

ABSTRACT
Increased demand for novel, highly effective vaccination strategies necessitates a better understanding of long-lived memory CD8 T cell differentiation. To achieve this understanding, we used the mouse model of acute lymphocytic choriomeningitis virus (LCMV) infection. We reexamined classical memory CD8 T cell subsets and performed in-depth, longitudinal analysis of their phenotype, transcriptional programming, and anatomic location within the spleen. All analyses were performed at multiple time points from 8 days to 1 year postinfection. Memory subsets are conventionally defined by their expression of KLRG1 and IL-7Rα, as follows: KLRG1+IL-7Rα− terminal effectors (TEs) and KLRG1−IL-7Rα+ memory precursors (MPs). But we also characterized a third KLRG1+IL-7Rα+ subset which we refer to as KLRG1+ MPs. In these analyses, we defined a comprehensive memory phenotype that is associated with higher levels of CD28 expression. We also demonstrated that MPs, KLRG1+ MPs, and TEs have distinct localization programs within the spleen. We found that MPs became preferentially enriched in the white pulp as early as 1 to 2 weeks postinfection, and their predominance in the white pulp was maintained throughout the course of a year. On the other hand, KLRG1+ MPs and TEs localized to the red pulp just as early, and they consistently localized to the red pulp thereafter. These findings indicate that location may be crucial for memory formation and that white pulp-derived signals may contribute to long-term memory survival. Achieving robust memory responses following vaccination may require more deliberate consideration of which memory phenotypes are induced, as well as where they traffic, as these factors could impact their longevity.
IMPORTANCE CD8 T cells play a critical role in viral immunity and it is important to understand how memory cells are formed and what processes lead to their long-term maintenance. Here, we use a mouse model of acute infection to perform an in-depth, longitudinal analysis of memory CD8 T cell differentiation, examining the phenotype and location of memory cells out to 1 year postinfection.
KEYWORDS: CD8 T cells, memory cell differentiation, viral infection, lymphocytic choriomeningitis virus, vaccines
INTRODUCTION
Development of CD8 T cell memory following vaccination is of great importance, as these cells are crucial for providing durable protection again secondary infection (1, 2). Memory CD8 T cells are able to undergo long-term self-renewal, and yet, they remain poised to rapidly recall effector functions upon antigen re-encounter (3, 4).
Following primary infection, naive antigen-specific CD8 T cells encounter antigen and become activated—undergoing proliferation and acquiring effector functions that enable them to kill virally infected cells (1, 2, 5, 6). This activated population is heterogeneous. The majority of cells which are KLRG1+IL-7Rα− are destined to die through apoptosis following clearance of the infection and are called “terminal effectors” (TEs) (7). A small subset (5% to 10%) survives—these “memory precursors” (MPs) express higher levels of IL-7Rα and lower levels of KLRG1 and seed the long-lived memory pool (8–10). These subsets are also referred to as short-lived effector cells and memory precursor effector cells (SLECs and MPECs), respectively (11). Importantly, IL-7Rα is critical for the survival of long-lived memory CD8 T cells (8).
The memory pool arising from KLRG1−IL-7Rα+ MPs can be subdivided based on cellular function and localization. Classically, memory CD8 T cells are divided into two populations based on their expression of the homing markers CD62L (L-selectin) and CCR7 (12). CD62LloCCR7lo cells are “effector memory” (Tem) and localize to inflamed tissues, while CD62LhiCCR7hi cells are “central memory” (Tcm) and preferentially home to secondary lymphoid organs (11–13). Both Tem and Tcm will recirculate through the blood to complete their respective surveillance cycles. An additional “tissue-resident” memory subset (Trm) does not circulate and provides protection exclusively from nonlymphoid compartments (14–17).
Memory CD8 T cells also have highly specialized localization programs within tissues—particularly within secondary lymphoid organs (18–20). Intrasplenic localization, for example, has been shown to correlate with memory phenotype. In some studies, Tem seem to be enriched in the red pulp (RP), while Tcm are enriched in the white pulp (WP) (20–22). Trm also have distinct expression profiles which enable their migration to and within specific tissue compartments (23, 24). Beyond homing capacity, these subsets are also distinguished by core functional properties. Tem and Trm are more terminally differentiated and cytotoxic, while Tcm are more fate permissive and have high proliferative and recall potential (11, 13, 25, 26).
Characteristics of these memory programs have been continually refined as new markers of function emerge. Recently, TCF-1 has become one of the most important markers of long-lived memory—a key transcription factor that drives self-renewal via Wnt signaling (27–32). CD28 is also of interest, as T cell receptor (TCR) signal strength has been shown to correlate with memory formation (25, 33, 34). Importantly, CD28 is expressed at high levels on long-lived memory CD8 T cells in humans (3, 35). Chemokine receptors (including CXCR3 and CX3CR1) have additionally been implicated in memory formation, as spatial organization has been shown to mediate survival and recall potential (36–40).
In this study, we provide a comprehensive, longitudinal analysis of the expression of established and putative memory markers across both time and space. We evaluated the expression of KLRG1 and IL-7Rα up to 1 year after acute lymphocytic choriomeningitis virus (LCMV) infection, and we performed phenotypic and transcriptional analyses of the subsets defined by these markers. We also performed spatial characterization of memory CD8 T cells within the spleen, tracking them in the RP and WP using intravenous (i.v.) anti-CD8α labeling (41). We were able to match phenotype and location in order to better characterize the functional compartmentalization of the spleen. We found that location may be an important determinant of long-lived memory. Taken together, our studies provide a deeper understanding of how memory CD8 T cells differentiate and survive following acute infection. Knowing which processes facilitate memory formation and dictate their longevity will be crucial in the development of novel therapeutic and vaccination strategies.
RESULTS
Longitudinal characterization of KLRG1/IL-7Rα subsets of CD8 T cells following acute infection.
In this study, we investigated the longitudinal responses of CD8 T cells following acute LCMV infection. We saw robust proliferation of virus-specific CD8 T cells that peaked on day 8, and then we monitored responses for 1 year by frequency (Fig. 1a) and absolute number (Fig. 1b). As shown previously, CD8 T cell responses against three LCMV epitopes varied according to their immunodominance, with NP396 eliciting the most robust response, followed by GP33 and GP276 (42–45).
FIG 1.

Characterization of LCMV-specific memory CD8 T cell subsets after acute infection. (a and b) Frequency (a) and absolute number (b) of tetramer+ CD8 T cells in the spleen after LCMV Armstrong infection of B6 mice (2 × 105 PFU, intraperitoneal [i.p.]). DbGP33+, DbGP276+, and DbNP396+ responses are shown. (c) Representative flow plots showing the frequency of GP33+ CD8 T cells and the expression of KLRG1 and IL-7Rα within them from 8 days to 1 year postinfection. (d to f) Summary of the frequency and absolute number of the indicated subpopulations within GP33+, GP276+ or NP396+ CD8 T cells. IL-7Rα+KLRG1− memory precursors (MPs, blue), IL-7Rα+KLRG1+ cells (KLRG1+ MPs, green), IL-7Rα-KLRG1+ terminal effectors (TEs, orange), IL-7Rα-KLRG1− double negative cells (DNs, red). Shown are the mean and SEM; n = 5 to 12 mice per time point.
