

Abstract
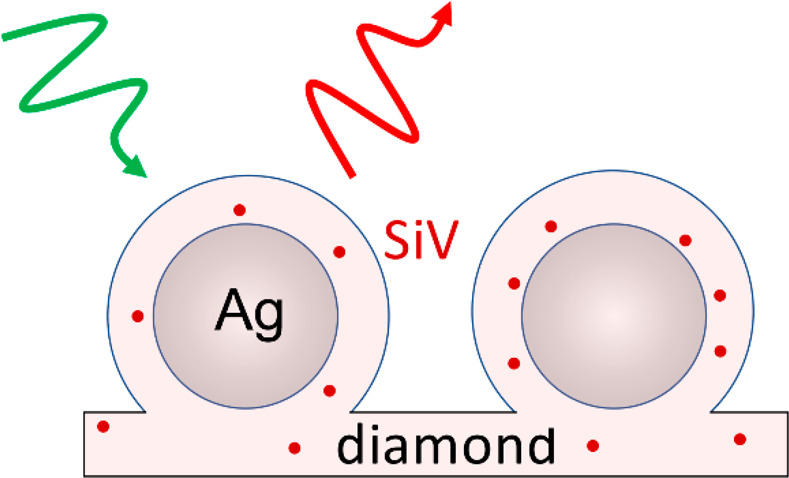
Silicon-vacancy (SiV) centers in diamond have attracted attention as highly stable fluorophores for sensing and as possible candidates for quantum information science. While prior studies have shown that the formation of hybrid diamond–metal structures can increase the rates of optical absorption and emission, many practical applications require diamond plasmonic structures that are stable in harsh chemical and thermal environments. Here, we demonstrate that Ag nanospheres, produced both in quasi-random arrays by thermal dewetting and in ordered arrays using electron-beam lithography, can be completely encapsulated with a thin diamond coating containing SiV centers, leading to hybrid core–shell nanostructures exhibiting extraordinary chemical and thermal stability as well as enhanced optical properties. Diamond shells with a thickness on the order of 20–100 nm are sufficient to encapsulate and protect the Ag nanostructures with different sizes ranging from 20 nm to hundreds of nanometers, allowing them to withstand heating to temperatures of 1000 °C and immersion in harsh boiling acid for 24 h. Ultrafast photoluminescence lifetime and super-resolution optical imaging experiments were used to study the SiV properties on and off the core–shell structures, which show that the SiV on core–shell structures have higher brightness and faster decay rate. The stability and optical properties of the hybrid Ag–diamond core–shell structures make them attractive candidates for high-efficiency imaging and quantum-based sensing applications.
Keywords: Diamond, Core−shell, Silicon-vacancy centers, Time-resolved photoluminescence, Cathodoluminescence
Introduction
Recent studies have shown that quantum emitters in diamond, such as the silicon vacancy (SiV) and nitrogen vacancy (NV) centers, have unique optical properties and spin coherence properties that make them attractive candidates for a range of applications including quantum sensing and quantum information processing and as fluorescence-based probes in complex chemical and physical environments.1−9 SiV centers, NV centers, and related color centers in diamond10 are particularly attractive as optical probes in harsh chemical environments and at elevated temperatures because the outstanding chemical and thermal stability of diamond provide a high degree of protection against degradation pathways.11,12 While the NV center has received a great deal of attention due to its long spin coherent times, it emits light into a broad band that can easily overlap with emission from nondiamond sources, making isolation of the NV center emission from background signals in complex matrices challenging.4,12−18 In contrast, the SiV center has a broad absorption spectrum extending from ∼460 to 620 nm but emits >70% of the photons into a narrow (∼1 nm wide) set of emission lines near 738 nm.19−28
Coupling color centers with plasmonic structures is a promising route to achieve higher photon generation rates and emission efficiencies.14,29−38 The wide wavelength tunability of metal plasmonics makes it possible to enhance the excitation and emission of diamond color centers separately or simultaneously through the Purcell effect. Several previous efforts of coupling diamond color centers to metal plasmonic effects have been conducted, demonstrating that introducing a plasmonic structure near a color center can enhance the observed photoluminescence (PL) intensity.14,29−37 While the introduction of metals on top of diamond can produce local enhancements of the electric field, many metals that are commonly used in plasmonic studies, such as Ag, are reactive and will rapidly corrode in aqueous media.39−42 Thus, the ability to chemically protect the nanostructures becomes an essential component for chemical and biological sensing.
Here we demonstrate that encapsulation of Ag nanoparticles with a thin diamond film containing SiV centers leads to the formation of optically emissive nanostructures with the outstanding chemical stability and optical properties of SiV centers in diamond. We demonstrate that the size of Ag core and the thickness of diamond shell can be controlled by changing the growth parameters. The extraordinary stability of the Ag-encapsulated diamond structures is demonstrated by their ability to withstand heating to 1000 °C (above the melting point of Ag) and immersion in boiling triacid H2SO4/HNO3/HClO4 = 1:1:1 v:v solution for 24 h with almost no change. While Ag nanoparticles can be produced in a quasi-random arrangement using simple thermal dewetting after deposition of a uniform Ag film on a diamond substrate, electron-beam lithography can produce these nanoparticles in ordered arrays. Time-resolved photoluminescence lifetime and photoluminescence imaging confirm that the emission rate and brightness from the SiV centers on Ag–diamond structures are increased. The fabrication and integration of the Ag nanoparticles coated with a diamond shell containing SiV centers provide a promising platform for ultrastable and bright emissive structure and are also of interest for studying the Ag–SiV interactions if combined with spatial control of color centers created by implantation techniques. Additionally, the Ag–diamond core–shell structures as ultrastable and bright optical probes have potential utility in a wide range of optical-based measurements including bioimaging and optical sensing in harsh chemical environments and/or at elevated temperatures and for enhancement of quantum sensing using the spin properties of quantum defects in diamond.
Methods
Sample Preparation
Diamond films with embedded Ag nanoparticles were fabricated by a three-step growth method using single-crystal Si (001) wafers as substrates. An initial diamond layer of ∼100 nm thickness was first grown by microwave plasma enhanced chemical vapor deposition (PECVD). In the second step, Ag nanoparticles were formed by deposition of a thin continuous Ag film onto the diamond substrate, which was then dewetted by placing the sample into the microwave chamber in the presence of pure hydrogen.43,44 Finally, a second diamond layer with embedded SiV centers was grown using conditions identical to those used in thegrowth of the first diamond layer. The presence of the Si wafer under the sample acted as the source of Si for the SiV centers.
The growth of diamond films was seeded using detonation nanodiamonds with a nominal 4–10 nm size distribution (Nanoscale and Amorphous Materials, Inc.); the nanodiamonds were hydrogen-terminated by heating in a fused silica tube in a Thermolyne tube furnace for 5 h in a pure H2 (>99.999%, Airgas) atmosphere at 500 °C. The H-terminated powder was then sonicated in ethanol for 4 h and centrifuged for 15 min at 14 500 rotations per minute. The supernatant was used as a seeding solution. Si substrates were cut into 1 × 1 cm2 pieces and cleaned by sonication with acetone and water for 10 min, respectively. To seed the diamond samples, a 1 mL aliquot of the nanodiamond suspension in ethanol was spin-coated (3000 rpm, 30 s) onto the substrate. Diamond thin films were grown using microwave plasma enhanced chemical vapor deposition (PECVD) in an AsTEX SDS 5010 Reactor that was modified in-house to provide additional sample heating capability and a thermocouple immediately below the substrate to measure the temperature. The pressure of the chamber was kept at 45 Torr during growth. The first layer diamond growth was performed using 200 standard cubic centimeters per minute (sccm) H2 and 3 sccm CH4 at a power of 800 W for 2 h, then transferred to an electron-beam metal evaporator and 40 nm Ag was deposited at a rate of 0.1 Å/s. This is followed by a 10 min H2 plasma treatment at 600 W and another diamond layer growth step using 200 sccm H2 and 3 sccm CH4 at a power of 800 W for 20 min. The other Ag–diamond structures with different Ag sizes and diamond thicknesses shown in Figure 3 were prepared by the same procedure by changing the Ag film deposition thickness or second diamond shell growth time.
