

Abstract
Cancer metastasis requires the transient activation of cellular programs enabling dissemination and seeding in distant organs1. Genetic, transcriptional and translational heterogeneity contributes to this dynamic process2,3. Metabolic heterogeneity has also been observed4, yet its role in cancer progression is less explored. Here, we discover that loss of phosphoglycerate dehydrogenase (PHGDH) potentiates metastatic dissemination. Specifically, we find that heterogeneous or low PHGDH expression in primary tumors of breast cancer patients is associated with decreased metastasis free survival time. In mice, circulating tumor cells and early metastatic lesions are enriched with PHGDH low cancer cells and silencing PHGDH in primary tumors increases metastasis formation. Mechanistically, PHGDH protein interacts with the glycolytic enzyme phosphofructokinase (PFK) and the loss of this interaction activates the hexosamine – sialic acid pathway, which provides precursors for protein glycosylation. Consequently, aberrant protein glycosylation including increased sialylation of integrin αvβ3 occurs, which potentiates cell migration and invasion. Inhibition of sialic acid metabolism counteracts the metastatic capacity of PHGDH low cancer cells. In conclusion, while the catalytic activity of PHGDH supports cancer cell proliferation, low PHGDH protein expression non-catalytically potentiates cancer dissemination and metastasis formation. Thus, the presence of PHDGH heterogeneity in primary tumors may be considered a sign of tumor aggressiveness.
Nonclonal tumor heterogeneity at the transcriptional and translational level promotes the plasticity of cancer cells by allowing transient activation of cellular programs1,5. These, in turn, enable cancer cells to dynamically change their phenotypical state and transition through the different steps of the metastatic cascade6. Yet, the role of non-clonal heterogeneity in the metabolism of cancer cells remains largely elusive. Here, we focused on the metabolic enzyme PHGDH, which is overexpressed and/or amplified in 70% of triple-negative breast cancers (TNBC) and some other cancers such as melanoma7,8 and its catalytic activity is known to be important for cancer proliferation9–12.
PHGDH heterogeneity indicates metastasis
We investigated intra-tumor heterogeneity in PHGDH protein expression in 129 predominately grade 2 and 3 invasive human breast ductal carcinomas that were treated by primary surgical resection (1988–2006)13. PHGDH was visualized using immunohistochemistry and intra-tumor heterogeneity and intensity was evaluated by a pathologist (Supplementary Table 1). We observed that 67% (87 out of 129) of the tumors showed a homogeneous high PHGDH expression, while 33% showed heterogeneous (29 out of 129) or low (13 out of 129) PHGDH expression (Extended Data Figure 1a). Homogeneous versus heterogeneous/low PHGDH expressing tumors were similarly distributed across the different tumor grades (Extended Data Figure 1b). Tumor stage (pT) was higher in homogeneous versus heterogeneous/low PHGDH-expressing tumors (Extended Data Figure 1c). Unexpectedly however, lymph node stage (pN) was significantly more advanced in heterogeneous/low PHGDH-expressing tumors compared to homogeneous PHGDH-expressing tumors (Extended Data Figure 1d). Accordingly, about 60% more patients with heterogeneous/low PHGDH-expressing tumors showed distant metastases (33%; 14 out of 42) compared to patients with homogeneous PHGDH-expressing tumors (20%; 17 out of 87; Extended Data Figure 1e). Median disease-free and metastasis-free survival time decreased by a factor of about 2.9 and 3.2, respectively, in patients with heterogeneous/low PHGDH-expressing tumors compared to patients with homogeneous PHGDH-expressing tumors (Figure 1a, b). Previously, low PHGDH protein expression was associated with shorter overall survival time14, which is consistent with a respective trend in our cohort (Extended Data Figure 1f). Thus, we concluded that heterogeneous and low PHGDH protein expression is indicative of metastasis in TNBC patients.
Figure 1. The presence of PHGDH low cancer cells in primary tumors indicates poor prognosis and PHGDH expression decreases in CTCs and early metastasis.
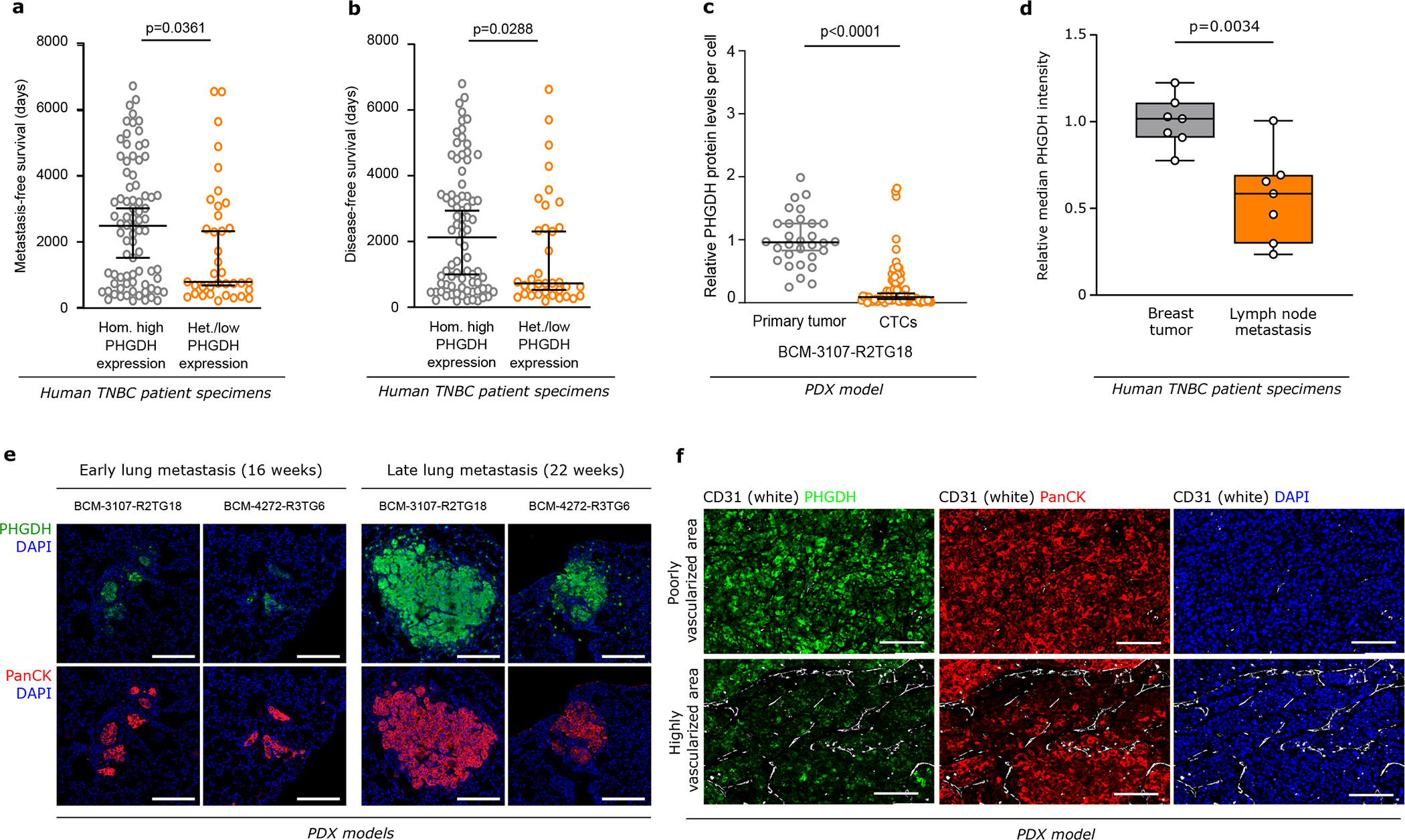
a-b. Metastasis-free (a) and disease-free (b) survival (days) of patients with TNBC divided by homogeneous high and heterogeneous/low PHGDH expression in primary tumors. The solid lines indicate the median, the whiskers indicate the 95% confidence interval. n=129; homogeneous high expression, n=87; heterogeneous expression, n=29; low expression, n=13. Unpaired nonparametric Mann-Whitney test, two-tailed.
c. PHGDH protein expression in circulating tumor cells (CTCs) compared to the respective primary tumors from orthotopic (m.f.p.) TNBC BCM-3107-R2TG18 PDX model, assessed by immunohistochemistry (6 mice, 30 randomly chosen microscopy fields for the primary tumors, 5 per mouse, 87 single CTCs). The solid lines indicate the median, the whiskers indicate the 95% confidence interval. Unpaired t test with Welch’s correction, two-tailed.
d. PHGDH protein expression in lymph node metastases and matched primary breast tumors from TNBC patients (n=7), assessed by immunohistochemistry. The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the minimum and maximum values. Unpaired t test with Welch’s correction, two-tailed.
e. Representative pictures of PHGDH protein expression in early (16 weeks) and late (22 weeks) lung metastases from orthotopic (m.f.p.) TNBC PDX models, assessed by immunohistochemistry. Green, PHGDH; red, pan-cytokeratin tumor marker; blue, DAPI nuclear staining. Scale bar 200 μm.
f. PHGDH protein expression in TNBC BCM-3107-R2TG18 PDX model in low or high vascularized primary tumor areas (defined by CD31 staining), assessed by immunohistochemistry. Green, Phgdh; red, pan-cytokeratin tumor marker; blue, DAPI nuclear staining. Scale bar 200 μm.
CTCs and early metastasis are PHGDH low
Like human invasive ductal carcinomas, mouse primary 4T1- and PDX-derived (BCM-5471) breast tumors displayed intra-tumor heterogeneity in PHGDH protein expression (Extended Data Figure 1g, 1h). PHGDH-low cancer cells were in a slow cycling state, as evidenced by the positive correlation between PHGDH expression and the proliferation markers Ki67 and phospho-histone H3 (PHH3; Extended Data Figure 1i, 1j, 1k). Moreover, in patients with TNBC breast cancer, a gene expression signature indicative of low PHGDH protein expression was inversely correlated with Ki67 expression and correlated with an epithelial-to mesenchymal-transition (EMT) signature (Extended Data Figure 1l).
Slow cycling tumor cells have been associated with metastasis initiation capacity15. Therefore, we asked whether average PHGDH expression changes during metastatic progression. We analyzed PHGDH protein expression in circulating tumor cells (CTCs) of three TNBC PDX mouse models (BCM-3107-R2TG18, BCM-3611-R3TG4 and BCM-4272-R3TG6) and compared it to the expression in the corresponding primary tumors. Strikingly, CTCs from all three PDX-models showed a 2.5- to 10.7-fold decrease in median PHGDH protein expression compared to the corresponding primary tumors (Figure 1c, Extended Data Figure 1m, 1n). Next, we investigated average PHGDH protein expression in early (small) metastatic lesions and compared them to the corresponding primary breast tumors. While primary 4T1 and EMT6.5 breast tumors as well as UM12 and M481 PDX melanomas showed on average high PHGDH protein expression, early lung metastases were defined by low average PHGDH protein expression (Extended Data Figure 2a, 2b, 2c, Supplementary Figure 1). Similarly, 63% (33 out of 52) of early lung metastases from the three different PDX breast tumors exhibited average low or no PHGDH protein expression compared to the corresponding primary tumors (Extended Data Figure 2d). Consistently, lymph node metastases from patients with breast cancer showed a lower average PHGDH protein expression compared to the matching primary breast tumors (Figure 1d, Extended Data Figure 2e, Supplementary Table 2). Unexpectedly, Phgdh mRNA expression did not decrease in accordance with protein expression (Extended Data Figure 2f).
Interestingly, we observed a correlation between PHGDH protein expression and lung metastases size in the 4T1 breast cancer mouse model and the Tyr::NrasQ61K/°; Ink4a−/−; Dsred murine melanoma model (Extended Data Figure 2g, 2h, Supplementary Figure 1). This suggests that PHGDH protein expression correlates with metastasis size and, potentially, proliferation. Thus, we analyzed PHGDH protein expression in lung metastases at two time points in two different PDX mouse models (BCM-3107-R2TG18 and BCM-4272-R3TG6). Remarkably, we discovered that early metastatic lesions had lower PHGDH protein expression compared to advanced metastatic lesions (Figure 1e). These data are consistent with the notion that average PHGDH protein expression is maintained at low levels during dissemination and increases when metastases proliferate.
Endothelial cells induce PHGDH loss
Next, we asked what induces the loss of PHGDH protein in cancer cells. We excluded microenvironmental conditions such as hypoxia and reoxygenation, depletion of serine, ATF4 activation or ER stress induction (Extended Data Figure 2i, 2j, Supplementary Figure 1). Therefore, we focused on stromal cells and particular endothelial cells, whose presence has been linked to tumor heterogeneity16. We co-cultured 4T1 and EMT6.5 cells with Bend3 immortalized mouse endothelial cells. Cancer and endothelial cells were distinguished based on cytokeratin (4T1, EMT6.5) or GFP (Bend3) expression. Strikingly, we observed that the presence of endothelial cells decreased PHGDH protein expression in both cell lines (Extended Data Figure 3a, 3b). Accordingly, 4T1 and EMT6.5 cells in co-culture with endothelial cells showed similar gene-set enrichment profiles than 4T1 cells silenced for Phgdh based on RNA sequencing (Extended Data Figure 3c). Moreover, we found that highly vascularized PDX tumor areas had low PHGDH protein expression and vice versa (Figure 1f). In line, co-injection of 4T1 breast cancer or Tyr::NrasQ61K/°; Ink4a−/−; Dsred melanoma cells with endothelial cells decreased PHGDH protein expression in the primary tumor and increased the metastatic area (Extended Data Figure3d, 3e, 3f). Thus, we concluded that cancer cells in highly vascularized tumor areas exhibit low PHGDH protein expression and vice versa.
Loss of PHGDH potentiates dissemination
Next, we asked whether loss of PHGDH protein expression increases cancer cell dissemination using time-lapse intravital imaging17,18 (Extended Data Figure 3g). We injected a mix of control 4T1 cells (expressing mTurquoise) and Phgdh-silenced cells (expressing Dendra) into the mammary fat pad of mice. Tumors of an approximate volume of 200 mm3 were surgically exposed and intravital images were acquired. Within each imaging field, the migratory behavior of the same number of randomly selected control and Phgdh-silenced cells was assessed. We found a significantly higher number of migratory cells in the Phgdh-silenced population than in the control population (Figure 2a). Furthermore, within the migratory population (>4 μm/h), Phgdh-silenced cells demonstrated a higher displacement compared to control cells (Figure 2b, Extended Data Figure 3h, Supplementary Video 1 and 2). Consequently, we analyzed the number of early metastatic lung lesions upon Phgdh silencing in 4T1 and EMT6.5 breast cancer mouse models. In line, we observed that Phgdh silencing increased the number of early metastatic lung lesions compared to control, (Figure 2c). Primary tumor weight did not significantly change (Supplementary Table 3). Next, we determined the rate of metastatic progression (area increase over time) of 4T1 lung metastases with and without Phgdh silencing. We observed that lung metastases with forced low PHGDH expression displayed a decreased rate of metastatic progression compared to control metastases that can regain PHGDH expression once successfully disseminated and seeded in the lung (Extended Data Figure 3i). Taken together, we found that Phgdh silencing increased in vivo dissemination and early metastatic seeding but hindered metastasis proliferation.
Figure 2: Low PHGDH expression promotes integrin αvβ3-mediated invasion and migration.
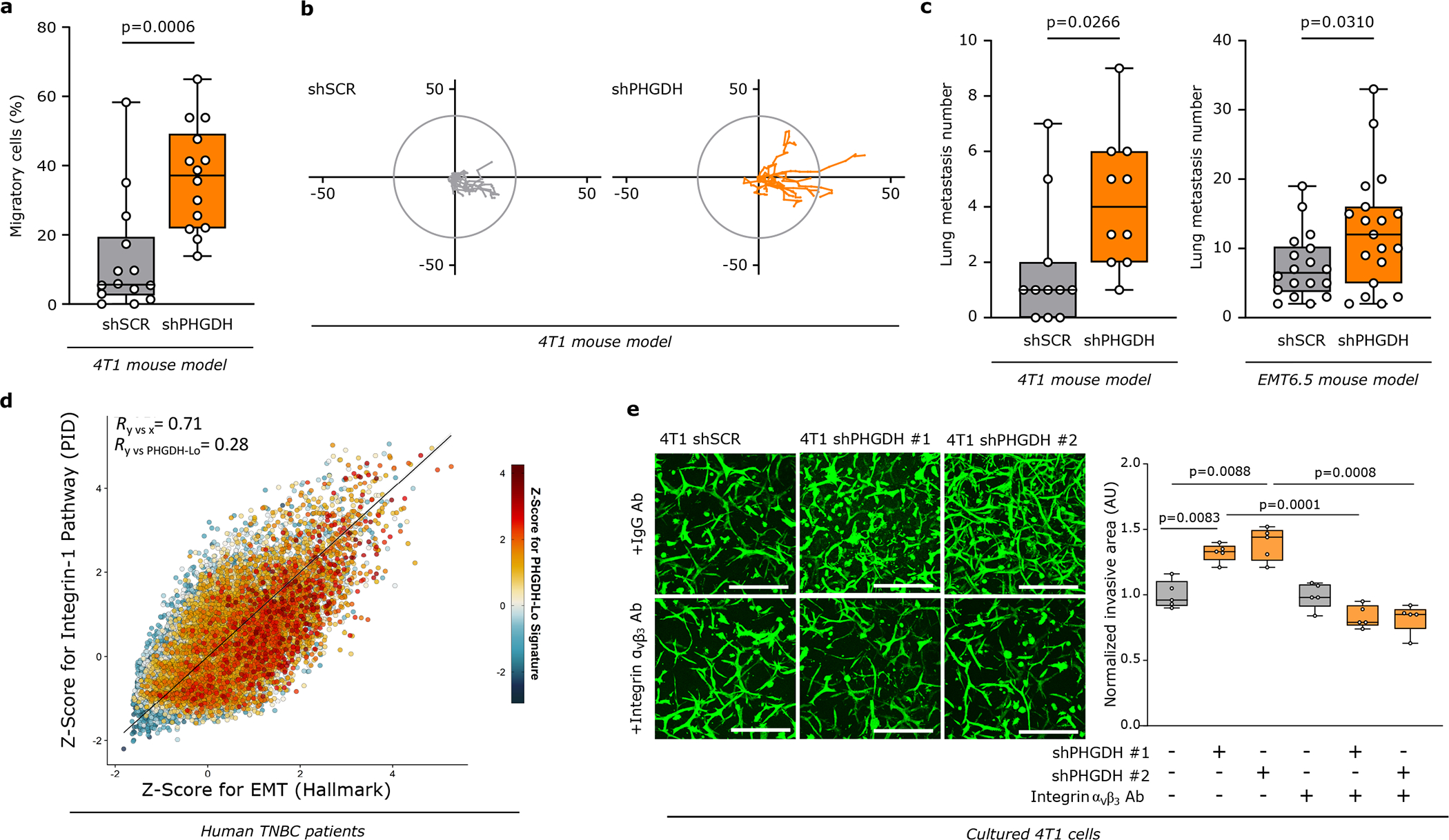
a. Percentage of migratory cells per migratory position (n=14) in primary tumors derived from orthotopically injected (m.f.p.) 4T1 (mixture of shSCR mTurquoise and shPHGDH Dendra) cells assessed by time-lapse intravital imaging (n=8). Unpaired t test with Welch’s correction, two-tailed.
b. Representative track length of migratory shSCR-mTurquoise (n=9 within one position) and shPHGDH-Dendra (n=10 within one position) cells in primary tumors from the orthotopic (m.f.p.) 4T1 mouse model, assessed by time-lapse intravital imaging.
c. Number of lung metastases per mouse in the orthotopic (m.f.p.) 4T1 (left panel) and EMT6.5 (right panel) mouse model, injected with either shSCR (n=11 and n=18) or shPHGDH cells (n=10 and n=19). Unpaired t test with Welch’s correction, two-tailed.
d. Correlation plot of GSVA-derived Z-scores for the Integrin-1 pathway, versus the scores for the Hallmark EMT gene signature, based on scRNA-seq data from primary tumors of 13 TNBC patients. Color code indicates the Z-Score for a gene expression signature indicative of low PHGDH protein expression. Total least-squares regression line and confidence intervals are overlaid on top, and Pearson correlation coefficient (R) values are shown on the top-left corners.
e. Invasive ability of 4T1 cells pre-treated (24h) with an antibody against integrin αvβ3 or control IgG (2.5 μg/ml) and upon Phgdh knockdown (shPHGDH) compared to control (shSCR) cells in a 3D matrix. The invasive area was stained with calcein green. Each dot represents a different microscopy field (n=5). Welch and Brown-Forsythe ANOVA with Dunnett’s multiple comparison.
