

Abstract
As a breakthrough strategy for cancer treatment, immunotherapy mainly consists of immune checkpoint inhibitors (ICIs) and other immunomodulatory drugs that provide a durable protective antitumor response by stimulating the immune system to fight cancer. However, due to the low response rate and unique toxicity profiles of immunotherapy, the strategies of combining immunotherapy with other therapies have attracted enormous attention. These combinations are designed to exert potent antitumor effects by regulating different processes in the cancer-immunity cycle. To date, immune-based combination therapy has achieved encouraging results in numerous clinical trials and has received Food and Drug Administration (FDA) approval for certain cancers with more studies underway. This review summarizes the emerging strategies of immune-based combination therapy, including combinations with another immunotherapeutic strategy, radiotherapy, chemotherapy, anti-angiogenic therapy, targeted therapy, bacterial therapy, and stroma-targeted therapy. Here, we highlight the rationale of immune-based combination therapy, the biomarkers and the clinical progress for these immune-based combination therapies.
Keywords: immunotherapy, immune checkpoint inhibitors, combination therapy, biomarkers, neoplasms
Introduction
Cancer treatment has progressed rapidly over the past 30 years, and has entered a new era of targeted therapy and immunotherapy, moving from traditional radiotherapy and chemotherapy. Especially for cancer immunotherapy, boosting the body’s immune system to fight cancer, was considered one of the top 10 breakthroughs in 2013 in the Science journal. Immunotherapies have exhibited remarkable and durable clinical efficacy in some refractory cancers and have been broadly applied across multiple cancer types.
However, given the complexity of the tumor immune microenvironment and the multifactorial mechanisms affecting the responses to immunotherapy, immunotherapy as monotherapy has not exhibited satisfactory efficacy in some clinical settings. Therefore, numerous studies have extensively investigated the interaction and crosstalk between immunotherapy and other therapies. Researchers are dedicated to improving the tolerance and response rate of immunologic agents, prolonging the response time, and strengthening the control of tumor progression through immune-based combination therapies.
In this article, we overview the emerging strategies of immune-based combination therapy, including combinations with another immunotherapeutic strategy, radiotherapy, chemotherapy, anti-angiogenic therapy, targeted therapy, bacterial therapy, and stroma-targeted therapy. We also highlight the rationale of immune-based combination therapy, the biomarkers and the clinical progress for these immune-based combination therapies.
Cancer-immunity cycle
The principle of immunotherapy is based on the cancer-immunity cycle, including the release and presentation of cancer cell antigens, the priming and activation of antigen-specific T cells, the transport and infiltration of T cells into tumors, and the recognition and killing of cancer cells by activated T cells [1]. Normally, this cycle is complete and continuous, and effectively defends against cancer cells without being restrained. However, immune evasion is a characteristic of malignant tumors. There are numerous mechanisms by which cancer cells evade immune recognition and avoid immune destruction, including poor immunogenicity of tumor antigens, immunosuppressive antigen presenting cells, poor T-cell priming, impaired T-cell trafficking, disordered neovascularization, low affinity for tumor antigens, and exhausted or dysfunctional T cells (Fig. 1). Once the immunity cycle is interrupted, cancer starts to occur and progress [1].
Fig. 1. Immune-based combination therapies convert cold tumors into hot tumors by regulating the cancer-immunity cycle.

Immunotherapy is based on the cancer-immunity cycle, including the release and presentation of tumor antigens, the priming and activation of T cells, the trafficking and infiltration of T cells into tumors, and the recognition and killing of tumor cells by activated T cells. However, multiple mechanisms lead to immune evasion. Numerous therapies, including chemotherapy, radiotherapy, targeted therapy, and anti-angiogenic therapy, all regulate the immune microenvironment and complement immunotherapy for stronger antitumor responses. TSA tumor-specific antigen, TLR toll-like receptors, DC dendritic cell, TKI tyrosine kinase inhibitor, CAR-T cell chimeric antigen receptor T cell, BsAbs bispecific antibodies, CAF cancer associated fibroblast, DAMPs damage-associated molecular patterns, APCs antigen presenting cells. Adapted from [1].
A comprehensive understanding of the mechanisms of the cancer-immunity cycle provides a theoretical basis for immunotherapy. In addition to immune checkpoint inhibitors (ICIs), other types of immunotherapies are also progressing rapidly, including cytokines, costimulatory receptor agonists, chimeric antigen receptor T (CAR-T) cells, cancer vaccines, oncolytic viruses, macrophage-targeted therapy, and agonists of the cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway, which play specific roles in different steps in the cancer-immunity cycle (Fig. 1) [2]. In addition, other conventional therapies, such as chemotherapy, radiotherapy, targeted therapy, and anti-angiogenic therapy, all participate in the regulation of the cancer-immunity cycle and synergistically promote antitumor responses when combined with immunotherapy (Fig. 1). Hence, immune-based combination therapies activate the immune system and strengthen antitumor responses to turn cold tumors into hot tumors, and this process deserves further study.
Immune checkpoint inhibitors
Immunotherapies mainly consist of ICIs, cytokines, costimulatory receptor agonists, adoptive T-cell transfer, oncolytic viruses, cancer vaccines, macrophage-targeted therapy, and cGAS-STING agonists, among which ICIs are the most widely used. Immune checkpoints are divided into costimulatory immune checkpoints and coinhibitory immune checkpoints. Inhibitory immune checkpoints are protective sites that inhibit immune overreactions and prevent autoimmune responses, and these checkpoints are often upregulated by tumors to cause immune evasion. Hence, immune checkpoint inhibitors (ICIs) promote the immune activation state and exert potent antitumor effects by blocking coinhibitory receptor-ligand interactions and rejuvenating dysfunctional or exhausted cytotoxic T cells (CTLs).
Currently, ICIs are mainly designed to target cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), programmed cell death protein 1/programmed cell death ligand 1 (PD-1/PD-L1), T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), lymphocyte activation gene-3 (LAG-3), T-cell immunoglobulin and ITIM domain (TIGIT), V-domain Ig suppressor of T-cell activation (VISTA), and B and T-cell lymphocyte attenuator (BTLA), among which PD-1/PD-L1 and CTLA-4 are the most widely used and prominent targets of immunotherapy [3]. To date, ICIs have exhibited remarkable and durable clinical efficacy in some refractory cancers, especially in melanoma. Nevertheless, many patients do not experience sufficiently effective or durable clinical benefits when treated with immunotherapy alone. Thus, it is urgent to explore the effectiveness of combination therapy.
Combination of two ICIs
Different immune checkpoints have specific spatiotemporal effects, such as CTLA-4 and PD-1/PD-L1 [4]. CTLA-4 is primarily involved in regulating T-cell activation in lymph nodes and inhibiting dendritic cell (DC) activity via regulatory T cells (Tregs), whereas PD-1 is mainly involved in inhibiting effector T-cell and natural killer (NK) cell activation and inducing Treg differentiation, typically in peripheral lymphatic tissues including tumor tissues. CTLA-4 suppresses T-cell proliferation at the initial stage of immune responses, whereas PD-1 inhibits preactivated T cells at a later stage of this response process [5]. Upon treatment, the anti-CTLA-4 antibody activates antigen-specific CD4+ T cells and clears Tregs in the tumor microenvironment (TME). Blocking PD-1/PD-L1 restores the cytotoxic function of CTLs inside the tumor [1]. Therefore, due to the different roles in immune responses, the combination of dual ICIs separately targeting CTLA-4 and PD-1/PD-L1 seems to represent a promising clinical treatment and has aroused extensive attention.
Indeed, the combinations of dual ICIs have acted synergistically against cancer in many clinical trials, leading to the FDA approval of these combinations in cancer treatment. For instance, the combination of ipilimumab (anti-CTLA-4) and nivolumab (anti-PD-1) has been approved by the FDA for the treatment of melanoma, renal cell carcinoma (RCC), colorectal cancer (CRC), hepatocellular carcinoma (HCC), non-small cell lung cancer (NSCLC), and malignant pleural mesothelioma (MPM) (Table 1). Melanoma was the first tumor approved for the treatment using a dual ICI combination. The latest data from the phase III CheckMate-067 study with a minimum follow-up of 6.5 years showed that nivolumab combined with ipilimumab resulted in a notably longer median overall survival (OS) (72.1 vs. 36.9 vs. 19.9 months), a longer median progression-free survival (PFS) (11.5 vs. 6.9 vs. 2.9 months), and increased the objective response rate (ORR) (58.3% vs. 44.9% vs. 19.0%) than nivolumab alone or ipilimumab alone [6]. Another phase III CheckMate-214 study was designed to compare the efficacy and safety of dual ICIs and sunitinib in the first line treatment of advanced clear-cell RCC [7]. The results showed that compared with sunitinib, the combination group showed an improved OS rate (75% vs. 60% at 18 months) and a higher ORR (42% vs. 27%). In another phase II CheckMate-142 study, the combination of ipilimumab and nivolumab showed robust and durable clinical benefit in metastatic microsatellite instability high (MSI-H)/deficient DNA mismatch repair (dMMR) CRC patients [8, 9]. Clinical trials combining anti-CLTA-4 and anti-PD-1 antibodies in more tumor types are actively underway. However, dual ICI therapy also has problems that need to be overcome, including an increased incidence of immune-related adverse events (irAEs) (Table 1), inability to delay early progression, and high cost.
Table 1.
Key clinical trials of the combination of different ICIs.
| Study | Indication | Phase | Treatment (n) | Efficacy endpoints | Grade 3/4 AEs | Reference | ||
|---|---|---|---|---|---|---|---|---|
| RR | PFS | OS | ||||||
| CheckMate-227 (NCT02477826) | NSCLC | 3 | Ipilimumab + nivolumab (583) | 35.9% | / | 17.1 m | 32.8% | [235] |
| Chemotherapy (583) | 30.0% | / | 13.9 m | 36.0% | ||||
| CheckMate-214 (NCT02231749) | RCC | 3 | Ipilimumab + nivolumab (550) | 42% | 11.6 m | 18-month OS rate 75% vs. 60% | 46% | [7] |
| Sunitinib (546) | 27% | 8.4 m | 63% | |||||
| CheckMate-067 (NCT01844505) | Melanoma | 3 | Nivolumab + ipilimumab (314) | 58.3% | 11.5 m | 72.1 | 59% | [6] |
| Nivolumab (316) | 44.9% | 6.9 m | 36.9 | 24% | ||||
| Ipilimumab (315) | 19.0% | 2.9 m | 19.9 | 28% | ||||
| CheckMate-069 (NCT01927419) | Melanoma | 2 | Nivolumab + ipilimumab (95) | 59% | 2-year PFS rate 51.3% vs. 12.0% | 2-year OS rate 63.8% vs. 53.6% | 54% | [236] |
| Ipilimumab (47) | 11% | 20% | ||||||
| CheckMate-142 (NCT02060188) | MMR/MSI-H CRC | 2 | Nivolumab + ipilimumab (119) | 55% | 1-year PFS rate 71% | 1-year OS rate 85% | 32% | [223] |
| ARCTIC (NCT02352948) | NSCLC | 3 | Durvaluma + tremelimumab (174) | 26% | 3.5 m | 11.5 m | 74% | [237] |
| Standard of care (118) | 8% | 3.5 m | 8.7 m | 57% | ||||
| CONDOR (NCT02319044) | HNSCC | 2 | Durvalumab + tremelimumab (133) | 7.8% | 2.0 m | 7.6 m | 15.8% | [238] |
| Durvalumab (67) | 9.2% | 1.9 m | 6.0 m | 12.3% | ||||
| Tremelimumab (67) | 1.6% | 1.9 m | 5.5 m | 16.9% | ||||
| DANUBE (NCT02516241) | Urothelial carcinoma | 3 | Durvalumab + tremelimumab (342) | 36% | 3.7 m | 15.1 m | 27% | [239] |
| Chemotherapy (344) | 49% | 6.7 m | 12.1 m | 60% | ||||
| NCT02919683 | Oral Cavity Squamous Cell Carcinoma | 2 | Nivolumab + ipilimumab (15) | 38% | / | / | 33.3% | [240] |
| Nivolumab (14) | 13% | / | / | 14.3% | ||||
| CheckMate-650 (NCT02985957) | Prostate Cancer | 2 | Nivolumab + ipilimumab (pre-chemotherapy; 45) | 25% | 5.5 m | 19.0 m | 17.6% | [205] |
| Nivolumab + ipilimumab (post-chemotherapy; 45) | 10% | 3.8 m | 15.2 m | 10.0% | ||||
| CheckMate-743 (NCT02899299) | Malignant pleural mesothelioma | 3 | Nivolumab + ipilimumab (303) | 32% | 6.8 m | 18.1 m | 30% | [241] |
| Chemotherapy (302) | 8% | 7.2 m | 14.1 m | 32% | ||||
Beyond PD-1/PD-L1 and CTLA-4, the clinical application of novel ICIs targeting other inhibitory receptors, including TIM-3, LAG-3, TIGIT, VISTA, and BTLA, is being explored. Preclinical studies have indicated the therapeutic potential of these second-generation ICIs to restrict tumor growth [10], especially in combination with anti-PD-1/PD-L1/CTLA-4 antibodies [11, 12]. For example, LAG-3, as one of the most promising ICI targets, is a negative regulator of CD4+ T-cell activation and T-cell function, while enhancing Treg activity by binding to major histocompatibility complex (MHC) class II molecules [13, 14]. LAG-3- and PD-1-positive CD8+ T cells have been identified with distinct phenotypes and functions, indicating the potential of dual ICI treatment [15]. Indeed, combinatorial anti-LAG-3/anti-PD-1 treatment sensitized mice resistant to a single drug and significantly inhibited tumor growth by synergistically boosting the immune system [16]. A recent phase III RELATIVITY-047 study showed that the combination of the LAG-3 inhibitor relatlimab and nivolumab improved the median PFS compared with nivolumab alone (10.1 vs. 4.6 months) [17]. TIGIT, which is specifically expressed on activated T cells and NK cells, induced immunosuppression by competing with CD226 to bind CD155 [18]. Considering that blocking TIGIT and PD-1/PD-L1 activates NK cells and T cells respectively, dual ICI therapy seems to be a promising combinatorial strategy. In fact, TIGIT and PD-L1 were coexpressed on tumor antigen-specific CD8+ T cells from melanoma patients, and coblockade promoted T-cell proliferation and function in various tumors [19, 20]. The CITYSCAPE trial involving 135 patients presented promising efficacy and well-tolerated toxicity of the TIGIT-inhibitor tiragolumab plus atezolizumab (anti-PD-L1) in PD-L1 positive NSCLC with a notable improvement in the ORR (66% vs. 24%) compared with atezolizumab alone [21]. More related studies are underway (NCT04080804, NCT03662659, NCT03680508, and NCT04139902) [22].
ICIs combined with another immunotherapeutic strategy
Cytokines
The most widely used cytokine in the past was traditional interleukin-2 (IL-2), which has now been replaced by bempegaldesleukin (BEMPEG), a novel CD122-preferential IL-2 pathway agonist. BEMPEG activates and stimulates the proliferation of NK cells and effector T cells by binding to the IL-2βγ receptor on these cell surfaces, thus playing a crucial role in the stimulation of the antitumor immune response [23, 24]. In patients with advanced solid tumors, BEMPEG treatment showed clinical benefit and tolerable safety although accompanied by increased expression of PD-L1, suggesting the rationality of ICI-based combination therapy [25]. In fact, combining IL-2 with PD-L1 blockade synergistically promoted CD8+ T-cell activation in a chronic inflammation mouse model, providing the basis for clinical trials [26]. Indeed, the combination of BEMPEG and nivolumab has reached some positive clinical outcomes in solid tumors. In 2019, the FDA granted breakthrough therapy designation to the combination of BEMPEG plus nivolumab for the treatment of treatment-naïve, unresectable or metastatic melanoma patients based on the encouraging preliminary phase II data of the PIVOT-02 study (NCT02983045) [27]. Recently, the PIVOT-02 study updated its latest data and showed the excellent antitumor activity and well-tolerated safety of the combination group with a 52.6% ORR in first-line metastatic melanoma and a 34.7% ORR in first-line RCC [28, 29]. Another phase III PIVOT IO 001 trial comparing BEMPEG plus nivolumab with nivolumab alone in malignant melanoma patients is in progress [30].
Costimulatory receptor agonist
A variety of costimulatory receptors participate in the tumor immune response, including 4-1BB (CD137), OX40 (CD134), and GITR (CD357). The use of agonists of these receptors has been reported to cause tumor regression in preclinical and clinical studies [31–34]. Considering that costimulatory agonists may upregulate PD-L1 and are insufficient to activate effector T cells as monotherapy, further evaluation of combination with ICIs is ongoing [35, 36]. Preclinical studies have shown that the combination of costimulatory agonists and ICIs has striking synergistic effects on immune responses in multiple cancers [37–39]. A phase Ib study found that the combination of utomilumab, a 4-1BB agonist and pembrolizumab (anti-PD-1) led to a 26.1% complete or partial response rate in patients with advanced solid tumors without dose-limiting toxicities [40], supporting further investigation. Similarly, sotigalimab, a CD40 agonistic monoclonal antibody, was tolerable and effective in metastatic pancreatic adenocarcinoma when combined with gemcitabine, nab-paclitaxel, and nivolumab in a phase Ib study [41]. Based on the supportive clinical results, sotigalimab obtained orphan drug designation granted by the FDA for the treatment of esophageal, gastroesophageal junction, and pancreatic cancer in 2021.
CAR-T cells
CAR-T cells are genetically modified T cells that are transduced with an engineered CAR fusion protein by means of a retrovirus or lentivirus into autologous T cells. CAR-T cells express a single-chain variable fragment (scFV) antigen-recognition domain, a CD3-derived T-cell activation domain, and a costimulatory domain (CD28, 4-1BB, or both) [42]. Thus, CARs recognize specific cell surface antigens and trigger T-cell activation in an MHC-dependent manner, as extensively studied in hematologic malignancies [43]. Although CAR-T cells infiltrate into the tumor and kill tumor cells, their function may be inhibited by an immunogenically silent TME partially induced by an upregulated immune checkpoint, which could be reversed by ICIs. Hence, the combination of ICIs and CAR-T cell therapy may produce synergistic effects. The enhanced antitumor immunity of the combination therapy observed in preclinical studies has supported further clinical research [44, 45]. A case published in 2017 reported that a refractory diffuse large B-cell lymphoma patient used pembrolizumab after failure of anti-CD19 CAR T-cell therapy. Following PD-1 blockade, the patient experienced multiple lesion regression with expansion of CART19 cells and TCRβ T-cell clones [46].