At day 8, the vast majority (~70%) of GP33+ CD8 T cells were TEs (Fig. 1c and d), while MPs constituted <10% of the total population. At this time point, roughly 10% of GP33+ CD8 T cells expressed both KLRG1 and IL-7Rα, which we refer to as “KLRG1+ MPs,” and 10% of GP33+ CD8 T cells expressed neither marker, which we refer to as “double-negatives” (DNs). By absolute number, the TEs exceeded KLRG1+ MPs and MPs by >10-fold. DNs constituted <10% of total GP33+ CD8 T cells after day 8, so we removed them from our analysis after this time point. Between days 8 and 60 postinfection, the relative dominance of TEs, KLRG1+ MPs, and MPs underwent a dramatic shift; after day 60, the populations grew more stable. On day 15, the proportion of TEs dropped to ~50% of the total GP33+ population, and their numbers contracted by ~10-fold. The proportion of KLRG1+ MPs increased to ~15%, while their numbers decreased by ~2- to 3-fold. The proportion of MPs doubled from day 8 to ~20%, and their numbers remained roughly the same. By day 30, MPs could also be broken down and referred to as Tem and Tcm, but we will continue to refer to them as a bulk MP population for simplicity. At day 30, there were roughly equivalent proportions and absolute numbers of TEs, KLRG1+ MPs, and MPs, but by day 60, we observed a clear dominance of the MP population. MPs began to constitute >50% of the total GP33+ population, while their numbers still remained roughly constant. TEs and KLRG1+ MPs continued to contract by absolute number—by ~10-fold and ~2-fold, respectively. From day 60 to 1 year postinfection, these trends continued but at a lower rate. At 1 year, MPs (now largely Tcm) reached >80% of the total GP33+ CD8 T cell population, while TEs had disappeared completely. Interestingly, KLRG1+ MPs still constituted ~10% to 15% of the population and, by absolute number, were present at ~5-fold lower levels than MPs. Altogether, following acute infection, TEs underwent a rapid period of contraction, by both number and frequency of total GP33+ CD8 T cells, while KLRG1+ MPs underwent a slower, more gradual contraction. MPs remained constant in numbers, achieving dominance in their proportion by 60 days postinfection. Their proportion among total GP33+ CD8 T cells continued to rise as time went on and the other populations diminished. These trends in the differentiation of memory GP33+ CD8 T cells were also observed for GP276+ and NP396+ CD8 T cells (Fig. 1e and f).
We next conducted a more in-depth analysis of IL-7Rα and KLRG1 expression within MPs, KLRG1+ MPs, TEs, and DNs (Fig. 2). Of course, IL-7Rα was expressed at highest levels within MPs, followed by KLRG1+ MPs, TEs, and DNs (Fig. 2a). Notably, as early as day 15 postinfection, MPs expressed higher levels of IL-7Rα than even naive cells. On the other hand, KLRG1 was always expressed most highly on TEs. Much like IL-7Rα, expression of CD62L was highest on MPs but exhibited bimodal distribution between days 30 and 60 postinfection. This presumably corresponded to the emergence of a heterogenous population of Tem and Tcm by day 30, and the eventual dominance of Tcm by 1 year postinfection.
FIG 2.
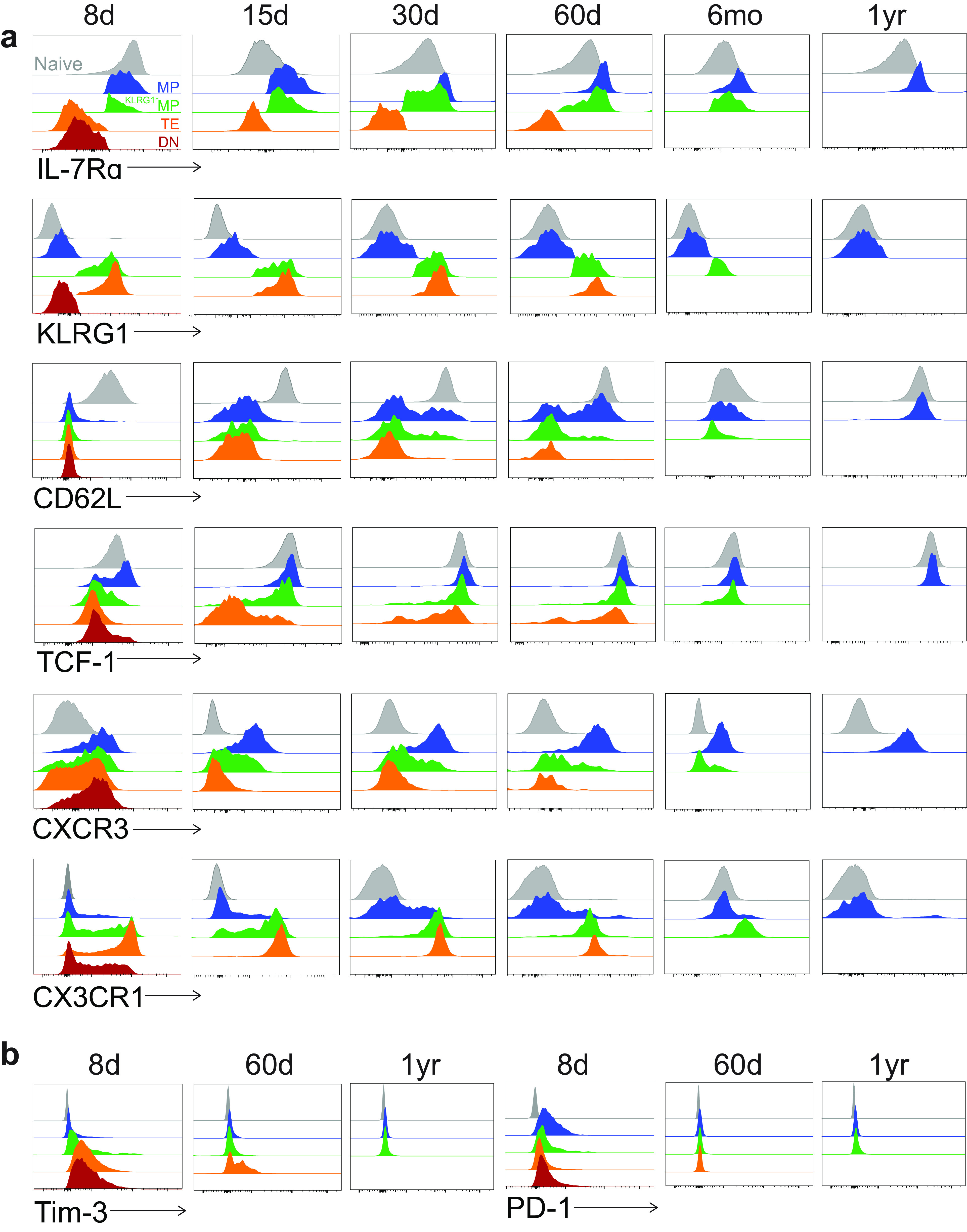
Phenotypic profile of KLRG1/IL-7Rα subsets among GP33+ CD8 T cells after acute infection. (a) Flow cytometry staining of memory markers within each of the four indicated subsets from 8 days to 1 year postinfection. All subsets are gated on GP33+ CD8 T cells. Naive CD44lo CD8 T cells are shown as gray histograms at the top of each plot. The DN subset quickly diminished after infection and so is not shown after day 8; the TE and KLRG1+ MP subsets also diminished after day 60 and so are not shown after that time point. (b) Flow cytometry staining of exhaustion markers within each of the four indicated subsets as in a. Representative flow plots are shown; n = 3 to 12 mice per time point.
We also evaluated the expression of other memory markers. TCF-1 signaling has been shown to promote the generation of long-lived memory CD8 T cells and enable recall potential (29, 32). In accordance, we found that TCF-1 levels were consistently highest in MPs and lowest in TEs. TCF-1 levels in MPs were consistently equivalent to those of naive CD8 T cells.
We also investigated the expression of the following two chemokine receptors: CXCR3 and CX3CR1. We found that naive CD8 T cells did not express CXCR3, while it was upregulated on day 8 in all four subsets and most highly within MPs. Following this result, expression remained consistently highest among MPs, followed by KLRG1+ MPs and TEs. These findings are consistent with prior studies, which suggest that CXCR3 may directly impact memory formation as it controls the positioning of CD8 T cells relative to antigen in the spleen (37, 46, 47). Our studies also suggest that CX3CR1 is associated with a more effector-like phenotype. From day 8 onward, CX3CR1 was expressed most highly on TEs, followed by KLRG1+ MPs, while MPs expressed much lower levels, and naive cells were uniformly CX3CR1−.
Finally, we examined the expression of two classic “exhaustion markers.” (Fig. 2b) Surprisingly, we found that both Tim-3 and PD-1 were expressed by some memory subsets early after acute infection. On day 8, Tim-3 was expressed most highly in the DN population, followed by TEs, KLRG1+ MPs, and MPs. On the other hand, PD-1 was expressed most highly in the KLRG1− populations (DNs and MPs), followed by KLRG1+ MPs and TEs. This finding is consistent with previous work which suggests that PD-1 also plays an inhibitory role during naive-to-effector CD8 T cell transitions (48). By day 60, TEs expressed the highest levels of Tim-3, followed by KLRG1+ MPs and MPs, but all populations became Tim-3− after 1 year. All memory subsets were PD-1− by day 60 and remained PD-1− out to 1 year postinfection.
