

Abstract
Antigen presentation machinery and professional antigen-presenting cells (APCs) are fundamental for an efficacious immune response against cancers, especially in the context of T cell–centric immunotherapy. Dendritic cells (DCs), the gold standard APCs, play a crucial role in initiating and maintaining a productive antigen-specific adaptive immunity. In recent decades, ex vivo–differentiated DCs from circulating CD14+ monocytes have become the reference for APC-based immunotherapy. DCs loaded with tumor-associated antigens, synthetic peptides, or RNA activate T cells with antitumor properties. This strategy has paved the way for the development of alternative antigen-presenting vaccination strategies, such as monocytes, B cells, and artificial APCs, that have shown effective therapeutic outcomes in preclinical cancer models. The search for alternative APC platforms was initiated by the overall limited clinical impact of DC vaccines, especially in indications such as gliomas, a primary brain tumor known for resistance to any immune intervention. In this Review, we navigate the APC immune therapeutics’ past, present, and future in the context of primary brain tumors.
Presentation of tumor-associated antigens (TAAs) via the major histocompatibility complex (MHC) class I and II (human leukocyte antigen [HLA] in humans) is fundamental for building a robust immune response and assuring the success of immunotherapies, including immune checkpoint blockade and immune cell–based immunotherapies (1, 2). MHC class I–mediated presentation of antigens (Ags) is fundamental to activation of granzyme- and perforin-producing cytotoxic CD8+ T cells. This process is vital for cytotoxic CD8+ T cells to target and kill undesirable cells such as virus-infected or cancerous cells. To promote an effective antitumor response, TAAs should be taken up and cross-presented by professional antigen-presenting cells (APCs), primarily dendritic cells (DCs), for the priming of naive CD8+ T cells (3). Subsequently, the TAA must be directly presented by tumor cells for recognition and killing by primed CD8+ T cells. Tumors develop multiple immune evasion mechanisms and reduce Ag presentation, including suppression of DC function and downregulation of HLA-I expression by tumor cells (4). Activation of CD4+ T cells by MHC class II (5), expressed preferentially by professional APCs such as DCs, macrophages, or B cells, also plays a fundamental role in mounting a therapeutic antitumor immune response. CD4+ T cells in brain tumors are best known for their protumoral effect driven by regulatory Foxp3+ T cells. However, effector CD4+ T helper cells can promote cytotoxic CD8+ T cell function via activation of DCs and regulate the myeloid compartment and tumor cells via secretion of immunomodulatory factors such as IFN-γ and TNF-α. In addition, CD4+ T helper cells can modulate the antitumoral humoral response by inducing plasmablast differentiation. CD4+ T cells are necessary to build a humoral response against tumor Ags by providing help via CD40 ligand signaling to CD40 on B cells to drive their differentiation and maturation into affinity-matured, class-switched plasma cells (6). Figure 1 summarizes MHC class I and II–mediated T cell activation and subsequent T cell subset differentiation.
Figure 1. MHC-dependent antigen presentation.

CD8+ (left) and CD4+ (right) T cell receptors are activated via antigens presented by MHC I and MHC II, respectively (first signal). Together with the costimulatory signal through CD28 engagement (second signal) and cytokines (third signal), this machinery can activate different T cells into different functional subsets (bottom rows of figure). For more details on naive CD4+ and CD8+ T cell differentiation and effector functions, see reviews (6, 141). AhR, aryl hydrocarbon receptor; BCL-6, B cell lymphoma 6; Eomes, eomesodermin; Foxp3, forkhead box P3; GATA3, GATA-binding protein 3; IRF4, interferon-regulatory factor 4; ROR, retinoic acid receptor–related orphan receptor; Tc, cytotoxic T cells; TCR, T cell receptor; Tfh, T follicular helper.
APCs in gliomas
High-grade malignant glioma and glioblastoma (GBM) are aggressive types of primary brain tumors that are almost universally fatal despite some progress in treatment and management. Most therapeutic benefit has been gained when the upfront treatment includes maximal safe resection followed by adjuvant multimodality chemotherapy (temozolomide) and radiotherapy. In clinical trials, the median progression-free survival is 5 to 7 months, and the median survival is 15 to 20 months. Better treatments and a more sustained efficacy are needed (7). These tumors are characterized by poor lymphocytic infiltration and a microenvironment preferentially populated by myeloid cells (8). Tumor-associated myeloid cells form a large and heterogeneous population of cells, including brain-resident microglia and bone marrow–derived macrophages, neutrophils, and DCs. Tumor-associated myeloid cells represent the primary APC compartment. Nonmyeloid cells such as B cells can rarely infiltrate gliomas and act as APCs. This section will briefly discuss the APC function of tumor-associated myeloid cells, DCs, and B cells and how gliomas inhibit their immune activation functions.
Macrophages and monocytes.
Bone marrow–derived myeloid cells, including macrophages and monocytes, represent the major immune cells infiltrating gliomas (9, 10). They have a vast immune and nonimmune effector function that ranges from thrombosis, phagocytosis, and debris clearance to Ag presentation and immunosuppression (11, 12). Initiation of the processing of TAAs to subsequently present to T cells via their MHC depends on the ability of macrophages and monocytes to engulf tumor cells. The interactions between tumor cells and macrophages/monocytes that regulate this engulfment are driven by “eat me” ligands, such as calreticulin, SLAMF7, opsonizing antibodies, or phosphatidylserine, and “don’t eat me” ligands, such as CD47, PD-L1, or MHC I (13). Tumor cells use this “don’t eat me” network to prevent phagocytosis, ensure their survival, and avoid antitumor T cell response. In addition, preclinical models of GBM have shown a limited impact of Ag presentation and Ag-specific T cell expansion by macrophages and monocytes (14). In GBM, myeloid cells are best known for their immunosuppressive function driven by a multifactorial network able to shut down the antitumor adaptive immune system. This network includes immunomodulatory factors (IL-10, TGF-β, IDO-1) (15–20), metabolic remodeling of the tumor microenvironment via the arginine pathway (21), and expression of suppressive molecules such as PD-L1 (22). Seminal work led by M. Suvà and I. Tirosh (23, 24), which explored intratumoral GBM diversity, unveiled the association of tumor-infiltrating macrophages with the mesenchymal-like (MES-like) state, one of the four malignant cellular states that define GBM heterogeneity. Further analysis of this association revealed a reciprocal interaction and underlined how macrophages induce the MES-like state via the secretion of oncostatin M. This work highlights the direct gliomagenesis effect of tumor-associated macrophages, which suggests that macrophages are plastic and multifunctional, and their role in supporting tumor growth goes beyond the well-documented immunosuppression.
Neutrophils.
Like macrophages and monocytes, neutrophils are tumor-infiltrating myeloid cells that can act as APCs upon maturation. In lung adenocarcinomas and squamous cell carcinomas, these APC-like neutrophils stimulate the proliferation of both CD4+ and CD8+ T cells in an MHC-dependent manner, and stimulate expression of the costimulatory molecules CD86, 4-1BB ligand, and OX40 ligand (25, 26). Secretion of lymphocyte-chemoattractant factors such as CXCL10, CCL2, CCL3, CXCL1, and CXCL2 further amplifies the potential of neutrophils to impact T cell immunity (27, 28). Both macrophages and neutrophils are highly susceptible to the immunosuppressive microenvironment of gliomas. For instance, gliomas recruit macrophages and monocytes (29) and rapidly convert them into glioma-supportive cells. This process involves the generation of specific metabolic niches, such as hypoxia (30, 31), or the production of metabolites with immunosuppressive capabilities, such as polyamines (21). Also, mutated IDH1 gliomas secrete immunomodulatory factors such as G-CSF that can inhibit the positive immune response of neutrophils (32). Because of the immunosuppressive and tumorigenic effect of glioma-associated macrophages and neutrophils, their inhibition or depletion are attractive therapeutic approaches (22, 32).
Microglia.
Microglia are resident myeloid cells of the central nervous system (CNS) with a known capacity to present Ags and activate cytotoxic T cells (33, 34). However, the immunosuppressive microenvironment of gliomas downregulates MHC expression, which limits their APC ability (35–39). Glioma cells also stimulate the secretion of IL-10 and inhibit the production of TNF-α by microglia, further promoting the suppression of the immune response (40).
