org/livestock-systems/global-distributions/pigs/), and the elevation data on the study area as estimated by the Shuttle Radar Topography Mission (https://www2.jpl.nasa.gov/srtm), as well as land cover variables (savannas, forests, croplands, urban areas) extracted from land cover data provided by the International Geosphere Biosphere Program (https://lpdaac.usgs.gov/products/mcd12q1v006/).
For investigation of the impact of environmental factors on the dispersal location and dispersal velocity of GETV lineages, we applied analytical approaches developed by Dellicour et al. (62, 90), respectively (see the supplemental material for a detailed description of these two approaches).
Data availability.
New GETV genomic and E2 gene sequence data in our study are available in GenBank and China National Genebank Database (CNGBdb) under accession numbers MZ736724 to MZ736801, and Project ID: CNP0003775.
ACKNOWLEDGMENTS
S.S., W.-T.H., J.Z., and Z.Y. are financially supported by the National Natural Science Foundation of Outstanding Youth Fund in China (NSFC grant no. 31922081), the National Key Research and Development Program of China (grant no. 2022YFC2604203), the Project of Sanya Yazhou Bay Science and Technology City (grant no. SCKJ-JYRC-2022-08), the 2021 Agricultural Research Outstanding Talents Training Program of the Ministry of Agriculture and Rural Affairs, and the Bioinformatics Center of Nanjing Agricultural University. S.D. acknowledges support from the Fonds National de la Recherche Scientifique (F.R.S.-FNRS, Belgium; grant no. F.4515.22) and from the Research Foundation—Flanders (Fonds voor Wetenschappelijk Onderzoek-Vlaanderen [FWO], Belgium; grant no. G098321N). S.D. and P.L. acknowledge funding from the European Union Horizon 2020 project MOOD (grant agreement no. 874850). P.L. acknowledges support by the European Research Council under the European Union’s Horizon 2020 research and innovation program (grant no. 725422-ReservoirDOCS) and the Research Foundation—Flanders (Fonds voor Wetenschappelijk Onderzoek-Vlaanderen; grant no. G066215N, G0D5117N, and 75 G0B9317N). M.A.S. and X.J. acknowledge support from the National Institutes of Health (grant no. U19 AI135995 and R01 AI153044), and M.V. acknowledges support from the German Research foundation (DFG; grant no. VE 141-15 and VE 141/18).
Footnotes
Supplemental material is available online only.
Contributor Information
Shuo Su, Email: shuosu@njau.edu.cn.
Colin R. Parrish, Cornell University
REFERENCES
- 1.Beigel JH, Farrar J, Han AM, Hayden FG, Hyer R, de Jong MD, Lochindarat S, Nguyen TK, Nguyen TH, Tran TH, Nicoll A, Touch S, Yuen KY, Writing Committee of the World Health Organization Consultation on Human Influenza A/H5 . 2005. Avian influenza A (H5N1) infection in humans. N Engl J Med 353:1374–1385. 10.1056/NEJMra052211. [DOI] [PubMed] [Google Scholar]
- 2.Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H, Zhang J, McEachern J, Field H, Daszak P, Eaton BT, Zhang S, Wang LF. 2005. Bats are natural reservoirs of SARS-like coronaviruses. Science 310:676–679. 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- 3.Cui J, Li F, Shi ZL. 2019. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 17:181–192. 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang LF, Anderson DE. 2019. Viruses in bats and potential spillover to animals and humans. Curr Opin Virol 34:79–89. 10.1016/j.coviro.2018.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun J, He WT, Wang L, Lai A, Ji X, Zhai X, Li G, Suchard MA, Tian J, Zhou J, Veit M, Su S. 2020. COVID-19: epidemiology, evolution, and cross-disciplinary perspectives. Trends Mol Med 26:483–495. 10.1016/j.molmed.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273. 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He W, Auclert LZ, Zhai X, Wong G, Zhang C, Zhu H, Xing G, Wang S, He W, Li K, Wang L, Han GZ, Veit M, Zhou J, Su S. 2019. Interspecies transmission, genetic diversity, and evolutionary dynamics of pseudorabies virus. J Infect Dis 219:1705–1715. 10.1093/infdis/jiy731. [DOI] [PubMed] [Google Scholar]
