

Abstract
The Werner Syndrome helicase, WRN, is a promising therapeutic target in cancers with microsatellite instability (MSI). Long‐term MSI leads to the expansion of TA nucleotide repeats proposed to form cruciform DNA structures, which in turn cause DNA breaks and cell lethality upon WRN downregulation. Here we employed biochemical assays to show that WRN helicase can efficiently and directly unfold cruciform structures, thereby preventing their cleavage by the SLX1‐SLX4 structure‐specific endonuclease. TA repeats are particularly prone to form cruciform structures, explaining why these DNA sequences are preferentially broken in MSI cells upon WRN downregulation. We further demonstrate that the activity of the DNA mismatch repair (MMR) complexes MutSα (MSH2‐MSH6), MutSβ (MSH2‐MSH3), and MutLα (MLH1‐PMS2) similarly decreases the level of DNA cruciforms, although the mechanism is different from that employed by WRN. When combined, WRN and MutLα exhibited higher than additive effects in in vitro cruciform processing, suggesting that WRN and the MMR proteins may cooperate. Our data explain how WRN and MMR defects cause genome instability in MSI cells with expanded TA repeats, and provide a mechanistic basis for their recently discovered synthetic‐lethal interaction with promising applications in precision cancer therapy.
Keywords: biochemistry, cancer, mismatch repair, synthetic lethality, Werner
Subject Categories: DNA Replication, Recombination & Repair
Biochemical assays define the molecular basis of synthetic lethality caused by loss of both Werner Syndrome helicase and MMR in cancers with microsatellite instability.

Introduction
Exploiting synthetic lethality represents a promising strategy in precision cancer therapy. A defect in one cellular pathway can make the respective cancer cells dependent on a second compensatory pathway for survival. Inhibition of the second pathway thus selectively kills the cancer cells, while having little or no effects on the surrounding healthy tissue (O'Neil et al, 2017). The first example of such an approach targeting DNA repair factors applied in cancer therapy is the treatment of selected breast, ovarian or endometrial cancers defective in homologous recombination (HR) with poly (ADP ribose) polymerase (PARP) inhibitors. These drugs trap PARP at sites of single‐stranded DNA breaks (SSBs), leading to replication‐associated double‐stranded DNA breaks (DSBs), which require homologous recombination for repair and cellular survival. Recombination deficiency thus represents a specific vulnerability of the cancer cells (Lord & Ashworth, 2017).
More recently, the Werner helicase (WRN) was found as a potential target in cancers exhibiting microsatellite instability (MSI) (Behan et al, 2019; Chan et al, 2019; Kategaya et al, 2019; Lieb et al, 2019). These cancers include both hereditary (Lynch syndrome) or sporadic cancers of the colorectum, stomach, ovary, and endometrium (Martinez‐Roca et al, 2022). MSI is defined as a small alteration in the number of short repetitive sequences, most commonly mononucleotide repeats. MSI arises from defects in postreplicative mismatch repair (MMR), and only becomes detectable after multiple cellular divisions (Boland & Goel, 2010; Pecina‐Slaus et al, 2020; Olave & Graham, 2021; Randrian et al, 2021). DNA polymerases replicating repetitive sequences are prone to slippage, leading to extrahelical loops in the replicated DNA. In healthy cells, loops that escaped the polymerase proofreading activity are normally identified and repaired by MMR. Depending on the size of the irregularity arising during DNA replication, the structures are either identified by the MMR recognition complex MutSα, a heterodimer consisting of MSH2 and MSH6, acting on mismatches and short loops, or MutSβ, a heterodimer of MSH2 and MSH3 that recognizes larger loops. Downstream of recognition, the MMR reaction involves the MutLα (MLH1‐PMS2) endonuclease, as well as exonuclease, DNA synthesis (polymerase), and ligation steps (Jiricny, 2006; Kunkel & Erie, 2015; Fishel, 2021).
The discovery of WRN dependency in MSI cells was an unexpected and seminal observation (Behan et al, 2019; Chan et al, 2019; Kategaya et al, 2019; Lieb et al, 2019). Werner is a helicase that belongs to the conserved RecQ family that includes five members in human cells. WRN has multiple but relatively poorly understood functions in DNA replication and repair, including homologous recombination and base excision repair pathways, as well as telomere protection (Ahn et al, 2004; Saydam et al, 2007; Sturzenegger et al, 2014; Aiello et al, 2019; Datta et al, 2021). Defects in WRN cause Werner's syndrome, characterized by accelerated aging and cancer predisposition (Oshima et al, 2017). However, WRN is not an essential protein, and the median survival of Werner's syndrome patients is 40–50 years. Therefore, short‐term WRN inhibition as a cancer therapy approach may not have a major impact on non‐cancerous surrounding cells, minimizing side effects. WRN inhibition may be particularly applicable to treat MSI tumors refractory to established therapies (Picco et al, 2021).
The synthetic lethality was not observed upon acute depletion of MMR factors and WRN in heathy cells (Chan et al, 2019; Lieb et al, 2019). WRN dependency was only found in MSI cancer cells, leading to a hypothesis that a prolonged MMR defect leads to a genomic scar, which triggers WRN dependency (Chan et al, 2019; Lieb et al, 2019). Subsequent sequencing data from the Nussenzweig laboratory revealed that the DNA breaks arising in MSI cells upon WRN depletion are almost exclusively localized to sites of extended TA repeats, shedding light on the likely identity of the genomic scar (van Wietmarschen et al, 2020, 2021). It was proposed that MMR deficiency leads to an expansion of TA repeats at multiple loci in genomic DNA. What makes specifically TA repeats unique in this context remains unclear. The observation was unexpected on yet another level: MSI caused by MMR deficiency typically leads to an alteration of mononucleotide repeats, usually shortening by a few repeat units, and not a major expansion (Hoang et al, 1997; Pecina‐Slaus et al, 2020; Olave & Graham, 2021; Randrian et al, 2021). A subset of MMR proteins was instead found to act pathologically in the expansion of trinucleotide repeats, underlying syndromes such as Fragile X or Huntington's disease (Schmidt & Pearson, 2016; Iyer & Pluciennik, 2021; Neil et al, 2021). However, in that case, the presence of MMR factors leads to the expansion of the trinucleotide repeats, and not their absence, as in the case of TA repeat expansion (Miller et al, 2020; Richard, 2021). The mechanism underlying the expansion of TA repeats in MSI cells (in the absence of MMR) is thus not known.
TA repeats are thought to fold into cruciform structures, which may stall replication forks (Kaushal et al, 2019). Stalled replication forks activate the ATR kinase that recruits WRN (Ammazzalorso et al, 2010), which may unwind the cruciforms (van Wietmarschen et al, 2020). In the absence of WRN or the phosphorylation cascade leading to its recruitment, the cruciform structures persist. These cruciform structures resemble Holliday junctions at their base, which are substrates for the SLX4‐associated structure‐specific nucleases MUS81‐EME1 and SLX1 (van Wietmarschen et al, 2020; Giaccherini & Gaillard, 2021). Accordingly they were shown to trigger aberrant DNA breakage dependent on MUS81 and SLX4 (Franchitto et al, 2008; van Wietmarschen et al, 2020). Although acute depletion of MMR and WRN was not reported to trigger lethality in cells without expanded TA repeats, a partial rescue of lethality was observed upon restoration of the missing MMR complexes in MSI cells in two independent studies (Chan et al, 2019; Lieb et al, 2019). These data are not explained by the current model and suggest that MMR proteins may also have an undefined direct protective function against the negative consequences of cruciform structures, beyond preventing the expansion of the TA repeats.
Here we used biochemical assays that allow us to monitor the stability of cruciform DNA and its processing by recombinant proteins in vitro. We found that TA repeats are particularly prone to form cruciforms, likely explaining why these sequences are preferentially broken in MSI cells upon WRN inhibition. We show that WRN, using its helicase function, can unfold cruciform structures leading to the formation of double‐stranded DNA, demonstrating that WRN is likely to have a direct role in the processing of these structures (van Wietmarschen et al, 2020). We further present that the MMR complexes, primarily MutSα (MSH2‐MSH6), MutSβ (MSH2‐MSH3) and MutLα (MLH1‐PMS2) similarly reduce the levels of cruciform structures in DNA. The underlying mechanism is different than that employed by WRN. MutSα and MutSβ may stabilize DNA in its double‐stranded form, exploiting dynamic structural transitions of the cruciform DNA. Furthermore, the WRN and MutLα (MLH1‐PMS2) act synergistically in cruciform unfolding, possibly supported by physical interactions identified previously (Saydam et al, 2007). The latter results may explain the partial rescue of lethality observed in MSI cells upon reintroduction of the missing MMR components (Chan et al, 2019; Lieb et al, 2019). Together, our data provide insights into the mechanisms underlying synthetic lethality in MSI cells upon WRN inhibition.
Results
WRN helicase unfolds cruciform DNA
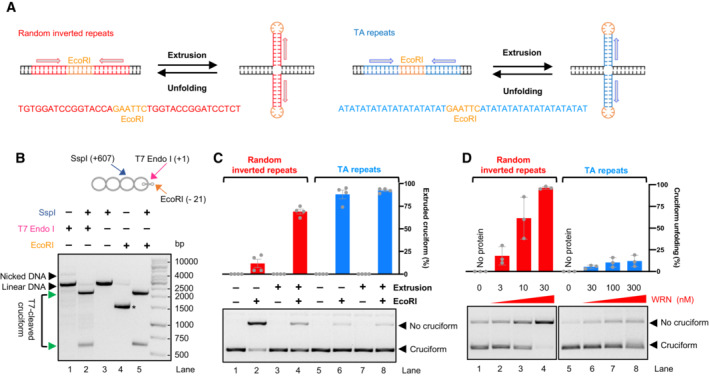
To study a potential function of WRN in cruciform DNA unfolding, we employed a pUC19‐derived substrate with inverted repeats of a random sequence bearing an EcoRI restriction site in between the repeats (Fig 1A). Upon DNA extrusion in negatively supercoiled plasmid (scDNA), the six nucleotides constituting the restriction site will be placed at the apex of the cruciform as a ssDNA hairpin, becoming uncleavable by EcoRI. The presence of the cruciform at the expected location was verified by DNA cleavage with T7 Endonuclease I, a Holliday junction resolvase, followed up by cutting with SspI (Fig EV1A). Therefore, with a simple restriction digest, we can monitor the proportion of the DNA molecules containing the cruciform (Fig 1B, bottom DNA band, scDNA refractory to EcoRI cutting, labeled as “Cruciform”) or not containing the cruciform structure (top DNA band, linearized by EcoRI, labeled as “No cruciform”).
Figure 1. WRN helicase unfolds cruciform DNA.
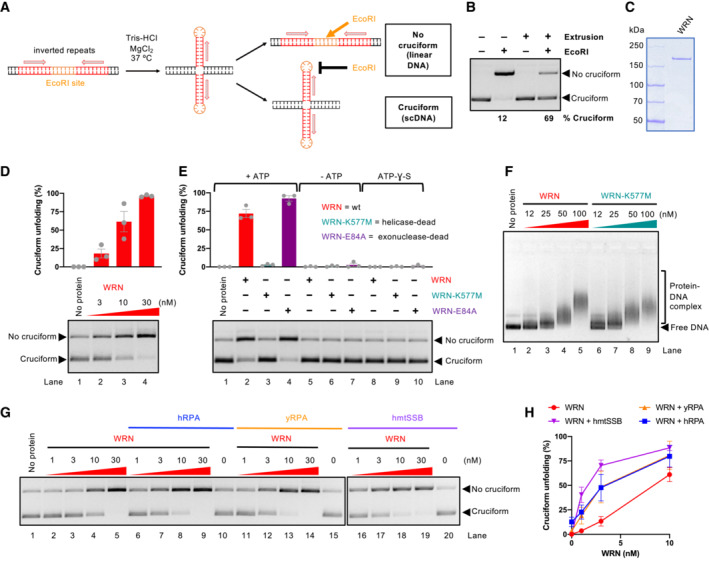
- A schematic representation of the cruciform detection assay.
