

Abstract
Background
Patient-derived organoid (PDO) models offer potential to transform drug discovery for inflammatory bowel disease (IBD) but are limited by inconsistencies with differentiation and functional characterization. We profiled molecular and cellular features across a range of intestinal organoid models and examined differentiation and establishment of a functional epithelial barrier.
Methods
Patient-derived organoids or monolayers were generated from control or IBD patient–derived colon or ileum and were molecularly or functionally profiled. Biological or technical replicates were examined for transcriptional responses under conditions of expansion or differentiation. Cell-type composition was determined by deconvolution of cell-associated gene signatures and histological features. Differentiated control or IBD-derived monolayers were examined for establishment of transepithelial electrical resistance (TEER), loss of barrier integrity in response to a cocktail of interferon (IFN)-γ and tumor necrosis factor (TNF)-α, and prevention of cytokine-induced barrier disruption by the JAK inhibitor, tofacitinib.
Results
In response to differentiation media, intestinal organoids and monolayers displayed gene expression patterns consistent with maturation of epithelial cell types found in the human gut. Upon differentiation, both colon- and ileum-derived monolayers formed functional barriers, with sustained TEER. Barrier integrity was compromised by inflammatory cytokines IFN-γ and TNF-α, and damage was inhibited in a dose-dependent manner by tofacitinib.
Conclusions
We describe the generation and characterization of human colonic or ileal organoid models capable of functional differentiation to mature epithelial cell types. In monolayer culture, these cells formed a robust epithelial barrier with sustained TEER and responses to pharmacological modulation. Our findings demonstrate that control and IBD patient-derived organoids possess consistent transcriptional and functional profiles that can enable development of epithelial-targeted therapies.
Keywords: organoids, cell differentiation, barrier function, tofacitinib
Graphical Abstract
Graphical Abstract.

Key Messages.
What is known?
Patient-derived organoids or monolayers replicate the intestinal epithelium in vitro. However, variability in transcriptional and functional responses to differentiation and activation stimuli has not been systematically characterized, limiting potential applications of organoid technology.
What is new here?
Intestinal organoids and monolayers responded consistently to differentiation and activation stimuli at the molecular, cellular, and functional levels. In monolayer models, a functional epithelial barrier was disrupted by a cocktail of IFN-γ and TNF-α and protected with tofacitinib.
How can this help with patient care?
Organoid models with reproducible transcriptomic and functional profiles can enable development of epithelial-targeted modulators for therapeutic applications.
Introduction
Inflammatory bowel diseases (IBD), encompassing Crohn’s disease (CD) and ulcerative colitis (UC), are debilitating gastrointestinal (GI) disorders characterized by epithelial damage and chronic inflammation. Current clinical practice in IBD is aimed at achieving mucosal healing, a predictor of long-term remission in IBD patients.1 Central to achieving this goal are repair and protection of the intestinal epithelial barrier, suggesting direct targeting of the epithelial layer as a therapeutic paradigm. Clinical pathologies and genetic association studies highlight points of intervention, including restoring the epithelial-produced mucus layer, strengthening of paracellular junctions, and increasing antimicrobial defenses.2 Furthermore, epithelial cell damage and death due to immune cell infiltration, inflammatory cytokines, and microbial ligands in IBD, suggest re-epithelialization and wound healing as prime therapeutic areas.3
To explore diverse epithelial targeting strategies in IBD, model systems to reproduce intestinal epithelial biology are needed. Previously, most epithelial research relied on cancer-derived nonprimary monocultures or nonhuman in vivo models, both of which lack the ability to accurately replicate the human intestinal niche.4 Recent breakthroughs in culturing intestinal stem cells allowed the emergence of patient-derived organoids (PDOs) as a promising patient-relevant human primary cell model.5,6 Patient-derived organoids are generated from crypt basal stem cells extracted from endoscopic biopsies or surgical resection material.7 In defined culture conditions, stem cells are propagated in vitro, typically in 3D cultures with a luminal-facing apical surface and external-facing basolateral surface.5 Organoid-derived cells can also be plated to form monolayers on permeable supports, allowing greater access to their apical surfaces. Differentiation of 3D or Transwell-plated monolayers toward epithelial lineages enables the recreation of physiologically relevant epithelial populations and responses to external stimuli. Patient-derived organoids and monolayers can be derived from tissues throughout the intestinal tract and largely recapitulate the organ from which they were derived at genetic, phenotypic, and histologic levels.6 These attributes have spurred researchers to use PDO and PDO-derived monolayers to explore the impact of IBD on the intestinal epithelium and seek new therapeutic targets.8 Patient-derived organoids and monolayers have also been used to predict patient responses to therapeutics, including examining effects of IBD therapies on the intestinal barrier.8
Although a great deal of progress has brought PDO and PDO-derived monolayer models to the forefront, there is still much we do not yet understand, which prevents these models from being widely adopted for drug discovery and industrial applications. For example, reproducibility of intestinal organoid or monolayer models from experiment-to-experiment or donor-to-donor or derived from distinct organ locations has not been adequately addressed. Furthermore, while the basic knowledge needed to differentiate stem cells into physiologically relevant epithelial lineages exists, differentiated cells are often insufficiently characterized at the molecular level. Moreover, how differentiated intestinal stem cells in 3D organoid models compare with 2D monolayers for cell-type diversity and transcriptional output remains an open question. To address these questions, we first generated PDO and monolayers derived from ileum and colon of a non-IBD control subject and define their transcriptional states. We characterize media for growth and cell differentiation and their impacts on development of PDO and monolayers. We next examine the transcriptional states of PDO and monolayers derived from IBD patients (IBD-PDOs). Lastly, we examine functional responses of monolayer models to barrier-damaging cytokines and the small molecule UC therapy Janus kinase (JAK) inhibitor tofacitinib. Collectively, our work seeks to demonstrate that PDO and monolayer models represent a research platform that may be applied to identification and characterization of the next generation of IBD therapies.
Methods
Organoid Establishment
Crypts were isolated from biopsies, embedded in Matrigel (Corning), and subsequently cultured in a stem cell expansion medium, colon normal medium (CNM)5 containing Wnt surrogate (Table S1). Organoid expansion was achieved by growth in CNM supplemented with a 50% Wnt3a-conditioned medium.5,9 For differentiation, stem cells were cultured for 3 days in CNM followed by differentiation in enterocyte differentiation medium (eDM)10 or combination differentiation medium (cDM)11 (Table S1). Subsequently, organoids were harvested for RNA analysis by quantitative reverse transcription real-time polymerase chain reaction (RT-qPCR), RNA-seq, or histology.
Organoid Biobank
Colon or ileum biopsies were obtained from “Surveillance for IBD-associated colorectal cancer” from the Department of Gastroenterology and Hepatology, University Medical Centre, Utrecht (Protocol ID: NL35053.041.11.) Selected IBD patients (age 18-70 years) had a diagnosis of UC or CD, with disease duration ≥8 years and inflammation of at least 30% of colonic mucosa at some point between diagnosis and inclusion (Table S2 and S3). Individuals above 18 years old undergoing diagnostic colonoscopy with no endoscopic abnormalities were included as non-IBD controls (Table S2 and S3). All material was obtained under a medical ethics review committee; and the Biobank review committee approved protocol from human intestinal tissue and University Medical Center Utrecht HUB-STEM. All patients signed informed consent for the IBD surveillance study and HUB-STEM protocol.