DCs.
DCs are typically not found in normal brain parenchyma but are present in the choroid plexus and meninges; this is suggestive of potential migratory pathways of peripheral DCs into the CNS (41–43). During chronic inflammatory diseases, acute infections, neurodegeneration, and cancer, DCs can migrate to the brain and spinal cord through either afferent lymphatics or high endothelial venules (44). The specific role of DCs in gliomas remains to be fully elucidated. Still, current studies suggest a complex interplay between DCs, microglia and macrophages, T cells, and tumor cells in the tumor microenvironment. One suggested role for DCs in this context is in recognizing and presenting tumor Ags in the brain or the tumor-draining deep cervical lymph nodes to elicit coordinated T cell–mediated responses (44). Through signal 1 and 2 costimulatory interactions, these DCs mobilize and stimulate the development of various effector T cells associated with immune defense, such as cytotoxic T cells and CD4+ T helper cells (45, 46). Indeed, the immunosuppressive milieu of gliomas is harsh on DCs. Recent explorations into the role of DCs in glioma progression have focused on homeostatic regulators of DC function, including Nrf, a redox-sensitive transcription factor that is involved in counteracting the effects of reactive oxygen species. The tumor microenvironment of GBM is thought to induce overexpression of Nrf in DCs, resulting in the suppression of DC maturation and the consequent decrease in effector T cell activation. The inhibition of Nrf2 pathways rescues the maturation of CD80+ and CD86+ DCs in a glioma-conditioned medium and partially restores the secretion of bioactive cytokines such as IL-12p70 (47).
Extracranial tumor Ag presentation occurs in peripheral lymph nodes. Activated T cells have been found in the deep cervical lymph nodes of rat GBM models (48). This activation is controlled by DCs that migrate from the CNS to the lymph nodes via the lymphatics (49). Alternatively, CNS- and tumor-associated Ags can move out of the CNS through perivascular spaces and be collected by resident DCs in cervical lymph nodes (50, 51).
B cells.
Mature B cells recognize Ags (soluble or cell-bound Ags) using their B cell receptor (BCR) and are activated to become antibody-producing cells. As part of the differentiation to plasmacytes, B cells use the MHC class II Ag presentation pathway to process BCR-bound and internalized protein Ags and present selected peptides in complex with MHC II to CD4+ T cells (52). Under pathological and inflammatory conditions, B cells can also cross-present exogenous Ags to CD8+ T cells via their MHC I (53, 54). The antitumor effect of B cells in cancers came from studies showing that their intratumoral density is associated with a good prognosis in breast cancer (55), colorectal cancer (56, 57), non–small cell lung cancer (58), head and neck cancer (59), ovarian cancer (60), biliary tract cancer (61), primary cutaneous melanoma (62), metastatic melanoma (63), and hepatocellular carcinoma (64). The analysis of 54 cohorts of 25 cancer types revealed that although the prognostic impact of tumor-infiltrating B cells was positive in 50% of the studies, it was deleterious in 9% and neutral in 41% (65). A few studies addressed the question of the role of regulatory B cells (Bregs), which have an immunosuppressive phenotype, in human cancers. The frequency of IL-10–producing Bregs correlated with shorter overall survival in bladder cancer patients (66) and in breast cancer (67), and the coexistence of Bregs with regulatory T cells correlated with shorter metastasis-free survival in breast cancer (68). These findings suggest that protumoral and antitumoral B cells might coexist. In neoadjuvant pembrolizumab (PD-1 blockade) treatment of soft-tissue sarcoma patients, the B cell signature was the best predictor of overall survival, even when combined with CD8, PD-1, or CTLA-4 signatures (69). In advanced metastatic melanoma, tertiary lymphoid structure (TLS) and B cell signatures, but not T cell signatures, predicted therapeutic responses to pembrolizumab and ipilimumab (CTLA-4 blockade). B cells in tumors of responding patients exhibited oligoclonal repertoires of the immunoglobulin (Ig) genes compared with nonresponding patients’ polyclonal B cell repertoires. Moreover, B cells and TLS densities increased during treatment only in responding patients (70). A TLS gene signature synergized with a T cell effector signature to predict responses to immune checkpoint inhibition (ICI) with PD-1 and CTLA-4 blockade (63). Antitumoral functions of B cells upon ICI therapy have been attributed to their differentiation into plasmablasts (71), the subsequent production of tumor-reactive antibodies (72–74), and T cell activation via Ag presentation (75, 76) or Ab-dependent complement activation (77). However, B cell–mediated APC immune function and T cell activation in primary brain tumors remain unclear owing to their rarity, as they represent less than 0.5% to 1% of the immune milieu (78–80). In addition to the low numbers in the tumor microenvironment, gliomas promote the conversion of B cells into immunosuppressive B cells that sustain tumorigenicity (81). More recently, work published by our groups suggested a more immunosuppressive function of glioma-infiltrating B cells, characterized by the suppression of CD8+ T cell activation in both GBM patients and glioma-bearing mice (79).
APCs as cellular immunotherapy
DC vaccines.
The primary goal of APC vaccination is to harness T cell antitumor immunity by the presentation of TAAs. DC vaccines were the first cell-based immunotherapy developed to treat cancers (82, 83). DCs in cancer immunotherapies can be explored by different approaches: (a) bulk tumor protein- and/or nucleic acid–based vaccines; (b) peptides targeting endogenous DCs; (c) ex vivo–generated DCs matured and loaded with tumor Ags; and (d) biomaterial-based platforms for the in situ recruitment and reprogramming of endogenous DCs (84, 85). Among the registered clinical trials performed with DC vaccines, the most common approach relies on the use of ex vivo–differentiated DCs from leukapheresis-isolated CD14+ monocytes (MoDCs) cultured in the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4 (86). Previous studies have highlighted the effectiveness of DC vaccination for gliomas in preclinical models and early-stage clinical trials (87–89). Preclinical work in rodents has shown that DCs pulsed with tumor-derived Ags elicited strong tumor-reactive T cell immunity and could prolong glioma-bearing animal survival (87, 90). These experimental proof-of-principle approaches paved the way for the development of several autologous DC vaccines pulsed with tumor lysates as a therapeutic for treating primary brain tumors (91–94). Pioneered by Liau et al., the first DC vaccines were tested in GBM patients using acid-eluted tumor Ags (95). These initial investigations by Liau and colleagues led to a phase II clinical trial (ClinicalTrials.gov NCT00045968). This randomized trial reported that DCVax-L, in combination with standard of care, could significantly extend the survival of patients with either newly diagnosed or recurrent GBM compared with historical controls. This randomized trial reported that DCVax-L in combination with standard of care could significantly extend the survival of patients with either newly diagnosed or recurrent GBM (96). Liau’s seminal work paved the way for further development of the field of DC vaccination in GBM.
As an alternative to using whole-tumor lysates, synthetic peptides can also be used to pulse DC vaccines. ICT-107 is a patient-specific DC-based immunotherapy for newly diagnosed GBM patients. ICT-107 DCs are pulsed with six synthetic TAAs (MAGE-1, HER-2, AIM-2, TRP-2, gp100, and IL-13Rα2) instead of a bulk tumor lysate approach seen in previous studies. ICT-107 was tested in a double-blind, placebo-controlled phase II trial to evaluate its safety and efficacy when administered in conjunction with the Stupp protocol for newly diagnosed GBM (97). This trial highlighted that prolonged overall survival (OS) correlated with the expression of four ICT-107–targeted Ags. Despite this encouraging result, the OS benefit was not confirmed in the later phase II trial (98). A phase III clinical trial was scheduled but was halted before reaching its primary outcome owing to insufficient financial resources.
Mitchell, Sampson, and colleagues showed that preconditioning of the vaccination site with tetanus/diphtheria (Td) toxoid (a potent Ag recall signal) can significantly improve the lymph node homing and efficacy of tumor Ag–specific DCs (99). DCs were pulsed with cytomegalovirus phosphoprotein 65 (pp65) RNA. This study (NCT00639639) was performed on six patients with newly diagnosed GBM and showed promising results with prolonged OS in comparison with the control group that received autologous lymphocytes. A follow-up clinical trial (NCT03615404) will test this DC vaccine approach in combination with GM-CSF as an adjuvant. Table 1 lists past and current clinical trials that use DCs as therapeutics in newly diagnosed GBM.