Altogether, we found that IL-7Rα-expressing classical MPs expressed high levels of CD62L, TCF-1, and CXCR3, while KLRG1-expressing classical TEs expressed high levels of CX3CR1. KLRG1+ MPs appeared to express intermediate levels of all the markers. Only early after acute infection were MPs expressing PD-1 and TEs expressing Tim-3.
CD28 expression correlates with a memory phenotype.
We next analyzed CD28 expression. We found that CD28 was uniformly upregulated in all GP33+ CD8 T cells by day 8 and to levels higher than even naive CD8 T cells (Fig. 3a). Following day 8, all GP33+ CD8 T cells continued to express higher levels of CD28 than naive cells, but CD28 was clearly expressed most highly on MPs, followed by KLRG1+ MPs and TEs. We next wondered whether CD28 expression specifically correlated with the expression of other memory-associated markers. Indeed, we found that CD28 could distinguish a classical MP phenotype (Fig. 3b). CD62L, TCF-1, and CXCR3 were all expressed at consistently higher levels within a distinct CD28hi population, while CX3CR1 was expressed at higher levels within a distinct CD28lo population. These findings were highly significant at multiple time points from 8 days to 1 year postinfection (Fig. 3c).
FIG 3.
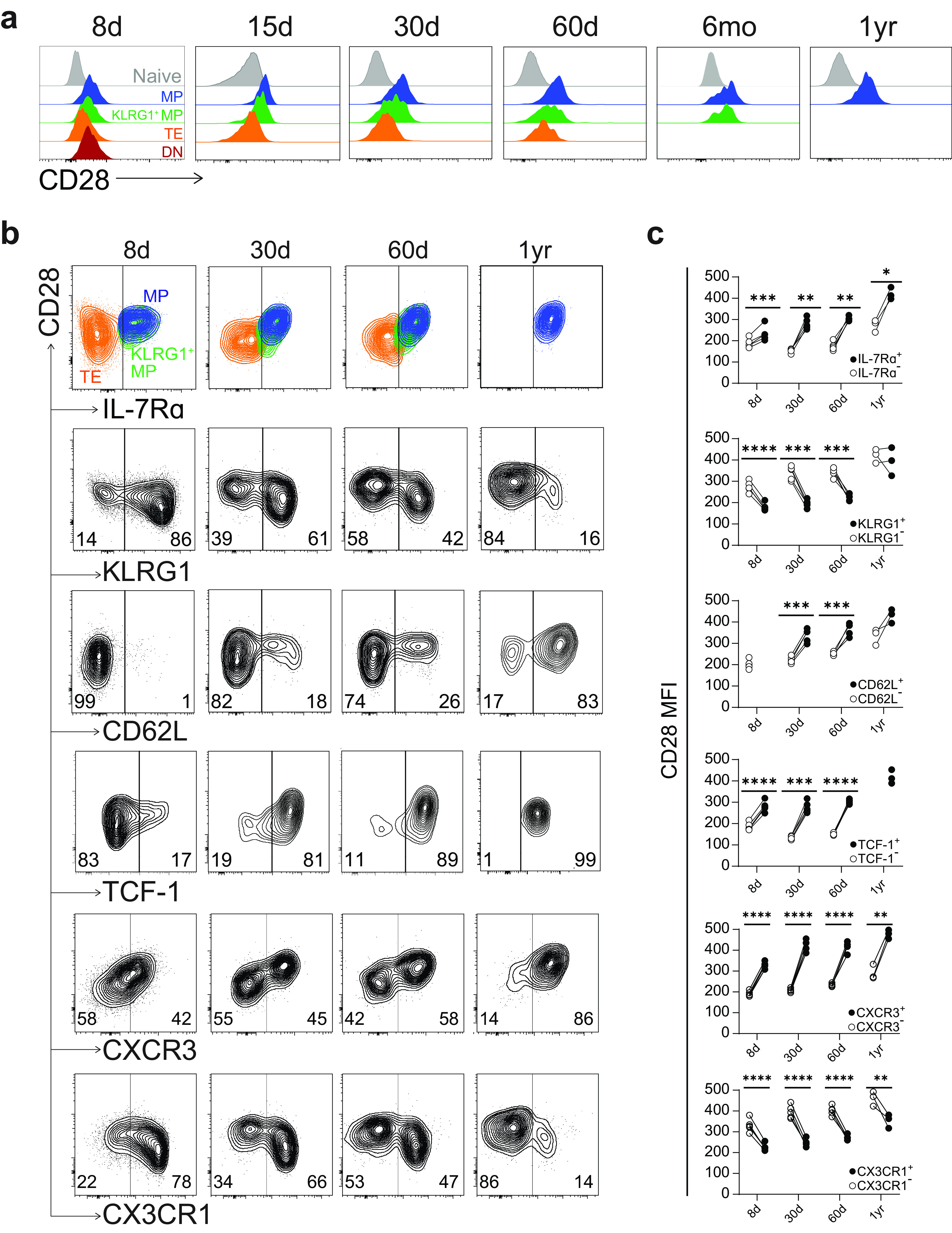
CD28 expression correlates with a memory phenotype. (a) Flow cytometry staining of CD28 within each of the four indicated subsets from 8 days to 1 year postinfection. All subsets are gated on GP33+ CD8 T cells. Naive CD44lo CD8 T cells are shown as gray histograms at the top of each plot. (b) Representative flow plots showing coexpression of IL-7Rα, KLRG1, CD62L, TCF-1, CXCR3, and CX3CR1 with CD28 on GP33+ CD8 T cells. (c) Quantification of CD28 MFI from b among marker+ and marker− GP33+ CD8 T cells. Values are from one representative experiment with 3 to 4 mice per time point. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001 (all determined using paired two-tailed t tests).
Transcriptional profiling of three KLRG1/IL-7Rα subsets following acute infection.
Having examined the surface-level phenotypes of MPs, KLRG1+ MPs, and TEs, we next sought to gain more insight into their fates and functions through transcriptomic analyses. We performed transcriptome sequencing (RNA-seq) on the three sorted populations (among GP33+ CD8 T cells) on day 50 postinfection (Fig. 4a). We also sorted CD44loCD62Lhi CD8 T cells from naive mice to analyze as controls.
FIG 4.