- Representative cruciform detection assay. Supercoiled DNA (cruciform) and linear DNA (no cruciform) were resolved on a 1% agarose gel, stained with GelRed. The % of cruciform‐containing molecules are expressed as averages; n = 3.
- Representative polyacrylamide gel showing purification of recombinant WRN. The gel was stained with Coomassie Brilliant blue.
- Cruciform unfolding assay with increasing WRN concentrations and cruciform consisting of random inverted repeats (random‐IR). Top, quantitation of cruciform unfolding. The amount of linear DNA from the “No protein” lane was subtracted from all other samples. Averages shown; n = 3 technical replicates; error bars, SEM.
- Cruciform unfolding assays as in (D) with WRN, WRN‐K577M, and WRN‐E84A (all used at 10 nM), carried out either in the presence or absence of ATP, or with ATP‐ƔS. Averages shown; n ≥ 3 technical replicates; error bars, SEM.
- Electrophoretic mobility shift assays with WRN and WRN‐K577M, using pUC19 with the random‐IR cruciform structure as a substrate.
- Representative cruciform unfolding assays with increasing concentrations of WRN, together with human RPA (hRPA, 30 nM), yeast S. cerevisiae RPA (yRPA, 30 nM), or human mitochondrial SSB (hmtSSB, 50 nM). pUC19 with the random‐IR cruciform structure was used as a substrate.
- Quantitation of assays such as in (G). The amount of linear DNA from the “No protein” lane was subtracted from all other samples. Averages shown; n ≥ 3 technical replicates; error bars, SEM.
Figure EV1. WRN helicase unfolds cruciform DNA structures.
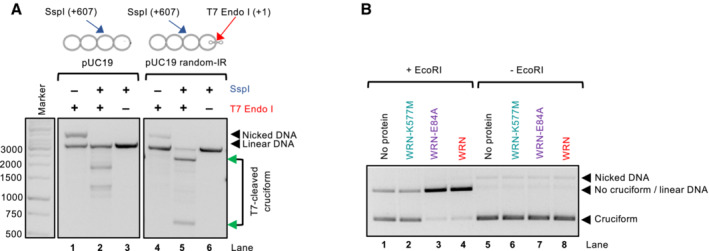
- Representative assay showing the activity of T7 Endonuclease I, which cleaves the 4‐way junction formed at the cruciform DNA site. Incubation with T7 Endonuclease I and SspI results in the production of bands at the expected positions of 607 and 2,119 bp, indicated by the green arrows. The SspI restriction site is 607 bp away from the cruciform site. A plasmid without the inverted repeats is cleaved less efficiently and produces bands of different sizes (lane 2), resulting from T7 Endonuclease I activity at other sites that spontaneously extrude in pUC19 DNA. The DNA molecules were resolved by standard electrophoresis on a 1% native agarose gel, stained with GelRed.
- Representative cruciform unfolding assay with WRN, WRN‐K577M, and WRN‐E84A (all used at 10 nM). The assay was incubated either with or without EcoRI and the DNA species were separated on a 1% native agarose gel, stained with GelRed.
Using purified recombinant WRN (Fig 1C), we observed that WRN unfolded the cruciform structure in a concentration‐dependent manner (Fig 1D). The WRN function in cruciform DNA unfolding required ATP hydrolysis and was dependent on the integrity of its ATPase site, while it did not involve WRN nuclease function (Fig 1E), in agreement with cellular data (Chan et al, 2019; Lieb et al, 2019). We noted that both wild type and helicase‐dead WRN bound DNA similarly (Fig 1F), arguing that the accessibility of DNA to EcoRI is not due to binding‐induced change in local DNA topology, as this would be common to both WRN variants. Likewise, we did not observe any nuclease activity upon incubation with either of the WRN variants (Fig EV1B), which rules out the hypothesis that the cruciform is indirectly destabilized by DNA relaxation, as the cruciform structures require negative supercoiling to be formed and maintained (Panayotatos & Wells, 1981; Mizuuchi et al, 1982; Lilley, 1983).
The cruciform unfolding by WRN was moderately stimulated not only by human replication protein A (RPA) but also by the non‐cognate yeast S. cerevisiae RPA or human mitochondrial SSB (Fig 1G and H), suggesting that cruciform processing by WRN involves a single‐stranded DNA (ssDNA) intermediate that is stabilized by the ssDNA‐binding proteins, without requirement for a physical interaction with WRN. Together, our experiments establish that the WRN helicase is directly capable of unfolding cruciform structures using its ATP hydrolysis‐driven motor activity.
WRN unfolding of cruciforms prevents DNA cleavage by the SLX1‐SLX4CCD complex
In MSI cells, the absence of WRN leads to MUS81 and SLX4‐dependent DNA breaks (van Wietmarschen et al, 2020). These data support a model where cruciform structures that escape the attention of WRN are cleaved by MUS81 in an unscheduled manner, leading to DNA breaks and consequent cellular lethality. In our reconstituted system, we observed that the cruciform DNA was highly susceptible to cleavage by the SLX1‐SLX4CCD complex (containing just the SLX1‐binding C‐terminal domain of SLX4), as expected (Fekairi et al, 2009). Upon incubation with WRN, DNA cleavage by SLX1‐SLX4CCD at the site of the cruciform was strongly reduced (Fig 2A and B), in agreement with the model in which WRN activity removes the substrate for cleavage by the structure specific nucleases. We observed that WRN reduced DNA cleavage also in conjunction with the non‐cognate T7 Endonuclease I (Fig EV2), suggesting that physical interaction between WRN and the nuclease is not essential for the DNA cleavage inhibition. These results show that WRN activity directly prevents DNA cleavage by the resolvase complex at DNA cruciform sites by unfolding cruciform DNA, underpinning a mechanism by which WRN prevents DNA fragmentation and lethality of MSI cells.
Figure 2. WRN unfolding of cruciforms prevents DNA cleavage by the SLX1‐SLX4CCD nuclease.
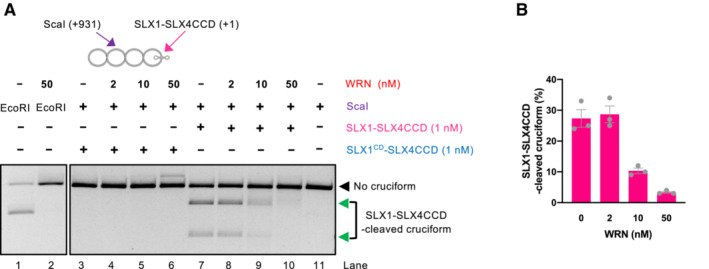
- Representative assays with cruciform DNA (consisting of random‐IR) either without or with WRN incubation prior to cruciform detection by SLX1‐SLX4CCD. The ScaI restriction site is 931 bp away from the cruciform site cleaved by SLX1‐SLX4CCD. Combined activity of ScaI and SMX results in 931 and 1,795 bp bands indicated by the green arrows. The intensity of these bands decreases upon WRN incubation, resulting predominantly in linear DNA cut by ScaI, suggesting that WRN removes the substrate for SLX1‐SLX4CCD. The catalytic dead version of SLX1‐SLX4CCD (SLX1CD‐SLX4CCD), where SLX1 carries an R41A mutations in its nuclease domain, has no activity on the cruciform DNA.
- Quantitation of assays such as in (A). Averages shown; n = 3 technical replicates; error bars, SEM.
Figure EV2. WRN helicase prevents cruciform DNA cleavage by T7 Endonuclease I.
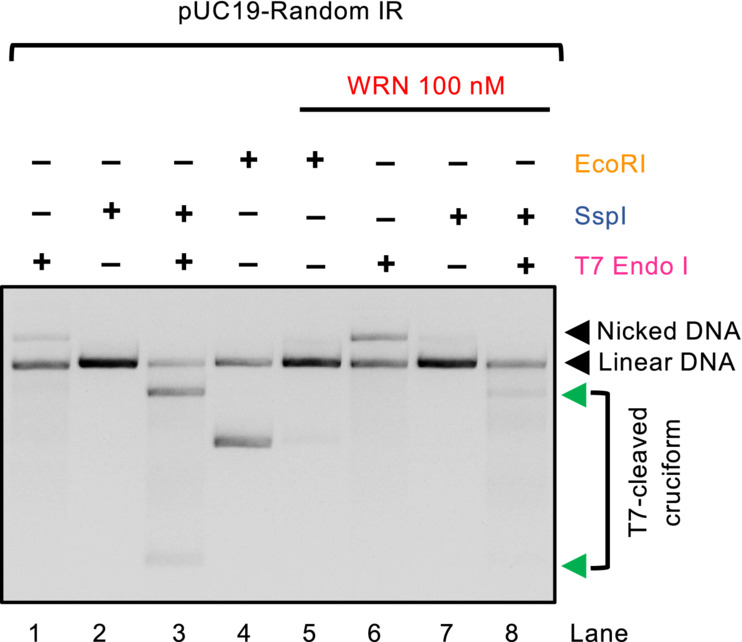
Representative assays detecting cruciform DNA (random‐IR) by T7 Endonuclease I, pre‐incubated or not with WRN. Preincubation with WRN reduces the intensity of bands resulting by cleavage with T7 Endonuclease I (607 and 2,119 bp, indicated by the green arrows), resulting in linearization by SspI. The DNA molecules were resolved by standard electrophoresis on a 1% native agarose gel, stained with GelRed.
AT repeats enhance cruciform DNA formation
Cellular experiments demonstrated that DNA breaks in MSI and WRN‐deficient cells preferentially occur at sites of extended TA repeats (van Wietmarschen et al, 2020). It was not clarified whether this observation reflects a preferential activity of WRN at TA cruciforms. Our initial reconstitution experiments were performed with a cruciform structure based on an inverted repeat of a synthetic origin consisting of a random DNA sequence (56% G/C content) (Figs 1 and 2). We next prepared DNA with TA repeats of the same length, which similarly included an EcoRI site in the center of the repeats (Fig 3A). The cruciform extrusion of the TA‐based substrate was confirmed by cutting the 4‐way junction at the base of the cruciform with T7 Endonuclease I, followed by SspI digestion, which yielded DNA fragments of the expected lengths (Fig 3B). We observed that supercoiled DNA was instead highly refractory to EcoRI cleavage due to the presence of the cruciform structure. However, upon incubation with both SspI and EcoRI, the EcoRI site became accessible (Fig 3B), in agreement with previous reports demonstrating that negative supercoiling is required for cruciform stability (Panayotatos & Wells, 1981; Mizuuchi et al, 1982; Lilley, 1983).
Figure 3. TA repeats enhance cruciform DNA formation.
- Comparison of cruciform substrates based on random inverted repeats (left) and TA repeats (right). Top, a schematic cartoon; Bottom, DNA sequence.
- Mapping of TA repeats cruciform structure. T7 Endonuclease I activity, followed by SspI, cleaves the cruciform within negatively supercoiled DNA, producing two bands of the expected size (607 and 2,119 bp, indicated by the green arrows, lane 2). Simultaneous incubation of the substrate with SspI and EcoRI allows EcoRI cutting at the TA repeats sequence (lane 5), while the extruded cruciform is refractory to EcoRI digestion (lane 4, the * indicates scDNA).
- Representative cruciform detection assays. Plasmid DNA was subjected to Cruciform extrusion procedure, where indicated. Supercoiled DNA (cruciform, refractory to EcoRI) and linear DNA (no cruciform, cleaved by EcoRI) were resolved on a 1% agarose gel, stained with GelRed. Top, quantitation of extruded cruciform in % with respect to the amount of total DNA that is present in the lane. Averages shown; n = 4 technical replicates; error bars, SEM.