Cancer vaccines
The weak immunogenicity of tumor cells contributes to immune evasion. Therefore, therapeutic cancer vaccines that stimulate patients’ own immune systems and produce antigen-specific immune responses have gained recognition. Currently, there are many cancer vaccines under preclinical and clinical studies, including Tedopi for lung cancer, ilixadencel for RCC, GVAX for pancreatic cancer, and PolyPEPI1018 for CRC [47]. Among them, the only vaccine approved for cancer treatment is sipuleucel-T for prostate cancer, which is an autologous dendritic cell preparation targeting prostatic acid phosphatase (PAP). Sipuleucel-T treatment for castration-resistant prostate cancer patients showed an OS benefit [48]. However, tumor-associated antigens (TAAs), as nonmutated self-antigens, have poor clinical effects with many adverse effects including central tolerance and overactive autoimmune toxicity. Therefore, cancer neoantigens, as a type of tumor-specific antigen (TSA), are the products of somatic mutations that exclusively exist on tumor cells but not on normal cells. TSA-targeted therapy has aroused widespread interest and has shown remarkable progress [47].
However, high PD-1 expression in antigen-specific T cells was observed when multiple cancers were treated with cancer vaccines [49, 50]. Cancer vaccines mainly affect the first three steps of the cancer-immunity cycle: tumor antigen release, tumor antigen presentation, and T-cell priming. However, the remaining steps and eventual efficacy still rely on the continuous activation of effector T cells in an immunologically hot TME. Therefore, the combination of cancer vaccines and ICIs may enhance clinical responses. To date, some phase I trials have shown a synergistic effect of ICI-based cancer vaccine therapy [51–54]. For example, a phase I study (KEYNOTE-603) found that the combination of mRNA-4157, a cancer neoantigen, with pembrolizumab contributed to encouraging clinical responses and neoantigen-specific CD8+ T-cell responses [55]. However, other trials reported unresponsive cases without expected improvement in OS and PFS [56–58]; thus, the conclusion is controversial. Therefore, more studies are planned to explore the efficacy, biomarkers, and optimal doses and regimens of ICI-based cancer vaccine therapy (NCT04300244, NCT03632941, and NCT03743298).
Oncolytic viral therapy
Oncolytic viruses are directly obtained or artificially modified from natural viruses, which exert antitumor effects mainly through dual mechanisms. First, oncolytic viruses preferentially infect, replicate in and damage tumor cells rather than normal cells, leading to immunogenic cell death (ICD) and the release of soluble tumor antigens. Second, oncolytic viruses bearing nonlytic vectors express therapeutic genes such as pro-inflammatory cytokines to generate antitumor effects. Talimogene laherparepvec (T-VEC), an attenuated HSV-1 that is engineered to express granulocyte-macrophage colony-stimulating factor (GM-CSF), is a first-in-class oncolytic viral immunotherapy [59]. The FDA has approved T-VEC for the treatment of unresectable, cutaneous, subcutaneous and nodal lesions in melanoma patients who experience recurrence after surgery based on the phase III OPTiM study. Final analyses of the OPTiM study showed that T-VEC provided a longer protective antitumor response than GM-CSF (median OS: 23.3 vs. 18.9 months; ORR: 19.0% vs. 1.4%) with well-tolerated safety [60].
Oncolytic viruses have been widely reported to promote tumor antigen release and T-cell recruitment, promisingly synergistically with ICIs to drive different steps of the cancer-immunity cycle. In addition, viral infection also upregulates the expression of immune checkpoint molecules, including CTLA-4 and PD-L1, which may sensitize tumors to ICIs. Therefore, the combination of ICIs and oncolytic viruses has attracted enormous attention. Preclinical studies have found that oncolytic viruses increased the sensitivity of tumor cells to ICIs in melanoma and triple-negative breast cancer (TNBC) [61, 62]. Recently, a randomized phase II study enrolling 198 patients showed that the combination of T-VEC and ipilimumab notably improved the ORR (39%) compared to ipilimumab alone (18%) without additional safety concerns in advanced unresectable melanoma [63], indicating the potential value of combined ICIs and viral therapy in cancer treatment.
Macrophage-targeted therapy
Macrophages, an important component in the TME, are highly plastic and can be simply divided into two types: M1 and M2. M1 macrophages have stronger tumor-killing and antigen-presenting abilities, which promote antitumor adaptive immunity. However, M2 macrophages, as the majority of tumor-associated macrophages (TAMs), tend to be anti-inflammatory and pro-tumor [64]. On the one hand, TAMs establish a tumor-supporting TME by producing growth factors, NF-κB, and pro-angiogenic factors, thereby promoting tumor progression and metastasis. On the other hand, TAMs with poor antigen-presenting ability establish an immunosuppressive TME and induce immune escape by releasing immunosuppressive factors (IL-1, IL-10, TNF-α), upregulating the expression of immunosuppressive molecules (PD-L1, MHC-I, CD80), and recruiting immunosuppressive cells [65]. Importantly, analysis of clinical tumor tissues revealed a correlation between PD-L1 expression and TAM infiltration [66, 67]. Hence, targeting TAMs has great potential for ICI-based combination therapy [68].
TAM-targeted therapies are mainly divided into three categories: (1) inhibition of TAM survival mainly by targeting colony stimulating factor 1 (CSF1) and CSF receptor (CSF1R); (2) inhibition of TAM recruitment by targeting CCL2, CCR2, and CXCR4; (3) reprogramming of TAMs by CD47 inhibitors, CD40 agonists, and TLR agonists [68]. Among them, the most studied target to inhibit TAM survival is the CSF1/CSF1R axis, which is crucial for TAM production, differentiation, and activation. Combining anti-PD-L1 and the CSF1R inhibitor pexidartinib has elicited potent antitumor activity by reducing TAM infiltration and activating CD8+ T cells in animal models of CRC, HCC, and esophageal adenocarcinoma [69, 70]. Recently, a phase Ib study (NCT02323191) revealed a manageable safety profile of anti-CSF1R emactuzumab combined with atezolizumab in advanced solid tumor patients with a 12.5% ORR for ICI-experienced NSCLC patients [71]. Similarly, the combination of AMG 820 (anti-CSF1R) and pembrolizumab showed an acceptable safety profile despite insufficient efficacy in adults with advanced solid tumors, which deserves further improvement [72]. For another TAM-targeting strategy, inhibition of TAM recruitment, the results from PICCASSO and COMBAT trials showed that combining pembrolizumab with maraviroc (anti-CCR5) or BL-8040 (anti-CXCR4) in colorectal and pancreatic cancer, respectively, achieved modest clinical outcomes [73, 74]. Currently, numerous clinical studies are assessing the combination of TAM-targeted therapy and immunotherapy, such as NCT02880371, NCT02452424, and NCT02777710.
cGAS-STING agonists
In addition to adaptive immunity, innate immunity also contributes to the cancer-immunity cycle. The cGAS-STING pathway is an important modifier and connector of the innate and adaptive immune systems. During tumorigenesis, exogenous DNA from tumor cells interacts with the cytosolic DNA sensor cGAS to catalyze adenosine triphosphate (ATP) and guanosine triphosphate (GTP) to produce 2ʹ,3ʹ-cyclic guanosine monophosphate-adenosine monophosphate (cGAMP), subsequently activating STING and inducing the release of type I interferons (IFNs) and proinflammatory cytokines [75]. These released factors further activate dendritic cells, promote the priming and activation of T cells and NK cells [76], and eventually generate strong antitumor responses. Indeed, the STING pathway is silenced in multiple cancers including pancreatic cancer [77], colon cancer [78], and melanoma [79], and activation of the cGAS-STING pathway effectively inhibits tumor metastasis. Considering the great potential of STING agonists in cancer treatment, several drugs have entered the clinical research stage, including ADU-S100 [75], E7766 [80], MK-1454 [81], BMS-986301, and SB-1128 [82].
However, preclinical studies have also presented the immunosuppressive functions of STING agonists in certain tumors. For example, activated STING enhanced Treg infiltration in HPV-related tongue squamous cell carcinoma [83]. STING agonists induced T-cell suppression and immune tolerance in an indoleamine 2,3-dioxygenase (IDO)-dependent manner in Lewis lung carcinoma (LLC) [84]. Moreover, a correlation between STING activation and upregulation of PD-L1 expression was observed in ovarian cancer mouse models [85]. The results from the MK-1454-001 trial showed a modest response in patients treated with MK-1454 alone [86]. Therefore, ICIs may block the pro-tumor effects of STING agonists and convert immunologically cold tumors into hot tumors, making combination therapy a better treatment strategy.
Indeed, preclinical studies have shown stronger antitumor efficacy when STING agonists are combined with ICIs [85, 87, 88]. For example, the most striking tumor shrinkage was observed in advanced MC38 and B16F10 mouse models treated with the combination of MK-1454 and the anti-PD-1 antibody mDX400 [81]. STING agonist-combined therapy has entered the clinical trial stage. The results from the phase I MK-1454-001 study found that the combination of MK-1454 and pembrolizumab produced encouraging efficiency with acceptable safety in solid tumors and lymphomas [86]. Other phase II studies investigating the efficacy and safety of STING agonists and pembrolizumab combination therapy in head and neck squamous cell carcinoma (HNSCC) are also underway (NCT04220866 and NCT03937141).
Immunotherapy combined with radiotherapy
Radiotherapy uses high doses of ionizing radiation to directly damage cell DNA, which kills cancer cells, shrinks tumors, and reduces tumor burden. Radiotherapy has complex effects on the immune system. On the one hand, radiotherapy causes ICD of tumor cells partially by upregulating the release of damage-associated molecular patterns (DAMPs), mainly calreticulin on the tumor cell surface, high mobility group box 1 (HMGB1), heat shock proteins (HSPs), and ATP. These released DAMPs promote DC maturation; improve the antigen-presenting ability of DCs; stimulate the release of IL-2, IL-4, and INF-γ; and eventually elicit a stronger antitumor response [89, 90]. In addition, radiotherapy could induce the secretion of inflammatory cytokines and chemokines, such as IFNs, IL-1β, and CXCL9, and facilitate the infiltration of DCs and effector T cells, thereby modulating the immune status of the TME. For instance, the increased release of tumor necrosis factor (TNF) induced by radiotherapy remarkably reduced the proportion of myeloid-derived suppressor cells (MDSCs) [91]. On the other hand, many studies have revealed the immunosuppressive role of radiotherapy. In addition to myelosuppression and peripheral blood loss, radiotherapy upregulates the expression of negative immune checkpoint ligands such as PD-L1, which may enhance the susceptibility of tumor cells to ICIs [91, 92]. Radiotherapy also induced the secretion of anti-immunogenic cytokines and chemokines such as TGF-β, IL-6, and CXCL12, and subsequently increased the tumor infiltration of immunosuppressive cells, including MDSCs, Tregs, and M2 macrophages [93]. In summary, these findings provide a theoretical basis for combined radiotherapy and immunotherapy.
Interestingly, a very few clinical cases and some animal models observed a phenomenon that local radiotherapy at the primary tumor site shrank tumors at distant metastases, which was called the abscopal effect of radiotherapy [94, 95]. Possible mechanisms include the distant transfer of activated effector T cells, improvements in DC function, and the release of multiple cytokines [96]. Studies have shown that the integrity of the host’s immune system determines the radiosensitivity of tumors and the incidence of abscopal effects. Recently, it was found that immunotherapy reversed immunosuppression and boosted the abscopal effect in multiple cancers including rectal cancer and prostate cancer [97–100]. Regarding biomarkers, using data from three institutional phase I/II studies, absolute lymphocyte count was reported to predict abscopal responses in patients receiving combined immunotherapy and radiotherapy [101].
In addition to abscopal responses, the combined application of radiotherapy and different immunotherapies (such as ICIs, cytokines, and costimulatory antibodies) has also been demonstrated to improve antitumor immunity responses in multiple cancers by boosting radiotherapy-induced immune activation and blocking the immunosuppressive effects of radiotherapy (Table 2). For example, a phase III PACIFIC study showed that adding durvalumab after chemoradiation therapy in stage III NSCLC patients improved the 24-month OS rate (66.3% vs. 55.6%, P = 0.005) and prolonged PFS (17.2 vs. 5.6 months, hazard ratio [HR] = 0.51) with manageable safety compared with those with placebo [102]. Another phase III study (CA184-043) enrolled 799 post-docetaxel metastatic castration-resistant prostate cancer patients to evaluate the activity of ipilimumab versus placebo following radiotherapy [103]. At a median follow-up of 2.4 years, compared with placebo treatment, the addition of ipilimumab to radiotherapy markedly improved OS rates at 2 years (25.2% vs. 16.6%), 3 years (15.3% vs. 7.9%), 4 years (10.1% vs. 3.3%), and 5 years (7.9% vs. 2.7%), indicating that radiotherapy and ICIs have a synergistic antitumor effect in prostate cancer.
Table 2.
Key clinical trials of immunotherapy combined with radiotherapy.
| Study | Indication | Phase | Treatment (n) | Efficacy endpoints | Grade 3/4 AEs | Reference | ||
|---|---|---|---|---|---|---|---|---|
| RR | PFS | OS | ||||||
| PACIFIC (NCT02125461) | NSCLC | 3 | Durvalumab + chemoradiation therapy (473) | 30.0% | 17.2 m | 24-month OS rate 66.3% vs. 55.6% | 30.5% | [102, 242] |
| Chemoradiation therapy (236) | 17.8% | 5.6 m | 26.1% | |||||
| PEMBRO-RT (NCT02492568) | NSCLC | 2 | Pembrolizumab + SBR (36) | 36% | 6.6 m | 15.9 m | / | [243] |
| Pembrolizumab (40) | 18% | 1.9 m | 7.6 m | / | ||||
| NCT02316002 | NSCLC | 2 | Pembrolizumab + LAT (45) | / | 19.1 m | 24-month OS rate 77.5% | / | [244] |
| NCT01807065 | Prostate cancer | 2 | Sipuleucel-T + RT (25) | / | 3.65 m | / | 4% | [245] |
| Sipuleucel-T (24) | / | 2.46 m | / | 0% | ||||
| CA184-043 (NCT00861614) | Prostate cancer | 3 | Ipilimumab + RT (399) | / | / | 5-year OS rate 7.9% vs. 2.7% | 59% | [103] |
| RT (400) | / | / | 41% | |||||
| NCT02704156 | Pancreatic cancer | 2 | Pembrolizumab + SBRT + trametinib (85) | / | 18.3 m | 24.9 m | 31% | [246] |
| SBRT + gemcitabine (85) | / | 15.6 m | 22.4 m | 20% | ||||
| PEMBRO-RT (NCT02492568) + MDACC (NCT02444741) | NSCLC | 1/2 | Pembrolizumab + RT (72) | 41.7% | 9.0 m | 19.2 m | / | [247] |
| Pembrolizumab (76) | 19.7% | 4.4 m | 8.7 m | / | ||||
| (NCT02730130) | TNBC | 2 | Pembrolizumab + RT (17) | 17.6% | 2.6 m | 7.6 m | 23.5% | [248] |
However, combination therapy only benefits some patients, and how to preselect these responders is very important. A review summarized the currently discovered biomarkers used to characterize the immune activation status after the combined application of radiotherapy and immunotherapy, and proposed an integrated model including the analysis of peripheral blood samples, histological specimens and medical imaging reports [104]. To achieve the best efficacy, more clinical trials are needed to determine the optimal time, doses, location, and fractionation of radiotherapy.
Immunotherapy combined with chemotherapy
Chemotherapy uses powerful chemicals to destroy rapidly growing tumor cells and has dual immune-modulating effects on the TME. It was historically considered that chemotherapy exhibited bone marrow toxicity, impaired antitumor immune cells, and induced immune tolerance and immunosuppression. However, numerous studies have gradually revealed the promotion of antitumor immune response by chemotherapy [105]. On the one hand, chemotherapy affects tumor immunity by directly modifying tumor cells, including enhancing the antigenicity of tumor cells (cyclophosphamide, gemcitabine, platinum, and paclitaxel) [105], inducing ICD of tumor cells to produce an antigen-specific immune response (anthracyclines and oxaliplatin) [106], upregulating the surface expression of MHC-I molecules and B7-1 (etoposide, topotecan, paclitaxel), and enhancing the sensitivity of tumor cells to cytotoxicity mediated by CTLs and NK cells (paclitaxel, cisplatin, doxorubicin) [107]. On the other hand, chemotherapy strengthens the immune response by targeting immune cells in the TME, including the inhibition of immunosuppressive cells (Treg, MDSCs, and M2 macrophages) and the activation of DCs, NK cells, and effector T cells (paclitaxel, doxorubicin, cisplatin) [108]. Considering the complexity of the TME, chemotherapy-induced antitumor immune responses may be attenuated, accompanied by a rebound of immunosuppressive effects, allowing the combination of ICIs to restore the tumor sensitivity to chemotherapy. In conclusion, these findings provide the rationale for the combination of chemotherapy and immunotherapy.
Numerous clinical trials have shown that immunotherapy combined with chemotherapy has a prognostic benefit compared with chemotherapy alone (Table 3), which led to FDA approval for cancer treatment. For example, the phase III KEYNOTE-189 study facilitated the FDA approval of the combination of pembrolizumab and chemotherapy for metastatic NSCLC patients without EGFR or ALK alterations [109, 110]. A total of 616 patients were randomized 2:1 to receive chemotherapy in combination with either pembrolizumab or placebo. The final analysis showed that combination therapy continued to improve the ORR (48.3% vs. 19.9%), and provided longer OS (22.0 vs. 10.6 months) and PFS (9.0 vs. 4.9 months) compared with chemotherapy alone regardless of PD-L1 expression [109]. Recently, the FDA announced the approval of pembrolizumab in combination with chemotherapy to treat unresectable recurrent/metastatic TNBC that expresses PD-L1 (comprehensive positive score [CPS] ≥ 10). This approval was based on the results of the phase III KEYNOTE-355 study, where pembrolizumab combined with chemotherapy produced a remarkable improvement in PFS versus placebo combined with chemotherapy (9.7 vs. 5.6 months, HR = 0.65) with manageable safety in TNBC patients with PD-L1 positivity [111].
Table 3.