Table 1. List of DC vaccine strategies in clinical trials for the treatment of newly diagnosed GBM.

Monocyte vaccines.
The overall encouraging preclinical and clinical results of DC vaccination in glioma patients motivated researchers to investigate complementary ways to achieve Ag presentation and subsequent antitumor T cell function successfully. Monocyte vaccines have been efficacious in triggering antitumor CD8+ T cell–mediated cytotoxic responses in preclinical glioma models (100). Huang et al. showed that tumor Ag–pulsed monocytes elicited a robust immune response and outperformed bone marrow–derived DCs when administered to glioma-bearing mice (100). In this report, the authors demonstrated that Ag-loaded monocytes do not activate CD8+ T cells directly but rather transfer Ag to endogenous splenic CD8+ cDCs to cross-prime naive CD8+ T cells. These results are consistent with previous reports that monocyte-derived cells do not trigger CTL responses directly but rather transfer Ag to lymphoid-resident CD8+ cDCs in murine models of viral infection (101–103), contact sensitization (104), and Ag phagocytosis (105). The group from Duke University initiated the DEMAND study in 2022 as the translational follow-up to this research study. The trial consists of a dose escalation study of monocyte Ag carrier cells for newly diagnosed GBM patients with unmethylated MGMT gene promoters. This phase I clinical trial (NCT04741984) uses engineered monocytes to express cytomegalovirus protein (MT-201-BM monocyte vaccine). Tie2-expressing monocytes (TEMs) were developed following a similar approach to that used by the Duke group, but not directly linked to the APC function. In 2008, De Palma et al. (106) explored the tumor tropism of TEMs and their potential for use as a carrier of IFN-α to tumors. This approach would inhibit the angiogenesis of tumors and the activation of innate and adaptive immunity. In 2019, Genenta Science initiated a phase I/II clinical trial (NCT03866109) using a single injection of autologous TEMs engineered to produce IFN-α (Temferon) in MGMT-unmethylated GBM patients.
Human umbilical vein endothelial cells.
Human umbilical vein endothelial cell (HUVEC) vaccines are less commonly investigated for the treatment of GBM. The presentation of the HUVEC Ag is believed to elicit anti-angiogenic cellular and humoral immune responses, thus inhibiting tumor growth (107–109). This is particularly important because bevacizumab has only shown limited clinical benefit in the setting of recurrent GBM (110). Thus far, clinical trials investigating HUVECs for recurrent GBM have shown them to be well tolerated and yielded encouraging early results (110, 111). Other tumor cell vaccine delivery techniques involve formalin fixation of the tumor cells before injection of the vaccine. It has been demonstrated that fixation with formalin allows for better tissue preservation, which allows for the most robust immune response against the tumor cells (112). The safety and efficacy of autologous formalin-fixed tumor vaccines (AFTVs) were tested in two clinical trials examining their use with only fractionated radiotherapy and chemoradiation in patients with newly diagnosed GBM (112, 113). Both trials demonstrated a tolerable safety profile and yielded a median OS greater than 19 months. These encouraging results prompted a prospective placebo-controlled phase IIb/III trial evaluating AFTV therapy in combination with standard chemoradiotherapy (UMIN Clinical Trials Registry UMIN10602; https://www.umin.ac.jp/ctr/). Although preliminary results confirmed the safety of AFTV therapy, this trial could not find a statistically significant difference in median progression-free survival between the two experimental arms.
Artificial APCs.
A considerable challenge in the therapeutic vaccination of GBM with DCs or monocytes is that the immune response elicited upon treatment must overcome the extreme immunosuppressive microenvironment. Gliomas and their microenvironment are highly immunosuppressive niches that lead to successful immune evasion. DC and monocyte vaccination may require the combination of additional therapeutic strategies to overcome the adverse effects of immunosuppression and immune checkpoint regulation. To overcome this limitation of autologous APCs, investigators have developed artificial APCs (aAPCs) as an alternative for both ex vivo and in vivo induction of tumor-reactive T cell immunity (114, 115). Artificial Ag presentation is less susceptible to the immunosuppressive effect of the tumor microenvironment.
Furthermore, aAPCs are an off-the-shelf approach that overcomes the challenges of autologous cell culture strategies (114, 116). There are three subcategories of aAPCs: cellular (allogeneic and xenogeneic), acellular (liposomes, magnetic beads, polystyrene beads, and biodegradable beads), and subcellular (lipid vesicles and exosomes). Table 2 summarizes current aAPC approaches in different cancer models.
Table 2. Different categories of artificial APCs and examples of their use as cancer treatments in clinical or preclinical settings.

Polystyrene bead–based aAPCs coated with MHC-peptide single-chain dimers or tetramers are an approach developed to stimulate tumor-specific T cells. In the context of gliomas, this APC nanoparticle was used to expand human HA-1–specific CD8+ T cells and to generate IL-13Rα2–specific CD8+ T cells to target glioma cells (117–120). An alternative approach using a similar rationale was developed to stimulate innate mucosal-associated invariant T (MAIT) cells. The aAPC design consisted of a polystyrene bead with a 5-OP-RU–loaded MR1 tetramer complex and anti-CD28 antibody (121). Subsequently, these activated MAIT cells were efficacious at killing human glioma cell lines. A limitation of aAPCs is the lack of tissue migratory capabilities (e.g., tumor) and the inability to cross-present Ags to CD8+ T cells. Altogether these limitations restrict the long-term maintenance of the antitumor immune response.
B cells, the next-generation APC immunotherapy
The B cell–based vaccine is a promising yet under-investigated approach to boosting anticancer immunity (122, 123). There are several advantages of B cells as vaccines over conventional APC vaccines, including: (a) they can act as both T cell activators and antibody producers (124); (b) mature B cells can be readily manufactured ex vivo; and (c) they have high mobility, which allows their homing to essential secondary lymphoid organs as well as tumor (125). However, not all B cells will show antitumor properties (79, 126–129), and only rare B cell subsets might be good candidates for cancer vaccines. In this section, we will navigate the possibility of using proinflammatory B cells as APC vaccines, their preclinical development, and their future translational potential.
Our research and the research of others indicate that specific B cell subpopulations hold the most potential for use in treating cancer. Specifically, studies on B cell–driven inflammation have revealed that a subset of B cells expressing the costimulatory marker 4-1BB ligand (4-1BBL, or CD137L) are especially effective in activating CD8+ T cell antitumor cytotoxicity. 4-1BBL is the single known ligand for 4-1BB (130), a TNF family costimulatory receptor that plays a fundamental role in activating Ag-experienced CD8+ T cells to establish long-term immunological memory (131, 132). Expression of 4-1BBL by B cells is achieved through B cell–mediated Ag presentation, T cell costimulation (4-1BBL and CD86), and cytokine production (TNF-α) (133, 134). This observation was further confirmed using 4-1BBL+ B cells from newly diagnosed glioma patients’ blood (135). It was observed that 4-1BBL+ B cells express proinflammatory cytokines and activation markers (TNF-α, IFN-γ, CD69, and CD86) and have a superior ability to activate autologous CD8+ T cells compared with 4-1BBL–negative B cells (135).
The B cell vaccine approach used 4-1BBL+ activated B cells from glioma-bearing mice (secondary lymphoid organs) or GBM patient–derived PBMCs as a source of B cell–based vaccine (BVax). To potentiate and stabilize the APC function, 4-1BBL+ B cells were further activated for a short time (48 hours) with CD40 and IFN-γ receptor (IFN-γR) activation (Figure 2) and pulsed with tumor protein lysates. CD40 ligation is a well-studied process that leads to B cell activation, proliferation, and enhancement of Ag-presenting and costimulatory functions (136), and ligand-associated activation of IFN-γR promotes the upregulation of costimulatory molecules such as CD86 in B cells (137). Unlike naive B cells, BVax could cross-present as potently as DCs in vitro. This agrees with a previous study that showed that cross-presentation by B cells activates autoimmune CD8+ T cells in type 1 diabetes (53). Most B cell–based vaccines use total circulating B cells (isolated using the CD20 or CD19 marker) and are activated ex vivo using CD40 agonism, Toll-like receptor ligands, and homeostatic cytokines such as IL-4 or IL-21 (138). Some studies have used CD27+ memory B cells (139). However, sorting Ag-experienced B cells (via 4-1BBL), and endowing them with potent APC function, can serve as a unique tool in B cell–based therapies.