- Representative cruciform unfolding assays with increasing WRN concentrations, using pUC19 with either random inverted repeats or TA repeats as a substrate. Top, quantitation of cruciform unfolding. The amount of linear DNA from the “No protein” lane was subtracted from all other samples. Averages shown; n = 3 technical replicates; error bars, SEM.
Next, we directly compared cruciform DNA formation with the random inverted repeat sequence. DNA with TA repeats was much more likely to adopt the cruciform conformation, even without employing an extrusion protocol (see Material and Methods; Fig 3C). Strikingly, the apparent activity of WRN on TA‐rich cruciform DNA was notably reduced (Fig 3D), although it is possible that WRN could still be highly active also on this structure, but due to a strong negative supercoiling the resulting dsDNA segment reverts immediately back to the cruciform conformation. We hypothesize that the preferential DNA cleavage at TA sites in MSI and WRN‐deficient cells might be due to the favored formation of cruciform DNA at these sites. Due to the low melting temperature of (TA)n sequences, cruciform DNA structures may form efficiently even in the weakly negatively supercoiled chromosomal DNA.
Cruciform DNA unfolding is not a shared activity of RecQ family helicases
We next compared the cruciform unfolding capacity of WRN with that of Bloom (BLM), a related human RecQ family helicase, as well as S. cerevisiae Sgs1 helicase from the same family (Fig 4A). Interestingly, in our minimal reconstituted system, BLM showed a cruciform unfolding capacity comparable to WRN, while Sgs1 was largely deficient (Fig 4B and C). Sgs1 and WRN did not notably differ in their DNA‐binding activities, and both proteins showed a moderate preference for binding cruciform DNA as opposed to linear plasmid DNA (Fig EV3A). Sgs1 is one of the most active helicases characterized to date (Cejka & Kowalczykowski, 2010). Under our conditions, Sgs1 showed a comparable activity to WRN in the unwinding of oligonucleotide‐based Holliday junctions (Fig EV3B and C), and notably higher activity in the unwinding of plasmid‐length dsDNA (Figs EV3D and 4D). Because Sgs1 is incapable to act on cruciform DNA, a vigorous helicase activity of a RecQ family helicase member does not correspond to cruciform unfolding activity, demonstrating a certain level of specificity for the WRN activity.
Figure 4. Cruciform DNA unfolding is not a shared activity of RecQ family helicases.
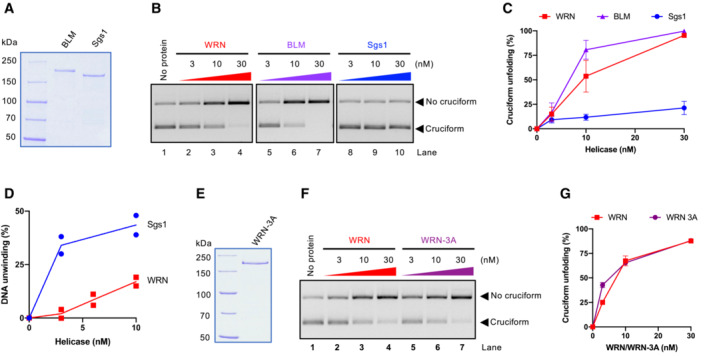
- Recombinant BLM and S. cerevisiae Sgs1 used in this study. The gel was stained with Coomassie Brilliant blue.
- Representative cruciform unfolding assay with increasing concentrations of WRN, BLM, and Sgs1. Cruciform DNA consisting of random inverted repeats was used.
- Quantitation of assays such as (B). Averages shown; n ≥ 3 technical replicates; error bars, SEM.
- Quantitation of DNA unwinding from experiments such shown in (EV4D). Averages and individual data points shown; n = 2 technical replicates.
- Recombinant WRN‐3A (S991A T1152A S1256A) used in this study. The gel was stained with Coomassie Brilliant blue.
- Representative cruciform unfolding assays with increasing concentrations of WRN and WRN‐3A. Cruciform DNA consisting of random inverted repeats was used.
- Quantitation of assays such as in (F). Averages shown; n = 3 technical replicates; error bars, SEM.
Figure EV3. Comparison of DNA binding and unwinding by WRN and Sgs1 DNA helicases.
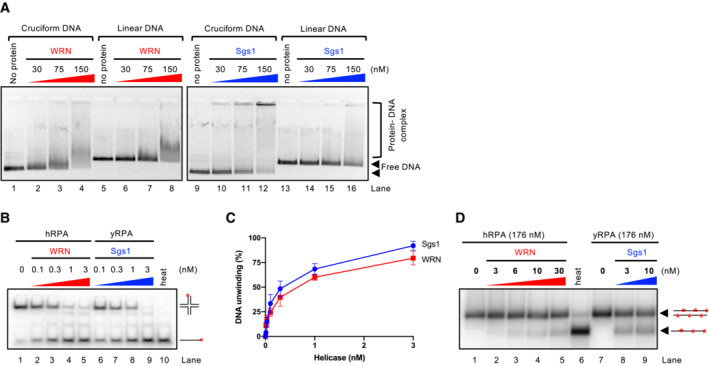
- Representative electrophoretic mobility shift assay with WRN and Sgs1, using either pUC19 harboring the random inverted repeat‐based cruciform structure (circular), or linearized pUC19. The samples were run on a 0.8% unstained native agarose gel. The gel was stained after the run with GelRed.
- Representative helicase assay with increasing concentrations of WRN and Sgs1, using an oligonucleotide‐based Holliday junction as a substrate. Reactions were supplemented with either human or yeast RPA (15 nM) and analyzed by 10% native acrylamide gel electrophoresis. Red asterisk indicates the position of the radioactive label.
- Quantitation of DNA unwinding from assays such as in (B). Averages shown; n ≥ 3; error bars, SEM.
- Representative helicase assay with increasing concentrations of WRN and Sgs1, using a 2.2 kbp‐long dsDNA substrate. Reactions were supplemented with either human or yeast RPA and analyzed by electrophoresis on a 1% agarose gel.
Our in vitro experiments revealed that in the minimal reconstituted system BLM had a cruciform unfolding activity similar to that of WRN. These results were unexpected because BLM is not required for survival of MSI cells (Chan et al, 2019; Lieb et al, 2019). The WRN function in cruciform unfolding is dependent on its recruitment to challenged replication forks, which may stall as they approach the cruciform structure (van Wietmarschen et al, 2020). WRN recruitment and nuclear foci formation require phosphorylation of its C‐terminal S/TQ sites by the ATR kinase (Ammazzalorso et al, 2010). A phosphorylation‐deficient WRN‐3A mutant fails to be recruited to forks and correspondingly does not support viability in MSI cells (van Wietmarschen et al, 2020). We have expressed and purified WRN‐3A (Fig 4E) and observed that its cruciform unfolding activity in vitro was indistinguishable from that of the wild‐type protein (Fig 4F and G). To function in cruciform unfolding in vivo, WRN recruitment to stalled forks is essential (van Wietmarschen et al, 2020). In the minimal reconstituted system, the need for recruitment is not a limiting step, which likely explains why BLM and WRN‐3A can unfold cruciforms in vitro, while being unable to do so in vivo. We conclude that the DNA unwinding activity of a RecQ family helicase does not directly correspond to its capacity to unfold cruciform DNA; however, cruciform unfolding in vitro is not a unique capacity of the WRN helicase. ATR phosphorylation‐dependent recruitment likely guarantees that WRN activity takes place specifically at sites of stalled replication forks.
WRN helicase core and RQC domains are responsible for cruciform DNA unfolding
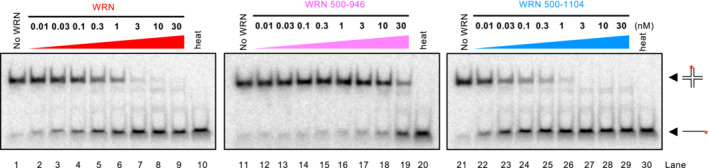
The RecQ helicase family members typically contain a conserved helicase core domain responsible for ATP hydrolysis, as well as RQC and HRDC domains (Chu & Hickson, 2009). The RQC domain is involved in the binding to branched DNA at the terminus of duplex DNA, and hence it is very important for DNA unwinding (Kitano et al, 2010). The function of the HRDC domain is less clear and was proposed to mediate protein–protein interactions (Kitano, 2014). To define which WRN domains are responsible for cruciform DNA unfolding, we prepared WRN truncations (Fig 5A and B), and compared unwinding of HJs and cruciform unfolding activities by these variants with the full‐length WRN protein (Figs 5C–E and EV4). We observed that WRN variant lacking the N‐terminus (exonuclease domain) and the C‐terminus (HRDC domain), but containing RQC was even more proficient in DNA unwinding compared to full‐length WRN, in agreement with previous reports (Harrigan et al, 2003). This WRN variant was correspondingly more efficient in cruciform DNA unfolding, indicating that the respective N‐ and C‐terminal domains are not involved in the metabolism of the cruciform structures. In contrast, the WRN core helicase domain lacking also the RQC domain was nearly deficient in unwinding of Holliday junctions, but still retained residual activity in cruciform DNA unfolding (Figs 5C–E and EV4). Our data indicating that the WRN RQC domain is more important for HJ unwinding compared to cruciform unfolding was unexpected, but in agreement with the results indicating that HJ unwinding activity does not correspond to cruciform unfolding, as is the case of Sgs1 (Figs 4B–D and EV3B–D). We conclude that the essential elements required for cruciform unfolding by WRN are dependent on its helicase core domain, and this activity is further stimulated by the RQC domain.
Figure 5. WRN helicase core and RQC domains are responsible for cruciform DNA unfolding.
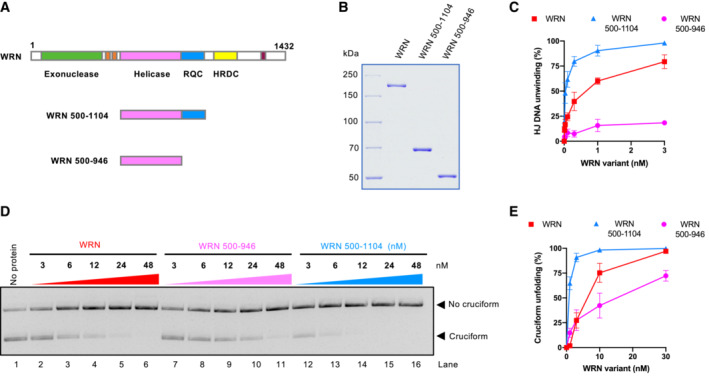
- Primary structure of WRN. Full‐length and truncation WRN variants are shown.
- Recombinant full‐length and truncation WRN variants used in this study are shown. The gel was stained with Coomassie Brilliant blue.
- Quantitation of DNA unwinding from assays such as shown in (EV5). Averages shown; n = 3 technical replicates; error bars, SEM.
- Cruciform unfolding assays with increasing concentrations of WRN variants.
- Quantitation of assays such as shown in (D). Averages shown; n = 3 technical replicates; error bars, SEM.
Figure EV4. DNA unwinding by WRN helicase variants.
Representative helicase assay with increasing concentrations of WRN variants, using an oligonucleotide‐based Holliday junction as a substrate. Reactions were supplemented with human RPA (15 nM) and analyzed by 10% native acrylamide gel electrophoresis. Red asterisks indicate the position of the radioactive label.
MutSα, MutSβ, and to a lesser degree MutLα MMR complexes unfold cruciform structures
Microsatellite instability over extended time periods was proposed to result in a genomic scar, represented by the expansion of TA repeats forming cruciform structures, which require WRN activity to prevent DSB formation (Chan et al, 2019). However, it was also reported that restoration of the respective missing MMR complex in MSI cells leads to a modest rescue of lethality induced by WRN knockdown (Chan et al, 2019; Lieb et al, 2019). The observed acute rescue effect cannot be explained by the genomic scar hypothesis because short‐term restoration of the MMR complexes would not affect the presence of the expanded TA repeats in the genomic DNA. However, the mechanism underlying the partial rescue effect was not clear. We thus aimed to test whether MMR complexes may also have a direct role in cruciform unfolding. To this point, we tested all three human MutS homolog complexes, including MutSα (MSH2‐MSH6), MutSβ (MSH2‐MSH3), and MutSγ (MSH4‐MSH5) as well as the three MutL homolog complexes MutLα (MLH1‐PMS2), MutLβ (MLH1‐PMS1), and MutLγ (MLH1‐MLH3) (Fig 6A and B).