3’RNAseq
The 3’RNA sequencing was performed by Single Cell Discoveries B.V. Fastq files were processed by QuickRNAseq12 (pipeline utilizing Hg38 and Gencode v24). Normalization and differential expression analyses were conducted using limma-voom.13 Genes with average normalized expression below 0 were removed. The RNA-seq data are available in GEO (https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE197698. Pathway analysis was performed using a hypergeometric test as implemented in TMOD package version 0.24 in R (https://CRAN.R-project.org/package=tmod).14 Analysis was performed with the Hallmark gene set from msigDB (version 6.0; http://www.gsea-msigdb.org/).15
Organoid-derived Epithelium Monolayers
Organoids were cultured for 3 to 4 days in CNM, dissociated to single cells using Accutase (ThermoFisher), washed, filtered (40 μm, PluriSelect), and resuspended in CNM supplemented with 10 μM of Rho kinase inhibitor (RhoKI, Abmole) at a density of 2 million cells/mL. 96-well Transwell inserts (Polyester 0.4 μm pore, Corning) were coated with Matrigel diluted 40x with ice-cold phosphate buffered saline (PBS; Ca2+ and Mg2+, ThermoFisher) for 1 hour at 37°C, before removing the residual PBS. Transwells were seeded with 100 μL of the cell suspension in the upper chamber and 300 μL of CNM with 10 μM RhoKI in the lower chamber and incubated at 37°C and 5% CO2. Media was changed 3 times per week. Room temperature–equilibrated monolayers were measured for TEER using a REMS AutoSampler (World Precision Instruments). Results were expressed as Ω·cm2 after subtracting background resistance values determined by Transwells containing no cells. Monolayers were kept in CNM until TEER values reached between 100 and 200 Ω·cm2, upon which the organoid medium was changed by replacing CNM with eDM, cDM (Table S1), or continued in CNM for several days depending on the experiment.
Cytokine-induced Barrier Injury
Established monolayers were treated with cytokines or cytokine cocktails at times indicated. Transepithelial electrical resistance was measured at baseline prior to cytokine addition. Interferon (IFN)-γ, tumor necrosis factor (TNF)-α, and/or interleukin (IL)-1α (R&D Systems) were applied to the apical and basal compartments. Tofacitinib was applied to the apical and basal compartments 1 hour prior to cytokine treatment. Transepithelial electrical resistance was measured at 6 and 24 hours postcytokine treatment.
Quantitative Reverse Transcriptase PCR
The RNA was isolated (RNeasy mini kit, Qiagen) and quantified (RNA HS Qubit Assay kit, ThermoFisher); and cDNA was synthesized from at least 200 ng of RNA with random hexamers (Superscript First-Strand Synthesis, ThermoFisher). Genomic DNA was removed (RNase free DNase, Qiagen), and RT-qPCR was performed (Sybr Green, ThermoFisher). Gene expression for LGR5 (Forward: 5’-ACACGTACCCACAGAAGCTC-3’, Reverse 5’-GGAATGCAGGCCACTGAAAC-3’), MUC2 (Forward 5’-AGGATCTGAAGAAGTGTGTCACTG-3’, Reverse 5’-TAATGGAACAGATGTTGAAGTGCT-3’), ALPI (Forward 5’-GGAGTTATCCTGCTCCCCAC-3’, Reverse 5’-CTAGGAGGTGAAGGTCCAACG-3’) were quantified and normalized to TBP (Forward 5’-ACGCCGAATATAATCCCAAGCG-3’, Reverse 5’-AAATCAGTGCCGTGGTTCGTG-3’).
Histology
Organoids and monolayers were washed in PBS and fixed at room temperature in 4% paraformaldehyde for 30 minutes and dehydrated by serial 25%, 50%, and 70% ethanol washes, followed by a wash in 100% ethanol and butanol. They were then embedded in paraffin (VWR), sectioned (4 μm) and mounted on glass slides. Slides were deparaffined with xylene (Klinipath), rehydrated by serial ethanol washes with a range from 100% to 25%, and stained for hematoxylin (VWR) and eosin (H&E; VWR) and Alcian blue (AB; Alfa Aesar). For antibodies, slides were incubated with peroxidase buffer for 15 minutes, heated with phosphate-citrate buffer for 20 minutes, and cooled for 30 minutes. Slides were blocked with 1% bovine serum albumin (VWR) for 30 minutes and stained overnight using anti-Ki67 (Monosan) or anti-MUC2 (Santa Cruz) antibodies. Secondary horse radish peroxidase antibodies to Ki67 or MUC2 were incubated for 1 hour or 30 minutes, respectively. Hematoxylin-stained slides were developed with 3,3’-diaminobenzidine (VWR Life Science) plus hydrogen peroxide (Merck) for 10 minutes.
Lucifer Yellow Permeability Assay
Lucifer yellow (LY) (60 μM; Sigma) was added to the apical compartment. Transwells were incubated at 37°C and 5% CO2 on an orbital shaker (50 rpm). Fluorescence of the basolateral compartment was measured by Spark multimode microplate reader (Tecan) with excitation wavelength of 400 nm and emission wavelength of 590 nm. Results were expressed as percent apparent permeability using the following formula:
Multiplex Permeability Assay
Monolayers were washed with Hank’s balanced salt solution (HBSS) with Ca2+ and Mg2+, and pH7.4 (Gibco). After refilling the basal compartments with HBSS, 100 μM of LY, FITC-Dex4, and TRITC-Dex40 (Sigma) in HBSS were added to the apical compartments. Transwells were incubated 37°C and 5% CO2 for 2 hours. Basal compartment fluorescence was measured using Synergy H4 (Biotek): for LY, 430 nm excitation wavelength and an emission wavelength of 530 nm were used; for FITC-Dex4, 490 nm excitation and 530 nm emission were used; and for TRITC-Dex40, 540 nm excitation and 577 nm emission were used. Results were expressed as percent permeability, normalizing to no treatment (0% permeability) or staurosporine (EMD Millipore; 100% permeability).
Viability
Medium was removed, and 100 μL of CellTiter-Glo (Promega) was added to each apical chamber and incubated with gentle shaking for 5 minutes, followed by room temperature incubation in the dark for 25 minutes. Luminescence was measured with Spark 10M (Tecan).
Results
Human Intestinal PDO From Control and IBD Donors Respond to Selected Media With Consistent Gene Expression and Physiological Cell-Type Diversity
To develop PDO models, we selected specific culture media to promote barrier function and cell-type diversity. For maintaining progenitor populations, we used CNM5 (Table S1). For conditions expected to promote robust barrier formation and to support enterocyte and goblet cell differentiation, we used eDM (Table S1),10 which is a Wnt3a-deficient medium supplemented with a small molecule Wnt secretion inhibitor (porcupine inhibitor C59) to block residual Wnt pathway activation. We developed combination differentiation medium11 (Table S1) to promote differentiation to cell types normally found in the human intestine, such as enterocyte, enterocyte progenitor, Paneth, goblet, enteroendocrine, transit amplifying (TA) and tuft cells, while retaining stem cells. The cDM medium has a unique formulation to achieve Notch and epithelial growth factor (EGF) pathway inhibition. The Wnt signaling was reduced but maintained through addition of 10% Wnt3a-conditioned medium.
To characterize these media, colon and ileum PDO derived from biopsies from a non-IBD donor (control-donor) were cultured under expansion conditions (CNM) or differentiated in eDM or cDM and examined. Histologic analysis revealed PDO in CNM possessed qualities of undifferentiated progenitors including positive staining for markers of proliferation (Ki67) and negative staining for markers associated with goblet cells, MUC2, and Alcian blue (Figure S1A and S1B). Differentiation of colon and ileum PDO with eDM or cDM was accompanied by development of columnar epithelial structure, indicative of enterocyte differentiation, reduced Ki67 staining, and positive staining for MUC2 and Alcian blue (Figure S1A and S1B). These data are consistent with eDM and cDM driving differentiation to mature human epithelium-like structures.