Figure 2. B cell vaccine generation.
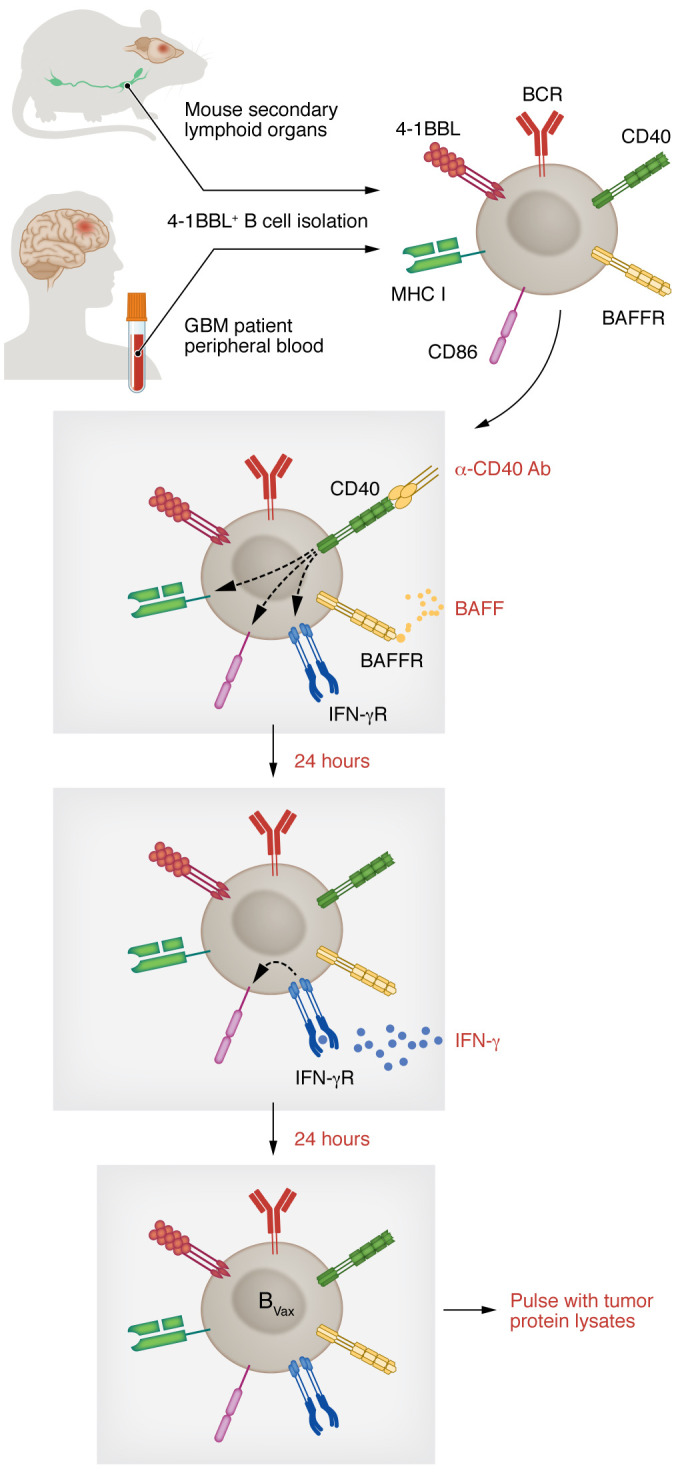
B cell–based vaccines (BVax) are produced from 4-1BBL+ B cells isolated from secondary lymphoid organs of tumor-bearing mice or GBM patients’ blood. B cells are activated ex vivo using CD40 agonism, the B cell survival factor BAFF (yellow), and IFN-γ (blue). After activation, B cells are pulsed with tumor lysates.
In the preclinical glioma model CT2-A, repeated administration of BVax and anti–PD-L1 allowed adoptively transferred CD8+ T cells to eradicate the tumor and prevent its regrowth upon reinjection in the opposite hemisphere in 50% of the treated mice after brain radiation and temozolomide treatment (GBM patient standard of care). Tumor eradication correlated with prominent infiltration of CD8+ T cells in the tumor cell injection sites. CD8+ T cells were also found in the choroid plexus, a structure that plays a fundamental role in CNS immunosurveillance via the cerebrospinal fluid–brain barrier (140). However, CD8+ T cells were also present in more distant sites like the cerebellum and pons, suggesting organ-wide surveillance to protect the CNS. Accordingly, CNS-infiltrating CD8+ T cells show an activated phenotype (characterized by the expression of IFN-γ and CD44) and the absence of inhibitory molecules such as PD-1 or TIGIT. These findings suggest that fully functional memory-like CD8+ T cells persist in the target organ. GBM patient–derived BVax is generated from the patient’s peripheral blood. This study used freshly resected patient tumors as a protein homogenate (tumor lysate) source. BVax were incubated with tumor lysate and tested for the ability to activate autologous CD8+ T cells in the absence of exogenous TCR stimulation. CD8+ T cells cultured with BVax pulsed with tumor lysates obtained from the same patient significantly expanded granzyme B–expressing CD8+ T cell numbers. This observation was almost exclusive to BVax pulsed with autologous tumor. We tested activated and expanded CD8+ T cells’ ability to kill autologous tumor cells via in vitro cytotoxicity assay. The results showed that CD8+ T cells activated via BVax potently kill glioma cells while sparing nontumor cells, in both newly diagnosed GBM and recurrent GBM biospecimens. These results support human BVax as promoting anti-GBM autologous CD8+ T cell activity.
In addition to the APC function, BVax differentiates into plasmablasts and produces tumor-reactive antibodies with therapeutic potential. While further studies are needed to elucidate the exact reactivity of BVax-derived IgG and its effector immune functions, it is undeniable that BVax represents a unique immunotherapy platform that merges both cellular (CD8+ T cell activation) and humoral (Ab production) function. Thus, the effectiveness of our approach relies on both cellular (CD8+ T cell activation) and humoral (Ab production) antitumor immune processes. These effector functions are unexplored in the brain tumor field and underexplored in cancer research in total. Figure 3 summarizes therapeutic BVax effector immune functions.
Figure 3. B cell vaccine immune effector functions.

BVax can exert an antitumor immune response via cellular immunity (activation of CD8+ T cells) and humoral immunity (production of tumor-reactive antibodies). PFN, perforin.
Conclusions
APC therapy in GBM was initiated more than two decades ago with the development of DC vaccines. Even though the therapeutic effect of this approach is inconclusive, a wealth of promising preclinical results urges us to continue developing APC therapies to boost the antitumor immune response, including novel cellular choices, such as B cells. The choice of the source and nature of the antigen, paired with tools to fight the glioma’s immunosuppressive microenvironment, is a key factor to be considered for future approaches. Treating preoperatively metastatic breast cancer and non–small cell lung cancer with ICI has proved that antitumoral immune responses can be generated if immunotherapy is administered while the tumor (and possibly draining lymph nodes) is present. Immunological response correlated with high rates of pathological response and improved long-term survival. Therefore, one might consider that the potential immunological impact of maximum versus partial resection, or even neoadjuvant immunotherapy treatment, in newly diagnosed GBM could improve immune therapies’ clinical outcomes.
Acknowledgments
The authors are supported by the National Cancer Institute (R37CA258426 to CLC; R35CA197725 and P50CA221747 to MSL), the National Institute of Neurological Disorders and Stroke (R01NS115955 to MSL), the Cancer Research Institute, the Malnati Brain Tumor Institute, and the American Brain Tumor Association.
Version 1. 02/01/2023
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2023, Lee-Chang et al. This is an open access article published under the terms of the Creative Commons Attribution 4.0 International License.
Reference information: J Clin Invest. 2023;133(3):e163449. https://doi.org/10.1172/JCI163449.