Figure 6. MutSα, MutSβ, and to a lesser degree MutLα MMR complexes unfold cruciform structures.
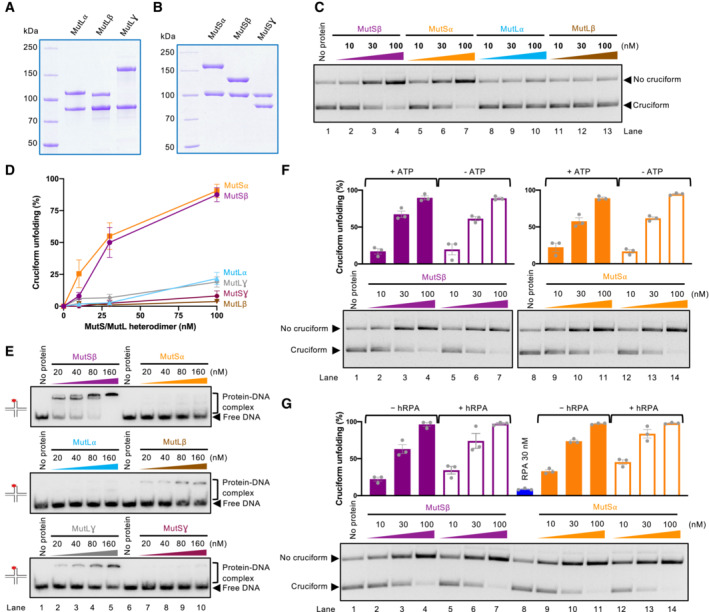
- MutL homolog heterodimers used in this study: MutLα (MLH1‐PMS2), MutLβ (MLH1‐PMS1), and MutLγ (MLH1‐MLH3).
- MutS homolog heterodimers used in this study: MutSα (MSH2‐MSH6), MutSβ (MSH2‐MSH3), and MutSγ (MSH4‐MSH5).
- Cruciform unfolding assays with MutSβ, MutS⍺, MutL⍺, and MutLβ. Cruciform DNA based on random inverted repeats was used.
- Quantitation of assays such as in (C) and (EV6A). Averages shown; n = 3 technical replicates; error bars, SEM.
- Representative electrophoretic mobility shift assays with the MutS and MutL homolog heterodimers, using an oligonucleotide‐based Holliday junction as a substrate. Red asterisk indicates the position of the radioactive label.
- Representative cruciform unfolding assays with MutSβ and MutS⍺, carried out without or with ATP, using the random‐IR cruciform as a substrate. Top, quantitation of cruciform unfolding. Averages shown; n = 3 technical replicates; error bars, SEM.
- Representative cruciform unfolding assays with MutSβ and MutS⍺, carried out without or with hRPA, using the random‐IR cruciform as a substrate. Top, quantitation of cruciform unfolding. Averages shown; n = 3 technical replicates; error bars, SEM.
Strikingly, we observed that the activity of MutSα, MutSβ, and to a much lesser degree MutLα and MutLγ led to the reduction of cruciform DNA observed in our assays (Fig 6C and D). The results were unexpected, because particularly MutSα and MutLα have not been implicated in the metabolism of cruciforms, at least prior to the seminal papers that described the synthetic lethality of WRN in MSI cells (Behan et al, 2019; Chan et al, 2019; Kategaya et al, 2019; Lieb et al, 2019; van Wietmarschen et al, 2020). In contrast, MutSγ and MutLγ complexes are involved in meiotic recombination and were found to bind Holliday junctions (Snowden et al, 2004; Ranjha et al, 2014), but these heterodimers showed only a minimal activity in cruciform DNA unfolding (Figs 6D and EV5A). MutLβ, a complex with the least understood function, did not show any activity (Fig 6C and D). We noted that none of the complexes nicked DNA under our conditions with magnesium (Fig EV5B), and the heterodimers at the concentration used (≤ 100 nM) did not affect the topology of plasmid DNA through DNA binding (Fig EV5C). Therefore, the apparent cruciform unfolding activity of the MutS and MutL homolog complexes cannot be explained by contaminating nuclease activities leading to DNA relaxation or DNA binding‐dependent changes in DNA topology. We also compared all MutS and MutL heterodimers for binding to an oligonucleotide‐based Holliday junction in the presence of a circular dsDNA competitor (Fig 6E). We observed that MutSβ was the most efficient, followed by MutLβ and meiotic MutLγ complexes, generally in agreement with previous reports (Snowden et al, 2004; Ranjha et al, 2014; Duroc et al, 2017; Young et al, 2020). Using linear pUC19, or supercoiled pUC19 bearing the AT cruciform, MutSβ but not MutSα showed preference for the cruciform DNA (Fig EV5D). We observed that the ability to stably bind HJs does not correspond to the capacity of the MutS and MutL homolog complexes to unfold cruciform structures (Fig 6D and E).
Figure EV5. Cruciform DNA binding by mismatch repair complexes.
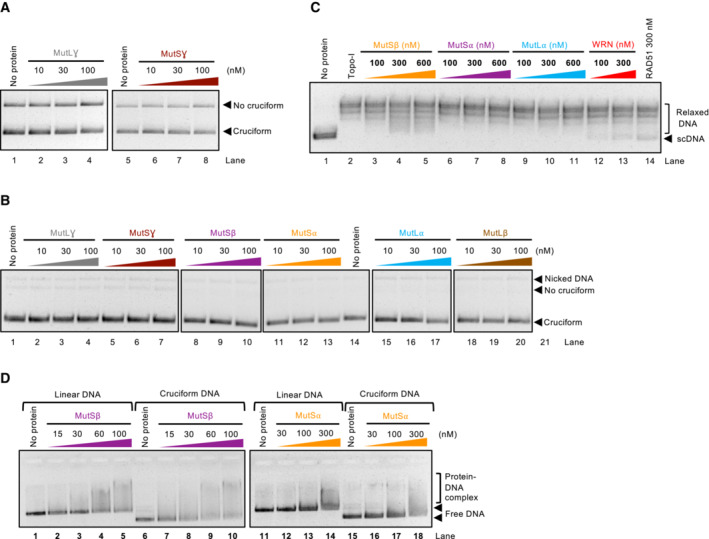
- Representative cruciform unfolding assay with MutLγ and MutSγ, using the random‐IR cruciform as a substrate.
- Representative cruciform unfolding assay with the MutL and MutS homolog heterodimers, carried out in the absence of EcoRI. The reactions indicate that the proteins do not cut DNA under the assay conditions.
- Representative Topoisomerase‐I‐coupled supercoiling assay with MutSβ, MutS⍺, MutL⍺, WRN, and RAD51. The reactions were analyzed by standard electrophoresis on a 1% native agarose gel. The gel was stained with GelRed after electrophoresis.
- Representative electrophoretic mobility shift assay with MutSβ and MutSα, using either circular pUC19 with the random inverted repeat cruciform structure, or linear pUC19 as a substrate. 0.8% unstained native agarose gel was used to separate the protein and DNA species.
Cruciform unfolding by the MMR complexes was independent of ATP (Fig 6F) and was in contrast to WRN not stimulated by RPA (Fig 6G), indicating that the reaction intermediates do not likely involve extended ssDNA. Accordingly, MutSβ or MutLα variants impaired in ATP binding and hydrolysis retained their apparent ability to unfold cruciform structures (Fig 7A–F), showing that the mechanism of cruciform unfolding by the MMR complexes is fundamentally different from the ATPase motor‐driven reaction catalyzed by WRN. Furthermore, mutation K255A in the MSH3 subunit of MutSβ disrupts the capacity of MutSβ to recognize and bind mismatched/looped DNA (Dufner et al, 2000), but does not affect the binding of DNA with the cruciform structure (Fig 7A–C). The MutSβ MSH3‐K255A mutant also retained its capacity to reduce the levels of cruciform DNA (Fig 7D), showing that apparent cruciform unfolding and mismatch recognition are distinct and separable functions. Holliday junctions including cruciform DNA are highly dynamic structures (McKinney et al, 2005). We propose that MutSα and MutSβ might stabilize DNA in its double‐stranded form, resulting from spontaneous and dynamic structural transitions of the cruciform DNA, rather than melting the cruciforms per se (see Discussion). Our data support a model in which MutSα, MutSβ, and MutLα play a direct role, separable from their functions in MMR, in the reduction of cruciform DNA structures, which may explain the partial rescue of WRN dependency in MSI cells upon restoration of MMR.
Figure 7. The activities of WRN and MMR proteins synergize in the unfolding of a TA cruciform.
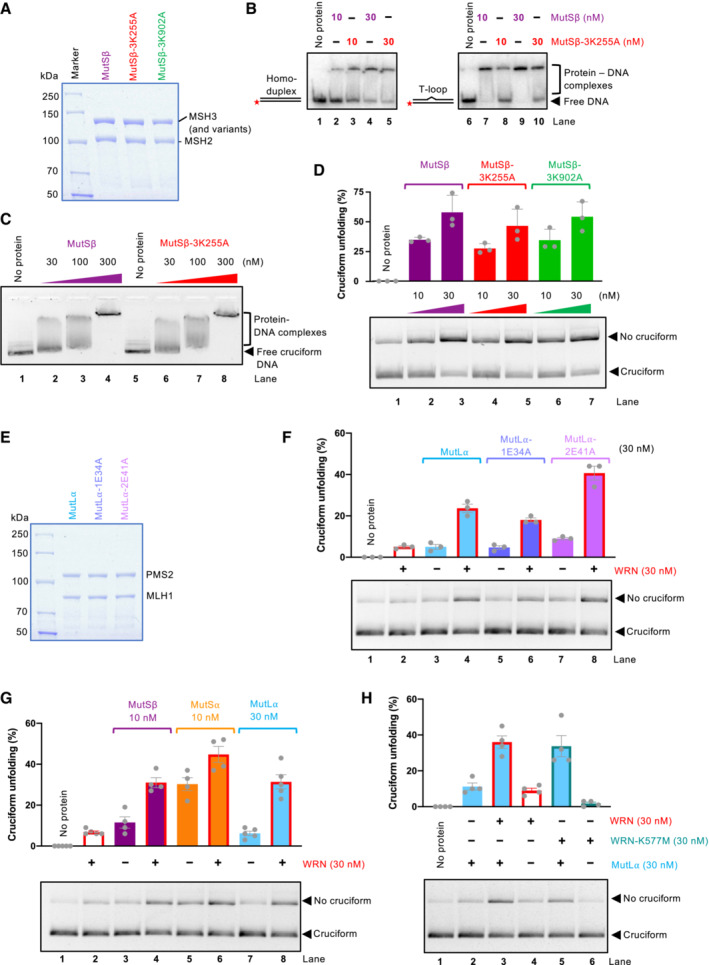
- Representative polyacrylamide gel showing recombinant MutSβ, MutSβ‐3K255A (MSH3 with mutation K255A, deficient in mismatch recognition), and MutSβ‐3K902A (MSH3 with mutation K902A, ATPase‐deficient). The gel was stained with Coomassie Brilliant blue.
- Electrophoretic mobility shift assay with MutSβ, MutSβ‐3K255A, and MutSβ‐3K902A, using either dsDNA (50 bp) or dsDNA bearing one extrahelical T, as a substrate. 6% native acrylamide gel was used.
- Electrophoretic mobility shift assays with MutSβ and MutSβ‐3K255A, using circular pUC19 with the AT repeat cruciform structure, as a substrate. 0.8% native agarose gel was used.