Patient-derived organoids from colon or ileum biopsies from a single control subject were grown in CNM, eDM, or cDM and molecularly characterized by RNA-seq. To evaluate variability, we examined 2 or 3 biological replicates (defined as complete experiments from independent thawing of the organoids to collection of the data) per growth condition. Cultures were replicated in triplicate (referred to as “technical replicates”) for each medium condition within each biological replicate for a total of 6 to 9 samples per medium. Samples were clustered by principal component analysis (PCA), with biological replicates and technical replicates clustering by culture medium and tissue origin with small intracluster distances (Figure 1A). Colon- and ileum-derived PDO showed a more similar profile when grown in CNM than when differentiated in either cDM or eDM, suggesting increased organ-specific identity with differentiation (Figure 1A). Changes in transcription between CNM- and eDM-grown cultures were more pronounced (average 3285 regulated genes; adj P < .01) than between CNM- and-cDM grown cultures (average 900 regulated genes; adj P < .01; Figure 1B).
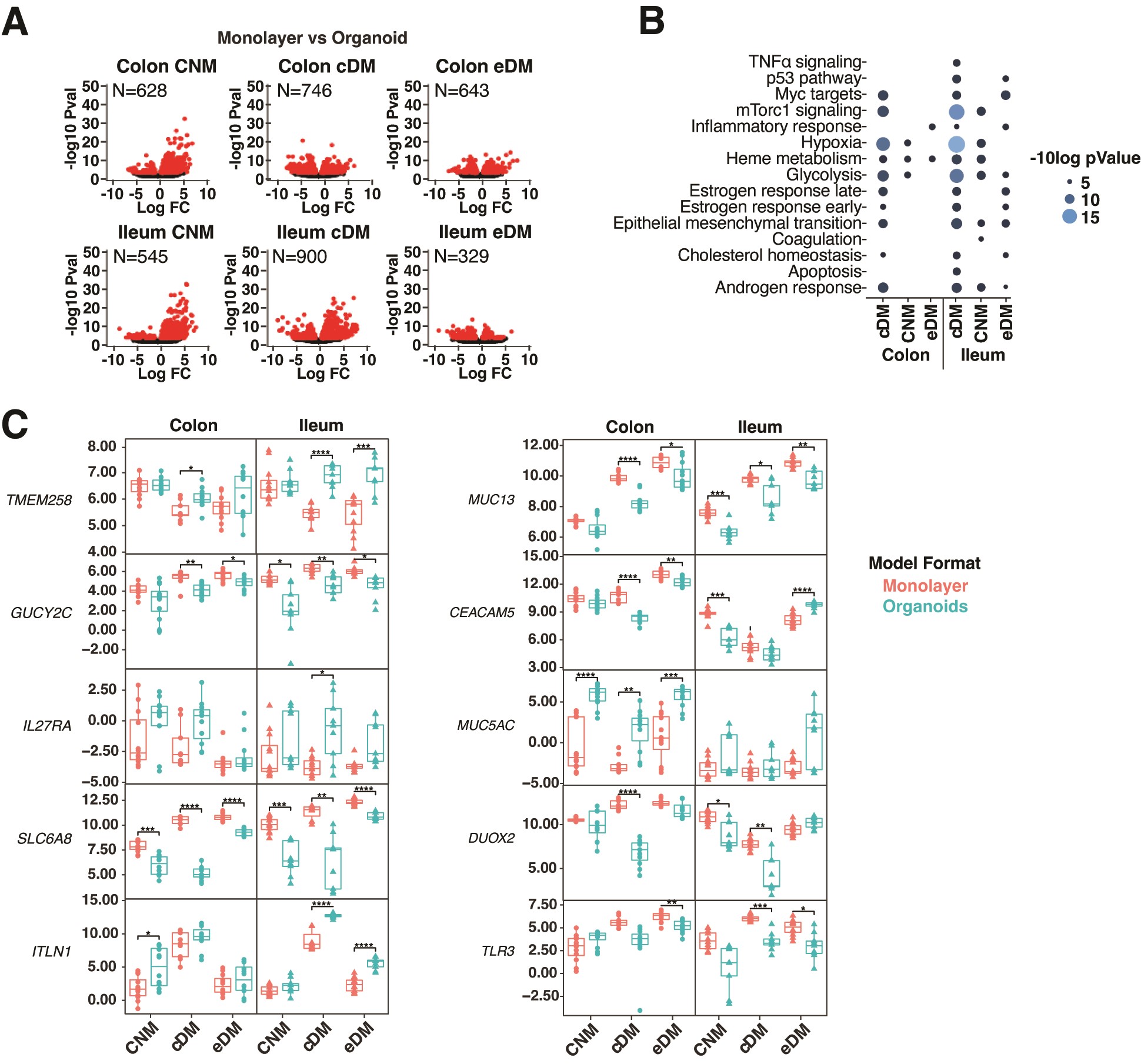
Figure 1.

PDO respond to selected media with consistent gene expression and physiological cell-type diversity. A, PCA analysis of biological replicates of control donor-derived PDO (upper panel) and IBD-PDOs (lower panel) grown in media specified. B, Control donor-derived and IBD-PDOs differentially expressed genes. Red symbols are significantly different between conditions. C, Heat map depicting expression of epithelial related genes. D, Epithelial cell-type signatures after differentiation are represented as fold-change relative to undifferentiated.
Many of the observed changes in transcription are likely attributed to changes in cellular populations within the organoid. Deconvolution of cell types using markers of known GI epithelial cell–associated transcripts10,16–18 revealed significant changes in cell populations (Figure 1C, 1D). Patient-derived organoids differentiated in eDM were enriched for markers of enterocytes and goblet cells, with reduced expression of genes associated with stem, Paneth, cycling TA and secretory TA cells. Patient-derived organoids differentiated with cDM had more physiological cell-type populations than with eDM, as evidenced by increased goblet and enterocyte signatures, as well as increased enteroendocrine, Paneth, and secretory TA signatures while maintaining stem and cycling TA signatures (Figure 1C and 1D). Expression of selected lineage-specific markers were confirmed by qPCR in an independent experiment. Both eDM and cDM promoted expression of goblet cell and enterocyte-associated genes, MUC2 and ALPI, but only CNM and cDM conditions retained expression of stem cell–associated LGR5 (Figure S1C).
To evaluate the ability of our media to promote differentiation of IBD-PDO, an organoid biobank was generated from 39 biopsies taken from 16 IBD patients. The IBD-PDOs were derived from inflamed, previously inflamed, and noninflamed regions of colon or ileum, some of which were obtained from the same individual (Table S2). The IBD-PDOs from inflamed regions expanded poorly or not at all (38% successful establishment of organoids) compared with those from previously inflamed or noninflamed regions (78% successfully expanded). Successfully expanded organoids from previously inflamed or noninflamed regions from 2 patients with UC and 3 patients with CD (Table S3) were selected for further characterization. The IBD-PDOs were maintained in CNM or differentiated in eDM or cDM and analyzed by RNA-seq transcriptional profiling. Individual IBD-PDOs showed a high level of transcriptional similarity and were clustered by medium and organ type, similar to what was observed for control donor organoids (Figure 1A). Cell-type analysis of IBD-PDOs reflected the control donor PDO following differentiation (Figure 1C and D). Furthermore, IBD-PDOs had the same magnitude of response to growth in specific media as control PDOs and similarly showed a more robust response to eDM compared to cDM (Figure 1B).
