

Abstract
Dysreglulated brain arachidonic acid (AA) metabolism is involved in chronic tissue inflammation and is influenced by apolipoprotein E4 (APOE4) genotype, the strongest genetic risk factor of late-onset Alzheimer’s disease (AD). Visualization of AA uptake and distribution in the brain can offer insight into neuroinflammation and AD pathogenesis. Here we present a novel synthesis and radiosynthesis of 20-[18F]-fluoroarachidonic acid ([18F]-FAA) for PET imaging using a convergent route and a one-pot, single-purification radiolabeling procedure, and demonstrate its brain uptake in human ApoE4 targeted replacement (ApoE4-TR) mice. By examining p38 phosphorylation in astrocytes, we found that fluorination of AA at the ω-position did not significantly alter its biochemical role in cells. The brain incorporation coefficient (K*) of [18F]-FAA was estimated via multiple methods by using an image-derived input function from the right ventricle of the heart as a proxy of the arterial input function and brain tracer concentrations assessed by PET-MR imaging. This new synthetic approach should facilitate the practical [18F]-FAA production and allow its translation into clinical use, making investigations of dysregulation of lipid metabolism more feasible in the study of neurodegenerative diseases.
Keywords: Arachidonic acid, fatty acid, PET/MR, brain imaging, ApoE4
1. INTRODUCTION
Lipoproteins influence the synthesis, metabolism, and storage of lipids to maintain brain lipid homeostasis [1]. One of the key proteins that regulate lipid metabolism in the brain is apolipoprotein E (ApoE) which exists as three common variants: E2, E3, and E4[2]. ApoE lipoproteins affect the transport of polyunsaturated fatty acids (PUFAs) such as arachidonic acid (AA, 20:4, ω−6) and docosahexaenoic acid (DHA, 22:6, ω−3) into the brain which are involved in the inflammatory response and inflammation resolution [1, 3, 4]. While AA can be a precursor to some anti-inflammatory lipid mediators, its release from membrane lipids by the activity of calcium-dependent phospholipase A2 (cPLA2) is associated with prostaglandin production and neuroinflammation [1, 5]. There is growing evidence that impairment of brain lipid metabolism, manifested by a higher AA/DHA ratio in the brain, contributes to neurodegenerative diseases such as Alzheimer’s disease (AD) [3, 6–8]. ApoE4 induces the activity of cPLA2 which potentially contributes to AD pathogenesis by promoting neuroinflammation [9, 10] and can be indirectly estimated by the increased ratio of circulating AA/DHA [3, 11]. Therefore, monitoring changes in PUFA metabolism by ApoE4 in the human brain may be useful for understanding AD pathogenesis.
Positron emission tomography (PET) using 20-[18F]fluoroarachidonic acid ([18F]-FAA) is a viable way to visualize the metabolism of AA. Estimation of the kinetics of AA brain incorporation has been performed in humans using an AA PET tracer based on carbon-11, [1-11C]-AA [12]. Labeling with positron radionuclide 18F is preferred due to its relatively long half-life (109.77 min) as compared to 11C (with a half-life of 20.38 min) with an improved spatial resolution [13]. An increase in brain AA uptake was shown in patients with Alzheimer’s dementia compared to age-matched controls by PET imaging [14]. Previous methods for the synthesis of 20-[18F/19F]fluoroarachidonic acid ([18F/19F]-FAA) have been reported with low yields and selectivity, making the probe not practical to use in clinical settings [13, 15]. Thus, a novel synthesis of [19F]-FAA by a convergent method toward a precursor for F-18 labeling has been developed, and the radiolabeling steps of [18F]-FAA were optimized. The new synthesis and radiosynthesis of [18F/19F]-FAA presented here will allow for the accurate evaluation of AA brain uptake with further implications for APOE allele-specific metabolic changes. Using dynamic PET and magnetic resonance (MR) imaging, we then estimated the incorporation coefficient (K*) representing the rate of the uptake of the probe into the mouse brain.
2. MATERIALS AND METHODS
2.1. General
All reagents and solvents were obtained from commercial sources and used without further purification unless otherwise noted. All reactions were performed under nitrogen gas unless otherwise mentioned. Thin-layer chromatography (TLC) was performed on precoated silica gel 60 Å plates with UV 254 indicator, and visualized with either UV light or potassium permanganate (KMnO4) staining solution. Purification was performed by either column chromatography with silica gel (60 Å, 230–400 mesh), or a CombiFlash Rf 200 automatic chromatography system (Teledyne ISCO) with prepacked silica gel cartridges. NMR spectra were recorded on a 400 MHz Varian Mercury or 500 MHz Varian VNMRS NMR spectrometer. Chemical shifts are reported in ppm (δ) downfield with the residual deuterated solvent as reference. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, td = triplet of doublets, tt = triplet of triplets, qd = quartet of doublets, dqd = doublet of quartet of doublets, m = multiplet), coupling constant (Hz), and integration. Low-resolution mass spectra were obtained on an MSQ Plus single quadrupole mass spectrometer in positive mode. The cone voltage was 75 V, and the probe temperature was 350 °C.
2.2. HPLC methods
Analytical reversed-phase high-performance liquid chromatography (HPLC) was carried out using a Thermo Scientific UltiMate 3000 HPLC system with a Phenomenex Luna C18(2) reversed-phase column (5 μm, 250 mm × 4.6 mm). The UV absorbance was recorded at 214 and 250 nm. Radioactivity was recorded using a model 105 radio-detector (Carroll & Ramsey Associates, Berkeley, CA). The mobile phases were A, H2O with 0.1% trifluoroacetic acid (TFA) and B, acetonitrile (MeCN) with 0.1% TFA. The flow rate was 1 mL/min. The gradient was as follows: 0–1 min, 100% A; 1–11 min, 0–100% B; 11–16.5 min, 100% B; 16.5–17.5 min, 100–0% B; and 17.5–20 min, 100% A.
Semipreparative HPLC (Method A): Purification of non-radioactive intermediates 8 and 9 was conducted on a second Thermo Scientific UltiMate 3000 HPLC system with a Phenomenex Luna C18(2) reversed-phase column (5 μm, 250 mm × 10 mm). The UV absorbance was recorded at 214 and 254 nm. The mobile phases were A, H2O with 0.1% TFA and B, MeCN with 0.1% TFA. The flow rate was 3 mL/min. The gradient was as follows: 0–3 min, 100% A; 3–20 min, 0–100% B; 20–27.5 min, 100% B; 27.5–30 min, 100–0% B; and 30–33 min, 100% A.
Semipreparative HPLC (Method B): Purification of radioactive compounds 11 and [18F]-FAA was performed on a Knauer HPLC system with an Azura P 6.1L HPG pump and BlueShadow 40D UV/VIS detector with a Phenomenex Luna C18(2) reversed-phase column (5 μm, 250 mm × 10 mm). The UV absorbance was recorded at 254 nm. Radioactivity was recorded using a model 101 radio-detector (Carroll & Ramsey Associates, Berkeley, CA). The mobile phases were A, H2O and B, MeCN. The flow rate was 3 mL/min. The gradient was as follows: 0–3 min, 100% A; 3–20 min, 0–100% B; 20–27.5 min, 100% B; 27.5–30 min, 100–0% B; and 30–40 min, 100% A.
2.3. Chemical synthesis
4-Chlorobut-2-yn-1-ol
To a solution of propargyl chloride (2.00 g, 26.8 mmol) in dry Et2O (50 mL) at −78 °C was added n-BuLi (2.5 M solution in tetrahydrofuran, 10.7 mL, 26.8 mmol, 1 equiv). After 2 h, the mixture was then added to a solution of paraformaldehyde (0.85 g, 28.19 mmol, 1.05 equiv) in Et2O (5.6 mL) at 0 °C and then stirred overnight while slowly warming to room temperature (RT). The reaction was quenched with ice water and extracted with Et2O (3 × 20 mL). The combined organic layers were washed with brine and dried over Na2SO4. The product was purified by column chromatography (0–20% ethyl acetate in hexanes) to afford the title compound as a light-yellow oil (1.19 g, 42.4% yield). 1H NMR (400 MHz, CDCl3) δ 4.32 (td, J = 2.0, 0.4 Hz, 2H), 4.18 (td, J = 2.0, 0.4 Hz, 2H), 1.80 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 84.62, 80.54, 51.06, 30.31.
Methyl 10-hydroxydeca-5,8-diynoate (2)
To a solution of 1 (651.70 mg, 5.17 mmol, 1.2 equiv) in dimethylformamide (DMF, 3.2 mL) at 0 °C was added Cs2CO3 (2.81 g, 8.61 mmol, 2 equiv), NaI (1.291 g, 8.61 mmol, 2 equiv), and CuI (1.64 g, 8.61 mmol, 2 equiv). The reaction was stirred for 30 min, then 4-chlorobut-2-yn-1-ol (450 mg, 4.3049 mmol, 1 equiv) in DMF (3.2 mL) was added and the reaction stirred overnight while slowly warming to RT. The reaction was quenched with ice water, washed with sat. aq. NH4Cl and Na2S2O3, then extracted with Et2O (3 × 3 mL). The combined organic layers were washed with water and brine then dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by column chromatography (0–50% ethyl acetate in hexanes) to obtain 2 as a pale-yellow oil (545.5 mg, 65.2% yield). 1H NMR (400 MHz, CDCl3) δ 4.25 (t, J = 2.2 Hz, 2H), 3.67 (s, 3H), 3.17 (p, J = 2.3 Hz, 2H), 2.43 (t, J = 7.4 Hz, 2H), 2.23 (tt, J = 6.9, 2.4 Hz, 2H), 1.87 − 1.75 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 173.72, 80.54, 79.75, 78.50, 74.43, 51.60, 51.21, 32.85, 23.77, 18.13, 9.81.
Methyl 10-bromodeca-5,8-diynoate (3)
To a solution of 2 (500 mg, 2.57 mmol) in dichloromethane (DCM) (13 mL) at 0 °C was added CBr4 (2.05 g, 6.69 mmol, 2.4 equiv). PPh3 (1.76 g, 6.69 mmol, 2.6 equiv) in DCM (8.5 mL) was added to the reaction which was then stirred for 4 h. The reaction mixture was evaporated to half its under reduced pressure and purified by column chromatography (0–13% ethyl acetate in hexanes) to afford 3 as an amber oil (527.8 mg, 79.7% yield). 1H NMR (400 MHz, CDCl3) δ 3.91 (t, J = 2.3, Hz, 2H), 3.68 (s, 3H), 3.20 (p, J = 2.4, 2H), 2.43 (t, J = 7.4 Hz, 2H), 2.24 (tt, J = 7.0, 2.5 Hz, 2H), 1.82 (p, J = 7.2 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 173.60, 81.82, 79.99, 75.36, 73.85, 51.57, 32.82, 23.77, 18.13, 14.76, 10.06.
10-(Trimethylsilyl)deca-6,9-diyn-1-ol (5)
To a solution of 6-heptyn-1-ol (300 mg, 2.67 mmol) in DMF (8 mL) at 0 °C was added Cs2CO3 (1.74 g, 5.35 mmol, 2 equiv), NaI (521.15 mg, 3.48 mmol, 1.3 equiv), and CuI (662.16 mg, 3.48 mol, 1.3 equiv) The reaction was stirred for 30 minutes, then a solution of 3-bromo-1-(trimethylsilyl)-1-propyne (613.45 mg, 3.21 mmol, 1.2 equiv) in DMF (23 mL) was added and the reaction stirred overnight while slowly warming to RT. The reaction was quenched with ice water and washed with sat. aq. NH4Cl and Na2S2O3, then extracted with Et2O (3 × 8 mL). The combined organic layers were washed with water and brine then dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by column chromatography (0–50% ethyl acetate in hexanes) and the compound 5 was obtained as a yellow oil (431.2 mg, 72.5% yield). 1H NMR (400 MHz, CDCl3) δ 3.65 (t, J = 6.5 Hz, 2H), 3.18 (t, J = 2.4 Hz, 2H), 2.21 − 2.17 (m, 2H), 1.61 − 1.41 (m, 6H), 0.15 (s, 9H), OH not observed; 13C NMR (101 MHz, CDCl3) δ 100.80, 84.77, 80.83, 73.65, 62.94, 32.34, 28.46, 25.05, 18.78, 10.96, 0.00.