- Cruciform unfolding assays with MutSβ, MutSβ‐3K255A, and MutSβ‐3K902A. Bottom, representative experiments; top, quantitation, averages shown; n = 3 technical replicates; error bars, SEM.
- Representative polyacrylamide gel showing recombinant MutLα, MutL⍺‐1E34A (MLH1 with mutation E34A, ATPase‐deficient), and MutL⍺‐2E41A (PMS2 with mutation E41A, ATPase‐deficient). The gel was stained with Coomassie Brilliant blue.
- Unfolding of TA cruciform by WRN, MutLα, and its ATPase‐deficient variants. Bottom, representative experiments; top, quantitation; averages shown; n = 3 technical replicates; error bars, SEM.
- Unfolding of TA cruciform by WRN and MMR proteins. Bottom, representative experiments; top, quantitation; averages shown; n = 4 technical replicates; error bars, SEM.
- Unfolding of TA cruciform by MutLα and wild‐type WRN or helicase dead WRN‐K577M. Bottom, representative experiments; top, quantitation, averages shown; n = 4 technical replicates; error bars, SEM.
The activities of WRN and MMR proteins synergize in the unfolding of TA cruciforms
Previously, WRN was found to physically interact with MutSα, MutSβ, and MutLα (Saydam et al, 2007). MutSα and MutSβ could stimulate unwinding of mismatched DNA by WRN (Saydam et al, 2007). Our observations that the MMR complexes and WRN proteins could reduce cruciform DNA structures prompted us to investigate whether these factors could stimulate each other. To this point, we employed the TA‐based cruciform, a more physiological substrate than the random sequence inverted repeat‐based structure used in the majority of our previous experiments. As noted above, WRN had only a minimal activity on the TA cruciform (30 nM WRN used, ~ 8% unfolding) (Fig 7G). Unexpectedly, the MMR complexes MutSα (MSH2‐MSH6) and MutSβ (MSH2‐MSH3) had a comparably higher apparent activity than WRN (~ 30 and 11%, respectively, with 10 nM protein concentration). When the MutSα and MutSβ proteins were combined with WRN, even higher unfolding was observed, ~ 45 and 31% with MutSα and MutSβ, respectively, which is somewhat higher than what would be expected from a simple additive effect. The greatest stimulation was observed when WRN was combined with MutLα (30 nM MutLα used). Individually, the proteins exhibited 8 and 14% unfolding, respectively, but when combined, 35% of TA cruciform was rendered double stranded and hence susceptible to EcoRI cleavage, showing a synergistic effect (Fig 7G).
To investigate further the mechanism underlying the synergy between MutLα and WRN in cruciform DNA unfolding, we used either wild‐type WRN or the ATPase‐dead WRN‐K577M mutant. We observed on the TA repeats based cruciform that the DNA unfolding activity of WRN per se was dependent on the integrity of its ATPase site, as WRN‐K577M alone showed no activity (Fig 7H). Interestingly, however, WRN‐K577M could still promote the reaction when used in conjunction with MutLα, underlying a further structural role of WRN in cruciform unfolding, when acting together with MutLα (Fig 7H). These experiments collectively demonstrate that the MMR complexes MutSα, MutSβ, and MutLα can reduce the levels of cruciform DNA structures, and may further cooperate with WRN to do so. By employing different mechanisms to unfold these structures, the combined activities of the ensemble are more than the sum of the individual activities. WRN and MMR proteins acting together in healthy cells may thus reduce the formation of cruciforms in genomic DNA, diminishing the threats to genome integrity.
Discussion
WRN was identified as a vulnerability in MSI cancers (Behan et al, 2019; Chan et al, 2019; Kategaya et al, 2019; Lieb et al, 2019). Upon WRN depletion in a majority of MSI cells, DNA undergoes fragmentation that maps to sites of extended TA repeats. According to the current model (van Wietmarschen et al, 2020, 2021), TA repeats undergo expansion in MMR‐deficient MSI cells, through an unknown mechanism. Expanded TA repeats then represent a genomic scar, which predisposes cells for the dependency on WRN. The extended TA repeats may fold into cruciform structures, which stall DNA replication, leading to WRN phosphorylation and recruitment at stalled replication forks. WRN was then proposed to unwind these cruciform structures, preventing aberrant DNA fragmentation (Chan et al, 2019). However, whether WRN acts directly, or as part of a larger complex, as well as the possible activities of MMR proteins without or with WRN on cruciform structures remained undefined.
Here we used biochemical assays to monitor the formation and the stability of cruciform DNA, and its processing by recombinant proteins recently implicated in their metabolism. We first observed that a plasmid containing TA repeats with a length of 46 nucleotides is much more likely to contain the cruciform structure than DNA with inverted repeats of a random synthetic sequence of the same length. The presence of the cruciform depends on the efficacy of DNA extrusion, which is thought to be driven by negative supercoiling, which in turn facilitates DNA melting, as well as on the stability of the resulting structure (Panayotatos & Wells, 1981; Mizuuchi et al, 1982; Lilley, 1983). TA‐rich sequences exhibit low melting temperature, thus promoting DNA melting and cruciform formation, which appears to be the rate‐limiting step in their formation. In genomic DNA that is only weakly negatively supercoiled, sequences with TA repeats may thus be preferentially prone to form the cruciform structures, potentially explaining why these repetitive sequences are uniquely prone to fragmentation (van Wietmarschen et al, 2020).
We next found that WRN was directly and effectively capable to unfold cruciform structures, depending on its helicase activity and ATP hydrolysis. Sgs1, the most active helicase from the RecQ family (Cejka & Kowalczykowski, 2010), was unexpectedly incapable to unwind cruciforms. Therefore, a vigorous helicase activity does not make the RecQ family helicase efficient in cruciform unfolding, demonstrating a certain level of specificity. However, BLM, a related human RecQ family helicase also unwound cruciforms in vitro, but does not promote survival of MSI cells (Chan et al, 2019). The discrepancy can be explained by the need for WRN recruitment to stalled replication forks. In cells, WRN recruitment depends on the phosphorylation of its C‐terminus by the ATR kinase (Ammazzalorso et al, 2010). Phosphorylation‐deficient WRN mutant fails to localize to DNA damage at sites of stalled replication, and consequently does not support the survival of MSI cells (van Wietmarschen et al, 2020). In the minimal reconstituted reactions, however, the need for WRN recruitment is bypassed. WRN variants that cannot be phosphorylated or completely lack the WRN C‐terminus, as well as the BLM helicase are able to support cruciform unfolding in vitro. However, in cells, these proteins do not localize to the sites of stalled replication and cannot function in cruciform unfolding in vivo.
The dependency on WRN was only observed in MMR‐deficient MSI cells with expanded TA repeats (van Wietmarschen et al, 2020). However, restoration of MMR in MSI cells was reported to partially rescue cell survival upon WRN depletion (Chan et al, 2019; Lieb et al, 2019). Therefore, MMR deficiency could have additional roles in the context of the synthetic lethality, beyond predisposing cells to WRN dependency by allowing the expansion of TA repeats. The partial rescue upon MMR restoration hints at an additional function of MMR proteins to prevent TA repeat‐dependent toxicity. To this point, we found that MMR complexes including MutSα, MutSβ, and MutLα can also apparently unfold cruciform structures, with MutSα and MutSβ being the most and MutLα being the least efficient, which may represent the additional function of MMR proteins in governing the toxicity of the cruciform structures (Fig 8A). Whereas cruciform unfolding by WRN was dependent on ATP and was stimulated by RPA, the activity of the MMR complexes was unaffected by either RPA or ATP, showing that the mechanisms of processing are fundamentally different. We show that the capacity of the MutSα, MutSβ, and MutLα complexes to reduce the levels of cruciform DNA is distinct and separable from their roles in MMR. Holliday junctions including cruciform DNA are highly dynamic structures that exist in various conformations (McKinney et al, 2005). In search for a mismatch, the MutSα and MutSβ complexes adopt a “praying hands” structure, which forms a thermal diffusion‐driven sliding clamp that scans dsDNA, independently of ATP (Kim et al, 2018). We propose that by tightly encircling dsDNA, the complexes may exploit the dynamic structure of the cruciform to stabilize DNA in its double‐stranded form. Therefore, the MMR complexes may not directly melt the cruciform structure, but rather stabilize the dsDNA form resulting from spontaneous transitions of the cruciform DNA.
Figure 8. Models for mismatch repair and WRN activities on cruciform structures.
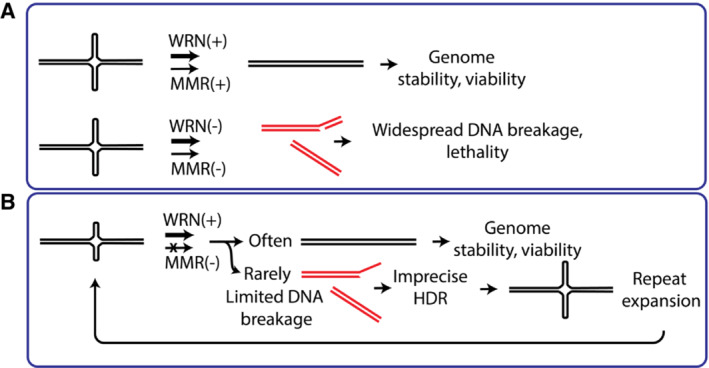
- Top: in wild‐type cells (MMR‐ and WRN‐proficient), cruciform DNA is efficiently unfolded, and does not cause any problems. WRN and MMR complex may act together or separately on cruciform DNA. Bottom: in MMR‐deficient (MSI) cells, inactivation of WRN triggers very frequent DNA breaks at cruciform sites, leading to cellular lethality.
- In MMR‐deficient (MSI+) but WRN proficient cells, large parts of DNA are generally stable. Only very few cruciform structures are formed in the absence of MMR and subject to breakage. Imprecise homology‐directed repair may lead to repeat expansion. DNA breaks are rare and do not result in lethality. Over many generations, cruciform structures extend at random genomic locations.
Interestingly, whereas WRN was more efficient than the MMR complexes on cruciforms arising from the inverted repeat with a random sequence, the MMR proteins were instead more efficient than WRN on TA‐based cruciforms. We reason that our observation may not accurately reflect the absolute respective activities of WRN and MMR proteins: it is possible that MMR complexes remain bound to dsDNA upon cruciform unfolding, which may prevent the re‐formation of the cruciform. TA sequences are more prone to form cruciforms even without applying an extrusion procedure (i.e., under our standard reaction conditions), which may explain the higher apparent activity of the MMR proteins on TA sequences. Nevertheless, when we combined WRN and the MMR complexes (in particular MutLα), we observed higher than additive effects in cruciform unfolding, suggesting that the factors may stimulate each other activities. In further support of an additional structural function, we observed that helicase‐dead WRN, which exhibited no activity per se, could still stimulate the cruciform unfolding by MutLα. The observed interplay will likely be dependent on direct physical interactions: WRN was reported to bind all the respective MMR complexes MutSα, MutSβ, and MutLα, and MutSα and MutSβ were shown to stimulate WRN helicase activities on mismatched DNA (Saydam et al, 2007).