Figure 3.

(a–d) Tunability of Ag core size by depositing the Ag layer with thicknesses of 20 (a), 35 (b), 40 (c), and 50 nm (d; , the Ag core sizes can range from 20 to 400 nm in diameter. All other growth parameters are the same as those in the Sample Preparation part in Methods except for (a), for which the second diamond layer growth was 5 min. (d–g) Tunability of diamond shell thicknesses by tuning the growth time of the second diamond layer to 10 (d), 20 (e), 30 (f), and 40 min (g); the diamond shell thicknesses can range from 20 to 100 nm. All other growth parameters are the same as those in the Sample Preparation part in Methods.
Thermal stability was evaluated by placing the samples into a custom-built vacuum chamber at a pressure of <1 × 10–5 Torr for 24 h at 1000 °C (10 °C/min ramp rate). Chemical stability was evaluated by immersing the samples in a boiling concentrated solution of H2SO4/HNO3/HClO4 (1:1:1 by volume) with reflux condensation for 24 h. To tune the size of Ag nanostructures, Ag films of different thicknesses were used while other growth parameters remained the same. To tune the thickness of the diamond shells, different durations of the second-layer growth were used while other growth parameters remained the same.
To make Ag–diamond patterned structures, the first layer of diamond was deposited using 200 sccm H2 and 3 sccm CH4 at a power of 800 W for 2 h. A layer of poly(methyl methacrylate) (PMMA) was spin-coated onto the diamond samples (500 rpm for 10 s followed by 4000 rpm for 1 min). The samples were then transferred to a baking plate to bake at 180 °C for 5 min. Another layer of PMMA was spin-coated onto the samples and baked at 180 °C for 5 min. Samples were transferred to a scanning electron microscope (LEO Supra55 VP) equipped with a beam-blanker and Nabity Nanometer Pattern Generation System for electron-beam writing capability. Patterns were made using the electron beam with 30 kV electron voltage. Exposed samples were immersed into a mixture of methyl isobutyl ketone and isopropanol (1:3 ratio by volume), allowed to develop for 25 s, rinsed with isopropanol, and then dried with nitrogen. The samples were transferred to an electron-beam metal evaporator, and 80 nm Ag was deposited at a rate of 0.1 Å/s. Note that for the subsequent lift-off process the Ag layer on the patterns will also be partially removed, resulting in a thinner Ag layer, so we used 80 nm Ag here but this would result in a final Ag thickness of ∼40 nm. After the deposition, the remaining PMMA was lifted off in acetone for 5 min, leaving behind the Ag patterns on the diamond. To fabricate D-Ag-D patterned structures, a 10 min H2 plasma treatment at 600 W was performed and another diamond layer was grown using 200 sccm H2 and 3 sccm CH4 at a power of 800 W for 20 min.
Characterization
Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) measurements were carried out using a Leo Supra 55 VP microscope. To best capture film morphology, an SE2 detector was used for imaging. Energy dispersive X-ray spectra were obtained on this same instrument using a ThermoFisher UltraDry Compact EDS detector. Focused ion beam (FIB) was conducted using a FEI Helios NanoLab 600i DualBeam SEM/FIB microscope. Transmission electron microscopy (TEM) characterization was performed using a Tecnai TF-30 microscope operating at an accelerating voltage of 300.0 kV. Emission spectra were collected using a 532 nm continuous-wave diode pumped solid-state laser (Opto Engine LLC, MLL-FN-532-500m), focusing 100 mW onto the sample using a 40× microscope objective. The photoluminescence was collected using the same lens, filtered using a dichroic mirror (Semrock, FF553-SDi01-25x36) and a 532 nm line-reject filter (Semrock, NF01-532U-25) and focused into the 25 μm slit of an Andor Shamrock 193i monochromator with a grating blazed at 760 nm and 150 lines/mm. The detector was an Andor iStar intensified CCD (DH334T-18F-03). All spectra presented here were calibrated using the 532 nm laser line. Differential imaging was performed using the same apparatus with a 100× objective microscope oil lenses (NA = 0.9). Super-resolution photoluminescence mapping was performed using a Leica SP8 Confocal and Super-Resolution Stimulated Emission Depletion (STED) microscope.
Cathodoluminescence imaging with corresponding secondary electron images was conducted with a Zeiss Gemini SUPRA 55 scanning electron microscope, where we mounted an aluminum diamond-turned parabolic mirror. The mirror collects the sample cathodoluminescence from a 1.3π solid angle and sends it to photomultiplier tubes through band pass filters and to a fiber-coupled spectrometer with a 300 mm focal length where a 300 lines/mm grating disperses the luminescence onto a UV-enhanced silicon CCD. We collected the images at acceleration voltages of 5 and 10 kV and a current of 700–900 pA. For microscope control and data acquisition, we used ScopeFoundry, the home-built software platform of the Molecular Foundry. For time-resolved photoluminescence (TRPL) measurements of the SiV in Ag–diamond sample, an ultrafast oscillator-amplifier system (Tsunami and Spitfire Pro, Spectra-Physics) produced near-infrared (NIR) pulses (800 nm, 1 ps duration) at a 1 kHz repetition rate. The NIR pulses fed an optical parametric amplifier (OPA) (OPA-800, Spectra-Physics) to create pulses as a wavelength of 532 nm. The beam illuminated the sample at an oblique incidence angle and with a spot size on the sample of approximately 70 μm. Optical emission was collected approximately normal to the sample surface, using a 630 nm long-pass filter (Corning 2-59) to reject scattered light. Optical emission from SiV centers at 738 nm was isolated with a monochromator (1 nm bandwidth). An avalanche photodiode (Micro Photon Devices PD-050-CTD) detected the monochromator output, and a time-correlator (Picoharp 300, PicoQuant) mapped the distribution of arrival times to generate the PL transient. The photon count rate was kept below 2.5% to prevent errors due to photon pile-up effects.
Results
Figure 1a is a schematic illustration of an as-prepared sample. A first layer of diamond was grown on a silicon wafer substrate, which also served as the source of Si for the SiV centers. After the Ag nanoparticles formed, another thinner layer of SiV-containing diamond was grown to surround the Ag while maintaining the spherical structure of Ag nanoparticles, forming Ag–diamond core–shell structures. During growth of the Ag–diamond structure, SiV centers were introduced into the diamond during growth, in both the diamond substrate layer and Ag–diamond shell layer. The existence of SiV centers was confirmed by measuring the PL spectrum of the Ag–diamond core–shell structures. Figure 1b shows a typical room-temperature PL spectrum when excited at 532 nm, exhibiting a narrow emission peak at 738 nm with full width at half-maximum (fwhm) of ∼9 nm that corresponds to the emission from SiV centers. A full PL spectrum of the Ag–diamond sample from 600 to 800 nm is shown in Figure S1. The Raman peak is at 572 nm, and there are some nitrogen-related peaks other than SiV, but no NV center related peaks are observed. The morphology of the Ag–diamond core–shell structure was characterized by SEM as shown in Figure 1c.
Figure 1.
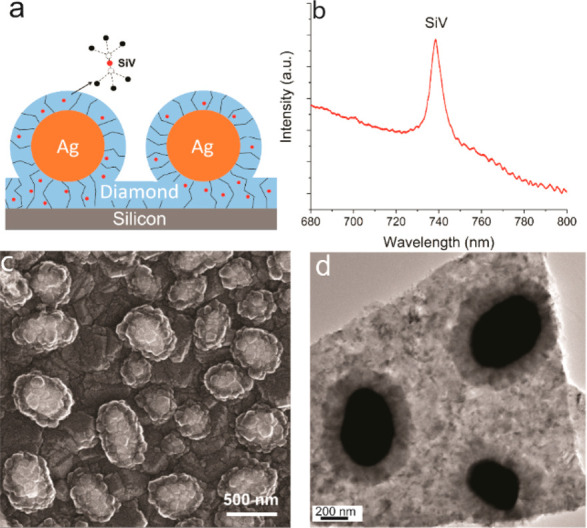
SiV–Ag structures. (a) Schematic illustration of the diamond-encapsulated Ag nanoparticles with embedded SiV centers. The SiV center consists of a Si atom between two adjacent carbon vacancies in the diamond lattice. (b) Room temperature photoluminescence spectrum of SiV centers. (c) SEM and (d) TEM images of the Ag–diamond core–shell structure, showing a columnar growth structure like that depicted in (a).
In this image, Ag structures appear bright because they scatter electrons better than diamond does. The Ag structures shown here are ∼300–500 nm in diameter; surrounding the Ag nanoparticles, the SEM images show a relatively uniform, thin shell of diamond approximately 50–100 nm thick. Both the diamond substrates and the diamond shell layers show a columnar grain morphology with a growth structure like that depicted in Figure 1a. Figure 1d (and Figure S2) shows TEM images of Ag–diamond core–shell structures that were grown on fused silica substrates in order to facilitate the removal of the sample from the underlying substrate. In the TEM images, the Ag nanoparticles are dark and the diamond shell is clearly visible. The Ag core size and diamond shell thickness measured in TEM are in good agreement with those obtained from SEM images. The TEM images reveal a columnar structure growing outward from the Ag core, suggesting that the diamond shell grows preferentially around the Ag nanoparticle surface. The Ag–diamond structures, shown in a tilted-view SEM in Figure S3a and cross-sectional FIB-SEM image in Figure S3b, are spherical and lying on a large piece of diamond which is the initially grown diamond substrate layer.
Unlike prior studies of Ag–diamond hybrid structures,30,31,35,36,45 in our studies, the Ag nanoparticles are coated entirely with diamond, yielding outstanding stability at high temperatures and in extremely corrosive environments. Figure 2a shows the results of a thermal stability test in which the Ag–diamond structures were placed in a custom-built vacuum tube furnace at a pressure of <1 × 10–5 Torr, and heated to 1000 °C for 24 h (note that the melting point of Ag is 960 °C). Figure 2a shows the SEM image of after this thermal treatment and cooling of the sample. Despite heating to high temperature, the SEM image shows no detectable changes in the morphology of the samples. The Ag core and diamond shell both remain the same sizes, and there is no evidence of diffusion of Ag or migration of the core–shell structure. This demonstrates that the Ag–diamond is a very stable structure as Ag is entirely encapsulated in the diamond shell and would not diffuse at such a high temperature or react with diamond. To test the chemical stability of the structures, we immersed the samples into a boiling concentrated acid H2SO4/HNO3/HClO4 = 1:1:1 v:v solution for 24 h (Caution! This solution is highly caustic.). As shown in Figure 2b (and Figure S5a for larger area SEM), after the acid treatment, nearly all the structures retained their intact core–shell structure. The boiling triacid solution used here is known for its ability to effectively remove all of the nondiamond material.46,47 The ability of the Ag–diamond structures to persist demonstrates that the outer diamond shell is of high quality and is able to protect the Ag core even under extremely corrosive conditions. The SiV emission before and after acid treatment does not show obvious changes. Close examination of the small number of particles in which Ag was etched away reveals an obvious pinhole that allows the creation of a hollow diamond shell structure. The corresponding SEM image and energy-dispersive X-ray spectroscopy (EDS) map in Figure S4 show no detectable Ag X-ray emission from the hollow diamond shell, demonstrating the complete removal of the Ag nanoparticle. Small pinholes in the diamond film likely arise during growth from the local concentration of graphite or amorphous carbon on this spot.
Figure 2.

SEM image of diamond-encapsulated Ag nanoparticles (a) after thermal annealing at 1000 °C under vacuum for 24 h and (b) after boiling concentrated triacid treatment for 24 h.
To verify, we performed a nitrogen implantation treatment on the Ag–diamond sample (1 × 1011 ions/cm2 dose and 7 keV implantation energy) and then performed the boiling acid treatment again. The nitrogen implantation creates more amorphous and graphitized carbon on the diamond shell, and, as a result, the harsh acid treatment leaves a significantly higher number of hollow diamond shells, as shown in Figure S5b. Intentional creation of nondiamond phase through implantation could be a promising route to prepare unusual diamond hollow spherical structures. Notably, however, on the nonimplanted films, the loss of the Ag core from any specific nanoparticle leaves the adjacent nanoparticles unaffected. Thus, while some small pinholes may result in degradation of a small number of embedded Ag nanoparticles under very harsh chemical conditions, the loss is limited to the nanoparticle immediately adjacent to the pinhole. As a result, the hollow diamond shell structure has the potential for localizing color centers in a thin thickness range.
The Ag–diamond structures reported here also demonstrate good tunability. As shown in Figure 3, by changing the growth conditions, the Ag core size and the diamond shell thickness can be precisely controlled. In Figure 3a–d, we show that the size of the Ag core could be tuned by tens of nanometers (a), to 200, 300, and 400 nm (b–d) by changing the Ag film deposition thickness before the dewetting process. The Ag film thicknesses used here are 20, 35, 40, and 50 nm, respectively, and it should be noted that when the Ag film thickness is larger than 50 nm, the shape of the Ag nanoparticles would be more elongated and inhomogeneous. The size distributions of the nanostructures with different Ag thicknesses are shown in Figure S6. By changing the second layer diamond growth time, as shown in Figure 3e–g, the thickness of the diamond shell coated on Ag nanoparticles can be precisely tuned, from as thin as ∼20 nm to 50, 80, and ∼100 nm. If the second layer diamond growth time keeps increasing, the diamond shell thickness will also keep growing and finally coalesce into a whole film, forming a diamond–Ag–diamond thin film sandwiched structure. These results demonstrate that the properties of Ag–diamond core–shell structure can potentially be controlled experimentally by altering the size of the Ag nanoparticle and the thickness of the diamond, which provides a platform with good tunability to study the light–matter interactions in a controllable way and tune the material responses as well. The simulated scattering spectrum of a Ag–diamond particle with a Ag core of 350 nm and diamond shell of 50 nm is shown in Figure S7 using Mie theory, showing that the plasmonic resonance is close to the SiV emission wavelength.
The 40 nm thickness Ag (∼300 nm sized Ag–diamond structure) was used for the optical measurements. We used ultrafast TRP) initiated by ∼1 ps excitation pulses at 532 nm to examine emission rate induced by the Ag nanoparticles. The direct observation of SiV–Ag interaction is limited by the diffraction limitation in optical PL microscope. Therefore, we compared the SiV PL lifetime of diamond samples with and without Ag nanoparticles (Ag–diamond and diamond sample, respectively). To make the pure diamond sample, the metal evaporation step of the synthesis was omitted.
Figure 4 shows TRPL decay curves from the Ag–diamond (red) and diamond (blue) samples. While both samples decay similarly at times beyond 1.5 ns, the Ag–diamond sample exhibits an additional fast decay component at shorter times. We attribute this fast decay component, present only in the Ag–diamond sample, to SiV centers that are close to the Ag nanoparticles, while the slow decay component, common to both samples, is attributed to SiV centers located far from the Ag nanoparticles. The fast and slow components can be clearly seen for the Ag–diamond sample, while for the pure diamond sample there is only a single decay component that is same as the slow component in the Ag–diamond. The existence of both varieties of SiV centers in the Ag–diamond sample is congruent with the diamond layer covering both the nanoparticles and the substrate, as shown in Figure 1a.
Figure 4.
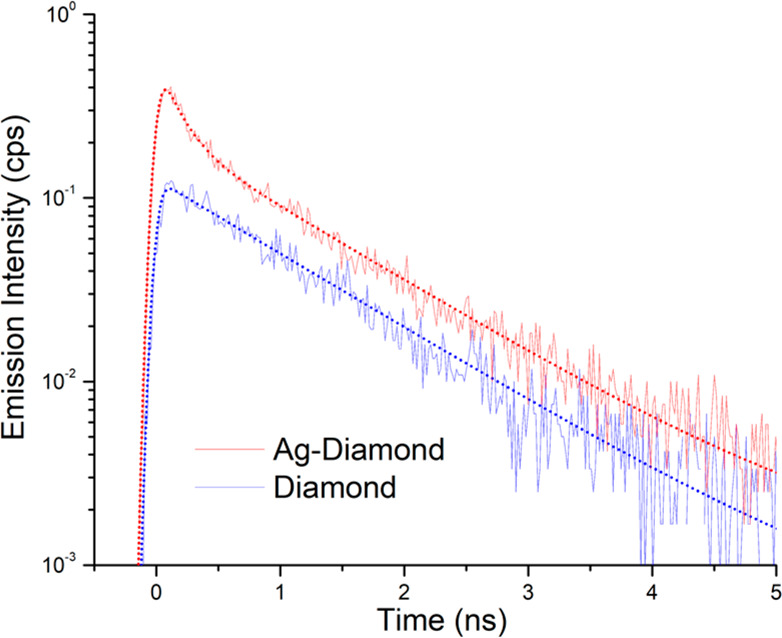
TRPL lifetime measurements of the SiV color center in the Ag–diamond (red) and diamond (blue) samples. SiV center lifetime curves (solid lines) in Ag–diamond and in diamond overlaid with corresponding fits (dotted lines) using eqs 3 and 1, respectively.
To isolate the effect of Ag nanoparticles on the PL decay for Ag–diamond, we modeled the TRPL as a two-component system of SiV centers located close to and far from the Ag nanoparticles. Both components are modeled as monoexponential decays:
| 1 |
| 2 |
where each decay has an amplitude, A, and time constant, τ. The two-component model ignores the variety of possible coupling arrangements between Ag and SiV color centers, but it is nonetheless useful for understanding this data. The pure diamond sample has no Ag nanoparticles and will decay according to eq 1, while the Ag sample will be a mixture of SiV centers close to and far from the Ag:
| 3 |
Ioff-Ag will describe a slow decay, and Ion-Ag will describe the fast decay. To fit the TRPL decays, we convolve the exponential decays with a Gaussian instrument response function of fwhm 110 ps. The fits are shown in Figure 4 as dotted lines. We find a decay time of SiV centers in plain diamond of τ1 = 1.07 ns, which is consistent with previously reported independent measurements.48−50 Meanwhile, the SiV centers on Ag have a shorter lifetime by a factor of ∼7× (τ2 = 160 ps). This shortened lifetime can be ascribed to the SiV enhanced emission rate due to the increased local density of states (LDOS) near Ag.
We also measured the optical emission as a function of excitation intensity. Figure S8a shows the excitation intensity dependence of PL in the Ag sample as well as the fits using eq 3. Both on-Ag (fast) and off-Ag (slow) components increase in prominence as fluence is increased, and there are no changes in the emission rate at different fluences. As a control, we also varied the excitation intensity of the pure diamond sample (see Figure S8b) and found the single exponential decay behavior to be consistent with the slow component (A1) in the Ag–diamond, which further demonstrates that the faster emission rate is caused by the Ag.
To demonstrate the controlled positioning of the Ag nanoparticles coated with diamond, we used electron-beam lithography to produce regular patterns of individual Ag–diamond core–shell structures separated by ∼2 μm (a long distance compared with the wavelength of light) and then characterized their optical properties using photoluminescence microscopy. The fabrication steps are shown in Figure 5a. We used e-beam lithography to create the pattern structure of Ag on the diamond film followed by a lift-off process. After a brief plasma treatment, each of the Ag patterns dewetted into a spherical shape. Another layer of diamond with embedded SiV centers was then grown on the sample. The SiV-containing diamond film encapsulates the Ag nanoparticles and also coats the region between the nanoparticles such that there a SiV centers both in the shell surrounding the Ag nanoparticles and also in the regions in between. We characterized the patterned structures using SEM, as shown in Figure 5b; here the bright dots of Ag in aligned patterns can be clearly observed and each Ag is encapsulated in a shell of diamond. Zoomed-in views like that shown in Figure 5c reveal that each dot is a well-shaped Ag–diamond core–shell structure, with a Ag core size of 300 nm and diamond shell thickness of 50 nm. We further confirmed the patterned structure by corresponding EDS mapping, shown in the Supporting Information (Figure S9). These measurements establish that the Ag–diamond structures are formed and have good structural uniformity. Autocorrelation analysis (Figure S10) for the patterned Ag–diamond array structure indicated a high degree of periodicity with identical peak-to-peak spacings. This shows that, even after the highly energetic plasma-enhanced diamond growth, the e-beam lithography patterned Ag still stays in the exact position, which is promising for the high-precision positioning of diamond-related defect enhancement with plasmonic structures.
Figure 5.
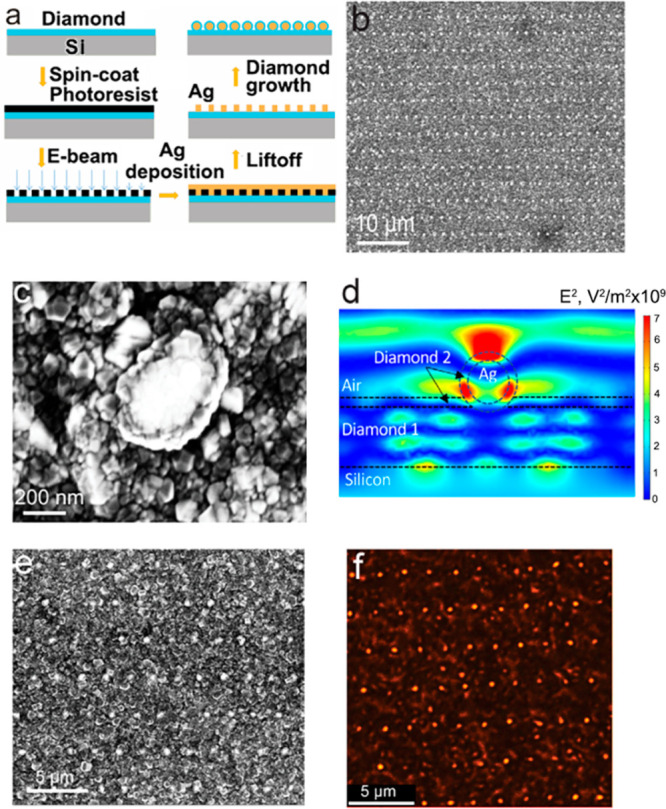
(a) Fabrication process of Ag–diamond patterned arrays. (b) SEM of Ag–diamond patterned array structure with a zoomed-out view and (c) a single core–shell structure. (d) Spatial distribution of mean-square electric field at 739 nm induced by a plane wave (1 V/m electric field) incident from the top. Diamond 1 and Diamond 2 refer to first layer and second layer diamond growth, respectively. Note that the Diamond 2 layer has only a small electric field across the entire horizontal width but has several regions of high magnitude within the shell structure. (e) SEM image and (f) corresponding high-resolution stimulated emission depletion (STED) microscopy images of lithographically patterned Ag nanoparticle arrays that were then coated with a SiV-containing diamond film. Note that growth of the SiV-containing diamond film forms a shell around the Ag nanoparticles and also coats the regions between the nanoparticles.
We hypothesize that the SiV centers in the diamond film directly coating the Ag nanoparticles would have a different emission intensity with respect to the SiV centers in the flatter regions between nanoparticles, and with the structure we can use simulation to estimate the effect of the structure on the emission. The plasmonic resonance and associated enhancement in the local electric field leads to an increased LDOS which can be quantified by a transition rate enhancement factor γ, that describes the decrease in the radiative lifetime of SiV and enhanced emission due to interaction with the adjacent Ag.51 Previous studies have used plasmonic structures to enhance fluorescence emission from nitrogen-vacancy (NV) centers. Gong et al.35 chemically linked Ag nanoparticles onto nanodiamonds containing NV centers and reported a faster emission rate from the NV centers. Lithographic techniques have also been used to implant NV centers into single-crystal diamond plates and then coat them with a layer of Ag, leading to enhancement of NV emission.31,45 Li et al. reported that the influence of the Ag film on NV center fluorescent lifetime depended on the thickness of the plasmonic Ag film and the associated plasmonic resonance, demonstrating coupling to the NV centers.45 To further understand the overall electromagnetic coupling associated with the Ag–diamond core–shell structures, we modeled the local electromagnetic field enhancements using Comsol Multiphysics, solving Maxwell’s equations in three dimensions for a structure simulating the ordered Ag–diamond patterned structures. Figure 5d shows a typical example, for a 150 nm radius Ag nanoparticle with the second diamond layer (“Diamond 2”) being 40 nm in thickness (thickness of the shell), a first-layer diamond (“Diamond 1”) 200 nm thick, and periodicity of 2 μm. These calculations determined the mean-square electric field in the diamond shell surrounding the nanoparticle and in the diamond film between the nanoparticles that result from a plane-wave electromagnetic field perpendicular to the surface. By microscopic reversibility, the ratio of mean-square electric fields in these two regions should also reflect differences in the probability of emission from ensembles of SiV centers. These calculations show that the mean square electric field in the shell region surrounding the nanoparticle is approximately 4 times higher than that in the underlying planar diamond regions, which indicate the structure is promising to enhance the SiV emission.
We used a STED microscope using 532 nm laser excitation to probe the local optical response of SiV signals in the Ag–diamond core–shell patterned structure. Figure 5e shows an SEM image of a patterned region, and Figure 5f shows the corresponding fluorescence image at a wavelength of 738 nm (±10 nm band pass). We note that while Figure 5e and f does not show exactly the same region of the sample, the periodic structures observed in each case are uniform over long distances and the photoluminescence image shows highly localized regions of intense emission that correspond to the locations of the diamond-coated Ag nanoparticles. To rule out Raman contributions to the photoluminescence image, we kept the same emission collection window and changed the excitation wavelength from 532 to 488 nm, which would shift the Raman signal to a lower wavelength but keep the SiV photoluminescence at 738 nm. The 488 nm laser excitation revealed no Raman signals at the same collection window, while the SiV photoluminescence was still captured. This confirms that the imaging is from the emission of the diamond and not from morphology-dependent spatial variations in scattering or other possible optical artifacts.
To obtain further insight into the Ag–diamond structures containing SiV centers, we studied the Ag–diamond core–shell structures by cathodoluminescence (CL) microscopy. Cathodoluminescence offers higher spatial resolution than can be achieved in purely optical measurements. Taking advantage of the spectral resolution of the CL setup, we performed hyperspectral CL imaging in which we obtained full spectra at each pixel in the scan. Figure 6a shows the SEM image at a 10 kV of acceleration voltage of a typical particle ∼500 nm in size. Figure 6b is the corresponding CL mapping of the particle in Figure 6a at 600–800 nm. The CL mapping shows higher emission when the incident electron beam is on the Ag nanoparticles compared with when it is on the regions in between. Figure 6c shows emission spectra from different spots on the particle and off the particle, respectively, and the spectra are normalized with the SiV intensity. Both on-particle and off-particle spectra show a strong, well-defined SiV emission zero-phonon line (ZPL) at 738 nm, in good agreement with the previous CL studies of SiV in nanodiamonds.52 The observation of the ZPL emission peak from the position of the NPs and from the region between the NPs shows that the SiV centers are distributed both on the Ag–diamond structure and the diamond substrate. The SiV emission peaks at locations on and off the particle have the same width and center emission wavelength, indicating that there are no detectable local strains affecting the SiV emission from the Ag–diamond structure. A broad luminescence background ranging from ∼580 to ∼800 nm is also present, which we attribute to luminescence from sp2-hybridized “graphitic” carbon at the grain boundaries and is the reason for the etched pinhole in Figure 2b. A comparison of the spectra measured off the particle and on the particle shows that the broad emission is more pronounced above the nanoparticles, suggesting that there may be increased sp2-hybridized carbon in the vicinity of the Ag nanoparticles.53
Figure 6.
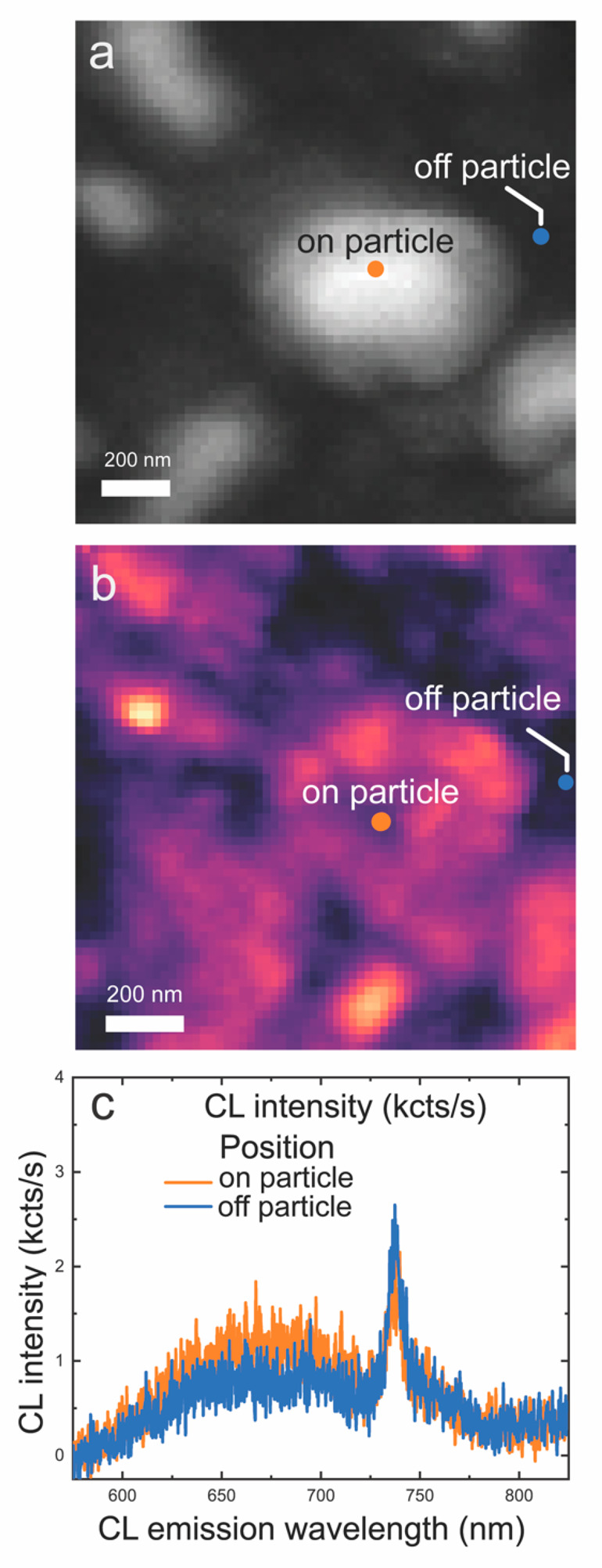
CL hyperspectral maps at 10 kV. (a) SEM image of a Ag–diamond core–shell structure (hyperspectral resolution). (b) Corresponding hyperspectral map of integrated 600–800 nm CL intensity. (c) CL spectra on and off the Ag particle extracted at the pixel positions indicated by dots in (a) and (b). CL spectra show a peak centered near 738 nm in wavelength corresponding to the SiV emission and a broader peak at lower energy.
The Ag–diamond core–shell structures offer a stable and tunable platform to controllably modify the SiV emission. While quantitative analysis of the enhancement is difficult due to the small size of the individual Ag–diamond structures and the fact that the SiV centers are distributed across the sample, this work demonstrates a new and stable Ag–diamond structure with bright SiV emission. Using lithographic patterning techniques, the SiV centers can be controllably implanted on the Ag–diamond core–shell structures and the thickness of the diamond shell could be controlled accordingly to study the interactions between the metal and SiV. Previous studies with hybrid structures of Ag plasmonics and diamond with color centers have shown that the direct exposure of the Ag to the local environment in different geometries is problematic for use in harsh environments, such as those required for chemical and biological sensing that involve the use of high salt concentrations and possibly oxidizing conditions under which Ag oxidizes to Ag+. While demonstrated here for SiV centers, the use of core–shell structures should also be applicable to a wider range of color centers that can be formed during growth, such as tin-vacancy (SnV) and germanium-vacancy (GeV) centers, or by postgrowth ion-implantation. By completely encapsulating the Ag core, the diamond shell serves a dual role as a host for the color centers and also as a protective layer. The outstanding chemical and thermal stability evidenced in Figure 2 demonstrates that the encapsulation of the Ag nanoparticles allows the use of this platform in extremely harsh chemical conditions. It is notable that the Ag–diamond structures survive extensive heating to 1000 °C, above the melting point of Ag.
Conclusion
We have demonstrated that microwave plasma chemical-vapor deposition methods can be used to form Ag–diamond core–shell structures in which Ag cores are completely encapsulated with thin diamond films with embedded SiV centers. These structures can be formed in quasi-random arrangements using simple dewetting of Ag or can be precisely patterned into ordered arrays using standard lithographic methods. The ability of these structures to resist extremely caustic chemical treatments and temperatures of 1000 °C, exceeding the melting point of Ag, suggests that they may find practical utility in a wide range of challenging environments. The hybrid Ag–diamond core–shell structures show an enhancement of optical emission, suggesting potential utility in applications such as bioimaging, nanophotonics, bright single-photon sources, nanothermometry, strain-sensing, as well as other quantum-based applications.
Acknowledgments
This material is based upon work supported by the National Science Foundation, DMR-1904106. Final data analysis, low-temperature measurements, and autocorrelation studies by S.L., E.T.Z., and N.M. were supported by the Department of Energy, Basic Energy Sciences (BES)-Materials Science and Engineering, DE-SC0020115 and the SLAC Laboratory Directed Research and Development (LDRD) program. S.L. and N.M. also acknowledge support from Equinor ASA. Special thanks to Professor J. Vuckovic, A. Rugar, and S. Sun from E. L. Ginzton Laboratory for their help and support on experiments and discussion. L.F., F.D.O., and A.W.-B. acknowledge support from the EFRC DOE Early Career Award. L.F. acknowledges support from the Swiss National Science Foundation through the Early Postdoc Mobility fellowship P2ELP2_184398. Cathodoluminescence work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Part of this work was performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under award ECCS-2026822.
Glossary
Abbreviations
- PL
photoluminescence
- SEM
scanning electron microscopy
- TEM
transmission electron microscopy
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmaterialsau.1c00027.
Additional SEM images on Ag–diamond structures and controlled samples; additional lifetime measurements on control samples; characterization of periodic arrays of diamond-coated Ag cores; additional hyperspectral and bandpass SEM CL maps (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Lang J.; Häußler S.; Fuhrmann J.; Waltrich R.; Laddha S.; Scharpf J.; Kubanek A.; Naydenov B.; Jelezko F. Long optical coherence times of shallow-implanted, negatively charged silicon vacancy centers in diamond. Appl. Phys. Lett. 2020, 116, 064001. 10.1063/1.5143014. [DOI] [Google Scholar]
- Lenzini F.; Gruhler N.; Walter N.; Pernice W. H. Diamond as a platform for integrated quantum photonics. Advanced Quantum Technologies 2018, 1, 1800061. 10.1002/qute.201800061. [DOI] [Google Scholar]
- Balasubramanian G.; Lazariev A.; Arumugam S. R.; Duan D.-w. Nitrogen-vacancy color center in diamond—emerging nanoscale applications in bioimaging and biosensing. Curr. Opin. Chem. Biol. 2014, 20, 69–77. 10.1016/j.cbpa.2014.04.014. [DOI] [PubMed] [Google Scholar]
- Jelezko F.; Wrachtrup J. Single defect centres in diamond: A review. Phys. Status Solidi A 2006, 203, 3207–3225. 10.1002/pssa.200671403. [DOI] [Google Scholar]
- Childress L.; Walsworth R.; Lukin M. Atom-like crystal defects. Phys. Today 2014, 67, 38. 10.1063/PT.3.2549. [DOI] [Google Scholar]
- Aharonovich I.; Neu E. Diamond nanophotonics. Adv. Opt. Mater. 2014, 2, 911–928. 10.1002/adom.201400189. [DOI] [Google Scholar]
- Mochalin V. N.; Shenderova O.; Ho D.; Gogotsi Y. The properties and applications of nanodiamonds. Nat. Nanotechnol. 2012, 7, 11–23. 10.1038/nnano.2011.209. [DOI] [PubMed] [Google Scholar]
- Lindner S.; Bommer A.; Muzha A.; Krueger A.; Gines L.; Mandal S.; Williams O.; Londero E.; Gali A.; Becher C. Strongly inhomogeneous distribution of spectral properties of silicon-vacancy color centers in nanodiamonds. New J. Phys. 2018, 20, 115002. 10.1088/1367-2630/aae93f. [DOI] [Google Scholar]
- Dei Cas L.; Zeldin S.; Nunn N.; Torelli M.; Shames A. I.; Zaitsev A. M.; Shenderova O. From fancy blue to red: Controlled production of a vibrant color spectrum of fluorescent diamond particles. Adv. Funct. Mater. 2019, 29, 1808362. 10.1002/adfm.201808362. [DOI] [Google Scholar]
- Iwasaki T.; Miyamoto Y.; Taniguchi T.; Siyushev P.; Metsch M. H.; Jelezko F.; Hatano M. Tin-Vacancy Quantum Emitters in Diamond. Phys. Rev. Lett. 2017, 119, 6. 10.1103/PhysRevLett.119.253601. [DOI] [PubMed] [Google Scholar]
- Munawar A.; Ong Y. R.; Schirhagl R.; Tahir M. A.; Khan W. S.; Bajwa S. Z. Nanosensors for diagnosis with optical, electric and mechanical transducers. RSC Adv. 2019, 9, 6793–6803. 10.1039/C8RA10144B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirhagl R.; Chang K.; Loretz M.; Degen C. L. Nitrogen-Vacancy Centers in Diamond: Nanoscale Sensors for Physics and Biology. Annu. Rev. Phys. Chem. 2014, 65, 83–105. 10.1146/annurev-physchem-040513-103659. [DOI] [PubMed] [Google Scholar]
- Robinson M. E.; Ng J. D.; Zhang H. L.; Buchman J. T.; Shenderova O. A.; Haynes C. L.; Ma Z. Q.; Goldsmith R. H.; Hamers R. J. Optically Detected Magnetic Resonance for Selective Imaging of Diamond Nanoparticles. Anal. Chem. 2018, 90, 769–776. 10.1021/acs.analchem.7b03157. [DOI] [PubMed] [Google Scholar]
- Bogdanov S. I.; Shalaginov M. Y.; Lagutchev A. S.; Chiang C.-C.; Shah D.; Baburin A. S.; Ryzhikov I. A.; Rodionov I. A.; Kildishev A. V.; Boltasseva A.; et al. Ultrabright room-temperature sub-nanosecond emission from single nitrogen-vacancy centers coupled to nanopatch antennas. Nano Lett. 2018, 18, 4837–4844. 10.1021/acs.nanolett.8b01415. [DOI] [PubMed] [Google Scholar]
- Dhomkar S.; Jayakumar H.; Zangara P. R.; Meriles C. A. Charge Dynamics in near-Surface, Variable-Density Ensembles of Nitrogen-Vacancy Centers in Diamond. Nano Lett. 2018, 18, 4046–4052. 10.1021/acs.nanolett.8b01739. [DOI] [PubMed] [Google Scholar]
- Tetienne J. P.; de Gille R. W.; Broadway D. A.; Teraji T.; Lillie S. E.; McCoey J. M.; Dontschuk N.; Hall L. T.; Stacey A.; Simpson D. A.; Hollenberg L. C. L. Spin properties of dense near-surface ensembles of nitrogen-vacancy centers in diamond. Phys. Rev. B: Condens. Matter Mater. Phys. 2018, 97, 085402. 10.1103/PhysRevB.97.085402. [DOI] [Google Scholar]
- Chang Y.-R.; Lee H.-Y.; Chen K.; Chang C.-C.; Tsai D.-S.; Fu C.-C.; Lim T.-S.; Tzeng Y.-K.; Fang C.-Y.; Han C.-C.; Chang H.-C.; Fann W. Mass production and dynamic imaging of fluorescent nanodiamonds. Nat. Nanotechnol. 2008, 3, 284. 10.1038/nnano.2008.99. [DOI] [PubMed] [Google Scholar]
- Chipaux M.; van der Laan K. J.; Hemelaar S. R.; Hasani M.; Zheng T. T.; Schirhagl R. Nanodiamonds and Their Applications in Cells. Small 2018, 14, 1704263. 10.1002/smll.201704263. [DOI] [PubMed] [Google Scholar]
- Hepp C.; Müller T.; Waselowski V.; Becker J. N.; Pingault B.; Sternschulte H.; Steinmüller-Nethl D.; Gali A.; Maze J. R.; Atatüre M.; et al. Electronic structure of the silicon vacancy color center in diamond. Phys. Rev. Lett. 2014, 112, 036405. 10.1103/PhysRevLett.112.036405. [DOI] [PubMed] [Google Scholar]
- Leifgen M.; Schröder T.; Gädeke F.; Riemann R.; Métillon V.; Neu E.; Hepp C.; Arend C.; Becher C.; Lauritsen K.; et al. Evaluation of nitrogen-and silicon-vacancy defect centres as single photon sources in quantum key distribution. New J. Phys. 2014, 16, 023021. 10.1088/1367-2630/16/2/023021. [DOI] [Google Scholar]
- Becker J. N.; Becher C. Coherence Properties and Quantum Control of Silicon Vacancy Color Centers in Diamond (Phys. Status Solidi A 11/2017). Phys. Status Solidi A 2017, 214, 1770170. 10.1002/pssa.201770170. [DOI] [Google Scholar]
- Pingault B.; Jarausch D.-D.; Hepp C.; Klintberg L.; Becker J. N.; Markham M.; Becher C.; Atatüre M. Coherent control of the silicon-vacancy spin in diamond. Nat. Commun. 2017, 8, 15579. 10.1038/ncomms15579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. L.; Ishiwata H.; Babinec T. M.; Radulaski M.; Müller K.; Lagoudakis K. G.; Dory C.; Dahl J.; Edgington R.; Soulière V.; Ferro G.; Fokin A. A.; Schreiner P. R.; Shen Z.-X.; Melosh N. A.; Vučković J. Hybrid Group IV Nanophotonic Structures Incorporating Diamond Silicon-Vacancy Color Centers. Nano Lett. 2016, 16, 212–217. 10.1021/acs.nanolett.5b03515. [DOI] [PubMed] [Google Scholar]
- Zhang J. L.; Sun S.; Burek M. J.; Dory C.; Tzeng Y.-K.; Fischer K. A.; Kelaita Y.; Lagoudakis K. G.; Radulaski M.; Shen Z.-X.; Melosh N. A.; Chu S.; Lončar M.; Vučković J. Strongly Cavity-Enhanced Spontaneous Emission from Silicon-Vacancy Centers in Diamond. Nano Lett. 2018, 18, 1360–1365. 10.1021/acs.nanolett.7b05075. [DOI] [PubMed] [Google Scholar]
- Zhang J. L.; Lagoudakis K. G.; Tzeng Y.-K.; Dory C.; Radulaski M.; Kelaita Y.; Fischer K. A.; Sun S.; Shen Z.-X.; Melosh N. A.; Chu S.; Vučković J. Complete coherent control of silicon vacancies in diamond nanopillars containing single defect centers. Optica 2017, 4, 1317–1321. 10.1364/OPTICA.4.001317. [DOI] [Google Scholar]
- Tzeng Y.-K.; Zhang J. L.; Lu H.; Ishiwata H.; Dahl J.; Carlson R. M. K.; Yan H.; Schreiner P. R.; Vučković J.; Shen Z.-X.; Melosh N.; Chu S. Vertical-Substrate MPCVD Epitaxial Nanodiamond Growth. Nano Lett. 2017, 17, 1489–1495. 10.1021/acs.nanolett.6b04543. [DOI] [PubMed] [Google Scholar]
- Häußler S.; Thiering G.; Dietrich A.; Waasem N.; Teraji T.; Isoya J.; Iwasaki T.; Hatano M.; Jelezko F.; Gali A.; Kubanek A. Photoluminescence excitation spectroscopy of SiV-and GeV-color center in diamond. New J. Phys. 2017, 19, 063036. 10.1088/1367-2630/aa73e5. [DOI] [Google Scholar]
- Neu E.; Agio M.; Becher C. Photophysics of single silicon vacancy centers in diamond: implications for single photon emission. Opt. Express 2012, 20, 19956–19971. 10.1364/OE.20.019956. [DOI] [PubMed] [Google Scholar]
- Aramesh M.; Cervenka J.; Roberts A.; Djalalian-Assl A.; Rajasekharan R.; Fang J.; Ostrikov K.; Prawer S. Coupling of a single-photon emitter in nanodiamond to surface plasmons of a nanochannel-enclosed silver nanowire. Opt. Express 2014, 22, 15530–15541. 10.1364/OE.22.015530. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Huck A.; Andersen U. L. Efficient coupling of a single diamond color center to propagating plasmonic gap modes. Nano Lett. 2013, 13, 1221–1225. 10.1021/nl304682r. [DOI] [PubMed] [Google Scholar]