Deca-6,9-diyn-1-ol (6)
To a solution of 5 (77.5 mg, 0.348 mmol) in THF (0.75 mL) was added tetra-n-butylammonium fluoride (TBAF, 1 M in THF, 0.383 mL, 0.383 mmol, 1.1 equiv) and glacial acetic acid (0.022 mL, 0.383 mmol, 1.1 equiv) simultaneously dropwise. The reaction stirred for 3 h then was quenched with ice water. Sat. aq. NH4Cl was added, and the reaction was extracted with Et2O (3 × 1 mL) and subsequently washed with water and brine then dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by column chromatography (0–30% ethyl acetate in hexanes), yielding 6 as a light-yellow oil (40.83 mg, 70.9% yield). 1H NMR (400 MHz, CDCl3) δ 3.64 (t, J = 6.5 Hz, 2H), 3.14 (qd, J = 2.4, 0.7 Hz, 2H), 2.20 − 2.17 (m, 2H), 2.05 (t, J = 2.7 Hz, 1H), 1.86 (s, 1H), 1.61 − 1.40 (m, 6H); 13C NMR (101 MHz, CDCl3) δ 80.97, 78.91, 73.24, 68.40, 62.81, 32.21, 28.36, 24.96, 18.62, 9.54.
Methyl 20-hydroxyicosa-5,8,11,14-tetraynoate (7)
To a solution of 3 (29.2 mg, 0.195 mmol, 1 equiv) in DMF (1 mL) at 0 °C was added Cs2CO3 (95.0 mg, 0.292 mmol, 1.5 equiv), NaI (35.0 mg, 0.23 mmol, 1.2 equiv), and CuI (44.4 mg, 0.23 mmol, 1.3 equiv) The reaction was stirred for 30 minutes, then 6 (50 mg, 0.195 mmol, 1 equiv) in DMF (1 mL) was added and the reaction was stirred overnight while slowly warming to RT. The reaction was quenched with ice water and washed with sat. aq. NH4Cl and Na2S2O3, then extracted with Et2O (3 × 8 mL). The combined organic layers were washed with water and brine then dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by a silica plug eluting with 25% ethyl acetate in hexanes to obtain 7 as a yellow oil (50.05 mg, 78.8% yield). 1H NMR (400 MHz, CDCl3) δ 3.68 (s, 3H), 3.65 (t, J = 6.6 Hz, 2H), 3.14 (m, 6H), 2.43 (t, J = 7.7 Hz, 2H), 2.25 − 2.18 (m, 4H), 2.03 (s, 1H), 1.81 (t, J = 7.5 Hz, 2H), 1.59 − 1.41 (m, 6H). MS (ESI) m/z: 327.31 (M + H+).
Methyl (5Z,8Z,11Z,14Z)-20-hydroxyicosa-5,8,11,14-tetraenoate (8)
To a solution of 7 (149.1 mg, 0.46 mmol) in methanol/pyridine (0.8/0.2 mL) in a foil-wrapped vial was added 2-methyl-2-butene (0.8 mL). Lindlar’s catalyst (60 mg) was added, and the reaction was stirred for 1 h under 1 atm H2. The reaction was filtered and washed with ethyl acetate. The filtrate was washed with 1 N aq. HCl (1 mL), water, and brine, then dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by column chromatography (15% ethyl acetate in hexanes) to obtain 8 (63.0 mg, 41.2% yield). The product was found after flash chromatography as a mixture of 8 and other reduction products. 1H NMR (400 MHz, CDCl3) δ 5.43 − 5.31 (m, 8H), 3.66 (s, 3H), 3.64 (t, J = 6.6 Hz, 2H), 2.85 − 2.76 (m, 6H), 2.32 (t, J = 7.5 Hz, 2H), 2.13 − 2.03 (m, 4H), 1.70 (p, J = 7.3 Hz, 2H), 1.61 − 1.53 (m, 2H), 1.41 − 1.35 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 174.10, 130.10, 128.92, 128.85, 128.46, 128.15, 127.93, 127.82, 127.12, 62.94, 51.49, 33.43, 32.68, 29.39, 27.17, 26.54, 25.62, 25.60, 25.39, 24.76. MS (ESI) m/z: 335.26 (M + H+).
Methyl (5Z,8Z,11Z,14Z)-20-(tosyloxy)icosa-5,8,11,14-tetraenoate (9)
To a solution of 8 (5.0 mg, 0.015 mmol) in DCM (0.5 mL) were added sequentially triethylamine (2.9 μL, 0.021 mmol, 1.4 equiv) and para-toluenesulfonyl chloride (freshly recrystallized from CHCl3 and hexane, 2.1 mg, 0.021 mmol, 1.4 equiv) at 0 °C. 4-Dimethylaminopyridine (DMAP, 0.2 mg, 0.0015 mmol, 0.1 equiv) was added and the reaction stirred for 6 h while slowly warming to room temperature. The reaction was quenched with ice water and extracted with DCM (3 × 1 mL). The combined organic layers were washed with water and brine, dried over MgSO4, filtered, and concentrated. The residue was purified by semipreparative reversed-phase HPLC using method A to obtain 9 as a colorless oil (3.0 mg, 41.1% yield). 1H NMR (400 MHz, CDCl3) δ 7.83 − 7.75 (m, 2H), 7.38 − 7.30 (m, 2H), 5.45 − 5.27 (m, 8H), 4.02 (t, J = 6.5 Hz, 2H), 3.66 (s, 3H), 2.86 − 2.74 (m, 6H), 2.45 (s, 3H), 2.39 − 2.26 (m, 2H), 2.16 − 2.05 (m, 4H), 1.76 − 1.62 (m, 4H), 1.31 (dqd, J = 7.2, 4.6, 1.6 Hz, 4H); 13C NMR (101 MHz, CDCl3) δ 144.62, 133.22, 129.79, 128.96, 128.82, 128.34, 128.23, 128.05, 127.88, 70.53, 51.46, 33.43, 28.91, 28.75, 26.93, 26.54, 25.60, 24.98, 24.76, 21.62. MS (ESI) m/z: 489.27 (M + H+).
Methyl (5Z,8Z,11Z,14Z)-20-fluoroicosa-5,8,11,14-tetraenoate (10)
To a solution of 9 (4.0 mg, 0.0082 mmol) in THF (0.5 mL) at 0 °C was added TBAF (1 M solution in THF, 0.033 mL, 0.033 mmol, 4 equiv) and the solution was stirred for 2 h. The solvent was evaporated, and the residue was dissolved in DCM (1 mL), washed with water and brine, dried over MgSO4, filtered, and concentrated. The residue was purified by column chromatography (0–2% ethyl acetate in hexanes) to give 10 as a colorless oil (2.0 mg, 72.6% yield). 1H NMR (400 MHz, CDCl3) δ 5.49 − 5.28 (m, 8H), 4.44 (dt, J = 47.3, 6.2 Hz, 2H), 3.67 (s, 3H), 2.89 − 2.74 (m, 6H), 2.32 (t, J = 7.5 Hz, 2H), 2.10 (dq, J = 13.4, 7.0 Hz, 4H), 1.77 − 1.64 (m, 4H), 1.41 (t, J = 3.5 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 174.05, 129.94, 128.94, 128.84, 128.42, 128.20, 128.14, 127.97, 127.96, 84.10 (d, J = 164.2 Hz), 51.47, 33.43, 30.31 (d, J = 19.5 Hz), 29.22, 27.08, 26.55, 25.63, 25.62, 25.60, 24.83 (d, J = 5.4 Hz), 24.77. 19F NMR (470 MHz, CDCl3) δ −218.06. MS (ESI) m/z: 337.25 (M + H+).
(5Z,8Z,11Z,14Z)-20-fluoroicosa-5,8,11,14-tetraenoic acid ([19F]-FAA, nonradiolabeled reference compound)
To a solution of 10 (2.0 mg, 0.0041 mmol, 1 equiv) in THF: water 5:1 (0.6 ml) was added LiOH (0.4 mg, 0.0164 mmol, 4 equiv). The mixture was stirred at RT for 24 h. 1 M aq. oxalic acid was added to acidify the reaction, and the THF was evaporated under reduced pressure. The aqueous mixture was extracted with DCM (3 × 1 mL) and the combined organic phases were washed with water and brine then dried over MgSO4, filtered, and concentrated. The residue was purified by column chromatography (0–2% ethyl acetate in hexanes) to give [19F]-FAA as a colorless oil (2.5 mg, 45.0% yield). 1H NMR (400 MHz, CDCl3) δ 5.52 − 5.28 (m, 8H), 4.44 (dt, J = 47.3, 6.1 Hz, 2H), 2.88 − 2.73 (m, 6H), 2.37 (t, J = 7.5 Hz, 2H), 2.14 (q, J = 7.1 Hz, 2H), 2.11 − 2.05 (m, 2H), 1.76 − 1.64 (m, 4H), 1.44 − 1.38 (m, 4H), OH not observed. 13C NMR (101 MHz, CDCl3) δ 178.60, 129.95, 129.03, 128.76, 128.43, 128.18, 128.15, 127.96 (2C), 84.13 (d, J = 164.1 Hz), 33.18, 30.31 (d, J = 19.3 Hz), 29.69, 29.22, 27.09, 26.45, 25.64, 25.62, 24.83 (d, J = 5.6 Hz), 24.51. 19F NMR (470 MHz, CDCl3) δ −218.01. MS (ESI) m/z: 323.24 (M + H+).
2.4. Radiosynthesis of [18F]-FAA (PET tracer)
[18F]-FAA was synthesized from the labeling precursor 9 and [18F]fluoride ion generated by the [18O] (p, n) [18F] nuclear reaction in a GE PETtrace 800 cyclotron. [18F]fluoride ion in [18O] water was transferred through a preconditioned (10 mL EtOH followed by 10 mL H2O) anion exchange cartridge (QMA). The retained [18F]fluoride was eluted into a V-vial with 0.4 mL 0.75 M aq. tetrabutylammonium carbonate solution. Anhydrous MeCN was added to the V-vial and the resulting solution of [18F]TBAF was azeotropically dried at 100 °C with nitrogen flow. A solution of 9 (ca. 1.5 mg) in 0.5 mL MeCN was added and the reaction mixture was heated at 85 °C for 20 min. To the vial was added 0.5 mL 2 M aq. KOH and the reaction was heated at 85 °C for 15 min and then acidified with 0.75 mL 1 M aq. HCl. The crude reaction mixture was purified by semipreparative reversed-phase HPLC (Method B) and the solvent was removed under reduced pressure to obtain radiochemically pure [18F]-FAA. The [18F]-FAA tracer was then dissolved in 0.75 mL 5 μM HEPES buffered saline containing 50 mg/mL of fatty acid-free bovine serum albumin (BSA) to use in the in vivo experiments.