How can TA repeats expand in MMR‐deficient cells? Canonical MSI phenotype typically leads to the shortening of mononucleotide repeat units, with the changes accumulating gradually over many cellular divisions (Hoang et al, 1997; Pecina‐Slaus et al, 2020; Olave & Graham, 2021; Randrian et al, 2021). Analysis of a single repeat, BAT‐26, can identify MSI phenotype in the majority of cell lines (Hoang et al, 1997). The alterations in the length of TA repeats that underlie WRN dependency are different (van Wietmarschen et al, 2020). First, TA repeats at different genomic locations expand in various cell lines, so the changes appear to be much more random with respect to the affected genomic locations. Furthermore, major increase in the number of repeat units was observed, which together suggests that TA repeat expansion in MMR‐deficient cells mechanistically differs from mononucleotide repeat shortening linked to DNA polymerase slippage. Our observations suggest that a lack of MMR complexes may partially stabilize cruciform structures. DNA breaks would then occur with higher frequency at these sites (possibility via the same cleavage mechanism mediated by the SLX4‐associated MUS81‐EME1 and SLX1 endonucleases), and repaired unfaithfully as a result of homologous recombination upon unequal pairing with the DNA template, resulting in copy number increase upon DNA synthesis (Fig 8B). These events would have to be rare, explaining why (i) TA repeat expansion takes many cell generations over months or years and (ii) the low number of breaks would not result in cellular toxicity. The newly identified and unexpected activity of MMR proteins to reduce the levels of cruciform DNA could thus explain both the expansion of TA repeats in MMR deficient but WRN proficient cells, as well as the partial rescue of survival in TA‐expanded cells upon restoration of MMR expression and WRN inhibition. However, more insights are needed to fully understand the metabolism of expanded TA repeats, which will be essential to predict mechanisms of potential therapy resistance.
Material and Methods
Cloning, expression, and purification of recombinant proteins
All expression vectors are available on request. Wild‐type human WRN, exonuclease‐dead WRN‐E84A, helicase‐dead WRN‐K577A, BLM, and S. cerevisiae Sgs1 were expressed in Spodoptera frugiperda 9 (Sf9) insect cells using a pFastBac1‐derived vector. The proteins were purified by affinity chromatography using the N‐terminal maltose‐binding protein (MBP)‐tag and the C‐terminal his‐tag (Cejka & Kowalczykowski, 2010; Cannavo & Cejka, 2014; Anand et al, 2016; Pinto et al, 2016; Cannavo et al, 2018). The WRN variants constructed in course of this study included phosphorylation‐deficient WRN‐3A (S991A T1152A S1256A) and WRN truncation mutants WRN 500–946 and WRN 500–1,104. The construct for the expression of WRN‐3A was obtained by cloning a synthetic WRN fragment bearing the S991A T1152A S1256A mutations between the unique NcoI and SphI restriction sites (DNA obtained from GenScript Biotech), into the pFB‐MBP‐WRN‐10Xhis, digested with NcoI and SphI. The constructs for the expression of truncated WRN variants (WRN 500–946 and WRN 500–1,104) were obtained by PCR amplification of the relevant WRN fragment, using primers: fw_WRN_ATPase domain_NheI and rev_WRN ATPase‐Helicase core_XhoI, or rev_WRN_RQCdomain_XhoI, respectively (Table EV1). The resulting PCR product was digested with NheI and XhoI, purified on QIAquick spin columns (Qiagen), and cloned into the same restriction sites in pFB‐MBP‐WRN‐10his plasmid: the sequence of full length WRN was removed in this step. The WRN‐3A mutant and the WRN truncations were expressed and purified as the wild‐type protein. All the purification steps were carried out at 4°C. The cell pellets were resuspended in three volumes of lysis buffer containing 50 mM Tris–HCl pH 7.5, 5 mM β‐mercaptoethanol (β‐ME), 1 mM phenylmethylsulfonylfluoride (PMSF), 1:400 (volume/volume) protease inhibitor cocktail (Sigma, P8340), 30 μg/ml leupeptin (Merck), 1 mM ethylenediaminetetraacetic acid (EDTA), and incubated for 20 min with stirring. Next, 1/2 volume of 50% glycerol was added (final concentration 16.7%), followed by 6.5% volume of 5 M NaCl (final concentration 325 mM), and the suspension was further incubated for 30 min with stirring. To obtain the soluble extract, the suspension was centrifuged at 48,000 g for 30 min. The soluble extract was transferred to a new tube containing pre‐equilibrated Amylose resin (New England Biolabs, 4 ml of resin per 1 l Sf9 cells) and incubated for 1 h with continuous agitation. The Amylose resin was collected by centrifugation at 2,000 g for 2 min. The resin was washed extensively, batchwise with ten resin volumes of Amylose wash buffer (50 mM Tris–HCl pH 7.5, 5 mM β‐ME, 300 mM NaCl, 1 mM PMSF, 10% glycerol), and subsequently on a disposable column with Amylose wash buffer containing 1 M NaCl. The last wash was carried out with Amylose wash buffer containing 300 mM NaCl. Protein was eluted with Amylose wash buffer containing 300 mM NaCl and 10 mM maltose (Sigma), and the total protein concentration was estimated using the Bradford assay (Bio‐Rad). To cleave off the MBP tag, the eluted sample was incubated with 1/5 (weight/weight) of PreScission protease (Anand et al, 2018) for 1 h. Next, the cleaved amylose eluate was supplemented with imidazole to a final concentration of 10 mM and incubated with pre‐equilibrated nickel‐nitriloacetic acid (NiNTA) resin (Qiagen) for 1 h with continuous agitation. The NiNTA resin was transferred to a disposable column and washed extensively with NiNTA wash buffer containing 1 M NaCl (50 mM Tris–HCl pH 7.5, 5 mM β‐ME, 1 M NaCl, 1 mM PMSF, 10% glycerol, 30 mM imidazole), followed by NiNTA wash buffer containing 150 mM NaCl (50 mM Tris–HCl pH 7.5, 5 mM β‐ME, 150 mM NaCl, 1 mM PMSF, 10% glycerol, 30 mM imidazole). Protein was eluted with NiNTA elution buffer (50 mM Tris–HCl pH 7.5, 5 mM β‐ME, 100 mM NaCl, 1 mM PMSF, 10% glycerol, 300 mM imidazole). Eluates containing protein, according to the Bradford assay, were pooled, aliquoted, frozen in liquid nitrogen, and stored at −80°C.
Human and yeast RPA were expressed in E. coli and purified using ÄKTA pure (GE Healthcare) with HiTrap Blue HP, HiTrap desalting, and HiTrap Q chromatography columns (all GE Healthcare; Anand et al, 2018). The human MSH2‐MSH3, MSH2‐MSH6, MLH1‐PMS1, and MLH1‐PMS2 heterodimers, as well as the corresponding mutants, were expressed in Sf9 insect cells. The following expression vectors were used: pFB‐FLAG‐PP‐hMSH2, pFB‐His‐hMSH3, pFB‐His‐hMSH6, pFB‐FLAG‐hMLH1co, pFB‐FLAG‐hMLH1co‐E34A, pFB‐hPMS1, pFB‐His‐hPMS2, pFB‐HisPMS2‐E41A (Dufner et al, 2000; Raschle et al, 2002; Cannavo et al, 2020). The sequence of MLH1 was codon‐optimized for expression in insect cells (Cannavo et al, 2020). To generate the MSH3‐K255A and K902A mutant constructs, the relevant residues were mutated by QuikChange II site‐directed mutagenesis kit (Agilent Technologies). To prepare pFB‐His‐hMSH3‐K255A, the pFB‐His‐hMSH3 vector was mutated with primers MSH3‐K255A‐FO and MSH3‐K255A‐RE. To prepare pFB‐His‐hMSH3‐K902A, the pFB‐His‐hMSH3 vector was mutated with primers MSH3‐K902A‐FO and MSH3‐K902A‐RE.
The insect cells were co‐infected by a combination of viruses to obtain the desired heterodimers, which were subsequently purified by affinity chromatography using the N‐terminal FLAG‐tag on MSH2 or MLH1, and the N‐terminal 6Xhis‐tag on MSH3, MSH6, or PMS2. All the purification steps were carried out at 4°C. The cell pellets were resuspended in three volumes of lysis buffer (50 mM Tris–HCl pH 7.5, 0.5 mM β‐ME, 1 mM PMSF, 1:400 [volume/volume] protease inhibitor cocktail [Sigma, P8340], 60 μg/ml leupeptin [Merck]) and incubated for 20 min with stirring. Next, 1/2 volume of 50% glycerol was added (final concentration 16.7%), followed by 6.5% volume of 5 M NaCl (final concentration 325 mM), and the suspension was further incubated for 30 min with stirring. To obtain the soluble extract, the suspension was centrifuged at 48,000 g for 30 min. The soluble extract was transferred to a new tube containing pre‐equilibrated NiNTA resin (Qiagen, 4 ml of resin per 1 l Sf9 cells) and incubated for 1 h with continuous agitation. The resin was collected by centrifugation at 2,000 g for 2 min, followed by extensive washing, at first, batchwise with 10 resin volumes of NiNTA wash buffer containing 300 mM NaCl (30 mM Tris–HCl pH 7.5, 0.5 mM β‐ME, 300 mM NaCl, 1 mM PMSF, 10% glycerol, supplemented with 20 mM imidazole), and subsequently on a disposable column. Elution was carried out with NiNTA elution buffer (50 mM Tris–HCl pH 7.5, 0.5 mM β‐ME, 150 mM NaCl, 0.5 mM PMSF, 10% glycerol, 250 mM imidazole), and the total protein concentration was estimated using the Bradford assay (Bio‐Rad). The eluate was diluted five times in NiNTA elution buffer lacking imidazole (30 mM Tris–HCl pH 7.5, 0.3 mM β‐ME, 150 mM NaCl, 0.5 mM PMSF, 10% glycerol) and incubated for 1 h with pre‐equilibrated αFLAG‐conjugated M2 agarose (Sigma), on a disposable column. Next, the resin was washed with NiNTA elution buffer without imidazole. Protein was eluted with FLAG elution buffer (30 mM Tris–HCl pH 7.5, 0.3 mM β‐ME, 150 mM NaCl, 0.5 mM PMSF, 10% glycerol, 200 μg/ml 3X FLAG peptide [GlpBio]). Eluates containing protein, according to the Bradford assay, were pooled, aliquoted, frozen in liquid nitrogen, and stored at −80°C. MLH1‐PMS1 was purified in a single step, using the N‐terminal FLAG‐tag on MLH1. The cell pellets were resuspended in three volumes of lysis buffer (50 mM Tris–HCl pH 7.5, 0.5 mM β‐ME, 1 mM EDTA, 1 mM PMSF, 1:400 [volume/volume] protease inhibitor cocktail [Sigma, P8340], 30 μg/ml leupeptin [Merck]) and incubated for 20 min with stirring. Next, 1/2 volume of 50% glycerol was added (final concentration 16.7%), followed by 6.5% volume of 5 M NaCl (final concentration 325 mM), and the suspension was further incubated for 30 min with stirring. To obtain the soluble extract, the suspension was centrifuged at 48,000 g for 30 min. The soluble extract was transferred to a new tube containing pre‐equilibrated αFLAG‐conjugated M2 agarose (Sigma, 1 ml of resin per 1 l Sf9 cells) and incubated for 1 h with continuous agitation. The resin was collected by centrifugation at 2,000 g for 2 min, followed by extensive washing, at first, batchwise with 10 resin volumes of wash buffer containing 300 mM NaCl (50 mM Tris–HCl pH 7.5, 0.5 mM β‐ME, 300 mM NaCl, 1 mM PMSF, 10% glycerol, 0.1% NP40 [Sigma]), and subsequently on a disposable column. The last wash was performed with wash buffer containing 100 mM NaCl. Elution was carried out with FLAG elution buffer (wash buffer containing 100 mM NaCl, supplemented with 200 μg/ml FLAG peptide [GlpBio]). Eluates containing protein, according to the Bradford assay, were pooled, aliquoted, frozen in liquid nitrogen, and stored at −80°C. The human MSH4‐MSH5 complex and the human MLH1‐MLH3 complex were expressed in Sf9 insect cells, and the purification protocol has been described in detail (Cannavo et al, 2020). Recombinant human SLX1‐SLX4CCD was provided by Pierre‐Henry L. Gaillard (Centre de Recherche en Cancérologie de Marseille).