Key clinical trials of immunotherapy combined with chemotherapy.
| Study | Indication | Phase | Treatment (n) | Efficacy endpoints | Grade 3/4 AEs | Reference | ||
|---|---|---|---|---|---|---|---|---|
| RR | PFS | OS | ||||||
| KEYNOTE-21 (NCT02039674) | NSCLC | 2 | Pembrolizumab + chemotherapy (60) | 58% | 24.5 m | 34.5 m | 39% | [112] |
| Chemotherapy (63) | 33% | 9.9 m | 21.1 m | 31% | ||||
| KEYNOTE-189 (NCT02578680) | NsqNSCLC | 3 | Pembrolizumab + chemotherapy (410) | 48.3% | 9.0 m | 22.0 m | 72.1% | [109, 110] |
| Chemotherapy (206) | 19.9% | 4.9 m | 10.6 m | 66.8% | ||||
| CA184-024 (NCT00324155) | Melanoma | 3 | Ipilimumab + dacarbazine (250) | / | / | 11.2 m | 56.3% | [249] |
| Dacarbazine (252) | / | / | 9.1 m | 27.5% | ||||
| CA184-041 (NCT00527735) | NSCLC | 2 | Concurrent ipilimumab + paclitaxel + carboplatin (68) | 32% | 5.1 m | 12.2 m | 15% | [250] |
| Phased ipilimumab + paclitaxel + carboplatin (68) | 21% | 4.1 m | 9.7 m | 20% | ||||
| Paclitaxel + carboplatin (68) | 14% | 4.2 m | 8.3 m | 6% | ||||
| CA184-041 (NCT00527735) | SCLC | 2 | Phased ipilimumab + paclitaxel/carboplatin (44) | 57% | 5.2 m | 12.9 m | 17% | [251] |
| Concurrent ipilimumab + paclitaxel/carboplatin (43) | 33% | 3.9 m | 9.1 m | 21% | ||||
| Paclitaxel/carboplatin (43) | 49% | 5.2 m | 9.9 m | 9% | ||||
| CA184-104 (NCT01285609) | NSCLC | 3 | Ipilimumab + chemotherapy (388) | 44% | 5.6 m | 13.4 m | 51% | [252] |
| Chemotherapy (361) | 47% | 5.6 m | 12.4 m | 35% | ||||
| KEYNOTE-522 (NCT03036488) | TNBC | 3 | Pembrolizumab + neoadjuvant chemotherapy (784) | 64.8% | / | / | 78.0% | [253] |
| Neoadjuvant chemotherapy (390) | 51.2% | / | / | 73.0% | ||||
| IMpassion130 (NCT02425891) | TNBC | 3 | Atezolizumab + Nab-paclitaxel (451) | 56.0% | 7.2 m | 21.3 m | 49% | [254, 255] |
| Nab-paclitaxel (451) | 45.9% | 5.5 m | 17.6 m | 43% | ||||
| IMpower133 (NCT02763579) | SCLC | 3 | Atezolizumab + carboplatin + etoposide (201) | 60.2% | 5.2 m | 12.3 m | 56.6% | [113] |
| Carboplatin + etoposide (202) | 64.4% | 4.3 m | 10.3 m | 56.1% | ||||
| IMpower150 (NCT02366143) | NsqNSCLC | 3 | Atezolizumab + bevacizumab + carboplatin + paclitaxel (356) | 63.5% | 8.3 m | 19.2 m | 55.7% | [194] |
| Bevacizumab + carboplatin + paclitaxel (336) | 48.0% | 6.8 m | 14.7 m | 47.7% | ||||
| KEYNOTE-407 (NCT02775435) | SqNSCLC | 3 | Pembrolizumab + chemotherapy (278) | 62.6% | 8.0 m | 17.1 m | 74.1% | [190, 256] |
| Chemotherapy (281) | 38.4% | 5.1 m | 11.6 m | 69.6% | ||||
| CASPIAN (NCT03043872) | SCLC | 3 | Durvalumab + tremelimumab + platinum-etoposide (268) | / | 4.9 m | 10.4 m | / | [257] |
| Durvalumab + platinum-etoposide (268) | / | 5.1 m | 12.9 m | / | ||||
| Platinum-etoposide (269) | / | 5.4 m | 10.5 m | / | ||||
| KEYNOTE-062 (NCT02494583) | Gastric/gastroesophageal junction (G/GEJ) cancer | 3 | Pembrolizumab + chemotherapy (257) | 48.6% | 6.9 m | 12.5 m | 73.2% | [258] |
| Pembrolizumab (256) | 14.8% | 2.0 m | 10.6 m | 16.9% | ||||
| Chemotherapy (250) | 37.2% | 6.4 m | 11.1 m | 69.3% | ||||
| KEYNOTE-355 (NCT02819518) | TNBC | 3 | Pembrolizumab + chemotherapy (566) | / | 9.7 m | / | 68% | [111] |
| Chemotherapy (281) | / | 5.6 m | / | 67% | ||||
| CameL (NCT03134872) | NsqNSCLC | 3 | Camrelizumab + chemotherapy (205) | 60.5% | 11.3 m | Not reached | 69% | [259] |
| Chemotherapy (207) | 38.6% | 8.3 m | 20.9 m | 47% | ||||
| KEYNOTE-361 (NCT02853305) | Urothelial cancer | 3 | Pembrolizumab + chemotherapy (351) | 54.7% | 8.3 m | 17.0 m | 87% | [260] |
| Chemotherapy (352) | 44.9% | 7.1 m | 14.3 m | 82% | ||||
| CheckMate-649 (NCT02872116) | G/GEJ cancer | 3 | Nivolumab + chemotherapy (789) | 60% | 7.7 m | 13.8 m | 59% | [193] |
| Chemotherapy (792) | 45% | 6.9 m | 11.6 m | 44% | ||||
| ESCORT-1st (CT03691090) | ESCC | 3 | Camrelizumab + chemotherapy (298) | 72.1% | 6.9 m | 15.3 m | 63.4% | [114] |
| Chemotherapy (298) | 62.1% | 5.6 m | 12.0 m | 67.7% | ||||
Regarding safety concerns, there is not much overlap in common and typical adverse events (AEs) between chemotherapy and immunotherapy. The AEs of chemotherapy mainly include bone marrow suppression, nausea, vomiting, mucositis, alopecia and neuropathy, whereas immunotherapy is usually well tolerated. A number of clinical trials, including KEYNOTE-21 [112], KEYNOTE-189 [109], IMpower133 [113], and ESCORT-1st [114], have indicated that the combination of immunotherapy and chemotherapy exhibits an acceptable safety profile and potent efficacy (Table 3). However, only some patients benefit from immune-based chemotherapy. It is still unclear how to identify which group will sensitively respond to the combination therapy. The impact of chemotherapy on the immune system is affected by multiple factors, including the types and doses of chemotherapy drugs, simultaneous or sequential regimen therapy, tumor burden, genome mutation and the expression levels of checkpoint inhibitors. Currently, many clinical studies are working on this issue, such as the INSIGNA study (NCT03793179) and KEYNOTE-975 study (NCT04210115).
Immunotherapy combined with anti-angiogenic therapy
The uncontrolled rapid growth of tumor cells is out of balance with the originally limited blood supply, which leads to intratumoral hypoxia and subsequent upregulation of hypoxia-inducible factor-1 (HIF-1) and vascular endothelial growth factor (VEGF). Under VEGF stimulation, sustained tumor angiogenesis occurs inside the tumor. However, the newly formed vessels are usually abnormal and malfunctional and contribute to the occurrence, progression, and metastasis of tumors [115]. Hence, anti-angiogenic therapy, as a special targeted therapy that normalizes the newly formed vessels, mainly includes three categories: anti-VEGF (bevacizumab), anti-VEGFR (ramucirumab, olaratumab), and tyrosine kinase inhibitors (TKIs, including apatinib, sorafenib, sunitinib, axitinib, and lenvatinib) [116].
Anti-angiogenic and immunotherapy both act on the TME. Accumulating evidence has shown that immune-vascular crosstalk exists and has a synergistic immune effect [117]. On the one hand, tumor angiogenesis results in immune suppression via numerous mechanisms. First, newly formed disordered tumor blood vessels directly make it difficult for CD8+ T cells to gather around and inside the tumor. Second, VEGF was reported to interrupt T-cell priming, exhaust CTLs, and inhibit DC maturation. Third, malformed tumor vasculature upregulates the PD-L1 expression of endothelial cells, promotes Treg differentiation, and induces TAMs to polarize into an immunosuppressive M2-like phenotype. Therefore, anti-angiogenic therapy normalizes intratumor blood vessels, makes T cells more likely to infiltrate, potentiates cancer immunity, and enhances the efficacy of other anti-cancer treatments [118]. For example, bevacizumab was found to increase the number of B cells and T cells, inhibit the immune infiltration of Tregs, and improve DC functions in the peripheral blood of metastatic CRC patients [119]. On the other hand, ICIs affect tumor angiogenesis partially by stimulating effector T cells and upregulating the release of IFN-γ [120, 121]. For example, activation of CD4+ T lymphocytes by combinatory inhibition of PD-1 and CTLA-4 was reported to promote the normalization of tumor blood vessels [121]. Other studies found that the differentiation and number of Tregs positively correlated with tumor angiogenesis [122, 123]. Overall, the preclinical results provided a rationale for the combination of immunotherapy and anti-angiogenic therapy in clinical trials.
Regarding RCC, there have been a number of phase III clinical trials of immunotherapy combined with anti-angiogenic therapy with impressive antitumor activity, including IMmotion151, KEYNOTE-426, JAVELIN Renal 101, BO17705, and KEYNOTE-581 (Table 4). For example, the KEYNOTE-426 study showed an advantage in the ORR (59.3% vs. 35.7%) and PFS (15.1 vs. 11.1 months) for pembrolizumab plus axitinib therapy with respect to sunitinib monotherapy [124]. Extended follow-up data showed that combination therapy obtained sustained clinical benefits in median OS (not reached vs. 35.7 months, HR = 0.68) and PFS (15.4 vs. 11.1 months, HR = 0.71) over sunitinib monotherapy [125]. The combination of immunotherapy and anti-angiogenic therapy has also made great progress in HCC. In 2020, the FDA approved atezolizumab plus bevacizumab as an initial treatment for unresectable or metastatic HCC, which was based on the data from the IMbrave150 study, a phase III, multicenter, open-label randomized trial [126]. A total of 501 patients were randomized 2:1 to receive either atezolizumab plus bevacizumab or sorafenib. The primary results showed that the atezolizumab-bevacizumab combination group resulted in better OS rates at 12 months (67.2% vs. 54.6%) and longer PFS (6.8 vs. 4.3 months) than sorafenib monotherapy [126]. Follow-up data showed that the combined therapy produced continued meaningful benefits in terms of patient-reported quality of life, functioning, and disease symptoms compared with sorafenib [127].
Table 4.
Key clinical trials of immunotherapy combined with anti-angiogenic therapy.
| Study | Indication | Phase | Treatment (n) | Efficacy endpoints | Grade 3/4 AEs | Reference | ||
|---|---|---|---|---|---|---|---|---|
| RR | PFS | OS | ||||||
| IMpower150 (NCT02366143) | NSCLC | 3 | Atezolizumab + bevacizumab + carboplatin + paclitaxel (394) | 56.4% | 10.2 m | Not estimable | 57% | [261] |
| Atezolizumab + carboplatin + paclitaxel (399) | 40.6% | 6.9 m | 18.7 m | 43% | ||||
| IMmotion150 (NCT01984242) | RCC | 2 | Atezolizumab + bevacizumab (101) | 32% | 11.7 m | / | 63% | [128] |
| Atezolizumab (103) | 25% | 6.1 m | / | 40% | ||||
| Sunitinib (101) | 29% | 8.4 m | / | 69% | ||||
| IMmotion151 (NCT02420821) | RCC | 3 | Atezolizumab + bevacizumab (454) | 37% | 11.2 m | 33.6 m | 40% | [262] |
| Sunitinib (461) | 33% | 8.4 m | 34.9 m | 54% | ||||
| IMbrave150 (NCT03434379) | HCC | 3 | Atezolizumab + bevacizumab (336) | 27.3% | 6.8 m | 19.2 m | 56.5% | [126, 263] |
| Sorafenib (165) | 11.9% | 4.3 m | 13.4 m | 55.1% | ||||
| KEYNOTE-146 (NCT02501096) | Endometrial cancer | 1b/2 | Lenvatinib+ pembrolizumab (108) | 38.9% | 21.2 m | 7.4 m | 66.9% | [264] |
| KEYNOTE-775 (NCT03517449) | Endometrial cancer | 3 | Pembrolizumab + Lenvatinib (413) | 30% | 6.6 m | 17.4 m | / | [265] |
| Chemotherapy (413) | 15% | 3.8 m | 12.0 m | / | ||||
| KEYNOTE-426 (NCT02853331) | RCC | 3 | Pembrolizumab + axitinib (432) | 59.3% | 15.1 m | Not reached | / | [124] |
| Sunitinib (429) | 35.7% | 11.1 m | 35.7 m | / | ||||
| JAVELIN Renal 101 (NCT02684006) | RCC | 3 | Avelumab + axitinib (442) | 52.5% | 13.8 | / | 71.2% | [129, 130, 266] |
| Sunitinib (444) | 27.3% | 8.4 | / | 71.5% | ||||
| EPOC1706 (NCT03609359) | Gastric cancer | 2 | Lenvatinib + pembrolizumab (29) | OR 20/29 | 7.1 m | / | 48% | [267] |
| BO17705E (NCT00738530) | RCC | 3 | Bevacizumab + interferon alfa (325) | 31% | 10.2 m | / | 29% | [268] |
| Interferon alfa (316) | 13% | 5.4 m | 19.8 m | 16% | ||||
| ORIENT-32 (NCT03794440) | HCC | 2/3 | Sintilimab + bevacizumab biosimilar (380) | 21% | 4.6 m | Not reached | / | [269] |
| Sorafenib (191) | 4% | 2.8 m | 10.4 m | / | ||||
| KEYNOTE-581 (NCT02811861) | RCC | 3 | Lenvatinib + pembrolizumab (355) | 71.0% | 23.9 m | 2-year OS rate 79.2% vs. 66.1% vs. 70.4% | 82.4% | [270] |
| Lenvatinib + everolimus (357) | 53.5% | 14.7 m | 83.1% | |||||
| Sunitinib (357) | 36.1% | 9.2 m | 71.8% | |||||
| NCT03603756 | ESCC | 2 | Camrelizumab + apatinib + chemotherapy (30) | 80.0% | 6.85 m | 19.43 m | 90.0% | [271] |
In addition, studies have also examined biomarkers that could predict the efficacy of combination therapy. Prognostic data from IMmotion150, a randomized phase II study of atezolizumab alone or combined with bevacizumab versus sunitinib in metastatic RCC patients showed that PFS was strongly associated with angiogenesis, T-effector/IFN-γ response, and myeloid inflammatory gene expression signatures (including PD-L1, CXCL9, IFN-γ), but not tumor mutation or neoantigen burden [128]. Similarly, a phase III JAVELIN Renal 101 trial showed that the combination of avelumab (anti-PD-L1) and axitinib resulted in a continuous improvement in PFS versus sunitinib in advanced RCC patients [129]. Further biomarker analysis found that PFS was closely correlated with new immunomodulatory and angiogenesis gene expression signatures (GESs), new mutation GESs, and certain human leukocyte antigen (HLA) types, but not PD-L1 expression or tumor mutation burden (TMB) [130] (Table 4). To date, no precise biomarkers have been identified that could accurately predict the efficacy of combination therapy, which deserves further investigation.
Immunotherapy combined with targeted therapy
Targeted therapy uses drugs to precisely identify and attack certain cancer cells and elicits rapid tumor regression without affecting normal cells by blocking oncogenes/driver genes in several malignancies, such as melanoma and breast cancer. Well-established therapeutic targets include BRAF, c-MET, epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2), anaplastic lymphoma kinase (ALK), the mitogen-activated protein kinase (MAPK) pathway, mechanistic target of rapamycin (mTOR), and poly (ADP-ribose) polymerase (PARP). Despite the quick and strong responses, most targeted therapies are not durable and exhibit a high incidence of drug resistance; thus, the long-term efficacy is unclear. Contrary to targeted therapy, immunotherapy is only beneficial in a small number of cancer patients with slow onset, but induces a highly durable immune response with acceptable toxicity, providing long-lasting protective antitumor immunity. Importantly, targeted therapy affects the cancer-immunity cycle, especially the first two steps, with the remaining steps still relying on immunotherapy [131]. Targeted therapy has been found to induce ICD in tumor cells, promote tumor antigen presentation, and initiate the cancer-immunity cycle, such as inhibitors targeting cyclin-dependent kinase 4 and 6 (CDK4/6) and phosphoinositide 3-kinase (PI3K) [131, 132]. Therefore, the combination of immunotherapy and targeted therapy may complement each other and achieve a fast and enduring antitumor response.
In clinical practice, the combination of immunotherapy and targeted therapy has made great progress in melanoma. BRAF mutations appear in half of malignant melanomas; these mutations activate the MAPK pathway and promote cancer progression. Vemurafenib and dabrafenib, two BRAF inhibitors, induce rapid and deep regression in melanoma patients and are approved by the FDA for the treatment of BRAF-mutant melanoma [133, 134]. Interestingly, numerous studies found that drugs targeting BRAF/MEK in melanoma enhanced antitumor immunity by promoting antigen presentation, enhancing the accumulation and function of effector T cells, and producing pro-inflammatory cytokines [135]. However, BRAF inhibitors also have immunosuppressive effects under certain circumstances. Elevated PD-1 and PD-L1 expression was found in melanoma patients previously treated with BRAF inhibitors in vivo and in BRAF inhibitor-resistant melanoma cell lines in vitro [136, 137], suggesting the potential therapeutic benefits of combining targeted therapy and immunotherapy. These findings have aroused enormous interest in immune-based combination strategies.
Recently, a phase III IMspire150 study investigating the safety and efficacy of immunotherapy in combination with targeted therapy in BRAF V600E/K-mutant metastatic melanoma achieved inspiring results [138]. In this study, when compared with targeted therapy of vemurafenib and cobimetinib (MEK inhibitor), the addition of atezolizumab to targeted therapy showed superiority in terms of PFS (15.1 vs. 10.6 months) with manageable safety (Table 5). However, recently, primary analysis from the phase II COLET study showed that cobimetinib plus atezolizumab and taxanes did not increase the ORR in advanced TNBC [139]. Another phase III HR-NBL1/SIOPEN study showed that the addition of IL-2 to dinutuximab beta (anti-GD2) did not improve the 3-year event-free survival rate (56% vs. 60%) but caused more combined toxicity in neuroblastoma compared to dinutuximab beta alone, indicating the controversial role of the combination therapy in different malignancies [140]. Indeed, one of the main challenges of immune-based targeted therapy is the combined drug toxicity and side effects, especially when two drugs are concurrently administered (Table 5). Therefore, many studies have been performed to assess the effects and safety of simultaneous or sequential regimens, and to exploit their synergistic effects in multiple cancers.
Table 5.