Contributor Information
Catalina Lee-Chang, Email: catalina.leechang@northwestern.edu.
Maciej S. Lesniak, Email: maciej.lesniak@northwestern.edu.
References
- 1.Mpakali A, Stratikos E. The role of antigen processing and presentation in cancer and the efficacy of immune checkpoint inhibitor immunotherapy. Cancers (Basel) 2021;13(1):134. doi: 10.3390/cancers13010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee MY, et al. Antigen processing and presentation in cancer immunotherapy. J Immunother Cancer. 2020;8(2):e001111. doi: 10.1136/jitc-2020-001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wculek SK, et al. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20(1):7–24. doi: 10.1038/s41577-019-0210-z. [DOI] [PubMed] [Google Scholar]
- 4.Vinay DS, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35(suppl):S185–S198. doi: 10.1016/j.semcancer.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 5.Tay RE, et al. Revisiting the role of CD4+ T cells in cancer immunotherapy—new insights into old paradigms. Cancer Gene Ther. 2021;28(1–2):5–17. doi: 10.1038/s41417-020-0183-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DuPage M, Bluestone JA. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat Rev Immunol. 2016;16(3):149–163. doi: 10.1038/nri.2015.18. [DOI] [PubMed] [Google Scholar]
- 7.Stupp R, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA. 2017;318(23):2306–2316. doi: 10.1001/jama.2017.18718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thorsson V, et al. The immune landscape of cancer. Immunity. 2018;48(4):812–830. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morimura T, et al. Monocyte subpopulations in human gliomas: expression of Fc and complement receptors and correlation with tumor proliferation. Acta Neuropathol. 1990;80(3):287–294. doi: 10.1007/BF00294647. [DOI] [PubMed] [Google Scholar]
- 10.Klemm F, et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell. 2020;181(7):1643–1660. doi: 10.1016/j.cell.2020.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ostuni R, et al. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol. 2015;36(4):229–239. doi: 10.1016/j.it.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Bronte V, Murray PJ. Understanding local macrophage phenotypes in disease: modulating macrophage function to treat cancer. Nat Med. 2015;21(2):117–119. doi: 10.1038/nm.3794. [DOI] [PubMed] [Google Scholar]
- 13.Feng M, et al. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer. 2019;19(10):568–586. doi: 10.1038/s41568-019-0183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malo CS, et al. Non-equivalent antigen presenting capabilities of dendritic cells and macrophages in generating brain-infiltrating CD8 + T cell responses. Nat Commun. 2018;9(1):633. doi: 10.1038/s41467-018-03037-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crane CA, et al. Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: implications for immunotherapy. Neuro Oncol. 2012;14(5):584–595. doi: 10.1093/neuonc/nos014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wainwright DA, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res. 2012;18(22):6110–6121. doi: 10.1158/1078-0432.CCR-12-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rashidi A, et al. GCN2 is essential for CD8+ T cell survival and function in murine models of malignant glioma. Cancer Immunol Immunother. 2020;69(1):81–94. doi: 10.1007/s00262-019-02441-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ravi VM, et al. T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat Commun. 2022;13(1):925. doi: 10.1038/s41467-022-28523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu A, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010;12(11):1113–1125. doi: 10.1093/neuonc/noq082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye XZ, et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J Immunol. 2012;189(1):444–453. doi: 10.4049/jimmunol.1103248. [DOI] [PubMed] [Google Scholar]
- 21.Miska J, et al. Polyamines drive myeloid cell survival by buffering intracellular pH to promote immunosuppression in glioblastoma. Sci Adv. 2021;7(8):eabc8929. doi: 10.1126/sciadv.abc8929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang P, et al. Therapeutic targeting of tumor-associated myeloid cells synergizes with radiation therapy for glioblastoma. Proc Natl Acad Sci U S A. 2019;116(47):23714–23723. doi: 10.1073/pnas.1906346116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neftel C, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. 2019;178(4):835–849. doi: 10.1016/j.cell.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hara T, et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell. 2021;39(6):779–792. doi: 10.1016/j.ccell.2021.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eruslanov EB, et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J Clin Invest. 2014;124(12):5466–5480. doi: 10.1172/JCI77053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singhal S, et al. Origin and role of a subset of tumor-associated neutrophils with antigen-presenting cell features in early-stage human lung cancer. Cancer Cell. 2016;30(1):120–135. doi: 10.1016/j.ccell.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mantovani A, et al. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11(8):519–531. doi: 10.1038/nri3024. [DOI] [PubMed] [Google Scholar]
- 28.Ponzetta A, et al. Neutrophils driving unconventional T cells mediate resistance against murine sarcomas and selected human tumors. Cell. 2019;178(2):346–360. doi: 10.1016/j.cell.2019.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang AL, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. 2016;76(19):5671–5682. doi: 10.1158/0008-5472.CAN-16-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo X, et al. Hypoxia promotes glioma-associated macrophage infiltration via periostin and subsequent M2 polarization by upregulating TGF-beta and M-CSFR. Oncotarget. 2016;7(49):80521–80542. doi: 10.18632/oncotarget.11825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brat DJ, et al. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res. 2004;64(3):920–927. doi: 10.1158/0008-5472.CAN-03-2073. [DOI] [PubMed] [Google Scholar]
- 32.Alghamri MS, et al. G-CSF secreted by mutant IDH1 glioma stem cells abolishes myeloid cell immunosuppression and enhances the efficacy of immunotherapy. Sci Adv. 2021;7(40):eabh3243. doi: 10.1126/sciadv.abh3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goddery EN, et al. Microglia and perivascular macrophages act as antigen presenting cells to promote CD8 T cell infiltration of the brain. Front Immunol. 2021;12:726421. doi: 10.3389/fimmu.2021.726421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moseman EA, et al. T cell engagement of cross-presenting microglia protects the brain from a nasal virus infection. Sci Immunol. 2020;5(48):eabb1817. doi: 10.1126/sciimmunol.abb1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taylor BC, Balko JM. Mechanisms of MHC-I downregulation and role in immunotherapy response. Front Immunol. 2022;13:844866. doi: 10.3389/fimmu.2022.844866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tran CT, et al. Differential expression of MHC class II molecules by microglia and neoplastic astroglia: relevance for the escape of astrocytoma cells from immune surveillance. Neuropathol Appl Neurobiol. 1998;24(4):293–301. doi: 10.1046/j.1365-2990.1998.00120.x. [DOI] [PubMed] [Google Scholar]
- 37.Schartner JM, et al. Impaired capacity for upregulation of MHC class II in tumor-associated microglia. Glia. 2005;51(4):279–285. doi: 10.1002/glia.20201. [DOI] [PubMed] [Google Scholar]
- 38.Zagzag D, et al. Downregulation of major histocompatibility complex antigens in invading glioma cells: stealth invasion of the brain. Lab Invest. 2005;85(3):328–341. doi: 10.1038/labinvest.3700233. [DOI] [PubMed] [Google Scholar]
- 39.de Charette M, et al. Turning tumour cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer. 2016;68:134–147. doi: 10.1016/j.ejca.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 40.Lisi L, et al. Expression of iNOS, CD163 and ARG-1 taken as M1 and M2 markers of microglial polarization in human glioblastoma and the surrounding normal parenchyma. Neurosci Lett. 2017;645:106–112. doi: 10.1016/j.neulet.2017.02.076. [DOI] [PubMed] [Google Scholar]
- 41.Matyszak MK, Perry VH. The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience. 1996;74(2):599–608. doi: 10.1016/0306-4522(96)00160-1. [DOI] [PubMed] [Google Scholar]
- 42.Serot JM, et al. Monocyte-derived IL-10-secreting dendritic cells in choroid plexus epithelium. J Neuroimmunol. 2000;105(2):115–119. doi: 10.1016/S0165-5728(99)00240-4. [DOI] [PubMed] [Google Scholar]
- 43.McMenamin PG. Distribution and phenotype of dendritic cells and resident tissue macrophages in the dura mater, leptomeninges, and choroid plexus of the rat brain as demonstrated in wholemount preparations. J Comp Neurol. 1999;405(4):553–562. doi: 10.1002/(SICI)1096-9861(19990322)405:4<553::AID-CNE8>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 44.D’Agostino PM, et al. Brain dendritic cells: biology and pathology. Acta Neuropathol. 2012;124(5):599–614. doi: 10.1007/s00401-012-1018-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Colton CA. Immune heterogeneity in neuroinflammation: dendritic cells in the brain. J Neuroimmune Pharmacol. 2013;8(1):145–162. doi: 10.1007/s11481-012-9414-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lanzavecchia A, Sallusto F. The instructive role of dendritic cells on T cell responses: lineages, plasticity and kinetics. Curr Opin Immunol. 2001;13(3):291–298. doi: 10.1016/S0952-7915(00)00218-1. [DOI] [PubMed] [Google Scholar]
- 47.Wang J, et al. Nrf2 suppresses the function of dendritic cells to facilitate the immune escape of glioma cells. Exp Cell Res. 2017;360(2):66–73. doi: 10.1016/j.yexcr.2017.07.031. [DOI] [PubMed] [Google Scholar]
- 48.Okamoto Y, et al. Cervical lymph nodes play the role of regional lymph nodes in brain tumour immunity in rats. Neuropathol Appl Neurobiol. 1999;25(2):113–122. doi: 10.1046/j.1365-2990.1999.00165.x. [DOI] [PubMed] [Google Scholar]
- 49.Hu X, et al. Meningeal lymphatic vessels regulate brain tumor drainage and immunity. Cell Res. 2020;30(3):229–243. doi: 10.1038/s41422-020-0287-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laman JD, Weller RO. Drainage of cells and soluble antigen from the CNS to regional lymph nodes. J Neuroimmune Pharmacol. 2013;8(4):840–856. doi: 10.1007/s11481-013-9470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simonds EF, et al. Deep immune profiling reveals targetable mechanisms of immune evasion in immune checkpoint inhibitor-refractory glioblastoma. J Immunother Cancer. 2021;9(6):e002181. doi: 10.1136/jitc-2020-002181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Batista FD, Harwood NE. The who, how and where of antigen presentation to B cells. Nat Rev Immunol. 2009;9(1):15–27. doi: 10.1038/nri2454. [DOI] [PubMed] [Google Scholar]
- 53.Marino E, et al. B-cell cross-presentation of autologous antigen precipitates diabetes. Diabetes. 2012;61(11):2893–2905. doi: 10.2337/db12-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Candolfi M, et al. B cells are critical to T-cell-mediated antitumor immunity induced by a combined immune-stimulatory/conditionally cytotoxic therapy for glioblastoma. Neoplasia. 2011;13(10):947–960. doi: 10.1593/neo.11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mahmoud SM, et al. The prognostic significance of B lymphocytes in invasive carcinoma of the breast. Breast Cancer Res Treat. 2012;132(2):545–553. doi: 10.1007/s10549-011-1620-1. [DOI] [PubMed] [Google Scholar]
- 56.Edin S, et al. The prognostic importance of CD20+ B lymphocytes in colorectal cancer and the relation to other immune cell subsets. Sci Rep. 2019;9(1):19997. doi: 10.1038/s41598-019-56441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berntsson J, et al. Expression of programmed cell death protein 1 (PD-1) and its ligand PD-L1 in colorectal cancer: relationship with sidedness and prognosis. Oncoimmunology. 2018;7(8):e1465165. doi: 10.1080/2162402X.2018.1465165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Germain C, et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med. 2014;189(7):832–844. doi: 10.1164/rccm.201309-1611OC. [DOI] [PubMed] [Google Scholar]
- 59.van Herpen CM, et al. Intratumoral rhIL-12 administration in head and neck squamous cell carcinoma patients induces B cell activation. Int J Cancer. 2008;123(10):2354–2361. doi: 10.1002/ijc.23756. [DOI] [PubMed] [Google Scholar]
- 60.Santoiemma PP, et al. Systematic evaluation of multiple immune markers reveals prognostic factors in ovarian cancer. Gynecol Oncol. 2016;143(1):120–127. doi: 10.1016/j.ygyno.2016.07.105. [DOI] [PubMed] [Google Scholar]
- 61.Goeppert B, et al. Prognostic impact of tumour-infiltrating immune cells on biliary tract cancer. Br J Cancer. 2013;109(10):2665–2674. doi: 10.1038/bjc.2013.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garg K, et al. Tumor-associated B cells in cutaneous primary melanoma and improved clinical outcome. Hum Pathol. 2016;54:157–164. doi: 10.1016/j.humpath.2016.03.022. [DOI] [PubMed] [Google Scholar]
- 63.Cabrita R, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577(7791):561–565. doi: 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- 64.Garnelo M, et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut. 2017;66(2):342–351. doi: 10.1136/gutjnl-2015-310814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wouters MCA, Nelson BH. Prognostic significance of tumor-infiltrating B cells and plasma cells in human cancer. Clin Cancer Res. 2018;24(24):6125–6135. doi: 10.1158/1078-0432.CCR-18-1481. [DOI] [PubMed] [Google Scholar]
- 66.Zirakzadeh AA, et al. Tumour-associated B cells in urothelial urinary bladder cancer. Scand J Immunol. 2020;91(2):e12830. doi: 10.1111/sji.12830. [DOI] [PubMed] [Google Scholar]
- 67.Murakami Y, et al. Increased regulatory B cells are involved in immune evasion in patients with gastric cancer. Sci Rep. 2019;9(1):13083. doi: 10.1038/s41598-019-49581-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ishigami E, et al. Coexistence of regulatory B cells and regulatory T cells in tumor-infiltrating lymphocyte aggregates is a prognostic factor in patients with breast cancer. Breast Cancer. 2019;26(2):180–189. doi: 10.1007/s12282-018-0910-4. [DOI] [PubMed] [Google Scholar]
- 69.Petitprez F, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577(7791):556–560. doi: 10.1038/s41586-019-1906-8. [DOI] [PubMed] [Google Scholar]
- 70.Helmink BA, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577(7791):549–555. doi: 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Griss J, et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat Commun. 2019;10(1):4186. doi: 10.1038/s41467-019-12160-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yeong J, et al. High densities of tumor-associated plasma cells predict improved prognosis in triple negative breast cancer. Front Immunol. 2018;9:1209. doi: 10.3389/fimmu.2018.01209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sharonov GV, et al. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat Rev Immunol. 2020;20(5):294–307. doi: 10.1038/s41577-019-0257-x. [DOI] [PubMed] [Google Scholar]
- 74.DeFalco J, et al. Non-progressing cancer patients have persistent B cell responses expressing shared antibody paratopes that target public tumor antigens. Clin Immunol. 2018;187:37–45. doi: 10.1016/j.clim.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 75.Rossetti RAM, et al. B lymphocytes can be activated to act as antigen presenting cells to promote anti-tumor responses. PLoS One. 2018;13(7):e0199034. doi: 10.1371/journal.pone.0199034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wennhold K, et al. CD86+ antigen-presenting B cells are increased in cancer, localize in tertiary lymphoid structures, and induce specific T-cell responses. Cancer Immunol Res. 2021;9(9):1098–1108. doi: 10.1158/2326-6066.CIR-20-0949. [DOI] [PubMed] [Google Scholar]
- 77.Lu Y, et al. Complement signals determine opposite effects of B cells in chemotherapy-induced immunity. Cell. 2020;180(6):1081–1097. doi: 10.1016/j.cell.2020.02.015. [DOI] [PubMed] [Google Scholar]