DNA substrate preparation
The cruciform‐containing dsDNA are derivatives of the pUC19 vector. pUC19 containing synthetic random inverted repeats (pUC19‐Random IR) was prepared by cloning the random inverted repeats (SacI‐Random IR‐XbaI, Table EV2) synthesized between SacI and XbaI, into pUC19 digested with the same restriction endonucleases. The original sequence of EcoRI was removed during this step, and was introduced again de novo flanked by the repeats. The random inverted repeat sequence corresponds to that used previously in vector pIRbke8mut (Rass et al, 2010). pUC19 containing TA repeats (pUC19‐TA IR) was created by cloning the TA repeats (KpnI‐TA IR‐BamHI, Table EV2) synthesized between KpnI and BamHI, into pUC19 digested with the same restriction endonucleases. The original sequence of EcoRI was removed during this step, and was introduced again de novo flanked by the repeats.
Cruciform extrusion (160 μl) was performed by incubating the plasmids in a reaction buffer containing 50 mM Tris–HCl pH 7.5, 0.5 mM magnesium chloride, 20 μg negatively supercoiled plasmid DNA, at 37°C for 90 min. The efficiency of cruciform extrusion was tested by digestion with EcoRI (extruded DNA was not cleavable by EcoRI). For each reaction (15 μl) 100 ng of DNA were digested with 5 units of EcoRI (New England Biolabs), diluted in CutSmart buffer (New England Biolabs), at 37°C for 60 min, and the products were separated by electrophoresis on a 1% native agarose gel, stained with GelRed (1:20,000, Biotium). The sequence of all oligonucleotides used for radioactively labeled DNA substrate preparation is listed in Table EV2. Oligonucleotide‐based DNA substrates were 32P‐labeled at the 3′ terminus using [α‐32P]dCTP (Perkin Elmer) and terminal transferase (New England Biolabs) according to the manufacturer's instructions and our previously‐described procedures (Pinto et al, 2018). Unincorporated nucleotides were removed using Micro Bio‐Spin P‐30 Tris chromatography columns (Bio‐Rad). The substrates were prepared by heating the oligonucleotides at 95°C and subsequent slow gradual cooling to reach room temperature, in Annealing buffer (10 mM Tris–HCL pH 8.0, 50 mM NaCl, 10 mM magnesium chloride). To prepare the HJ substrate, the oligonucleotides PC1253, PC1254, PC1255, and PC1256 were used. To prepare the dsDNA, the oligonucleotides X12‐3, X12‐4C, and X12‐4_extraT were used. The randomly labeled 2.2‐kbp‐long substrate was prepared amplifying the human NBS1 gene by PCR from pFB‐MBP‐NBS1‐his plasmid using Phusion high‐fidelity DNA polymerase (New England Biolabs) and the NBS1_F and NBS1_R primers (Anand et al, 2018). 66 nM [α‐32P]dCTP was added to the PCR reaction together with the standard dNTPs concentration (200 μM each). The PCR reaction product was purified using the QIAquick PCR purification kit (Qiagen) and Chroma Spin TE‐200 columns (Clontech). Upon purification, the DNA was quantitated by comparing the radioactive DNA amplificate with known amounts of a corresponding cold PCR product on an agarose gel, stained with GelRed (Biotium).
Cruciform unfolding assay
Cruciform unfolding assays (15 μl) were carried out in a reaction buffer containing 25 mM Tris–HCl pH 7.5, 2 mM ATP, 1 mM dithiothreitol (DTT), 1 mM phosphoenolpyruvate (Sigma), 0.1 mg/ml BSA (New England Biolabs), 80 U/ml pyruvate kinase (Sigma), 2 mM magnesium acetate, and 100 ng of extruded DNA pro reaction. Where indicated, ATP‐γ‐S (Cayman) was used instead of ATP. Unless specified otherwise, the final concentration of NaCl was adjusted in the reactions to 25 mM, accounting for salt brought into the reactions with protein storage or protein dilution buffer. The reactions were assembled and supplemented with the relevant proteins on ice and incubated at 37°C for 20 min. Reactions were subsequently taken to room temperature and 5 units/sample of EcoRI in CutSmart buffer (New England Biolabs) were added, unless indicated otherwise. Reactions were then incubated at 37°C for 30 min. The reactions were loaded on the gel upon the addition of 3.5 μl of DNA loading dye (10 mM Tris–HCl pH 8, 30% glycerol, bromophenol blue). The products were separated by electrophoresis in 1% native agarose gels in the presence of GelRed (1:20,000, Biotium). Images of gels were acquired at InGenius3 (GeneSys). The results were quantified using ImageJ and expressed as % of cruciform unfolding; any linear DNA (no cruciform) present in control (no protein) reactions was referred to as 0% cruciform unfolding, and subtracted as a background from all other samples. Graphs were plotted using Prism software (Prism 9, GraphPad). Reactions with T7 endonuclease were carried out similarly as described above. The samples were incubated at 37°C for 10 min upon WRN addition, and subsequently T7‐Endonuclease I (New England Biolabs) together with the respective restriction endonucleases were added to the reactions, and further incubated at 37°C for 60 min. Similarly, the reactions with SLX1‐SLX4CCD (10 μl, in cruciform unfolding assay buffer) were incubated at 37°C for 10 min upon WRN addition, the nuclease was added and the reaction buffer was adjusted to a final volume of 15 μl, and a final concentration of 42 mM Tris‐acetate pH 7.5, 0.5 mM manganese chloride, 1.2 mM DTT. Reactions were incubated at 37°C for 45 min. Subsequently, 5 units of ScaI (New England Biolabs), diluted in CutSmart buffer (New England Biolabs), were added and incubated for at 37°C for 30 min. The reaction was terminated by adding 1 μl Proteinase K (14–22 mg/ml, Roche) and 5 μl 0.2% stop solution (150 mM EDTA, 0.2% SDS, 30% glycerol, bromophenol blue) and incubated at 37°C for 10 min, and the samples were analyzed as above.
Helicase assays
DNA unwinding assays (15 μl) were performed in a reaction buffer consisting of 25 mM Tris‐acetate pH 7.5, 2 mM magnesium acetate, 2 mM ATP, 1 mM DTT, 0.1 mg/ml BSA, 1 mM phosphoenolpyruvate, 80 U/ml pyruvate kinase, and 25 mM NaCl, with 0.5 nM of oligonucleotide‐based DNA substrate (in molecules). Either human or yeast RPA was included as indicated to saturate ssDNA. Recombinant proteins were added on ice, as indicated. Unless specified otherwise, reactions were incubated at 30°C for 30 min and stopped by adding 5 μl of 2% stop solution (150 mM EDTA, 2% SDS, 30% glycerol, bromophenol blue) and 1 μl of proteinase K (14–22 mg/ml, Roche) and incubated at 37°C for 10 min. To avoid re‐annealing of the substrate, the 2% stop solution was supplemented with a 20‐fold excess of the unlabeled oligonucleotide with the same sequence as the 32P labeled one. The products were separated by 10% polyacrylamide gel electrophoresis, dried on 17 CHR chromatography paper (Whatman), exposed to storage phosphor screens (GE Healthcare), and scanned by a Typhoon 9500 phosphorimager (GE Healthcare). Helicase assays with the 2.2 kb long substrate were analyzed by running the samples by electrophoresis in 1% agarose. Gels were dried on DE81 chromatography paper (Whatman), exposed to storage phosphor screens (GE Healthcare), and scanned using the Typhoon Phosphor Imager FLA 9500 (GE Healthcare). The bands were quantified using ImageJ and data were plotted using Prism software (Prism 9, GraphPad).
Electrophoretic mobility shift assays
The DNA‐binding reactions with plasmid DNA (15 μl) were carried out in a binding buffer containing 25 mM Tris‐acetate pH 7.5, 1 mM DTT, 0.1 mg/ml BSA, 3 mM EDTA, and 25 mM NaCl, with 100 ng of the plasmid DNA substrate. The indicated concentrations of recombinant proteins were added as the last components. The reactions were assembled and incubated on ice for 15 min, followed by the addition of 5 μl EMSA loading dye (50% glycerol, 0.01% bromophenol blue). The products were separated on 0.8% native agarose gels. Post electrophoresis, gels were stained in water containing GelRed (1:20,000) with gentle agitation for 45 min and imaged at InGenius3 (GeneSys). The oligonucleotide‐based DNA‐binding reactions (15 μl) were carried out in the binding buffer described above, with 0.5 nM (in molecules) radioactively labeled substrate. The reactions were supplemented with a competitor dsDNA (1.5 ng/μl of pUC19). The reactions were assembled in ice, incubated at 37°C for 30 min, followed by the addition of 5 μl EMSA loading dye. The products were separated on a native 6% polyacrylamide gel. Gels were dried on 17CHR chromatography paper (Whatman), exposed to storage phosphor screens (GE Healthcare), and scanned using the Typhoon Phosphor Imager FLA 9500 (GE Healthcare).
Topoisomerase I‐coupled supercoiling assay
Topoisomerase I‐coupled DNA supercoiling assays (15 μl) were performed in a reaction buffer consisting of 25 mM Tris–HCl pH 7.5, 2 mM magnesium chloride, 2.5 mM ATP, 1 mM DTT, 0.1 mg/ml BSA, and 50 mM NaCl, with 100 ng/reaction of covalently closed pUC19‐Random IR as a substrate. The reactions were assembled in ice, and topoisomerase I (16.5 nM) was added to the samples to induce the relaxation of the substrate by incubation at 37°C for 10 min. Recombinant proteins were subsequently added at room temperature, as indicated. Reactions were incubated at 37°C for 30 min and terminated by the addition of 555 mM NaCl, followed by incubation at 37°C for 10 min with 5 μl of 2% stop solution (150 mM EDTA, 2% SDS, 30% glycerol, bromophenol blue) and 1 μl of proteinase K (14–22 mg/ml, Roche). The samples were analyzed by electrophoresis on 1% native agarose gels, unstained. The gels were post‐stained with water containing GelRed (1:20,000, Biotium) with gentle agitation for 45 min. Images of gels were acquired at InGenius3 (GeneSys). The results were quantified using ImageJ, and graphs were plotted using Prism software (Prism 9, GraphPad).
Author contributions
Petr Cejka: Conceptualization; supervision; funding acquisition; methodology; writing – original draft; project administration; writing – review and editing. Valentina Mengoli: Conceptualization; data curation; formal analysis; investigation; methodology; writing – original draft; writing – review and editing. Ilaria Ceppi: Investigation; writing – review and editing. Aurore Sanchez: Investigation; writing – review and editing. Elda Cannavo: Investigation; writing – review and editing. Swagata Halder: Investigation; writing – review and editing. Sarah Scaglione: Resources; writing – review and editing. Pierre‐Henri Gaillard: Resources; supervision; writing – original draft; writing – review and editing. Peter J McHugh: Resources; writing – original draft; writing – review and editing. Nathalie Riesen: Resources; writing – original draft; writing – review and editing. Piergiorgio Pettazzoni: Resources; funding acquisition; writing – original draft; writing – review and editing.
Disclosure and competing interests statement
Piergiorgio Pettazzoni and Nathalie Riesen are employees of Hoffmann‐La Roche LTD. All other authors declare no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
PDF+
Acknowledgements
Valentina Mengoli is a recipient of a Roche Postdoctoral Fellowship. The Cejka laboratory is further supported by grants from the Swiss National Science Foundation (310030_205199), European Research Council (681630 and 101018257), and Swiss Cancer League (KFS‐5397‐08‐2021). Related work in the laboratory of PHG was supported by Institut National du Cancer (INCA PLBIO 2019‐152). We thank members of the Cejka laboratory for comments on the manuscript. The plasmids pFB‐HisPMS2, pFB‐His‐hMSH6, pFB‐His‐hMSH3, and pFB‐HisPMS2‐E41A were a kind gift of Prof. Josef Jiricny.
The EMBO Journal (2023) 42: e111998
Data availability
This study includes no data deposited in external repositories.