Key clinical trials of immunotherapy combined with targeted therapy.
| Study | Indication | Phase | Treatment (n) | Efficacy endpoints | Grade 3/4 AEs | Reference | ||
|---|---|---|---|---|---|---|---|---|
| RR | PFS | OS | ||||||
| COMBI-i (NCT02967692) | Melanoma | 3 | Spartalizumab + dabrafenib + trametinib (36) | 78% | 23 m | 24-month OS rate 74% | 72% | [272] |
| KEYNOTE-252 (NCT02752074) | Melanoma | 3 | Pembrolizumab + epacadostat (354) | 34% | 4.7 m | 12-month OS rate 74.4% vs. 74.1% | 22% | [273] |
| Pembrolizumab (352) | 32% | 4.9 m | 17% | |||||
| IMspire150 (NCT02908672) | Melanoma | 3 | Atezolizumab + vemurafenib/cobimetinib (256) | 66% | 15.1 m | / | 79% | [138] |
| vemurafenib/cobimetinib (258) | 65% | 10.6 m | / | 73% | ||||
| COLET (NCT02322814) | TNBC | 2 | Atezolizumab + cobimetinib + chemotherapy (32) | 34.4% | 3.8 m | 11.0 m | / | [139] |
| Cobimetinib + chemotherapy (47) | 38.3% | 5.5 m | 16.0 m | / | ||||
| NCT03082534 | HNSCC | 2 | Pembrolizumab + cetuximab (33) | 45% | 6.5 m | 18.4 m | / | [274] |
| MEDIOLA (NCT02734004) | Breast cancer | 1/2 | Durvalumab + olaparib (34) | 63.3% | 8.2 m | 21.5 m | 32% | [147] |
| PEMDAC (NCT02697630) | Uveal melanoma | 2 | Pembrolizumab + entinostat (29) | 14% | 2.1 m | 13.4 m | 66% | [275] |
| EV-103 (NCT03288545) | Urothelial cancer | 1/2 | Pembrolizumab + enfortumab vedotin (45) | 73.3% | 12.3 m | / | / | [153] |
| HR-NBL1/SIOPEN (NCT01704716) | Neuroblastoma | 3 | IL-2 + dinutuximab beta (206) | / | 5-year EFS rate 57% vs. 53% | 5-year OS rate 62% vs. 63% | / | [140] |
| Dinutuximab beta (200) | / | / | ||||||
| NCT02484404 | Ovarian cancer | 2 | Durvalumab + olaparib (35) | 14% | 3.9 m | / | / | [276] |
| KEYNOTE-162 (NCT02657889) | Ovarian carcinoma | 1/2 | pembrolizumab + niraparib (62) | 18% | 3.4 m | / | / | [148] |
In addition to the above conventional targeted drugs, novel drugs with new targets have shown remarkable efficacy. PARPs are a family of related enzymes that share the ability to catalyze the transfer of ADP-ribose to target proteins, contributing to the detection and repair of DNA damage [141, 142]. Importantly, germline mutations in BRCA1/2 are present in ovarian cancer, breast cancer, pancreatic cancer and prostate cancer, making these tumors particularly sensitive to PARP inhibitors (PARPi). PARPi treatment, including olaparib, rucaparib, and niraparib, has been approved for clinical cancer treatment as monotherapy. Regarding immune effects, PARPi enhance tumor antigenicity and promote intratumoral T-cell infiltration by activating the STING pathway in multiple cancers [143, 144]. Importantly, PARPi upregulated PD-L1 expression in breast cancer cell lines and mouse tumor models, which may sensitize tumor cells to ICIs [145]. Indeed, anti-PD-L1 treatment promoted PARPi efficacy by restoring antitumor immunity, providing evidence for ICI-based PARPi therapy [145, 146]. The results from a phase I/II study (MEDIOLA) reported encouraging antitumor activity for the combination of olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer [147]. This study reported a potent disease control rate of 80% at 12 weeks of combination therapy without additional safety concerns. Other studies have also achieved positive results with more clinical trials underway (NCT04034927, NCT02571725, NCT03824704, NCT04169841) [147, 148].
In addition, some antibody–drug conjugates (ADCs) are also being developed and explored. ADCs consist of a humanized monoclonal antibody (mAb) conjugated to the cytotoxic payload via a chemical linker that recognizes and targets tumor antigens specifically [149]. For example, enfortumab vedotin connects the microtubule-disrupting agent monomethyl auristatin E (MMAE) and anti-Nectin-4 mAb, thereby targeting and killing solid tumors highly expressing the cell adhesion molecule Nectin-4. Preclinical and clinical studies have shown its potent therapeutic effects in urothelial cancer and bladder cancer [150–152]. When considering combination therapy to improve its efficacy, a phase I/II study showed the promising activity and durability of enfortumab vedotin plus pembrolizumab as a first-line treatment for locally advanced or metastatic urothelial carcinoma, with a 73.3% ORR and a manageable safety profile [153]. Further evaluation of ICI-based ADC therapy is ongoing.
Immunotherapy combined with bacterial therapy
Bacteria‐assisted cancer therapy, as a new antitumor approach, has shown great promise in cancer treatment via unique mechanisms [154]. First, some bacterial strains, such as Salmonella, selectively target tumor hypoxia and nutritious regions and directly kill host tumor cells by secreting exotoxins and competing for nutrients [155]. These apoptotic tumor cells release DAMPs, induce ICD, and enhance antitumor immune responses. Second, some bacteria and their metabolites can convert the TME from immunosuppressive to immunostimulatory [156–158]. For example, bacterial delivery of Staphylococcus aureus increased the release of pro-inflammatory cytokines including IL-12, IFN-γ, and TNF-α; enhanced tumor infiltration of CTLs and activated NK cells; and finally caused tumor regression and necrosis in murine melanoma tumors [159, 160]. Third, with the aid of genetic engineering, bacteria are genetically modified to deliver specific genes that code immunogenic cytokines, cytotoxic proteins, anticancer agents, and tumor‐specific antigens (bacterial DNA vaccines), allowing for individualized treatment [161].
Although many advances in bacterial cancer therapy in preclinical studies have been achieved, clinical progress remains slow. Current studies mainly stay in phase I with the lack of large-scale and well-designed clinical trials. Some tough challenges exist, including innate toxicity, short half-life, and DNA instability of bacteria [162]. To overcome the limitations of monotherapy and effectively eradicate tumors, the efficacy and safety of bacterial therapy in combination with conventional cancer treatments have been broadly explored [163]. Accumulating evidence has shown that bacterial therapy affects the immune response, providing the rationale for the combination of immunotherapy and bacteriotherapy. Indeed, immune-based combination therapy was reported to maintain tumor targeting, reduce bacterial toxicity by reducing the dose, and evoke antitumor immune responses through immunomodulatory factors. Although the regulation of PD-L1 expression by Salmonella is controversial, the combination of Salmonella and anti-PD-1 treatment synergistically inhibits tumor growth in CRC and melanoma tumor models [164, 165]. Similarly, the combination of Salmonella VNP20009 coated with the photothermal material polydopamine and an anti-PD-1 peptide showed robust antitumor immune responses and effectively eliminated tumors in a mouse melanoma model [166].
Some clinical trials of combination therapy are in progress. For instance, a phase I study (NCT03435952) investigated the highest tolerable dose of Clostridium novyi-NT spores in combination with pembrolizumab in patients with advanced solid tumors. Another phase I/II study (NCT02291055) explored the efficacy and safety of ADXS11-001 (live attenuated Listeria monocytogenes bacterium) combined with durvalumab in cervical cancer or HPV+ head and neck cancer. The NCT03750071 study evaluating the efficacy and safety of VXM01 (a cancer vaccine based on live attenuated Salmonella typhimurium) combined with avelumab in glioblastoma patients is in progress. Clinical studies assessing the combination of bacterial consortia and ICIs, such as NCT03775850, NCT03595683, and NCT03637803, are underway.
In addition, recent studies have revealed that the gut microbiota and its metabolites also affect the incidence of ICI-associated colitis [167]. Therefore, the addition of bacterial therapy to ICIs may reduce ICI toxicity and improve patient outcomes [168]. In fact, there was a case report of using fecal microbiota transplantation (FMT) to relieve refractory ICI-associated colitis [169]. In this case, two patients developed severe colitis after receiving anti-CTLA-4 and anti-PD-1 treatment. Surprisingly, after treatment with FMT from healthy donors in the two patients, the abundance and composition of gut microbiota were remodeled, intestinal inflammation was reduced, and the clinical symptoms were completely or mostly relieved. These cases provide evidence that bacteria‐assisted therapy may abrogate irAEs and improve health-related quality of life in ICI-treated patients. We hope that in the near future, genetically engineered bacteria will be more clinically practical either in monotherapy or combination therapy.
Immunotherapy combined with stroma-targeted therapy
The dynamic interaction between cancer cells and the complex TME affects antitumor immune responses and regulates cancer progression. Many studies have explored the role of the TME in immunotherapy resistance, aiming to develop novel strategies specifically targeting the TME. Cancer-associated fibroblasts (CAFs), as the most abundant stromal cells, suppress antitumor immunity in a variety of solid tumors via multiple mechanisms [170]. For example, activation of TGF-β signaling in CAFs inhibited immune responses to atezolizumab by restricting T-cell infiltration in patients with metastatic urothelial cancer [171]. Fibroblast activation protein (FAP), as one of the widely used CAF markers, regulates tumor progression by exerting protease function and signal transduction. On the one hand, FAP promotes the reconstruction of the extracellular matrix by degrading fibronectin and rearranging collagen in the TME, guiding tumor cells to invade along the fiber. On the other hand, FAP is involved in the signal regulation of multiple cytokines including TGF-β, VEGF, stromal cell derived factor-1 (SDF-1), and platelet-derived growth factor (PDGF), which inhibit the recruitment and function of immune cells and establish an immunosuppressive TME suitable for aggressive tumors. For instance, CCL2 secreted by FAP+ CAFs enhanced the recruitment of MDSCs by binding to CCR2 on the surface of MDSCs, thereby inducing immunosuppression in HCC [172]. These observations make FAP a promising target for stroma-targeted therapy. Sibrotuzumab, an anti-FAP mAb, inhibited the proliferation, invasion, and metastasis of cancer cells in preclinical studies. However, in phase I and II trials of sibrotuzumab alone, only a few patients had stable disease control and the expected ORR was not reached [173, 174].
Dissatisfaction with CAF-targeted monotherapy leads to further exploration of combinations with other anti-cancer therapies, including immunotherapy. In fact, preclinical data have indicated that combining CAF-targeted therapy and ICIs serves as a good strategy. Tranilast inhibits the growth, function, and collagen synthesis of CAFs and is considered a CAF-targeted therapy [175]. In vitro studies found that the combination of tranilast and DC-based vaccines synergistically improved systemic antitumor immune responses and effectively suppressed tumor growth in various tumor-bearing mouse models compared with monotherapy [176]. In addition to directly targeting CAFs, blockade of CAF-secreted immunosuppressive factors also enhances the efficacy of immunotherapy and has promising clinical value. Preclinical studies have found that ablation of FAP+ fibroblasts or its secreted CXCL12 protein increased the tumor sensitivity to immunotherapy in KPC mice [177–179]. Similarly, blockade of CAF-derived WNT2 markedly enhanced the therapeutic efficacy of an anti-PD-1 antibody by activating DCs and improving the cytotoxicity of splenic T cells in both oral squamous cell carcinoma (OSCC) and CRC allograft tumors [180]. In addition, some novel combination treatments are entering clinical trials. M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β, showed encouraging efficacy and endurable safety in a phase I study (NCT02517398) [181]. Simlukafusp alfa (FAP-IL2v) is a novel immunocytokine consisting of an anti-FAP antibody and an IL-2 variant that achieves the combination of CAF-targeted therapy and immunotherapy to some extent. Recent evidence has shown that FAP-IL2v potentiates the efficacy of different immunotherapies including anti-PD-1 and agonistic CD40 antibodies [182]. The efficacy and safety of FAP-IL2v as monotherapy or combined with immunotherapy are currently being investigated in some phase I studies (NCT03386721, NCT02627274, NCT03063762, and NCT03875079).
Biomarkers in immune-based combination therapy
Despite promising results in the application of immunotherapy in some cancers, numerous patients are non-responders. Therefore, exploring more precise biomarkers is a critical issue to improve the efficacy of immunotherapy alone or combined with other conventional therapies. The identification of composite biomarkers has been recognized as one of the top 10 challenges of immunotherapy proposed by the Immunity journal in 2020 [2]. To date, several recognized biomarkers of immunotherapy have been identified, including the expression of PD-L1 in tumor cells and immune cells, tumor infiltrating T cells, tumor mutation burden, immune cells and cytokines in peripheral blood, gut microbiota, and circulating tumor DNA [183] (Fig. 2). Biomarkers for immune-based combination therapy are also being actively explored. Unfortunately, there are still no reliable biomarkers that can accurately predict the efficacy of immunotherapy. In addition, occasionally inconsistent clinical outcomes make the conclusion more controversial.
Fig. 2. Potential biomarkers for immunotherapy response prediction.
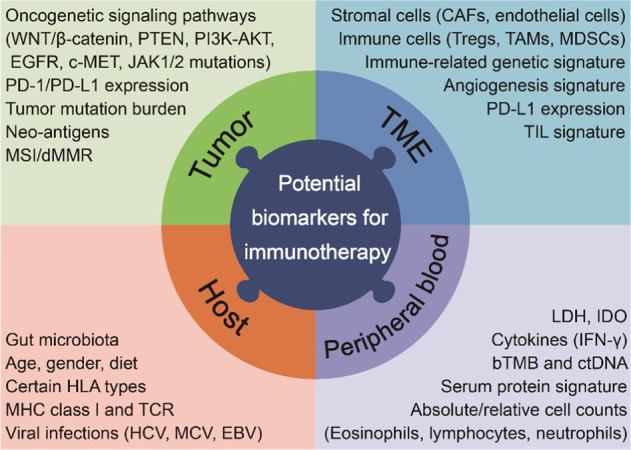
Biomarkers are mainly divided into four categories: tumor, the tumor microenvironment (TME), host factor, and serum/circulating factors from peripheral blood. TIL tumor infiltrating lymphocyte, LDH lactate dehydrogenase, IDO indoleamine 2,3-dioxygenase, ctDNA circulating tumor DNA, TCR T cell receptor.
PD-L1
PD-L1 is one of the most widely applied biomarkers for predicting the efficacy of immunotherapy either as monotherapy or in combination. ICIs have been found to be more effective in tumor patients with high PD-L1 expression in many clinical trials including phase III KEYNOTE-024 [184] and IMpower110 [185] studies. However, other studies found that in PD-L1 positive NSCLC patients, the application of immunotherapy did not improve OS compared with chemotherapy, such as the CheckMate-026 [186] and MYSTIC [187] studies. ICIs are even reported to be effective in some PD-L1-negative patients but ineffective in some PD-L1-positive patients.
The predictive value of PD-L1 on the efficacy of immune-based combination therapy has also been extensively explored; however, a definite conclusion on the combination of dual ICIs has not been drawn. For example, the phase III CheckMate-227 study found that the 4-year OS rates of the nivolumab combined with ipilimumab group were 29% and 24% in advanced NSCLC patients with PD-L1 ≥ 1% or <1, respectively, regardless of PD-L1 expression [188]. However, clinical data from the CITYSCAPE study1 CITYSCAPE found that in NSCLC patients with high levels of PD-L1 (tumor proportion score [TPS] ≥ 50%), anti-TIGIT tiragolumab combined with atezolizumab had notably improved the ORR (66% vs. 24%) and PFS (not reached vs. 4.11 months, HR = 0.30) compared with atezolizumab alone [21]. These improvement effects were not observed in those with low PD-L1 expression (TPS < 50%). For immunotherapy combined with chemotherapy, data from KEYNOTE-189 [109], IMpower130 [189], KEYNOTE-407 [190], RATIONALE-307 [191], and CheckMate-9LA [192] studies showed that PD-L1 expression cannot predict the efficacy of combined therapy. However, the results from the CheckMate-649 study found that the combination of nivolumab and chemotherapy resulted in significant improvements in OS and PFS versus chemotherapy alone in gastric/gastro-esophageal junction (G/GEJ) and esophageal adenocarcinoma patients with PD-L1 CPS ≥ 5 [193].
When considering immunotherapy combined with anti-angiogenic therapy, a subgroup analysis of the IMpower150 study showed that in the atezolizumab plus bevacizumab plus chemotherapy group, the HRs of patients with high PD-L1 (HR = 0.39, 95% CI 0.25–0.60), low PD-L1 (HR = 0.56, 95% CI 0.41–0.77), and negative PD-L1 (HR = 0.77, 95% CI 0.61–0.99) expression were sequentially ascending compared with bevacizumab plus chemotherapy group, indicating that patients with high PD-L1 expression have better responses in combination therapy [194]. Overall, more studies are needed to determine the role of PD-L1 in efficacy prediction. Moreover, PD-L1 as a biomarker has other limitations that need to be overcome. The PD-L1 expression pattern has spatiotemporal specificity [195]. Due to the inducible and dynamic expression characteristics, PD-L1 is expressed differently in different tumor lesions and different treatment stages, which may confuse the true status of PD-L1 expression [196, 197]. The detection of PD-L1 expression by different platforms and reagents did not reach good concordance [198, 199].
TMB
Tumor mutation burden (TMB) generally refers to the number of non-synonymous somatic mutations per megabase of the tumor genome. To some extent, TMB reflects the ability of tumor cells to produce neoantigens, which correlates with antitumor immune responses. TMB was first discovered in 2014 to be associated with clinical response to anti-CTLA-4 agents in melanoma [200]. In 2015, high TMB was found to predict the response to anti-PD-L1 treatment in NSCLC [201]. Several subsequent phase III studies confirmed the value of TMB for predicting efficacy in NSCLC [186, 202]. The CheckMate-227 study found that for patients with a high TMB (≥10 mut/Mb), dual ICI therapy versus chemotherapy induced longer PFS (7.2 vs. 5.5 months) and higher ORR (45% vs. 27%) in patients with stage IV or recurrent NSCLC [202]. However, no improvement in the ORR and PFS with combination therapy was observed in patients with a low TMB, suggesting the role of TMB as a predictive biomarker [202]. Indeed, this finding has been verified in other tumors. A meta-analysis found a significant correlation between TMB and ORR with anti-PD-1/PD-L1 therapy with a correlation coefficient of 0.74 across 27 tumor types [203]. In the KEYNOTE-158 study, which included multiple advanced solid tumors treated with pembrolizumab, patients with a high TMB (TMB-H) had a higher ORR than patients with a low TMB (29% vs. 6%) [204]. Therefore, the FDA approved pembrolizumab monotherapy for patients with advanced TMB-H solid tumors who have progressed after prior therapy. Regarding combinations regiments including immunotherapy, some studies reported good performance of TMB in the prediction of efficacy, such as combinations with another ICI [205–208], chemotherapy [209], and anti-angiogenic therapy [210].
However, other studies found no significant correlation between TMB status and immunotherapy efficacy in a variety of tumors, either as a single agent or in combination [128, 211–215], indicating the controversial role of TMB as a biomarker of immunotherapy. In addition to the uncertain predictive effect, TMB also has other limitations [216]. First, TMB is affected by various factors, including race, gender, tumor type, tumor genetic factors, TME status, and external carcinogens. TMB was observed to be relatively high in melanoma and CRC, and low in pancreatic cancer [203]. Therefore, it is of great importance to determine the optimal cut-off values in each cancer type. Second, the detection accuracy of TMB is affected by sample quality and the detection and analytical methods. Whole-exome sequencing (WES) is the gold standard for TMB detection. Due to its high cost, WES has been gradually replaced by low-cost targeted next-generation sequencing (NGS) gene panels. However, a multi-institutional study found that the consistency of NGS panel-TMB and WES-TMB in different laboratories varied greatly, with Spearman’s coefficients ranging from 0.77 to 0.96, indicating the challenge of TMB standardization [217]. Third, considering that tumor tissue-TMB (tTMB) has problems of inaccessibility and heterogeneity, blood-TMB (bTMB) has emerged as a potential alternative. In NSCLC, bTMB was reported to be closely correlated with tTMB and predicted response to atezolizumab [218, 219]. More studies are needed to confirm the role of bTMB in predicting the clinical benefit of immunotherapy.
Microsatellite instability (MSI)
MSI refers to the insertion or deletion of short repetitive DNA sequences due to deficient DNA mismatch repair (dMMR) during DNA replication, which is especially deeply studied in CRC. MSI is divided into high MSI (MSI-H), low MSI (MSI-L), and stable MSI (MSS). Many studies have demonstrated that CRC patients with high MSI (MSI-H) have a stable response to PD-1 blockade with fewer adverse events [220, 221]. Extended studies further found that dMMR predicted the response of solid tumors to anti-PD-L1 treatment [222], which further contributed to the approval of pembrolizumab for the treatment of MSI-H or dMMR in adults and children with unresectable or metastatic solid tumors. This is the first antitumor therapy based on the biomarkers of genome-wide characterization of MSI, not the source of the tumor or the expression and mutation status of specific genes. In regard to combination therapy, ipilimumab plus nivolumab was also approved for the treatment of metastatic melanoma, advanced RCC and metastatic CRC with dMMR/MSI-H on the basis of some trials, including the CheckMate-142 study [8, 223]. Nevertheless, the current understanding of MSI in different cancer types remains unclear. A recent study examined more than 20,000 microsatellite loci in 18 cancer types and found that the average number of MSI sites varied across cancer types, with fewer sites noted in thyroid cancers and more sites in CRC and endometrial cancer [224]. This heterogeneity complicates the use of MSI as a biomarker for immunotherapy. When detecting MMR-related proteins by immunohistochemistry staining, false-positives may occur due to the presence of nonfunctional proteins, which reduces the detection accuracy.
Gut microbiota
The predictive value of gut microbiota on the efficacy of immunotherapy is worthy of in-depth study. Due to technological advances in NGS, the impact of gut microbiota on the host immune system has been extensively investigated and some studies have identified gut microbiota as a novel biomarker in the immunotherapy of certain cancers, especially melanoma. Researchers from the University of Chicago analyzed the stools of metastatic melanoma patients prior to anti-PD-1 antibody treatment and found a striking association between commensal microbial composition and clinical response. Further animal experiments indicated that transplantation of germ-free mice with feces from responders restrained tumor growth, promoted T-cell activation, and improved the efficacy of anti-PD-L1 therapy [225]. Similarly, a study on the gut microbiome of 112 melanoma patients undergoing anti-PD-1 treatment observed differences in the diversity and composition of the gut microbiota between responders and non-responders. For example, the Ruminococcaceae bacteria and Faecalibacterium genus were abundant in responders, whereas Bacteroidales was enriched in feces of non-responders. FMT experiments verified that stools from responders enhanced antitumor immunity [226], which was further confirmed in another study [227]. The relevance of gut microbiota and anti-PD-1 therapy efficacy has also been revealed in other cancer types including NSCLC, RCC, urothelial carcinoma, and HCC [228, 229].
Only a few studies have reported the predictive values of gut microbiota in the efficacy and toxicity of immune-based combination therapy by analyzing human samples. For dual anti-PD-L1/CTLA-4 combination therapy, analysis of the gut microbiome from 77 melanoma patients receiving combination therapy found a correlation between commensal species such as P. distasonis and antitumor responses. Moreover, dual ICI-induced toxicity was also associated with specific gut microbiota signatures, including an increased abundance of Bacteroides intestinalis [230]. Regarding immune-based targeted therapy, a recent study found that Agathobacter and Blautia species may represent potential biomarkers for prognosis in patients with mCRC and NSCLC treated with cetuximab plus avelumab. Significantly increased abundances of Agathobacter and Blautia were found in the gut microbiota of five long-term responding patients [231]. At present, the clinical evidence for this issue remains limited and deserves further exploration.
The enteric microbiota is a promising biomarker correlating with the efficacy of immunotherapy either alone or in combination. The regulation of gut microbiota by adjusting diet, applying antibiotics, supplementing probiotics, and FMT, is expected to promote immune responses, reduce irAEs, and improve prognosis, which may play an important role in precision medicine in the near future [232]. Nevertheless, there are still some limitations. Despite the declining cost of next-generation sequencing, large-scale clinical application of microbial sequencing remains difficult, which places an economic burden on some patients. Inconsistent results from different studies regarding the impact of the gut microbiota on immunotherapy have been noted; thus, further large-scale clinical cohorts are needed.
Overall, biomarkers of different tumors have been reported to be heterogeneous, suggesting that biomarkers need to be subdivided by tumor types or even subtypes [233]. A single biomarker is obviously inefficient for wide application to multiple cancers with high accuracy. Comprehensive integration of multiparametric biomarkers is expected to establish more precise clinical medicine. More prospective studies are needed to reinforce the predictive value of the existing biomarkers, and to build a multi-factor predictive model to screen responders. At present, there are several ongoing clinical trials exploring more precise biomarkers in immune-based combination therapy. These studies analyzed peripheral blood mononuclear cells (PBMCs), established patient-derived xenograft (PDX) models and tumor cell lines from fresh tumor tissues, sequenced the genome of fresh or frozen tissues, and performed immunohistochemistry staining of solid tumors. Finally, the correlation between the above results and the clinical outcomes were analyzed and multiparametric computational models were constructed. For instance, a study proposed a machine learning model to predict ICI efficacy by comprehensively analyzing multiple biological factors, which predicted OS and PFS with high sensitivity and specificity in multiple cancers [234].
Conclusions
As a breakthrough technology, immunotherapy has made rapid and significant progress and brought hope to many advanced cancer patients. Nevertheless, immunotherapy still faces certain limitations and urgent problems that need to be overcome, such as the low response rates in most solid tumors and unique adverse events. Immune-based combination therapies synergistically convert cold tumors into hot tumors and achieve better antitumor effects than monotherapy. In combination strategies, it is important to determine the optimal dosage, time span and order of drug administration to achieve the best curative effect. In addition, drug safety and pharmacoeconomics must also be taken into full consideration. Another serious problem involves accurately identifying responders to immunotherapy, so that ineffective populations would be free from cost burdens and side effects. Numerous studies are actively underway to explore more precise and effective biomarkers through comprehensive integration of multi-parametric indicators, which is full of expectation.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81972276, 82173040), and Fundamental Research Funds for the Central Universities (226202200165).
Author contributions
Formulation of conception and design: JJN, WZ; creation of the initial draft: JJN; revision of the draft: ZZZ, MJG, and JYC; oversight and critical revising: WZ.
Competing interests
The authors declare no competing interests.
References
- 1.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 2.Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity. 2020;52:17–35. doi: 10.1016/j.immuni.2019.12.011. [DOI] [PubMed] [Google Scholar]
- 3.Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol. 2021;16:223–49. doi: 10.1146/annurev-pathol-042020-042741. [DOI] [PubMed] [Google Scholar]
- 4.Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell. 2017;170:1120–33. doi: 10.1016/j.cell.2017.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev. 2008;224:166–82. doi: 10.1111/j.1600-065X.2008.00662.x. [DOI] [PubMed] [Google Scholar]
- 6.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Grob J-J, Rutkowski P, Lao CD, et al. CheckMate 067: 6.5-year outcomes in patients (pts) with advanced melanoma. J Clin Oncol. 2021;39:9506. doi: 10.1200/JCO.2021.39.15_suppl.9506. [DOI] [Google Scholar]
- 7.Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378:1277–90. doi: 10.1056/NEJMoa1712126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lenz HJ, Van Cutsem E, Luisa Limon M, Wong KYM, Hendlisz A, Aglietta M, et al. First-line nivolumab plus low-dose ipilimumab for microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: the phase II checkMate 142 Study. J Clin Oncol. 2021;40:161–70. doi: 10.1200/JCO.21.01015. [DOI] [PubMed] [Google Scholar]
- 9.Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–91. doi: 10.1016/S1470-2045(17)30422-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. 2016;44:989–1004. doi: 10.1016/j.immuni.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207:2187–94. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zelba H, Bedke J, Hennenlotter J, Mostbock S, Zettl M, Zichner T, et al. PD-1 and LAG-3 dominate checkpoint receptor-mediated T-cell inhibition in renal cell carcinoma. Cancer Immunol Res. 2019;7:1891–9. doi: 10.1158/2326-6066.CIR-19-0146. [DOI] [PubMed] [Google Scholar]
- 13.Baixeras E, Huard B, Miossec C, Jitsukawa S, Martin M, Hercend T, et al. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J Exp Med. 1992;176:327–37. doi: 10.1084/jem.176.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Macon-Lemaitre L, Triebel F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology. 2005;115:170–8. doi: 10.1111/j.1365-2567.2005.02145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grosso JF, Goldberg MV, Getnet D, Bruno TC, Yen HR, Pyle KJ, et al. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J Immunol. 2009;182:6659–69. doi: 10.4049/jimmunol.0804211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72:917–27. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tawbi HA, Schadendorf D, Lipson EJ, Ascierto PA, Matamala L, Castillo Gutierrez E, et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med. 2022;386:24–34. doi: 10.1056/NEJMoa2109970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manieri NA, Chiang EY, Grogan JL. TIGIT: a key inhibitor of the cancer immunity cycle. Trends Immunol. 2017;38:20–8. doi: 10.1016/j.it.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 19.Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J Clin Invest. 2015;125:2046–58. doi: 10.1172/JCI80445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology. 2018;7:e1466769. doi: 10.1080/2162402X.2018.1466769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horvath L, Pircher A. ASCO 2020 non-small lung cancer (NSCLC) personal highlights. Memo. 2021;14:66–9. doi: 10.1007/s12254-020-00673-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Solomon BL, Garrido-Laguna I. TIGIT: a novel immunotherapy target moving from bench to bedside. Cancer Immunol Immunother. 2018;67:1659–67. doi: 10.1007/s00262-018-2246-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, Kirk PB, et al. NKTR-214, an engineered cytokine with biased IL2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clin Cancer Res. 2016;22:680–90. doi: 10.1158/1078-0432.CCR-15-1631. [DOI] [PubMed] [Google Scholar]
- 24.Bempegaldesleukin Stimulates Immune Response. Cancer Discov. 2019; 9: OF1. [DOI] [PubMed]
- 25.Bentebibel SE, Hurwitz ME, Bernatchez C, Haymaker C, Hudgens CW, Kluger HM, et al. A first-in-human study and biomarker analysis of NKTR-214, a novel IL2Rbetagamma-biased cytokine, in patients with advanced or metastatic solid tumors. Cancer Discov. 2019;9:711–21. doi: 10.1158/2159-8290.CD-18-1495. [DOI] [PubMed] [Google Scholar]
- 26.West EE, Jin HT, Rasheed AU, Penaloza-Macmaster P, Ha SJ, Tan WG, et al. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J Clin Invest. 2013;123:2604–15. doi: 10.1172/JCI67008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diab A, Tannir NM, Bentebibel SE, Hwu P, Papadimitrakopoulou V, Haymaker C, et al. Bempegaldesleukin (NKTR-214) plus nivolumab in patients with advanced solid tumors: phase I dose-escalation study of safety, efficacy, and immune activation (PIVOT-02) Cancer Discov. 2020;10:1158–73. doi: 10.1158/2159-8290.CD-19-1510. [DOI] [PubMed] [Google Scholar]
- 28.Diab A, Tykodi SS, Daniels GA, Maio M, Curti BD, Lewis KD, et al. Bempegaldesleukin plus nivolumab in first-line metastatic melanoma. J Clin Oncol. 2021;39:2914–25. doi: 10.1200/JCO.21.00675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tannir NM, Cho DC, Diab A, Sznol M, Bilen MA, Balar AV, et al. Bempegaldesleukin plus nivolumab in first-line renal cell carcinoma: results from the PIVOT-02 study. J Immunother Cancer. 2022;10:e004419. doi: 10.1136/jitc-2021-004419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khushalani NI, Diab A, Ascierto PA, Larkin J, Sandhu S, Sznol M, et al. Bempegaldesleukin plus nivolumab in untreated, unresectable or metastatic melanoma: Phase III PIVOT IO 001 study design. Future Oncol. 2020;16:2165–75. doi: 10.2217/fon-2020-0351. [DOI] [PubMed] [Google Scholar]
- 31.Mayes PA, Hance KW, Hoos A. The promise and challenges of immune agonist antibody development in cancer. Nat Rev Drug Discov. 2018;17:509–27. doi: 10.1038/nrd.2018.75. [DOI] [PubMed] [Google Scholar]
- 32.Pourakbari R, Hajizadeh F, Parhizkar F, Aghebati-Maleki A, Mansouri S, Aghebati-Maleki L. Co-stimulatory agonists: an insight into the immunotherapy of cancer. Excli J. 2021;20:1055–85. doi: 10.17179/excli2021-3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013;73:7189–98. doi: 10.1158/0008-5472.CAN-12-4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duhen R, Ballesteros-Merino C, Frye AK, Tran E, Rajamanickam V, Chang SC, et al. Neoadjuvant anti-OX40 (MEDI6469) therapy in patients with head and neck squamous cell carcinoma activates and expands antigen-specific tumor-infiltrating T cells. Nat Commun. 2021;12:1047. doi: 10.1038/s41467-021-21383-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zappasodi R, Sirard C, Li Y, Budhu S, Abu-Akeel M, Liu C, et al. Rational design of anti-GITR-based combination immunotherapy. Nat Med. 2019;25:759–66. doi: 10.1038/s41591-019-0420-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang R, Gao C, Raymond M, Dito G, Kabbabe D, Shao X, et al. An integrative approach to inform optimal administration of OX40 agonist antibodies in patients with advanced solid tumors. Clin Cancer Res. 2019;25:6709–20. doi: 10.1158/1078-0432.CCR-19-0526. [DOI] [PubMed] [Google Scholar]
- 37.Buchan SL, Fallatah M, Thirdborough SM, Taraban VY, Rogel A, Thomas LJ, et al. PD-1 blockade and CD27 stimulation activate distinct transcriptional programs that synergize for CD8+ T-cell-driven antitumor immunity. Clin Cancer Res. 2018;24:2383–94. doi: 10.1158/1078-0432.CCR-17-3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma Y, Li J, Wang H, Chiu Y, Kingsley CV, Fry D, et al. Combination of PD-1 inhibitor and OX40 agonist induces tumor rejection and immune memory in mouse models of pancreatic cancer. Gastroenterology. 2020;159:306–19.e12. doi: 10.1053/j.gastro.2020.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diggs LP, Ruf B, Ma C, Heinrich B, Cui L, Zhang Q, et al. CD40-mediated immune cell activation enhances response to anti-PD-1 in murine intrahepatic cholangiocarcinoma. J Hepatol. 2021;74:1145–54. doi: 10.1016/j.jhep.2020.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tolcher AW, Sznol M, Hu-Lieskovan S, Papadopoulos KP, Patnaik A, Rasco DW, et al. Phase Ib study of utomilumab (PF-05082566), a 4-1BB/CD137 agonist, in combination with pembrolizumab (MK-3475) in patients with advanced solid tumors. Clin Cancer Res. 2017;23:5349–57. doi: 10.1158/1078-0432.CCR-17-1243. [DOI] [PubMed] [Google Scholar]
- 41.O’Hara MH, O’Reilly EM, Varadhachary G, Wolff RA, Wainberg ZA, Ko AH, et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: an open-label, multicentre, phase 1b study. Lancet Oncol. 2021;22:118–31. doi: 10.1016/S1470-2045(20)30532-5. [DOI] [PubMed] [Google Scholar]
- 42.Sermer D, Brentjens R. CAR T-cell therapy: full speed ahead. Hematol Oncol. 2019;37:95–100. doi: 10.1002/hon.2591. [DOI] [PubMed] [Google Scholar]
- 43.Holstein SA, Lunning MA. CAR T-cell therapy in hematologic malignancies: a voyage in progress. Clin Pharmacol Ther. 2020;107:112–22. doi: 10.1002/cpt.1674. [DOI] [PubMed] [Google Scholar]
- 44.Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126:3130–44. doi: 10.1172/JCI83092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19:5636–46. doi: 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- 46.Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood. 2017;129:1039–41. doi: 10.1182/blood-2016-09-738245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blass E, Ott PA. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol. 2021;18:215–29. doi: 10.1038/s41571-020-00460-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 49.Kinkead HL, Hopkins A, Lutz E, Wu AA, Yarchoan M, Cruz K, et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight. 2018;3:e122857. doi: 10.1172/jci.insight.122857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu GZ, Lynn GM, Jacobson O, Chen K, Liu Y, Zhang HM, et al. Albumin/vaccine nanocomplexes that assemble in vivo for combination cancer immunotherapy. Nat Commun. 2017;8:1954. doi: 10.1038/s41467-017-02191-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lopez JS, Camidge R, Iafolla M, Rottey S, Schuler M, Hellmann M, et al. Abstract CT301: A phase Ib study to evaluate RO7198457, an individualized Neoantigen Specific immunoTherapy (iNeST), in combination with atezolizumab in patients with locally advanced or metastatic solid tumors. Cancer Res. 2020;80:CT301-CT. doi: 10.1158/1538-7445.AM2020-CT301. [DOI] [Google Scholar]
- 52.Burris HA, Patel MR, Cho DC, Clarke JM, Gutierrez M, Zaks TZ, et al. A phase I multicenter study to assess the safety, tolerability, and immunogenicity of mRNA-4157 alone in patients with resected solid tumors and in combination with pembrolizumab in patients with unresectable solid tumors. J Clin Oncol. 2019;37(15_suppl):2523. doi: 10.1200/JCO.2019.37.15_suppl.2523. [DOI] [Google Scholar]
- 53.Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222–6. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 54.Ott PA, Hu-Lieskovan S, Chmielowski B, Govindan R, Naing A, Bhardwaj N, et al. A phase Ib trial of personalized neoantigen therapy plus anti-PD-1 in patients with advanced melanoma, non-small cell lung cancer, or bladder cancer. Cell. 2020;183:347–62. doi: 10.1016/j.cell.2020.08.053. [DOI] [PubMed] [Google Scholar]
- 55.III HAB. Patel MR, Cho DC, Clarke JM, Gutierrez M, Zaks TZ, et al. A phase 1, open-label, multicenter study to assess the safety, tolerability, and immunogenicity of mRNA-4157 alone in subjects with resected solid tumors and in combination with pembrolizumab in subjects with unresectable solid tumors (Keynote-603) J Glob Oncol. 2019;5:93. doi: 10.1200/JGO.2019.5.suppl.93. [DOI] [Google Scholar]
- 56.Wang J, Mamuti M, Wang H. Therapeutic vaccines for cancer immunotherapy. ACS Biomater Sci Eng. 2020;6:6036–52. doi: 10.1021/acsbiomaterials.0c01201. [DOI] [PubMed] [Google Scholar]
- 57.Sarnaik AA, Yu B, Yu D, Morelli D, Hall M, Bogle D, et al. Extended dose ipilimumab with a peptide vaccine: immune correlates associated with clinical benefit in patients with resected high-risk stage IIIc/IV melanoma. Clin Cancer Res. 2011;17:896–906. doi: 10.1158/1078-0432.CCR-10-2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsujikawa T, Crocenzi T, Durham JN, Sugar EA, Wu AA, Onners B, et al. Evaluation of cyclophosphamide/GVAX pancreas followed by listeria-mesothelin (CRS-207) with or without nivolumab in patients with pancreatic cancer. Clin Cancer Res. 2020;26:3578–88. doi: 10.1158/1078-0432.CCR-19-3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kohlhapp FJ, Kaufman HL. Molecular pathways: mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clin Cancer Res. 2016;22:1048–54. doi: 10.1158/1078-0432.CCR-15-2667. [DOI] [PubMed] [Google Scholar]
- 60.Andtbacka RHI, Collichio F, Harrington KJ, Middleton MR, Downey G, hrling K, et al. Final analyses of OPTiM: a randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III-IV melanoma. J Immunother Cancer. 2019;7:145. doi: 10.1186/s40425-019-0623-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6:226ra32. doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bourgeois-Daigneault MC, Roy DG, Aitken AS, El Sayes N, Martin NT, Varette O, et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple- negative breast cancer to immune checkpoint therapy. Sci Transl Med. 2018;10:eaao1641. doi: 10.1126/scitranslmed.aao1641. [DOI] [PubMed] [Google Scholar]
- 63.Chesney J, Puzanov I, Collichio F, Singh P, Milhem MM, Glaspy J, et al. Randomized, open-label phase II study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. 2018;36:1658–67. doi: 10.1200/JCO.2017.73.7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55. doi: 10.1016/S1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 65.Li XL, Liu R, Su X, Pan YS, Han XF, Shao CS, et al. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol Cancer. 2019;18:177. doi: 10.1186/s12943-019-1102-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harada K, Dong XC, Estrella JS, Correa AM, Xu Y, Hofstetter WL, et al. Tumor-associated macrophage infiltration is highly associated with PD-L1 expression in gastric adenocarcinoma. Gastric Cancer. 2018;21:31–40. doi: 10.1007/s10120-017-0760-3. [DOI] [PubMed] [Google Scholar]
- 67.Tsukamoto M, Imai K, Ishimoto T, Komohara Y, Yamashita YI, Nakagawa S, et al. PD-L1 expression enhancement by infiltrating macrophage-derived tumor necrosis factor-alpha leads to poor pancreatic cancer prognosis. Cancer Sci. 2019;110:310–20. doi: 10.1111/cas.13874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xiang X, Wang J, Lu D, Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct Target Ther. 2021;6:75. doi: 10.1038/s41392-021-00484-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Omstead AN, Paskewicz M, Gorbunova A, Zheng P, Salvitti MS, Mansoor R, et al. CSF-1R inhibitor, pexidartinib, sensitizes esophageal adenocarcinoma to PD-1 immune checkpoint blockade in a rat model. Carcinogenesis. 2022;bgac043. 10.1093/carcin/bgac043. [DOI] [PubMed]
- 70.Shi G, Yang Q, Zhang Y, Jiang Q, Lin Y, Yang S, et al. Modulating the tumor microenvironment via oncolytic viruses and CSF-1R inhibition synergistically enhances anti-PD-1 immunotherapy. Mol Ther. 2019;27:244–60. doi: 10.1016/j.ymthe.2018.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gomez-Roca C, Cassier P, Zamarin D, Machiels JP, Luis Perez Gracia J. Stephen Hodi F, et al. Anti-CSF-1R emactuzumab in combination with anti-PD-L1 atezolizumab in advanced solid tumor patients naive or experienced for immune checkpoint blockade. J Immunother Cancer. 2022;10:e004076. doi: 10.1136/jitc-2021-004076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Razak AR, Cleary JM, Moreno V, Boyer M, Calvo Aller E, Edenfield W, et al. Safety and efficacy of AMG 820, an anti-colony-stimulating factor 1 receptor antibody, in combination with pembrolizumab in adults with advanced solid tumors. J Immunother Cancer. 2020;8:e001006. doi: 10.1136/jitc-2020-001006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haag GM, Springfeld C, Grun B, Apostolidis L, Zschabitz S, Dietrich M, et al. Pembrolizumab and maraviroc in refractory mismatch repair proficient/microsatellite-stable metastatic colorectal cancer - The PICCASSO phase I trial. Eur J Cancer. 2022;167:112–22. doi: 10.1016/j.ejca.2022.03.017. [DOI] [PubMed] [Google Scholar]
- 74.Bockorny B, Semenisty V, Macarulla T, Borazanci E, Wolpin BM, Stemmer SM, et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat Med. 2020;26:878–85. doi: 10.1038/s41591-020-0880-x. [DOI] [PubMed] [Google Scholar]
- 75.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015;11:1018–30. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, Raulet DH. Tumor-derived cGAMP triggers a sting-mediated interferon response in non-tumor cells to activate the NK cell response. Immunity. 2018;49:754. doi: 10.1016/j.immuni.2018.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu W, Kim GB, Krump NA, Zhou Y, Riley JL, You J. Selective reactivation of STING signaling to target Merkel cell carcinoma. Proc Natl Acad Sci USA. 2020;117:13730–9. doi: 10.1073/pnas.1919690117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 2016;14:282–97. doi: 10.1016/j.celrep.2015.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xia T, Konno H, Barber GN. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 2016;76:6747–59. doi: 10.1158/0008-5472.CAN-16-1404. [DOI] [PubMed] [Google Scholar]
- 80.Kim DS, Endo A, Fang FG, Huang KC, Bao X, Choi HW, et al. E7766, a macrocycle-bridged stimulator of interferon genes (STING) agonist with potent pan-genotypic activity. ChemMedChem. 2021;16:1740–3. doi: 10.1002/cmdc.202100068. [DOI] [PubMed] [Google Scholar]
- 81.Chang W, Altman MD, Lesburg CA, Perera SA, Piesvaux JA, Schroeder GK, et al. Discovery of MK-1454: a potent cyclic dinucleotide stimulator of interferon genes agonist for the treatment of cancer. J Med Chem. 2022;65:5675–89. doi: 10.1021/acs.jmedchem.1c02197. [DOI] [PubMed] [Google Scholar]
- 82.Ding CY, Song ZL, Shen AC, Chen TT, Zhang A. Small molecules targeting the innate immune cGAS-STING-TBK1 signaling pathway. Acta Pharm Sin B. 2020;10:2272–98. doi: 10.1016/j.apsb.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ding L, Huang XF, Dong GJ, Hu EL, Chen S, Wang TT, et al. Activated STING enhances Tregs infiltration in the HPV-related carcinogenesis of tongue squamous cells via the c-jun/CCL22 signal. Bba-Mol Basis Dis. 2015;1852:2494–503. doi: 10.1016/j.bbadis.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 84.Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, Arbab AS, et al. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res. 2016;76:2076–81. doi: 10.1158/0008-5472.CAN-15-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grabosch S, Bulatovic M, Zeng FTZ, Ma TZ, Zhang LX, Ross M, et al. Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene. 2019;38:2380–93. doi: 10.1038/s41388-018-0581-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Harrington K, Brody J, Ingham M, Strauss J, Cemerski S, Wang M, et al. LBA15Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann Oncol. 2018;29(suppl_8):VIII712. doi: 10.1093/annonc/mdy424.015. [DOI] [Google Scholar]
- 87.Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, Curran MA. Intratumoral STING activation with T-cell checkpoint modulation generates systemic antitumor immunity. Cancer Immunol Res. 2017;5:676–84. doi: 10.1158/2326-6066.CIR-17-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shae D, Becker KW, Christov P, Yun DS, Lytton-Jean AKR, Sevimli S, et al. Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat Nanotechnol. 2019;14:269. doi: 10.1038/s41565-018-0342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203:1259–71. doi: 10.1084/jem.20052494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L, et al. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. 2008;15:3–12. doi: 10.1038/sj.cdd.4402269. [DOI] [PubMed] [Google Scholar]
- 91.Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. 2014;124:687–95. doi: 10.1172/JCI67313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barker HE, Paget JT, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer. 2015;15:409–25. doi: 10.1038/nrc3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC. Using immunotherapy to boost the abscopal effect. Nat Rev Cancer. 2018;18:313–22. doi: 10.1038/nrc.2018.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;366:925–31. doi: 10.1056/NEJMoa1112824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Abuodeh Y, Venkat P, Kim S. Systematic review of case reports on the abscopal effect. Curr Probl Cancer. 2016;40:25–37. doi: 10.1016/j.currproblcancer.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 96.Rodriguez-Ruiz ME, Vanpouille-Box C, Melero I, Formenti SC, Demaria S. Immunological mechanisms responsible for radiation-induced abscopal effect. Trends Immunol. 2018;39:644–55. doi: 10.1016/j.it.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Park SS, Dong H, Liu X, Harrington SM, Krco CJ, Grams MP, et al. PD-1 restrains radiotherapy-induced abscopal effect. Cancer Immunol Res. 2015;3:610–9. doi: 10.1158/2326-6066.CIR-14-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Reynders K, Illidge T, Siva S, Chang JY, De Ruysscher D. The abscopal effect of local radiotherapy: using immunotherapy to make a rare event clinically relevant. Cancer Treat Rev. 2015;41:503–10. doi: 10.1016/j.ctrv.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dudzinski SO, Cameron BD, Wang J, Rathmell JC, Giorgio TD, Kirschner AN. Combination immunotherapy and radiotherapy causes an abscopal treatment response in a mouse model of castration resistant prostate cancer. J Immunother Cancer. 2019;7:218. doi: 10.1186/s40425-019-0704-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ji D, Song C, Li Y, Xia J, Wu Y, Jia J, et al. Combination of radiotherapy and suppression of Tregs enhances abscopal antitumor effect and inhibits metastasis in rectal cancer. J Immunother Cancer. 2020;8:e000826. doi: 10.1136/jitc-2020-000826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen D, Verma V, Patel RR, Barsoumian HB, Cortez MA, Welsh JW. Absolute lymphocyte count predicts abscopal responses and outcomes in patients receiving combined immunotherapy and radiation therapy: analysis of 3 phase 1/2 trials. Int J Radiat Oncol Biol Phys. 2020;108:196–203. doi: 10.1016/j.ijrobp.2020.01.032. [DOI] [PubMed] [Google Scholar]
- 102.Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N Engl J Med. 2018;379:2342–50. doi: 10.1056/NEJMoa1809697. [DOI] [PubMed] [Google Scholar]
- 103.Fizazi K, Drake CG, Beer TM, Kwon ED, Scher HI, Gerritsen WR, et al. Final analysis of the ipilimumab versus placebo following radiotherapy phase III trial in postdocetaxel metastatic castration-resistant prostate cancer identifies an excess of long-term survivors. Eur Urol. 2020;78:822–30. doi: 10.1016/j.eururo.2020.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Grassberger C, Ellsworth SG, Wilks MQ, Keane FK, Loeffler JS. Assessing the interactions between radiotherapy and antitumour immunity. Nat Rev Clin Oncol. 2019;16:729–45. doi: 10.1038/s41571-019-0238-9. [DOI] [PubMed] [Google Scholar]
- 105.Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity. 2013;39:74–88. doi: 10.1016/j.immuni.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 106.Wang Q, Ju X, Wang J, Fan Y, Ren M, Zhang H. Immunogenic cell death in anticancer chemotherapy and its impact on clinical studies. Cancer Lett. 2018;438:17–23. doi: 10.1016/j.canlet.2018.08.028. [DOI] [PubMed] [Google Scholar]
- 107.Ramakrishnan R, Huang C, Cho HI, Lloyd M, Johnson J, Ren X, et al. Autophagy induced by conventional chemotherapy mediates tumor cell sensitivity to immunotherapy. Cancer Res. 2012;72:5483–93. doi: 10.1158/0008-5472.CAN-12-2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Galluzzi L, Humeau J, Buque A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol. 2020;17:725–41. doi: 10.1038/s41571-020-0413-z. [DOI] [PubMed] [Google Scholar]
- 109.Rodriguez-Abreu D, Powell SF, Hochmair MJ, Gadgeel S, Esteban E, Felip E, et al. Pemetrexed plus platinum with or without pembrolizumab in patients with previously untreated metastatic nonsquamous NSCLC: protocol-specified final analysis from KEYNOTE-189. Ann Oncol. 2021;32:881–95. doi: 10.1016/j.annonc.2021.04.008. [DOI] [PubMed] [Google Scholar]
- 110.Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378:2078–92. doi: 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 111.Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im SA, Yusof MM, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet. 2020;396:1817–28. doi: 10.1016/S0140-6736(20)32531-9. [DOI] [PubMed] [Google Scholar]
- 112.Awad MM, Gadgeel SM, Borghaei H, Patnaik A, Yang JCH, Powell SF, et al. Long-term overall survival From KEYNOTE-021 cohort G: pemetrexed and carboplatin with or without pembrolizumab as first-line therapy for advanced nonsquamous NSCLC. J Thorac Oncol. 2021;16:162–8. doi: 10.1016/j.jtho.2020.09.015. [DOI] [PubMed] [Google Scholar]
- 113.Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med. 2018;379:2220–9. doi: 10.1056/NEJMoa1809064. [DOI] [PubMed] [Google Scholar]
- 114.Luo H, Lu J, Bai Y, Mao T, Wang J, Fan Q, et al. Effect of camrelizumab vs placebo added to chemotherapy on survival and progression-free survival in patients with advanced or metastatic esophageal squamous cell carcinoma: The ESCORT-1st randomized clinical trial. JAMA. 2021;326:916–25. doi: 10.1001/jama.2021.12836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91:1071–121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Viallard C, Larrivee B. Tumor angiogenesis and vascular normalization: alternative therapeutic targets. Angiogenesis. 2017;20:409–26. doi: 10.1007/s10456-017-9562-9. [DOI] [PubMed] [Google Scholar]
- 117.Huang Y, Kim BYS, Chan CK, Hahn SM, Weissman IL, Jiang W. Improving immune-vascular crosstalk for cancer immunotherapy. Nat Rev Immunol. 2018;18:195–203. doi: 10.1038/nri.2017.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol. 2018;15:325–40. doi: 10.1038/nrclinonc.2018.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, et al. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013;73:539–49. doi: 10.1158/0008-5472.CAN-12-2325. [DOI] [PubMed] [Google Scholar]
- 120.Kammertoens T, Friese C, Arina A, Idel C, Briesemeister D, Rothe M, et al. Tumour ischaemia by interferon-gamma resembles physiological blood vessel regression. Nature. 2017;545:98–102. doi: 10.1038/nature22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T, et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature. 2017;544:250–4. doi: 10.1038/nature21724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475:226–30. doi: 10.1038/nature10169. [DOI] [PubMed] [Google Scholar]
- 123.Carretero R, Sektioglu IM, Garbi N, Salgado OC, Beckhove P, Hammerling GJ. Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of CD8+ T cells. Nat Immunol. 2015;16:609–17. doi: 10.1038/ni.3159. [DOI] [PubMed] [Google Scholar]
- 124.Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380:1116–27. doi: 10.1056/NEJMoa1816714. [DOI] [PubMed] [Google Scholar]