- 78.Kmiecik J, et al. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol. 2013;264(1–2):71–83. doi: 10.1016/j.jneuroim.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 79.Lee-Chang C, et al. Myeloid-derived suppressive cells promote B cell-mediated immunosuppression via transfer of PD-L1 in glioblastoma. Cancer Immunol Res. 2019;7(12):1928–1943. doi: 10.1158/2326-6066.CIR-19-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Domingues PH, et al. Immunophenotypic identification and characterization of tumor cells and infiltrating cell populations in meningiomas. Am J Pathol. 2012;181(5):1749–1761. doi: 10.1016/j.ajpath.2012.07.033. [DOI] [PubMed] [Google Scholar]
- 81.Han S, et al. Glioma cell-derived placental growth factor induces regulatory B cells. Int J Biochem Cell Biol. 2014;57:63–68. doi: 10.1016/j.biocel.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 82.Hsu FJ, et al. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med. 1996;2(1):52–58. doi: 10.1038/nm0196-52. [DOI] [PubMed] [Google Scholar]
- 83.Dhodapkar MV, et al. Rapid generation of broad T-cell immunity in humans after a single injection of mature dendritic cells. J Clin Invest. 1999;104(2):173–180. doi: 10.1172/JCI6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity. 2013;39(1):38–48. doi: 10.1016/j.immuni.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Calmeiro J, et al. Biomaterial-based platforms for in situ dendritic cell programming and their use in antitumor immunotherapy. J Immunother Cancer. 2019;7(1):238. doi: 10.1186/s40425-019-0716-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Constantino J, et al. Antitumor dendritic cell-based vaccines: lessons from 20 years of clinical trials and future perspectives. Transl Res. 2016;168:74–95. doi: 10.1016/j.trsl.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 87.Liau LM, et al. Treatment of intracranial gliomas with bone marrow-derived dendritic cells pulsed with tumor antigens. J Neurosurg. 1999;90(6):1115–1124. doi: 10.3171/jns.1999.90.6.1115. [DOI] [PubMed] [Google Scholar]
- 88.Rutkowski S, et al. Surgery and adjuvant dendritic cell-based tumour vaccination for patients with relapsed malignant glioma, a feasibility study. Br J Cancer. 2004;91(9):1656–1662. doi: 10.1038/sj.bjc.6602195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liau LM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11(15):5515–5525. doi: 10.1158/1078-0432.CCR-05-0464. [DOI] [PubMed] [Google Scholar]
- 90.Heimberger AB, et al. Bone marrow-derived dendritic cells pulsed with tumor homogenate induce immunity against syngeneic intracerebral glioma. J Neuroimmunol. 2000;103(1):16–25. doi: 10.1016/S0165-5728(99)00172-1. [DOI] [PubMed] [Google Scholar]
- 91.Wheeler CJ, et al. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res. 2008;68(14):5955–5964. doi: 10.1158/0008-5472.CAN-07-5973. [DOI] [PubMed] [Google Scholar]
- 92.Yamanaka R, et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase I/II trial. Clin Cancer Res. 2005;11(11):4160–4167. doi: 10.1158/1078-0432.CCR-05-0120. [DOI] [PubMed] [Google Scholar]
- 93.Fadul CE, et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J Immunother. 2011;34(4):382–389. doi: 10.1097/CJI.0b013e318215e300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vik-Mo EO, et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother. 2013;62(9):1499–1509. doi: 10.1007/s00262-013-1453-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liau LM, et al. Treatment of a patient by vaccination with autologous dendritic cells pulsed with allogeneic major histocompatibility complex class I-matched tumor peptides. Case Report. Neurosurg Focus. 2000;9(6):e8. doi: 10.3171/foc.2000.9.6.9. [DOI] [PubMed] [Google Scholar]
- 96.Liau LM, et al. Association of autologous tumor lysate-loaded dendritic cell vaccination with extension of survival among patients with newly diagnosed and recurrent glioblastoma: a phase 3 prospective externally controlled cohort trial. JAMA Oncol. doi: 10.1001/jamaoncol.2022.5370. [published online November 17, 2022]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Phuphanich S, et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother. 2013;62(1):125–135. doi: 10.1007/s00262-012-1319-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wen PY, et al. A randomized double-blind placebo-controlled phase II trial of dendritic cell vaccine ICT-107 in newly diagnosed patients with glioblastoma. Clin Cancer Res. 2019;25(19):5799–5807. doi: 10.1158/1078-0432.CCR-19-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mitchell DA, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519(7543):366–369. doi: 10.1038/nature14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Huang MN, et al. Antigen-loaded monocyte administration induces potent therapeutic antitumor T cell responses. J Clin Invest. 2020;130(2):774–788. doi: 10.1172/JCI128267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wakim LM, Bevan MJ. Cross-dressed dendritic cells drive memory CD8+ T-cell activation after viral infection. Nature. 2011;471(7340):629–632. doi: 10.1038/nature09863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Allan RS, et al. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 2006;25(1):153–162. doi: 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 103.Belz GT, et al. Distinct migrating and nonmigrating dendritic cell populations are involved in MHC class I-restricted antigen presentation after lung infection with virus. Proc Natl Acad Sci U S A. 2004;101(23):8670–8675. doi: 10.1073/pnas.0402644101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Le Borgne M, et al. Dendritic cells rapidly recruited into epithelial tissues via CCR6/CCL20 are responsible for CD8+ T cell crosspriming in vivo. Immunity. 2006;24(2):191–201. doi: 10.1016/j.immuni.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 105.Qu C, et al. MHC class I/peptide transfer between dendritic cells overcomes poor cross-presentation by monocyte-derived APCs that engulf dying cells. J Immunol. 2009;182(6):3650–3659. doi: 10.4049/jimmunol.0801532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.De Palma M, et al. Tumor-targeted interferon-alpha delivery by Tie2-expressing monocytes inhibits tumor growth and metastasis. Cancer Cell. 2008;14(4):299–311. doi: 10.1016/j.ccr.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 107.Wei YQ, et al. Immunotherapy of tumors with xenogeneic endothelial cells as a vaccine. Nat Med. 2000;6(10):1160–1166. doi: 10.1038/80506. [DOI] [PubMed] [Google Scholar]
- 108.Zhou L, et al. Assessment of in vivo anti-tumor activity of human umbilical vein endothelial cell vaccines prepared by various antigen forms. Eur J Pharm Sci. 2018;114:228–237. doi: 10.1016/j.ejps.2017.12.020. [DOI] [PubMed] [Google Scholar]
- 109.Chen XY, et al. Vaccination with viable human umbilical vein endothelial cells prevents metastatic tumors by attack on tumor vasculature with both cellular and humoral immunity. Clin Cancer Res. 2006;12(19):5834–5840. doi: 10.1158/1078-0432.CCR-06-1105. [DOI] [PubMed] [Google Scholar]
- 110.Tanaka M, et al. Human umbilical vein endothelial cell vaccine therapy in patients with recurrent glioblastoma. Cancer Sci. 2013;104(2):200–205. doi: 10.1111/cas.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Okaji Y, et al. Pilot study of anti-angiogenic vaccine using fixed whole endothelium in patients with progressive malignancy after failure of conventional therapy. Eur J Cancer. 2008;44(3):383–390. doi: 10.1016/j.ejca.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 112.Ishikawa E, et al. Clinical trial of autologous formalin-fixed tumor vaccine for glioblastoma multiforme patients. Cancer Sci. 2007;98(8):1226–1233. doi: 10.1111/j.1349-7006.2007.00518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Muragaki Y, et al. Phase I/IIa trial of autologous formalin-fixed tumor vaccine concomitant with fractionated radiotherapy for newly diagnosed glioblastoma. Clinical article. J Neurosurg. 2011;115(2):248–255. doi: 10.3171/2011.4.JNS10377. [DOI] [PubMed] [Google Scholar]
- 114.Sunshine JC, Green JJ. Nanoengineering approaches to the design of artificial antigen-presenting cells. Nanomedicine (Lond) 2013;8(7):1173–1189. doi: 10.2217/nnm.13.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Turtle CJ, Riddell SR. Artificial antigen-presenting cells for use in adoptive immunotherapy. Cancer J. 2010;16(4):374–381. doi: 10.1097/PPO.0b013e3181eb33a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kim JV, et al. The ABCs of artificial antigen presentation. Nat Biotechnol. 2004;22(4):403–410. doi: 10.1038/nbt955. [DOI] [PubMed] [Google Scholar]