References
- Ahn B, Harrigan JA, Indig FE, Wilson DM 3rd, Bohr VA (2004) Regulation of WRN helicase activity in human base excision repair. J Biol Chem 279: 53465–53474 [DOI] [PubMed] [Google Scholar]
- Aiello FA, Palma A, Malacaria E, Zheng L, Campbell JL, Shen B, Franchitto A, Pichierri P (2019) RAD51 and mitotic function of mus81 are essential for recovery from low‐dose of camptothecin in the absence of the WRN exonuclease. Nucleic Acids Res 47: 6796–6810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammazzalorso F, Pirzio LM, Bignami M, Franchitto A, Pichierri P (2010) ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J 29: 3156–3169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R, Ranjha L, Cannavo E, Cejka P (2016) Phosphorylated CtIP functions as a Co‐factor of the MRE11‐RAD50‐NBS1 endonuclease in DNA end resection. Mol Cell 64: 940–950 [DOI] [PubMed] [Google Scholar]
- Anand R, Pinto C, Cejka P (2018) Methods to study DNA end resection I: recombinant protein purification. Methods Enzymol 600: 25–66 [DOI] [PubMed] [Google Scholar]
- Behan FM, Iorio F, Picco G, Goncalves E, Beaver CM, Migliardi G, Santos R, Rao Y, Sassi F, Pinnelli M et al (2019) Prioritization of cancer therapeutic targets using CRISPR‐Cas9 screens. Nature 568: 511–516 [DOI] [PubMed] [Google Scholar]
- Boland CR, Goel A (2010) Microsatellite instability in colorectal cancer. Gastroenterology 138: e2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, Cejka P (2014) Sae2 promotes dsDNA endonuclease activity within Mre11‐Rad50‐Xrs2 to resect DNA breaks. Nature 514: 122–125 [DOI] [PubMed] [Google Scholar]
- Cannavo E, Johnson D, Andres SN, Kissling VM, Reinert JK, Garcia V, Erie DA, Hess D, Thoma NH, Enchev RI et al (2018) Regulatory control of DNA end resection by Sae2 phosphorylation. Nat Commun 9: 4016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, Sanchez A, Anand R, Ranjha L, Hugener J, Adam C, Acharya A, Weyland N, Aran‐Guiu X, Charbonnier JB et al (2020) Regulation of the MLH1‐MLH3 endonuclease in meiosis. Nature 586: 618–622 [DOI] [PubMed] [Google Scholar]
- Cejka P, Kowalczykowski SC (2010) The full‐length Saccharomyces cerevisiae Sgs1 protein is a vigorous DNA helicase that preferentially unwinds Holliday junctions. J Biol Chem 285: 8290–8301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EM, Shibue T, McFarland JM, Gaeta B, Ghandi M, Dumont N, Gonzalez A, McPartlan JS, Li T, Zhang Y et al (2019) WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 568: 551–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu WK, Hickson ID (2009) RecQ helicases: Multifunctional genome caretakers. Nat Rev Cancer 9: 644–654 [DOI] [PubMed] [Google Scholar]
- Datta A, Biswas K, Sommers JA, Thompson H, Awate S, Nicolae CM, Thakar T, Moldovan GL, Shoemaker RH, Sharan SK et al (2021) WRN helicase safeguards deprotected replication forks in BRCA2‐mutated cancer cells. Nat Commun 12: 6561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufner P, Marra G, Raschle M, Jiricny J (2000) Mismatch recognition and DNA‐dependent stimulation of the ATPase activity of hMutSalpha is abolished by a single mutation in the hMSH6 subunit. J Biol Chem 275: 36550–36555 [DOI] [PubMed] [Google Scholar]
- Duroc Y, Kumar R, Ranjha L, Adam C, Guerois R, Md Muntaz K, Marsolier‐Kergoat MC, Dingli F, Laureau R, Loew D et al (2017) Concerted action of the MutLbeta heterodimer and Mer3 helicase regulates the global extent of meiotic gene conversion. Elife 6: e21900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekairi S, Scaglione S, Chahwan C, Taylor ER, Tissier A, Coulon S, Dong MQ, Ruse C, Yates JR 3rd, Russell P et al (2009) Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 138: 78–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishel R (2021) Mismatch repair: choreographing accurate strand excision. Curr Biol 31: R293–R296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchitto A, Pirzio LM, Prosperi E, Sapora O, Bignami M, Pichierri P (2008) Replication fork stalling in WRN‐deficient cells is overcome by prompt activation of a MUS81‐dependent pathway. J Cell Biol 183: 241–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaccherini C, Gaillard PH (2021) Control of structure‐specific endonucleases during homologous recombination in eukaryotes. Curr Opin Genet Dev 71: 195–205 [DOI] [PubMed] [Google Scholar]
- Harrigan JA, Opresko PL, von Kobbe C, Kedar PS, Prasad R, Wilson SH, Bohr VA (2003) The Werner syndrome protein stimulates DNA polymerase beta strand displacement synthesis via its helicase activity. J Biol Chem 278: 22686–22695 [DOI] [PubMed] [Google Scholar]
- Hoang JM, Cottu PH, Thuille B, Salmon RJ, Thomas G, Hamelin R (1997) BAT‐26, an indicator of the replication error phenotype in colorectal cancers and cell lines. Cancer Res 57: 300–303 [PubMed] [Google Scholar]
- Iyer RR, Pluciennik A (2021) DNA mismatch repair and its role in Huntington's disease. J Huntingtons Dis 10: 75–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiricny J (2006) The multifaceted mismatch‐repair system. Nat Rev Mol Cell Biol 7: 335–346 [DOI] [PubMed] [Google Scholar]
- Kategaya L, Perumal SK, Hager JH, Belmont LD (2019) Werner syndrome helicase is required for the survival of cancer cells with microsatellite instability. iScience 13: 488–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal S, Wollmuth CE, Das K, Hile SE, Regan SB, Barnes RP, Haouzi A, Lee SM, House NCM, Guyumdzhyan M et al (2019) Sequence and nuclease requirements for breakage and healing of a structure‐forming (AT)n sequence within fragile site FRA16D. Cell Rep 27: e1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Fishel R, Lee JB (2018) Coordinating multi‐protein mismatch repair by managing diffusion mechanics on the DNA. J Mol Biol 430: 4469–4480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano K (2014) Structural mechanisms of human RecQ helicases WRN and BLM. Front Genet 5: 366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano K, Kim SY, Hakoshima T (2010) Structural basis for DNA strand separation by the unconventional winged‐helix domain of RecQ helicase WRN. Structure 18: 177–187 [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA (2015) Eukaryotic mismatch repair in relation to DNA replication. Annu Rev Genet 49: 291–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb S, Blaha‐Ostermann S, Kamper E, Rippka J, Schwarz C, Ehrenhofer‐Wolfer K, Schlattl A, Wernitznig A, Lipp JJ, Nagasaka K et al (2019) Werner syndrome helicase is a selective vulnerability of microsatellite instability‐high tumor cells. Elife 8: e43333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley DM (1983) Dynamic, sequence‐dependent DNA structure as exemplified by cruciform extrusion from inverted repeats in negatively supercoiled DNA. Cold Spring Harb Symp Quant Biol 47: 101–112 [DOI] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A (2017) PARP inhibitors: synthetic lethality in the clinic. Science 355: 1152–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Roca A, Giner‐Calabuig M, Murcia O, Castillejo A, Soto JL, Garcia‐Heredia A, Jover R (2022) Lynch‐like syndrome: potential mechanisms and management. Cancers (Basel) 14: 1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney SA, Freeman AD, Lilley DM, Ha T (2005) Observing spontaneous branch migration of Holliday junctions one step at a time. Proc Natl Acad Sci USA 102: 5715–5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CJ, Kim GY, Zhao X, Usdin K (2020) All three mammalian MutL complexes are required for repeat expansion in a mouse cell model of the fragile X‐related disorders. PLoS Genet 16: e1008902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuuchi K, Mizuuchi M, Gellert M (1982) Cruciform structures in palindromic DNA are favored by DNA supercoiling. J Mol Biol 156: 229–243 [DOI] [PubMed] [Google Scholar]
- Neil AJ, Hisey JA, Quasem I, McGinty RJ, Hitczenko M, Khristich AN, Mirkin SM (2021) Replication‐independent instability of Friedreich's ataxia GAA repeats during chronological aging. Proc Natl Acad Sci USA 118: e2013080118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olave MC, Graham RP (2021) Mismatch repair deficiency: the what, how and why it is important. Genes Chromosomes Cancer 61: 314–321 [DOI] [PubMed] [Google Scholar]
- O'Neil NJ, Bailey ML, Hieter P (2017) Synthetic lethality and cancer. Nat Rev Genet 18: 613–623 [DOI] [PubMed] [Google Scholar]
- Oshima J, Sidorova JM, Monnat RJ Jr (2017) Werner syndrome: clinical features, pathogenesis and potential therapeutic interventions. Ageing Res Rev 33: 105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panayotatos N, Wells RD (1981) Cruciform structures in supercoiled DNA. Nature 289: 466–470 [DOI] [PubMed] [Google Scholar]
- Pecina‐Slaus N, Kafka A, Salamon I, Bukovac A (2020) Mismatch repair pathway, genome stability and cancer. Front Mol Biosci 7: 122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picco G, Cattaneo CM, van Vliet EJ, Crisafulli G, Rospo G, Consonni S, Vieira SF, Rodriguez IS, Cancelliere C, Banerjee R et al (2021) Werner helicase is a synthetic‐lethal vulnerability in mismatch repair‐deficient colorectal cancer refractory to targeted therapies, chemotherapy, and immunotherapy. Cancer Discov 11: 1923–1937 [DOI] [PubMed] [Google Scholar]
- Pinto C, Kasaciunaite K, Seidel R, Cejka P (2016) Human DNA2 possesses a cryptic DNA unwinding activity that functionally integrates with BLM or WRN helicases. Elife 5: e18574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto C, Anand R, Cejka P (2018) Methods to study DNA end resection II: biochemical reconstitution assays. Methods Enzymol 600: 67–106 [DOI] [PubMed] [Google Scholar]
- Randrian V, Evrard C, Tougeron D (2021) Microsatellite instability in colorectal cancers: carcinogenesis, neo‐antigens, immuno‐resistance and emerging therapies. Cancers (Basel) 13: 3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjha L, Anand R, Cejka P (2014) The Saccharomyces cerevisiae Mlh1‐Mlh3 heterodimer is an endonuclease that preferentially binds to Holliday junctions. J Biol Chem 289: 5674–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschle M, Dufner P, Marra G, Jiricny J (2002) Mutations within the hMLH1 and hPMS2 subunits of the human MutLalpha mismatch repair factor affect its ATPase activity, but not its ability to interact with hMutSalpha. J Biol Chem 277: 21810–21820 [DOI] [PubMed] [Google Scholar]
- Rass U, Compton SA, Matos J, Singleton MR, Ip SC, Blanco MG, Griffith JD, West SC (2010) Mechanism of Holliday junction resolution by the human GEN1 protein. Genes Dev 24: 1559–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard GF (2021) The startling role of mismatch repair in trinucleotide repeat expansions. Cell 10: 1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saydam N, Kanagaraj R, Dietschy T, Garcia PL, Pena‐Diaz J, Shevelev I, Stagljar I, Janscak P (2007) Physical and functional interactions between Werner syndrome helicase and mismatch‐repair initiation factors. Nucleic Acids Res 35: 5706–5716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MHM, Pearson CE (2016) Disease‐associated repeat instability and mismatch repair. DNA Repair (Amst) 38: 117–126 [DOI] [PubMed] [Google Scholar]
- Snowden T, Acharya S, Butz C, Berardini M, Fishel R (2004) hMSH4‐hMSH5 recognizes Holliday junctions and forms a meiosis‐specific sliding clamp that embraces homologous chromosomes. Mol Cell 15: 437–451 [DOI] [PubMed] [Google Scholar]
- Sturzenegger A, Burdova K, Kanagaraj R, Levikova M, Pinto C, Cejka P, Janscak P (2014) DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long‐range DNA end resection in human cells. J Biol Chem 289: 27314–27326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wietmarschen N, Sridharan S, Nathan WJ, