

Abstract
A Candida albicans mutant with mutations in the N-acetylglucosamine (GlcNAc) catabolic pathway gene cluster, including the GlcNAc-6-phosphate deacetylase (DAC1), glucosamine-6-phosphate deaminase (NAG1), and GlcNAc kinase (HXK1) genes, was not able to grow on amino sugars, exhibited highly attenuated virulence in a murine systemic candidiasis model, and was less adherent to human buccal epithelial cells in vitro. No germ tubes were formed by the mutant after induction with GlcNAc, but the mutant exhibited hyperfilamentation under stress-induced filamentation conditions. In addition, the GlcNAc catabolic pathway played a vital role in determining the colony phenotype. Our results imply that this pathway is very important because of its diverse links with pathways involved in virulence and morphogenesis of the organism.
Candida albicans, which causes life-threatening superficial and systemic candidiasis in immunocompromised hosts, can invade and colonize human mucosal surfaces rich in amino sugars (29, 44). The unique ability of the pathogenic Candida species to utilize amino sugars like N-acetylglucosamine (GlcNAc) and glucosamine (GlcN) as alternate carbon sources (37) led us to surmise that there is a correlation between such specific adaptation and the virulence of the organism. Besides inducing enzymes of the catabolic pathway, GlcNAc can also induce cellular morphogenesis in C. albicans, and dimorphism is suspected to be an important aspect of manifestation of infection (21, 24). The amino sugar catabolic pathway in C. albicans was elucidated previously (3, 27, 31, 36), and the terminal enzyme of the GlcNAc catabolic pathway, glucosamine-6-phosphate deaminase (encoded by NAG1), was cloned in our laboratory (22, 28) in order to investigate the importance of this pathway. Here we describe disruption of the GlcNAc catabolic pathway and establish direct correlations of this pathway with virulence, adhesion, and morphogenesis of C. albicans. All animal experiments in this study were performed in accordance with the rules of the Institutional Animals Ethics Committee (JNU-IAEC code 9/1999).
Both copies of NAG1 were disrupted in Ura− C. albicans wild-type strain CAF3-1 (13) by the Ura-blaster technique (13). The targeting construct for disruption was made in parental genomic clone pED4 (22), in which the 1.94-kb NcoI fragment housing the NAG1 open reading frame, part of the bidirectional promoter (22), and the downstream region was replaced with the 4.01-kb BamHI-BglII fragment from pCUB6 (13) containing the hisG-URA3-hisG cassette (Fig. 1A). CAF3-1 was transformed by the spheroplast method (39) with the 5.97-kb SalI fragment derived from the targeting construct. Transformants were selected on synthetic minimal medium (SD medium) plates (0.67% yeast nitrogen base [YNB] without amino acids, 2% dextrose, 2% agar) to obtain Ura+ transformants. After confirmation of disruption by Southern analysis (Fig. 1B), a Ura+ transformant (N-2) was screened for Ura-cured segregants on 5-FOA plates (4). A Ura− mutant (N-2-1) (Fig. 1B) was transformed with the 5.97-kb SalI fragment to disrupt the second copy of NAG1 by using a similar process in order to generate the homozygous mutants N-2-1-6 (Ura+) and N-2-1-6-1 (Ura−). For Southern analysis of genotypes, genomic DNAs isolated (18) from the transformants were digested with SalI (1 to 2 μg of DNA), and hybridization was performed at 42°C by using a 32P-labeled 3.91-kb SalI fragment from pED4 as the probe. The genotypes of all of the mutants generated in this study are shown in Table 1.
FIG. 1.

Disruption of GlcNAc catabolic pathway gene cluster in C. albicans. (A) Schematic diagram showing the genomic organization of the DAC1NAG1HXK1 gene cluster in the 3.91-kb SalI fragment of pED4. The restriction sites are indicated as follows: S, SalI; N, NcoI; BH, BamHI; Bg, BglII. The 1.94-kb NcoI fragment was replaced by the 4.01-kb hisG-URA3-hisG cassette from pCUB6. The resultant 5.97-kb SalI fragment was used as the disruption cassette. (B) Southern blot of SalI-digested genomic DNA from wild-type and mutant derivatives, showing sequential disruption of both copies of the cluster for wild-type strain CAF3-1 (DAC1NAG1HXK1/DAC1NAG1HXK1 ura3), N-2 (DAC1NAG1HXK1/dac1nag1hxk1 URA3), N-2-1 (DAC1NAG1HXK1/dac1nag1hxk1 ura3), N-2-1-6 (dac1nag1hxk1/dac1nag1hxk1 URA3), and N-2-1-6-1 (dac1nag1hxk1/dac1nag1hxk1 ura3). When the first allele of DAC1NAG1HXK1 was disrupted, the 3.91-kb SalI fragment of the undisrupted allele gave rise to a 5.97-kb SalI fragment as a result of integration of the disruption cassette by homologous recombination, as seen in N-2. Curing of URA3 resulted in loss of the hisG-URA3 fragment and in a smaller, 3-kb SalI fragment, as seen in N-2-1. Integration of the 5.97-kb SalI disruption cassette in N-2-1 resulted in two SalI fragments, which were 5.97 and 3 kb long, as seen in N-2-1-6. Curing of URA3 in this homozygous mutant resulted in two 3-kb SalI fragments. (C) hisG-URA3-hisG cassette inserted at the NcoI site downstream of NAG1, shown in construct P-33. (D) Southern blot of SalI-digested genomic DNA of N-2-1-6-1+P-33 (DAC1NAG1hxk1/dac1nag1hxk1 URA3). The 7.9-kb SalI cassette of P-33 was integrated into N-2-1-6-1. (E) Southern blot of SalI-digested genomic DNA of final revertant P-4. The 3.91-kb SalI fragment of pED4 was integrated into N-2-1-6. In panels B, D, and E, the 3.91-kb SalI fragment from pED4 was the probe. In panels D and E, the 3.91-kb SalI fragment from wild-type CAF3-1 was the marker. (F) Growth of wild type and mutants on glucose and amino sugars at 30°C. Sector 1, SC5314 (wild type, Ura+); sector 2, N-2 (heterozygous mutant); sector 3, N-2-1-6 (homozygous mutant); sector 4, N-2-1-6-1+P-33 (hxk1 mutant); sector 5, P-4 (heterozygous revertant). Note that the hxk1 mutant was not able to grow on GlcN.