For panels a, c, and e, the solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the minimum and maximum values.
Loss of PHGDH activates integrin αvβ3
Next, we confirmed that also in vitro Phgdh silencing increased migration and invasion (Extended Data Fig. 3j–k, 4a) as well as molecular markers of cancer cell plasticity (Extended Data Figure 4b–h). Consistently, Phgdh or PHGDH overexpression resulted in a decreased invasive area in Matrigel-collagen I compared to control in 4T1 and MDA-MB-231 breast cancer cells (Extended Data Figure 4i, 4j). Based on these data we concluded that PHGDH status modulates in vitro migration and invasion. To define the underlying mechanism, we applied gene-set enrichment analysis to RNA sequencing and proteomics data from cultured 4T1 cancer cells upon Phgdh silencing, and to the RNA sequencing data from 4T1 and EMT6.5 cells co-culture with Bend3 endothelial cells. Considering all three datasets simultaneously, we found a strong enrichment for gene/protein sets indicative of an altered EMT, matrix metalloproteinase (MMP) activity, extracellular matrix (ECM) remodeling, glycoprotein processing, integrin signaling, osteopontin (Opn also known as Spp1) signaling via AP-1, and KRAS signaling (Extended Data Figure 5a). Conversely, many of these gene-sets were downregulated in cultured MDA-MB-231 cells upon PHGDH overexpression (Extended Data Figure 5b). Accordingly, in TNBC tumors from patients, single cell-level enrichment scores for gene sets indicative of integrin signaling, Opn signaling via AP-1, MMP activity, extracellular matrix degradation, EMT and a gene expression signature indicative of low PHGDH protein expression were all correlated (Figure 2d, Extended Data Figure 5c). Interestingly, integrin function and in particular integrin αvβ3 function has been related to EMT19,20, KRAS signaling21, OPN signaling via AP-122 and MMP22 signatures. Thus, we hypothesized that low PHGDH expression induces cell invasion via integrin αvβ3. Accordingly, activity of MMP3, which is well described to bind to integrin αvβ322, was increased in invading 4T1 cells upon loss of PHGDH (Extended Data Figure 5d). Next, we inhibited integrin αvβ3 function using a blocking antibody and found impaired invasion of Phgdh silenced but not control 4T1 and EMT6.5 cells (Figure 2e, Extended Data Figure 5e). Thus, we concluded that low PHGDH expression induces cell invasion via integrin αvβ3 function.
Loss of PHGDH increases sialylation
Integrin αvβ3 is sialylated22, which is the covalent addition of sialic acid to the terminal end of a protein, known to support migration and invasion23–25. Sialic acid is produced by the hexosamine-sialic acid pathway, which branches from glycolysis at the level of fructose-6-phosphate (Extended Data Figure 6a). We hypothesized that the hexosamine-sialic acid pathway is induced by Phgdh silencing leading to increased sialylation of integrin αvβ3. We determined the activity of the hexosamine-sialic acid pathway in Phgdh silenced 4T1 cells using dynamic 13C tracer analysis26. We found an increased carbon flux through the hexosamine-sialic acid pathway and increased levels of the main metabolites upon Phgdh silencing in 4T1 cells (Figure 3a, Extended Data Figure 6b, Supplementary Table 4). Conversely, overexpression of PHGDH in MDA-MB-231 cells led to decreased sialic acid pathway flux and metabolite levels (Extended Data Figure 6c, 6d, Supplementary Table 5). Based on these data, we concluded that low PHGDH expression activates the hexosamine-sialic acid pathway in breast cancer cells. Consequently, we hypothesized that this increases protein sialylation, which is a form of N-glycosylation. Thus, we removed N-glycosylation from 4T1 cells using tunicamycin (0.05 μg/ml, 72 hours)27 (Extended Data Figure 6e). Subsequently, we washed out tuncamycin and monitored β-1,4-GlcNAc- and sialic acid-linked proteins using confocal imaging based on fluorescent wheat germ agglutinin (WGA)28. We found that Phgdh-silenced cancer cells recovered faster their protein glycosylation showing that PHGDH loss facilitates protein glycosylation (Extended Data Figure 6f). Moreover, Phgdh-silenced cancer cells were more sensitive to tunicamycin treatment (Extended Data Figure 6g).
Figure 3: Low PHGDH expression promotes sialic acid metabolism and consequently protein sialylation.
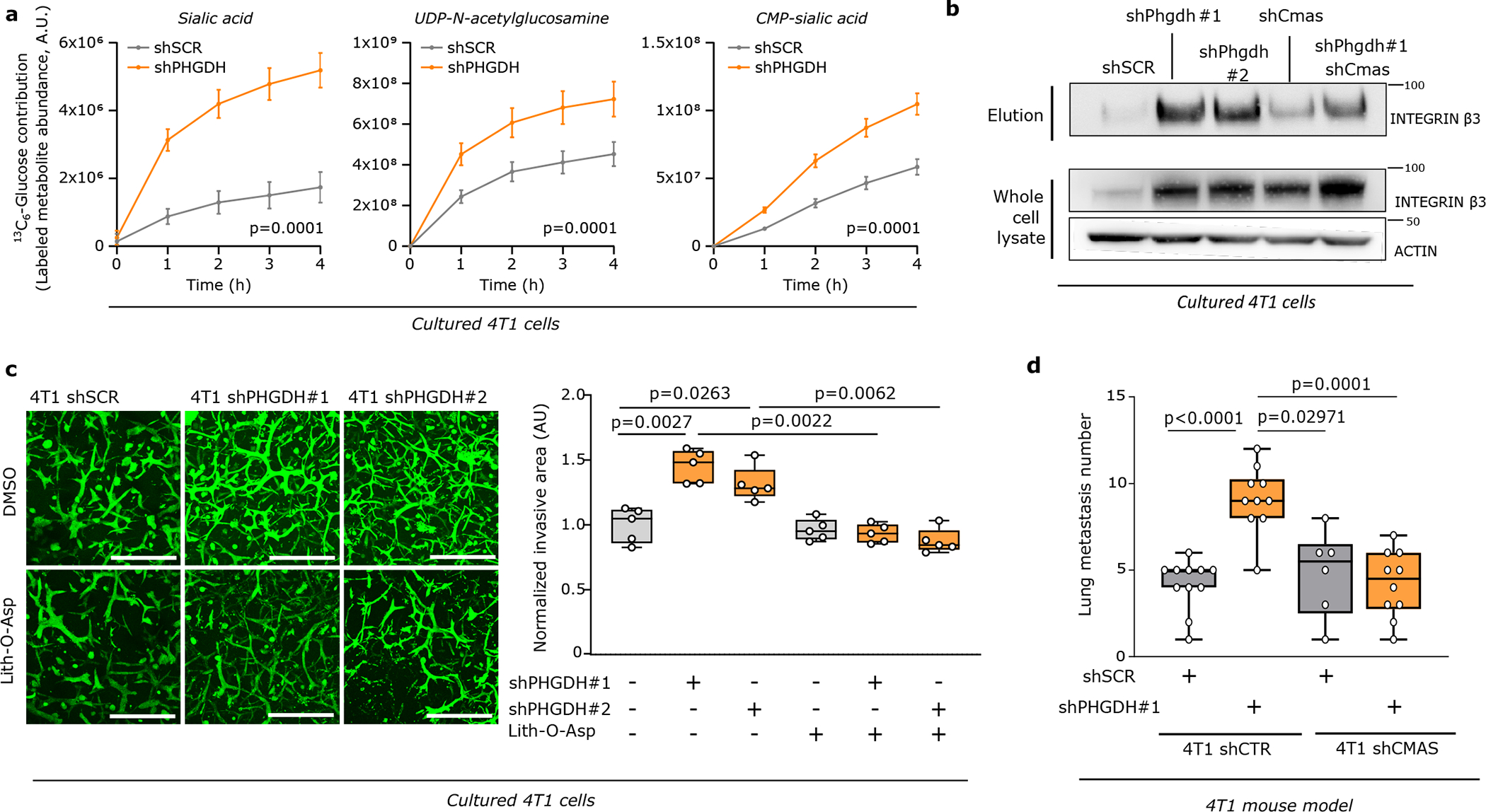
a. Dynamic labeling of 4T1 cells with 13C6-glucose showing 13C incorporation into sialic acid, UDP-N-acetylglucosamine and CMP-sialic acid upon Phgdh knockdown. Two-way ANOVA. Error bars represent standard deviation (s.d.) from mean (n=3 independent samples).
b. Protein levels of glycosylated integrin β3 (elution) after WGA-mediated isolation of β-1,4-GlcNAc- and sialic acid-linked proteins from total lysates of 4T1 cells upon Phgdh (4T1 shPHGDH), Cmas, and double Phgdh and Cmas knockdown compared to control cells (4T1 shSCR). Total levels of integrin β3 from the whole cell lysate and actin as loading control are shown. A representative experiment is shown (n=3 independent experiments).
c. Invasive Capacity of 4T1 cells upon Phgdh knockdown (shPHGDH) compared to control (shSCR) cells after 48h of pretreatment with the sialytransferase inhibitor Lith-O-Asp (30 μM) in a 3D matrix. The invasive area was stained with calcein green. Representative images are depicted in the left panel (scale bar 500 μm), quantification in the right panel. Each dot represents a different microscopy field (n=5). The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the minimum and the maximum values. Welch and Brown-Forsythe ANOVA with Dunnett’s multiple comparison.
d. Number of lung metastases per mouse injected (m.f.p.) with either 4T1 cells with functional (shSCR, n=11) or inactive (Cmas knockdown, n=6) sialic acid pathway, upon Phgdh knockdown (shPHGDH, n=10). The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the smallest and the largest values. Welch and Brown-Forsythe ANOVA with Dunnett’s multiple comparison.
Next, we focused on integrin αvβ3 sialylation. We isolated all β-1,4-GlcNAc- and sialic acid-linked proteins from 4T1 cells and immunoblotted for integrin αvβ3. Strikingly, we recovered more integrin αvβ3 in Phgdh-silenced cells, showing that it was more glycosylated upon loss of PHGDH (Figure 3b, Extended Data Figure 6h, i). Consistently, blocking the hexosamine-sialic acid pathway by silencing N-acylneuraminate cytidylyltransferase (Cmas) (Extended Data Figure 6a) prevented the increase in integrin αvβ3 glycosylation upon loss of PHGDH in 4T1 (Figure 3b, Extended Data Figure 6h). Hence, we concluded that the hexosamine-sialic acid pathway couples low PHGDH expression to integrin αvβ3 sialylation.
Loss of sialylation decreases metastasis
Next, we investigated whether targeting sialylation counteracts the increased metastatic dissemination induced by loss of PHGDH. Treatment with tunicamycin, the sialylation inhibitor Lith-O-Asp29, and combined loss of CMAS and PHGDH decreased the Phgdh silencing-induced invasive capacity of 4T1 cancer cells to the approximate level of control cells in the same conditions (Figure 3c, Extended Data Figure 7a–d). Moreover, Cmas overexpression increased the invasive capacities of 4T1 and EMT6.5 cells (Extended Data Figure 7e). Next, we injected 4T1 cells with combined Cmas and Phgdh loss as well as their respective controls, into the mammary fat pad of mice. We observed that control 4T1 cells and sole Cmas knockout/knockdown cells resulted in similar numbers of early lung metastatic lesions, whereas, as expected, loss of PHGDH alone increased the number of early lung metastatic lesions (Figure 3d, Extended Data Figure 7f, Supplementary Table 3). Strikingly, combined CMAS and PHGDH loss greatly reduced the number of early lung metastatic lesions to the approximate level of control 4T1 cells (Figure 3d, Extended Data Figure 7f). Collectively, these data demonstrate that protein sialylation mechanistically links low PHGDH protein expression to metastatic dissemination.
Dissemination depends on PHGDH protein
Next, we asked how the loss of PHGDH directs flux into the hexosamine-sialic acid pathway. Upon Phgdh silencing the glycolytic metabolite fructose bisphosphate (FBP) was consistently reduced in 4T1 and EMT6.5 cells (Extended Data Figure 8a, Supplementary Table 6). This suggests decreased activity of phospofructokinase (PFK), which competes with the hexosamine pathway for fructose 6-phosphate (F6P) (Extended Data Figure 6a). Thus, we assessed PFK activity based on localization30 and dynamic 13C6-glucose tracing. We found that upon Phgdh silencing less PFKP was located at actin filaments (Figure 4a) and FBP, but not F6P, 13C-labeling was reduced (Extended Data Figure 8b, SupplementaryTable 7), which is consistent with reduced PFK activity. This prompted us to infer whether PHGDH protein interacts with PFK protein. Consistently, we found a protein interaction of PHGDH with PFKP, which was confirmed upon Phgdh silencing (Figure 4b, Extended Data Figure 8c). Thus, we concluded that PHGDH interacts with PFKP and that PFK activity is reduced upon PHGDH silencing.
Figure 4: PHGDH interacts with PFKP and loss of this interaction non-catalytically drives metastatic dissemination.
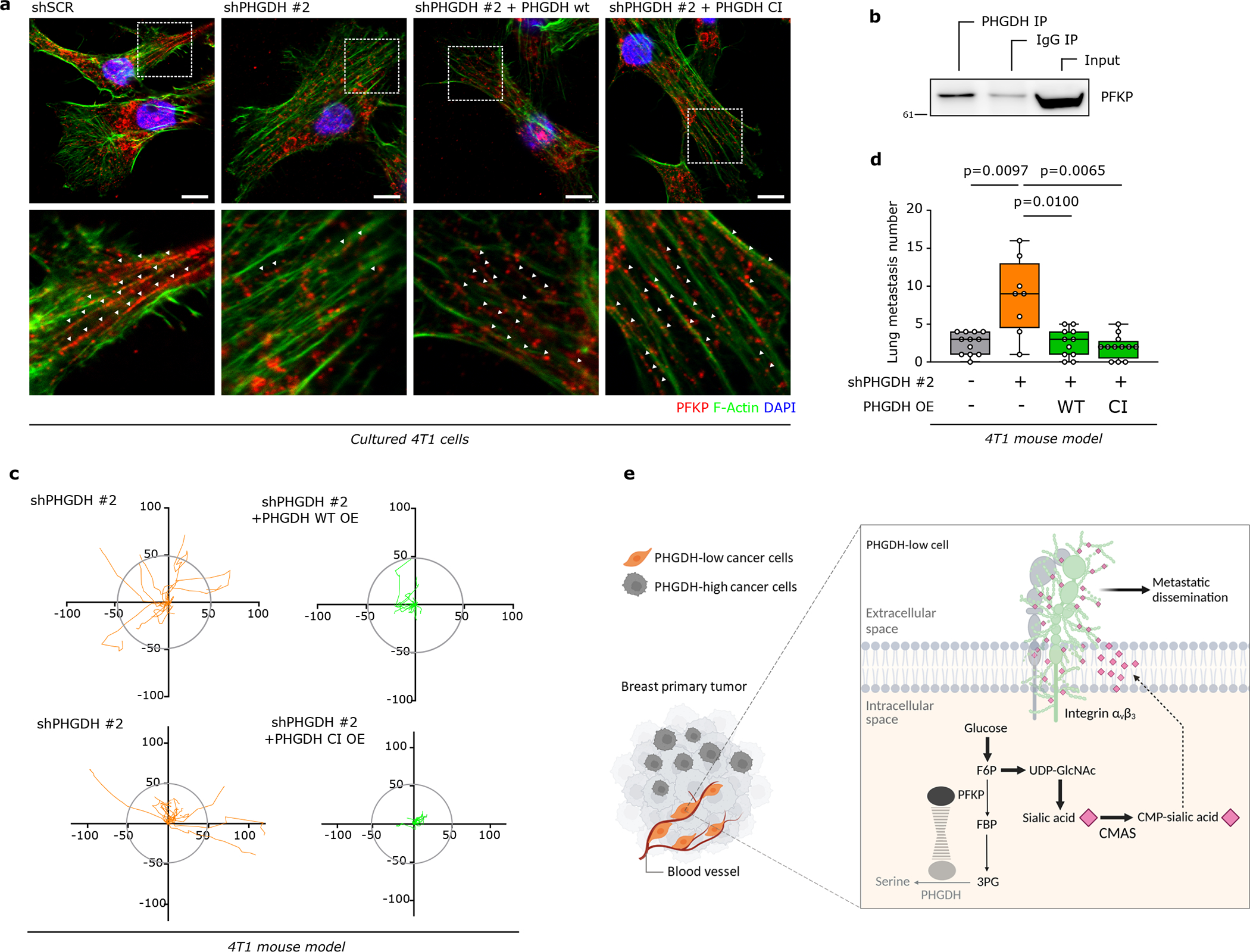
a. PFKP localization in 4T1 control (shSCR) and Phgdh knockdown (shPHGDH) cells and Phgdh-silenced 4T1 cells with wildtype (shPHGDH +wt OE) or catalytic inactive overexpression (shPHGDH + CI OE) assessed by immunofluorescence. Green, PHGDH; red, PFKP; blue, DAPI nuclear staining. A representative experiment is shown (n=3 independent experiments).
b. Interaction of PHGDH with PFKP in 4T1 mouse model assessed by co-immunoprecipitation of PHGDH. A representative experiment is shown (n=3 independent experiments).
c. Tracks in representative migratory position for 4T1 shPHGDH-Dendra and shPHGDH with overexpression of PHGDH-wildtype-mTurquoise or PHGDH-catalytic inactive-mTurquoise measured by time-lapse intravital imaging.
d. Number of lung metastases per mouse injected (m.f.p.) with either 4T1 control cells (n=12), Phgdh knockdown (shPHGDH, n=8), and Phgdh knockdown cells with wildtype (shPHGDH WT, n=11) or catalytic inactive Phgdh overexpression (CI OE, n=12). The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the minimum and maximum values. Unpaired t test with Welch’s correction, two-tailed.
e. Schematic representation of the mechanism by which PHGDH-low cells increase metastatic dissemination from the primary tumor.
Based on the identified protein interaction we hypothesized that loss of PHGDH protein rather than catalytic activity was required for metastatic dissemination. Accordingly, PHGDH heterogeneity in primary TNBC PDX tumors (BCM-3107-R2TG18) did not correlate with the expression of the other serine biosynthesis enzymes phosphoserine aminotransferase 1 (PSAT1) and phosphoserine phosphatase (PSPH), which were both rather homogeneously expressed (Extended Data Figure 8d).
Next, we used a PHGDH catalytic inhibitor PH-755 (1 μM)10,31, which does not affect protein expression (Extended Data Figure 8e, f, Supplementary Table 8) and we performed mutagenesis in the catalytic site of Phgdh to prevent catalytic activity combined with subsequent overexpression of the catalytic inactive or the respective wildtype Phgdh. We found that PH-755 treatment of control 4T1 cells did not phenocopy the metabolic profiles of Phgdh-silenced 4T1 cells (Extended Data Figure 8g, h, Supplementary Tables 8, 9). Moreover, wildtype and catalytic inactive overexpression both rescued metabolite profiles, PFK localization, and integrin αvβ3 glycosylation induced by Phgdh silencing (Figure 4a, Extended Data Figure 9a–d, Supplementary Tables 5, 9). Accordingly, the invasion ability of 4T1 cells was not altered by PH-755 treatment or α-ketoglutarate supplementation (product of PSAT1)9 (Extended Data Figure 9e). However, the overexpression of wildtype and catalytic inactive Phgdh in 4T1 cells silenced for Phgdh and in MDA-MB-231 cells decreased invasion (Extended Data Figure 9f, g), while Psat1 and Psph knockout did not alter invasion (Figure 9i). Thus, this shows that inhibition of the catalytic activity of PHGDH does not induce invasion in vitro.
Next, we injected a mix of Phgdh-silenced cells (expressing Dendra) and Phgdh-silenced cells overexpressing wildtype or catalytic inactive Phgdh (expressing mCerulean) into the mammary fat pad of mice and performed intravital imaging. We found a reduction in in vivo migratory cells in the Phgdh-silenced population overexpressing wildtype or catalytic inactive Phgdh compared to the control Phgdh-silenced population (Extended Data Figure 9i). Furthermore, in each imaging region, Phgdh-silenced cells overexpressing wildtype or catalytic inactive Phgdh demonstrated a lower displacement compared to control Phgdh-silenced cells (Figure 4c, Supplementary Video 3 and 4). Subsequently, we determined the number of early lung metastatic lesions (Supplementary Table 3). Strikingly, tumors from Phgdh-silenced, but not Phgdh-silenced 4T1 cells overexpressing catalytic inactive or wildtype Phgdh, exhibited an increased number of early lung metastatic lesions compared to control 4T1 cells (Figure 4d). Based on these data we concluded that loss of PHGDH protein, but not of its catalytic function, drives metastatic dissemination.
There is an extensive body of literature showing that increased PHGDH activity is important for (cancer cell) proliferation32–34. Accordingly, average high PHGDH gene expression has been associated with poor prognosis in multiple tumor types32. Our findings add a new level of understanding to the biology of PHGDH by showing that heterogeneous and low PHGDH protein expression is associated with increased metastasis in breast cancer patients. This finding does not contradict previous data but highlights the importance of differential regulation of gene and protein expression. Furthermore, tumors with increased or amplified PHGDH gene expression may be more aggressive because they can switch between proliferation and metastatic dissemination by modulating PHGDH protein expression resulting in intra-tumor heterogeneity.
Importantly, the mechanism we identified depends on a non-catalytic function of PHGDH. Moonlighting functions of PHGDH are highly understudied, with only two additional reports suggesting that high PHGDH expression stabilizes the pro-tumor transcription factor forkhead box M1 (FOXM1) in glioma cells35 and promotes proliferation through an interaction with translation initiation factors eIF4A1 and eIF4E in pancreatic cancer cells36. Strikingly, we describe – for the first time – a functional relevance for low PHGDH expression, which potentiates metastatic dissemination (Figure 4e). Thus, our study may ignite further research in this new area.
RESOURCE AVAILABILITY
Lead Contact
Further requests for resources should be directed to the lead contact, Sarah-Maria Fendt (sarah-maria.fendt@kuleuven.be).
Materials Availability
This study did not generate new unique reagents, except of genetically manipulated cell lines based on commercially available constructs. Reagents generated in this study will be made available on request through the lead author or the collaboration partner that generated the resource, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data Availability
The authors declare that all the other data supporting the findings of this study are available within the paper and its supplementary information files, and from the corresponding author upon reasonable request. Gel source images are available in Supplementary Figure 1. Transcriptomic data are available at the Gene Expression Omnibus (GEO) repository, accession code GSE198380. Source data for in vivo experiments are provided with this paper.
Code Availability
All the custom code used in the study is available from the corresponding author on reasonable request.
METHODS
Cell Culture
Human HEK293T epithelial cells, MDA-MB-231 breast adenocarcinoma cells and murine 4T1 mammary gland cancer cells were obtained from ATCC. Murine EMT6.5 mammary gland cancer cells were provided by R. Anderson (Peter MacCallum Cancer Center). 4T1 cells with CMAS knock-out (4T1 CMAS KO) and the respective control cells (4T1 CTR) were previously described and characterized37. HEK293T and MDA-MB-231 cells were cultured in high glucose (4.5 g/L) Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum (Life Technologies), 1% penicillin (final concentration of 50 U/mL) (Life Technologies) and 1% streptomycin (final concentration of 50μg/mL) (Life Technologies). Bend3 cells were cultured in high glucose (4.5 g/L) Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies) supplemented with sodium pyruvate (final concentration of 1mM) (Life Technologies), HEPES, 10% heat-inactivated fetal bovine serum (Life Technologies), 1% penicillin (final concentration of 50 U/mL) (Life Technologies) and 1% streptomycin (final concentration of 50μg/mL) (Life Technologies). 4T1 and EMT6.5 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 Medium (Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum (Life Technologies), 1% penicillin (final concentration of 50 U/mL) (Life technologies) and 1% streptomycin (final concentration of 50μg/mL) (Life Technologies). The heat-inactivation of the fetal bovine serum was performed at 55°C for 45 minutes. Puromycin dihydrochloride (Life Technologies) and Hygromycin B (Life Technologies) were added to the growth medium for selection of knock-down and overexpression cell lines, respectively. All cell lines were confirmed to be mycoplasma free, based on routine testing with MycoAlert Mycoplasma Detection Kit (Lonza). MDA-MB-231 was validated by DNA fingerprinting.
Cell-permeable α-ketoglutarate (dimethyl alpha-ketoglutarate, Sigma-Aldrich) was used at a concentration of 1 mM. The PHGDH inhibitor PH-755 was obtained from RAZE Therapeutics and used at a final concentration of 1 μM.
Knock-down and overexpression strategies
PHGDH knock-down #1 in 4T1 cells was generated using the plasmid pLKO-shRNA2 vector expressing the shRNA against Phgdh obtained from Peter Carmeliet’s lab (VIB/KU Leuven)38. The plasmids pLKO expressing the shRNA sequences for PHGDH knock-down #2 was obtained from BCCM (Belgian co-ordinated collections of micro-organisms). The same plasmid expressing a non-targeting shRNA sequence was used as control. Lentiviral particles were produced in HEK293T cells. Transduction of 4T1 cells was performed overnight and the medium was replaced the next day. Cells were selected with puromycin (2 μg/mL for 4T1 cells and 1 μg/mL for EMT6.5, 4T1 CMAS KO and 4T1 CTR cells). The overexpression of PHGDH in MDA-MB-231 cells was achieved using the plasmid pLHCX expressing the PHGDH cDNA obtained from the lab of Prof. Matthew Vander Heiden (MIT). The empty plasmid pLHCX was used as a control. Retroviral particles were produced in HEK293T cells. Transduction of MDA-MB-231 cells was performed overnight with freshly prepared virus and the medium was replaced the next day. Cells were selected with hygromycin (400 μg/mL). Polyclonal cells were selected for 1–2 weeks. Knock-down, knock-out and overexpression of PHGDH, PSAT1, PSPH and CMAS were validated by western blot analysis (Supplementary Figure 1, Extended Data Figure 7). Moreover, serine de novo biosynthesis was assessed based on 13C glucose incorporation into serine upon modulation of PHGDH, PSPH and PSAT1 (Extended Data Figure 7). 4T1 shSCR mTurquoise and 4T1 shPHGDH Dendra were obtained by using the plasmids pLenti-SFFV-H2B-Tq and pLKO-UBC-H2B-Dendra, respectively. Lentiviral particles were produced in HEK293T cells. Transduction of 4T1 cells was performed overnight and the medium was replaced the next day. mTurquoise- and Dendra-positive cells were FACS-sorted.
Protein extraction and western blot analysis
Cells were collected in dPBS and lysed in RIPA lysis and extraction buffer (Thermo Scientific, 89901) supplemented with protease (Merck Sigma, 5892970001) and phosphatase (Merck Sigma, 4906845001) inhibitors. Frozen tissue samples were placed in 1.5 mL tubes with sufficient amount of RIPA buffer and grinded with a tissue lyser to help the lysis of the tissue.
Extracted proteins were quantified using a Pierce BCA Protein Assay Kit (Thermo Scientific, 23225). Subsequently, 10–40 μg of proteins were loaded on a precast gel NuPAGE Novex 4–12% Bis-Tris (Thermo Scientific, NP0336BOX). Proteins were then transferred onto a nitrocellulose membrane using an iBlot2 dry blotting system with iBlot2 transfer stacks (Thermo Scientific, IB301031). Membranes were incubated for 1h at room temperature in a blocking solution of 5% milk in TRIS Buffer Saline 0.05% Tween (TBS-T). Subsequently, membranes were incubated overnight at 4 °C with primary antibodies against Phospho-SRC (Y416) (Cell Signaling Technology, 6943), SRC (Cell Signaling Technology, 2123), Phospho-p38 kinase (T180/Y182) (Cell Signaling Technology, 4511), p38 kinase (Cell Signaling Technology), β-actin (Merck Sigma, A5441), PHGDH (Merck Sigma, HPA021241), PSAT1 (Bio-techne, H00029968-A01), Integrin β3 (Cell signaling, 13166), CMAS (Merk Sigma, HPA039905), PFKL (Abcam, EPR11904), PFKM (Abcam, EPR10734(B)), PFKP (Abcam, EPR17314), or ALDOA (Novus Biologicals, NBP1-87488). All primary antibodies were used in a 1:1,000 dilution in 5% bovine serum albumin in TBS-T, except for PHGDH (1:3,000 dilution), β-Actin (1:10,000 dilution) and ALDOA (1:500 dilution). The following day, membranes were incubated with HRP-linked secondary antibodies anti-rabbit (Cell Signaling Technology, 7074S) or mouse (Cell Signaling Technology, 7076S) used 1:4,000 in 5% milk in TBS-T. Bound antibodies were visualized using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific, 34095) or SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Scientific, 34580). Images were acquired using an ImageQuant LAS 4000 (GE Healthcare). Images were quantified using the software ImageQuant TL (GE Healthcare). The signals were normalized on the loading control β-Actin.
Co-immunoprecipitation (IP)
Cells were collected in dPBS and lysed in CoIP lysis buffer (10 mM Tris-HCl pH 7.6, 140 mM NaCl, 5 mM EDTA, 0.5% Nonidet P-40) supplemented with protease (Merck Sigma, 5892970001) and phosphatase (Merck Sigma, 4906845001) inhibitors. Whole cell extracts (500 μg per IP) were immunoprecipitated for 1h using a PHGDH-specific antibody (Merck Sigma, HPA021241) complexed with Dynabeads Protein A (Life Technologies). After three washes with CoIP buffer, the isolated immunocomplexes were subjected to western blot analysis as described above.
β-1,4-GlcNAc- and sialic acid-linked protein isolation
Glycoprotein Isolation Kit WGA (Thermo Scientific) was used to isolate the glycoproteins rich in N-acetylglucosamine and sialic acid from whole cell lysates according to the manufacturer’s protocol. Briefly, cellular pellets were extracted twice, first with RIPA buffer and after with RIPA + 0.1% triton X100. Extracted proteins were quantified using a Pierce BCA Protein Assay Kit (Thermo Scientific). Samples containing 1 mg of protein per condition were processed through the WGA Lectin Resin column. After glycoprotein capture, the flow-through was collected for western blot probing. Eluted sample protein concentration was determined using Qubit protein assay.
RNA isolation and RT-PCR
Total RNA was isolated with TRI reagent (Life Technologies). Quality and quantity of the isolated RNA were measured with a NanoDrop One Microvolume UV-Vis Spectrophotometer (Thermo Scientific). RNA was reverse transcribed into cDNA using a qScript cDNA Synthesis Kit (Quantabio). The relative levels of transcripts compared to the control RPL19 were determined by qPCR using PerfeCTa SYBR Green SuperMix, Low ROX (Quantabio) and specific primers on a 7500 Fast Real Time PCR System (Applied Biosystems, Life Technologies). Amplification was performed at 95 °C for 10 min, followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C.
The primer list is provided in Supplementary Table 10.
Bulk RNAseq analysis
For each condition of interest (4T1 shSCR/shPHGDH, 4T1/EMT6.5 mono-culture/co-culture with bEnd3, and MDA-MB-231 CTR/PHGDH OE), total RNA from freshly collected cells was extracted with TRI reagent (Life Technologies). For 4T1/EMT6.5 co-cultured with Bend3 cells, tumor and endothelial cells were separated by FACS sorting prior RNA extraction. Quality and quantity of the isolated RNA was measured with a NanoDrop One Microvolume UV-Vis Spectrophotometer (Thermo Scientific). RNAseq libraries were prepared from 1 μg of total RNA per sample using the KAPA Stranded mRNA Sequencing Kit (Roche). In short, poly-A containing mRNA was purified from total RNA using oligo(dT) magnetic beads and fragmented into 200–500 bp pieces using divalent cations at 94°C for 8 min. The cleaved RNA fragments were copied into first-strand cDNA. After second-strand cDNA synthesis, fragments were A-tailed and indexed adapters were ligated. The products were purified and enriched by PCR to create the final cDNA libraries. After quantification with qPCR, the resulting libraries were sequenced on a HiSeq4000 (Illumina) using a flow cell generating 1×50bp single-end reads. The resulting reads were trimmed for adaptors and low quality basecalls using Trim Galore! (v0.5.0), mapped to the mm10 genome assembly using HISAT2 (v2.1.0) (PMID: 25751142) and quantified using default settings with featureCounts (v1.6.4) (PMID: 14668230) as implemented in the nf-core rnaseq pipeline (v1.3).
Differential expression analysis between each pair of conditions of interest was performed using the DESeq2 R package (v1.24.0). Resulting log2(fold change) values were ranked and used as input for pre-ranked gene-set enrichment analysis (GSEA), using the fgseaMultilevel method implemented in the fgsea R package (v1.19.3), with a p-value calculation boundary (eps) of 0. GSEA was performed on a collection of 2972 curated gene sets consisting in all HALLMARK, PID, Reactome, Biocarta, KEGG, and WikiPathways gene sets included in the Broad Institute Molecular Signature Database (https://www.gsea-msigdb.org/). The corresponding mouse (for 4T1/EMT6.5 cells) or human (for MDA-MB-231 cells) gene-set collections were obtained using the msigdbr R package (v7.4.1). Signature gene sets containing those genes most highly upregulated upon Phgdh knock-down (relative to control) and/or co-culture with endothelial cells (relative to mono-culture), determined as described in the section Gene signature definition below, as well as a custom-made gene set for OPN signaling via AP-1 (see Supplementary Table 11), were added to the above mentioned curated gene-set collection. Only gene sets containing between 3 and 1000 genes were retained for GSEA. Transcriptomic data are available at the Gene Expression Omnibus (GEO) repository, accession code GSE198380.
Proteomics
Cell lysis, digestion and TMT labeling were performed as described39. In short, 10 million cells were lysed in 3 ml SDS lysis buffer (5% SDS, 100 mM Tris-HCl pH 7.6) at 95°C for 4 minutes. Protein concentration was determined by Pierce BCA protein assay (Thermo Fisher Scientific). 100 μg of protein was used for subsequent reduction with 5 mM TCEP, alkylation with 15 mM iodoacetamide and quenching with 10 mM DTT. Protein lysates were purified by methanol-chloroform precipitation. The resulting protein pellets were resuspended in 40 mM HEPES pH 8.4 and digested with trypsin (10 μg) for 16 hours at 37°C. Peptide concentration was measured with Pierce BCA assay.
10 μg of each peptide preparations was dissolved in 25 μl of Hepes (40 mM, pH 8.4) and incubated with 40 μg of one of 15 amino reactive TMTpro Label Reagents (Thermo Fisher Scientific) for 1h at ambient temperature. Excess TMT label was quenched by addition of 6 μl 5% hydroxylamine and incubation for 15 min at ambient temperature. Labelled peptide samples were then mixed and freeze-dried. TMT-labeled mixtures were dissolved in 1 ml 10 mM ammonium bicarbonate and fractionated on 1cc C18 SPE cartridges (Oasis HLB, Waters) using 5%, 10%, 15%, 20%, 25% and 35% acetonitrile in 10 mM ammonium bicarbonate.
TMT-labeled peptides were dissolved in water/formic acid (100/0.1 v/v) and subsequently analyzed in duplicate by on- line C18 nanoHPLC MS/MS with a system consisting of an Ultimate3000nano gradient HPLC system (Thermo, Bremen, Germany), and an Exploris480 mass spectrometer (Thermo). Fractions were injected onto a cartridge precolumn (300 μm × 5 mm, C18 PepMap, 5 μm, 100 A, and eluted via a homemade analytical nano-HPLC column (50 cm × 75 μm; Reprosil-Pur C18-AQ 1.9 um, 120 A (Dr. Maisch, Ammerbuch, Germany). The gradient was run from 2% to 40% solvent B (20/80/0.1 water/acetonitrile/formic acid (FA) v/v) in 120 min. The nano-HPLC column was drawn to a tip of ~10 μm and acted as the electrospray needle of the MS source. The mass spectrometer was operated in data-dependent MS/MS mode for a cycle time of 3s, with a HCD collision energy at 36 V and recording of the MS2 spectrum in the orbitrap, with a quadrupole isolation width of 1.2 Da. In the master scan (MS1) the resolution was 120,000, the scan range 350–1600, at standard AGC target @maximum fill time of 50 ms. A lock mass correction on the background ion m/z=445.12 was used. Precursors were dynamically excluded after n=1 with an exclusion duration of 45 s, and with a precursor range of 20 ppm. Charge states 2–5 were included. For MS2 the first mass was set to 110 Da, and the MS2 scan resolution was 45,000 at an AGC target of auto. Post-analysis processing was done with Maxquant 1.6.17.0 using all default settings, and using the Uniprot Mus musculus database (55398 entries).
The ensuing protein count matrices were restricted to include only proteins with unambiguously assigned coding gene IDs, in order to enable comparison to the RNAseq data. Differential expression analysis between each pair of conditions of interest was then performed using the DEP R package (v1.6.1). Resulting log2(fold change) values were ranked and used as input for pre-ranked GSEA, identically as described above for RNAseq data.
Gene signature definition
Gene signatures containing those genes most highly upregulated upon Phgdh knock-down (shPHGDH) relative to control (shSCR), or upon co-culture with endothelial cells relative to mono-culture, were determined based on the DESeq2 (for RNAseq data) or DEP (for Proteomics) differential expression analysis results for the corresponding data sets. Specifically, the following cutoffs were used for each data set: (a) shPHGDH vs shSCR (RNAseq): Fold-change > 2, FDR < 0.001; (b) shPHGDH vs shSCR (Proteomics): Fold-change > 1.2, p-value < 0.001; (c) Co-Culture vs Mono-Culture (RNAseq): Fold-change > 2, p-value < 0.01. A gene expression signature indicative of low PHGDH protein expression was further determined by intersecting the RNAseq-based signatures for both Phgdh knock-down (a) and co-culture (c). This signature was specifically used for performing correlation analysis on scRNA-seq patient data.
Integrated RNAseq/Proteomics gene-set enrichment analysis
For integrated gene-set enrichment analysis, GSEA results based on RNAseq/Proteomics data for 4T1 cells upon Phgdh knock-down (relative to control), and on RNAseq data for 4T1/EMT6.5 cells in co-culture with bEnd3 endothelial cells (relative to mono-culture), were first combined, and further restricted to just those gene sets showing positive normalized enrichment scores (NES) in all three data sets (for a total of 786 gene sets). NES values for each of these gene sets were then averaged over the three data sets, and the resulting mean NES values were used as ranking metric to select the top 50 most commonly-upregulated gene sets overall, as shown in Extended Figure 5a–b.
Single-cell RNAseq analysis
Single-cell RNAseq data for primary breast tumors of 13 TNBC patients was obtained from Bassez et al. Nat Med 27, 820–832 (2021) (DOI: 10.1038/s41591-021-01323-8). The raw count matrix, comprising a total of 24390 cells and 23150 genes (after restricting to only TNBC patients), was first subject to size-factor and variance-stabilizing normalization using the Monocle R package (v2.99.3). Gene-set enrichment scores for every cell, and for each of the gene sets included in the gene-set collection previously used for bulk RNAseq data, were then determined based on this normalized count matrix, using the GSVA R package (v1.38.2) with default parameters. GSVA scores for each gene-set were then converted to Z-Scores across all cells, for the purpose of plotting score correlations and performing total least-squares regression, as shown in Figure 2d and Extended Figure 1l, 5c.
Orthotopic mouse models
Breast cancer
All animal experiments were approved by the local authorities in compliance with all relevant ethical regulations. For injection models, mice were randomized before injection of cancer cells. All samples were analyzed blinded. Sample size was determined using power calculations with B = 0.8 and P < 0.05 based on preliminary data and in compliance with the 3R system: Replacement, Reduction, Refinement.
Six-week-old female BALB/c (Envigo) mice were inoculated in the mammary fat pad with 1×106 cancer cells, in 50 μL PBS using a 29G syringe. Mice were euthanized 18–21 days after cell injection, except for experiments in Extended Data Fig. 1i in which tumors were resected one week post-injection and mice were euthanized 3, 4 or 5 weeks post-injection. At the end of the experiment, primary tumors were dissected and weighted.
At the end of every in vivo experiment, mice were sacrificed by injecting approximately 50 μL of a 60 mg/mL Dolethal (pentobarbital sodium) solution (Vetoquinol). Primary tumors, lung metastases and lung healthy tissue were dissected and washed in ice-cold saline, placed into pre-labelled bags and frozen using a liquid nitrogen-cooled Biosqueezer (Biospec Products). The bags were then placed in liquid nitrogen until all collections were finished and finally stored at −80 °C until further processing.
Melanoma
Primary melanoma tumor from Tyr::N-RasQ61K;Ink4a−/− (Tyr::CreERT2) animals was dissociated into small pieces using forceps and scissors. Tissue was digested using collagenase I (2mg/ml, Sigma Aldrich, cat. C0130) and IV (2mg/ml, Sigma Aldrich, cat. C5138) mix for 20 min at 37°C followed by a Trypsin (Trypsin-EDTA 0.05%, ThermoFisher Scientific, cat. 25300054) digestion for 5 min at 37°C. Single cells were separated from remaining tissue using a 40 μm cell strainer and cultured in vitro using Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum and 100 μg/mL Penicillin/Streptomycin.
Tyr::N-Ras+/Q61K;Ink4a−/− mouse melanoma cells stably expressing dsRed-encoding lentiviruses and Bend3 immortalized endothelial cells (ECs) stably expressing GFP encoding lentiviruses were mixed in a ratio 1:4 (105 melanoma:4×105 ECs) and resuspended in matrigel (5mg/ml; Thermofisher scientific, cat. 356255). Then, cells were injected subcutaneously in the back skin of Foxn1nu mice. Mice were sacrificed where tumors and organs were collected 25 days upon melanoma initiation. Tumor volume was monitored with a caliper and the volume was calculated using the following formula: V = (π/6)*length*width*height.
Mice were housed in filter top cages and IVC cages. Housing and experimental animal procedures were approved by the Institutional Animal Care and Research Advisory Committee of KU Leuven, Belgium. The animal study complies with ethical regulations and was approved by the KU Leuven ethics committee. Humane end points were determined as follows: tumor size of 1.8 cm3, loss of ability to ambulate, labored respiration, surgical infection or weight loss over 10% of initial body weight. Mice were monitored and upon detection of one of the previous mentioned symptoms, the animal was euthanized.
Patient-derived xenografts (PDX) tumor models
Breast cancer
All animal experiments were performed under IACUC approved protocols at Baylor College of Medicine (Houston, TX). Triple negative breast cancer PDX models40 were screened via IHC for expression of PHGDH in the tumor, lung, liver, and brain. Four models (BCM-5471, BCM-3107, BCM-3611 and BCM-4272) were chosen based on the positivity for PHGDH in the primary tumor. For three of the models (BCM-3107, BCM-3611 and BCM-4272), fresh PDX tumor tissue fragments were transplanted into the cleared fourth fat-pad (right abdominal) of four-week-old SCID/Beige mice41.
In a first cohort of 6 mice, when tumors reached a size of ~500 mm3 (length × width2 × 0.5), the animals were sacrificed and blood was collected and processed for circulating tumor cell (CTC) isolation. A portion of the tumor was fixed overnight in 10% neutral buffered formalin and placed in 70% ethanol until paraffin embedding (FFPE) and the remainder was snap frozen in liquid nitrogen. In a second cohort of 4 mice, when tumors reached a size of ~500mm3 (length × width2 × 0.5), tumor resection surgeries were performed to remove the primary tumor. Two months after tumor resection, animals were sacrificed.
At the terminal endpoint for each cohort, lungs were collected fixed overnight in 10% neutral buffered formalin and placed in 70% ethanol until paraffin embedding (FFPE).
All patient tissue samples used for PDXs were collected at the McGill University Health Centre and Jewish General Hospital after obtaining informed consent under REB-approved protocols and in accordance with the McGill University Health Centre research ethics board (SUR-2000-966) guidelines. Mice for PDXs were maintained and treated in accordance with the McGill University Health Centre research ethics board (SUR-99-780) and the Rosalind & Morris Goodman Cancer Institute of McGill University Facility Animal Care Committee (2014–7514) guidelines. Upon surgery or biopsy, excess patient breast tumour tissue was collected and transported from the hospitals to the laboratory in an ice-cold transport medium (DMEM/F12, 50 μg/ml gentamicin, 1×penicillin– streptomycin, 2.5 μg/ml fungizone). Transported samples were then cut into small fragments (1 mm3) using blades and transplanted into the fourth mammary fat pad of 5 to 7-week-old of NOD scid gamma (NSG) mice (The Jackson Laboratory) under sterile conditions. After surgery, mice were palpated weekly, and tumours were measured twice per week using calipers. Tumor volume was calculated by the following formula: (length × width2)/2. Tumor was harvested for biochemical analyses or in vivo passaging before or when reaching the endpoint (>20 mm in the largest dimension or the volume of 2000 mm3). Excess tumor fragments were cryopreserved for later passaging by freezing in cryotubes containing FBS with 10% DMSO at −80°C in a CoolCell container (Biocision). The banking of human specimens and associated clinical data are approved by the MUHC research ethics board (study approval SUR-2000-966 and SUR-99-780). All patient data and biological samples were obtained from patients at the RI-MUHC and Jewish General Hospital after obtaining informed consent.
Melanoma
specimens were obtained with informed consent from all patients according to protocols approved by the Institutional Review Board (IRB) of the University of Michigan Medical School (IRBMED approvals HUM00050754 and HUM0005008542) and the University of Texas Southwestern Medical Center (IRB approval 102010-051). Single-cell suspensions were obtained by dissociating tumors mechanically with a scalpel on ice. Cells were filtered through a 40-μm cell strainer to remove clumps.
All mouse experiments complied with all relevant ethical regulations and were performed according to protocols approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center (protocol 2016-101360). For all experiments, the maximum permitted tumor diameter was 2.5 cm and this limit was not exceeded in any experiment. For all experiments, mice were kept on normal chow and fed ad-libitum. Melanoma cell suspensions were prepared for injection in staining medium (L15 medium containing bovine serum albumin (1 mg/ml), 1% penicillin–streptomycin and 10 mM HEPES (pH 7.4) with 25% high-protein Matrigel (354248; BD Biosciences)).
Patient-derived melanomas were transplanted into 4-to-8-week-old male and female NOD.CB17-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice. Subcutaneous injections were performed in the right flank of mice in a final volume of 50 μl using 100 cells per injection for human melanoma cells. Subcutaneous tumor diameters were measured weekly with calipers until any tumor in the mouse cohort reached 2.5 cm in its largest diameter. At that point, all mice in the cohort were euthanized, per approved protocol, for collection of subcutaneous tumors and macrometastatic nodules.
To generate the melanoma cell line, primary tumor from Tyr::N-RasQ61K;Ink4a−/− (Tyr::CreERT2) animals was dissociated into small pieces using forceps and scissors. Tissue was digested using collagenase I (2 mg/ml, Sigma Aldrich, cat. C0130) and IV (2mg/ml, Sigma Aldrich, cat. C5138) mix for 20 min at 37°C followed by a Trypsin (Trypsin-EDTA 0.05%, ThermoFisher Scientific, cat. 25300054) digestion for 5 min at 37°C. Single cells were separated from remaining tissue using a 40μm cell strainer and cultured in vitro using Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum and 100 μg/mL Penicillin/Streptomycin. For the co-injection of melanoma and Endothelial Cells in vivo, Tyr::N-Ras+/Q61K;Ink4a−/− mouse melanoma cells stably expressing dsRed-encoding lentiviruses and Bend3 immortalized endothelial cells (ECs) stably expressing GFP encoding lentiviruses were mixed in a ratio 1:4 (105 melanoma:4×105 ECs) and resuspended in matrigel (5 mg/ml; Thermofisher scientific, cat. 356255). Then, cells were injected subcutaneously in the back skin of Foxn1nu mice. Mice were sacrificed where tumors and organs were collected 25 d upon melanoma initiation.
Collection of Circulating Tumor Cells
Whole blood collected from the PDX-bearing mice was diluted 1:40 in PBS. Diluted blood was processed using VTX-1 microscale vortices technology (Vortex BioSciences) to collect live circulating tumor cells directly onto microscope slides containing a FlexiPERM ring. Slides were placed in a 4-well dish, spun, and placed in a 37°C incubator for one hour to allow cells to attach. Cells were then fixed with 4% PFA. Two washes with PBS were used to remove the PFA and the slides were allowed to air dry prior to staining.
Histopathology and immunohistochemistry (IHC)
Immunohistochemical analyses were performed on human breast cancer tissues from the tissue archives of the Institute of Pathology of the LMU Munich. Grade 2/3 invasive ductal carcinomas of the breast, not otherwise specified (NOS), and treated by primary surgical resection between 1988 and 2006 were investigated. Samples were selected for triple-negative breast cancers based on routine immunophenotypical profiling for hormone receptors (ER/PR) and Her2-neu expression. Tumor samples had been anonymized and analyzed according to the local ethics committee regulations (19–914 KB). Disease-free survival was calculated as the time in days between primary tumor resection and recurrence (local or metastatic). Metastasis-free survival was calculated as the time in days between primary tumor resection and metastatic recurrence. Only patients with a minimum follow-up time of 6 months were included in the analysis. Tissue samples from representative lesions were collected and fixed in 4% paraformaldehyde for 24 h and then processed for paraffin embedding (HistoStar™ Embedding Workstation). Sections of 4 μm of thickness obtained from the paraffin-embedded tissues (Thermo Scientific Microm HM355S microtome) were mounted on Superfrost™ Plus Adhesion slides (Thermo Scientific) and routinely stained with hematoxylin and eosin (H&E, Diapath #C0302 and #C0362) for histopathological examination. PHGDH staining was evaluated based on clinical pathology standards. Homogeneous high tumors were defined as 100% PHGDH and intensity larger than zero. Heterogenous tumors were defined as overall PHGDH intensity larger than zero but PHGDH-positive area lower than 100%. Low tumors were defined as intensity of zero.
Hematoxylin and eosin staining
Hematoxylin and eosin staining was used to identify the cancerous lesions in the lung of the different mouse models. During dissection, the lungs were gently infused via the trachea with 10% neutral buffered formalin. Next, samples were embedded in paraffin and sliced in 7 μm thick sections that were stained with hematoxylin and eosin. Images were acquired on a Zeiss Axio Scan.Z1 using a 20× objective and ZEN 2 software and analyzed using the ZEN Blue software (Zeiss). In order to focus on the early stages of the metastatic process, only mice bearing lung metastases ≤0.1 mm2 (4T1) or ≤0.4 mm2 (EMT6.5) were analyzed. The rate of metastatic progression was calculated over a 3-day period, comparing the average metastatic burden (MB) in the lung (measured as total metastasis area) of 10 mice per group at 18 and 21 d post orthotopic injection with either 4T1 shSCR or 4T1 shPHGDH cells. The following formula was used: rate of metastatic progression = (ln(MB21days) − ln(MB18days) / ln(2) × time.
Immunofluorescence with and without tyramide signal amplification.
Tissue sections of 4 μm were deparaffinized and hydrated in distilled water, followed by 23 min of heat-induced epitope retrieval (HIER) in AR6 buffer (Perkin Elmer, AR6001KT) using the 2100 Antigen Retriever (Aptum Biologics Ltd). After a cooldown of 15 min in milliQ water the endogenous peroxidase activity of the samples was blocked by a 20 min incubation in 0.3 % hydrogen peroxide in methanol. The tissue was then blocked for 30 min using TNB blocking buffer (0.1 M TRIS-HCl, pH 7.5; 0.15 M NaCl; 0.5% TSA Blocking Reagent (PerkinElmer, FP1012 or FP1020)). Pan-cytokeratin (panCK) was detected by incubating the tissue with anti-panCK antibody (mouse, Agilent, M351501-2, 1:100 in TNB blocking buffer) for 30 min. Antibody detection was followed by 45 min of incubation with goat anti-mouse-biotin (DAKO, E0433, 1:200 in TNB blocking buffer) and subsequent incubation with Streptavidin-HRP Conjugate (PerkinElmer, NEL750001EA, 1:100 in TNB blocking buffer). Signal detection was performed by 8 min incubation with the PerkinElmer TSA Plus Cyanine 3 kit (PerkinElmer, NEL744E001KT, 1:50 in amplification buffer from the Cyanine 3 kit). For the detection of PHGDH, the tissue was first blocked for 30 min using a blocking buffer (TBS with 1% BSA (VWR, 22013, Bovine Serum Albumin (BSA), fraction V, Biotium (50 g))) with 10% normal goat serum (Invitrogen, 10000C) and then incubated with anti-PHGDH antibody (rabbit, Merck Sigma, HPA021241, 1:3,000 in TNB blocking buffer) for 30 min. The PHGDH antibody was visualized by incubating the sections with goat anti-rabbit Alexa Fluor 647 antibody (Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody Alexa Fluor 647, Lifetech, A21245, 1:200 in TNB blocking buffer) for 45 min followed by DAPI staining for 5 min (Spectral DAPI, Akoya Biosciences, FP1490, 2 drops per 1 ml TBST). The slides were then mounted using ProLong Diamond Antifade Mountant (Thermo Fisher, P36961).
For triple stainings for PHGDH, panCK and PHH3, the following antibodies were used: anti-panCK (mouse, 1:1,000; DAKO, M3515, Clone AE1/AE3), anti-PHH3 (rabbit, Cell Signaling Technology, #9701S), and anti-PHGDH antibody (rabbit, 1:10,000 in TNB blocking buffer, Merck Sigma, HPA021241). The PerkinElmer Opal 4-Color Manual IHC Kit (PerkinElmer/Akoya, NEL810001KT) was used for the tyramide signal amplification according to the manufacturer’s protocol. For introduction of the secondary-HRP the Envision+/HRP goat anti-Rabbit (Dako Envision+ Single Reagents, HRP, Rabbit, Code K4003) was used for antibody raised in rabbit (PH3 and PHGDH) and the OPAL Polymer HRP Ms+Rb (Akoya/Perkin Elmer, ARH1001EA) was used for the antibody raised in mouse (panCK). The slides were then mounted using ProLong Diamond Antifade Mountant (Thermo Fisher, P36961). The various proteins were detected by first using the OPAL 570 (panCK), then OPAL 690 (PHH3), and at last OPAL 520 (PHGDH) reagents according to the manufacturer’s protocol.
Microscope image acquisition and image processing.
Images were acquired on a Zeiss Axio Scan.Z1 using a x20 objective and ZEN 2 software. For exporting images the ZEN 2 software (Zeiss) and the software package QuPath (Version: 0.1.2,43) were used. QuPath was also used for automatic cell detection using the DAPI channel and for subsequent creation of a detection classifier using all 55 given parameters resulting in the classification of panCK-positive cells within the whole slide.
Opal Multiplex Fluorescent Immunohistochemistry of PDX FFPE tissues
PDX tissues were fixed with 10% formalin for 24 hours, paraffin-embedded and sectioned at 4 μm. FFPE sections were deparaffinized in xylenes, rehydrated in ethanol and underwent heat- mediated antigen retrieval in 10 mM citrate buffer pH6.0 (Vector Laboratories H-3300) or EDTA buffer pH9.0 (Vector Laboratories H-3301) for 10 min in a pressure cooker. Slides were cooled and blocked with Power Block (BioGenex HK083-50K) for 5 min at room temperature and incubated with the primary antibody for 30 min at room temperature, followed by washing with TBS/T (3×3 min). Slides were then incubated with secondary antibody-HRP (Cell Signalling 8125, 8114) for 30 min at room temperature and washed with TBS/T (3×3 min). Slides were stained with Opal fluorophore working solution (Akoya Biosciences) for 10 min. This was then followed by heat-mediated antibody stripping to remove the primary and secondary antibodies in order to repeat additional rounds for labelling with other primary antibodies. Slides were then counterstained with 1 ng/ml DAPI (Molecular Probes D-1306) for 10 minutes at room temperature and mounted with coverslips using ImmunMount mounting medium (Thermo Fisher Scientific 9990402). All the primary antibodies used are listed in Supplementary Table 12. The antibody specificity and dilution were tested before the multiplex assay. Slides were imaged using AxioScan Z1 scanner (Zeiss) and analyzed using Zen software (Zeiss) and HALO (indica labs). H&E staining of tissue areas represented in the figures are shown in Supplementary Figure 2.
Imaging Mass Cytometry (IMC)
Tissue preparation and staining
Carrier-free antibodies were labelled with stable metal isotopes using the MaxPar labeling kit (Fluidigm) following the manufacturer’s instructions. Antibodies were quantified photometrically and stored at 4 °C.
Formalin-Fixed Paraffin-Embedded (FFPE) tumors were fixed in 10% NBF overnight and embedded in paraffin. 5um sections were baked at 60 °C for 2 h before dewaxing in xylene for 20 min. Samples were then rehydrated in descending grades of ethanol (100%, 95%, 89%, 70%) 5 min each. Antigen retrieval was performed in Tris-EDTA HIER buffer pH 9.2 at 96 °C for 30 min. Samples were cooled at room temperature before blocking. FFPE sections were blocked in TBS/0.3% Triton X-100/3% BSA for 1 h. Samples were incubated overnight at 4 °C in primary antibody panel at 5 μg/mL each diluted in TBS/0.1% Triton X-100/1% BSA in a humid chamber. The antibody panel used is described below. Samples were washed three times in TBS and once in milliQ water before nuclei staining with MaxPar Intercalator-Ir (Fluidigm) diluted 1:400 in TBS for 15 min. Samples were washed once in milliQ water for 5 min and air dried before imaging. Respective DNA staining is presented in Supplementary Figure 3. All the antibodies used are provided in Supplementary Table 12.
Data acquisition
IMC acquisitions were performed on a Fluidigm Hyperion instrument connected to a Fluidigm Helios mass cytometer. Laser frequency was 200 Hz with a raster scanning resolution of 1 μm (XY). The abundances of each isotope per pixel were measured and converted in image by the Fluidigm Cytof software.
Data analysis – Single cell segmentation and quantification
IMC images were segmented into single cells following using the packages imctools, Ilastik 1.2.244 and CellProfiler 3.1.545 as described in the IMCSegmentationPipeline46 available at https://github.com/BodenmillerGroup/ImcSegmentationPipeline. In brief, imctools was used to convert MCD and *txt files obtained from imaging into TIFF format. Supervised pixel classification was done with Ilastik using a combination of nuclear, cytoplasm and cell membrane markers to generate probability maps of single cells (nuclei, cytoplasm/membrane and background). Probability maps were then used to generate a segmentation mask using CellProfiler. Finally, segmentation masks and TIFF images for all channels of interest were overlaid to filter out outlier pixels and extract single-cell measurements for each channel.
Data analysis – Clustering and correlation analysis
Single-cell mean intensities for each channel were analyzed using the R package Seurat 3.2 (http://satijalab.org/seurat/). Cell outliers corresponding to hot pixels were filtered out. The remaining cells were scaled and clustered using a graph-based approach. For high-dimensional clustering all measured channels were used. For visualization, high-dimensional single-cell data were reduced to two dimensions using UMAP. Tumor cells were selected by a negative selection of clusters corresponding to immune cells (CD45+), stromal cells (aSMA+) and endothelial cells (CD31+ and/or CD34+). Data distribution was tested for normality. Because all variables were not normally distributed, correlation of expression between PHGDH and Ki67 in tumor cells was investigated with the nonparametric Spearman rank correlation. Differences were considered significant at p<0.05.
Time-lapse intravital microscopy
NOD-scid Il2rynullB2mnull (NSG-B2M) mice were obtained from Jackson Laboratories. All animal experiments were approved by the Animal Welfare Committee of the Netherland Cancer Institute (NKI), in accordance with national guidelines. All animals were maintained in the animal department of the NKI, housed in individually ventilated cage (IVC) systems under specific pathogen-free conditions and received food and water ad libitum. Sample size was determined using power calculations with B = 0.8 and P < 0.05 based on preliminary data and in compliance with the 3R system: Replacement, Reduction, Refinement.
Time-lapse intravital imaging of primary tumor
The cell populations to compare ((1) 4T1 shCRTL- mTurquosie 4T1 shPDGDH-Dendra cells; (2) 4T1 shPHDGH-Dendra 4T1 shPDGDH-mTurquosie +wt OE cells; (3) 4T1 shPHDGH-Dendra 4T1 shPDGDH-mTurquosie +CD OE cells) were mixed in 100 μl Matrigel (Corning, cat. no. 356231) and injected into the 4th mammary fat pad (50,000 cells per injection) of recipient NSG mice. Experiments were performed on 8 NSG mice and time-laps intravital imaging was performed on tumors of about 200 mm3. During the entire procedure mice were sedated using isoflurane inhalation anesthesia (~1.0% isoflurane/compressed air mixture) and received 200 μl sterile PBS by subcutaneous injection before the surgery. The tumors were surgically exposed and mice were then placed in a custom designed imaging box on the microscope while kept under constant anesthesia, with the imaging box and the microscope adjusted to 34.5°C using a climate chamber. Intravital images were acquired using an inverted Leica SP8 Dive system equipped with a MaiTai eHP DeepSee laser (Spectra-Physics) and Insight X3 (Spectra-Physics) and 4 HyD-RLD detectors. mTurquosie and Dendra were simultaneously excited at 820 nm (Mai Tai) and 960 nm (Insight ×3), respectively, and singles including second harmonic generation (Collagen I, stroma) were detected using the hybrid detector. Different positions within the tumor were acquired as three-dimensional tile scans with 5 μm Z-steps. Images were collected every 30 min for a period of 7 to 9 h during which the mouse was kept sedated and alive, constantly hydrated with subcutaneous infusion of glucose and electrolytes (NutriFlex special 70/240, Braun, 100 μl/h). All images were collected at 12 bit and acquired with a 25x water immersion objective with a free working distance of 2.40 mm (HC FLUOTAR L 25x/0.95 W VISIR 0.17).
Post-processing and analysis of time-lapse intravital microscopy
The time-lapse three-dimensional positions were corrected for XYZ-drift using the Huygens Object Stabilizer module (Scientific Volume Imaging). The correctness of the XY correction was visually inspected and manually adjusted using ImageJ if required. Migration of randomly picked individual cells was analyzed using the MTrack2 plugin in ImageJ (http://imagej.net/MTrack2). Only cells that could be followed over a minimum period of 4 h were included in the analysis. Cells were defined as being migratory when the mean displacement per hour was greater than 4 μm. For track length analysis, only cells which could be followed over the whole duration of time-lapse imaging were included in the analysis. Due to different microenvironments, migration was different for each imaging position. Therefore migration characteristics were always compared in mTurquoise cells and Dendra2 cells that are located in the same imaging position (and therefore microenvironment).
N-glycosylation staining after tunicamycin pre-treatment
4T1 shSCR and shPHGDH#2 cells were treated for 72 h with 0.05 ug/ml tunicamycin (Merk Life Science, T7765). Tunicamycin was washed out and the cells still attached to the flask were harvested and seeded – at 1×105, 8×104, 4×104 per well for the 3 time points – on 10 mm round glass coverslip previously coated with fibronectin bovine plasma (1:1 in PBS) (Merk Life Science, F1141) and placed in 12-wells plates. At each time point cells adhering to coverslip were incubated for 10 min at 37°C with wheat germ agglutinin (WGA) Alexa Fluor 555 (Life Technologies, W32464) at final concentration of 5 μg/ml in Hank’s balanced salt solution (HBSS) without phenol red. Samples were then fixed in paraformaldehyde 4% in PBS for 20 min at RT and permeabilized in Triton X-100 0,2% in PBS for 15 min at RT. At last samples were incubated with 1 μg/ml of DAPI (Life Technologies, D1306) for 5 min at RT. Imaging was performed on a Leica TCS SP8 X confocal microscope equipped with a White Light Laser and UV lamp and a HCX PL APO CS 63x/1.40 OIL objective. Images were acquired and processed with LAS X software (Leica).
Immunofluorescence analysis
4T1 shSCR, shPHGDH#2, shPHGDH#2 + PHGDH WT and shPHGDH#2 + PHGDH CD cells were seeded on 10 mm round glass coverslips, previously coated with fibronectin bovine plasma (1:1 in PBS) and placed in 12-wells plate, at 5×104 cells/well. Cells were cultured for 24 h and then fixed in paraformaldehyde 4% in PBS for 20 min at RT and permeabilized in Triton X-100 0.2% in PBS for 15 min at RT. In the case of PFKP and actin immunofluorescence, cells were permeabilized with digitonin to ensure a complete removal of the PFKP fraction not bound to actin filaments. Samples were incubated with a blocking buffer of bovine serum albumin (BSA) 1% in PBS for 15 minutes at RT and washed twice in BSA 0.5% in PBS for 5 minutes. Afterwards, samples were incubated with Alexa Fluor 488 Phalloidin and anti-PfkP primary antibodies diluted in BSA 0.5% in PBS for 30 min at RT. Samples were washed three times in BSA 0.5% in PBS for 10 min and incubated with secondary antibody Alexa Fluor 555 diluted in BSA 0.5% in PBS for 30 min at RT. Samples were washed three times in BSA 0.5% in PBS for 10 min and were incubated with 1 μg/ml of DAPI for 5 min at RT. At last coverslips were washed in MilliQ water and mounted on a microscope slide using the Fluorescence Mounting Medium (Dako) (Agilent, S3023). Imaging was performed on a Leica TCS SP8 X confocal microscope equipped with a White Light Laser and UV lamp and a HCX PL APO CS 63x/1.40 OIL objective. Images were acquired and processed with LAS X software (Leica). The information of the used antibodies is provided in Supplementary Table 12.
Transwell migration assay
4T1 mTq and EMT6.5 mTq cells, both shSCR and shPHGDH, were seeded in ClearView 96-well Cell Migration Plates (Essen BioScience) at 4×103 cells/well. Where indicated in the figure legends, transwells were pre-coated with a confluent monolayer of either primary human umbilical vein endothelial cells (HUVECs) or primary lymphatic endothelial cells (LECs). Time-lapse monitoring of cancer cell migration was performed over a period of 48 h using the IncuCyte ZOOM Live-Cell Imaging Instrument equipped with the IncuCyte Chemotaxis Cell Migration Software Module (Essen BioScience). The number of migrated cancer cells, defined as mTq-positive cells, was determined in each well by analyzing the time-lapse data with the Incucyte Zoom software (Essen BioScience).
In vitro invasion assay
Cancer cells were tested for in vitro invasion either as loose cells (4T1, EMT6.5 and MDA-MB-231) or spheroids (4T1). For the invasion assay with loose cells, 50,000 cells were embedded in a 50:50 mix of growth factor-reduced Matrigel (BD Biosciences) and collagen I (Life Technologies) and seeded onto 35 mm glass bottom culture dishes (MatTek). Cells were allowed to invade for 48 h (EMT6.5) or 72 h (4T1 and MDA-MB-231), then they were stained with calcein green (Life Technologies) for 1h, washed with PBS and immediately imaged. For tunicamycin (Merk Life Science, T7765), anti-Integrin αV/β3 therapeutic antibody (Santa Cruz, sc-7312) and Lith-O-Asp (MedChemExpress, HY-112415) treatments, cells were pretreated (0.05 μg/ml 72h, 2.5 μg/ml 24h, and 30 μM 48h respectively) and then seeded for invasion assay. For the invasion assay with spheroids, 6,000 cancer cells were grown in hanging drops in full medium for 72 h, then the spheroids were collected, embedded in a 50:50 mix of growth factor-reduced Matrigel (BD Biosciences) and collagen I (Life Technologies) and seeded onto 35 mm glass bottom culture dishes (MatTek). Cells were allowed to invade for 72h, then they were stained with calcein green (Life Technologies) for 1 h, washed with PBS and immediately imaged. Imaging was performed on a Leica TCS SP8 X confocal microscope equipped with a White Light Laser and a HCX PL APO CS 10x/0.40 DRY objective. Images were acquired as three-dimensional scans with 10 μm Z-steps and processed with LAS X software (Leica) to obtain maximum projection images. Quantification of invasive area and invasive distance was performed on the maximum projection images using the FIJI47 distribution of ImageJ248.
MMP-3 activity assay
MMP-3 activity was measured using the MMP-3 activity Assay Kit (Abcam ab118972) in cell culture media from 4T1 cells seeded into in vitro invasion assay as previously described. following the manufacturer’s protocol. To directly measure MMP-3 activity, 50 ul of media was transferred to a 96-well black plate. After reacting with the MMP-3 substrate (prepared according to the protocol), the plate was read at Ex/Em = 325/393 nm twice in 1 h.
Metabolite measurements
All labeling experiments were performed in media with 10% dialyzed serum for the indicated time points. Metabolites for the subsequent mass spectrometry analysis were prepared by quenching the cells in liquid nitrogen followed by a cold two-phase methanol-water-chloroform extraction49. Phase separation was achieved by centrifugation at 4°C. The methanol-water phase containing polar metabolites was separated and dried using a vacuum concentrator. Dried metabolite samples were stored at −80°C. The protein interphase was also dried down and dissolved in 200 μl 0.2 M potassium hydroxide, and the protein concentration was then quantified using the Pierce BCA Protein Assay Kit (Thermo Fisher).
Gas chromatography-mass spectrometry (GC-MS)
Serine and glycine were measured with gas chromatography-mass spectrometry. Polar metabolites were derivatized for 90 min at 37°C with 20 μl of 20 mg per ml methoxyamine (Merck Sigma, 226904) in pyridine (Merck Sigma, 270970). Subsequently, 15 μL of N-(tert-butyldimethylsilyl)-N-methyl-trifluoroacetamide, with 1 % tert-butyldimethylchlorosilane were added to 7.5 μL of each derivative and incubated for 60 min at 60°C (Merck Sigma, 375934)49. Isotopologue distributions and metabolite concentrations were measured with a 7890 A GC system (Agilent Technologies) combined with a 5975C Inert MS system (Agilent Technologies). 1 μl of sample was injected into a DB35MS column in split mode (ratio 1 to 3) using an inlet temperature of 270°C. The carrier gas was helium with a flow rate of 1 ml/min. Upon injection, the GC oven was set at 100°C for 1 min and then increased to 105°C at 2.5°C/min and with a gradient of 2.5°C/ min finally to 320 °C at 22°C/min. The measurement of metabolites was performed under electron impact ionization at 70 eV using a selected-ion monitoring (SIM) mode. Isotopologue distributions were extracted from the raw ion chromatograms using a custom Matlab M-file, which applies consistent integration bounds and baseline correction to each ion50. In addition, we corrected for naturally occurring isotopes51 and metabolite abundances were normalized by protein content. All labeling fractions were transformed into natural abundance-corrected mass distribution vectors (MDVs)26.
Liquid chromatography-mass spectrometry (LC-MS)
For the detection of metabolites by LC-MS, a Dionex UltiMate 3000 LC System (Thermo Scientific) with a thermal autosampler set at 4°C, coupled to a Q Exactive Orbitrap mass spectrometer (Thermo Scientific) was used. Samples were resuspended in 50 μL of water and a volume of 10 μl of sample was injected on a C18 column (Acquity UPLC HSS T3 1.8 μm 2.1×100 mm). The separation of metabolites was achieved at 40 °C with a flow rate of 0.25 ml/min. A gradient was applied for 40 min (solvent A: 10mM Tributyl-Amine, 15 mM acetic acid – solvent B: Methanol) to separate the targeted metabolites (0 min: 0% B, 2 min: 0% B, 7 min: 37% B, 14 min: 41% B, 26 min: 100% B, 30 min: 100% B, 31 min: 0% B; 40 min: 0% B. The MS operated in negative full scan mode (m/z range: 70–1050 and 300–700 from 5 to 25 min) using a spray voltage of 4.9 kV, capillary temperature of 320 °C, sheath gas at 50.0, auxiliary gas at 10.0. Data was collected using the Xcalibur software (Thermo Scientific) and analyzed with Matlab for the correction of protein content and natural abundance, but also to determine the isotopomer distribution using the method developed by Fernandez et al, 199651. Metabolites abundances were corrected by protein content or cell number.
Statistical analysis
Statistical data analysis was performed using GraphPad Prism version 8.0 (GraphPad Software) on n ≥ 3 biologically independent replicates. Details on statistical tests and post-tests are presented in the figure legends. P values lower than 10−15 are indicated as p<0.0001. Sample size for all experiments was chosen empirically. Independent experiments were pooled and analyzed together whenever possible as detailed in figure legends. Data are presented as mean ± s.d.,or median ± 95% Confidence Interval, as indicated in the figure legends.
Extended Data
Extended Data Figure 1: Circulating tumor cells and early metastatic lesions exhibit low PHGDH expression.
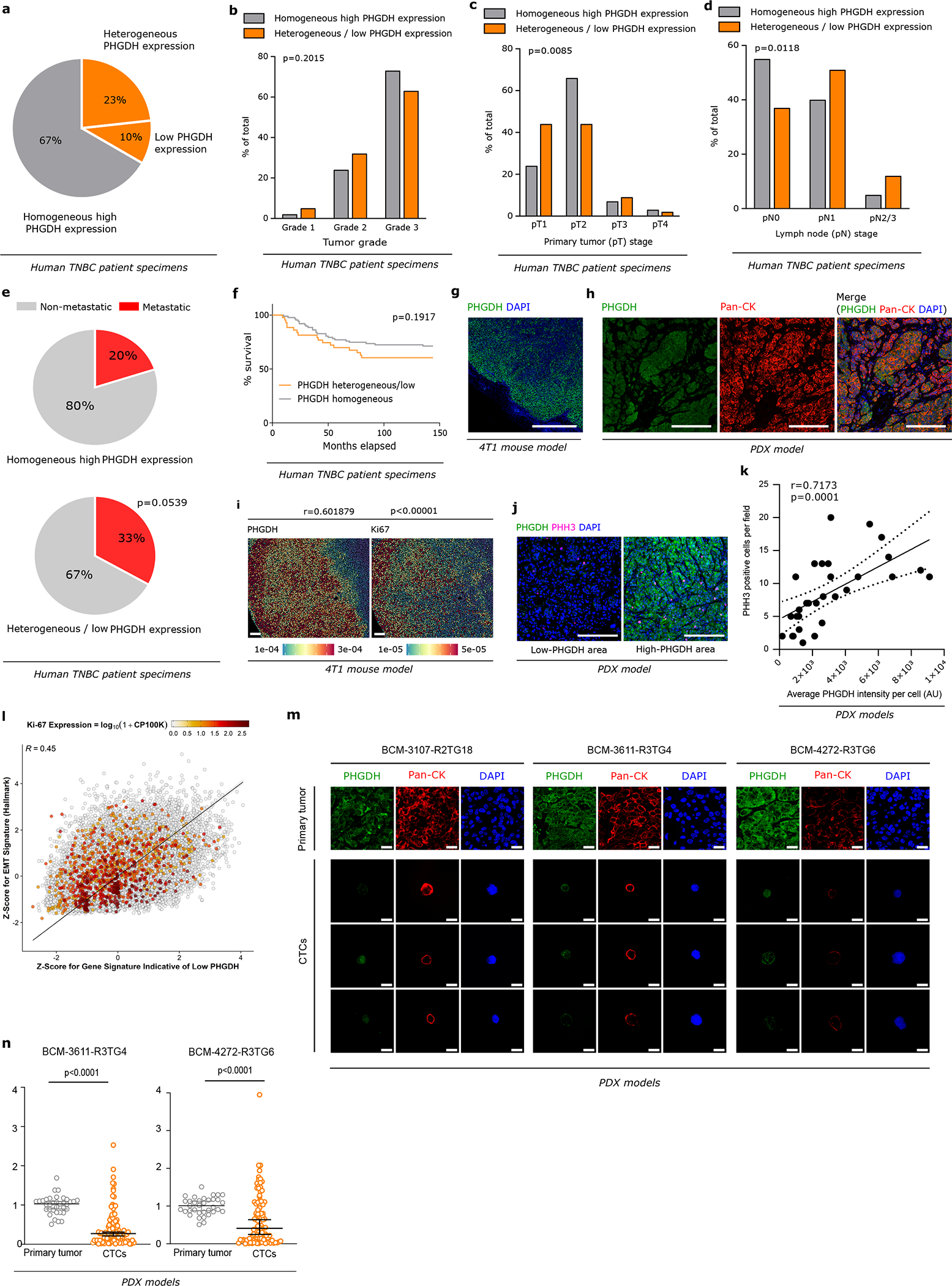
a. Distribution of PHGDH expression in human TNBC primary tumor specimens. PHGDH expression was assessed by immunohistochemistry (n=129).
b. Comparison of the tumor grade in human TNBC primary tumors with homogeneous high and heterogeneous/low PHGDH expression (n=126). Chi-squared test.
c. Comparison of the tumor stage (pT) in human TNBC primary tumors with homogeneous high and heterogeneous/low PHGDH expression (n=129). Chi-squared test.
d. Comparison of the lymph node stage (pN) in human TNBC primary tumors with homogeneous high and heterogeneous/low PHGDH expression (n=128). Chi-squared test.
e. Metastasis occurrence in TNBC patients bearing primary tumors with homogeneous high (17 out of 87) or heterogeneous/low (14 out of 42) PHGDH expression (n=129). Fisher’s exact test, two-sided.
f. Kaplan-Mayer curve comparing the % of survival of TNBC patients with heterogeneous/low and homogeneous PHGDH protein expression in the primary tumor (n=129). Mantel-Cox test.
g. Representative picture of PHGDH protein heterogeneity in the primary tumor from orthotopic (mammary fat pad, m.f.p.) 4T1 mouse model, assessed by immunohistochemistry. Green, PHGDH; blue, DAPI nuclear staining. Scale bar 1 mm.
h. Representative picture of PHGDH protein heterogeneity in the primary tumor from orthotopic (m.f.p.) TNBC PDX model, assessed by immunohistochemistry. Green, PHGDH; red, pan-cytokeratin tumor marker; blue, DAPI nuclear staining. Scale bar 200 μm.
i. Distribution and correlation of PHGDH and Ki67 protein expression in the primary tumor from orthotopic (m.f.p.) 4T1 mouse model assessed by imaging mass cytometry. Nonparametric Spearman rank correlation, two-tailed. Scale bar 400 μm.
j-k. Correlation of PHGDH and phospho-histone H3 (PHH3) protein expression in the primary tumor from orthotopic (m.f.p.) TNBC PDX model assessed by immunohistochemistry (j). The pooled analysis of 3 different PDX models, on 9 randomly chosen microscopy fields for each model, is shown in (k). Nonparametric Spearman rank correlation, two-tailed.
l. Correlation plot between GSVA-based Z-scores for a gene expression signature indicative of low PHGDH and one indicative of EMT, with the color code indicating Ki67 normalized expression levels (CP100k = counts per 100k reads), based on single cell RNA sequencing data for primary tumors of 13 TNBC patients.
m. Representative images of the expression levels of PHGDH protein in circulating tumor cells (CTCs) compared to the respective primary tumors from TNBC PDX models, assessed by immunohistochemistry. Green, PHGDH; red, pan-cytokeratin tumor marker; blue, DAPI nuclear staining. Scale bar 25 μm.
n. Expression levels of PHGDH protein in circulating tumor cells (CTCs) compared to the corresponding primary tumors from TNBC PDX models, assessed by immunohistochemistry (complementary to Figure 1c). Analysis performed on PDX models BCM-3611-R3TG4 (7 mice, 35 randomly chosen microscopy fields for the primary tumors, 5 per mouse, 104 single CTCs) and BCM-4272-R3TG6 (7 mice, 35 randomly chosen microscopy fields for the primary tumors, 5 per mouse, 101 single CTCs). The solid lines indicate the median, the whiskers indicate the 95% confidence interval. Unpaired t test with Welch’s correction, two-tailed.
Extended Data Figure 2. PHGDH protein levels but not mRNA expression is low in early metastatic lesions compared to primary tumors and advanced metastatic lesions.
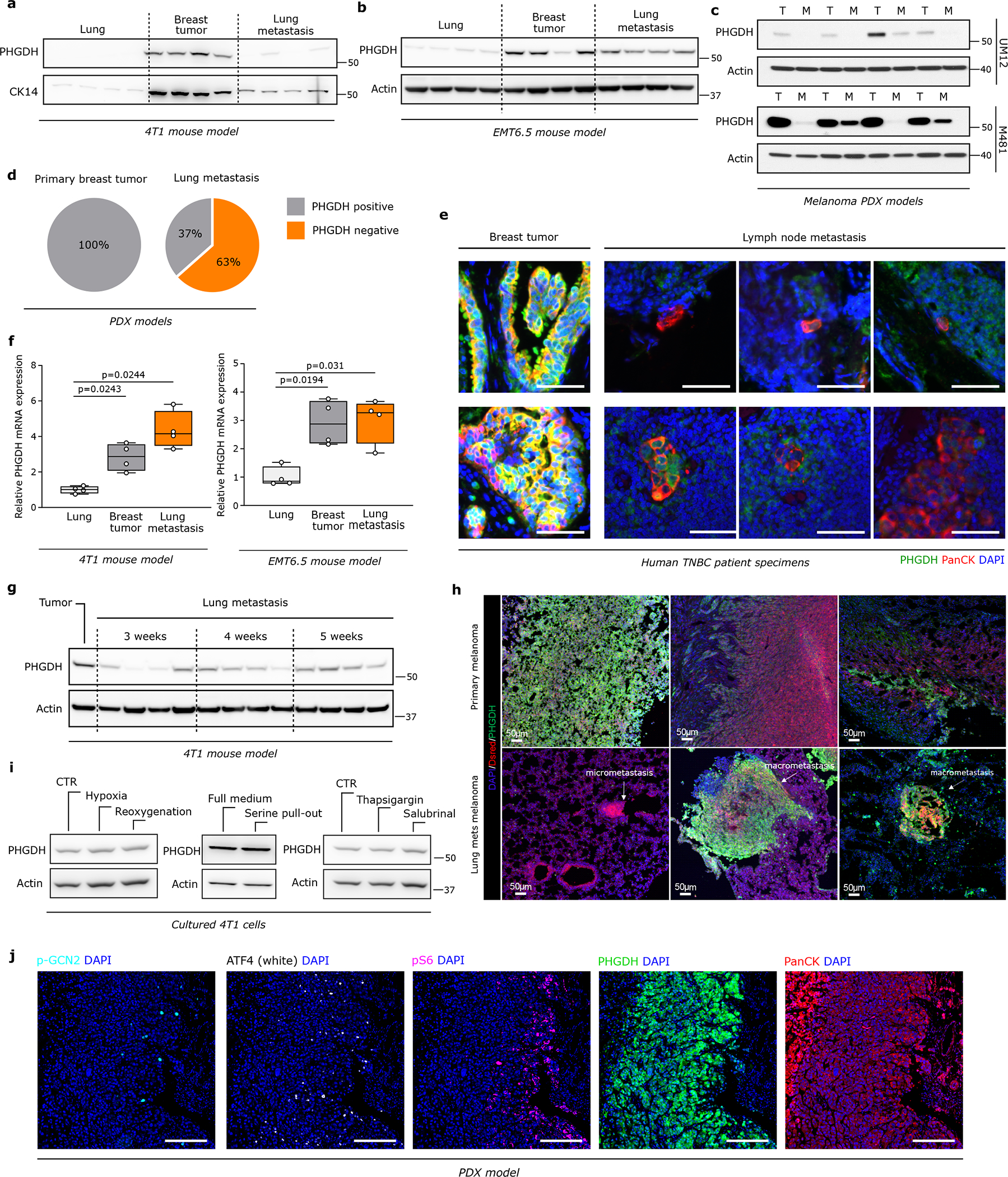
a-b. Western blot of PHGDH in lungs, primary breast tumors and lung metastases from orthotopic (m.f.p.) 4T1 (n=4) (a) and EMT6.5 (n=4) (b) mouse models.
c. Western blot analysis of PHGDH in primary tumors and liver metastases from two different orthotopic (subcutaneous) melanoma PDX models (n=4).
d. Positivity to PHGDH in primary breast tumors (n=9) and early lung metastases (~16 weeks after primary tumor initiation; n=52) from TNBC PDX models assessed by immunohistochemistry.
e. Representative images of PHGDH protein expression in lymph node metastases and matching primary breast tumors from TNBC patients assessed by immunohistochemistry. Green, PHGDH; red, pan-cytokeratin tumor marker; blue, DAPI nuclear staining. Scale bar 50 μm.
f. Relative change in Phgdh gene expression in lungs, primary breast tumors and lung metastases from orthotopic (m.f.p.) 4T1 and EMT6.5 mouse models (n=4). The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the minimum and maximum values. Welch and Brown-Forsythe ANOVA with Dunnett’s multiple comparison.
g. Western blot of PHGDH in lung metastases from orthotopic (m.f.p.) 4T1 mouse model (n=4), at 3, 4 and 5 weeks after injection of the cancer cells.
h. Representative pictures of PHGDH protein expression in primary and metastatic melanoma mouse model (Tyr::N-Ras+/Q61K;Ink4a−/−). Green, PHGDH; red, dsRed tumor cell marker; blue, DAPI nuclear staining.
i. Western blot of PHGDH levels in 4T1 cells upon hypoxia and reoxygenation, serine pull-out compared to full medium, and treatment with salubrinal (ATF4 activation) or thapsigargin (ER stress induction). One representative experiment is shown (n=3).
j. Representative pictures of expression levels of PHGDH and ATF4 activation markers in TNBC PDX model, assessed by multiplex immunofluorescence. Turquoise, p-GCN2; White, ATF4; Pink, pS6; Green, PHGDH; red, pan-cytokeratin tumor marker; blue, DAPI nuclear staining. Scale bar 200 μm.
Extended Data Figure 3. Proximity to endothelial cells induces loss of PHGDH in cancer cells.
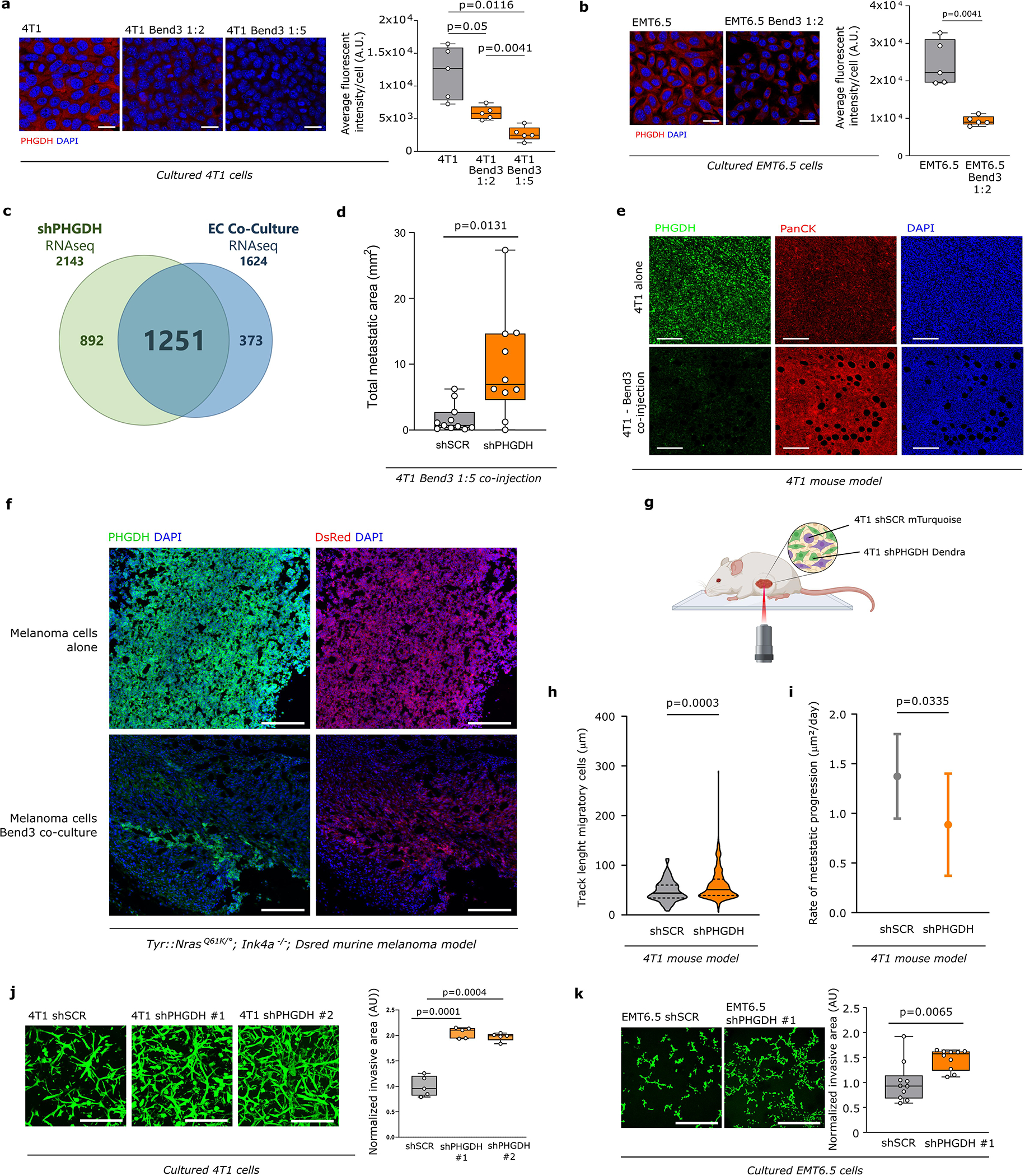
a-b. PHGDH expression in 4T1 (1:2 and 1:5 ratio) and EMT6.5 cells (1:2 ratio) co-cultured with Bend3 immortalized mouse endothelial cells based on immunofluorescence. Left panel, representative pictures (scale bar 50 μm), red, PHGDH; blue, DAPI nuclear staining. The image represented was selected to show only 4T1/EMT6.5 cells based on pan-cytokeratin expression in cancer cells and GFP expression in Bend3 cells. Right panel, fluorescent intensity quantification (n=4 independent samples). Solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the minimum and maximum values. Welch and Brown-Forsythe ANOVA with Dunnett’s multiple comparison (a) and unpaired t test with Welch’s correction, two-tailed (b).
c. Venn diagram depicting the number of overlapping enriched gene sets, based on Gene Set Enrichment Analysis (GSEA) in RNAseq data from 4T1 Phgdh knock-down (shPHGDH) compared to control cells (left), versus enriched gene sets in 4T1 and EMT6.5 cells co-cultured (1:2 ratio) with Bend3 immortalized mouse endothelial cells (EC Co-Culture) compared to mono-cultured 4T1 or EMT6.5 cells (right). The numbers at the top represent the total enriched gene-sets found in the corresponding data set.
d. Lung metastatic area in mice co-injected with 4T1 cancer cells and Bend3 endothelial cells (ratio 1:2) or 4T1 cells alone in the mammary fat pad (n≥10). The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the minimum and maximum values. Unpaired t test with Welch’s correction, two-tailed.
e. PHGDH protein expression in 4T1 primary tumor of mice injected with cancer cells alone or co-injected with Bend3 immortalized mouse endothelial cells assessed by immunohistochemistry. Green, Phgdh; red, pan-cytokeratin tumor marker; blue, DAPI nuclear staining. Scale bar 200 μm.
f. Representative pictures of PHGDH protein expression in primary melanoma model (Tyr::N-Ras+/Q61K;Ink4a−/−) from mice injected with melanoma cells alone or co-injected with melanoma and Bend 3 endothelial cells (ratio 1:2). Green, PHGDH; red, dsRed tumor cell marker; blue, DAPI nuclear staining. Scale bar 200 μm.
g. Schematic representation of the time-lapse intravital imaging experiment setup.
h. Track length of migratory 4T1 shSCR-mTurquoise and 4T1 shPHGDH-Dendra in primary tumors from orthotopic (m.f.p.) 4T1 mouse model assessed by time-lapse intravital imaging. The solid lines indicate the median, the dashed lines indicate the 25th and 75th percentiles. Unpaired t test with Welch’s correction, two-tailed.
i. Rate of metastatic progression of lung metastases from mice injected with either 4T1 shSCR (n=10 per time point) or 4T1 shPHGDH cells (n=10 per time point). Error bars represent standard deviation (s.d.) from mean. Unpaired t test with Welch’s correction, two-tailed.
j-k. Invasive capacity of 4T1 and EMT6.5 cells upon Phgdh knock-down (shPHGDH) compared to control (shSCR) cells in a 3D matrix. Invasion was assessed by measuring the invasive area of cancer cells stained with calcein green. Representative images are depicted in the left panel (scale bar 500 μm), quantification in the right panel. Each dot represents a different, randomly selected microscopy field (n=5 for (j), n=10 for (k)). The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the minimum and maximum values. Welch and Brown-Forsythe ANOVA with Dunnett’s multiple comparison (j) and Unpaired t test with Welch’s correction, two-tailed (k).
Extended Data Figure 4. Loss of PHGDH induces a partial EMT and the expression of markers indicating altered cell-cell or cell matrix interactions in cancer cells.
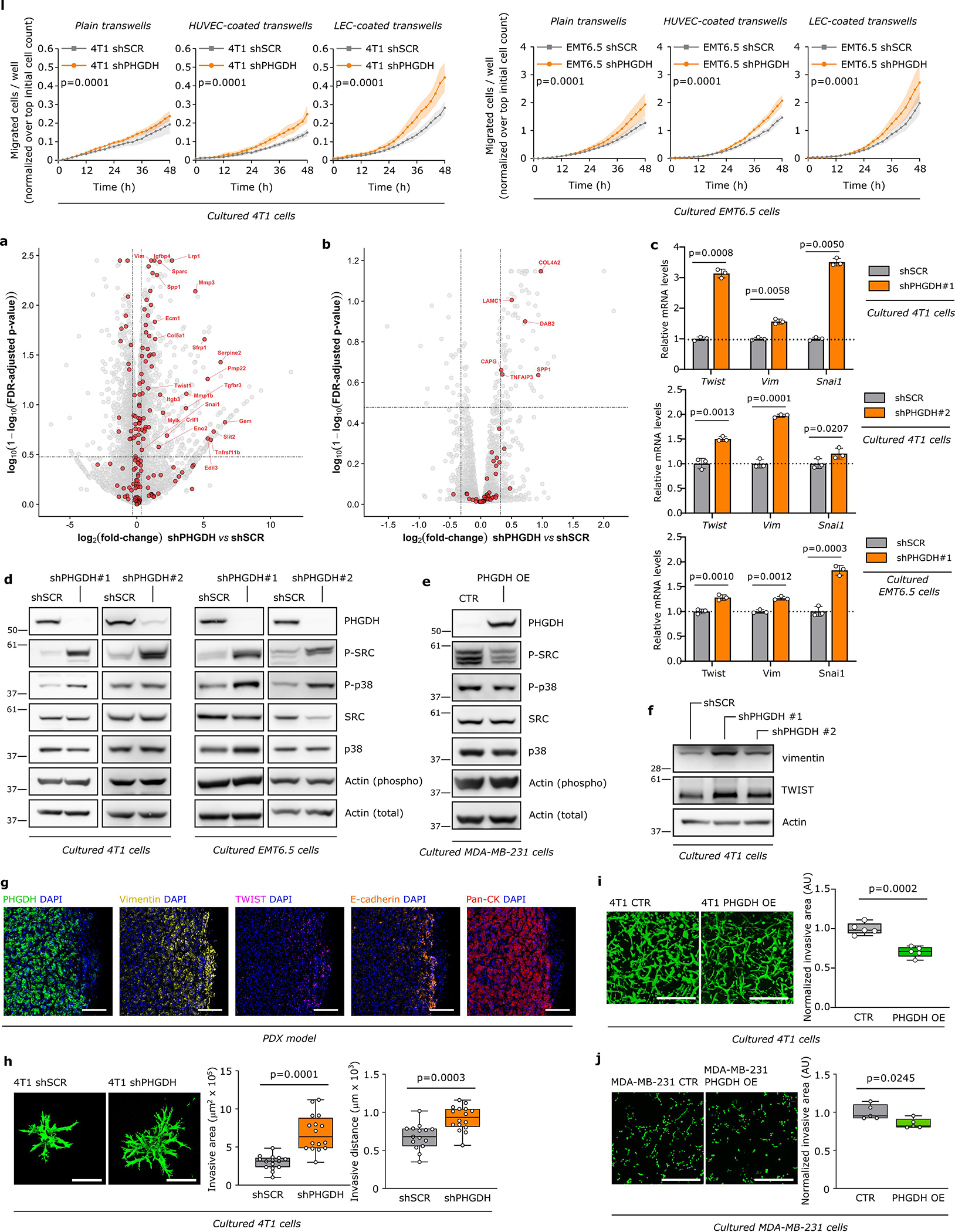
a. Migratory ability of 4T1 and EMT6.5 cells upon Phgdh knockdown (shPHGDH) compared to control (shSCR) cells. Migration was assessed in plain transwells and in transwells coated with either vascular endothelial cells (HUVECs) or lymphatic endothelial cells (LECs) (n=3). Two-way ANOVA.b-c. Volcano plots showing genes (b) or proteins (c) upregulated or downregulated upon loss of PHGDH based on RNAseq (b) or proteomics (c) data for 4T1 cells with Phgdh knockdown (shPHGDH) relative to control cells (shSCR). The cutoffs represent p-values of 0.01 and absolute fold-changes of 1.25. Genes (b) or proteins (c) indicative of EMT are shown in red, with labels included for select genes/proteins above the respective p-value/fold-change cutoffs.
c. Relative change in Twist, vimentin (Vim) and Snai1 gene expression upon PHGDH knockdown in 4T1 and EMT6.5 cells. Error bars represent standard deviation (s.d.) from mean (n=3). Unpaired t test with Welch’s correction, two-tailed.
d. Western blot of phosphorylated proto-oncogene tyrosine-protein kinase Src (c-Src) and p38 mitogen-activated protein kinase upon Phgdh knockdown in 4T1 and EMT6.5 cells. One representative experiment is shown (n=3).
e. Western blot of Vimentin and Twist in 4T1 cells upon Phgdh knockdown (shPHGDH) compared to control (shSCR). One representative experiment is shown (n=3).
f. Western blot of phosphorylated proto-oncogene tyrosine-protein kinase Src (c-SRC) and p38 mitogen-activated protein kinase upon Phgdh overexpression in MDA-MB-231 cells. One representative experiment is shown (n=3).
g. Representative pictures of PHGDH and EMT markers protein expression in TNBC PDX model assessed by multiplex immunofluorescence. Green, PHGDH; Yellow, VIMENTIN; Pink, TWIST; Orange, E-CADHERIN; red, pan-cytokeratin tumor marker; blue, DAPI nuclear staining. Scale bar 200 μm.
h. Invasive area and distance of 4T1 cells upon Phgdh knockdown (shPHGDH) compared to control (shSCR) cells (n=15 independent samples). The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the minimum and maximum values. Unpaired t test with Welch’s correction, two-tailed.
i-j. Invasive capacity of 4T1 and MDA-MB-231 cells upon PHGDH overexpression (PHGDH OE) compared to control (CTR) cells in a 3D matrix. The invasive area of cancer cells was stained with calcein green. Representative images are depicted in the left panel (scale bar 500 μm), quantification in the right panel. Each dot represents a different microscopy field (n=5). The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the minimum and maximum values. Unpaired t test with Welch’s correction, two-tailed.
Extended Data Figure 5. Modulation of PHDGH expression alters gene expression signatures related to metastasis formation.
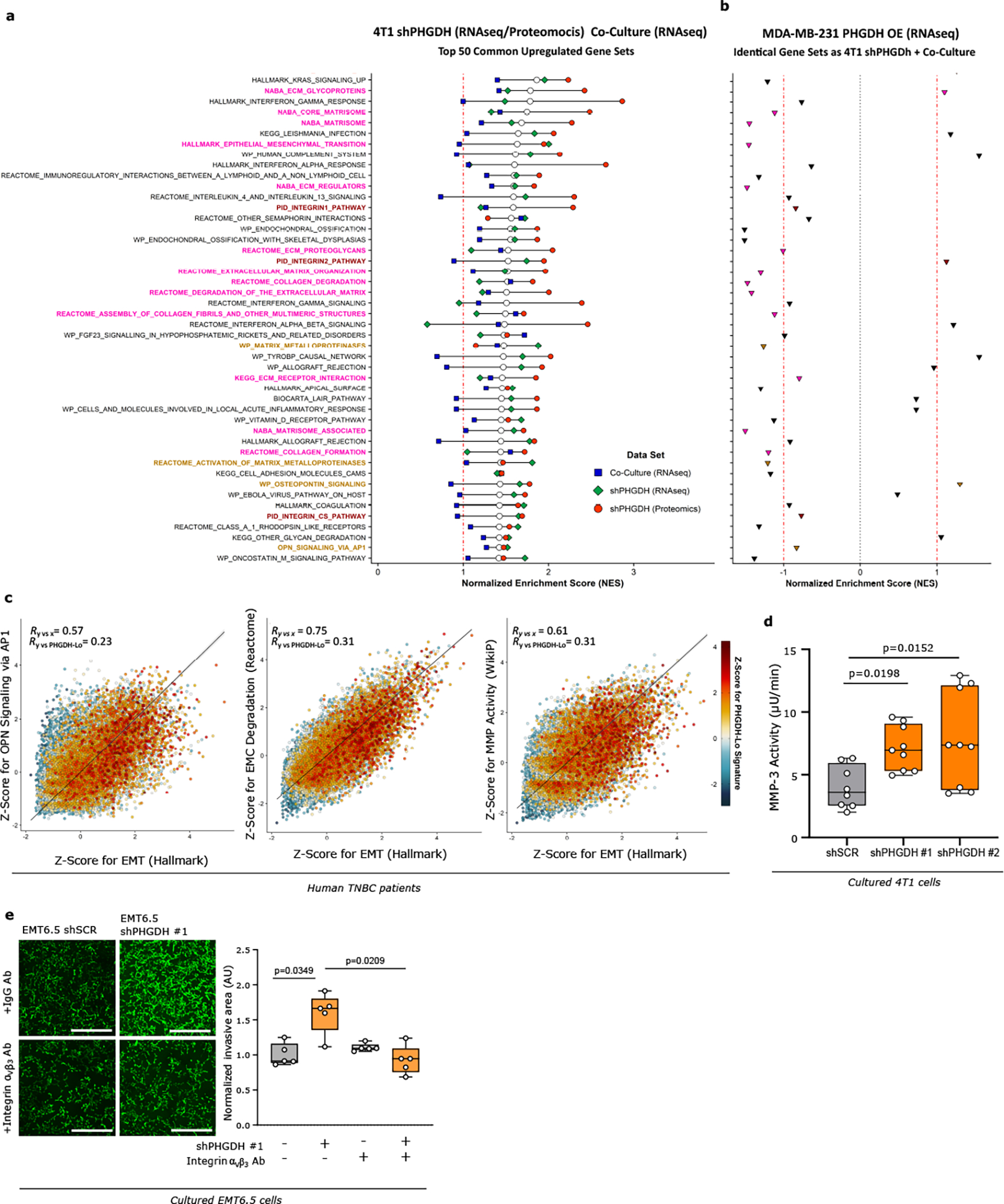
a. GSEA showing the top 50 commonly upregulated gene sets upon integration of the RNAseq/Proteomics data sets for 4T1 cells upon Phgdh knockdown (shPHGDH) compared to control cells, and the RNAseq data set for 4T1 or EMT6.5 cells co-cultured with Bend 3 immortalized mouse endothelial cells, compared to mono-cultured 4T1 or EMT6.5 cells. Normalized enrichment scores (NES) for each data set are indicated by the colored symbols, as defined in the plot legend. Gene sets are ranked based on their average NES among all three data sets, indicated by the white dots, with those gene sets with the highest mean NES shown on top. The red dash-dotted line indicates a NES of 1. The gene-set entries on the y-axis include three single-sample signatures comprising the most differentially upregulated genes in each of the three data sets (color-coded identically to the plot legend), as well as a signature consisting in the intersection of those for the Phgdh knockdown and co-culture RNAseq data (color-coded in orange), indicative of low Phgdh protein expression. The remaining entries on the y-axis are color-coded based on their belonging to one of the following categories: EMT and ECM remodeling (pink), integrin signaling (burgundy), OPN/AP-1 signaling and MMP activity (light brown), or other (black).
b. GSEA results based on RNAseq data for MDA-MB-231 cells upon PHGDH overexpression (PHGDH OE) compared to control cells for the identical gene sets as in a. The red dash-dotted lines indicate a NES of ±1, whereas the black dotted line indicates a NES of 0. Data points are color-coded according to the same color scheme used for the respective gene-set entries in a.
c. Correlation plots of GSVA-derived Z-scores for 3 of the top 50 hits found upon integration of the RNAseq/Proteomics/Co-Culture data sets (see Extended Figure 5a) versus the scores for the Hallmark EMT gene signature (on the x-axis). Data were obtained from RNAseq of scRNA-seq data for primary tumors of 13 TNBC patients. The color code indicates the Z-Score for a gene expression signature indicative of low PHGDH protein expression. Total least-squares regression lines and confidence intervals are overlaid on top of each plot, with the corresponding Pearson correlation coefficient (R) values shown on the top-left corners.
d. Activity of extracellularly secreted MMP-3 (μU/min) in cell culture media collected from invasion assays of 4T1 cells upon Phgdh knockdown (shPHGDH) or control (shSCR) cells after 72h of seeding (n=3 independent experiments). The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the minimum and maximum values. One-way ANOVA with Dunn’s multiple comparison.
e. Invasive capacity of EMT6.5 cells pre-treated (24h) with an antibody against integrin αvβ3 or control IgG (2.5 μg/ml) and upon Phgdh knockdown (shPHGDH) compared to control (shSCR) cells in a 3D matrix. The invasive area was stained with calcein green. Each dot represents a different microscopy field (n=5). The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the minimum and maximum values. Welch and Brown-Forsythe ANOVA with Dunnett’s multiple comparison.
Extended Data Figure 6. Low PHGDH protein expression increases sialic acid metabolism and promotes glycosylation.

a. Schematic representation of glycolysis and its branching metabolic pathways. Enzymes are depicted in bold, pathway names in italics. Solid lines represent single reactions, dashed lines recapitulate multiple reactions.
b. Metabolite abundances of sialic acid, UDP-N-acetylglucosamine (UDP-GlcNAc) and CMP-sialic acid upon Phgdh knock-down in 4T1 cells (n=15). The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the minimum and the maximum values. Unpaired t test with Welch’s correction, two-tailed.
c. Dynamic 13C6 glucose labeling of MDA-MB-231 cells showing 13C incorporation into sialic acid, UDP-N-acetylglucosamine and CMP-sialic acid upon PHGDH overexpression (PHGDH OE) (n=3). Error bars represent s.d. from mean. Two-way ANOVA.
d. Metabolite abundances of sialic acid, UDP-N-acetylglucosamine and CMP-sialic acid upon PHGDH overexpression (PHGDH OE) in MDA-MB-231 cells (n=15). The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the smallest and the largest values. Unpaired t test with Welch’s correction, two-tailed.
e. Sialic acid/GlcNAc-containing-proteins isolated from whole cell lysate of 4T1 cells upon Phgdh knock-down (shPHGDH) or control (shSCR) cells treated with tunicamycin (0.05 μg/ml) or DMSO for 72h. Total isolated Sialic acid/GlcNAc-linked proteins were quantifying using Qubit™ Protein Assay Kit (n=2 independent experiments). Error bars represent s.d. from mean.
f. Levels of β-1,4-GlcNAc- and sialic acid-linked residues in 4T1 Phgdh knockdown (shPHGDH) and control (shSCR) cells after 72h of tunicamycin pretreatment (0.05 μg/ml) measured at 7, 24 and 48h after tunicamycin removal using wheat germ agglutinin (WGA) staining (n=3). Red, WGA β-1,4-GlcNAc- and sialic acid-linked proteins; blue, DAPI nuclear staining. Error bars represent standard deviation (s.d.) from mean. Unpaired t test with Welch’s correction, two-tailed. Scale bar 20 μm.
g. Cell viability upon tunicamycin (0.05 μg/ml) and PHGDH inhibitor (PH755, 1 μM) treatment (36h) in 4T1 cells upon Phgdh knock-down (shPHGDH) or control (shSCR) (n=5). Error bars represent s.d. from mean. One-way ANOVA with Turkey’s multiple comparison test.
h. Protein expression levels of glycosylated integrin β3 (elution) after WGA-mediated isolation of β-1,4-GlcNAc- and sialic acid-linked proteins from total lysates of 4T1 cells upon Phgdh knock-down (4T1 shPHGDH), Cmas knockout, and double Phgdh and Cmas gene inactivation, compared to control cells (4T1 shSCR). Total levels of integrin β3 from the whole cell lysate and actin as loading control are shown. Experiments were performed in triplicate, and one representative experiment is shown.
i. β-1,4-GlcNAc- and sialic acid-linked proteins isolated from whole cell lysate of 4T1 cells upon Phgdh and Cmas knockdown (shPHGDH, shCMAS), Cmas knockout (KO) or control (shSCR) cells treated with tunicamycin (0.05 μg/ml) or DMSO for 72h (n=2independent experiments). Error bars represent s.d. from mean.
Extended Data Figure 7. Low PHGDH protein expression promotes invasion and metastasis via protein sialylation.

a. Invasive capacity of 4T1 cells upon Phgdh knockdown (shPHGDH) compared to control (shSCR) cells pretreated with tunicamycin (0.05 μg/ml) for 72h prior seeding in a 3D matrix. The invasive area was stained with calcein green. Each dot represents a different microscopy field (n=5). The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the minimum and maximum values. Welch and Brown-Forsythe ANOVA with Dunnett’s multiple comparison.
b-e. Invasive ability of 4T1 cells with functional (CTR or shSCR) or inactive (Cmas KO or shCMAS) sialic acid pathway upon Phgdh knockdown (shPHGDH) compared to control (shSCR) cells(b, d); EMT6.5 cells upon Phgdh knock-down (shPHGDH) compared to control (shSCR) cells after 48h of pretreatment with the sialytransferase inhibitor Lith-O-Asp (30 μM) (c); and 4T1 and EMT6.5 cells upon CMAS overexpression (CMAS OE) compared to control (CTR) cells (e) in a 3D matrix. The invasive area was stained with calcein green. Representative images are depicted in the left panel (scale bar 500 μm), quantification in the right panel. Each dot represents a different microscopy field (n=5). The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the smallest and the largest values. Welch and Brown-Forsythe ANOVA with Dunnett’s multiple comparison.
f. Number of lung metastases per mouse in the orthotopically injected (m.f.p.) mice with 4T1 cells with either functional (CTR, n=12) or inactive (Cmas KO, n=10) sialic acid pathway, with (shPHGDH, n=10) or without (shSCR, n=10) concomitant Phgdh knockdown. The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the smallest and the largest values. Welch and Brown-Forsythe ANOVA with Dunnett’s multiple comparison.
Extended Data Figure 8. Loss of PHGDH protein decreases PKF activity and increases carbon flux into the sialic acid pathway.

a. Changes in metabolite abundance upon Phgdh knockdown in 4T1 and EMT6.5 cells. Data represent fold changes compared to non-silenced cells (shSCR) (n=3). H6P, hexose-6-phosphate; FBP, fructose bisphosphate; DHAP, dihydroxyacetone phosphate; GAP, glyceraldehyde 3-phosphate; xPG, 2/3-phosphoglycerate; PEP, phosphoenolpyruvate. Unpaired t test two-tailed. *indicates statistically significant as follows: FBP, p=0.0042 (4T1), p=0.0012 (EMT6.5); PEP, p=0.0031 (4T1), p=0.034 (EMT6.5); Pyruvate, p=2.88e−11 (4T1).
b. Fractional contribution of 13C6-glucose to glycolytic intermediates in 4T1 upon Phgdh knockdown (shPHGDH) compared to control (shSCR) cells (n=3). Error bars represent s.d. from mean. Two-way ANOVA.
c. Interaction of PHGDH with PFKP in 4T1 control (shSCR) and Phgdh knockdown (shPHGDH) cells assessed by co-immunoprecipitation of PHGDH. A representative experiment is shown (n=3 independent experiments).
d. Protein expression levels of PHGDH, PSAT and PSPH in TNBC PDX model (BCM-3107-R2TG18) assessed by multiplex immunofluorescence. Green, PHGDH; Turquoise, PSAT; Pink, PSPH; red, pan-cytokeratin tumor marker; blue, DAPI nuclear staining. Scale bar 200 μm.
e. Protein expression levels of PHGDH upon treatment with the PHGDH catalytic inhibitor PH-775 (1 μM) for 72h. One representative experiment is shown (n=3).
f. Inhibition of de novo serine biosynthesis assessed through measurement of serine m+3 labeling after incubation of 4T1 cells for 24h in culture medium containing 13C6-glucose upon treatment with the PHGDH catalytic inhibitor PH-775 (1 μM) for 72h. Error bars represent s.d. from mean. Unpaired t test with Welch’s correction, two-tailed.
g. Dynamic labeling using 13C6-glucose of 4T1 cells showing 13C incorporation into sialic acid upon treatment with the PHGDH catalytic inhibitor PH-775 (1 μM) for 72h (n=3). Error bars represent s.d. from mean. Two-way ANOVA.
h. Metabolite abundance of sialic acid upon treatment with the PHGDH catalytic inhibitor PH-775 (1 μM) for 72h in 4T1 cells. The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the smallest and the largest values (n=15). Unpaired t test with Welch’s correction, two-tailed.
G6P, glucose-6-phosphate; F6P, fructose-6-phosphate; H6P, hexose-6-phosphate; FBP, fructose bisphosphate; DHAP, dihydroxyacetone phosphate; xPG, 2/3-phosphoglycerate; PEP, phosphoenolpyruvate.
Extended Data Figure 9. Low PHGDH protein expression, but not low catalytic activity, promotes sialic acid synthesis and drives metastatic dissemination.

a-b. Measurement of serine m+3 labeling enrichment after incubation of (a) MDA-MB-231 control (CTR), PHGDH (PHGDH OE) overexpressing and catalytic inactive PHGDH (PHGDH CI OE) overexpressed cells or (b) 4T1 cells upon Phgdh knockdown (shPHGDH), Phgdh-silencing with wildtype (shPHGDH + wt OE) and catalytic inactive overexpression (shPHGDH + CI OE) for 24h in culture medium containing 13C6-glucose (n=3). Error bars represent s.d. from mean. One-way ANOVA with Holm-Sidak’s multiple comparison test.
c. Dynamic labeling using 13C6-glucose and total abundance of sialic acid in 4T1 cells upon Phgdh knockdown (shPHGDH), Phgdh-silenced cells with wildtype (shPHGDH + wt OE) and catalytic inactive overexpression (shPHGDH + CI OE) cells. Left panel, p values refer to comparison of shSCR or shPHGDH + CI OE vs shPHGDH (n=3). For sialic acid abundance, the solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the minimum and maximum values (n=6). One-way ANOVA with Holm-Sidak’s multiple comparison test.
d. Levels of glycosylated integrin β3 (elution) after WGA-mediated isolation of β-1,4-GlcNAc- and sialic acid-linked proteins from total lysates of 4T1 control (shSCR), Phgdh knockdown (shPHGDH) cells, and Phgdh-silenced 4T1 cells with wildtype (shPHGDH +wt OE) and catalytic inactive (shPHGDH + CI OE) overexpression. The last two samples correspond to tunicamycin treated (48h, 0.05 μg/ml) 4T1 control (shSCR) and Phgdh knockdown (shPHGDH). Total levels of integrin β3 from the whole cell lysate and actin as housekeeper are shown. One representative experiment is shown (n=3).
e. Invasive capacityof 4T1 cells upon treatment with the PHGDH catalytic inhibitor PH-775 (1 μM) for 72h and rescue with cell-permeable α-ketoglutarate (αKG, 1 mM) in a 3D matrix. The invasive area was stained with calcein green. Representative images are depicted in the left panel (scale bar 500 μm), quantification in the right panel. Each dot represents a different, randomly selected microscopy field. The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the smallest and the largest values (n=5). One-way ANOVA with Tukey’s multiple comparison test.
f-h. Invasive ability of 4T1 control (shSCR), Phgdh knockdown (shPHGDH) cells and Phgdh-silenced 4T1 cells with wildtype (shPHGDH +wt OE) and catalytic inactive overexpression (shPHGDH + CI OE) (f); MDA-MB-231 control (CTR), PHGDH wildtype overexpression (OE) and catalytic inactive overexpression (CI OE) cells (g); 4T1 control (SCR KO), Psat (PSAT KO) and Psph knockout cells (PSPH KO) (h) in 3D matrix. The invasive area was stained with calcein green. Representative images are depicted in the left panel (scale bar 500 μm), quantification in the right panel. Each dot represents a different microscopy field (n=5). The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the minimum and maximum values. Welch and Brown-Forsythe ANOVA with Dunnett’s multiple comparison.
i. Percentage of migratory cells per migratory position (n=11) in the primary tumor of the orthotopic (m.f.p.) 4T1 mouse model assessed by time-lapse intravital imaging. Mice were injected with a mixture of 4T1 shPHGDH-Dendra and 4T1 shPHGDH with overexpression of wildtype (shPHGDH + wt OE) (n=8) or catalytic inactive (shPHGDH + wt CI) PHGDH-mTurquoise (n=8). The solid lines indicate the median, the boxes extend to the 25th and 75th percentiles, the whiskers span the minimum and the maximum values. Unpaired t test with Welch’s correction, two-tailed.
Extended Data Figure 10. Validation of genetic modification of breast cancer cells.

a. Protein expression levels of PHGDH and CMAS in 4T1 cells used in Figures 2e, 3a, 3b, 3c, 3d, and Extended Data Figures 3j, 3l, 4a, 4b, 4c, 4d, 4e, 4h, 5a, 5b, 5e, 6b, 6e, 6f, 6g, 6h, 6i, 7a, 7b, 7d, 7f, 8b, 8c and 9e.
b. Protein expression levels of PHGDH and CMAS in 4T1 and EMT6.5 cells used in Extended Data Figure Figures 2j, 4c
c. Protein expression levels of PHGDH in MDA-MB-231 and 4T1 cells used in Figures 4c, 4c and Extended Data Figures 4f, 4j,6c, 6d, 9a–f and 9i.
d. Relative Phgdh expression in MDA-MB-231 and 4T1 cells detailed in c (n=3 independent samples). Error bars represent standard deviation (s.d.) from mean.
e. Psat and Psph gene inactivation measured by a decrease in 13C6-Glucose incorporation into serine in 4T1 cells used in Extended Data Figure 9g (n=3 independent samples). Error bars represent standard deviation (s.d.) from mean.
f. Relative Phgdh expression and invasion ability in 4T1 cells used in Figures 2a, 2b, 4c, and Extended Data Figures 3h, 3i and 9i (n=3independent samples). Error bars represent standard deviation (s.d.) from mean.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Matt Vander Heiden (MIT) for providing the PHGDH overexpression plasmid. We would like to thank Pawel Bieniasz-Krzywiec (VIB-KU Leuven) for his help with the transwell migration assay and Vincent van Hoef (VIB Bioinformatics Core Facility) for his advice on the RNAseq analysis. We would like to thank Raze Therapeutics for providing us with the PHGDH inhibitor PH-755. The breast tissue and data bank at the Goodman Cancer Research Centre-Research Institute of McGill University Health Cantre (MUHC) is supported by the Database and Tissue Bank Axis of the Réseau de Recherche en Cancer of the Fonds de Recherche du Québec-Santé and the Québec Breast Cancer Foundation and certified by the Canadian Tumor Repository Network (CTRNet). Ilustrations in Figure 4e and Extended Data Figure 4g were created with BioRender.com.
FUNDING
MR has received consecutive postdoctoral fellowships from FWO and Stichting tegen Kanker and an Early Access Grant from the VIB Technology Watch Team Program. PA-M has received funding from Marie Curie Actions and the Beug Foundation. AMC has received a fellowship form Boehringer Ingelheim. GD and GR have received consecutive PhD fellowships from Kom op tegen Kanker and FWO and AV from FWO. HFA has received funding from the Stichting tegen Kanker and King Boudewijn Foundation. JF-G has received consecutive postdoctoral fellowships from FWO. JvR and LB were funded by Cancer Genomics Netherlands and Doctor Josef Steiner Foundation. TGPG acknowledges funding from the Barbara and Wilfried Mohr Foundation, the Matthias-Lackas Foundation, the Dr. Leopold and Carmen Ellinger Foundation, the Dr. Rolf M. Schwiete Foundation, the German Cancer Aid (DKH-70112257, DKH-70114111), the Gert and Susanna Mayer Foundation, the Boehringer Ingelheim Foundation, and the SMARCB1 association. SYL was supported by the National Cancer Institute of the National Institutes of Health under Award Number R01CA270136 and the METAvivor Early Career Investigator Grant. MP is grateful for the support from the Quebec Cancer Consortium and the financial support from the Ministère de l’Économie et de l’Innovation du Québec through the Fonds d’accélérations des collaborations en santé. The Patient-derived Xenograft and Advanced In Vivo Models Core at Baylor College received funding from CPRIT Core Facility Award (RP170691) and P30 Cancer Center Support Grant (NCI-CA125123). S-MF acknowledges funding from the European Research Council under the ERC Consolidator Grant Agreement n. 771486–MetaRegulation, FWO – Research Projects (G088318N), KU Leuven – FTBO, King Baudouin Foundation, Beug Foundation and Fonds Baillet Latour.
Footnotes
DECLARATION OF INTERESTS
S-MF has received funding from Bayer AG, Merck and Black Belt Therapeutics, has consulted for Fund+ and is in the advisory board of Alesta Therapeutics. TGPG has consulted for Boehringer Ingelheim. MTL is an uncompensated President, CEO, and Limited Partner of StemMed Ltd. and an uncompensated Manager in StemMed Holdings LP, its General Partner. He is a founder and equity holder in Tvardi Therapeutics Inc., and a faculty member at Baylor College of Medicine. LED is a compensated employee at Baylor College of Medicine. All other authors declare no competing interests.
REFERENCES MAIN TEXT
- 1.Gupta PB, Pastushenko I, Skibinski A, Blanpain C & Kuperwasser C Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell stem cell 24, 65–78, doi: 10.1016/j.stem.2018.11.011 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vitale I, Shema E, Loi S & Galluzzi L Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nature medicine 27, 212–224, doi: 10.1038/s41591-021-01233-9 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Lüönd F, Tiede S & Christofori G Breast cancer as an example of tumour heterogeneity and tumour cell plasticity during malignant progression. British journal of cancer 125, 164–175, doi: 10.1038/s41416-021-01328-7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergers G & Fendt SM The metabolism of cancer cells during metastasis. Nature reviews. Cancer 21, 162–180, doi: 10.1038/s41568-020-00320-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prasetyanti PR & Medema JP Intra-tumor heterogeneity from a cancer stem cell perspective. Molecular Cancer 16, 41, doi: 10.1186/s12943-017-0600-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pastushenko I & Blanpain C EMT Transition States during Tumor Progression and Metastasis. Trends in Cell Biology 29, 212–226, doi: 10.1016/j.tcb.2018.12.001 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Locasale JW et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nature genetics 43, 869–874 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Possemato R et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350, doi: 10.1038/nature10350 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rinaldi G et al. In Vivo Evidence for Serine Biosynthesis-Defined Sensitivity of Lung Metastasis, but Not of Primary Breast Tumors, to mTORC1 Inhibition. Molecular Cell 81, 386–397, doi: 10.1016/j.molcel.2020.11.027 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ngo B et al. Limited Environmental Serine and Glycine Confer Brain Metastasis Sensitivity to PHGDH Inhibition. Cancer discovery, CD-19–1228, doi: 10.1158/2159-8290.CD-19-1228 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geeraerts SL et al. Repurposing the antidepressant sertraline as SHMT inhibitor to suppress serine/glycine synthesis addicted breast tumor growth. Molecular Cancer Therapeutics, molcanther.0480.2020, doi: 10.1158/1535-7163.MCT-20-0480 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pacold ME et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nature chemical biology 12, 452–458, doi: 10.1038/nchembio.2070 http://www.nature.com/nchembio/journal/v12/n6/abs/nchembio.2070.html#supplementary-information (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmidt JM et al. Stem-cell-like properties and epithelial plasticity arise as stable traits after transient Twist1 activation. Cell Rep 10, 131–139, doi: 10.1016/j.celrep.2014.12.032 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Noh S, Kim DH, Jung WH & Koo JS Expression levels of serine/glycine metabolism-related proteins in triple negative breast cancer tissues. Tumour Biol 35, 4457–4468, doi: 10.1007/s13277-013-1588-z (2014). [DOI] [PubMed] [Google Scholar]
- 15.Pascual G et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541, 41–45, doi: 10.1038/nature20791 http://www.nature.com/nature/journal/v541/n7635/abs/nature20791.html#supplementary-information (2017). [DOI] [PubMed] [Google Scholar]
- 16.Oshimori N, Oristian D & Fuchs E TGF-β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 160, 963–976, doi: 10.1016/j.cell.2015.01.043 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Margarido AS, Bornes L, Vennin C & van Rheenen J Cellular Plasticity during Metastasis: New Insights Provided by Intravital Microscopy. Cold Spring Harbor Perspectives in Medicine 10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beerling E, Oosterom I, Voest E, Lolkema M & van Rheenen J Intravital characterization of tumor cell migration in pancreatic cancer. IntraVital 5, e1261773, doi: 10.1080/21659087.2016.1261773 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kariya Y, Oyama M, Suzuki T & Kariya Y αvβ3 Integrin induces partial EMT independent of TGF-β signaling. Communications Biology 4, 490, doi: 10.1038/s42003-021-02003-6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mori S et al. Enhanced Expression of Integrin αvβ3 Induced by TGF-β Is Required for the Enhancing Effect of Fibroblast Growth Factor 1 (FGF1) in TGF-β-Induced Epithelial-Mesenchymal Transition (EMT) in Mammary Epithelial Cells. PloS one 10, e0137486, doi: 10.1371/journal.pone.0137486 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seguin L et al. An integrin β3–KRAS–RalB complex drives tumour stemness and resistance to EGFR inhibition. Nature Cell Biology 16, 457–468, doi: 10.1038/ncb2953 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bellahcène A, Castronovo V, Ogbureke KU, Fisher LW & Fedarko NS Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): multifunctional proteins in cancer. Nature reviews. Cancer 8, 212–226, doi: 10.1038/nrc2345 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janik ME, Lityoska A & Vereecken P Cell migration—The role of integrin glycosylation. Biochimica et Biophysica Acta (BBA) - General Subjects 1800, 545–555, doi: 10.1016/j.bbagen.2010.03.013 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Poched E et al. Aberrant Glycosylation of αvβ3 Integrin is Associated with Melanoma Progression. Anticancer Research 35, 2093 (2015). [PubMed] [Google Scholar]
- 25.Kremser ME et al. Characterisation of alpha3beta1 and alpha(v)beta3 integrin N-oligosaccharides in metastatic melanoma WM9 and WM239 cell lines. Biochimica et biophysica acta 1780, 1421–1431, doi: 10.1016/j.bbagen.2008.07.011 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Buescher JM et al. A roadmap for interpreting 13C metabolite labeling patterns from cells. Current Opinion in Biotechnology 34, 189–201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elbein AD The Use of Glycosylation Inhibitors to Study Glycoconjugate Function. Cell Surface and Extracellular Glycoconjugates, 119–180, doi: 10.1016/B978-0-12-589630-6.50009-5 (1993). [DOI] [Google Scholar]
- 28.Sakai N, Insolera R, Sillitoe RV, Shi S-H & Kaprielian Z Axon sorting within the spinal cord marginal zone via Robo-mediated inhibition of N-cadherin controls spinocerebellar tract formation. J Neurosci 32, 15377–15387, doi: 10.1523/JNEUROSCI.2225-12.2012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen JY et al. A novel sialyltransferase inhibitor suppresses FAK/paxillin signaling and cancer angiogenesis and metastasis pathways. Cancer research 71, 473–483, doi: 10.1158/0008-5472.Can-10-1303 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Sola-Penna M, Da Silva D, Coelho WS, Marinho-Carvalho MM & Zancan P Regulation of mammalian muscle type 6-phosphofructo-1-kinase and its implication for the control of the metabolism. IUBMB Life 62, 791–796, doi: 10.1002/iub.393 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez AE et al. Serine Metabolism Supports Macrophage IL-1β Production. Cell metabolism 29, 1003–1011.e1004, doi: 10.1016/j.cmet.2019.01.014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao X, Fu J, Du J & Xu W The Role of D-3-Phosphoglycerate Dehydrogenase in Cancer. Int J Biol Sci 16, 1495–1506, doi: 10.7150/ijbs.41051 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma C et al. The alternative activity of nuclear PHGDH contributes to tumour growth under nutrient stress. Nature Metabolism 3, 1357–1371, doi: 10.1038/s42255-021-00456-x (2021). [DOI] [PubMed] [Google Scholar]
- 34.Baksh SC et al. Extracellular serine controls epidermal stem cell fate and tumour initiation. Nature Cell Biology 22, 779–790, doi: 10.1038/s41556-020-0525-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J et al. Phosphoglycerate dehydrogenase induces glioma cells proliferation and invasion by stabilizing forkhead box M1. J Neurooncol 111, 245–255, doi: 10.1007/s11060-012-1018-x (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma X, Li B, Liu J, Fu Y & Luo Y Phosphoglycerate dehydrogenase promotes pancreatic cancer development by interacting with eIF4A1 and eIF4E. Journal of experimental & clinical cancer research : CR 38, 66–66, doi: 10.1186/s13046-019-1053-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
REFERENCES METHODS
- 37.Teoh ST, Ogrodzinski MP, Ross C, Hunter KW & Lunt SY Sialic Acid Metabolism: A Key Player in Breast Cancer Metastasis Revealed by Metabolomics. Frontiers in Oncology 8, 174, doi: 10.3389/fonc.2018.00174 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vandekeere S et al. Serine Synthesis via PHGDH Is Essential for Heme Production in Endothelial Cells. Cell metabolism 28, 573–587.e513, doi: 10.1016/j.cmet.2018.06.009 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Paulo JA & Gygi SP Nicotine-induced protein expression profiling reveals mutually altered proteins across four human cell lines. Proteomics 17, doi: 10.1002/pmic.201600319 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X et al. A Renewable Tissue Resource of Phenotypically Stable, Biologically and Ethnically Diverse, Patient-Derived Human Breast Cancer Xenograft Models. Cancer research 73, 4885, doi: 10.1158/0008-5472.CAN-12-4081 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Au - Lv X et al. Orthotopic Transplantation of Breast Tumors as Preclinical Models for Breast Cancer. JoVE, e61173, doi:doi: 10.3791/61173 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quintana E et al. Human Melanoma Metastasis in NSG Mice Correlates with Clinical Outcome in Patients. Science Translational Medicine 4, 159ra149, doi: 10.1126/scitranslmed.3004599 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bankhead P et al. QuPath: Open source software for digital pathology image analysis. Scientific Reports 7, 16878, doi: 10.1038/s41598-017-17204-5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berg S et al. ilastik: interactive machine learning for (bio)image analysis. Nature Methods 16, 1226–1232, doi: 10.1038/s41592-019-0582-9 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Carpenter AE et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7, R100, doi: 10.1186/gb-2006-7-10-r100 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zanotelli Vito, ndamond, & Strotton Merrick. (2020, May 24). BodenmillerGroup/ImcSegmentationPipeline: IMC Segmentation Pipeline (Version v0.9). Zenodo. 10.5281/zenodo.3841961. [DOI] [Google Scholar]
- 47.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682, doi: 10.1038/nmeth.2019 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rueden CT et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, 529, doi: 10.1186/s12859-017-1934-z (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Gorsel M, Elia I & Fendt S-M 13C tracer analysis and metabolomics in 3D cultured cancer cells Methods in Molecular Biology 1862 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Young JD, Walther JL, Antoniewicz MR, Yoo H & Stephanopoulos G An elementary metabolite unit (EMU) based method of isotopically nonstationary flux analysis. Biotechnology and bioengineering 99, 686–699, doi: 10.1002/bit.21632 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Fernandez CA, Des Rosiers C, Previs SF, David F & Brunengraber H Correction of 13C Mass Isotopomer Distributions for Natural Stable Isotope Abundance. Journal of Mass Spectrometry 31, 255–262, doi: (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all the other data supporting the findings of this study are available within the paper and its supplementary information files, and from the corresponding author upon reasonable request. Gel source images are available in Supplementary Figure 1. Transcriptomic data are available at the Gene Expression Omnibus (GEO) repository, accession code GSE198380. Source data for in vivo experiments are provided with this paper.