PDO-derived Monolayers From Control and IBD Donors Respond to Selected Media With Consistent Gene Expression and Possess Physiological Cell-type Signatures and Mature Epithelial Features
To characterize the ability of our media to promote barrier formation and maintain transcriptional reproducibility on permeable supports, control donor colon and ileum PDOs, maintained in CNM, were digested, plated on Transwells, and allowed to form monolayers. Monolayers were maintained on CNM or differentiated at 4 to 6 days postplating with eDM or cDM. Barrier formation was monitored by examining transepithelial electrical resistance (TEER), which was increased over time in both colon and ileum monolayers with differentiation (Figure S2A and S2B), indicative of increased tight junction formation and epithelial barrier function. Histologic analysis of CNM-cultured monolayers revealed positive staining for Ki67 and absence of MUC2 or Alcian blue staining (Figure S2C), indicative of progenitor populations. Monolayers in eDM or cDM drove development of columnar epithelial morphology and loss of Ki67 staining, suggesting differentiation to enterocytes (Figure S2C).11 Colon monolayers in eDM or cDM11 and ileum monolayers (Figure S2C) in cDM were positive for MUC2 and Alcian blue staining, suggesting goblet cell differentiation under these conditions.
Monolayers derived from IBD-PDOs were molecularly profiled by RNA-seq. As with 3D organoids, PCA analysis of transcripts showed a high level of similarity between biological and technical replicates with differences mainly associated with organ source and growth medium (Figure 2A). Moreover, differentiation of the monolayers in eDM had a more profound effect on transcription than differentiation in cDM for both colon- and ileum-derived cultures (Figure 2B), which was independent of whether cultures were derived from control or diseased tissue.
Figure 2.

PDO-derived monolayers display consistent responses to media for gene expression and physiological cell-type signatures. A, PCA analysis of biological replicates of control donor-derived monolayers (upper panel) and IBD-PDO-derived monolayers (lower panel) grown in media specified. B, Control donor- and IBD-PDO-derived monolayers differentially expressed genes. Red symbols are significantly different between conditions. C, Heat map depicting expression of epithelial cell-type related genes. D, Epithelial cell-type signatures after differentiation are represented as fold-change relative to undifferentiated.
Differentiation with eDM and cDM enriched signatures for enterocyte and goblet cells, as well as TA1 and TA2, which was accompanied by a reduction in signatures for cycling TA and stem cells (Figure 2C and 2D). Changes in expression of cell markers were consistent, independent of whether the cultures were derived from control or IBD donors and independent of whether the cells were originally isolated from colon or ileum tissue. qPCR analysis of independent samples confirmed increased ALPI and MUC2 expression with reduced LGR5 expression in eDM and cDM treated cultures (Figure S2D).
PDO respond differently to plating in organoid vs monolayer formats. Transcriptional responses to plating in organoid and monolayer formats were examined. Principal component analysis of transcripts revealed that samples clustered by format, which was notable for differentiated cultures (Figure 3A). Non-IBD organoid and monolayer cultures showed similar cell signatures (Figure 1D and 2D), but dramatically different transcriptional profiles (Figure 3B). This was also observed for IBD-derived cultures (Figure S3A). Differences between organoids and monolayers were greatest for colon or ileum cultures differentiated in cDM compared with other media (Figure 3B and S3A). In contrast, IBD-PDOs ileum cultures differentiated in eDM exhibited the fewest transcriptional differences between organoids and monolayers (Figure S3A). We observed significant differences between ileum-derived organoid and monolayer formats differentiated in cDM for mammalian target of rapamycin complex 1 (mTorc1) signaling, hypoxia, and glycolysis pathway genes in both control (Figure 3C) and IBD-derived organoids and monolayers (Figure S3B). Several notable genes were differentially regulated between the organoid and monolayer formats. Genes with lower expression in organoids compared with monolayers—regardless of media, organ-type, or donor—include those encoding guanylate cyclase GUCY2C, cell adhesion molecule CEACAM5, innate receptor TLR3, creatine transporter SLC6A8, redox regulator DUOX2, and transmembrane mucus protein MUC13 (Figure 3D and S3C). We also observed genes with higher expression in organoids compared with monolayers including those encoding ER stress regulator TMEM258 (IBD-PDO ileum cultured in cDM and eDM), mucus protein MUC5AC (colon cultured in cDM and eDM), soluble lectin ITLN1 (colon or ileum cultured in cDM and ileum in eDM), and IL27 receptor subunit Il27RA (ileum cultured in CNM and ileum or colon cultured in cDM; Figure 3D and S3C).
Figure 3.

Intestinal cells respond differently to plating in organoid vs monolayer formats. A, PCA analysis of organoid and monolayer formats. B, Genes differentially expressed between monolayers and organoids. Red symbols are significantly different between conditions (FDR < .01). C, Differentially expressed pathways between monolayers and organoids. D, Normalized expression of selected epithelial genes. Box plots denote the average and standard deviation of 3 technical replicates of 2 to 3 independent biological replicates per condition. Relevant statistics were performed using ttest with Bonferroni multiple comparison correction (*P < .05, **P < .01, ***P < .001, ****P < .0001).
Intestinal Monolayers Can Be Applied to Model Epithelial Barrier Injury
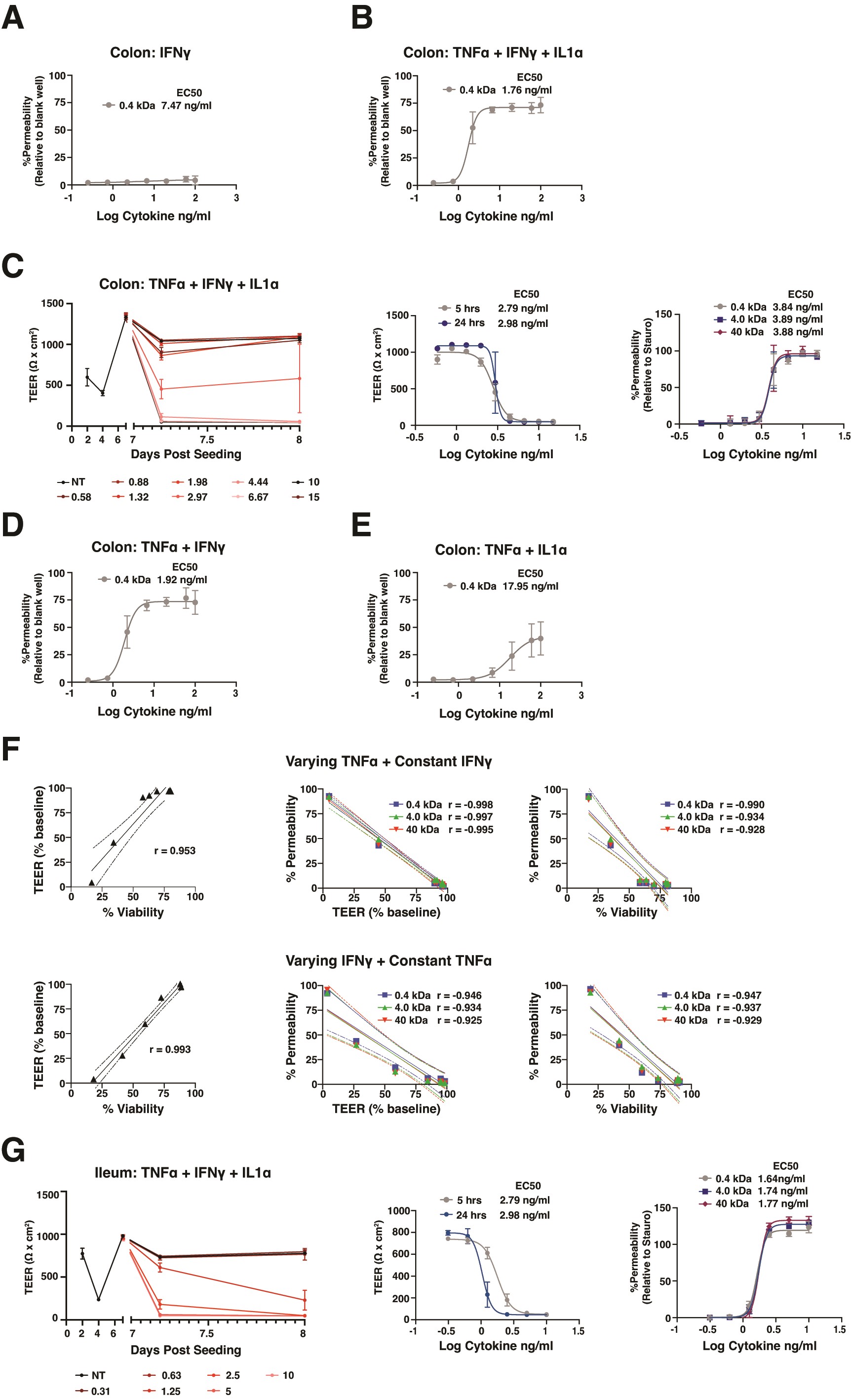
To explore the utility of PDO-derived monolayers for examining intestinal barrier injury, we stimulated colon-derived monolayers from a control donor with a range of concentrations of IBD-relevant pro-inflammatory cytokines, including IFN-γ, TNF-α, and/or IL-1α,19 and examined TEER as a measure of barrier integrity. We focused on monolayers maintained in cDM, which comprised differentiated human epithelium with robust TEER development. Monolayers exhibited mild, dose-dependent loss of TEER in response to increasing concentrations of IFN-γ compared with unstimulated monolayers (Figure 4A). Furthermore, stimulated monolayers remained impermeable to 0.4 kDa Lucifer yellow, suggesting intact paracellular junctions (Figure S4A). These results are consistent with previous reports demonstrating that stimulation with IFN-γ alone induces mild barrier damage in polarized epithelial models within the first 24 hours.20 To mimic an inflammatory milieu, colon monolayers were stimulated with concentrations of a cocktail of IFN-γ +TNF-α + IL-1α. We observed a marked dose-dependent reduction in TEER with a half-maximal effect (EC50) of 1-3 ng/mL (Figure 4B) in response to this cocktail compared with unstimulated monolayers. Monolayers stimulated with concentrations of IFN-γ + TNF-α + IL-1α greater than EC50 exhibited complete TEER loss by 24 hours poststimulation (Figure 4B). Furthermore, reduction in TEER in response to IFN-γ + TNF-α + IL-1α was concomitant with increased paracellular permeability to 0.4 kDa Lucifer yellow or 4 and 40 kDa dextran (Figure S4B and C), consistent with complete barrier loss and gross epithelial damage at concentrations above EC50. These results were generated by HUB researchers (Figure 4B and S4B) and independently replicated by Pfizer researchers (Figure S4C).
Figure 4.

Intestinal monolayers can be applied to model epithelial barrier injury. A-E, Control PDO-derived monolayers were monitored for TEER before and after stimulation with cytokines listed. E, Viability and permeability relative to wells treated with staurosporine were measured at 24 hours following cytokine addition. Cultures were differentiated with cDM media on day 5 and cytokines were administered on day 10. (A-D) The concentration of cytokine indicates the amount of each individual cytokine in the cocktail. E, The “varying” cytokine concentrations in the cocktail are reflected on the x axis and “constant” cytokines were held at 5 ng/mL in the cocktail. Nonlinear regression analysis and EC50 values were calculated at 5- or 24-hours postcytokine stimulation. Points represent the average of 3 wells (A-D) or 2 wells (E), and error bars indicate the standard deviation. Relevant statistics were performed using 1-way ANOVA with Tukey’s multiple comparison test. Statistics refer to differences between cytokine treatment and no treatment groups.
Differentiated colon monolayers stimulated with combinations of IFN-γ + TNF-α or TNF-α + IL-1α induced a dose-dependent loss of barrier between 5 hours and 24 hours (EC50 of 0.93 and 3.0 ng/mL, respectively; Figure 4C and 4D). The IFN-γ + TNF-α promoted early barrier damage with complete TEER loss in response to high doses of cytokines at 5 hours poststimulation (Figure 4C) and were highly permeable to Lucifer yellow (Figure S4D), showing similar responses as observed with IFN-γ + TNF-α + IL-1α. In contrast, TNF-α + IL-1α promoted delayed TEER loss observed mostly at the 24-hour time point (Figure 4D) and were less permeable to Lucifer yellow compared with cultures stimulated with IFN-γ + TNF-α (Figure S4D and S4E).
The acute sensitivity of monolayers to IFN-γ + TNF-α prompted us to explore the relative contribution of these cytokines to barrier permeability and loss of cell viability. Colon monolayers were challenged for 24 hours with varying concentrations of TNF-α, while holding IFN-γ at a constant concentration of 5 ng/mL (chosen based on the minimum concentration to see an effect on TEER with IFN-γ alone, Figure 4A) and examined for barrier function and viability. Monolayers were also challenged using varying concentrations of IFN-γ while holding TNF-α at 5 ng/mL. As expected, 5 ng/mL IFN-γ alone reduced TEER by approximately 6% compared with vehicle control, whereas 5 ng/mL TNF-α did not reduce TEER (Figure 4E). Individual cytokines had little to no impact on permeability or viability (Figure 4E). Titrating either cytokine in the presence of a constant dose of the other resulted in a dose-dependent loss of TEER, coinciding with permeabilization of the barrier to 0.4 kDa Lucifer yellow, 4 and 40 kDa dextran, and loss of viability (Figure 4E). Viability and TEER were highly correlated, and viability or TEER displayed a strong inverse correlation with permeability to fluorescent molecules (Figure S4F). Half-maximal effects for barrier function and cell viability were equivalent for each cytokine combination and were within the same range as when cytokines were held at the same concentration (Figure 1C). Responses to cytokine cocktails at or near EC50 varied widely, with some wells exhibiting complete loss of TEER, which coincided with permeability to large molecules, and a >50% loss in viable cells, whereas other wells retained barrier function and had <50% loss in viable cells. These findings highlight the acute sensitivity of colon monolayers to IFN-γ + TNF-α.
Next, we explored the translatability of the assay to ileum-derived monolayers. Ileum monolayers derived from a control donor and differentiated in cDM showed an IFN-γ + TNF-α + IL-1α-induced loss of TEER that was dose-dependent with an EC50 of 1 ng/mL at 24 hours poststimulation (Figure S4G). As with colon, this combination induced dose-dependent permeability to 0.4 kDa Lucifer yellow as well as 4 and 40 kDa dextran, suggesting complete loss of barrier at the 3 highest doses of cytokines (Figure S4G).
Monolayers Derived From Control and IBD-PDOs Model Responses to Tofacitinib
We determined the response of PDO monolayers to a known IBD clinical therapy using TEER, permeability, and viability as readouts. Control donor-derived colon and ileum monolayers differentiated with cDM were pretreated with a range of concentrations of the Janus kinase inhibitor, tofacitinib,21 prior to stimulation with a cocktail of IFN-γ + TNF-α + IL-1α (1 ng/mL each). For both colon- and ileum-derived monolayers, tofacitinib dose-dependently rescued the cytokine-induced TEER loss (24 hours: half maximal inhibitory concentration [IC50] of 2.0 and approximately 6.6 μM, respectively; Figure 5A and 5B) and paracellular permeability to 0.4 kDa Lucifer yellow (Figure S5A and S5B). Additionally, tofacitinib reduced viability loss in colon monolayers (Figure S5A). In ileum monolayers, tofacitinib also reduced permeability to the larger dextran molecules (4 and 40 kDa; Figure S5B). We note the significant role of IFN-γ and TNF-α in inducing barrier loss in monolayers, as signaling in response to IL-1α minimally contributed to the damage at the concentrations tested (Figure 4B and 4C).
Figure 5.

Monolayers derived from control and IBD-PDOs can be applied to model responses to IBD therapies. Monolayers were pretreated with various concentrations of tofacitinib 1 hour prior to stimulation with inflammatory cytokines. A, Colon and (B) ileum monolayers derived from control donors or (C and D) colon derived from 2 UC patients, or (E) ileum derived from a CD patient were monitored for changes in TEER. Nonlinear regression analysis and IC50 values were calculated at 5 or 24 hours postcytokine stimulation. Points represent the average of 3 wells and error bars indicate the standard deviation. Relevant statistics were performed using 1-way ANOVA with Tukey’s multiple comparison test. Statistics refer to differences between tofacitinib treatment and no treatment groups.
Monolayers derived from IBD-PDOs have similar properties as monolayers derived from control donors. Differentiation of IBD-PDOs with cDM achieved stable TEER consistent with monolayers derived from the control donor (Figure 5C-E). Stimulation of these monolayers with 1 ng/mL IFN-γ + TNF-α + IL-1α induced barrier damage including loss of TEER (Figure 5C-E) and increased permeability and loss of viability (Figure S5C and E). Pretreatment with tofacitinib at concentrations near the IC50, defined in control-donor derived organoids, protected monolayers from cytokine-induced loss of TEER (Figure 5C-E) and increased permeability and loss of viability (Figure S5C and E). As might be expected, EC50 for cytokine responses varied between donors. One colon PDO-derived monolayer from a patient with UC exhibited loss of TEER and reduced viability in response to 1 ng/mL of cytokines but showed little increase in permeability compared with other IBD donor-derived monolayers (Figure 5C and S5C). Of note, findings on the impact of tofacitinib on protection of ileum-derived monolayers from cytokine-induced damage were independently replicated by both HUB (Figure 5E and S5E) and Pfizer (Figure 5B and S5B) researchers.
Discussion
In this study, we highlight the utility and reproducibility of PDOs and PDO-derived monolayers for producing gene expression signatures associated with epithelial differentiation and modeling agents that damage or protect the epithelial barrier. Culture media and conditions were defined to support differentiation of epithelial cell types, whether derived from colon or ileum, from control donors or IBD patients, or plated in 3D or monolayer culture. Principal component analysis of the transcriptome revealed that technical and biological replicates have a high level of reproducibility within experiments and from batch to batch in cultures derived from both colon and ileum tissue. Importantly, gene expression patterns from either organoid or monolayer cultures were consistent between cultures derived from IBD and control donors. Our findings agree with previous studies that have explored the effect of organ type, format, and donor on transcriptional variability.22 Beyond this, our study explores 2 distinct compositions of differentiation media, including the unique formulation of cDM. We also introduce cell-type deconvolution to evaluate the impact of media, organ-type, donor, or format on cell-type signatures. The media choices described herein reliably produced distinct differentiation states and cell-type proportions in both organoid and monolayer formats.
Colon normal medium has high levels of Wnt3a and mimics the Wnt-rich crypt base while promoting stem cell amplification and development of TA and Paneth cell signatures. Differentiation in eDM was used to model the upper crypt, which is characterized by reduced Wnt and Notch signaling with enrichment in enterocytes and goblet cells.23 Differentiation in cDM models features of both the base and upper crypt in a single culture, enabled by modulation of Wnt3a to maintain expression of stem cell markers in the culture.24 Both eDM and cDM media promote cells that are morphologically and functionally similar with columnar epithelial morphology and robust epithelial marker expression, mucus staining, and TEER development. However, eDM and cDM differ at the level of cell-type signatures, with cDM-differentiated organoids possessing a cell-type profile most reflective of human epithelial tissue. Importantly, all 3 media generate cells with gene expression signatures of cell types identified by single cell RNA-seq of UC biopsies.16 Based on our findings, we suggest cDM as the preferred media formulation to support modeling of human biology and barrier function in vitro.
We considered the importance of organoid vs monolayer formats on gene transcription. Hundreds of differentially expressed genes were identified between monolayers and organoids. Differentiation in eDM resulted in the lowest number of differentially expressed genes among the 3 media and had the most similar cell-type signatures when differentiated in monolayers and organoids. In contrast, monolayers grown in cDM had reduced apparent cell-type diversity compared with organoids grown in the same media. The reasons for this are unclear but may reflect a requirement for 3D architecture, mechanical stress, access to nutrients, or oxygen concentrations within the organoid.22 Additionally, cDM-grown cultures had significant differences in gene expression of pathways associated with mTorc1 signaling, hypoxia, and glycolysis in monolayers compared with organoids. The reasons behind this were not apparent, as these pathways are involved in a multitude of epithelial processes including proliferation, ER-stress responses, and epithelial junction development.25,26 Differences between monolayers and organoids were also observed at the individual gene level, including expression of IBD-relevant genes such as DUOX2, GUCY2C, ITLN1, and TMEM258.27–30 Some genes were differentially expressed in monolayers vs organoids regardless of the differentiation media chosen, suggesting that gene expression is influenced by cellular organization and architecture. The consequences of differential expression of IBD-associated genes were not examined in our study but could have implications for cellular responses to stimuli or therapies. Collectively, our analyses highlight transcriptional differences between organoid and monolayer platforms that can guide model selection in functional applications.
We also introduce cDM-grown monolayers as reproducible models for examining disease-relevant stimuli and barrier-protective IBD therapies. We optimized readouts for TEER, permeability, and cell viability using well-known barrier insult agents IFN-γ, TNF-α, and IL-1α.19 We observed that the pro-inflammatory cocktail of IFN-γ + TNF-α promoted acute early damage compared with TNF-α + IL-1α, suggesting the importance of IFN-γ + TNF-α synergy in promoting barrier damage in our assays. These findings are consistent with observations in cancer-derived monolayers.31 Cocktails containing IFN-γ + TNF-α + IL-1α or IFN-γ + TNF-α resulted in complete barrier disruption and permeability to large molecules accompanied by loss of viability. In contrast, previous studies using T84, Caco-2 and iPSC-derived organoid models suggest IFN-γ + TNF-α-induced loss of tight junction stability and TEER is independent of loss of viability.32,33 Studies have also reported considerable viability loss induced by IFN-γ + TNF-α in T84 and HT29 models as well as intestinal stem cell–derived organoids from control and IBD patients at concentrations similar to those used in this study.20,34–36 Why these cell models respond differently to IFN-γ + TNF-α has not been addressed but could be related to magnitude of response, such that the acute sensitivity of primary intestinal cell models to IFN-γ + TNF-α precludes the ability to resolve effects on tight junctions. Further research will be needed to fully understand these responses to stimuli in vitro.
Barrier damage, permeability, and loss of viability induced by IFN-γ + TNF-α + IL-1α could be blocked by the JAK kinase inhibitor tofacitinib. These findings are consistent with previous studies that showed inhibition of JAK/STAT signaling downstream of IFN-γ was sufficient to inhibit the synergy between IFN-γ and TNF-α.36,37 Although responses to cytokines varied between donors, tofacitinib predictably blocked barrier-damaging effects. Importantly, experiments demonstrating barrier damage or protection with tofacitinib were independently replicated by groups of researchers within HUB and Pfizer, highlighting the reproducibility of our models. Collectively, our data support the use of cDM-differentiated epithelial monolayers in modeling responses to barrier damaging agents or protective therapies.
Although conventional cell lines such as T84, Caco2, or HT29 could be used to demonstrate the effects of cytokines or tofacitinib on intestinal barrier,36,37 we note these cell lines lack the 3D structure of PDOs and physiological cell-type diversity of PDOs and PDO-derived monolayers. Our results suggest differences between monolayers and 3D culture may influence gene expression profiles, suggesting that examination of model format on responses to stimuli is fertile ground for further investigation. Future studies could also leverage organoids to examine the impact of barrier modulators on individual cell types.
We obtained PDOs and IBD-PDOs from a range of tissues including inflamed, previously inflamed, and noninflamed. However, success in generating long-term cultures with organoids from inflamed regions was greatly reduced compared with organoids from previously inflamed or noninflamed regions, which were chosen to establish a robust biobank to perform our study. Previous studies concluded that IBD phenotypes are not retained in organoids after long-term culture.38 Addition of inflammatory stimuli could reestablish features of the disease in IBD-PDOs,39 suggesting PDOs from previously inflamed or noninflamed regions may be leveraged to examine the impact of therapies in healthy vs IBD patients. Further analysis will be needed to demonstrate the impact of inflammatory cytokines on total gene expression in cDM differentiated control and IBD-PDOs.
Recent studies suggest IBD-PDOs and non-IBD-PDOs have distinct phenotypes with respect to transcriptional signatures39 and baseline permeability.40 However, transcriptional variation in our study was more strongly associated with growth media, culture conditions, and donor variability than with disease status (Figure 3A). Moreover, control- and IBD-PDOs consistently formed monolayers with stable TEER that were impermeable to 0.4 kDa Lucifer yellow (Figure 5 and S5). The factors driving differences between the studies are unclear; however, we note that our study represents a small sample size and was not designed to detect disease-associated differences. Furthermore, as our study shows, phenotypic responses in organoids are highly influenced by the media and method used to culture them, which vary between studies. Future studies with larger sample sizes will be needed to evaluate the impact of IBD on PDOs.
In summary, we characterize conditions to promote epithelial differentiation paradigms in PDOs and PDO-derived monolayers for modeling stem cells, lower crypt, and upper crypt populations. Responses to media were consistent in both 3D organoid and monolayer formats, whereas transcriptional differences between these models may reflect distinctions in functional responses or activation state. In accordance with this, we were able to model epithelial barrier generation, disruption by cytokines, and protection with tofacitinib. Collectively, our findings suggest the applicability of PDOs and PDO-derived monolayers in IBD drug discovery and enable further exploration of damage and repair mechanisms in these models.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Graphical abstract was created with BioRender.com. Relevant statistics were performed using GraphPad Prism version 9.0.0 for MacOS, GraphPad Software, San Diego, California, USA (www.graphpad.com).
Glossary
Abbreviations
- CNM
colon normal medium
- cDM
combination differentiation medium
- CD
Crohn’s disease
- eDM
enterocyte differentiation medium
- IBD
inflammatory bowel disease
- PDO
patient-derived organoids
- TEER
transepithelial electrical resistance
- TA
transit amplifying
- UC
ulcerative colitis
Contributor Information
Scott A Jelinsky, Pfizer Worldwide Research, Development, and Medical, Cambridge, MA, USA.
Merel Derksen, HUB Organoids, Utrecht, the Netherlands.
Eric Bauman, Pfizer Worldwide Research, Development, and Medical, Cambridge, MA, USA.
Carla S Verissimo, HUB Organoids, Utrecht, the Netherlands.
Wies T M van Dooremalen, HUB Organoids, Utrecht, the Netherlands.
Jamie Lee Roos, HUB Organoids, Utrecht, the Netherlands.
Celia Higuera Barón, HUB Organoids, Utrecht, the Netherlands.
Celia Caballero-Franco, Pfizer Worldwide Research, Development, and Medical, Cambridge, MA, USA.
Bryce G Johnson, Pfizer Worldwide Research, Development, and Medical, Cambridge, MA, USA.
Michelle G Rooks, Pfizer Worldwide Research, Development, and Medical, Cambridge, MA, USA.
Johanna Pott, HUB Organoids, Utrecht, the Netherlands.
Bas Oldenburg, Department of Gastroenterology and Hepatology, University Medical Centre Utrecht, Utrecht, the Netherlands.
Robert G J Vries, HUB Organoids, Utrecht, the Netherlands.
Sylvia F Boj, HUB Organoids, Utrecht, the Netherlands.
Marion T Kasaian, Pfizer Worldwide Research, Development, and Medical, Cambridge, MA, USA.
Farzin Pourfarzad, HUB Organoids, Utrecht, the Netherlands.
Charles V Rosadini, Pfizer Worldwide Research, Development, and Medical, Cambridge, MA, USA.
Author Contributions
S.J.: conceptualization, lead; data curation, lead; formal analysis, lead; investigation, lead; methodology, lead; validation, lead; writing of original draft, lead; review and editing, lead
M.D.: conceptualization, lead; data curation, lead; formal analysis, lead; investigation, lead; methodology, lead; validation, lead; writing of original draft, lead; review and editing, lead
E.B.: conceptualization, supporting; data curation, supporting; formal analysis, supporting; investigation, supporting; methodology, supporting; resources, supporting; review and editing, supporting
C.V.: conceptualization, supporting; data curation, supporting; formal analysis, supporting; investigation, supporting; methodology, supporting; resources, supporting; review and editing, supporting
W.T.M.v.D.: conceptualization, supporting; data curation, lead; formal analysis, supporting; investigation, lead; methodology, lead; resources, supporting; review and editing, supporting
J.L.R.: conceptualization, supporting; data curation, lead; formal analysis, supporting; investigation, supporting; methodology, supporting; resources, supporting; review and editing, supporting
C.H.B. conceptualization, supporting; data curation, supporting; formal analysis, supporting; investigation, supporting; methodology, supporting; resources, supporting; review and editing, supporting
C.C.F.: conceptualization, supporting; formal analysis, supporting; investigation, supporting; methodology, supporting; resources, supporting; review and editing, supporting
B.J.: conceptualization, supporting; formal analysis, supporting; investigation, supporting; methodology, supporting; resources, supporting; review and editing, supporting
M.R.: conceptualization, supporting; formal analysis, supporting; investigation, supporting; methodology, supporting; resources, supporting; review and editing, supporting
J.P.: formal analysis, supporting; investigation, supporting; methodology, supporting; resources, supporting; review and editing, supporting
B.O.: conceptualization, supporting; investigation, supporting; methodology, supporting; resources, lead; review and editing, supporting
R.V.: conceptualization, supporting; formal analysis, supporting; investigation, supporting; methodology, supporting; resources, supporting; supervision, supporting; review and editing, supporting
S.B.: conceptualization, supporting; formal analysis, supporting; investigation, supporting; methodology, supporting; resources, supporting; supervision, supporting; review and editing, supporting
M.K.: conceptualization, supporting; formal analysis, supporting; investigation, supporting; methodology, supporting; resources, supporting; supervision, supporting; review and editing, supporting
F.P.: conceptualization, lead; data curation, lead; formal analysis, lead; investigation, lead; methodology, lead; project administration, lead; resources, lead; supervision, lead; validation, lead; original draft, lead; review and editing, lead
C.R.: conceptualization, lead; data curation, lead; formal analysis, lead; investigation, lead; methodology, lead; project administration, lead; resources, lead; supervision, lead; validation, lead; original draft, lead; review and editing, lead
Conflicts of Interest
S.J., E.B., C.C.F., B.J., M.R., M.K., and C.R. are employed by Pfizer, Inc., which funded the study. All other authors declare no conflicts.
Data Availability
The authors confirm that transcriptional data associated with this study are available through a public database. All other material supporting the conclusions are available in the article. Clinical material or live organoids are not available for distribution.
References
- 1. Peyrin-Biroulet L, Ferrante M, Magro F, et al. ; Scientific Committee of the European Crohn's and Colitis Organization. Results from the 2nd scientific workshop of the ECCO. I: impact of mucosal healing on the course of inflammatory bowel disease. J Crohns Colitis 2011;5:477-483. [DOI] [PubMed] [Google Scholar]
- 2. Martini E, Krug SM, Siegmund B, Neurath MF, Becker C.. Mend your fences: the epithelial barrier and its relationship with mucosal immunity in inflammatory bowel disease. Cell Mol Gastroenterol Hepatol. 2017;4:33-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sommer K, Wiendl M, Müller TM, et al. Intestinal mucosal wound healing and barrier integrity in IBD-crosstalk and trafficking of cellular players. Front Med (Lausanne) 2021;8:643973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. O’Connell L, Winter DC, Aherne CM.. The role of organoids as a novel platform for modeling of inflammatory bowel disease. Front Pediatr. 2021;9:624045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sato T, Stange DE, Ferrante M, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011;141:1762-1772. [DOI] [PubMed] [Google Scholar]
- 6. Clevers H. Modeling development and disease with organoids. Cell 2016;165:1586-1597. [DOI] [PubMed] [Google Scholar]
- 7. Jung P, Sato T, Merlos-Suárez A, et al. Isolation and in vitro expansion of human colonic stem cells. Nat Med. 2011;17:1225-1227. [DOI] [PubMed] [Google Scholar]
- 8. Günther C, Winner B, Neurath MF, Stappenbeck TS.. Organoids in gastrointestinal diseases: from experimental models to clinical translation. Gut 2022;71:1892-1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Willert K, Brown JD, Danenberg E, et al. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 2003;423:448-452. [DOI] [PubMed] [Google Scholar]
- 10. Yin X, Farin HF, van Es JH, Clevers H, Langer R, Karp JM.. Niche-independent high-purity cultures of Lgr5+ intestinal stem cells and their progeny. Nat Methods. 2014;11:106-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van Dooremalen WTM, Au - Derksen M, Au - Roos JL, et al. Organoid-derived epithelial monolayer: a clinically relevant in vitro model for intestinal barrier function. JoVE. 2021;(173):e62074. [DOI] [PubMed] [Google Scholar]
- 12. Zhao S, Xi L, Quan J, et al. QuickRNASeq lifts large-scale RNA-seq data analyses to the next level of automation and interactive visualization. BMC Genomics. 2016;17:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Law CW, Chen Y, Shi W, Smyth GK.. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weiner J 3rd, Domaszewska T.. Tmod: an R package for general and multivariate enrichment analysis. PeerJ Preprints. 2016;4:e2420v1. [Google Scholar]
- 15. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P.. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smillie CS, Biton M, Ordovas-Montanes J, et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 2019;178:714-730.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Es JH, Sato T, van de Wetering M, et al. Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol. 2012;14:1099-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sasaki N, Sachs N, Wiebrands K, et al. Reg4+ deep crypt secretory cells function as epithelial niche for Lgr5+ stem cells in colon. Proc Natl Acad Sci USA. 2016;113:E5399-E5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Capaldo CT, Nusrat A.. Cytokine regulation of tight junctions. Biochim Biophys Acta. 2009;1788:864-871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bruewer M, Luegering A, Kucharzik T, et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. 2003;171:6164-6172. [DOI] [PubMed] [Google Scholar]
- 21. Hodge JA, Kawabata TT, Krishnaswami S, et al. The mechanism of action of tofacitinib - an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2016;34:318-328. [PubMed] [Google Scholar]
- 22. Criss ZK, 2nd, Bhasin N, Di Rienzi SC, et al. Drivers of transcriptional variance in human intestinal epithelial organoids. Physiol Genomics. 2021;53:485-508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beumer J, Clevers H.. Cell fate specification and differentiation in the adult mammalian intestine. Nat Rev Mol Cell Biol. 2021;22:39-53. [DOI] [PubMed] [Google Scholar]
- 24. VanDussen KL, Marinshaw JM, Shaikh N, et al. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut 2015;64:911-920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Foerster EG, Mukherjee T, Cabral-Fernandes L, et al. How autophagy controls the intestinal epithelial barrier. Autophagy. 2022;18:86-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Muenchau S, Deutsch R, de Castro IJ, et al. Hypoxic environment promotes barrier formation in human intestinal epithelial cells through regulation of microRNA 320a expression. Mol Cell Biol. 2019;39(14):e00553-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Barrett JC, Hansoul S, Nicolae DL, et al. ; NIDDK IBD Genetics Consortium. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955-962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Graham DB, Lefkovith A, Deelen P, et al. TMEM258 Is a component of the oligosaccharyltransferase complex controlling ER stress and intestinal inflammation. Cell Rep 2016;17:2955-2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hayes P, Dhillon S, O’Neill K, et al. Defects in NADPH oxidase genes NOX1 and DUOX2 in very early onset inflammatory bowel disease. Cell Mol Gastroenterol Hepatol 2015;1:489-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lu Y, Zhou L, Liu L, et al. Serum omentin-1 as a disease activity marker for Crohn’s disease. Dis Markers. 2014;2014:162517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR.. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol. 2005;166:409-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gleeson JP, Estrada HQ, Yamashita M, et al. Development of physiologically responsive human iPSC-derived intestinal epithelium to study barrier dysfunction in IBD. Int J Mol Sci . 2020;21(4):1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zolotarevsky Y, Hecht G, Koutsouris A, et al. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology 2002;123:163-172. [DOI] [PubMed] [Google Scholar]
- 34. Deem RL, Shanahan F, Targan SR.. Triggered human mucosal T cells release tumour necrosis factor-alpha and interferon-gamma which kill human colonic epithelial cells. Clin Exp Immunol. 1991;83:79-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nava P, Koch S, Laukoetter MG, et al. Interferon-gamma regulates intestinal epithelial homeostasis through converging beta-catenin signaling pathways. Immunity 2010;32:392-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Woznicki JA, Saini N, Flood P, et al. TNF-α synergises with IFN-γ to induce caspase-8-JAK1/2-STAT1-dependent death of intestinal epithelial cells. Cell Death Dis. 2021;12:864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sayoc-Becerra A, Krishnan M, Fan S, et al. The JAK-inhibitor tofacitinib rescues human intestinal epithelial cells and colonoids from cytokine-induced barrier dysfunction. Inflamm Bowel Dis. 2020;26:407-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ojo BA, VanDussen KL, Rosen MJ.. The promise of patient-derived colon organoids to model ulcerative colitis. Inflamm Bowel Dis. 2022;28:299-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Arnauts K, Verstockt B, Ramalho AS, Vermeire S, Verfaillie C, Ferrante M.. Ex vivo mimicking of inflammation in organoids derived from patients with ulcerative colitis. Gastroenterology 2020;159:1564-1567. [DOI] [PubMed] [Google Scholar]
- 40. Angus HC, Urbano PC, Laws GA, et al. An autologous colonic organoid-derived monolayer model to study immune: bacterial interactions in Crohn’s disease patients. Clin Transl Immunol. 2022;11:e1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that transcriptional data associated with this study are available through a public database. All other material supporting the conclusions are available in the article. Clinical material or live organoids are not available for distribution.