TABLE 1.
C. albicans strains used in this work
| Strain | Parent | Genotype | Source or reference |
|---|---|---|---|
| SC5314 (wild type, Ura+) | URA3/URA3 | 16 | |
| CAF3-1 (wild type, Ura−) | CAF2-1 | Δura3::imm434/Δura3::imm434 | 13 |
| N-2 (heterozygous mutant, Ura+, DAC1NAG1HXK1/dac1nag1hxk1) | CAF3-1 | DAC1NAG1HXK1/Δdac1-proΔnag1Δhxk1::hisG-URA3-hisGΔura3/Δura3 | This study |
| N-2-1 (heterozygous mutant, Ura−) | N-2 | DAC1NAG1HXK1/Δdac1-proΔnag1Δhxk1::hisG Δura3/Δura3 | This study |
| N-2-1-6 (homozygous mutant, Ura+, dac1nag1hxk1/dac1nag1hxk1) | N-2-1 | Δdac1-proΔnag1Δhxk1::hisG/Δdac1-proΔnag1Δhxk1::hisG-URA3-hisGΔura3/ Δura3 | This study |
| N-2-1-6-1 (homozygous mutant, Ura−) | N-2-1-6 | Δdac1-proΔnag1Δhxk1::hisG/Δdac1-proΔnag1Δhxk1::hisG Δura3/Δura3 | This study |
| N-2-1-6-1+P-33 (hexokinase mutant, Ura+, dac1nag1hxk1/DAC1NAG1hxk1) | N-2-1-6-1 | Δdac1-proΔnag1Δhxk1::hisG/DAC1NAG1Δhxk1::hisG-URA3-hisG Δura3/Δura3 | This study |
| P-4 (revertant, heterozygous, Ura+, dac1nag1hxk1/DAC1NAG1HXK1) | N-2-1-6 | Δdac1-proΔnag1Δhxk1::hisG-URA3-hisG/DAC1NAG1HXK1 Δura3/Δura3 | This study |
When homozygous mutant N-2-1-6 was checked for growth on glucose (SD medium), GlcNAc (0.67% YNB, 2% GlcNAc, 2% agar), and GlcN (0.67% YNB, 2% GlcN, 2% agar) at 30°C, it was not able to grow on amino sugars (Fig. 1F), while no growth defect was observed in the case of the Ura+ parental control (SC5314) (16) and heterozygous mutant (N-2) strains. Growth at 37°C exhibited a similar pattern (data not shown).
In the process of NAG1 disruption, the GlcNAc-6-phosphate deacetylase (DAC1) gene was also functionally impaired because it shares a bidirectional promoter with NAG1 (Fig. 1A). In order to create a revertant, a construct was made by inserting the hisG-URA3-hisG cassette at the NcoI site downstream of NAG1 in pED4 (Fig. 1C). The 7.9-kb SalI fragment from recombinant P-33 was integrated into the genome of homozygous mutant N-2-1-6-1 by using the spheroplast method of transformation, and it was checked by Southern analysis (Fig. 1D). The 3.91-kb SalI fragment from pED4 was used as the probe. The revertant generated, N-2-1-6-1+P-33, with functional DAC1 and NAG1 genes, failed to restore growth on GlcNAc (Fig. 1F), indicating that the region downstream of NAG1 contained a gene important for catabolism. A BLAST homology search of the National Center for Biotechnology Information website disclosed the presence of a hexokinase (HXK1) gene in the cluster along with DAC1 and NAG1 (22). Although clusters of functionally related genes are less prevalent in eukaryotes, it has often been reported that genes for dispensable metabolic pathways in fungi are organized in clusters. Our data establish, for the first time, that there is a gene cluster in C. albicans (22). The inability of the hxk1 mutant (N-2-1-6-1+P-33) to grow on GlcNAc suggests that HXK1 is the GlcNAc kinase gene. This mutant surprisingly did not grow on GlcN. It has been hypothesized that GlcN is phosphorylated by a different kinase (43), but the failure of the hxk1 mutant to grow on GlcN suggests that the same kinase is responsible for phosphorylation of both GlcNAc and GlcN. The inability of the homozygous mutant to grow on GlcNAc and GlcN also established that this is the sole pathway for utilization of amino sugars. To restore function, revertant P-4, which was heterozygous for the DAC1, NAG1, and HXK1 genes, was created by integrating the SalI fragment from pED4 into the genome of N-2-1-6. Transformants were selected on GlcNAc plates, and integration was confirmed by Southern analysis (Fig. 1E). P-4 restored growth on amino sugars (Fig. 1F).
The GlcNAc catabolic pathway of C. albicans is very similar to that of Escherichia coli, in which both GlcNAc and GlcN can induce the Nag regulon (30). Besides E. coli, utilization of GlcNAc has also been reported for other pathogenic bacteria, such as Klebsiella pneumoniae and Vibrio cholerae. Therefore, development of the amino sugar catabolic pathway during evolution could be a common feature of many pathogens.
In addition to its role as a carbon and nitrogen source, GlcNAc can induce cellular morphogenesis in C. albicans (34). After induction with 2.5 mM GlcNAc at 37°C in salt base (0.335% YNB, 0.45% NaCl) (38), homozygous mutant N-2-1-6 stayed in the yeast form, and there was a total lack of formation of germ tubes; in contrast, wild-type strain SC5314 formed profuse germ tubes (Fig. 2). Heterozygous mutant N-2 and revertant P-4 exhibited no defect in germ tube formation and formed elongated germ tubes similar to those of SC5314 (Fig. 2). Formation of germ tubes is accompanied by heavy aggregation of cells (34), but unlike the wild-type, heterozygous mutant, and revertant strains, the homozygous mutant failed to form aggregates after induction with GlcNAc, as determined visually (data not shown).
FIG. 2.

Morphology of GlcNAc catabolic pathway mutants under different hypha-inducing conditions. Wild-type (SC5314), heterozygous mutant (N-2), homozygous mutant (N-2-1-6), and heterozygous revertant (P-4) strains were induced for filamentation under different conditions. After induction with 2.5 mM GlcNAc, the homozygous mutant exhibited a complete lack of germ tube formation, while SC5314, N-2, and P-4 formed germ tubes. N-2-1-6 was hyperfilamentous on SLAD and Spider medium plates. A novel colony morphology displayed by the homozygous mutant on a Spider medium plate is shown. N-2 and P-4, which are heterozygous for the catabolic pathway genes, failed to exhibit an intermediate phenotype. The original magnifications are indicated.
Since transport of GlcNAc inside cells is not necessary for germ tube induction (35), the total lack of germ tube formation by the mutant is an interesting phenomenon. We hypothesize that disruption of the pathway probably disturbed the cell surface receptor(s) responsible for reception or transmission of signals. It would be interesting to identify the link between the catabolic pathway and cellular signaling.
It has been suspected for a long time that dimorphism is a mechanism of virulence (21). The effect of the disruption on colony morphology was studied by using previously described media, such as SLAD (17) and Spider medium (23). Cells of SC5314, N-2, N-2-1-6, and P-4 were grown in SD medium at 30°C for 2 days, counted with a hemocytometer, plated on SLAD plates (0.17% YNB without amino acids and ammonium sulfate, 2% dextrose, 2% Bacto Agar [Difco], 50 μM ammonium sulfate) at a concentration of 80 to 100 cells per plate, and incubated at 37°C for 10 days. For induction on Spider medium plates (1% nutrient broth, 1% mannitol, 0.2% K2HPO4, 1.35% Bacto Agar; pH 7.2 after autoclaving), the cells were grown in Spider medium for 5 days at 30°C, counted, plated at a concentration of 80 to 100 cells per plate, and incubated at 37°C for 7 days.
Homozygous mutant N-2-1-6 exhibited a novel pattern of filamentation consisting of very extensive ramified filaments on SLAD plates (Fig. 2). A dramatic change in colony phenotype was observed on Spider medium plates that had extensive filamentation; there was an unusual irregular wrinkled colony surface consisting of entangled hyphae (Fig. 2). The hyperfilamentation displayed by N-2-1-6 on SLAD and Spider medium plates established, for the first time, the role of the GlcNAc signaling pathway in morphogenesis of C. albicans under stress-induced conditions. However, heterozygous mutant N-2 and revertant P-4 did not produce an intermediate filamentation pattern, and they exhibited a highly compromised state of filamentation, suggesting that gene dosage is important for morphogenesis of C. albicans under certain inducing conditions, a phenomenon that was observed in previous studies (14).
Hyperfilamentation could be the result of derepression of a repressor like RBF1 (20) or TUP1 (5); mutants with mutations in these genes exhibit excessive filamentation. Alternatively, the pathway could act coordinately with components of the mating hyphal mitogen-activated protein kinase pathway (11, 25), with Efg1p, an essential regulator of filamentation that acts downstream of the Ras-cAMP-Tpk2p pathway (24, 38, 40), or with other pathway components involved in morphogenesis and virulence (8). In addition, the GlcNAc catabolic pathway also plays a role in determining colony surface morphology that could be mediated by one of the signaling pathways mentioned above.
To investigate the role of the GlcNAc catabolic pathway in the virulence of C. albicans, hematogenously disseminated candidiasis was studied in the BALB/c mouse model (1, 9, 11). Female mice (age, 8 to 10 weeks; body weight, 18 to 20 g) were used in groups of eight per dose per strain. Wild-type (SC5314), homozygous mutant (N-2-1-6), and revertant (P-4) cells were grown on YPD (1% yeast extract, 2% peptone, 2% dextrose) plates at 30°C for 48 h, washed twice with phosphate-buffered saline (PBS), and counted, and an aliquot plated on YPD was grown overnight to determine the number of CFU. Doses containing 106 CFU per 200 μl of PBS were prepared, and mice were injected intravenously through the lateral tail vein. When the homozygous mutant was used, none of the mice died by day 25, but when the wild type was used, all of the mice died by day 11. The virulence of the revertant was less than that of the wild-type strain, and 25% of the mice survived to the end of the experiment (Fig. 3A). The doses of the homozygous mutant used in the experiments and the length of the period for which survival of the mice was monitored indicated that the virulence of the homozygous mutant was highly attenuated.
FIG. 3.

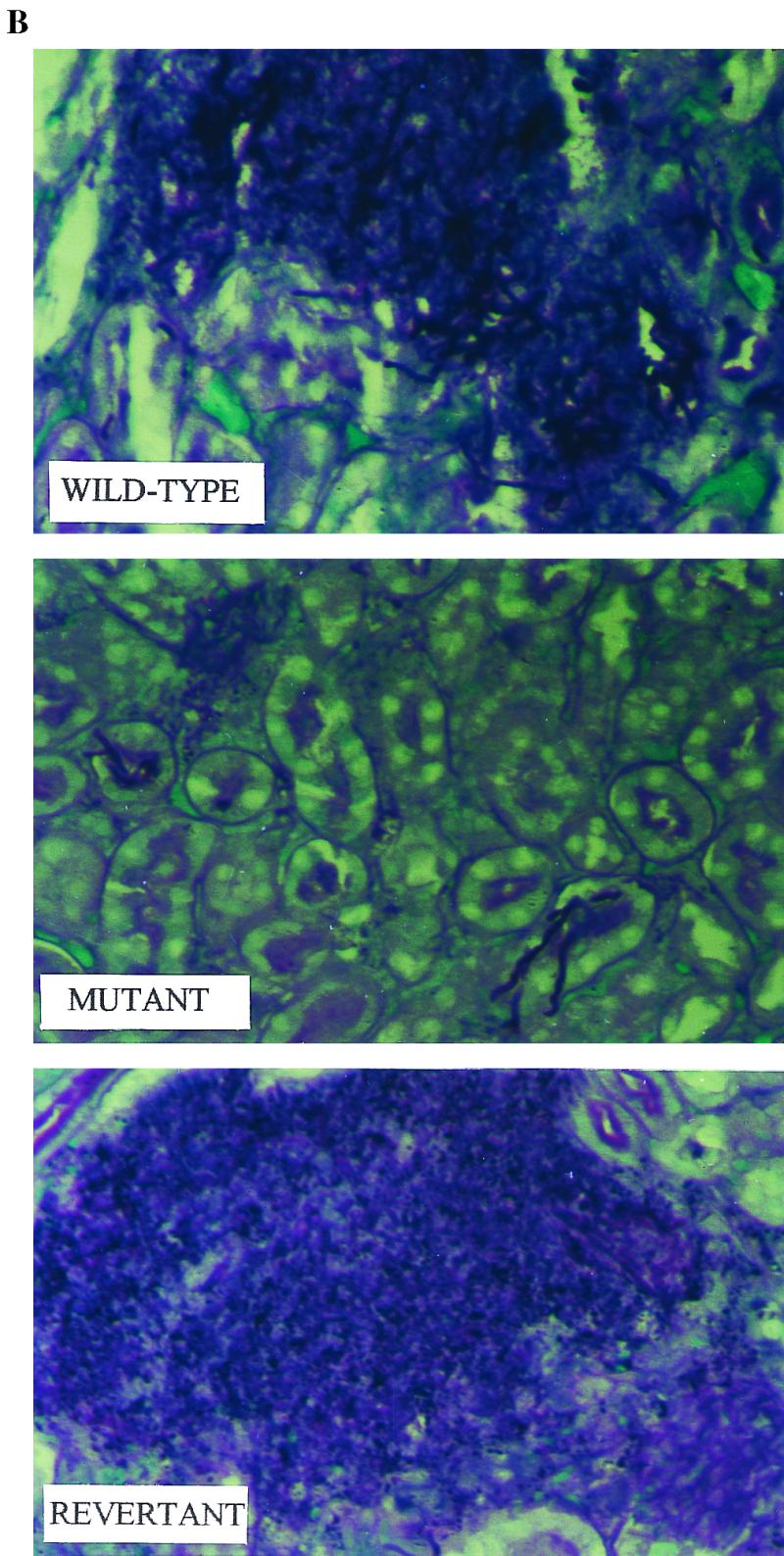
Disruption of the GlcNAc catabolic pathway results in loss of virulence in the murine systemic candidiasis model. (A) When inocula consisting of 1 × 106 blastospores were used, all of the mice inoculated with the wild type died by day 11, 25% of the mice inoculated with the heterozygous revertant survived, and all of the mice inoculated with the homozygous mutant were still alive on day 25. A log rank test revealed that the overall differences in survival among the strains were statistically significant (P = 0.0014). Symbols: ●, wild-type strain SC5314; ∗, homozygous mutant N-2-1-6; □, revertant P-4. (B) Histology of kidneys 24 h postinfection. Mice were challenged with 106 blastospores of the SC5314 (wild-type), N-2-1-6 (homozygous mutant), and P-4 (revertant) strains. Sections (thickness, 6 μm) were stained with periodic acid-Schiff reagent. Kidneys that were infected with the wild-type and revertant strains were fully infested with fungus and had large necrotic areas, while few Candida cells were observed when the mutant was used. Note that the mutant was able to form hyphae. Magnification, ×400.
C. albicans in systemic infections is known to infect vital organs, and for some unexplained reason kidneys are the most conspicuously affected organ. The fungal loads recovered from mouse kidneys infected with wild-type, homozygous mutant, and revertant strains are shown in Table 2. Mice were divided into groups of four animals per strain per time point, and an inoculum consisting of 106 CFU in 200 μl of PBS was injected into the lateral tail vein of each mouse. Chloroformed mice were euthanatized by cervical dislocation after 24, 48, and 72 h. The kidneys were removed, weighed, and crushed in sterile mortars with sterile pestles in 5 ml of PBS. Dilutions were prepared, and aliquots were plated on YED-chloramphenicol plates (1% yeast extract, 2% dextrose, 2% agar, 5 μg of chloramphenicol per ml) and incubated at 30°C for 2 days. Colonies were counted, the numbers of CFU per gram of tissue were determined, and the values were expressed in terms of log10 CFU. The mice infected with wild-type and revertant strains had very high fungal loads in their kidneys 24 h postinfection, as shown by the high number of CFU that rapidly increased (Table 2). The mice inoculated with the homozygous mutant had a strikingly low fungal burden on the first day, and the number of organisms recovered after 72 h was lower (Table 2). The trend towards renal clearance observed in mice infected with the homozygous mutant clearly indicates that a functional GlcNAc catabolic pathway in C. albicans is essential for colonization of the target organs.
TABLE 2.
Fungal loads in kidneys
| Strain | Log10 CFU (mean ± SD)a
|
||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| SC5314 | 5.93 ± 0.07 | 6.10 ± 0.16 | 6.46 ± 0.06 |
| N-2-1-6 | 3.71 ± 0.13 | 3.99 ± 0.28 | 2.91 ± 0.13 |
| P-4 | 5.75 ± 0.33 | 5.96 ± 0.07 | 6.18 ± 0.13 |
Four samples per group per time point were taken.
To find out more about the behavior of the homozygous mutant in vivo, histopathological studies were conducted by using 106 CFU with groups of four mice per strain. Kidneys removed 24 h postinfection were fixed in 10% formaldehyde–PBS, sectioned (thickness, 6 μm) in paraffin blocks, and stained with periodic acid-Schiff reagent by using conventional procedures. The sections revealed huge focal collections of Candida cells in the case of wild-type and revertant strains (Fig. 3B). In contrast, the area infected by the mutant strain was very small, but the mutant formed mycelia like those of the wild type (Fig. 3B). From the results we determined that the loss of virulence was not due to a defect in filamentation.
The possibility that virulence was lost because of retarded growth of the homozygous mutant was ruled out by determining the growth rates of the wild-type, homozygous mutant, and revertant strains in YPD at 30°C, in 2× YPD at 37°C (24), and in serum at 37°C (in vitro) by measuring the rates of glucose consumption (15). No significant difference in the growth rates was observed in any case (data not shown).
The virulence of Candida species is closely correlated with the ability of the organisms to adhere to cells in vitro (7, 33). We speculated that the initial low numbers of fungus cells recovered from kidneys infected with the homozygous mutant could have been due to a lower ability of the mutant to adhere to endothelial cells in vivo, resulting in rapid clearance from the blood. We examined the abilities of SC5314, N-2-1-6, and P-4 to adhere to human buccal epithelial cells (HBEC) in vitro by performing a visual assay (2). The strains were grown on YPD plates at 30°C for 2 days and washed with 0.02 M PBS (0.02 M NaCl, 0.15 M Na2HPO4; pH 7.2), and the cells were counted. HBEC from three male and female volunteers (ages, 28 to 31 years) were washed with 0.02 M PBS and counted. A total of 107 Candida cells were incubated with 105 HBEC in 0.02 M PBS at 37°C for 1 h. The cells were then passed through 12-μm-pore-size filters obtained from SPI Supplies and washed with 0.02 M PBS. The contents of the filters were transferred to microscope slides and stained with crystal violet. The total number of Candida cells adhering to 100 HBEC was determined. The experiment was repeated three times, and a statistical analysis (standard error of the mean) was done with GraphPad Prism 2.01 software. The number of C. albicans cells that adhered per HBEC was considerably lower in the case of the homozygous mutant (42.7% reduction compared to the wild type) (Fig. 4), which partially accounts for the loss of virulence.
FIG. 4.

Adherence assay: average number of Candida cells adhering per HBEC when the SC5314 (wild-type), N-2-1-6 (homozygous mutant), and P-4 (revertant) strains were used. The data represent means ± standard errors of the means based on 100 HBEC and three independent experiments.
Our results demonstrate that C. albicans requires a functional GlcNAc catabolic pathway to become successfully established in systemic infections. We considered the many factors that are responsible for the virulence of this organism and examined the possible alterations that might have resulted in attenuation of virulence in a homozygous mutant. Although there is no compelling evidence that filamentation plays a definite role in virulence, it is widely believed that the two events are correlated, as many mutants defective in in vitro filamentation are also less virulent in systemic infections (11, 24). However, C. albicans cells recovered from lesions have both yeast and filamentous forms, suggesting that both forms are important for virulence. It has been observed that an efg1 cph1 double mutant that is not filamentous under most in vitro induction conditions and exhibits highly attenuated virulence in mouse systemic infections retains the ability to form filaments at sites of infection (24, 32). On the other hand, a cpp1 mutant is hyperfilamentous in vitro but is not hypervirulent; rather, it displays attenuated virulence in mouse systemic infections (10). Our studies show that a homozygous mutant with a mutation in the GlcNAc catabolic pathway is highly attenuated in terms of virulence, although there is no defect in in vivo filamentation. Whether filamentation plays an important role in virulence remains a moot point, but from our results it appears that the GlcNAc catabolic pathway may not be important for filamentation in serum. This was further shown by in vitro filamentation of the homozygous mutant in serum, which exhibited no defect (data not shown). There is no known interaction of this pathway with RAS1, which is important for filamentation in serum (12) and possibly under many other filament-inducing conditions. Filamentation may not be an important attribute in conferring virulence to the organism but could be a process that is regulated concurrently with many factors that play important roles in virulence.
The possibility that the GlcNAc catabolic pathway is important as a source of carbon in vivo can be ruled out because glucose is available to the cells in serum. In addition, a recent report showed that a GlcNAc concentration of less than 10 μM in serum was not sufficient to support growth of the organism (26). The importance of the GlcNAc catabolic pathway probably lies in the diverse link that it establishes with mechanisms involved in production of different virulent factors, which could be adhesins (41) or hydrolytic enzymes (19, 42). The possibility that there is a defect in cell wall or cell surface structure which leads to a loss of virulence should also be considered since GlcNAc is polymerized into chitin, an integral component of the cell wall (6). It would be very interesting to investigate the prospective routes of the GlcNAc catabolic pathway regulating virulence and morphogenetic signaling in C. albicans.
Acknowledgments
Praveen Singh and Sharmistha Ghosh contributed equally to this work.
We thank William A. Fonzi for providing plasmid pCUB6 and strains SC5314 and CAF3-1.
This work was supported by a grant from the Department of Biotechnology, India.
REFERENCES
- 1.Alonso-Monge R, Navarro-Garcia F, Molero G, Diez-Orejas R, Gustin M, Pla J, Sanchez M, Nombela C. Role of the mitogen-activated protein kinase Hog1p in morphogenesis and virulence of Candida albicans. J Bacteriol. 1999;181:3058–3068. doi: 10.1128/jb.181.10.3058-3068.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bailey A, Wadsworth E, Calderone R. Adherence of Candida albicans to human buccal epithelial cells: host-induced protein synthesis and signaling events. Infect Immun. 1995;63:569–572. doi: 10.1128/iai.63.2.569-572.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhattacharya A, Puri M, Datta A. Induction of N-acetylglucosamine kinase in yeast. Biochem J. 1974;141:593–595. doi: 10.1042/bj1410593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boeke J D, LaCroute F, Fink G R. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet. 1984;197:345–346. doi: 10.1007/BF00330984. [DOI] [PubMed] [Google Scholar]
- 5.Braun B R, Johnson A D. Control of filament formation in Candida albicans by the transcriptional repressor TUP1. Science. 1997;277:105–109. doi: 10.1126/science.277.5322.105. [DOI] [PubMed] [Google Scholar]
- 6.Braun P C, Calderone R A. Chitin synthesis in Candida albicans: comparison of yeast and hyphal forms. J Bacteriol. 1978;133:1472–1477. doi: 10.1128/jb.133.3.1472-1477.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calderone R A. Molecular interactions at the interface of Candida albicans and host cells. Arch Med Res. 1993;24:275–279. [PubMed] [Google Scholar]
- 8.Calera J A, Calderone R. Histidine kinase, two-component signal transduction proteins of Candida albicans and the pathogenesis of candidosis. Mycoses. 1999;42:49–53. [PubMed] [Google Scholar]
- 9.Calera J A, Zhao X J, Calderone R. Defective hyphal development and avirulence caused by a deletion of the SSK1 response regulator gene in Candida albicans. Infect Immun. 2000;68:518–525. doi: 10.1128/iai.68.2.518-525.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Csank C, Makris C, Meloche S, Schröppel K, Röllinghoff M, Dignard D, Thomas D Y, Whiteway M. Derepressed hyphal growth and reduced virulence in a VH1 family-related protein phosphatase mutant of the human pathogen Candida albicans. Mol Biol Cell. 1997;8:2539–2551. doi: 10.1091/mbc.8.12.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Csank C, Schröppel K, Leberer E, Harcus D, Mohamed O, Meloche S, Thomas D Y, Whiteway M. Roles of the Candida albicans mitogen-activated protein kinase homolog, Cek1p, in hyphal development and systemic candidiasis. Infect Immun. 1998;66:2713–2721. doi: 10.1128/iai.66.6.2713-2721.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng Q, Summers E, Guo B, Fink G. Ras signaling is required for serum-induced hyphal differentiation in Candida albicans. J Bacteriol. 1999;181:6339–6346. doi: 10.1128/jb.181.20.6339-6346.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fonzi W A, Irwin M Y. Isogenic strain construction and gene mapping in Candida albicans. Genetics. 1993;134:717–728. doi: 10.1093/genetics/134.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gale C A, Bendel C M, McClellan M, Hauser M, Becker J M, Berman J, Hostetter M K. Linkage of adhesion, filamentous growth, and virulence in Candida albicans to a single gene, INT1. Science. 1998;279:1355–1358. doi: 10.1126/science.279.5355.1355. [DOI] [PubMed] [Google Scholar]
- 15.Garrigues J C, Cadet de Fontenay G, Linas M D, Lagente M, Seguela J P. New in vitro assay based on glucose consumption for determining intraconazole and amphotericin B activities against Aspergillus fumigatus. Antimicrob Agents Chemother. 1994;38:2857–2862. doi: 10.1128/aac.38.12.2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gillum A M, Tsay E Y H, Kirsch D R. Isolation of the Candida albicans gene for orotidine-5′-phosphate decarboxylase by complementation of S. cerevisiae ura3 and E. coli pyrF mutations. Mol Gen Genet. 1984;198:179–182. doi: 10.1007/BF00328721. [DOI] [PubMed] [Google Scholar]
- 17.Gimeno C J, Ljungdahl P O, Styles C A, Fink G R. Unipolar cell divisions in the yeast S. cerevisiae lead to filamentous growth: regulation by starvation and RAS. Cell. 1992;68:1077–1090. doi: 10.1016/0092-8674(92)90079-r. [DOI] [PubMed] [Google Scholar]
- 18.Hoffman C S, Winston F. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene. 1987;57:267–272. doi: 10.1016/0378-1119(87)90131-4. [DOI] [PubMed] [Google Scholar]
- 19.Hube B, Monod M, Schofeild D A, Brown A J P, Gow N A R. Expression of seven members of gene family encoding secretory aspartyl proteinases in Candida albicans. Mol Microbiol. 1994;14:87–99. doi: 10.1111/j.1365-2958.1994.tb01269.x. [DOI] [PubMed] [Google Scholar]
- 20.Ishii N, Yamamoto M, Yoshihara F, Arisawa M, Aoki Y. Biochemical and genetic characterization of Rbf1p, a putative transcription factor of Candida albicans. Microbiology. 1997;143:429–435. doi: 10.1099/00221287-143-2-429. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi S D, Cutler J E. Candida albicans hyphal formation and virulence: is there a clearly defined role? Trends Microbiol. 1998;6:92–94. doi: 10.1016/s0966-842x(98)01218-9. [DOI] [PubMed] [Google Scholar]
- 22.Kumar M J, Jamaluddin M S, Natarajan K, Kaur D, Datta A. The inducible N-acetylglucosamine catabolic pathway gene cluster in Candida albicans: discrete N-acetylglucosamine-inducible factors interact at the promoter of NAG1. Proc Natl Acad Sci USA. 2000;97:14218–14223. doi: 10.1073/pnas.250452997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu H, Köhler J, Fink G R. Suppression of hyphal formation in Candida albicans by mutation of a STE12 homolog. Science. 1994;266:1723–1726. doi: 10.1126/science.7992058. [DOI] [PubMed] [Google Scholar]
- 24.Lo H-J, Köhler J R, DiDomenico B, Loebenberg D, Cacciapuoti A, Fink G R. Nonfilamentous C. albicans mutants are avirulent. Cell. 1997;90:939–949. doi: 10.1016/s0092-8674(00)80358-x. [DOI] [PubMed] [Google Scholar]
- 25.Malathi K, Ganesan K, Datta A. Identification of a putative transcription factor in Candida albicans that can complement the mating defect of Saccharomyces cerevisiae ste12 mutants. J Biol Chem. 1994;269:22945–22951. [PubMed] [Google Scholar]
- 26.Mio T, Kokado M, Arisawa M, Yamada-Okabe H. Reduced virulence of Candida albicans mutants lacking the GNA1 gene encoding glucosamine-6-phosphate acetyltransferase. Microbiology. 2000;146:1753–1758. doi: 10.1099/00221287-146-7-1753. [DOI] [PubMed] [Google Scholar]
- 27.Natarajan K, Rai Y P, Datta A. Induction of N-acetyl-d-glucosamine catabolic enzymes and germinative response in Candida albicans. Biochem Int. 1984;9:735–744. [PubMed] [Google Scholar]
- 28.Natarajan K, Datta A. Molecular cloning and analysis of the NAG1 cDNA coding for glucosamine-6-phosphate deaminase from Candida albicans. J Biol Chem. 1993;268:9206–9214. [PubMed] [Google Scholar]
- 29.Odds F C. Candida and candidosis: a review and bibliography. 2nd ed. London, United Kingdom: Bailliere Tindall; 1988. [Google Scholar]
- 30.Plumbridge J A. Induction of the nag regulon of Escherichia coli by N-acetylglucosamine and glucosamine: role of the cyclic AMP-catabolite activator protein complex in expression of the regulon. J Bacteriol. 1990;172:2728–2735. doi: 10.1128/jb.172.5.2728-2735.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rai Y P, Datta A. Induction of N-acetylglucosamine-6-phosphate deacetylase in yeast. Indian J Biochem Biophys. 1982;19:285–287. [PubMed] [Google Scholar]
- 32.Riggle P J, Andrutis K A, Chen X, Tzipori S R, Kumamoto C A. Invasive lesions containing filamentous forms produced by a Candida albicans mutant that is defective in filamentous growth in culture. Infect Immun. 1999;67:3649–3652. doi: 10.1128/iai.67.7.3649-3652.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rotrosen D, Calderone R A, Edwards J E., Jr Adherence of Candida species to host tissues and plastic surfaces. Rev Infect Dis. 1986;8:73–85. doi: 10.1093/clinids/8.1.73. [DOI] [PubMed] [Google Scholar]
- 34.Shepherd M G, Yin C Y, Ram S P, Sullivan P A. Germ tube induction in Candida albicans. Can J Microbiol. 1980;26:21–26. doi: 10.1139/m80-004. [DOI] [PubMed] [Google Scholar]
- 35.Shepherd M G, Sullivan P A. Candida albicans germ-tube formation with immobilized GlcNAc. FEMS Microbiol Lett. 1983;17:167–170. [Google Scholar]
- 36.Singh B, Datta A. Induction of N-acetylglucosamine-catabolic pathway in spheroplasts of Candida albicans. Biochem J. 1979;178:427–431. doi: 10.1042/bj1780427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh B, Datta A. Regulation of N-acetylglucosamine uptake in yeast. Biochim Biophys Acta. 1979;557:248–258. doi: 10.1016/0005-2736(79)90107-x. [DOI] [PubMed] [Google Scholar]
- 38.Sonneborn A, Bockmuhl D P, Gerads M, Kurpanek K, Sanglard D, Ernst J F. Protein kinase A encoded by TPK2 regulates dimorphism of Candida albicans. Mol Microbiol. 2000;35:386–396. doi: 10.1046/j.1365-2958.2000.01705.x. [DOI] [PubMed] [Google Scholar]
- 39.Srikantha T, Morrow B, Schroppel K, Soll D R. The frequency of integrative transformation at phase-specific genes of Candida albicans correlates with their transcriptional state. Mol Gen Genet. 1995;246:342–352. doi: 10.1007/BF00288607. [DOI] [PubMed] [Google Scholar]
- 40.Stoldt V R, Sonneborn A, Leuker C E, Ernst J F. Efg1p, an essential regulator of morphogenesis of the human pathogen Candida albicans, is a member of a conserved class of bHLH proteins regulating morphogenetic processes in fungi. EMBO J. 1997;16:1982–1991. doi: 10.1093/emboj/16.8.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sundstrom P. Adhesins in Candida albicans. Curr Opin Microbiol. 1999;2:353–357. doi: 10.1016/S1369-5274(99)80062-9. [DOI] [PubMed] [Google Scholar]
- 42.Takahashi M, Banno Y, Nozawa Y. Secreted Candida albicans phospholipase: purification and characterization of two forms of lysophospholipase-transacylase. J Med Vet Mycol. 1991;29:193–204. [PubMed] [Google Scholar]
- 43.Torosantucci A, Cassone A. Induction and morphogenesis of chlamydospores in an agerminative variant of Candida albicans. Sabouraudia. 1983;21:49–57. doi: 10.1080/00362178385380081. [DOI] [PubMed] [Google Scholar]
- 44.Wey S B, Mori M, Pfaller M A, Woolson R F, Wenzel R P. Risk factors for hospital-acquired candidemia. A matched case-control study. Arch Intern Med. 1989;149:2349–2353. [PubMed] [Google Scholar]