- 8.He WT, Ji X, He W, Dellicour S, Wang S, Li G, Zhang L, Gilbert M, Zhu H, Xing G, Veit M, Huang Z, Han GZ, Huang Y, Suchard MA, Baele G, Lemey P, Su S. 2020. Genomic epidemiology, evolution, and transmission dynamics of porcine deltacoronavirus. Mol Biol Evol 37:2641–2654. 10.1093/molbev/msaa117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ang BSP, Lim TCC, Wang L. 2018. Nipah virus infection. J Clin Microbiol 56. 10.1128/JCM.01875-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dawood FS, Jain S, Finelli L, Shaw MW, Lindstrom S, Garten RJ, Gubareva LV, Xu X, Bridges CB, Uyeki TM, Novel Swine-Origin Influenza Investigation Team . 2009. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med 360:2605–2615. 10.1056/NEJMoa0903810. [DOI] [PubMed] [Google Scholar]
- 11.Mena I, Nelson MI, Quezada-Monroy F, Dutta J, Cortes-Fernández R, Lara-Puente JH, Castro-Peralta F, Cunha LF, Trovão NS, Lozano-Dubernard B, Rambaut A, van Bakel H, García-Sastre A. 2016. Origins of the 2009 H1N1 influenza pandemic in swine in Mexico. Elife 5:e16777. 10.7554/eLife.16777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Q, Wang X, Xie C, Ding S, Yang H, Guo S, Li J, Qin L, Ban F, Wang D, Wang C, Feng L, Ma H, Wu B, Zhang L, Dong C, Xing L, Zhang J, Chen H, Yan R, Wang X, Li W. 2020. A novel human acute encephalitis caused by pseudorabies virus variant strain. Clin Infect Dis 73:e3690–e3700. 10.1093/cid/ciaa987. [DOI] [PubMed] [Google Scholar]
- 13.Bragg L, Tyson GW. 2014. Metagenomics using next-generation sequencing. Methods Mol Biol 1096:183–201. 10.1007/978-1-62703-712-9_15. [DOI] [PubMed] [Google Scholar]
- 14.Miao Q, Ma Y, Wang Q, Pan J, Zhang Y, Jin W, Yao Y, Su Y, Huang Y, Wang M, Li B, Li H, Zhou C, Li C, Ye M, Xu X, Li Y, Hu B. 2018. Microbiological diagnostic performance of metagenomic next-generation sequencing when applied to clinical practice. Clin Infect Dis 67:S231–S240. 10.1093/cid/ciy693. [DOI] [PubMed] [Google Scholar]
- 15.Kalantar KL, Carvalho T, de Bourcy CFA, Dimitrov B, Dingle G, Egger R, Han J, Holmes OB, Juan YF, King R, Kislyuk A, Lin MF, Mariano M, Morse T, Reynoso LV, Cruz DR, Sheu J, Tang J, Wang J, Zhang MA, Zhong E, Ahyong V, Lay S, Chea S, Bohl JA, Manning JE, Tato CM, DeRisi JL. 2020. IDseq—an open source cloud-based pipeline and analysis service for metagenomic pathogen detection and monitoring. Gigascience 9:giaa111. 10.1093/gigascience/giaa111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lemey P, Rambaut A, Bedford T, Faria N, Bielejec F, Baele G, Russell CA, Smith DJ, Pybus OG, Brockmann D, Suchard MA. 2014. Unifying viral genetics and human transportation data to predict the global transmission dynamics of human influenza H3N2. PLoS Pathog 10:e1003932. 10.1371/journal.ppat.1003932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Müller NF, Dudas G, Stadler T. 2019. Inferring time-dependent migration and coalescence patterns from genetic sequence and predictor data in structured populations. Virus Evol 5:vez030. 10.1093/ve/vez030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dellicour S, Lequime S, Vrancken B, Gill MS, Bastide P, Gangavarapu K, Matteson NL, Tan Y, Du Plessis L, Fisher AA, Nelson MI, Gilbert M, Suchard MA, Andersen KG, Grubaugh ND, Pybus OG, Lemey P. 2020. Epidemiological hypothesis testing using a phylogeographic and phylodynamic framework. Nat Commun 11:5620. 10.1038/s41467-020-19122-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gill MS, Lemey P, Faria NR, Rambaut A, Shapiro B, Suchard MA. 2013. Improving Bayesian population dynamics inference: a coalescent-based model for multiple loci. Mol Biol Evol 30:713–724. 10.1093/molbev/mss265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gill MS, Lemey P, Bennett SN, Biek R, Suchard MA. 2016. Understanding past population dynamics: Bayesian coalescent-based modeling with covariates. Syst Biol 65:1041–1056. 10.1093/sysbio/syw050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lemey P, Rambaut A, Drummond AJ, Suchard MA. 2009. Bayesian phylogeography finds its roots. PLoS Comput Biol 5:e1000520. 10.1371/journal.pcbi.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lemey P, Rambaut A, Welch JJ, Suchard MA. 2010. Phylogeography takes a relaxed random walk in continuous space and time. Mol Biol Evol 27:1877–1885. 10.1093/molbev/msq067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pybus OG, Suchard MA, Lemey P, Bernardin FJ, Rambaut A, Crawford FW, Gray RR, Arinaminpathy N, Stramer SL, Busch MP, Delwart EL. 2012. Unifying the spatial epidemiology and molecular evolution of emerging epidemics. Proc Natl Acad Sci USA 109:15066–15071. 10.1073/pnas.1206598109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacquot M, Nomikou K, Palmarini M, Mertens P, Biek R. 2017. Bluetongue virus spread in Europe is a consequence of climatic, landscape and vertebrate host factors as revealed by phylogeographic inference. Proc Biol Sci 284:20170919. 10.1098/rspb.2017.0919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brunker K, Lemey P, Marston DA, Fooks AR, Lugelo A, Ngeleja C, Hampson K, Biek R. 2018. Landscape attributes governing local transmission of an endemic zoonosis: rabies virus in domestic dogs. Mol Ecol 27:773–788. 10.1111/mec.14470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rawle DJ, Nguyen W, Dumenil T, Parry R, Warrilow D, Tang B, Le TT, Slonchak A, Khromykh AA, Lutzky VP, Yan K, Suhrbier A. 2020. Sequencing of historical isolates, K-mer mining and high serological cross-reactivity with Ross River virus argue against the presence of Getah virus in Australia. Pathogens 9:848. 10.3390/pathogens9100848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumanomido T, Wada R, Kanemaru T, Kamada M, Hirasawa K, Akiyama Y. 1988. Clinical and virological observations on swine experimentally infected with Getah virus. Vet Microbiol 16:295–301. 10.1016/0378-1135(88)90033-8. [DOI] [PubMed] [Google Scholar]
- 28.Xing C, Jiang J, Lu Z, Mi S, He B, Tu C, Liu X, Gong W. 2020. Isolation and characterization of Getah virus from pigs in Guangdong province of China. Transbound Emerg Dis 10.1111/tbed.13567. [DOI] [PubMed] [Google Scholar]
- 29.Zhai YG, Wang HY, Sun XH, Fu SH, Wang HQ, Attoui H, Tang Q, Liang GD. 2008. Complete sequence characterization of isolates of Getah virus (genus Alphavirus, family Togaviridae) from China. J Gen Virol 89:1446–1456. 10.1099/vir.0.83607-0. [DOI] [PubMed] [Google Scholar]
- 30.Li YY, Liu H, Fu SH, Li XL, Guo XF, Li MH, Feng Y, Chen WX, Wang LH, Lei WW, Gao XY, Lv Z, He Y, Wang HY, Zhou HN, Wang GQ, Liang GD. 2017. From discovery to spread: the evolution and phylogeny of Getah virus. Infect Genet Evol 55:48–55. 10.1016/j.meegid.2017.08.016. [DOI] [PubMed] [Google Scholar]
- 31.Liu H, Zhang X, Li LX, Shi N, Sun XT, Liu Q, Jin NY, Si XK. 2019. First isolation and characterization of Getah virus from cattle in northeastern China. BMC Vet Res 15:320. 10.1186/s12917-019-2061-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu G, Ou J, Ji J, Ren Z, Hu X, Wang C, Li S. 2019. Emergence of Getah virus infection in horse with fever in China, 2018. Front Microbiol 10:1416. 10.3389/fmicb.2019.01416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li L, Guo X, Zhao Q, Tong Y, Fan H, Sun Q, Xing S, Zhou H, Zhang J. 2017. Investigation on mosquito-borne viruses at Lancang River and Nu River watersheds in Southwestern China. Vector Borne Zoonotic Dis 17:804–812. 10.1089/vbz.2017.2164. [DOI] [PubMed] [Google Scholar]
- 34.Shi N, Li LX, Lu RG, Yan XJ, Liu H. 2019. Highly pathogenic swine Getah virus in blue foxes, Eastern China, 2017. Emerg Infect Dis 25:1252–1254. 10.3201/eid2506.181983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, Fu S, Guo X, Li X, Li M, Wang L, Gao X, Lei W, Cao L, Lu Z, He Y, Wang H, Zhou H, Liang G. 2019. Serological survey of Getah virus in domestic animals in Yunnan Province, China. Vector Borne Zoonotic Dis 19:59–61. 10.1089/vbz.2018.2273. [DOI] [PubMed] [Google Scholar]
- 36.Liang Y, Zhang L, Liu Y, Dong Y, Wang Z, Wang J, Liang G. 2010. The screening of Getah virus IgM antibody to patients with fever from She county in Hebei province. Chinese J Health Lab Technol 20:2562–2563. (In Chinese.) [Google Scholar]
- 37.Suhrbier A. 2019. Rheumatic manifestations of chikungunya: emerging concepts and interventions. Nat Rev Rheumatol 15:597–611. 10.1038/s41584-019-0276-9. [DOI] [PubMed] [Google Scholar]
- 38.Taylor KG, Paessler S. 2013. Pathogenesis of Venezuelan equine encephalitis. Vet Microbiol 167:145–150. 10.1016/j.vetmic.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 39.Crosby B, Crespo ME. 2022. Venezuelan equine encephalitis. In StatPearls. StatPearls Publishing LLC, Treasure Island, FL. [PubMed] [Google Scholar]
- 40.Lu G, Chen R, Shao R, Dong N, Liu W, Li S. 2020. Getah virus: an increasing threat in China. J Infect 80:350–371. 10.1016/j.jinf.2019.11.016. [DOI] [PubMed] [Google Scholar]
- 41.Ren T, Min X, Mo Q, Wang Y, Wang H, Chen Y, Ouyang K, Huang W, Wei Z. 2022. Construction and characterization of a full-length infectious clone of Getah virus in vivo. Virol Sin 37:348–357. 10.1016/j.virs.2022.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li F, Zhang B, Xu Z, Jiang C, Nei M, Xu L, Zhao J, Deng H, Sun X, Zhou Y, Zhu L. 2022. Getah virus infection rapidly causes testicular damage and decreases sperm quality in male mice. Front Vet Sci 9:883607. 10.3389/fvets.2022.883607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Voss JE, Vaney MC, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, Thompson A, Bricogne G, Rey FA. 2010. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468:709–712. 10.1038/nature09555. [DOI] [PubMed] [Google Scholar]
- 44.Holmes AC, Basore K, Fremont DH, Diamond MS. 2020. A molecular understanding of alphavirus entry. PLoS Pathog 16:e1008876. 10.1371/journal.ppat.1008876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fox JM, Long F, Edeling MA, Lin H, van Duijl-Richter MKS, Fong RH, Kahle KM, Smit JM, Jin J, Simmons G, Doranz BJ, Crowe JE, Jr, Fremont DH, Rossmann MG, Diamond MS. 2015. Broadly neutralizing alphavirus antibodies bind an epitope on E2 and inhibit entry and egress. Cell 163:1095–1107. 10.1016/j.cell.2015.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Powell LA, Miller A, Fox JM, Kose N, Klose T, Kim AS, Bombardi R, Tennekoon RN, Dharshan de Silva A, Carnahan RH, Diamond MS, Rossmann MG, Kuhn RJ, Crowe JE, Jr.. 2020. Human mAbs broadly protect against arthritogenic alphaviruses by recognizing conserved elements of the Mxra8 receptor-binding site. Cell Host Microbe 28:699–711.e7. 10.1016/j.chom.2020.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ragonnet-Cronin M, Hodcroft E, Hué S, Fearnhill E, Delpech V, Brown AJ, Lycett S, UK HIV Drug Resistance Database . 2013. Automated analysis of phylogenetic clusters. BMC Bioinformatics 14:317. 10.1186/1471-2105-14-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kass RE, Raftery AE. 1995. Bayes factors. J Am Statist Assoc 90:773–795. 10.1080/01621459.1995.10476572. [DOI] [Google Scholar]
- 49.Azar SR, Campos RK, Bergren NA, Camargos VN, Rossi SL. 2020. Epidemic alphaviruses: ecology, emergence and outbreaks. Microorganisms 8:1167. 10.3390/microorganisms8081167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guth S, Hanley KA, Althouse BM, Boots M. 2020. Ecological processes underlying the emergence of novel enzootic cycles: arboviruses in the neotropics as a case study. PLoS Negl Trop Dis 14:e0008338. 10.1371/journal.pntd.0008338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ren T, Mo Q, Wang Y, Wang H, Nong Z, Wang J, Niu C, Liu C, Chen Y, Ouyang K, Huang W, Wei Z. 2020. Emergence and phylogenetic analysis of a Getah virus isolated in Southern China. Front Vet Sci 7:552517. 10.3389/fvets.2020.552517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rattanatumhi K, Prasertsincharoen N, Naimon N, Kuwata R, Shimoda H, Ishijima K, Yonemitsu K, Minami S, Supriyono, Tran NTB, Kuroda Y, Tatemoto K, Virhuez MM, Hondo E, Rerkamnuaychoke W, Maeda K, Phichitraslip T. 2021. A serological survey and characterization of Getah virus in domestic pigs in Thailand, 2017–2018. Transbound Emerg Dis 69:913–918. 10.1111/tbed.14042. [DOI] [PubMed] [Google Scholar]
- 53.Althouse BM, Hanley KA. 2015. The tortoise or the hare? Impacts of within-host dynamics on transmission success of arthropod-borne viruses. Philos Trans R Soc Lond B Biol Sci 370:20140299. 10.1098/rstb.2014.0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Michie A, Dhanasekaran V, Lindsay MDA, Neville PJ, Nicholson J, Jardine A, Mackenzie JS, Smith DW, Imrie A. 2020. Genome-scale phylogeny and evolutionary analysis of Ross River virus reveals periodic sweeps of lineage dominance in Western Australia, 1977–2014. J Virol 94:e01234-19. 10.1128/JVI.01234-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen CL, Hasan SS, Klose T, Sun Y, Buda G, Sun C, Klimstra WB, Rossmann MG. 2020. Cryo-EM structure of eastern equine encephalitis virus in complex with heparan sulfate analogues. Proc Natl Acad Sci USA 117:8890–8899. 10.1073/pnas.1910670117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Button JM, Qazi SA, Wang JC, Mukhopadhyay S. 2020. Revisiting an old friend: new findings in alphavirus structure and assembly. Curr Opin Virol 45:25–33. 10.1016/j.coviro.2020.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang N, Zhai X, Li X, Wang Y, He WT, Jiang Z, Veit M, Su S. 2020. Attenuation of getah virus by a single amino acid substitution at residue 253 of the E2 protein that might be part of a new heparan sulfate binding site on alphaviruses. J Virol 96:e0175121. 10.1128/jvi.01751-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang R, Kim AS, Fox JM, Nair S, Basore K, Klimstra WB, Rimkunas R, Fong RH, Lin H, Poddar S, Crowe JE, Jr, Doranz BJ, Fremont DH, Diamond MS. 2018. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 557:570–574. 10.1038/s41586-018-0121-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brault AC, Powers AM, Holmes EC, Woelk CH, Weaver SC. 2002. Positively charged amino acid substitutions in the e2 envelope glycoprotein are associated with the emergence of Venezuelan equine encephalitis virus. J Virol 76:1718–1730. 10.1128/jvi.76.4.1718-1730.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou F, Wang A, Chen L, Wang X, Cui D, Chang H, Wang C. 2020. Isolation and phylogenetic analysis of Getah virus from a commercial modified live vaccine against porcine reproductive and respiratory syndrome virus. Mol Cell Probes 53:101650. 10.1016/j.mcp.2020.101650. [DOI] [PubMed] [Google Scholar]
- 61.Wu H, Lu L, Meng F, Guo Y, Qy L. 2017. Reports on national surveillance of mosquitoes in China, 2006–2015. Chin J Vector Biol & Control 28:409–415. [Google Scholar]
- 62.Dellicour S, Troupin C, Jahanbakhsh F, Salama A, Massoudi S, Moghaddam MK, Baele G, Lemey P, Gholami A, Bourhy H. 2019. Using phylogeographic approaches to analyse the dispersal history, velocity and direction of viral lineages—application to rabies virus spread in Iran. Mol Ecol 28:4335–4350. 10.1111/mec.15222. [DOI] [PubMed] [Google Scholar]
- 63.He WT, Hou X, Zhao J, Sun J, He H, Si W, Wang J, Jiang Z, Yan Z, Xing G, Lu M, Suchard MA, Ji X, Gong W, He B, Li J, Lemey P, Guo D, Tu C, Holmes EC, Shi M, Su S. 2022. Virome characterization of game animals in China reveals a spectrum of emerging pathogens. Cell 185:1117–1129.e8. 10.1016/j.cell.2022.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsetsarkin KA, Weaver SC. 2011. Sequential adaptive mutations enhance efficient vector switching by Chikungunya virus and its epidemic emergence. PLoS Pathog 7:e1002412. 10.1371/journal.ppat.1002412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. 2015. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 1:vev003. 10.1093/ve/vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Holmes EC, Worobey M, Rambaut A. 1999. Phylogenetic evidence for recombination in dengue virus. Mol Biol Evol 16:405–409. 10.1093/oxfordjournals.molbev.a026121. [DOI] [PubMed] [Google Scholar]
- 69.Boni MF, Posada D, Feldman MW. 2007. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 176:1035–1047. 10.1534/genetics.106.068874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Padidam M, Sawyer S, Fauquet CM. 1999. Possible emergence of new geminiviruses by frequent recombination. Virology 265:218–225. 10.1006/viro.1999.0056. [DOI] [PubMed] [Google Scholar]
- 71.Gibbs MJ, Armstrong JS, Gibbs AJ. 2000. Sister-scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 16:573–582. 10.1093/bioinformatics/16.7.573. [DOI] [PubMed] [Google Scholar]
- 72.Smith JM. 1992. Analyzing the mosaic structure of genes. J Mol Evol 34:126–129. 10.1007/BF00182389. [DOI] [PubMed] [Google Scholar]
- 73.Martin D, Rybicki E. 2000. RDP: detection of recombination amongst aligned sequences. Bioinformatics 16:562–563. 10.1093/bioinformatics/16.6.562. [DOI] [PubMed] [Google Scholar]
- 74.Sabir JS, Lam TT, Ahmed MM, Li L, Shen Y, Abo-Aba SE, Qureshi MI, Abu-Zeid M, Zhang Y, Khiyami MA, Alharbi NS, Hajrah NH, Sabir MJ, Mutwakil MH, Kabli SA, Alsulaimany FA, Obaid AY, Zhou B, Smith DK, Holmes EC, Zhu H, Guan Y. 2016. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 351:81–84. 10.1126/science.aac8608. [DOI] [PubMed] [Google Scholar]
- 75.Kosakovsky Pond SL, Poon AFY, Velazquez R, Weaver S, Hepler NL, Murrell B, Shank SD, Magalis BR, Bouvier D, Nekrutenko A, Wisotsky S, Spielman SJ, Frost SDW, Muse SV. 2020. HyPhy 2.5—a customizable platform for evolutionary hypothesis testing using phylogenies. Mol Biol Evol 37:295–299. 10.1093/molbev/msz197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL. 2012. Detecting individual sites subject to episodic diversifying selection. PLoS Genet 8:e1002764. 10.1371/journal.pgen.1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kosakovsky Pond SL, Frost SD. 2005. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22:1208–1222. 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- 78.Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL, Scheffler K. 2013. FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Mol Biol Evol 30:1196–1205. 10.1093/molbev/mst030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smith MD, Wertheim JO, Weaver S, Murrell B, Scheffler K, Kosakovsky Pond SL. 2015. Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol Biol Evol 32:1342–1353. 10.1093/molbev/msv022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tavaré S. 1986. Some probabilistic and statistical problems in the analysis of DNA sequences. American Mathematical Society, Providence, RI. [Google Scholar]
- 82.Yang Z. 1994. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J Mol Evol 39:306–314. 10.1007/BF00160154. [DOI] [PubMed] [Google Scholar]
- 83.Rambaut A, Lam TT, Max Carvalho L, Pybus OG. 2016. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol 2:vew007. 10.1093/ve/vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. 2018. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol 4:vey016. 10.1093/ve/vey016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ayres DL, Cummings MP, Baele G, Darling AE, Lewis PO, Swofford DL, Huelsenbeck JP, Lemey P, Rambaut A, Suchard MA. 2019. BEAGLE 3: improved performance, scaling, and usability for a high-performance computing library for statistical phylogenetics. Syst Biol 68:1052–1061. 10.1093/sysbio/syz020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. 2018. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol 67:901–904. 10.1093/sysbio/syy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Drummond AJ, Ho SY, Phillips MJ, Rambaut A. 2006. Relaxed phylogenetics and dating with confidence. PLoS Biol 4:e88. 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dellicour S, Rose R, Pybus OG. 2016. Explaining the geographic spread of emerging epidemics: a framework for comparing viral phylogenies and environmental landscape data. BMC Bioinformatics 17:82. 10.1186/s12859-016-0924-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dellicour S, Rose R, Faria NR, Lemey P, Pybus OG. 2016. SERAPHIM: studying environmental rasters and phylogenetically informed movements. Bioinformatics 32:3204–3206. 10.1093/bioinformatics/btw384. [DOI] [PubMed] [Google Scholar]
- 90.Dellicour S, Rose R, Faria NR, Vieira LFP, Bourhy H, Gilbert M, Lemey P, Pybus OG. 2017. Using viral gene sequences to compare and explain the heterogeneous spatial dynamics of virus epidemics. Mol Biol Evol 34:2563–2571. 10.1093/molbev/msx176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental text, Fig. s1 to S6, and Tables S1 and S2. Download jvi.01091-22-s0001.pdf, PDF file, 6.0 MB (6MB, pdf)
Data Availability Statement
New GETV genomic and E2 gene sequence data in our study are available in GenBank and China National Genebank Database (CNGBdb) under accession numbers MZ736724 to MZ736801, and Project ID: CNP0003775.