2.5. Partition coefficient
The 1-octanol–phosphate-buffered saline (PBS) partition coefficient was measured at RT and the value was designated as Log P. A solution of [18F]-FAA (370 KBq) in 10 μL of PBS (pH = 7.4) was added to a microcentrifuge tube containing 500 μL of PBS (pH 7.4) and 500 μL of 1-octanol. The mixture was vortexed for 3 min and then centrifuged (12,500 rpm) for 10 min. The PBS and 1-octanol layers (150 μL of each layer) were pipetted into separate gamma counter test tubes. The radioactivity was determined using a PerkinElmer 2480 WIZARD automatic gamma counter (PerkinElmer Inc., Waltham, MA). The partition coefficients of 1-octanol-to-PBS were calculated as Log P = log ([organic-phase cpm]/[aqueous-phase cpm]). Measurements were carried out in quintuplicate.
2.6. Formulation stability
A solution of approximately 370 MBq of [18F]-FAA in 0.25 mL 5 μM HEPES buffered saline containing 50 mg/mL fatty-acid free BSA was divided into 8 aliquots and incubated at RT for 2, 4, 6, and 8 h. At each time point, the sample was diluted with water and analyzed by HPLC to determine the purity of the tracer. Measurements were performed in duplicate.
2.7. Stability in mouse serum
A solution of approximately 7.4 MBq of [18F]-FAA in 250 μL mouse serum was shaken at 37 °C for 1, 2, 4, 6, or 8 h, after which 100 μL MeCN was added. The mixture was vortexed for 3 min and centrifuged for 10 min (12,500 rpm). The supernatant was analyzed by HPLC. Measurements were performed in duplicate.
2.8. p38 phosphorylation
Immortalized mouse astrocytes derived from human ApoE3-TR mice were maintained in Dulbecco’s modified Eagle’s medium (DMEM/F12, Corning, MT10090CV) supplemented with 10% FBS, 1 mM geneticin (Thermo Fisher, 10131035), 1 mM sodium pyruvate (Thermo Fisher, 11360070), and 1% anti-anti. After treating with AA or [19F]-FAA, the astrocytes were lysed using 1x RIPA buffer (Cell Signaling Technology, CST 9806) containing a protease inhibitor cocktail (Sigma, P8340) and phosphatase inhibitor cocktail (Sigma, P0044). The cell lysates were then centrifuged at 14,000 × g for 15 min at 4 °C and supernatants were collected for Western blotting. Proteins from the cell lysates were separated using 12% TGX™ FastCast™ Acrylamide gels (Bio-Rad, 1610175) and then transferred onto nitrocellulose membranes (Bio-Rad, 1,704,270). After blocking with 5% fat-free milk (Bio-Rad, 1,706,404) for 30 min at RT, the membranes were incubated overnight with the primary antibody in 5% BSA at 4 °C. Then, the membranes were washed (3 × 5 min) with Tris Buffered Saline- 0.1% Tween-20 (TBS-T) and incubated with HRP conjugated secondary antibody for 30 min at RT. After washing with TBS-T (3 × 5 min), the membranes were imaged with a Fujifilm LAS-4000 imager system, and protein was detected using Chemiluminescent HRP substrate (Millipore, WBKLS0500). Gel Quant NET software was used for the analysis of densitometric quantification. The following antibodies and dilution factors were used: p38 antibody (CST, 9212) (1:1000), phospho-p38 antibody (CST, 4511) (1:1000, GAPDH antibody (CST, 5174) (1:1000), β-tubulin antibody (CST, 2146) (1:1000), HRP-linked anti-mouse IgG (CST, 7076) (1:2000), HRP-linked anti-rabbit IgG (CST, 7074) (1: 2000).
2.9. Animal model
All animal studies were approved by the Institutional Animal Care and Use Committee of the University of Southern California (USC). The wild-type C57BL/6J and humanized ApoE4-TR mice (16-month-old), weighing 29–38 g, used in the study were bred in the USC animal facility. Animals were housed with standardized 12 h light and dark cycles and had access to food and water ad libitum. Vivarium temperature was maintained between 22 and 24 °C and humidity was maintained between 50 and 60%.
2.10. In vivo uptake and stability
2.10.1. Injection and tissue extraction
Two C57BL/6J mice, anesthetized with isoflurane, were injected with ~15 MBq of the tracer through a tail vein. Blood samples were collected from the submandibular vein of each mouse under anesthesia at 10- and 30-min post-injection. At 45 min post-injection, the mouse was dissected under anesthesia, and urine was extracted from the bladder. Then, about 500 μL of blood was extracted from the inferior vena cava and the mouse was euthanized by decapitation. The brain was extracted from the skull within 1 min and kept on ice until processing.
2.10.2. Folch extraction
The following procedure was performed either on plasma obtained by centrifugation of the blood, on whole urine, or on homogenized brain tissue, adapting a method in the literature [16]. To the tissue was added 7 mL of 2:1 (v/v) chloroform/methanol at 0 °C. The mixture was sonicated for 20 s, and 40% aq. urea (1.75 mL) and 5% aq. H2SO4 (1.75 mL) were added. After sonicating for 20 s, the mixture was centrifugated at 1800 × g for 10 min. Organic, aqueous, and interphase (pellet) fractions were isolated and 18F radioactivity was determined with a gamma counter.
2.11. Effect of DOI administration
The administration of (+/−)-2,5-dimethoxy-4-iodophenyl-2-aminopropane (DOI), partial 5-HT2A/2C agonist, is known to increase AA uptake into the brain [23] and was used to validate the method of estimating [18F]-FAA brain uptake. One C57BL/6J mouse was first injected with the [18F]-FAA tracer through a tail vein cannula, then injected with 100 μL of a saline solution containing DOI (1.5 mg/kg) through the same cannula 25 min after delivery of the tracer. Another mouse as control was subjected to the same imaging protocol but only saline was injected after the tracer delivery.
2.12. PET and MR imaging acquisition
PET and MR images were acquired using a 7T MRI scanner with an integrated PET scanner (MR solutions, Guildford, UK). Mice were anesthetized with isoflurane (1.5–2%) and respiration was monitored using a pneumatic pillow. Body temperature was maintained at 37 °C using a heated animal holder (Minerve, France). Two-dimensional Fast Spin Echo (FSE) transverse T1-weighted MR images were acquired to define anatomy with the following parameters: TE/TR = 11 ms/3556 ms, slice thickness = 1 mm, 1 average, echo train length = 4, field of view = 72 mm × 72 mm, matrix size = 512 × 256, flip angle = 90°. For PET, a list mode acquisition protocol was obtained from 2 min before to 45 min after bolus injection of [18F]-FAA through the tail vein via a cannula (9.0 ± 1.9 MBq, range 6.1 – 10.7 MBq). Image data were corrected for intrascan radioactivity decay, detector nonuniformity, and random coincident noise. Three-dimensional ordered subsets expectation-maximization (3D-OSEM) reconstruction was performed, resulting in a 0.28 × 0.28 × 0.28 mm3 voxel dimension, 34 mm field of view, 64 subsets, and 4 iterations. The acquisition protocol was as follows: 4 frames of 30 s, 12 frames of 10 s, 6 frames of 30 s, 5 frames of 60 s, and 7 frames of 300 s.
2.13. PET/MR imaging analysis
PET/MR images were processed using VivoQuant 2.0 software for Windows 64-bit, (Invicro, Inc., Boston, MA, USA, www.vivoquant.com). PET and MR images were co-registered using a PET/MR phantom, which consisted of a 1 cm PE-60 tube filled with 10 μL of ~40 kBq [18F]-FAA solution. Three-dimensional regions of interest (ROIs) were defined through manual segmentation using the MRI image as the anatomical reference and activity concentrations were quantified by extracting the mean radioactivity over time and the maximum uptake value per ROI. ROIs included in the analysis are whole brain, muscle, liver, and kidneys. Percent injected dose per gram (%ID/g) for the brain, liver, kidney, muscle, and bone were calculated from the injected dose and the weight of the animal from the frame at 20–25 min and at 40–45 min.
2.14. PET tracer uptake kinetics
A). Image-derived input function
To obtain an appropriate image-derived input function (IDIF) to be used as a proxy of the arterial input function, an ROI was placed within the right ventricle cavity (as a surrogate for an artery) using the respiratory-gated MRI scan for anatomical reference. A time-activity curve (TAC) for the blood pool was obtained by calculating the concentration of the tracer within the ROI for frames 4–34, corresponding to 0–45 min post-injection. A TAC for the brain was also obtained from the ROI in the cerebrum. To correct the heart TAC concentration [Cheart(t)] for spillover from the surrounding myocardium, the method by Zhou et al. was implemented [17]. Briefly, this involved a linear fit of the proportion function [P(t), Equation 1] based on the dose-normalized venous blood activities and the interpolated raw PET-based heart activities. This effectively corrects the heart TAC based on the shape of the venous input function to obtain the value of the IDIF at each frame [CIDIF(t)].
| (1) |
B). Calculation of K* by the area under the IDIF curve (AUC)
We evaluated three approaches to estimate the regional incorporation coefficients (K*) as a measure of the kinetics of tracer incorporation into the brain. In the first approach, K* values were calculated following the method by DeGeorge et al. [18] where the radioactivity in the brain ROI at the last time point (42.5 min) is divided by the IDIF integrated over time from 0 to 42.5 min (Equation 2).
| (2) |
C). Calculation of K* by the irreversible two-tissue compartment model
In the second approach, kinetic modeling of the brain TAC data was performed using MATLAB custom numerical fitting routines on MATLAB (version R2021a). First, either the heart TAC or the spillover-corrected IDIF was fitted into an 8-parameter model, which includes a linear interpolation of the data before the curve’s peak, and the sum of three decreasing exponentials after the peak. Then, kinetic micro-parameters (K1, k2, and k3) for the irreversible two-tissue compartment model (2TCM-IRR) were estimated from the brain TAC using the fitted IDIF as a proxy for the arterial input function. The value of K* in this model, which indicates the net influx rate of irreversible tracer into the brain tissue, was calculated for each set of mice dynamic PET data from the estimated micro-parameters following Equation 3.
| (3) |
D). Calculation of K* by the Patlak plot
In the third approach, the method for tracers with irreversible kinetics by Patlak et al. [19] was adapted. The ratio between the brain activity and the IDIF versus the IDIF integrated over time was plotted. Then, linear regression was performed on the last seven time points (corresponding to time t* = 10 min post-injection onward) following Equation 4. The slope of the line corresponds to the estimated K* value.
| (4) |
3. RESULTS
3.1. Synthesis
The synthetic routes used to prepare the non-radiolabeled and radiolabeled compounds are shown in Scheme 1. We utilized a convergent route previously published by Hwang et al., using multiple copper-mediated terminal alkyne-alkyl halide couplings to synthesize an intermediate 8 [20]. The synthesis of [19F]-FAA from 9 was performed according to the procedure by Pichika et al. [13].
Scheme 1.
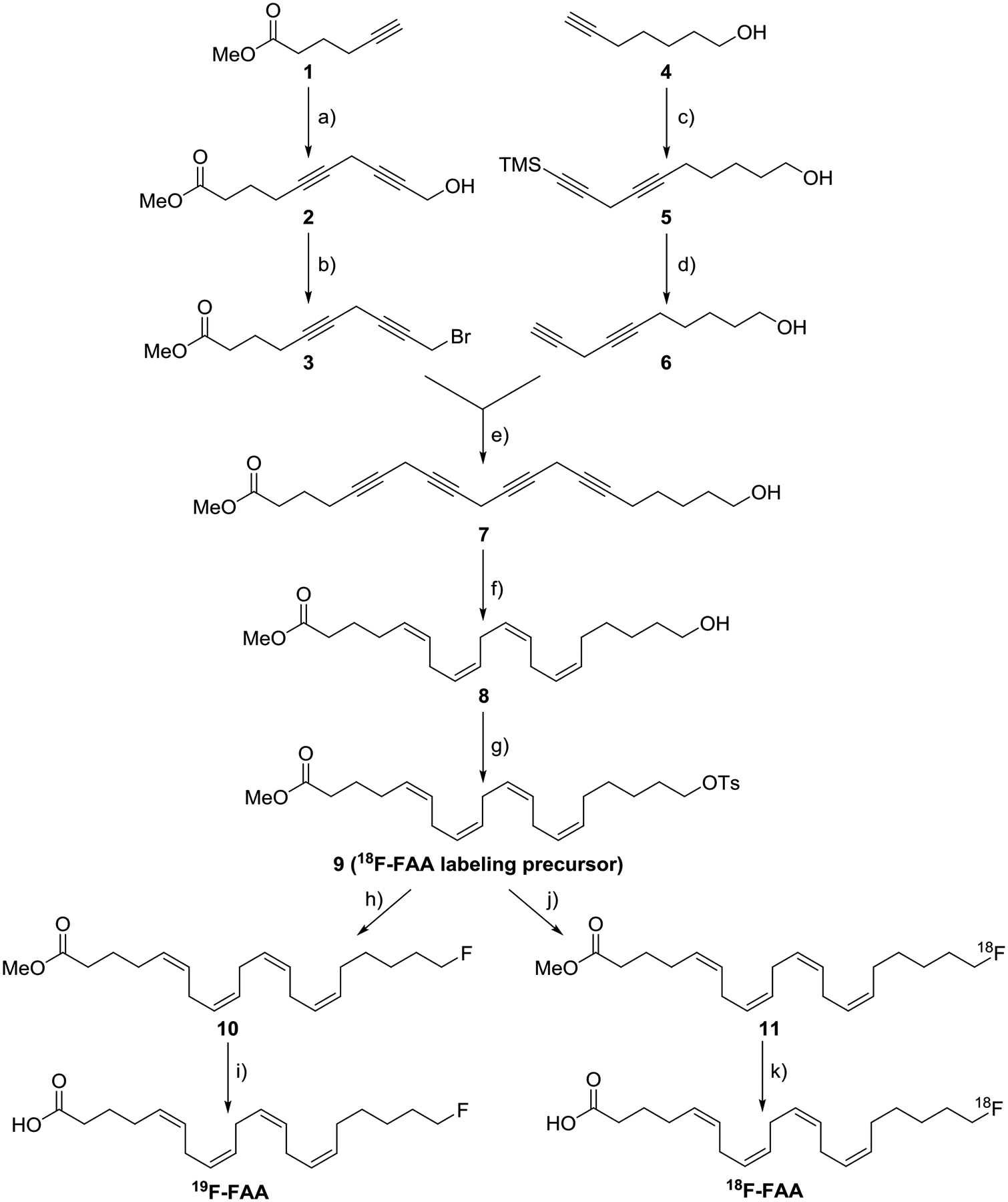
Synthesis of radiolabeling precursor 9, 20-fluoroarachidonic acid ([19F]-FAA) and [18F]-FAA. a) 4-chlorobytyn-1-ol, CuI, NaI, Cs2CO3, DMF, 0 °C→ RT, 65.2% yield. b) CBr4, PPh3, DCM, 0 °C, 4h, RT, 79.7% yield. c) 3-bromo-1-(TMS)-1-propyne, CuI, NaI, Cs2CO3, DMF, 0 °C→RT, 72.5% yield. d) TBAF, AcOH, 0 °C, 70.9% yield. e) CuI, NaI, Cs2CO3, DMF, 0 °C→RT, 78.8% yield. f) H2, Lindlar’s catalyst, MeOH/pyridine/EtOAc 4:1:4, 41.2% yield. g) NEt3, TsCl, DMAP, DCM, 0 °C→RT, 41.1% yield. h) TBAF, THF, 0 °C, 72.6% yield. i) LiOH, THF/H2O 5:1, RT, 45.0% yield. j) 18F−/TBAHCO3, 85 °C, MeCN. k) KOH, 85 °C.
To prepare the required tetrayne compound 7 by convergent synthesis, compounds 3 and 6 were separately synthesized in parallel. After the synthesizing 5 from 4, the removal of trimethylsilyl group to obtain compound 6 was achieved through the simultaneous addition of TBAF and acetic acid. Coupling of 1 and 4-chlorobutyn-1-ol to give 2 with the same copper-catalysis conditions was followed by bromination using Appel conditions to give 3. The coupling of compounds 3 and 6 was achieved with the copper catalysis conditions. Due to the instability of the skipped-conjugated tetrayne 7, the compound was immediately purified through a short plug of silica gel before using in the following reaction.
Partial hydrogenation of tetrayne 7 to the corresponding all-Z skipped alkene scaffold of 8 was achieved using Lindlar’s catalyst in 4:1:4 MeOH/pyridine/2-methyl-2-butene. The crude product contained a mixture of different degrees of hydrogenated compounds, with the desired tetraene 8 as a major product (~60%), determined by HPLC (Figure S1). It should be noted that reaction times greater than 1 h only increased over-hydrogenated compounds rather than increasing the amount of the desired tetraene. After confirming the presence of the desired tetraene 8 in the crude product, it was carried over to the next step without purification due to the presence of inseparable undesired products. Crude compound 8 was subjected to tosylation of the ω-hydroxyl group to give a mixture of tosylates 9 (which includes over and under-reduced products) similar to the starting alcohol 8 (Figure S2) [13]. The crude reaction mixture was purified by reverse-phase semipreparative HPLC to afford the pure tetraene 9 (Figure S3). Nucleophilic fluorination of the tosylate 9 with TBAF gave compound 10, which was hydrolyzed to obtain cold fluorinated compound standard [19F]-FAA.
3.2. Radiosynthesis
[18F]-FAA was successfully synthesized by radiofluorination of 9 using the general procedure from Nagatsugi et al, with some modifications [15]. The first stage of the one-pot procedure, [18F]fluoride displacement of the tosylate precursor, was achieved by reacting 9 with [18F]TBAF at 85 °C for 20 min. Formation of [18F]-FAA methyl ester (11) was confirmed by radio-HPLC (Figure 1A). Aqueous KOH was added directly to the crude reaction mixture for hydrolysis, and after 15 min the product was formed (Figure 1B). Purification by semipreparative HPLC (Figure S4) afforded [18F]-FAA in high radiochemical purity (>99%, Figure 1C), with 1–7% decay-corrected radiochemical yields. The total synthesis time was about 180 min, including the HPLC purification time. Specific activity was determined to be 27.5 – 1147.78 GBq/μmol.
Figure 1.
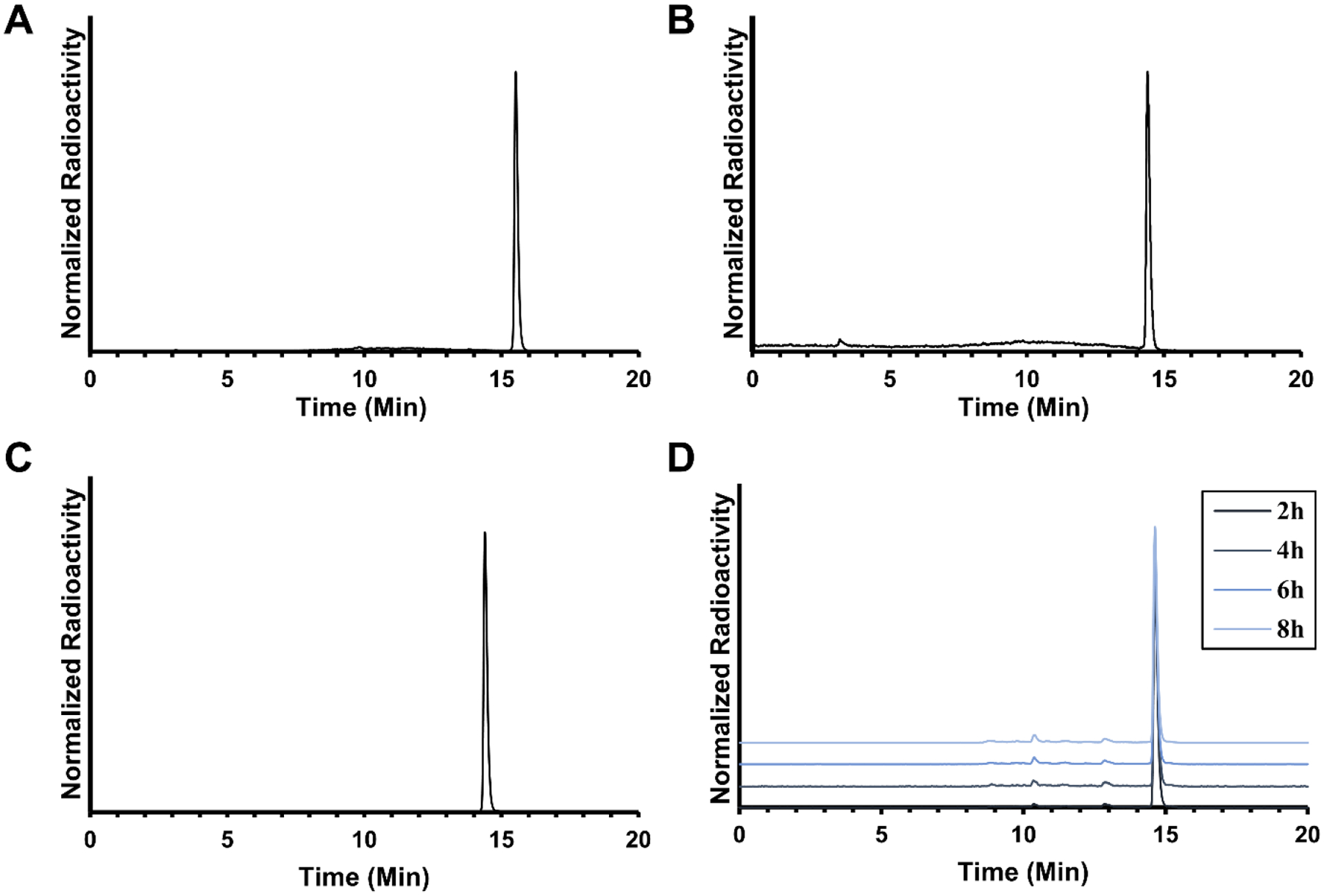
Analytical HPLC chromatograms of (A) crude reaction mixture of methyl 20-[18F]-fluoroarachidonate 11, (B) crude reaction mixture of [18F]-FAA, (C) [18F]-FAA after semipreparative HPLC, and (D) stability of [18F]-FAA in 5 μM HEPES buffered saline containing 50 mg/mL BSA standing for 2, 4, 6, and 8 h at room temperature.
3.3. LogP and tracer stability
The partition coefficient of [18F]-FAA was determined to measure its lipophilicity. A greater positive partition coefficient indicates higher lipophilicity, while a greater negative value indicates higher hydrophobicity. Greater lipophilicity is one factor that can predict the likelihood of a molcule to cross the blood-brain barrier, and a negative value could mean the tracer would be a poor means of brain imaging. The octanol-PBS partition coefficient (LogP) of [18F]-FAA was measured by gamma counter to be 1.81 ± 0.08. There is often elapsed time between when the tracer is formulated and the time of injection, and over time, the tracer might be subject to defluorination or other forms of degradation due to air, water, light, or components of the formulation. [18F]-FAA was incubated in the formulation used for injection to determine whether it will be suitable for injection after time has elapsed i.e. significant degradation of the parent compound has not occurred. After analyzing the stability of the tracer in the formulation used for animal imaging (5 μM HEPES in saline containing 50 mg/mL BSA) by HPLC at 2, 4, 6, and 8 h after formulation (Figure 1D), the percent purity of the tracer was determined to be 96.20 ± 0.16, 95.27 ± 0.32, 93.82 ± 0.01, and 92.41 ± 0.50 at 2, 4, 6, and 8 h, respectively. The stability of [18F]-FAA in mouse serum was evaluated to assess tracer decomposition in vivo. The stability of the tracer in mouse serum at 37 °C was analyzed by HPLC at 1, 2, 4, 6, and 8 h, (Figure S5). The tracer showed very little appreciable decomposition, with >98% purity at all timepoints.
3.4. In vitro assay
AA is known to stimulate the phosphorylation of p38 mitogen-activated protein kinase [21]. To assess if fluorination of the terminal carbon of AA alters its biological activity, the ability of [19F]-FAA to phosphorylate p38 was compared to AA. Cells were treated with the nonradiolabeled standard [19F]-FAA (as a surrogate of [18F]-FAA) or AA and the phosphorylation of p38 was assesssed. Western blot for the expression of phospho-p38 in astrocytes [22] after treatment with either AA or [19F]-FAA for 30 min revealed that the p38 phosphorylation levels of cell lysates (Figure 2) showed a similar increase after both AA and [19F]-FAA treatment relative to vehicle control. This effect was dose-dependent at 15 μM, 30 μM, and 60 μM for both compounds and there was no significant difference between either treatment group.
Figure 2.
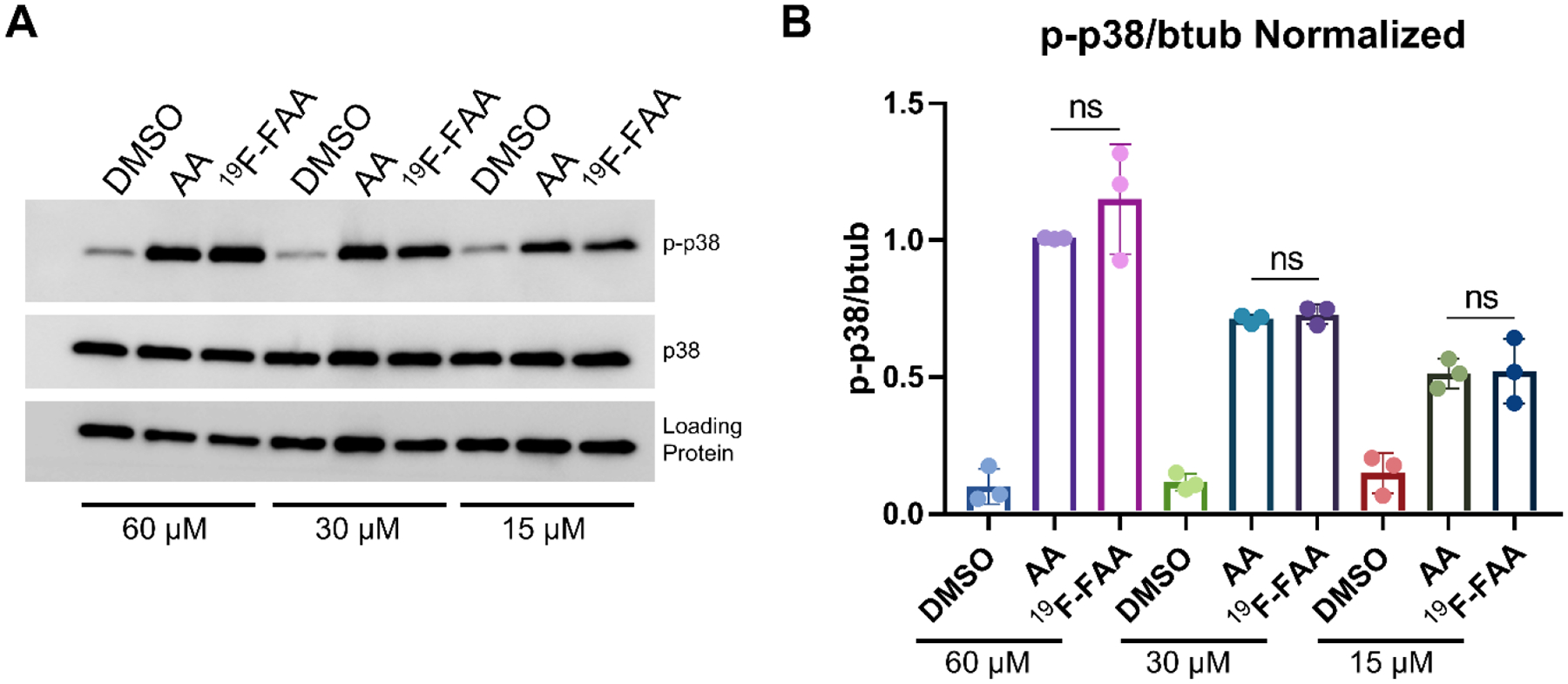
Western blot (A) and quantitation (B) of p38 phosphorylation in astrocytes treated with AA and [19F]-FAA. ns = not significant. Loading protein = β tubulin or GAPDH.
3.5. Effect of DOI administration
Previously, it was reported that AA K* increases after administration of subcutaneous 1.5 mg/kg DOI (by 17–65%, depending on specific brain region) [14]. Using the K* estimation described above, we estimated the effect of DOI administration on the K* value and found a 75% higher K* for the mouse injected with i.v. DOI than the mouse injected with saline (11.0 vs 6.3 μL mL−1 min−1, for the spillover-corrected K* by the Patlak method, Figure 3).
Figure 3.
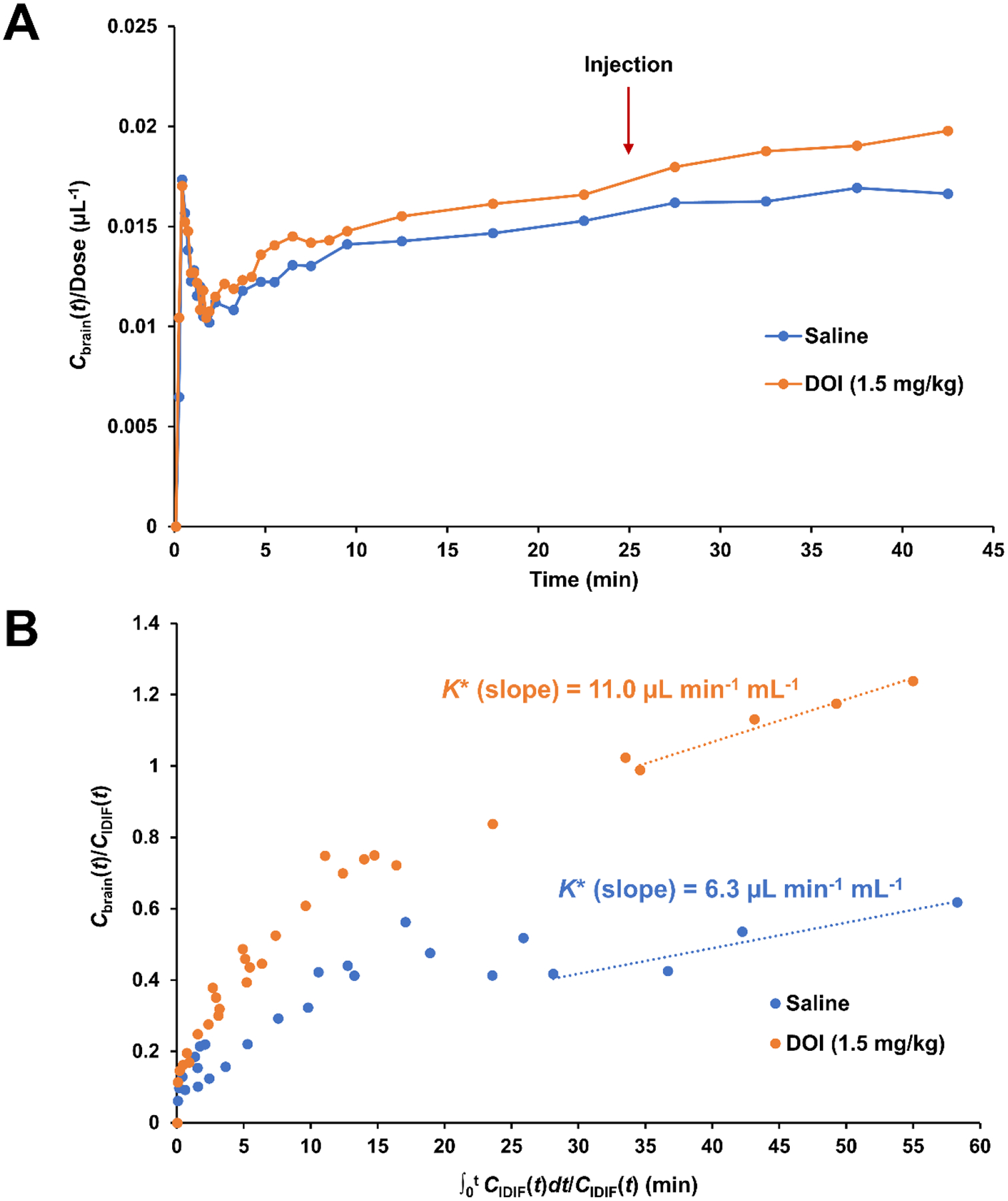
(A) Time-activity curves (TAC) of [18F]-FAA for ROI in the in the brains of mice injected with either intravenous saline (n=1) or DOI (1.5 mg/kg, n=1) 25 min after administration of [18F]-FAA, normalized to the dose and expressed in Bq mL−1 kBq−1. (B) Patlak plots for the brain ROI. Linear regression was performed for time points after 25 min to obtain K* as the slope.
3.6. PET-MR imaging
The in vivo distribution of [18F]-FAA in ApoE4-TR mice was evaluated by PET/MR imaging. Immediately after radiosynthesis, [18F]-FAA was prepared as an infusate in HEPES-buffered saline containing fatty acid-free BSA, then injected as a bolus through the tail vein into human ApoE4-TR mice (n = 5). Based on the %ID/g values determined for the mice in each ROI, relatively high uptake was observed in the liver and kidneys 20–25 min post-injection (Figure 4 and Table 1) with about 0.5% of the total injected tracer having reached the brain at 40–45 min. The concentration of the tracer in the brain over time (Figure 5A) was also observed as a sharp increase within 2 min but the initial spike was then followed by a slow, steady increase, even up to 45 min post-injection. By taking an ROI in the right ventricle cavity (as an approximation of the sampling of the blood pool), the concentration of the tracer in the heart (Figure 5B) was observed as a large spike that dissipated within 2 min. Radioactivity was also observed to slowly increase in bone, indicating the defluorination may occur from the parent tracer.
Figure 4.
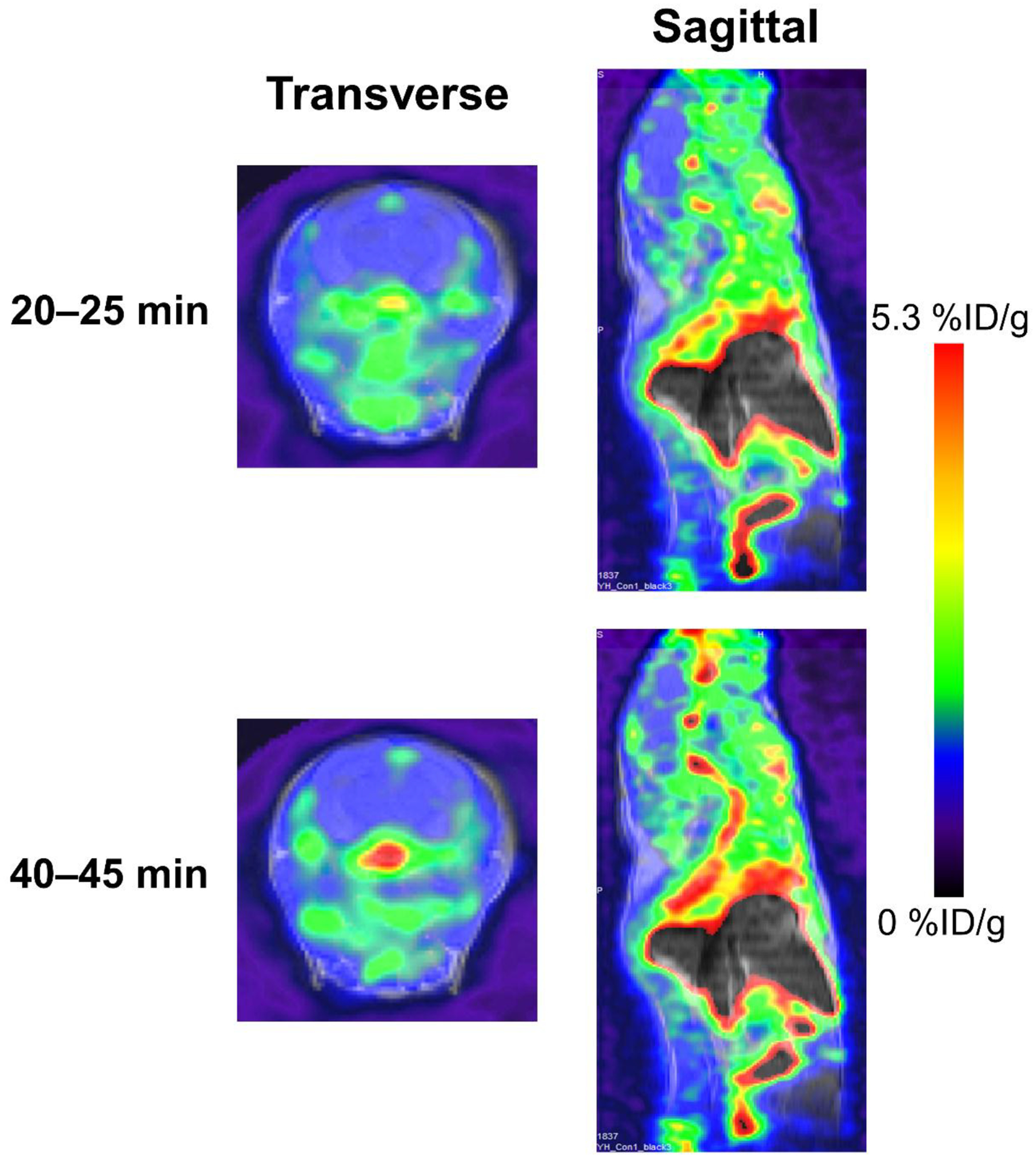
Representative PET/MR images of ApoE4-TR mouse at 20–25 minute frame and 40–45 minute frames.
Table 1.
Percent injected dose per gram (%ID/g) for the tissue ROIs.
| Tissue | 20–25 Min | 40–45 Min | ||
|---|---|---|---|---|
| Mean %ID/g | Max. %ID/g | Mean %ID/g | Max. %ID/g | |
| Brain | 1.32 ± 0.24 | 2.65 ± 0.52 | 1.54 ± 0.28 | 3.08 ± 0.61 |
| Liver | 10.46 ± 1.50 | 17.03 ± 1.99 | 9.77 ± 1.64 | 15.76 ± 2.19 |
| Kidneys | 5.91 ± 1.39 | 8.98 ± 1.50 | 5.45 ± 1.30 | 8.02 ± 1.19 |
| Muscle | 1.28 ± 0.31 | 1.71 ± 0.38 | 1.38 ± 0.47 | 1.79 ± 0.61 |
| Bone | 2.71 ± 0.85 | 3.23 ± 0.96 | 3.44 ± 0.64 | 4.23 ± 0.69 |
Values are expressed as mean ± S.D. (n = 5).
Figure 5.
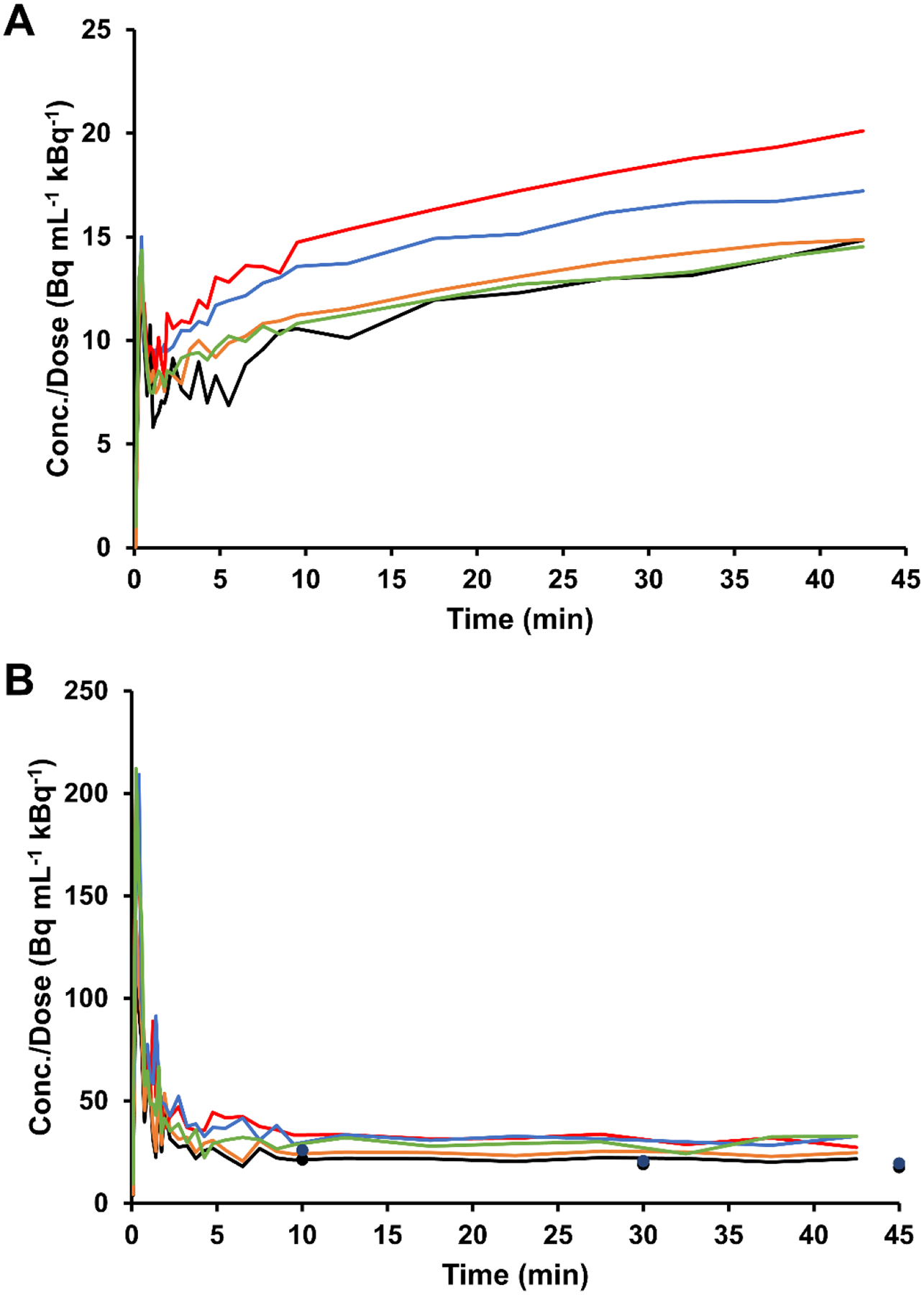
Time-activity curve (TAC) for ROIs in the mouse brain (A) (n = 5) and right ventricle (B) (n = 5) post-injection of [18F]-FAA, normalized to the dose and expressed in Bq mL−1 kBq−1. Tracer concentrations obtained from venous blood sampling at 10, 30, and 45 min in a subset of mice are shown in the heart TAC graph as points.
3.7. Biodistribution
The activity of blood samples and brain tissue from mice injected with [18F]-FAA was measured to determine if there was a suitable amount of tracer remaining in circulation and reaching the brain during the time frame used for imaging. Folch extraction was performed on blood and brain tissue to determine the radioactive species contributing to the activity as a quantitative measure of the in vivo stability of [18F]-FAA. This method relies on partitioning hydrophilic and hydrophobic compounds in tissues, i.e. [18F]-fluoride and small metabolites of [18F]-FAA, and [18F]-FAA and lipid metabolites, respectively. We first injected mice with the tracer and then obtained blood and tissues, and partitioned the radioactive components into three fractions—organic, aqueous, and interphase (pellet) (Table 2). We found that the pure tracer separated predominantly (~98%) into the organic fraction. In the blood samples, we found that a considerable portion of the tracer was converted to hydrophilic metabolites after 10 min. The partitioning of the radioactivity was similar in the brain with about 39% found in the organic portion. For the urine samples, the radioactive compounds partitioned to the aqueous fraction to an even larger extent, as expected for the clearance of hydrophilic metabolites from the blood.
Table 2.
Radioactivity in tissues after Folch extraction.
| Tissue | % Total Radioactivity | ||
|---|---|---|---|
| Aqueous | Organic | Interphase | |
| Blood (10 min) | 59.3 ± 9.1 | 36.5 ± 11.4 | 4.1 ± 2.3 |
| Blood (30 min) | 56.6 ± 5.2 | 39.5 ± 7.6 | 3.8 ± 2.4 |
| Blood (45 min) | 54.6 ± 6.6 | 39.5 ± 9.3 | 5.9 ± 2.7 |
| Urine | 84.4 ± 5.3 | 12.7 ± 3.3 | 2.85 ± 2.0 |
| Brain | 54.8 ± 3.7 | 35.2 ± 2.2 | 1.5 ± 0.6 |
Values are expressed as mean ± S.D. (n = 2).
3.8. Kinetic modeling
Using the IDIFs, and the brain TACs, we were able to calculate the K* values for each mouse using three methods, as shown in Table 3. Using the uncorrected IDIF (based on the right ventricle ROI), the obtained K* values were significantly different with each estimation method used (AUC, Patlak, or 2TCM-IRR). Generally, the K* obtained from the AUC of the IDIF was higher than those from the two other methods. After correction of the IDIF for spillover due to uptake of the tracer in the myocardium, a more accurate proxy of the actual input function was obtained (Figure 6) which matched up more closely with the venous input function. The K* values calculated from these new IDIFs approached similar values across estimation methods. The final values obtained were close to K* values estimated by PET using [1-11C]-AA [14], but were found to be lower than K* values reported via autoradiography using [1-14C]-AA in wild-type mice [23] and rats [18, 24] or [1-11C]-AA in rats [25], which ranged from 30 to 60 μL min−1 mL−1 (depending on the particular brain region of interest).
Table 3.
Uptake coefficient (K*) values calculated for ApoE4-TR mice (n = 5) through three methods using uncorrected and corrected IDIFs.
| Method | K* values from this work | Previous Work | ||
|---|---|---|---|---|
| Uncorrected Heart IDIFa | Spillover-corrected IDIFb | [1-14C]-AA Autoradiographyc | [1-11C]-AA PETd | |
| AUC | 11.52 ± 1.20 | 15.38 ± 2.29 | 31.7–86.4 (mice) [23] 31.2–93.0 (rats) [24] |
- |
| 2TCM-IRR | 6.75 ± 1.35 | 11.66 ± 1.31 | - | - |
| Patlak Plot | 5.29 ± 0.91 | 9.49 ± 0.82 | - | 6.9 ± 0.8 (Control) 8.7 ± 1.4 (AD) [14] |
The uncorrected IDIF used for K* calculation used was obtained by raw heart TAC.
Spillover-corrected IDIFs were obtained from the raw heart TAC, and corrected for spillover from the myocardium using the shape of the population-based venous input function.
K* was determined on awake rodents when they were sacrificed 20 min after infusion of the tracer.
K* was determined in healthy human controls and AD patients after correction for the partial volume effect.
Figure 6.
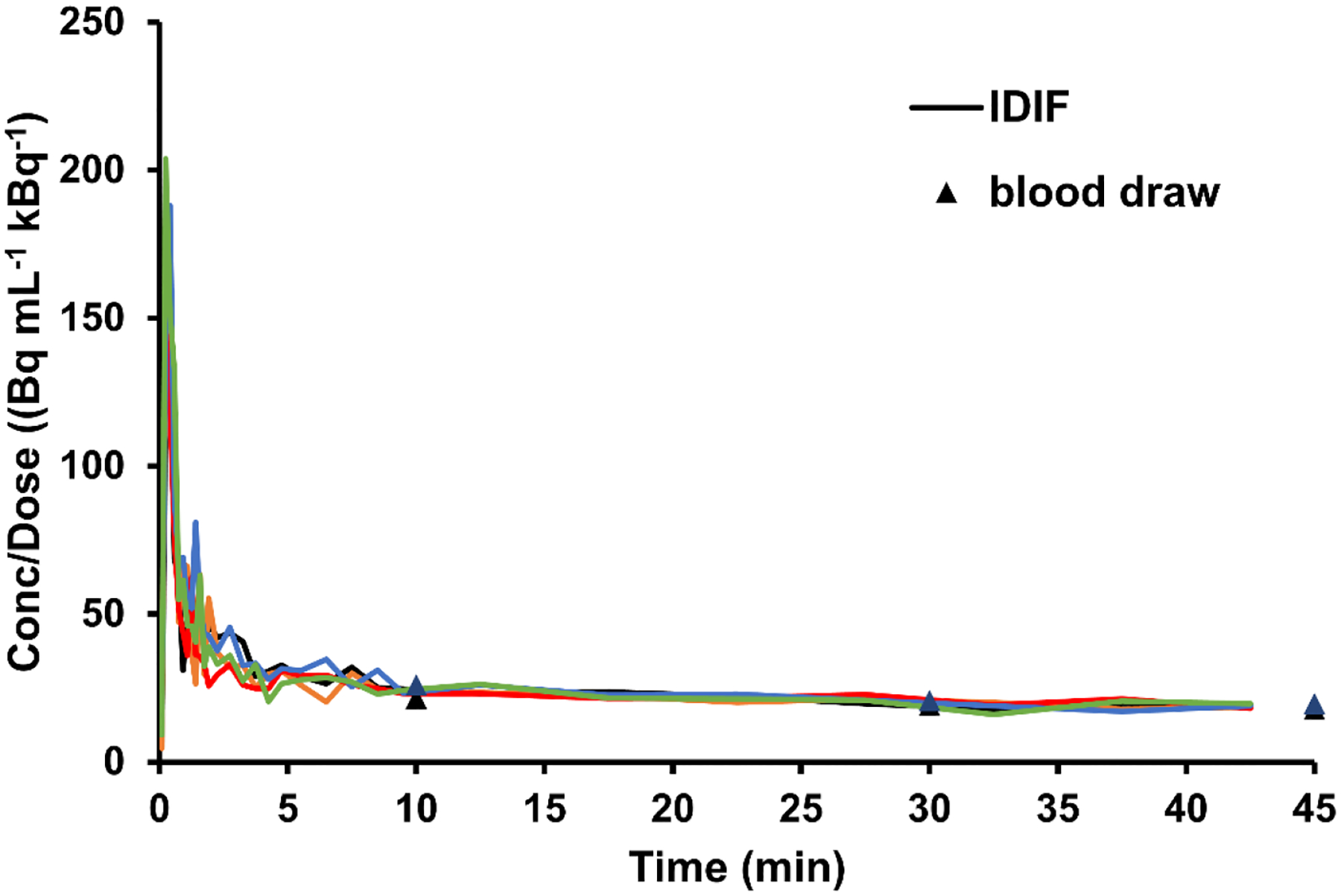
Spillover-corrected IDIF, derived from the heart TAC, normalized to the tracer dose. Correction was made by a linear fit of the proportion function (the ratio between the measured apparent concentrations in the heart ROI and the venous blood) over time for each set of mouse PET data. TAC: Time-activity curve, IDIF: Image derived input function.
DISCUSSION
Using [18F]-FAA as a tracer of AA uptake and metabolism is useful in studying disorders involving brain lipid dysfunction such as AD. By improving previous methods of [18F]-FAA synthesis, we obtained higher yields of the compound. This has implications in the clinical setting by enhancing the resolution of current PET scans due to higher uptake of the probe into the brain and thus providing a more accurate assessment for imaging of neuroinflammation [13].
Our synthetic route toward [18F]-FAA was based on the method by Hwang, et al. for the synthesis of ω-hydroxy PUFAs, including AA and DHA [20]. 20-Hydroxyarachidonic acid methyl ester (8) is a late-stage intermediate in the synthesis, and since it is easily converted to tosylate 9, we chose to use this convergent route to construct the skipped tetraene scaffold of [18F]-FAA. The key steps of the synthesis are the repeated copper-catalyzed alkyne couplings and the Z-selective partial reduction of the tetrayne scaffold.
We found tetrayne 7 to be unstable, and extensive purification or storage before led to extremely poor yields. Filtration through a short silica plug was found to be sufficient and improved the yield to 78.8%. The reduction of the tetrayne scaffold of 7 to the all-Z-skipped tetraene 8 was performed by hydrogenation with Lindlar’s catalyst. The tetraene 8 was obtained as a mixture with other hydrogenation products that could not be separated with column chromatography. The under- and over-reduced products could be removed by HPLC separation, but we first subjected the alcohol to tosylation before HPLC purification to increase the stability. Although HPLC purification is required, the byproducts are fully separable, unlike the Wittig reaction in Pichika et al [13]. Additionally, two steps in the olefination route have yields that are not quantified and are reported as low, while all steps in our synthetic route have yields of at least 41% [13].
For the radiosynthesis of [18F]-FAA, the totally synthetic time from receiving [18F]fluoride ion in [18O] water to the tracer ready for formulation was about 180 min. This is shorter than synthetic time reported by Nagatsugi et al., which was 200 min starting with already dried [18F]TBAF for both fluorination and hydrolysis steps, which both require HPLC purification [15]. Our radiosynthesis method is a one-pot procedure requiring only one HPLC purification at the end of the synthesis, making it more amenable to transfer to an automatic synthesis module. The specific activity was also up to 1000 times higher than reported [15].
We found that fluorination at the ω-position preserved the ability of the fatty acid to induce p38 phosphorylation in a dose-dependent manner. This suggests that the chemical modification of AA did not significantly alter its biochemical role in cells. To study the uptake of [18F]-FAA in vivo, the compound was synthesized for injection. After synthesis, the probe was found to have minimal to no defluorination in the formulation and was ≥95% stable at least 4 h after synthesis, making it acceptable for injection into the tail vein of the mouse. Serum stability is an indication of probable in vivo stability, and the probe showed ≥99% purity in mouse serum at 37 °C after 2 h. After injection in mice, however, we observed the metabolism of the fatty acid tracer into hydrophilic compounds.
PET-MR images show brain uptake of [18F]-FAA which is expected in ApoE4-TR mice, as well as liver and kidney uptake due to metabolic clearance pathways. The brain uptake increases over time as the tracer clears from the blood, and as such timepoint selection will be important in further studies. Bone uptake was observed by PET suggesting defluorination in vivo to produce free [18F]-fluoride. This is not surprising given that AA is metabolized by cytochrome P450 omega hydroxylases at the ω-position to produce the 20-hydroxy derivative and other lipid mediators.
Evaluating the uptake of the tracer into the brain involves the proper selection of the model that accurately describes the transfer of the tracer from the blood to brain tissue. PET-MR imaging allows for non-invasive estimation of the tracer K* into the brain tissue through several methods, each of them with advantages and disadvantages. The first method, involving the AUC of the IDIF, has been used extensively in autoradiography to estimate K*. This method does not involve monitoring the levels of radioactivity in the brain over time and is a highly simplified implementation of the Patlak model of tracer uptake (i.e., it is prone to overestimating K*). In the 2TCM-IRR method, the K* is estimated via a kinetic compartment model and considering both the levels of radioactivity in the brain and the IDIF curve over time. This model assumes that the activity in the brain ROI is arising only from tracer that is being uptaken into brain tissue. However, the PET measure in the brain ROI also contains a portion of radioactivity arising from tracer in the vasculature, due to the spatial resolution of PET. To account for this portion of radioactivity coming from the brain vasculature, the percent of the volume of the blood in the brain which contributes to brain volume (the Vb value) has been estimated to bed between 0.5%–6%, and has been subtracted from the ROI TACs before applying the 2TCM-IRR model [26]. Not accounting for Vb in the K* calculation using 2TCM-IRR would tend to overestimate K*. Alternatively, Vb could also be estimated during modeling by using 2TCM-IRR by adding Vb to the free parameters to be estimated. The third method we investigated, the Patlak method needs as well vasculature correction as well but is a linear approach that is faster and more robust to noise in the PET data than the nonlinear 2TCM-IRR. Dynamic PET, unlike autoradiography, has the advantage of being able to implement various uptake models to estimate the kinetics of the tracer transfer from the blood to the brain.
It should be noted that, in absence of arterial blood sampling for determination of the actual metabolite-corrected arterial input function to the brain, the kinetic values obtained by modeling the uptake of the tracer into the brain using the heart-derived IDIF are likely underestimations of the true values. This is because although the IDIFs obtained from the image processing were corrected for spillover, we did not account for the metabolites which would include [18F]-fluoride that were present in the blood which we observed by Folch extraction. The significant portion of the total radioactivity found in the aqueous fraction after Folch extraction suggests that the parent tracer is rapidly metabolized into water-soluble by-products in vivo, much like the [14C]-AA [27] or [11C]-AA counterpart [12]. We did not perform further analysis of the organic fraction from the blood or brain extract, but a previous study by Nagatsugi et al. with the same tracer in rats showed that a sizeable portion of the tracer in the brain was incorporated into phospholipids after 30 min [28]. This will affect the model used to estimate the amount of free tracer in the blood. Thus, the true concentrations of the free and unmetabolized tracer are lower than in the corrected IDIFs and cannot be ruled out. There is also the possibility that the observed difference in the K* values we obtained and those reported for 1-[11C]-arachidonic acid is due to the differential uptake of unmodified AA and the fluorinated compound due to modification at the ω-position which may affect recognition by fatty acid transporters. In PET studies of glucose uptake using 2-deoxy-2-[18F]fluoroglucose (FDG), the uptake of d-glucose is inferred from the observed metabolic rate for FDG using the lumped constant [29]. While the metabolism of [18F]-FAA has been investigated previously, K* had not been estimated in the in vivo study [13]. In the absence of metabolic information (such as the percent of parent fraction and the lumped constant), it may still however be possible to monitor relative differences in the K* based on genotype or treatments that would alter the uptake of PUFAs using only spillover corrected IDIFs.
The ApoE4 variant is proposed as a major genetic risk factor for AD due to structural changes resulting in different functional abilities for the binding and transportation of cholesterol and phospholipids [30, 31]. These isoform-specific changes in lipid metabolism show an imbalance in AA/DHA levels which may be related to the pathological effects seen in AD. Utilization of an improved [18F]-FAA synthesis can expand on current PET imaging capabilities to accurately assess AA metabolism in healthy subjects as well as evaluate imbalances in AA levels found in AD patients with the ApoE4 variant. Monitoring the uptake of the [18F]-FAA tracer in the brain has the potential to be used as a biomarker of neuroinflammation in AD before the onset of cognitive symptoms.
In summary, we have synthesized [18F]-FAA with high specific activity and radiochemical purity. The tracer behaves similarly to AA based on serum stability and in vitro studies. We validated an increase in brain uptake of [18F]-FAA in C57BL/6J mice after DOI injection, and our preliminary in vivo studies in ApoE4-TR mice showed a time-dependent increase in brain uptake which was used to calculate K* by three different methods, allowing us to estimate the K* of AA. The use of the corrected IDIF to calculate K* also affords a translatable method to evaluate uptake kinetics in other organisms. Measuring the differential uptake of AA by PET will allow for further insight into the effect of ApoE isoform on neuroinflammation and neurodegenerative diseases, identifying and selecting participants with increased brain AA uptake to guide interventions, such as cPLA2 inhibitors, targeting neuroinflammation.
Supplementary Material
Highlights.
An increase in brain arachidonic acid (AA) uptake is a marker of calcium dependent phospholipase A2 activation and neuroinflammation.
A novel translational synthesis approach of 20-[18F]-fluoroarachidonic acid ([18F]-FAA) for PET imaging is presented.
[18F]-FAA showed bioequivalent signaling properties to AA in cells.
The brain incorporation coefficient (K*) of [18F]-FAA was estimated via multiple methods in ApoE4 targeted replacement mice using the image derived input function.
The application of [18F]-FAA PET imaging to humans has relevance to identify and guide the effectiveness of treatments focused on neuroinflammation in neurodegenerative diseases.
Funding
HNY is supported by RF1AG076124, RF1AG078362, R01AG067063, R01AG054434, R01AG055770, R21AG056518, and P30AG066530 from the National Institute on Aging, GC-201711-2014197 from the Alzheimer’s Drug Discovery Foundation (ADDF), and generous donations from the Vranos and Tiny Foundations and from Ms. Lynne Nauss. KC is supported by the Department of Radiology at the University of Southern California.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
The Supplementary Material for this article can be found online at: https://supplementary-material
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Southern California.
Declaration of Competing Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
References
- [1].Sanchez-Mejia RO, Mucke L, Phospholipase A2 and arachidonic acid in Alzheimer’s disease, Biochim Biophys Acta, 1801 (2010) 784–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yassine HN, Finch CE, APOE alleles and diet in brain aging and Alzheimer’s disease, Frontiers in Aging Neuroscience, 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Abdullah L, Evans JE, Emmerich T, Crynen G, Shackleton B, Keegan AP, Luis C, Tai L, LaDu MJ, Mullan M, Crawford F, Bachmeier C, APOE ε4 specific imbalance of arachidonic acid and docosahexaenoic acid in serum phospholipids identifies individuals with preclinical mild cognitive ompairment/Alzheimer’s disease, Aging (Albany NY), 9 (2017) 964–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Duro MV, Ebright B, Yassine HN, Lipids and brain inflammation in APOE4-associated dementia, Curr Opin Lipidol, 33 (2022) 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zimmer ER, Leuzy A, Benedet AL, Breitner J, Gauthier S, Rosa-Neto P, Tracking neuroinflammation in Alzheimer’s disease: the role of positron emission tomography imaging, Journal of neuroinflammation, 11 (2014) 120–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Di Paolo G, Kim TW, Linking lipids to Alzheimer’s disease: cholesterol and beyond, Nat Rev Neurosci, 12 (2011) 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Patrick RP, Role of phosphatidylcholine-DHA in preventing APOE4-associated Alzheimer’s disease, Faseb j, 33 (2019) 1554–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yassine HN, Rawat V, Mack WJ, Quinn JF, Yurko-Mauro K, Bailey-Hall E, Aisen PS, Chui HC, Schneider LS, The effect of APOE genotype on the delivery of DHA to cerebrospinal fluid in Alzheimer’s disease, Alzheimer’s research & therapy, 8 (2016) 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ebright B, Assante I, Poblete RA, Wang S, Duro MV, Bennett DA, Arvanitakis Z, Louie SG, Yassine HN, Eicosanoid lipidome activation in post-mortem brain tissues of individuals with APOE4 and Alzheimer’s dementia, Alzheimer’s Research & Therapy, 14 (2022) 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang S, Li B, Solomon V, Fonteh A, Rapoport SI, Bennett DA, Arvanitakis Z, Chui HC, Sullivan PM, Yassine HN, Calcium-dependent cytosolic phospholipase A2 activation is implicated in neuroinflammation and oxidative stress associated with ApoE4, Molecular Neurodegeneration, 17 (2022) 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tomaszewski N, He X, Solomon V, Lee M, Mack WJ, Quinn JF, Braskie MN, Yassine HN, Effect of APOE genotype on plasma docosahexaenoic acid (DHA), eicosapentaenoic acid, arachidonic acid, and hippocampal volume in the Alzheimer’s disease cooperative study-sponsored DHA clinical trial, J Alzheimers Dis, 74 (2020) 975–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Giovacchini G, Chang MC, Channing MA, Toczek M, Mason A, Bokde AL, Connolly C, Vuong BK, Ma Y, Der MG, Doudet DJ, Herscovitch P, Eckelman WC, Rapoport SI, Carson RE, Brain incorporation of [11C]arachidonic acid in young healthy humans measured with positron emission tomography, J Cereb Blood Flow Metab, 22 (2002) 1453–1462. [DOI] [PubMed] [Google Scholar]
- [13].Pichika R, Taha AY, Gao F, Kotta K, Cheon Y, Chang L, Kiesewetter D, Rapoport SI, Eckelman WC, The synthesis and in vivo pharmacokinetics of fluorinated arachidonic acid: implications for imaging neuroinflammation, J Nucl Med, 53 (2012) 1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Esposito G, Giovacchini G, Liow JS, Bhattacharjee AK, Greenstein D, Schapiro M, Hallett M, Herscovitch P, Eckelman WC, Carson RE, Rapoport SI, Imaging neuroinflammation in Alzheimer’s disease with radiolabeled arachidonic acid and PET, J Nucl Med, 49 (2008) 1414–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nagatsugi F, Hokazono J, Sasaki S, Maeda M, Synthesis of 20-[18F] fluoroarachidonic acid: A potential phospholipid metabolic agent, Journal of Labelled Compounds and Radiopharmaceuticals, 34 (1994) 1121–1127. [Google Scholar]
- [16].DeGrado TR, Bhattacharyya F, Pandey MK, Belanger AP, Wang S, Synthesis and preliminary evaluation of 18F-dluoro-4-thia-oleate as a PET probe of fatty acid oxidation, Journal of Nuclear Medicine, 51 (2010) 1310. [DOI] [PubMed] [Google Scholar]
- [17].Zhou S, Chen K, Reiman EM, Li DM, Shan B, A method for generating image-derived input function in quantitative 18F-FDG PET study based on the monotonicity of the input and output function curve, Nucl Med Commun, 33 (2012) 362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].DeGeorge JJ, Noronha JG, Bell J, Robinson P, Rapoport SI, Intravenous injection of [1-14C]arachidonate to examine regional brain lipid metabolism in unanesthetized rats, Journal of Neuroscience Research, 24 (1989) 413–423. [DOI] [PubMed] [Google Scholar]
- [19].Patlak CS, Blasberg RG, Fenstermacher JD, Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data, J Cereb Blood Flow Metab, 3 (1983) 1–7. [DOI] [PubMed] [Google Scholar]
- [20].Hwang SH, Wagner K, Xu J, Yang J, Li X, Cao Z, Morisseau C, Lee KS, Hammock BD, Chemical synthesis and biological evaluation of ω-hydroxy polyunsaturated fatty acids, Bioorg Med Chem Lett, 27 (2017) 620–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hii CST, Huang ZH, Bilney A, Costabile M, Murray AW, Rathjen DA, Der CJ, Ferrante A, Stimulation of p38 ohosphorylation and activity by arachidonic acid in HeLa cells, HL60 promyelocytic leukemic cells, and human neutrophils: Evidence for cell type-specific activation of mitogen-activated protein kinases, Journal of Biological Chemistry, 273 (1998) 19277–19282. [DOI] [PubMed] [Google Scholar]
- [22].Evans J, Ko Y, Mata W, Saquib M, Eldridge J, Cohen-Gadol A, Leaver HA, Wang S, Rizzo MT, Arachidonic acid induces brain endothelial cell apoptosis via p38-MAPK and intracellular calcium signaling, Microvascular Research, 98 (2015) 145–158. [DOI] [PubMed] [Google Scholar]
- [23].Basselin M, Fox MA, Chang L, Bell JM, Greenstein D, Chen M, Murphy DL, Rapoport SI, Imaging elevated brain arachidonic acid signaling in unanesthetized serotonin transporter (5-HTT)-deficient mice, Neuropsychopharmacology, 34 (2009) 1695–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rapoport SI, In vivo fatty acid incorporation into brain phosholipids in relation to plasma availability, signal transduction and membrane remodeling, Journal of Molecular Neuroscience, 16 (2001) 243–261. [DOI] [PubMed] [Google Scholar]
- [25].Basselin M, Villacreses NE, Lee HJ, Bell JM, Rapoport SI, Chronic lithium administration attenuates up-regulated brain arachidonic acid metabolism in a rat model of neuroinflammation, J Neurochem, 102 (2007) 761–772. [DOI] [PubMed] [Google Scholar]
- [26].Pathak AP, Kim E, Zhang J, Jones MV, Three-dimensional imaging of the mouse neurovasculature with magnetic resonance microscopy, PLoS One, 6 (2011) e22643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Murphy EJ, Rosenberger TA, Patrick CB, Rapoport SI, Intravenously injected [1-14C]arachidonic acid targets phospholipids, and [1-14C]palmitic acid targets neutral lipids in hearts of awake rats, Lipids, 35 (2000) 891–898. [DOI] [PubMed] [Google Scholar]
- [28].Nagatsugi F, Hokazono J, Sasaki S, Maeda M, 20-[18F]Fluoroarachidonic acid: tissue biodistribution and incorporation into phospholipids, Biological & Pharmaceutical Bulletin, 19 (1996) 1316–1321. [DOI] [PubMed] [Google Scholar]
- [29].Graham MM, Muzi M, Spence AM, O’Sullivan F, Lewellen TK, Link JM, Krohn KA, The FDG lumped constant in normal human brain, Journal of Nuclear Medicine, 43 (2002) 1157–1166. [PubMed] [Google Scholar]
- [30].Yassine HN, Croteau E, Rawat V, Hibbeln JR, Rapoport SI, Cunnane SC, Umhau JC, DHA brain uptake and APOE4 status: a PET study with [1-11C]-DHA, Alzheimer’s research & therapy, 9 (2017) 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lanfranco MF, Ng CA, Rebeck GW, ApoE lipidation as a therapeutic target in Alzheimer’s disease, International journal of molecular sciences, 21 (2020) 6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.