- 117.Schilbach K, et al. Cytotoxic minor histocompatibility antigen HA-1-specific CD8+ effector memory T cells: artificial APCs pave the way for clinical application by potent primary in vitro induction. Blood. 2005;106(1):144–149. doi: 10.1182/blood-2004-07-2940. [DOI] [PubMed] [Google Scholar]
- 118.Oosten LE, et al. Artificial antigen-presenting constructs efficiently stimulate minor histocompatibility antigen-specific cytotoxic T lymphocytes. Blood. 2004;104(1):224–226. doi: 10.1182/blood-2003-07-2461. [DOI] [PubMed] [Google Scholar]
- 119.Jiang X, et al. HLA tetramer based artificial antigen-presenting cells efficiently stimulate CTLs specific for malignant glioma. Clin Cancer Res. 2007;13(24):7329–7334. doi: 10.1158/1078-0432.CCR-07-1025. [DOI] [PubMed] [Google Scholar]
- 120.Sahin A, et al. Development of third generation anti-EGFRvIII chimeric T cells and EGFRvIII-expressing artificial antigen presenting cells for adoptive cell therapy for glioma. PLoS One. 2018;13(7):e0199414. doi: 10.1371/journal.pone.0199414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Priya R, Brutkiewicz RR. MR1 tetramer-based artificial APCs expand MAIT cells from human peripheral blood that effectively kill glioblastoma cells. Immunohorizons. 2021;5(6):500–511. doi: 10.4049/immunohorizons.2100003. [DOI] [PubMed] [Google Scholar]
- 122.Kim EK, et al. Enhanced antitumor immunotherapeutic effect of B-cell-based vaccine transduced with modified adenoviral vector containing type 35 fiber structures. Gene Ther. 2014;21(1):106–114. doi: 10.1038/gt.2013.65. [DOI] [PubMed] [Google Scholar]
- 123.Schultze JL, et al. CD40-activated human B cells: an alternative source of highly efficient antigen presenting cells to generate autologous antigen-specific T cells for adoptive immunotherapy. J Clin Invest. 1997;100(11):2757–2765. doi: 10.1172/JCI119822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wennhold K, et al. Using antigen-specific B cells to combine antibody and T cell-based cancer immunotherapy. Cancer Immunol Res. 2017;5(9):730–743. doi: 10.1158/2326-6066.CIR-16-0236. [DOI] [PubMed] [Google Scholar]
- 125.Gonzalez NK, et al. In vitro and in vivo imaging of initial B-T-cell interactions in the setting of B-cell based cancer immunotherapy. Oncoimmunology. 2015;4(9):e1038684. doi: 10.1080/2162402X.2015.1038684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tsou P, et al. The emerging role of B cells in tumor immunity. Cancer Res. 2016;76(19):5597–5601. doi: 10.1158/0008-5472.CAN-16-0431. [DOI] [PubMed] [Google Scholar]
- 127.Garaud S, et al. Tumor infiltrating B-cells signal functional humoral immune responses in breast cancer. JCI Insight. 2019;5(18):e129641. doi: 10.1172/jci.insight.129641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bruno TC, et al. Antigen-presenting intratumoral B cells affect CD4+ TIL phenotypes in non-small cell lung cancer patients. Cancer Immunol Res. 2017;5(10):898–907. doi: 10.1158/2326-6066.CIR-17-0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nielsen JS, et al. CD20+ tumor-infiltrating lymphocytes have an atypical CD27- memory phenotype and together with CD8+ T cells promote favorable prognosis in ovarian cancer. Clin Cancer Res. 2012;18(12):3281–3292. doi: 10.1158/1078-0432.CCR-12-0234. [DOI] [PubMed] [Google Scholar]
- 130.Goodwin RG, et al. Molecular cloning of a ligand for the inducible T cell gene 4-1BB: a member of an emerging family of cytokines with homology to tumor necrosis factor. Eur J Immunol. 1993;23(10):2631–2641. doi: 10.1002/eji.1830231037. [DOI] [PubMed] [Google Scholar]
- 131.Uno T, et al. Eradication of established tumors in mice by a combination antibody-based therapy. Nat Med. 2006;12(6):693–698. doi: 10.1038/nm1405. [DOI] [PubMed] [Google Scholar]
- 132.Melero I, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med. 1997;3(6):682–685. doi: 10.1038/nm0697-682. [DOI] [PubMed] [Google Scholar]
- 133.Lee-Chang C, et al. Aging converts innate B1a cells into potent CD8+ T cell inducers. J Immunol. 2016;196(8):3385–3397. doi: 10.4049/jimmunol.1502034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lee-Chang C, et al. Accumulation of 4-1BBL+ B cells in the elderly induces the generation of granzyme-B+ CD8+ T cells with potential antitumor activity. Blood. 2014;124(9):1450–1459. doi: 10.1182/blood-2014-03-563940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lee-Chang C, et al. Activation of 4-1BBL+ B cells with CD40 agonism and IFNγ elicits potent immunity against glioblastoma. J Exp Med. 2021;218(1):e20200913. doi: 10.1084/jem.20200913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Foy TM, et al. gp39-CD40 interactions are essential for germinal center formation and the development of B cell memory. J Exp Med. 1994;180(1):157–163. doi: 10.1084/jem.180.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Braun D, et al. IFN-alpha/beta enhances BCR-dependent B cell responses. Int Immunol. 2002;14(4):411–419. doi: 10.1093/intimm/14.4.411. [DOI] [PubMed] [Google Scholar]
- 138.Wennhold K, et al. B cell-based cancer immunotherapy. Transfus Med Hemother. 2019;46(1):36–46. doi: 10.1159/000496166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jourdan M, et al. Characterization of human FCRL4-positive B cells. PLoS One. 2017;12(6):e0179793. doi: 10.1371/journal.pone.0179793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wilson EH, et al. Trafficking of immune cells in the central nervous system. J Clin Invest. 2010;120(5):1368–1379. doi: 10.1172/JCI41911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.St Paul M, Ohashi PS. The roles of CD8(+) T cell subsets in antitumor immunity. Trends Cell Biol. 2020;30(9):695–704. doi: 10.1016/j.tcb.2020.06.003. [DOI] [PubMed] [Google Scholar]
- 142.Butler MO, et al. Long-lived antitumor CD8+ lymphocytes for adoptive therapy generated using an artificial antigen-presenting cell. Clin Cancer Res. 2007;13(6):1857–1867. doi: 10.1158/1078-0432.CCR-06-1905. [DOI] [PubMed] [Google Scholar]
- 143.Hirano N, et al. Engagement of CD83 ligand induces prolonged expansion of CD8+ T cells and preferential enrichment for antigen specificity. Blood. 2006;107(4):1528–1536. doi: 10.1182/blood-2005-05-2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhong XS, et al. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. 2010;18(2):413–420. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Laport GG, et al. Adoptive transfer of costimulated T cells induces lymphocytosis in patients with relapsed/refractory non-Hodgkin lymphoma following CD34+-selected hematopoietic cell transplantation. Blood. 2003;102(6):2004–2013. doi: 10.1182/blood-2003-01-0095. [DOI] [PubMed] [Google Scholar]
- 146.Rapoport AP, et al. Molecular remission of CML after autotransplantation followed by adoptive transfer of costimulated autologous T cells. Bone Marrow Transplant. 2004;33(1):53–60. doi: 10.1038/sj.bmt.1704317. [DOI] [PubMed] [Google Scholar]
- 147.Durai M, et al. In vivo functional efficacy of tumor-specific T cells expanded using HLA-Ig based artificial antigen presenting cells (aAPC) Cancer Immunol Immunother. 2009;58(2):209–220. doi: 10.1007/s00262-008-0542-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Xiao P, et al. Engineering nanoscale artificial antigen-presenting cells by metabolic dendritic cell labeling to potentiate cancer immunotherapy. Nano Lett. 2021;21(5):2094–2103. doi: 10.1021/acs.nanolett.0c04783. [DOI] [PubMed] [Google Scholar]
- 149.Zappasodi R, et al. The effect of artificial antigen-presenting cells with preclustered anti-CD28/-CD3/-LFA-1 monoclonal antibodies on the induction of ex vivo expansion of functional human antitumor T cells. Haematologica. 2008;93(10):1523–1534. doi: 10.3324/haematol.12521. [DOI] [PubMed] [Google Scholar]
- 150.Zitvogel L, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med. 1998;4(5):594–600. doi: 10.1038/nm0598-594. [DOI] [PubMed] [Google Scholar]
- 151.Wolfers J, et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med. 2001;7(3):297–303. doi: 10.1038/85438. [DOI] [PubMed] [Google Scholar]