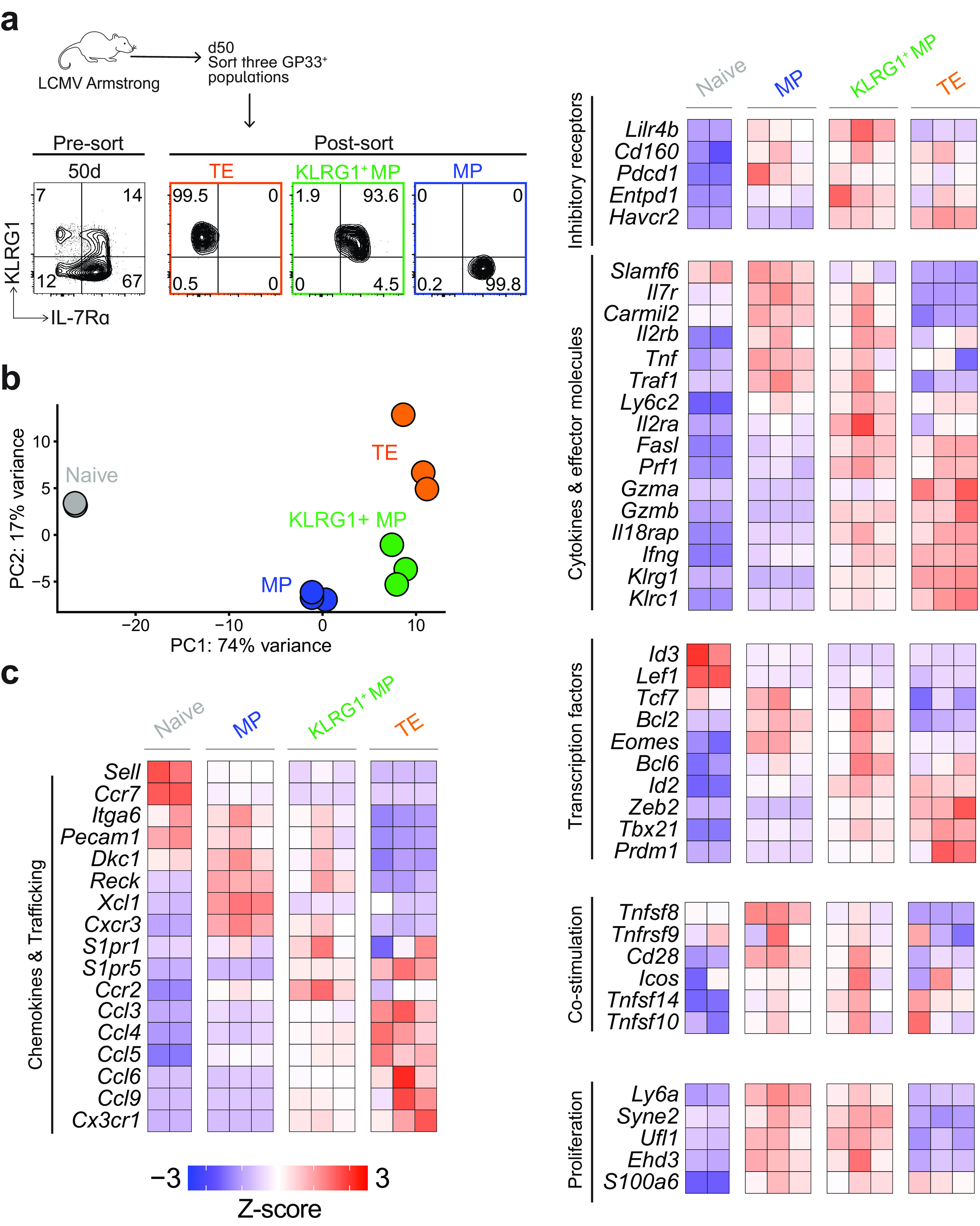
Transcriptional profile of three KLRG1/IL-7Rα CD8 T cell subsets after acute infection. (a) RNA was isolated from three sorted subsets (MPs, KLRG1+ MPs, and TEs) and submitted for RNA-seq. Representative flow plots showing purity (>93%) of the three subsets before and after sorting. Each subset was sorted from bulk GP33+ CD8 T cells at day 50 postinfection. Three sets of three mice were pooled, and then the three subsets were isolated from each pooled sample. (b) Principal-component analysis (PCA) identified two principal components that explained 91% of variance between the samples. Each sample is plotted on these two components. (c) Heatmap showing Z-scores of top DEGs and other genes of interest across the three memory subsets.
We first performed principal-component analysis (PCA) to broadly compare the three subsets and naive cells (Fig. 4b). Two principal components were identified that were responsible for 91% of variance in gene expression among the samples. The first principal component (PC1) explained 74% of the variance and showed that the three subsets were much more similar to each other than to naive cells. On the other hand, PC2 explained 17% of the total variance and revealed that the three subsets were also distinct from one another.
Naive CD8 T cells expressed the highest levels of Lef1, Id3, Ccr7, and Sell (Fig. 4c). Following LCMV infection, MPs and KLRG1+ MPs were distinct but expressed many markers in parallel, while TEs expressed more subset-specific genes. MPs and KLRG1+ MPs both expressed higher levels of Il7r, Bcl2, and Eomes than naive cells, as well as higher levels of some proliferation-associated proteins (Syne2, Ufl1, and Ehd3) and some effector molecules (Tnf, Traf1, and Carmil2). As for unique genes, MPs had the highest levels of Cxcr3, Xcl1, Reck, Tnfsf8, and Cd28, while KLRG1+ MPs had the highest levels of S1pr1, Ccr2, Lilr4b, Entpd1, and Il2ra. The expression profile of TEs was dominated by effector-like genes (Klrg1, Prf, Gzma, Gzmb, Il18rap, and Ifng), effector-programming transcription factors (Zeb2 and Tbx1), inhibitory receptors (Havcr2), and multiple chemokine and trafficking molecules (Cx3cr1, Ccl3, Ccl4, Ccl5, Ccl6, Ccl9, and S1pr5). Notably, KLRG1+ MPs also expressed intermediate levels of these genes.
Among antigen-specific CD8 T cells, MPs and TEs had the highest number of differentially expressed genes (DEGs) between them (2,002) (Fig. 5a), while KLRG1+ MPs differentially expressed 291 and 1,237 genes compared with MPs and TEs, respectively (Fig. 5b and c). Importantly, transcriptional levels of key memory markers (Fig. 5d) matched the surface expression profiling in Fig. 2 and 3.
FIG 5.
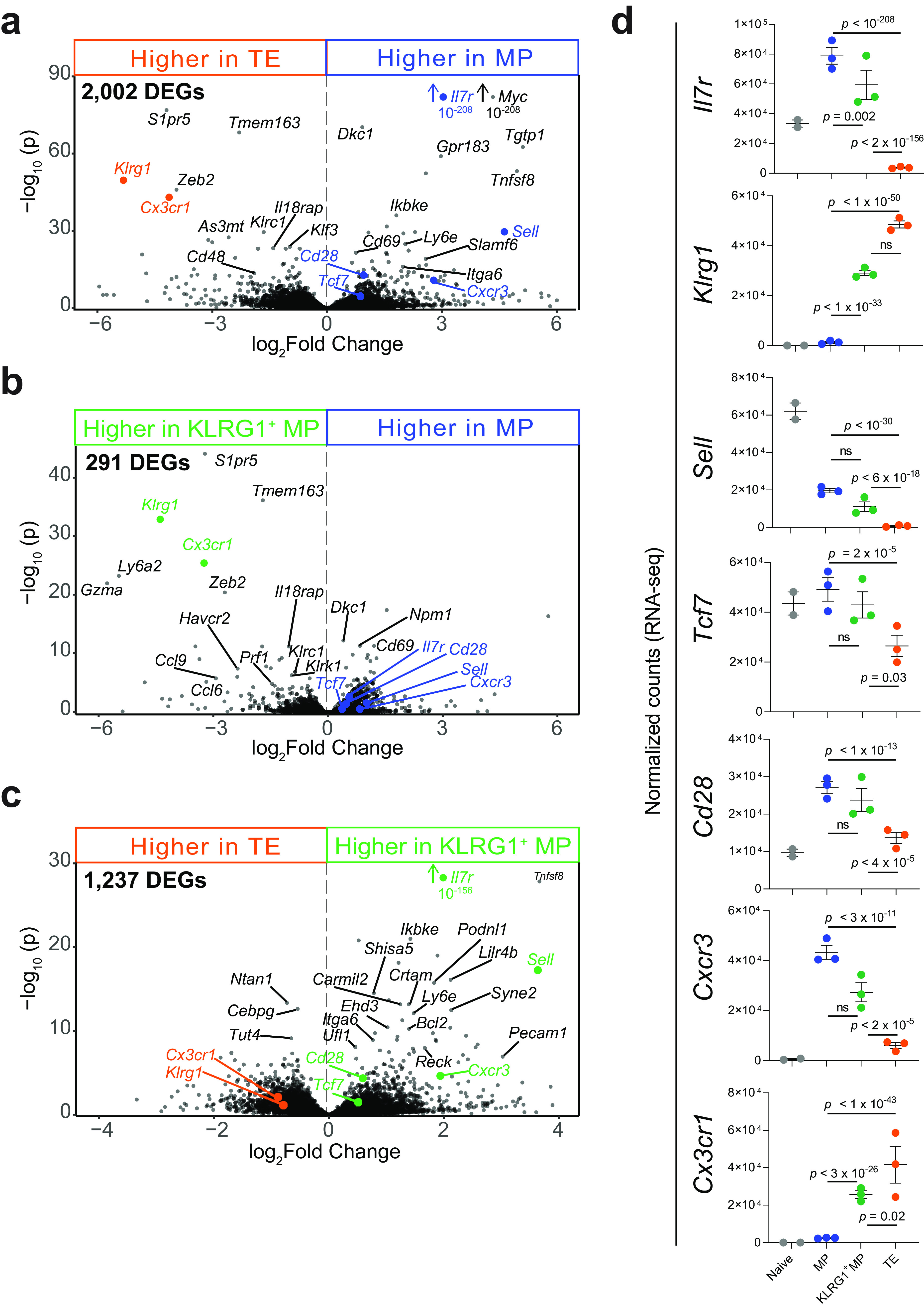
Expression of key memory markers among three KLRG1/IL-7Rα CD8 T cell subsets after acute infection. RNA was isolated from MPs, KLRG1+ MPs, and TEs (among GP33+ CD8 T cells) and submitted for RNA-seq, as in Fig. 4. (a to c) Volcano plots showing DEGs between each of the three memory subsets (P < 0.05 was used as a cutoff). Compared subsets are shown at the top of each plot. Numbers of DEGs are shown in the top left corner of each plot. (d) Normalized counts of key memory markers (P values are from Wald’s test, performed by “DESeq2” in R).
Taken together, MPs and TEs were highly transcriptionally distinct—MPs had proliferative and stem-like capacity, while TEs were terminally differentiated. In contrast, KLRG1+ MPs had a transcriptional profile which was roughly intermediate between those of MPs and TEs.
Longitudinal anatomic location of three KLRG1/IL-7Rα subsets within the spleen.
We next sought to investigate how KLRG1 and IL-7Rα expression may be specifically tied to anatomic location and how the location of memory CD8 T cell subsets may ultimately determine their fate. We examined these questions out to 1 year postinfection, specifically within the spleen.
To evaluate localization within the spleen, we performed i.v. injection of fluorescently conjugated anti-CD8α at 3 min prior to euthanasia of the mouse (41) (Fig. 6a). The injected antibody enters circulation and does not penetrate beyond the vasculature, so this technique enables the discrimination of blood-borne cells and tissue-localized cells (49). As such, CD8 T cells within the peripheral blood mononuclear cells (PBMCs) stained nearly 100% i.v.+ (Fig. 6b). In the spleen, the RP is highly vascularized, while the WP is not. Instead, the WP is comprised of lymphoid aggregates: periarteriolar lymphatic sheaths (PALS) surrounded by T cell zones and B cell zones. The RP is thus accessible to the injected antibody and stains largely i.v.+, while the WP is not accessible to the antibody and stains largely i.v.− (41). As is well-known, naive CD44lo CD8 T cells specifically home to T cell zones within the WP, and we found that they were, in fact, highly protected from i.v. labeling, staining >70% i.v.− (Fig. 6c). Naive CD8 T cells within infected mice also remained embedded at this proportion within the WP, and this population was used to determine the placement of the gates between the RP and WP for all experiments.
FIG 6.

Intravascular (i.v.) labeling reveals the location of GP33+ CD8 T cells in the spleen after acute infection. (a) Experimental scheme. Labeling was performed as described previously (41). Mice were infected with 2 × 105 PFU of LCMV Armstrong and sacrificed at multiple time points from 8 days to 1 year postinfection. At 3 min prior to sacrifice, mice received 3 μg of anti-CD8α i.v. that was conjugated to a fluorophore, which labeled all CD8 T cells in circulation and in the splenic RP. CD8 T cells in the tissues and the splenic WP were inaccessible. (b) GP33+ CD8 T cells within the PBMCs gated on i.v. labeling. If >95% of PBMCs were i.v.+, i.v. labeling was considered successful. (c) Naive CD44lo CD8 T cells within the spleen, gated on i.v. labeling. CD8 T cells were presumed to be in the RP if they stained i.v.+ or in the WP if they stained i.v.−. Naive CD44lo CD8 T cells were always used to establish the gates between i.v.+ and i.v.− tetramer+ CD8 T cells in the spleen. (d and e) Analysis of the location of GP33+ CD8 T cells in the spleen from 8 days to 1 year postinfection; representative flow plots (d) and summary plot (e) of the frequency of GP33+ CD8 T cells within the RP or WP. Individual mice are shown; n = 5 to 22 per time point. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001 (all determined using two-way ANOVA with Sidak’s test for multiple comparisons).
On day 8 postinfection, GP33+ CD8 T cells were roughly equally distributed throughout the RP and WP of the spleen, with a slight majority in the WP, although there was some variability among the samples (Fig. 6d and e). By day 15, there was a significant preference in the localization of the GP33+ CD8 T cells to the RP, as ~70% to 90% of GP33+ CD8 T cells stained i.v.+ at this time point. At day 30, a similar frequency of GP33+ CD8 T cells was observed in the RP. This preference continued until at least day 90, gradually becoming less dramatic with only ~50% to 70% of GP33+ CD8 T cells in the RP on days 60 and 90. By 6 months postinfection, GP33+ CD8 T cells had undergone complete reorganization. A significant majority (~50% to 80%) now appeared in the WP. At 1 year postinfection, the GP33+ CD8 T cells ultimately lost this WP preference, becoming more widely dispersed between the RP and WP once again, with no significant majority in either compartment. These patterns of localization were consistent among GP276+ and NP396+ CD8 T cells.
Finally, we investigated the heterogeneity of tetramer+ CD8 T cells undergoing these localization programs. We assessed the location of each KLRG1/IL-7Rα subset from 8 days to 1 year postinfection, hypothesizing that IL-7Rα and KLRG1 expression might delineate WP versus RP-associated cells, as has been suggested previously (21). Thus, we examined IL-7Rα and KLRG1 expression on GP33+ CD8 T cells using i.v. labeling (Fig. 7a and b). We found that MPs were skewed toward the WP as early as 1 week postinfection, with a significant majority persisting in the WP over the course of 1 year.
FIG 7.
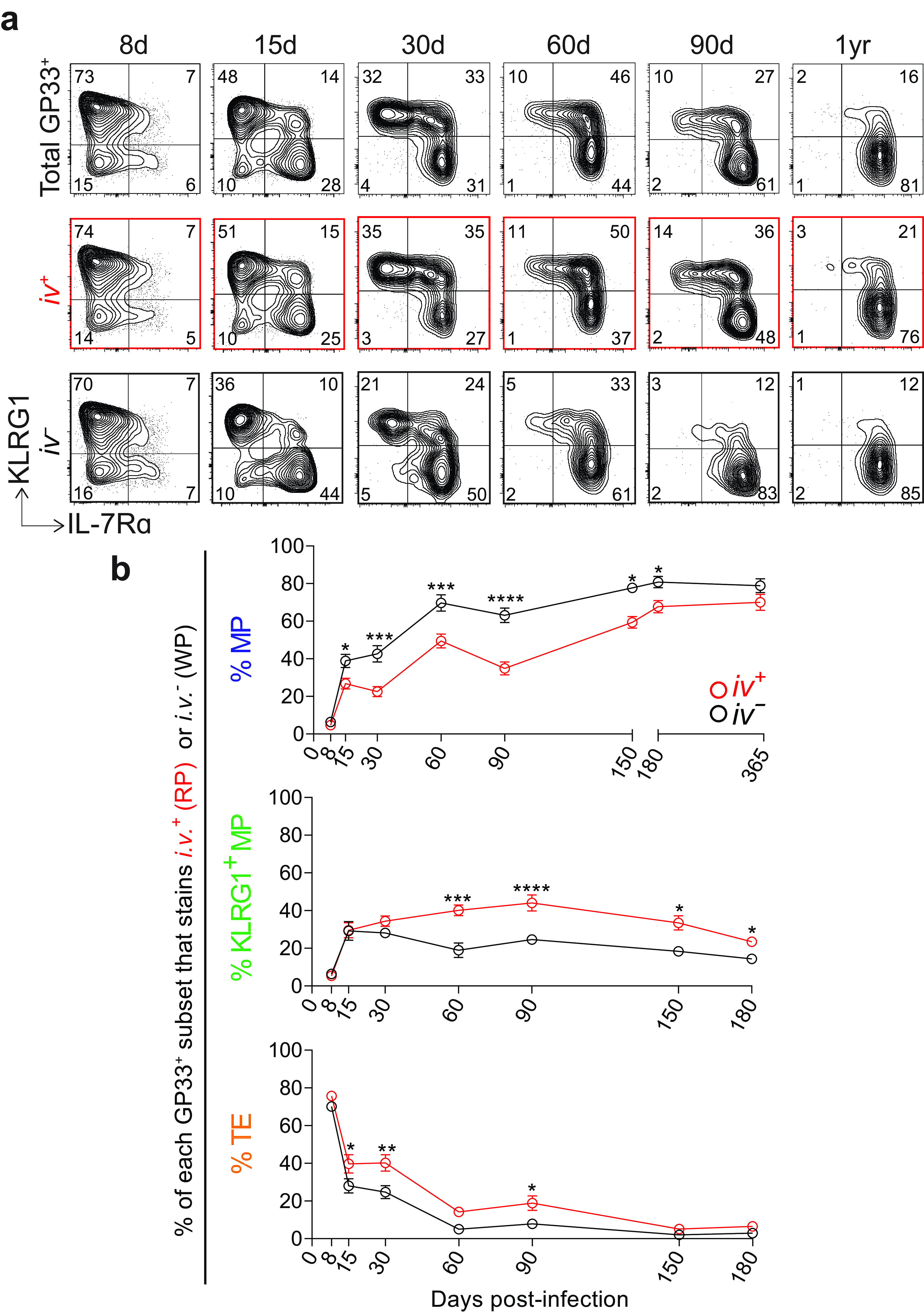
MPs localize to the splenic white pulp as early as 1 to 2 weeks after acute infection. (a) Representative flow plots showing KLRG1 and IL-7Rα expression on total GP33+ CD8 T cells in the spleen (top row). Below, GP33+ CD8 T cells are further gated by i.v. labeling, either i.v.+ (RP) (middle row) or i.v.− (WP) (bottom row). (b) Summary of the frequency of each memory subset within GP33+ CD8 T cells in the RP or WP. Shown are the mean and SEM; n = 5 to 9 mice per time point. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001 (all determined using two-way ANOVA with Sidak’s test for multiple comparisons).
On day 8 postinfection, at the peak of the CD8 T cell response, the majority of GP33+ CD8 T cells were TEs, and these TEs were in the RP in a slight majority. At day 15, MPs and KLRG1+ MPs were increasing in their overall proportion of the GP33+ population. Among GP33+ CD8 T cells in the WP, ~40% of were MPs, while this frequency was halved in the RP. Also at day 15, a slight majority of KLRG1+ MPs were in the RP, and a significant majority of TEs were in the RP. From 30 days to 6 months postinfection, stark differences emerged in the compartmentalization of each memory subset. In the WP, MPs were consistently found at nearly twice the frequency as in the RP. On the other hand, KLRG1+ MPs and TEs were consistently found at nearly twice the frequency in the RP versus those in the WP. By 1 year postinfection, the majority of GP33+ CD8 T cells were MPs and KLRG1+ MPs, and there was still a modestly higher frequency of MPs in the WP and KLRG1+ MPs in the RP.
DISCUSSION
It is well-established that KLRG1 and IL-7Rα together define the populations of CD8 T cells which persist or die following infection. TEs contract, while MPs survive and seed the long-lived memory pool. During this period of contraction and differentiation, we observed a third KLRG1+ MP population which actually accounted for ~5% to 40% of memory CD8 T cells at any given time. Our results show that KLRG1+ MPs possess a unique phenotype and functional program. This population has a surface expression profile which is intermediate between that of MPs and TEs, but their transcriptional program suggests they are more similar to MPs than to TEs. In contrast, KLRG1+ MPs appear to localize within the spleen in a manner most similar to TEs—within the RP. We speculate that TEs could actually give rise to KLRG1+ MPs, which may in turn give rise to MPs. That is, a subset of TEs with higher expression of IL-7Rα could selectively survive and seed the KLRG1+ MP pool, and KLRG1+ MPs could subsequently downregulate KLRG1 to become MPs. It is also possible that KLRG1+ MPs simply die, as do TEs, and the MP population dominates by default.
Our results show that CD28 is expressed by long-lived memory CD8 T cells out to 1 year post-acute infection. This finding is consistent with others’ studies in both mice and humans (3, 35, 50–53). We found that CD28 expression correlated with the expression of other memory-associated markers, including IL-7Rα, CD62L, TCF-1, and CXCR3, and inversely correlated with the expression of effector-associated markers, including KLRG1 and CX3CR1.
We also investigated the patterns of memory CD8 T cell localization following clearance of an acute viral infection. We examined their location within the spleen using i.v. labeling and characterized their location-specific phenotypes; these findings are summarized in Fig. 8. We found that bulk LCMV-specific CD8 T cells, although equally distributed on day 8, displayed a “preference” for the RP until day 90. We posit that this i.v.+ population is actually comprised of memory CD8 T cells (Tem and Tcm) which are in the midst of their canonical circulation, between the vasculature and secondary lymphoid organs.
FIG 8.

Summary of the anatomical distribution of memory CD8 T cell subsets over time. On day 8 following acute infection, the majority of antigen-specific CD8 T cells are TEs, and they are primarily localized within the RP. After, TEs begin to contract, while KLRG1+ MPs and MPs increase in their proportion. MPs quickly dominate within the WP—as early as 1 to 2 weeks following infection. KLRG1+ MPs also become enriched within the RP. By day 60, the majority of antigen-specific CD8 T cells are MPs and these are primarily localized within the WP. TEs and KLRG1+ MPs are significantly enriched within the RP.
As early as 1 week postinfection, we observed preferential localization of MPs to the WP and TEs/KLRG1+ MPs to the RP. This finding is consistent with that of previous studies from Jung et al. (21) and Seo et al. (22). But we continued our investigation significantly longer than those studies—out to 1 year postinfection—because we sought to track the precise longitudinal relationship between memory phenotype and location. Remarkably, the anatomic preferences established on day 8 postinfection held steady for ~1 year following infection. This result suggests that there are specific signals which regulate early localization following infection and that early homing may be crucial for driving the long-lived memory program. In particular, positioning within the T cell zones of the WP may regulate exposure to antigen as well as costimulatory signaling. Of course, it will also be necessary to determine whether these localization kinetics change in different contexts of acute infection, such as after respiratory, enteric, or epidermal infection.
Together, these data reexamine the relationship between memory CD8 T cell phenotype and location. We have uncovered novel characteristics of memory CD8 T cell subsets, and we have comprehensively tracked their localization within the spleen over an extended period of time. Our findings suggest that memory CD8 T cell function may be inextricably tied to their location. Designing vaccines with highly effective memory responses may require a more thorough consideration of the phenotype of memory CD8 T cells elicited and how these cells traffic within the spleen.
MATERIALS AND METHODS
Mice and viruses.
All animal experiments were performed in accordance with Emory University Institutional Animal Care and Use Committee. C57BL/6J female mice were purchased from Jackson Laboratory. Mice were infected with the LCMV Armstrong strain (2 × 105 PFU, intraperitoneally [i.p.]).
Cell isolation.
Once isolated from mice, spleens were smashed through 70-μm nylon cell strainers to remove clumps. Cells were spun down at 2,000 rpm for 10 min, and the supernatant was removed. After resuspending the pellet, cells were incubated at room temperature with 2 mL of Ack lysis buffer for 2 min in order to lyse the red blood cells (RBCs). After incubation, an additional 35 mL of RPMI + 5% fetal bovine serum (FBS) was added to dilute the Ack lysis buffer, and the cells were spun down at 2,000 rpm for 10 min. The resulting cell pellet was resuspended in phosphate-buffered saline (PBS) + 2% FBS + 0.5 mM EDTA, and cells were transferred to a 96-well round-bottom plate for staining.
Intravascular labeling.
Intravascular labeling was performed as described previously (41). Anti-CD8α conjugated to PE or Alexa Fluor 647 was prepared for injection (3 μg of antibody in 300 μL of PBS), and 300 μL of the antibody dilution was injected i.v. into each mouse via the tail vein. After 3 min, mice were bled via heparinized capillary tubes and then sacrificed using isoflurane. Spleens were collected in RPMI + 5% FBS and immediately smashed through 70-μm nylon cell strainers to remove clumps.
Flow cytometry.
Surface staining was performed by incubating cells with fluorochrome-conjugated antibodies against CD8, CD4, CD19, PD-1, CD44, IL-7Rα, KLRG1, CD62L, CD28, CXCR3, and CX3CR1 (BD Bioscience, eBioscience, and Biolegend). Cells were incubated on ice for 30 min in PBS + 2% FBS + 0.5 mM EDTA. The transcription factor TCF-1 was stained intracellularly with the eBioscience Foxp3/transcription factor fixation/permeabilization kit (Thermofisher). For detecting LCMV-specific CD8 T cell responses, peptides bound to major histocompatibility complex (pMHC) tetramers were prepared against LCMV-GP33, LCMV-GP276, and LCMV-NP396 proteins, as described previously (42, 54). Cell viability was determined with the live/dead fixable aqua or near-infrared (IR) dead cell stain kit (Invitrogen). Samples were acquired on an LSR II or Symphony flow cytometer (BD Biosciences), and data were analyzed with FlowJo software (TreeStar).
Bulk RNA sequencing and analysis.
Samples were stained with extracellular antibodies for 30 min, washed with PBS + 2% FBS, and sorted on a FACSAria II instrument (BD Biosciences). The following three populations of GP33+ CD8 T cells were sorted in triplicate: IL-7Rα+KLRG1-, IL-7Rα+KLRG1+, and IL-7Rα-KLRG1+. RNA was then isolated from each sample using the All-Prep DNA/RNA micro kit (Qiagen) and then submitted to the Emory Yerkes Nonhuman Primate Genomics Core. Reads from RNA-seq were aligned to the mm10 genome (accessed through Ensembl [55]) with STAR version 2.7. A cutoff of >3,000 counts per gene was applied to all samples.
Data analysis.
All experiments were analyzed using Prism 9 (GraphPad Software). Statistical differences were assessed using paired two-tailed t tests or two-way analysis of variance (ANOVA) with Sidak’s test for multiple comparisons. All summary plots indicate means ± standard error of the mean (SEM). In all cases *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; and ****, P ≤ 0.0001. The number of mice analyzed at each time point ranged from 3 to 22. The sample size for individual experiments is denoted in the figure legend.
Data availability.
All RNA-seq data has been deposited in NCBI’s Gene Expression Omnibus and is accessible through GEO Series accession number GSE218777.
ACKNOWLEDGMENTS
We thank Robert Karaffa and Kametha Fife for assistance with cell sorting in the Emory University School of Medicine Flow Cytometry Core. We also thank Kathryn Pellegrini in the Yerkes Nonhuman Primate Genomics Core for assistance with RNA-seq.
Funding was provided by National Institutes of Health grant R01AI030048 (R.A.) and Achievement Rewards for College Scientists Standard Scholarship (A.L.G.).
Conceptualization, A.L.G., H.T.K., R.S.A., and R.A.; Methodology, A.L.G., H.T.K., and R.A.; Investigation, A.L.G., W.H.H., R.M.V., E.A., A.W., and D.T.M.; Visualization, A.L.G., W.H.H., D.J.M., and R.A.; Funding acquisition, R.A.; Project administration, R.A.; Supervision, R.A.; Writing – original draft, A.L.G., W.H.H., and R.A.; Writing – review & editing, A.L.G. and R.A.
Contributor Information
Rafi Ahmed, Email: rahmed@emory.edu.
Stacey Schultz-Cherry, St. Jude Children's Research Hospital.
REFERENCES
- 1.Kaech SM, Hemby S, Kersh E, Ahmed R. 2002. Molecular and functional profiling of memory CD8 T cell differentiation. Cell 111:837–851. 10.1016/s0092-8674(02)01139-x. [DOI] [PubMed] [Google Scholar]
- 2.Wherry EJ, Ahmed R. 2004. Memory CD8 T-cell differentiation during viral infection. J Virol 78:5535–5545. 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, Li KW, Youngblood BA, Abdelsamed HA, McGuire DJ, Cohen KW, Alexe G, Nagar S, McCausland MM, Gupta S, Tata P, Haining WN, McElrath MJ, Zhang D, Hu B, Greenleaf WJ, Goronzy JJ, Mulligan MJ, Hellerstein M, Ahmed R. 2017. Origin and differentiation of human memory CD8 T cells after vaccination. Nature 552:362–367. 10.1038/nature24633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A, Araki K, West EE, Ghoneim HE, Fan Y, Dogra P, Davis CW, Konieczny BT, Antia R, Cheng X, Ahmed R. 2017. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature 552:404–409. 10.1038/nature25144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dutton RW, Bradley LM, Swain SL. 1998. T cell memory. Annu Rev Immunol 16:201–223. 10.1146/annurev.immunol.16.1.201. [DOI] [PubMed] [Google Scholar]
- 6.Kaech SM, Cui W. 2012. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol 12:749–761. 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Homann D, Teyton L, Oldstone MBA. 2001. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. 8. Nat Med 7:913–919. 10.1038/90950. [DOI] [PubMed] [Google Scholar]
- 8.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. 2003. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol 4:1191–1198. 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 9.Wherry EJ, Teichgräber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R. 2003. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol 4:225–234. 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 10.Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. 2008. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med 205:625–640. 10.1084/jem.20071641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaech SM, Wherry EJ. 2007. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity 27:393–405. 10.1016/j.immuni.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. 1999. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401:708–712. 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 13.Martin MD, Badovinac VP. 2018. Defining memory CD8 T cell. Front Immunol 9:2692. 10.3389/fimmu.2018.02692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, Lucas PJ, Artis D, Wherry EJ, Hogquist K, Vezys V, Masopust D. 2012. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol 188:4866–4875. 10.4049/jimmunol.1200402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masopust D, Vezys V, Wherry EJ, Barber DL, Ahmed R. 2006. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J Immunol 176:2079–2083. 10.4049/jimmunol.176.4.2079. [DOI] [PubMed] [Google Scholar]
- 16.Mueller SN, Zaid A, Carbone FR. 2014. Tissue-resident T cells: dynamic players in skin immunity. Front Immunol 5:332. 10.3389/fimmu.2014.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosato PC, Beura LK, Masopust D. 2017. Tissue resident memory T cells and viral immunity. Curr Opin Virol 22:44–50. 10.1016/j.coviro.2016.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dauner JG, Williams IR, Jacob J. 2008. Differential microenvironment localization of effector and memory CD8 T cells. J Immunol 180:291–299. 10.4049/jimmunol.180.1.291. [DOI] [PubMed] [Google Scholar]
- 19.Khanna KM, McNamara JT, Lefrançois L. 2007. In situ imaging of the endogenous CD8 T cell response to infection. Science 318:116–120. 10.1126/science.1146291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Potsch C, Vöhringer D, Pircher H. 1999. Distinct migration patterns of naive and effector CD8 T cells in the spleen: correlation with CCR7 receptor expression and chemokine reactivity. Eur J Immunol 29:3562–3570. . [DOI] [PubMed] [Google Scholar]
- 21.Jung YW, Rutishauser RL, Joshi NS, Haberman AM, Kaech SM. 2010. Differential localization of effector and memory CD8 T cell subsets in lymphoid organs during acute viral infection. J Immunol 185:5315–5325. 10.4049/jimmunol.1001948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seo Y-J, Jothikumar P, Suthar MS, Zhu C, Grakoui A. 2016. Local cellular and cytokine cues in the spleen regulate in situ T cell receptor affinity, function, and fate of CD8+ T cells. Immunity 45:988–998. 10.1016/j.immuni.2016.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, Heath WR, Carbone FR, Gebhardt T. 2012. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci USA 109:7037–7042. 10.1073/pnas.1202288109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schenkel JM, Fraser KA, Masopust D. 2014. Cutting edge: resident memory CD8 T cells occupy frontline niches in secondary lymphoid organs. J Immunol 192:2961–2964. 10.4049/jimmunol.1400003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarkar S, Teichgraber V, Kalia V, Polley A, Masopust D, Harrington LE, Ahmed R, Wherry EJ. 2007. Strength of stimulus and clonal competition impact the rate of memory CD8 T cell differentiation. J Immunol 179:6704–6714. 10.4049/jimmunol.179.10.6704. [DOI] [PubMed] [Google Scholar]
- 26.Martin MD, Kim MT, Shan Q, Sompallae R, Xue H-H, Harty JT, Badovinac VP. 2015. Phenotypic and functional alterations in circulating memory CD8 T cells with time after primary infection. PLoS Pathog 11:e1005219. 10.1371/journal.ppat.1005219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Escobar G, Mangani D, Anderson AC. 2020. T cell factor 1: a master regulator of the T cell response in disease. Sci Immunol 5:eabb9726. 10.1126/sciimmunol.abb9726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kratchmarov R, Magun AM, Reiner SL. 2018. TCF1 expression marks self-renewing human CD8+ T cells. Blood Adv 2:1685–1690. 10.1182/bloodadvances.2018016279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Willinger T, Freeman T, Herbert M, Hasegawa H, McMichael AJ, Callan MFC. 2006. Human naive CD8 T cells down-regulate expression of the WNT pathway transcription factors lymphoid enhancer binding factor 1 and transcription factor 7 (T cell factor-1) following antigen encounter in vitro and in vivo. J Immunol 176:1439–1446. 10.4049/jimmunol.176.3.1439. [DOI] [PubMed] [Google Scholar]
- 30.Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, Anderson SM, Handon R, Kenyon E, Elkahloun A, Wu W, Lang PA, Gattinoni L, McGavern DB, Schwartzberg PL. 2016. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol 1:eaai8593. 10.1126/sciimmunol.aai8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao D-M, Yu S, Zhou X, Haring JS, Held W, Badovinac VP, Harty JT, Xue H-H. 2010. Constitutive activation of Wnt signaling favors generation of memory CD8 T cells. J Immunol 184:1191–1199. 10.4049/jimmunol.0901199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou X, Yu S, Zhao D-M, Harty JT, Badovinac VP, Xue H-H. 2010. Differentiation and persistence of memory CD8+ T cells depend on T cell factor 1. Immunity 33:229–240. 10.1016/j.immuni.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linsley PS, Ledbetter JA. 1993. The role of the CD28 receptor during T cell responses to antigen. Annu Rev Immunol 11:191–212. 10.1146/annurev.iy.11.040193.001203. [DOI] [PubMed] [Google Scholar]
- 34.Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. 2016. CD28 costimulation: from mechanism to therapy. Immunity 44:973–988. 10.1016/j.immuni.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akondy RS, Monson ND, Miller JD, Edupuganti S, Teuwen D, Wu H, Quyyumi F, Garg S, Altman JD, Del Rio C, Keyserling HL, Ploss A, Rice CM, Orenstein WA, Mulligan MJ, Ahmed R. 2009. The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. J Immunol 183:7919–7930. 10.4049/jimmunol.0803903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Groom JR, Luster AD. 2011. CXCR3 in T cell function. Exp Cell Res 317:620–631. 10.1016/j.yexcr.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu JK, Kagari T, Clingan JM, Matloubian M. 2011. Expression of chemokine receptor CXCR3 on T cells affects the balance between effector and memory CD8 T-cell generation. Proc Natl Acad Sci USA 108:E118–E127. 10.1073/pnas.1101881108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurachi M, Kurachi J, Suenaga F, Tsukui T, Abe J, Ueha S, Tomura M, Sugihara K, Takamura S, Kakimi K, Matsushima K. 2011. Chemokine receptor CXCR3 facilitates CD8+ T cell differentiation into short-lived effector cells leading to memory degeneration. J Exp Med 208:1605–1620. 10.1084/jem.20102101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Böttcher JP, Beyer M, Meissner F, Abdullah Z, Sander J, Höchst B, Eickhoff S, Rieckmann JC, Russo C, Bauer T, Flecken T, Giesen D, Engel D, Jung S, Busch DH, Protzer U, Thimme R, Mann M, Kurts C, Schultze JL, Kastenmüller W, Knolle PA. 2015. Functional classification of memory CD8+ T cells by CX3CR1 expression. Nat Commun 6:8306. 10.1038/ncomms9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, Garg R, de la Torre JC, von Andrian UH. 2016. The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity 45:1270–1284. 10.1016/j.immuni.2016.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, Masopust D. 2014. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc 9:209–222. 10.1038/nprot.2014.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murali-Krishna K, Altman JD, Suresh M, Sourdive DJD, Zajac AJ, Miller JD, Slansky J, Ahmed R. 1998. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity 8:177–187. 10.1016/S1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 43.van der Most RG, Sette A, Oseroff C, Alexander J, Murali-Krishna K, Lau LL, Southwood S, Sidney J, Chesnut RW, Matloubian M, Ahmed R. 1996. Analysis of cytotoxic T cell responses to dominant and subdominant epitopes during acute and chronic lymphocytic choriomeningitis virus infection. J Immunol 157:5543–5554. [PubMed] [Google Scholar]
- 44.van der Most RG, Murali-Krishna K, Whitton JL, Oseroff C, Alexander J, Southwood S, Sidney J, Chesnut RW, Sette A, Ahmed R. 1998. Identification of Db- and Kb-restricted subdominant cytotoxic T-cell responses in lymphocytic choriomeningitis virus-infected mice. Virology 240:158–167. 10.1006/viro.1997.8934. [DOI] [PubMed] [Google Scholar]
- 45.Blattman JN, Wherry EJ, Ha S-J, van der Most RG, Ahmed R. 2009. Impact of epitope escape on PD-1 expression and CD8 T-cell exhaustion during chronic infection. J Virol 83:4386–4394. 10.1128/JVI.02524-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Groom JR, Richmond J, Murooka TT, Sorensen EW, Sung JH, Bankert K, von Andrian UH, Moon JJ, Mempel TR, Luster AD. 2012. CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity 37:1091–1103. 10.1016/j.immuni.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wherry EJ, Ha S-J, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. 2007. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27:670–684. 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 48.Ahn E, Araki K, Hashimoto M, Li W, Riley JL, Cheung J, Sharpe AH, Freeman GJ, Irving BA, Ahmed R. 2018. Role of PD-1 during effector CD8 T cell differentiation. Proc Natl Acad Sci USA 115:4749–4754. 10.1073/pnas.1718217115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anderson KG, Sung H, Skon CN, Lefrancois L, Deisinger A, Vezys V, Masopust D. 2012. Cutting edge: intravascular staining redefines lung CD8 T cell responses. J Immunol 189:2702–2706. 10.4049/jimmunol.1201682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boesteanu AC, Katsikis PD. 2009. Memory T cells need CD28 costimulation to remember. Semin Immunol 21:69–77. 10.1016/j.smim.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Borowski AB, Boesteanu AC, Mueller YM, Carafides C, Topham DJ, Altman JD, Jennings SR, Katsikis PD. 2007. Memory CD8+ T cells require CD28 costimulation. J Immunol 179:6494–6503. 10.4049/jimmunol.179.10.6494. [DOI] [PubMed] [Google Scholar]
- 52.Fuse S, Zhang W, Usherwood EJ. 2008. Control of memory CD8+ T cell differentiation by CD80/CD86-CD28 costimulation and restoration by IL-2 during the recall response. J Immunol 180:1148–1157. 10.4049/jimmunol.180.2.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suresh M, Whitmire JK, Harrington LE, Larsen CP, Pearson TC, Altman JD, Ahmed R. 2001. Role of CD28-B7 interactions in generation and maintenance of CD8 T cell memory. J Immunol 167:5565–5573. 10.4049/jimmunol.167.10.5565. [DOI] [PubMed] [Google Scholar]
- 54.Altman JD, Moss PAH, Goulder PJR, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM. 1996. Phenotypic analysis of antigen-specific T lymphocytes. Science 274:94–96. 10.1126/science.274.5284.94. [DOI] [PubMed] [Google Scholar]
- 55.Cunningham F, Allen JE, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Austine-Orimoloye O, Azov AG, Barnes I, Bennett R, Berry A, Bhai J, Bignell A, Billis K, Boddu S, Brooks L, Charkhchi M, Cummins C, Da Rin Fioretto L, Davidson C, Dodiya K, Donaldson S, El Houdaigui B, El Naboulsi T, Fatima R, Giron CG, Genez T, Martinez JG, Guijarro-Clarke C, Gymer A, Hardy M, Hollis Z, Hourlier T, Hunt T, Juettemann T, Kaikala V, Kay M, Lavidas I, Le T, Lemos D, Marugán JC, Mohanan S, Mushtaq A, Naven M, Ogeh DN, Parker A, Parton A, Perry M, Piližota I, Prosovetskaia I, et al. 2022. Ensembl 2022. Nucleic Acids Res 50:D988–D995. 10.1093/nar/gkab1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All RNA-seq data has been deposited in NCBI’s Gene Expression Omnibus and is accessible through GEO Series accession number GSE218777.