- 125.Powles T, Plimack ER, Soulieres D, Waddell T, Stus V, Gafanov R, et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol. 2020;21:1563–73. doi: 10.1016/S1470-2045(20)30436-8. [DOI] [PubMed] [Google Scholar]
- 126.Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382:1894–905. doi: 10.1056/NEJMoa1915745. [DOI] [PubMed] [Google Scholar]
- 127.Galle PR, Finn RS, Qin S, Ikeda M, Zhu AX, Kim TY, et al. Patient-reported outcomes with atezolizumab plus bevacizumab versus sorafenib in patients with unresectable hepatocellular carcinoma (IMbrave150): an open-label, randomised, phase 3 trial. Lancet Oncol. 2021;22:991–1001. doi: 10.1016/S1470-2045(21)00151-0. [DOI] [PubMed] [Google Scholar]
- 128.McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med. 2018;24:749–57. doi: 10.1038/s41591-018-0053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Choueiri TK, Motzer RJ, Rini BI, Haanen J, Campbell MT, Venugopal B, et al. Updated efficacy results from the JAVELIN Renal 101 trial: first-line avelumab plus axitinib versus sunitinib in patients with advanced renal cell carcinoma. Ann Oncol. 2020;31:1030–9. doi: 10.1016/j.annonc.2020.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Motzer RJ, Robbins PB, Powles T, Albiges L, Haanen JB, Larkin J, et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: biomarker analysis of the phase 3 JAVELIN Renal 101 trial. Nat Med. 2020;26:1733–41. doi: 10.1038/s41591-020-1044-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhu S, Zhang T, Zheng L, Liu H, Song W, Liu D, et al. Combination strategies to maximize the benefits of cancer immunotherapy. J Hematol Oncol. 2021;14:156. doi: 10.1186/s13045-021-01164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548:471–5. doi: 10.1038/nature23465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kim G, McKee AE, Ning YM, Hazarika M, Theoret M, Johnson JR, et al. FDA approval summary: vemurafenib for treatment of unresectable or metastatic melanoma with the BRAFV600E mutation. Clin Cancer Res. 2014;20:4994–5000. doi: 10.1158/1078-0432.CCR-14-0776. [DOI] [PubMed] [Google Scholar]
- 134.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–65. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 135.Petroni G, Buque A, Zitvogel L, Kroemer G, Galluzzi L. Immunomodulation by targeted anticancer agents. Cancer Cell. 2021;39:310–45. doi: 10.1016/j.ccell.2020.11.009. [DOI] [PubMed] [Google Scholar]
- 136.Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res. 2013;19:598–609. doi: 10.1158/1078-0432.CCR-12-2731. [DOI] [PubMed] [Google Scholar]
- 137.Atefi M, Avramis E, Lassen A, Wong DJ, Robert L, Foulad D, et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res. 2014;20:3446–57. doi: 10.1158/1078-0432.CCR-13-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gutzmer R, Stroyakovskiy D, Gogas H, Robert C, Lewis K, Protsenko S, et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;395:1835–44. doi: 10.1016/S0140-6736(20)30934-X. [DOI] [PubMed] [Google Scholar]
- 139.Brufsky A, Kim SB, Zvirbule Z, Eniu A, Mebis J, Sohn JH, et al. A phase II randomized trial of cobimetinib plus chemotherapy, with or without atezolizumab, as first-line treatment for patients with locally advanced or metastatic triple-negative breast cancer (COLET): primary analysis. Ann Oncol. 2021;32:652–60. doi: 10.1016/j.annonc.2021.01.065. [DOI] [PubMed] [Google Scholar]
- 140.Ladenstein R, Potschger U, Valteau-Couanet D, Luksch R, Castel V, Yaniv I, et al. Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018;19:1617–29. doi: 10.1016/S1470-2045(18)30578-3. [DOI] [PubMed] [Google Scholar]
- 141.Li H, Liu ZY, Wu N, Chen YC, Cheng Q, Wang J. PARP inhibitor resistance: the underlying mechanisms and clinical implications. Mol Cancer. 2020;19:107. doi: 10.1186/s12943-020-01227-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Francica P, Rottenberg S. Mechanisms of PARP inhibitor resistance in cancer and insights into the DNA damage response. Genome Med. 2018;10:101. doi: 10.1186/s13073-018-0612-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sen T, Rodriguez BL, Chen LM, Della Corte CM, Morikawa N, Fujimoto J, et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019;9:646–61. doi: 10.1158/2159-8290.CD-18-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, et al. PARP inhibitor efficacy depends on CD8+ T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Disco. 2019;9:722–37. doi: 10.1158/2159-8290.CD-18-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23:3711–20. doi: 10.1158/1078-0432.CCR-16-3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, et al. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res. 2019;79:311–9. doi: 10.1158/0008-5472.CAN-18-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Domchek SM, Postel-Vinay S, Im SA, Park YH, Delord JP, Italiano A, et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020;21:1155–64. doi: 10.1016/S1470-2045(20)30324-7. [DOI] [PubMed] [Google Scholar]
- 148.Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, et al. Single-arm phases 1 and 2 trial of niraparib in combination with pembrolizumab in patients with recurrent platinum-resistant ovarian carcinoma. JAMA Oncol. 2019;5:1141–9. doi: 10.1001/jamaoncol.2019.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chau CH, Steeg PS, Figg WD. Antibody-drug conjugates for cancer. Lancet. 2019;394:793–804. doi: 10.1016/S0140-6736(19)31774-X. [DOI] [PubMed] [Google Scholar]
- 150.Rosenberg JE, O’Donnell PH, Balar AV, McGregor BA, Heath EI, Yu EY, et al. Pivotal trial of enfortumab vedotin in urothelial carcinoma after platinum and anti-programmed death 1/programmed death ligand 1 therapy. J Clin Oncol. 2019;37:2592–600. doi: 10.1200/JCO.19.01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Challita-Eid PM, Satpayev D, Yang P, An ZL, Morrison K, Shostak Y, et al. Enfortumab vedotin antibody-drug conjugate targeting nectin-4 is a highly potent therapeutic agent in multiple preclinical cancer models. Cancer Res. 2016;76:3003–13. doi: 10.1158/0008-5472.CAN-15-1313. [DOI] [PubMed] [Google Scholar]
- 152.Targeting nectin-4 in bladder cancer. Cancer Discov. 2017; 7: OF3. https://aacrjournals.org/cancerdiscovery/article/7/8/OF3/6501/Targeting-Nectin-4-in-Bladder-CancerTargeting. [DOI] [PubMed]
- 153.Rosenberg JE, Flaig TW, Friedlander TW, Milowsky MI, Srinivas S, Petrylak DP, et al. Study EV-103: preliminary durability results of enfortumab vedotin plus pembrolizumab for locally advanced or metastatic urothelial carcinoma. J Clin Oncol. 2020;38:441. doi: 10.1200/JCO.2020.38.6_suppl.441. [DOI] [Google Scholar]
- 154.Cheng WY, Wu CY, Yu J. The role of gut microbiota in cancer treatment: friend or foe? Gut. 2020;69:1867–76. doi: 10.1136/gutjnl-2020-321153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chen W, Wang Y, Qin M, Zhang X, Zhang Z, Sun X, et al. Bacteria-driven hypoxia targeting for combined biotherapy and photothermal therapy. ACS Nano. 2018;12:5995–6005. doi: 10.1021/acsnano.8b02235. [DOI] [PubMed] [Google Scholar]
- 156.Avogadri F, Martinoli C, Petrovska L, Chiodoni C, Transidico P, Bronte V, et al. Cancer immunotherapy based on killing of salmonella-infected tumor cells. Cancer Res. 2005;65:3920–7. doi: 10.1158/0008-5472.CAN-04-3002. [DOI] [PubMed] [Google Scholar]
- 157.Lin D, Feng X, Mai B, Li X, Wang F, Liu J, et al. Bacterial-based cancer therapy: an emerging toolbox for targeted drug/gene delivery. Biomaterials. 2021;277:121124. doi: 10.1016/j.biomaterials.2021.121124. [DOI] [PubMed] [Google Scholar]
- 158.Stern C, Kasnitz N, Kocijancic D, Trittel S, Riese P, Guzman CA, et al. Induction of CD4+ and CD8+ anti-tumor effector T cell responses by bacteria mediated tumor therapy. Int J Cancer. 2015;137:2019–28. doi: 10.1002/ijc.29567. [DOI] [PubMed] [Google Scholar]
- 159.St Jean AT, Swofford CA, Panteli JT, Brentzel ZJ, Forbes NS. Bacterial delivery of Staphylococcus aureus alpha-hemolysin causes regression and necrosis in murine tumors. Mol Ther. 2014;22:1266–74. doi: 10.1038/mt.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.al-Ramadi BK, Adeghate E, Mustafa N, Ponery AS, Fernandez-Cabezudo MJ. Cytokine expression by attenuated intracellular bacteria regulates the immune response to infection: the Salmonella model. Mol Immunol. 2002;38:931–40. doi: 10.1016/S0161-5890(02)00020-2. [DOI] [PubMed] [Google Scholar]
- 161.Zhou S, Gravekamp C, Bermudes D, Liu K. Tumour-targeting bacteria engineered to fight cancer. Nat Rev Cancer. 2018;18:727–43. doi: 10.1038/s41568-018-0070-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mughal MJ, Kwok HF. Multidimensional role of bacteria in cancer: Mechanisms insight, diagnostic, preventive and therapeutic potential. Semin Cancer Biol. 2021. S1044-579X(21)00181-4. 10.1016/j.semcancer.2021.06.011. [DOI] [PubMed]
- 163.Lou X, Chen Z, He Z, Sun M, Sun J. Bacteria-mediated synergistic cancer therapy: small microbiome has a big hope. Nanomicro Lett. 2021;13:37. doi: 10.1007/s40820-020-00560-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zhao T, Feng Y, Guo M, Zhang C, Wu Q, Chen J, et al. Combination of attenuated Salmonella carrying PD-1 siRNA with nifuroxazide for colon cancer therapy. J Cell Biochem. 2020;121:1973–85. doi: 10.1002/jcb.29432. [DOI] [PubMed] [Google Scholar]
- 165.Zhao T, Wei T, Guo J, Wang Y, Shi X, Guo S, et al. PD-1-siRNA delivered by attenuated Salmonella enhances the antimelanoma effect of pimozide. Cell Death Dis. 2019;10:164. doi: 10.1038/s41419-019-1418-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chen WF, Guo ZF, Zhu YN, Qiao N, Zhang ZR, Sun X. Combination of bacterial-photothermal therapy with an anti-PD-1 peptide depot for enhanced immunity against advanced cancer. Adv Funct Mater. 2020;30:1906623. doi: 10.1002/adfm.201906623. [DOI] [Google Scholar]
- 167.Dubin K, Callahan MK, Ren BY, Khanin R, Viale A, Ling LL, et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun. 2016;7:10391. doi: 10.1038/ncomms10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chang AE, Golob JL, Schmidt TM, Peltier DC, Lao CD, Tewari M. Targeting the gut microbiome to mitigate immunotherapy-induced colitis in cancer. Trends Cancer. 2021;7:583–93. doi: 10.1016/j.trecan.2021.02.005. [DOI] [PubMed] [Google Scholar]
- 169.Wang Y, Wiesnoski DH, Helmink BA, Gopalakrishnan V, Choi K, DuPont HL, et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat Med. 2018;24:1804–8. doi: 10.1038/s41591-018-0238-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhao F, Evans K, Xiao C, DeVito N, Theivanthiran B, Holtzhausen A, et al. Stromal fibroblasts mediate anti-PD-1 resistance via MMP-9 and dictate TGFbeta inhibitor sequencing in melanoma. Cancer Immunol Res. 2018;6:1459–71. doi: 10.1158/2326-6066.CIR-18-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang YL, et al. TGF beta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, et al. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res. 2016;76:4124–35. doi: 10.1158/0008-5472.CAN-15-2973. [DOI] [PubMed] [Google Scholar]
- 173.Scott AM, Wiseman G, Welt S, Adjei A, Lee FT, Hopkins W, et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res. 2003;9:1639–47. [PubMed] [Google Scholar]
- 174.Hofheinz RD, al-Batran SE, Hartmann F, Hartung G, Jager D, Renner C, et al. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie. 2003;26:44–8. doi: 10.1159/000069863. [DOI] [PubMed] [Google Scholar]
- 175.Ohshio Y, Hanaoka J, Kontani K, Teramoto K. Tranilast inhibits the function of cancer-associated fibroblasts responsible for the induction of immune suppressor cell types. Scand J Immunol. 2014;80:408–16. doi: 10.1111/sji.12242. [DOI] [PubMed] [Google Scholar]
- 176.Ohshio Y, Teramoto K, Hanaoka J, Tezuka N, Itoh Y, Asai T, et al. Cancer-associated fibroblast-targeted strategy enhances antitumor immune responses in dendritic cell-based vaccine. Cancer Sci. 2015;106:134–42. doi: 10.1111/cas.12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22:851–60. doi: 10.1038/nm.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. 2013;110:20212–7. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330:827–30. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- 180.Huang TX, Tan XY, Huang HS, Li YT, Liu BL, Liu KS, et al. Targeting cancer-associated fibroblast-secreted WNT2 restores dendritic cell-mediated antitumour immunity. Gut. 2022;71:333–44. [DOI] [PMC free article] [PubMed]
- 181.Strauss J, Heery CR, Schlom J, Madan RA, Cao L, Kang ZG, et al. Phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGF beta, in advanced solid tumors. Clin Cancer Res. 2018;24:1287–95. doi: 10.1158/1078-0432.CCR-17-2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Waldhauer I, Gonzalez-Nicolini V, Freimoser-Grundschober A, Nayak TK, Fahrni L, Hosse RJ, et al. Simlukafusp alfa (FAP-IL2v) immunocytokine is a versatile combination partner for cancer immunotherapy. MAbs. 2021;13:1913791. doi: 10.1080/19420862.2021.1913791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17:e542–e51. doi: 10.1016/S1470-2045(16)30406-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Aguilar EJ, Ricciuti B, Gainor JF, Kehl KL, Kravets S, Dahlberg S, et al. Outcomes to first-line pembrolizumab in patients with non-small-cell lung cancer and very high PD-L1 expression. Ann Oncol. 2019;30:1653–9. doi: 10.1093/annonc/mdz288. [DOI] [PubMed] [Google Scholar]
- 185.Herbst RS, Giaccone G, de Marinis F, Reinmuth N, Vergnenegre A, Barrios CH, et al. Atezolizumab for first-line treatment of PD-L1-selected patients with NSCLC. N Engl J Med. 2020;383:1328–39. doi: 10.1056/NEJMoa1917346. [DOI] [PubMed] [Google Scholar]
- 186.Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med. 2017;376:2415–26. doi: 10.1056/NEJMoa1613493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Rizvi NA, Cho BC, Reinmuth N, Lee KH, Luft A, Ahn MJ, et al. Durvalumab with or without tremelimumab vs standard chemotherapy in first-line treatment of metastatic non-small cell lung cancer the MYSTIC phase 3 randomized clinical trial. JAMA Oncol. 2020;6:661–74. doi: 10.1001/jamaoncol.2020.0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Paz-Ares LG, Ramalingam SS, Ciuleanu TE, Lee JS, Urban L, Caro RB, et al. First-line nivolumab plus ipilimumab in advanced NSCLC: 4-year outcomes from the randomized, open-label, phase 3 checkmate 227 part 1 trial. J Thorac Oncol. 2022;17:289–308. doi: 10.1016/j.jtho.2021.09.010. [DOI] [PubMed] [Google Scholar]
- 189.West H, McCleod M, Hussein M, Morabito A, Rittmeyer A, Conter HJ, et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20:924–37. doi: 10.1016/S1470-2045(19)30167-6. [DOI] [PubMed] [Google Scholar]
- 190.Paz-Ares L, Vicente D, Tafreshi A, Robinson A, Soto Parra H, Mazieres J, et al. A randomized, placebo-controlled trial of pembrolizumab plus chemotherapy in patients with metastatic squamous NSCLC: protocol-specified final analysis of KEYNOTE-407. J Thorac Oncol. 2020;15:1657–69. doi: 10.1016/j.jtho.2020.06.015. [DOI] [PubMed] [Google Scholar]
- 191.Wang J, Lu S, Yu XM, Hu YP, Sun YP, Wang ZJ, et al. Tislelizumab plus chemotherapy vs chemotherapy alone as first-line treatment for advanced squamous non-small-cell lung cancer a phase 3 randomized clinical trial. JAMA Oncol. 2021;7:709–17. doi: 10.1001/jamaoncol.2021.0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Reck M, Ciuleanu T-E, Cobo M, Schenker M, Zurawski B, Janoski de Menezes J, et al. First-line nivolumab (NIVO) plus ipilimumab (IPI) plus two cycles of chemotherapy (chemo) versus chemo alone (4 cycles) in patients with advanced non-small cell lung cancer (NSCLC): Two-year update from CheckMate 9LA. J Clin Oncol. 2021;39:9000. doi: 10.1200/JCO.2021.39.15_suppl.9000. [DOI] [Google Scholar]
- 193.Janjigian YY, Shitara K, Moehler M, Garrido M, Salman P, Shen L, et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet. 2021;398:27–40. doi: 10.1016/S0140-6736(21)00797-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378:2288–301. doi: 10.1056/NEJMoa1716948. [DOI] [PubMed] [Google Scholar]
- 195.Haragan A, Field JK, Davies MPA, Escriu C, Gruver A, Gosney JR. Heterogeneity of PD-L1 expression in non-small cell lung cancer: Implications for specimen sampling in predicting treatment response. Lung Cancer. 2019;134:79–84. doi: 10.1016/j.lungcan.2019.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Vilain RE, Menzies AM, Wilmott JS, Kakavand H, Madore J, Guminski A, et al. Dynamic changes in PD-L1 expression and immune infiltrates early during treatment predict response to PD-1 blockade in melanoma. Clin Cancer Res. 2017;23:5024–33. doi: 10.1158/1078-0432.CCR-16-0698. [DOI] [PubMed] [Google Scholar]
- 197.Li D, Chen R, Wang YW, Fornace AJ, Jr, Li HH. Prior irradiation results in elevated programmed cell death protein 1 (PD-1) in T cells. Int J Radiat Biol. 2018;94:488–94. doi: 10.1080/09553002.2017.1400192. [DOI] [PubMed] [Google Scholar]
- 198.McLaughlin J, Han G, Schalper KA, Carvajal-Hausdorf D, Pelekanou V, Rehman J, et al. Quantitative assessment of the heterogeneity of PD-L1 expression in non-small-cell lung cancer. JAMA Oncol. 2016;2:46–54. doi: 10.1001/jamaoncol.2015.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hirsch FR, McElhinny A, Stanforth D, Ranger-Moore J, Jansson M, Kulangara K, et al. PD-L1 immunohistochemistry assays for lung cancer: results from phase 1 of the blueprint PD-L1 IHC assay comparison project. J Thorac Oncol. 2017;12:208–22. doi: 10.1016/j.jtho.2016.11.2228. [DOI] [PubMed] [Google Scholar]
- 200.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–99. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378:2093–104. doi: 10.1056/NEJMoa1801946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377:2500–1. doi: 10.1056/NEJMc1713444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21:1353–65. doi: 10.1016/S1470-2045(20)30445-9. [DOI] [PubMed] [Google Scholar]
- 205.Sharma P, Pachynski RK, Narayan V, Flechon A, Gravis G, Galsky MD, et al. Nivolumab plus ipilimumab for metastatic castration-resistant prostate cancer: preliminary analysis of patients in the checkmate 650 trial. Cancer Cell. 2020;38:489–99. doi: 10.1016/j.ccell.2020.08.007. [DOI] [PubMed] [Google Scholar]
- 206.Rozeman EA, Hoefsmit EP, Reijers ILM, Saw RPM, Versluis JM, Krijgsman O, et al. Survival and biomarker analyses from the OpACIN-neo and OpACIN neoadjuvant immunotherapy trials in stage III melanoma. Nat Med. 2021;27:256–63. doi: 10.1038/s41591-020-01211-7. [DOI] [PubMed] [Google Scholar]
- 207.Forschner A, Battke F, Hadaschik D, Schulze M, Weissgraeber S, Han CT, et al. Tumor mutation burden and circulating tumor DNA in combined CTLA-4 and PD-1 antibody therapy in metastatic melanoma - results of a prospective biomarker study. J Immunother Cancer. 2019;7:180. doi: 10.1186/s40425-019-0659-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Perets R, Bar J, Rasco DW, Ahn MJ, Yoh K, Kim DW, et al. Safety and efficacy of quavonlimab, a novel anti-CTLA-4 antibody (MK-1308), in combination with pembrolizumab in first-line advanced non-small-cell lung cancer. Ann Oncol. 2021;32:395–403. doi: 10.1016/j.annonc.2020.11.020. [DOI] [PubMed] [Google Scholar]
- 209.Forde PM, Anagnostou V, Sun Z, Dahlberg SE, Kindler HL, Niknafs N, et al. Durvalumab with platinum-pemetrexed for unresectable pleural mesothelioma: survival, genomic and immunologic analyses from the phase 2 PrE0505 trial. Nat Med. 2021;27:1910–20. doi: 10.1038/s41591-021-01541-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Huang X, He M, Peng H, Tong C, Liu Z, Zhang X, et al. Genomic profiling of advanced cervical cancer to predict response to programmed death-1 inhibitor combination therapy: a secondary analysis of the CLAP trial. J Immunother Cancer. 2021;9:e002223. doi: 10.1136/jitc-2020-002223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Tolaney SM, Barroso-Sousa R, Keenan T, Li TY, Trippa L, Vaz-Luis I, et al. Effect of eribulin with or without pembrolizumab on progression-free survival for patients with hormone receptor-positive, erbb2-negative metastatic breast cancer: a randomized clinical trial. Jama Oncol. 2020;6:1598–605. doi: 10.1001/jamaoncol.2020.3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Chen X, Wu X, Wu H, Gu Y, Shao Y, Shao Q, et al. Camrelizumab plus gemcitabine and oxaliplatin (GEMOX) in patients with advanced biliary tract cancer: a single-arm, open-label, phase II trial. J Immunother Cancer. 2020;8:e001240. doi: 10.1136/jitc-2020-001240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Leighl NB, Laurie SA, Goss GD, Hughes BGM, Stockler M, Tsao MS, et al. CCTG BR34: a randomized phase 2 trial of durvalumab and tremelimumab with or without platinum-based chemotherapy in patients with metastatic NSCLC. J Thorac Oncol. 2022;17:434–45. doi: 10.1016/j.jtho.2021.10.023. [DOI] [PubMed] [Google Scholar]
- 214.Garassino M, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, Speranza G, et al. OA04.06 evaluation of TMB in KEYNOTE-189: pembrolizumab plus chemotherapy vs placebo plus chemotherapy for nonsquamous NSCLC. J Thorac Oncol. 2019;14:S216–S7. doi: 10.1016/j.jtho.2019.08.427. [DOI] [Google Scholar]
- 215.Langer C, Gadgeel S, Borghaei H, Patnaik A, Powell S, Gentzler R, et al. OA04.05 KEYNOTE-021: TMB and outcomes for carboplatin and pemetrexed with or without pembrolizumab for nonsquamous NSCLC. J Thorac Oncol. 2019;14:S216. doi: 10.1016/j.jtho.2019.08.426. [DOI] [Google Scholar]
- 216.Jardim DL, Goodman A, Gagliato DD, Kurzrock R. The challenges of tumor mutational burden as an immunotherapy biomarker. Cancer Cell. 2021;39:154–73. doi: 10.1016/j.ccell.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Merino DM, McShane L, Butler M, Funari VA, Hellmann MD, Chaudhary R, et al. TMB standardization by alignment to reference standards: Phase II of the Friends of Cancer Research TMB Harmonization Project. J Clin Oncol. 2019;37:2624. doi: 10.1200/JCO.2019.37.15_suppl.2624. [DOI] [Google Scholar]
- 218.Gandara DR, Paul SM, Kowanetz M, Schleifman E, Zou W, Li Y, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med. 2018;24:1441. doi: 10.1038/s41591-018-0134-3. [DOI] [PubMed] [Google Scholar]
- 219.Wang ZJ, Duan JC, Cai SL, Han M, Dong H, Zhao J, et al. Assessment of blood tumor mutational burden as a potential biomarker for immunotherapy in patients with non-small cell lung cancer with use of a next-generation sequencing cancer gene panel. JAMA Oncol. 2019;5:696–702. doi: 10.1001/jamaoncol.2018.7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Mandal R, Samstein RM, Lee KW, Havel JJ, Wang H, Krishna C, et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science. 2019;364:485–91. doi: 10.1126/science.aau0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Andre T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. N Engl J Med. 2020;383:2207–18. doi: 10.1056/NEJMoa2017699. [DOI] [PubMed] [Google Scholar]
- 222.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36:773–9. doi: 10.1200/JCO.2017.76.9901. [DOI] [PubMed] [Google Scholar]
- 224.Hause RJ, Pritchard CC, Shendure J, Salipante SJ. Classification and characterization of microsatellite instability across 18 cancer types. Nat Med. 2016;22:1342–50. doi: 10.1038/nm.4191. [DOI] [PubMed] [Google Scholar]
- 225.Matson V, Fessler J, Bao R, Chongsuwat T, Zha YY, Alegre ML, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359:104. doi: 10.1126/science.aao3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103. doi: 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science. 2021;371:602–9. doi: 10.1126/science.abb5920. [DOI] [PubMed] [Google Scholar]
- 228.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–7. doi: 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 229.Zheng Y, Wang T, Tu X, Huang Y, Zhang H, Tan D, et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J Immunother Cancer. 2019;7:193. doi: 10.1186/s40425-019-0650-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Andrews MC, Duong CPM, Gopalakrishnan V, Iebba V, Chen WS, Derosa L, et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat Med. 2021;27:1432–41. doi: 10.1038/s41591-021-01406-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Martini G, Ciardiello D, Dallio M, Famiglietti V, Esposito L, Corte CMD, et al. Gut microbiota correlates with antitumor activity in patients with mCRC and NSCLC treated with cetuximab plus avelumab. Int J Cancer. 2022;151:473–80. doi: 10.1002/ijc.34033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Fessler J, Matson V, Gajewski TF. Exploring the emerging role of the microbiome in cancer immunotherapy. J Immunother Cancer. 2019;7:108. doi: 10.1186/s40425-019-0574-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Banchereau R, Leng N, Zill O, Sokol E, Liu G, Pavlick D, et al. Molecular determinants of response to PD-L1 blockade across tumor types. Nat Commun. 2021;12:3969. doi: 10.1038/s41467-021-24112-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Chowell D, Yoo SK, Valero C, Pastore A, Krishna C, Lee M, et al. Improved prediction of immune checkpoint blockade efficacy across multiple cancer types. Nat Biotechnol. 2022;40:499–506. doi: 10.1038/s41587-021-01070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N Engl J Med. 2019;381:2020–31. doi: 10.1056/NEJMoa1910231. [DOI] [PubMed] [Google Scholar]
- 236.Hodi FS, Chesney J, Pavlick AC, Robert C, Grossmann KF, McDermott DF, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016;17:1558–68. doi: 10.1016/S1470-2045(16)30366-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Planchard D, Reinmuth N, Orlov S, Fischer JR, Sugawara S, Mandziuk S, et al. ARCTIC: durvalumab with or without tremelimumab as third-line or later treatment of metastatic non-small-cell lung cancer. Ann Oncol. 2020;31:609–18. doi: 10.1016/j.annonc.2020.02.006. [DOI] [PubMed] [Google Scholar]
- 238.Siu LL, Even C, Mesia R, Remenar E, Daste A, Delord JP, et al. Safety and efficacy of durvalumab with or without tremelimumab in patients with PD-L1-low/negative recurrent or metastatic HNSCC the phase 2 CONDOR randomized clinical trial. JAMA Oncol. 2019;5:195–203. doi: 10.1001/jamaoncol.2018.4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Powles T, van der Heijden MS, Castellano D, Galsky MD, Loriot Y, Petrylak DP, et al. Durvalumab alone and durvalumab plus tremelimumab versus chemotherapy in previously untreated patients with unresectable, locally advanced or metastatic urothelial carcinoma (DANUBE): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2020;21:1574–88. doi: 10.1016/S1470-2045(20)30541-6. [DOI] [PubMed] [Google Scholar]
- 240.Schoenfeld JD, Hanna GJ, Jo VY, Rawal B, Chen YH, Catalano PS, et al. Neoadjuvant nivolumab or nivolumab plus ipilimumab in untreated oral cavity squamous cell carcinoma: a phase 2 open-label randomized clinical trial. JAMA Oncol. 2020;6:1563–70. doi: 10.1001/jamaoncol.2020.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Baas P, Scherpereel A, Nowak AK, Fujimoto N, Peters S, Tsao AS, et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): a multicentre, randomised, open-label, phase 3 trial. Lancet. 2021;397:375–86. doi: 10.1016/S0140-6736(20)32714-8. [DOI] [PubMed] [Google Scholar]
- 242.Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after chemoradiotherapy in stage iii non-small-cell lung cancer. N Engl J Med. 2017;377:1919–29. doi: 10.1056/NEJMoa1709937. [DOI] [PubMed] [Google Scholar]
- 243.Theelen W, Peulen HMU, Lalezari F, van der Noort V, de Vries JF, Aerts J, et al. Effect of pembrolizumab after stereotactic body radiotherapy vs pembrolizumab alone on tumor response in patients with advanced non-small cell lung cancer: results of the PEMBRO-RT phase 2 randomized clinical trial. JAMA Oncol. 2019;5:1276–82. doi: 10.1001/jamaoncol.2019.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Bauml JM, Mick R, Ciunci C, Aggarwal C, Davis C, Evans T, et al. Pembrolizumab after completion of locally ablative therapy for oligometastatic non-small cell lung cancer: a phase 2 trial. JAMA Oncol. 2019;5:1283–90. doi: 10.1001/jamaoncol.2019.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Twardowski P, Wong JYC, Pal SK, Maughan BL, Frankel PH, Franklin K, et al. Randomized phase II trial of sipuleucel-T immunotherapy preceded by sensitizing radiation therapy and sipuleucel-T alone in patients with metastatic castrate resistant prostate cancer. Cancer Treat Res Commun. 2019;19:100116. doi: 10.1016/j.ctarc.2018.100116. [DOI] [PubMed] [Google Scholar]
- 246.Zhu X, Cao Y, Liu W, Ju X, Zhao X, Jiang L, et al. Stereotactic body radiotherapy plus pembrolizumab and trametinib versus stereotactic body radiotherapy plus gemcitabine for locally recurrent pancreatic cancer after surgical resection: an open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2021;22:1093–102. doi: 10.1016/S1470-2045(21)00286-2. [DOI] [PubMed] [Google Scholar]
- 247.Theelen W, Chen D, Verma V, Hobbs BP, Peulen HMU, Aerts J, et al. Pembrolizumab with or without radiotherapy for metastatic non-small-cell lung cancer: a pooled analysis of two randomised trials. Lancet Respir Med. 2021;9:467–75. doi: 10.1016/S2213-2600(20)30391-X. [DOI] [PubMed] [Google Scholar]
- 248.Ho AY, Barker CA, Arnold BB, Powell SN, Hu ZI, Gucalp A, et al. A phase 2 clinical trial assessing the efficacy and safety of pembrolizumab and radiotherapy in patients with metastatic triple-negative breast cancer. Cancer. 2020;126:850–60. doi: 10.1002/cncr.32599. [DOI] [PubMed] [Google Scholar]
- 249.Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–26. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 250.Lynch TJ, Bondarenko I, Luft A. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study (vol 30, pg 2046, 2012) J Clin Oncol. 2012;30:3654. doi: 10.1200/JCO.2011.38.4032. [DOI] [PubMed] [Google Scholar]
- 251.Reck M, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol. 2013;24:75–83. doi: 10.1093/annonc/mds213. [DOI] [PubMed] [Google Scholar]
- 252.Govindan R, Szczesna A, Ahn MJ, Schneider CP, Gonzalez Mella PF, Barlesi F, et al. Phase III trial of ipilimumab combined with paclitaxel and carboplatin in advanced squamous non-small-cell lung cancer. J Clin Oncol. 2017;35:3449–57. doi: 10.1200/JCO.2016.71.7629. [DOI] [PubMed] [Google Scholar]
- 253.Schmid P, Cortes J, Pusztai L, McArthur H, Kummel S, Bergh J, et al. Pembrolizumab for early triple-negative breast cancer. N Engl J Med. 2020;382:810–21. doi: 10.1056/NEJMoa1910549. [DOI] [PubMed] [Google Scholar]
- 254.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379:2108–21. doi: 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]
- 255.Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21:44–59. doi: 10.1016/S1470-2045(19)30689-8. [DOI] [PubMed] [Google Scholar]
- 256.Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gumus M, Mazieres J, et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med. 2018;379:2040–51. doi: 10.1056/NEJMoa1810865. [DOI] [PubMed] [Google Scholar]
- 257.Goldman JW, Dvorkin M, Chen YB, Reinmuth N, Hotta K, Trukhin D, et al. Durvalumab, with or without tremelimumab, plus platinum-etoposide versus platinum-etoposide alone in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): updated results from a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2021;22:51–65. doi: 10.1016/S1470-2045(20)30539-8. [DOI] [PubMed] [Google Scholar]
- 258.Shitara K, Van Cutsem E, Bang YJ, Fuchs C, Wyrwicz L, Lee KW, et al. Efficacy and safety of pembrolizumab or pembrolizumab plus chemotherapy vs chemotherapy alone for patients with first-line, advanced gastric cancer: The KEYNOTE-062 phase 3 randomized clinical trial. JAMA Oncol. 2020;6:1571–80. doi: 10.1001/jamaoncol.2020.3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Zhou C, Chen G, Huang Y, Zhou J, Lin L, Feng J, et al. Camrelizumab plus carboplatin and pemetrexed versus chemotherapy alone in chemotherapy-naive patients with advanced non-squamous non-small-cell lung cancer (CameL): a randomised, open-label, multicentre, phase 3 trial. Lancet Respir Med. 2021;9:305–14. doi: 10.1016/S2213-2600(20)30365-9. [DOI] [PubMed] [Google Scholar]
- 260.Powles T, Csoszi T, Ozguroglu M, Matsubara N, Geczi L, Cheng SY, et al. Pembrolizumab alone or combined with chemotherapy versus chemotherapy as first-line therapy for advanced urothelial carcinoma (KEYNOTE-361): a randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22:931–45. doi: 10.1016/S1470-2045(21)00152-2. [DOI] [PubMed] [Google Scholar]
- 261.Reck M, Mok TSK, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir Med. 2019;7:387–401. doi: 10.1016/S2213-2600(19)30084-0. [DOI] [PubMed] [Google Scholar]
- 262.Rini BI, Powles T, Atkins MB, Escudier B, McDermott DF, Suarez C, et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open-label, phase 3, randomised controlled trial. Lancet. 2019;393:2404–15. doi: 10.1016/S0140-6736(19)30723-8. [DOI] [PubMed] [Google Scholar]
- 263.Finn RS, Qin SK, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. IMbrave150: Updated overall survival (OS) data from a global, randomized, open-label phase III study of atezolizumab (atezo) plus bevacizumab (bev) versus sorafenib (sor) in patients (pts) with unresectable hepatocellular carcinoma (HCC) J Clin Oncol. 2021;39(3_suppl):267. doi: 10.1200/JCO.2021.39.3_suppl.267. [DOI] [Google Scholar]
- 264.Makker V, Taylor MH, Aghajanian C, Oaknin A, Mier J, Cohn AL, et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer. J Clin Oncol. 2020;38:2981–92. doi: 10.1200/JCO.19.02627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Makker V, Colombo N, Casado Herraez A, Santin AD, Colomba E, Miller DS, et al. Lenvatinib plus pembrolizumab for advanced endometrial cancer. N Engl J Med. 2022;386:437–48. doi: 10.1056/NEJMoa2108330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380:1103–15. doi: 10.1056/NEJMoa1816047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Kawazoe A, Fukuoka S, Nakamura Y, Kuboki Y, Wakabayashi M, Nomura S, et al. Lenvatinib plus pembrolizumab in patients with advanced gastric cancer in the first-line or second-line setting (EPOC1706): an open-label, single-arm, phase 2 trial. Lancet Oncol. 2020;21:1057–65. doi: 10.1016/S1470-2045(20)30271-0. [DOI] [PubMed] [Google Scholar]
- 268.Escudier B, Pluzanska A, Koralewski P, Ravaud A, Bracarda S, Szczylik C, et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet. 2007;370:2103–11. doi: 10.1016/S0140-6736(07)61904-7. [DOI] [PubMed] [Google Scholar]
- 269.Ren Z, Xu J, Bai Y, Xu A, Cang S, Du C, et al. Sintilimab plus a bevacizumab biosimilar (IBI305) versus sorafenib in unresectable hepatocellular carcinoma (ORIENT-32): a randomised, open-label, phase 2-3 study. Lancet Oncol. 2021;22:977–90. doi: 10.1016/S1470-2045(21)00252-7. [DOI] [PubMed] [Google Scholar]
- 270.Motzer R, Alekseev B, Rha SY, Porta C, Eto M, Powles T, et al. Lenvatinib plus pembrolizumab or everolimus for advanced renal cell carcinoma. N Engl J Med. 2021;384:1289–300. doi: 10.1056/NEJMoa2035716. [DOI] [PubMed] [Google Scholar]
- 271.Zhang B, Qi L, Wang X, Xu J, Liu Y, Mu L, et al. Phase II clinical trial using camrelizumab combined with apatinib and chemotherapy as the first-line treatment of advanced esophageal squamous cell carcinoma. Cancer Commun. 2020;40:711–20. doi: 10.1002/cac2.12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Dummer R, Lebbe C, Atkinson V, Mandala M, Nathan PD, Arance A, et al. Combined PD-1, BRAF and MEK inhibition in advanced BRAF-mutant melanoma: safety run-in and biomarker cohorts of COMBI-i. Nat Med. 2020;26:1557–63. doi: 10.1038/s41591-020-1082-2. [DOI] [PubMed] [Google Scholar]
- 273.Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 2019;20:1083–97. doi: 10.1016/S1470-2045(19)30274-8. [DOI] [PubMed] [Google Scholar]
- 274.Sacco AG, Chen R, Worden FP, Wong DJL, Adkins D, Swiecicki P, et al. Pembrolizumab plus cetuximab in patients with recurrent or metastatic head and neck squamous cell carcinoma: an open-label, multi-arm, non-randomised, multicentre, phase 2 trial. Lancet Oncol. 2021;22:883–92. doi: 10.1016/S1470-2045(21)00136-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Ny L, Jespersen H, Karlsson J, Alsen S, Filges S, All-Eriksson C, et al. The PEMDAC phase 2 study of pembrolizumab and entinostat in patients with metastatic uveal melanoma. Nat Commun. 2021;12:5155. doi: 10.1038/s41467-021-25332-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Lampert EJ, Zimmer A, Padget M, Cimino-Mathews A, Nair JR, Liu Y, et al. Combination of PARP inhibitor olaparib, and PD-L1 inhibitor durvalumab, in recurrent ovarian cancer: a proof-of-concept phase II study. Clin Cancer Res. 2020;26:4268–79. doi: 10.1158/1078-0432.CCR-20-0056. [DOI] [PMC free article] [PubMed] [Google Scholar]