- Choy J. T.; Hausmann B. J.; Babinec T. M.; Bulu I.; Khan M.; Maletinsky P.; Yacoby A.; Lončar M. Enhanced single-photon emission from a diamond-silver aperture. Nat. Photonics 2011, 5, 738. 10.1038/nphoton.2011.249. [DOI] [Google Scholar]
- Szenes A.; Bánhelyi B.; Szabó L. Z.; Szabó G.; Csendes T.; Csete M. Enhancing diamond color center fluorescence via optimized plasmonic nanorod configuration. Plasmonics 2017, 12, 1263–1280. 10.1007/s11468-016-0384-1. [DOI] [Google Scholar]
- Kumar S.; Davydov V. A.; Agafonov V. N.; Bozhevolnyi S. I. Excitation of nanowire surface plasmons by silicon vacancy centers in nanodiamonds. Opt. Mater. Express 2017, 7, 2586–2596. 10.1364/OME.7.002586. [DOI] [Google Scholar]
- Szenes A.; Bánhelyi B.; Szabó L. Z.; Szabó G.; Csendes T.; Csete M. Improved emission of SiV diamond color centers embedded into concave plasmonic core-shell nanoresonators. Sci. Rep. 2017, 7, 13845. 10.1038/s41598-017-14227-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J.; Steinsultz N.; Ouyang M. Nanodiamond-based nanostructures for coupling nitrogen-vacancy centres to metal nanoparticles and semiconductor quantum dots. Nat. Commun. 2016, 7, 11820. 10.1038/ncomms11820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schietinger S.; Barth M.; Aichele T.; Benson O. Plasmon-enhanced single photon emission from a nanoassembled metal- diamond hybrid structure at room temperature. Nano Lett. 2009, 9, 1694–1698. 10.1021/nl900384c. [DOI] [PubMed] [Google Scholar]
- Bulu I.; Babinec T.; Hausmann B.; Choy J. T.; Loncar M. Plasmonic resonators for enhanced diamond NV-center single photon sources. Opt. Express 2011, 19, 5268–5276. 10.1364/OE.19.005268. [DOI] [PubMed] [Google Scholar]
- Bradac C.; Gao W.; Forneris J.; Trusheim M. E.; Aharonovich I. Quantum nanophotonics with group IV defects in diamond. Nat. Commun. 2019, 10, 5625. 10.1038/s41467-019-13332-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova O. S.; Zamborini F. P. Size-Dependent Electrochemical Oxidation of Silver Nanoparticles. J. Am. Chem. Soc. 2010, 132, 70. 10.1021/ja908780g. [DOI] [PubMed] [Google Scholar]
- Opilik L.; Dogan U.; Szczerbinski J.; Zenobi R. Degradation of silver near-field optical probes and its electrochemical reversal. Appl. Phys. Lett. 2015, 107, 091109. 10.1063/1.4929880. [DOI] [Google Scholar]
- Oates T. W. H.; Losurdo M.; Noda S.; Hinrichs K. The effect of atmospheric tarnishing on the optical and structural properties of silver nanoparticles. J. Phys. D: Appl. Phys. 2013, 46, 145308. 10.1088/0022-3727/46/14/145308. [DOI] [Google Scholar]
- Fafarman A. T.; Hong S. H.; Oh S. J.; Caglayan H.; Ye X. C.; Diroll B. T.; Engheta N.; Murray C. B.; Kagan C. R. Air-Stable, Nanostructured Electronic and Plasmonic Materials from Solution-Processable, Silver Nanocrystal Building Blocks. ACS Nano 2014, 8, 2746–2754. 10.1021/nn406461p. [DOI] [PubMed] [Google Scholar]
- Li S.; Bandy J.; Hamers R. J. Tunable coloration of diamond films by encapsulation of plasmonic Ag nanoparticles. Diamond Relat. Mater. 2018, 89, 190–196. 10.1016/j.diamond.2018.09.003. [DOI] [Google Scholar]
- Li S.; Bandy J. A.; Hamers R. J. Enhanced photocatalytic activity of diamond thin films using embedded Ag nanoparticles. ACS Appl. Mater. Interfaces 2018, 10, 5395–5403. 10.1021/acsami.7b13821. [DOI] [PubMed] [Google Scholar]
- Li D.-F.; Li C.-H.; Zhou L.-M.; Zheng Y.; Zhao B.-W.; Li S.; Zhao N.; Chen X.-D.; Guo G.-C.; Sun F.-W. Thickness dependent surface plasmon of silver film detected by nitrogen vacancy centers in diamond. Opt. Lett. 2018, 43, 5587–5590. 10.1364/OL.43.005587. [DOI] [PubMed] [Google Scholar]
- D’Evelyn M. P. S. L. M.; Rawles R. E.. Surface cleaning, topopgraphy, and temperature measurements of single crystal diamond. In Diamond, SiC and Nitride Wide Bandgap Semiconductors, Carter C. H., Gildenblat G., Nakamura S., Nemanich R. J., Eds.; Materials Research Society: Pittsburgh, PA, 1994; Vol. 339, pp 89–94 [Google Scholar]
- Prins J. F. Preparation of Ohmic Contacts to Semiconducting Diamond. J. Phys. D: Appl. Phys. 1989, 22, 1562–1564. 10.1088/0022-3727/22/10/026. [DOI] [Google Scholar]
- Lagomarsino S.; Flatae A. M.; Sciortino S.; Gorelli F.; Santoro M.; Tantussi F.; De Angelis F.; Gelli N.; Taccetti F.; Giuntini L.; et al. Optical properties of silicon-vacancy color centers in diamond created by ion implantation and post-annealing. Diamond Relat. Mater. 2018, 84, 196–203. 10.1016/j.diamond.2018.03.010. [DOI] [Google Scholar]
- Neu E.; Hepp C.; Hauschild M.; Gsell S.; Fischer M.; Sternschulte H.; Steinmüller-Nethl D.; Schreck M.; Becher C. Low-temperature investigations of single silicon vacancy colour centres in diamond. New J. Phys. 2013, 15, 043005. 10.1088/1367-2630/15/4/043005. [DOI] [Google Scholar]
- Jantzen U.; Kurz A. B.; Rudnicki D. S.; Schäfermeier C.; Jahnke K. D.; Andersen U. L.; Davydov V. A.; Agafonov V. N.; Kubanek A.; Rogers L. J.; et al. Nanodiamonds carrying silicon-vacancy quantum emitters with almost lifetime-limited linewidths. New J. Phys. 2016, 18, 073036. 10.1088/1367-2630/18/7/073036. [DOI] [Google Scholar]
- Lourenço-Martins H.; Kociak M.; Meuret S.; Treussart F.; Lee Y. H.; Ling X. Y.; Chang H.-C.; Galvão Tizei L. H. Probing Plasmon-NV0 Coupling at the Nanometer Scale with Photons and Fast Electrons. ACS Photonics 2018, 5, 324–328. 10.1021/acsphotonics.7b01093. [DOI] [Google Scholar]
- Zhang H.; Aharonovich I.; Glenn D. R.; Schalek R.; Magyar A. P.; Lichtman J. W.; Hu E. L.; Walsworth R. L. Silicon-vacancy color centers in nanodiamonds: cathodoluminescence imaging markers in the near infrared. Small 2014, 10, 1908–1913. 10.1002/smll.201303582. [DOI] [PubMed] [Google Scholar]
- Zaitsev A. M.Optical properties of diamond: A data handbook; Springer Science & Business Media: 2013. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.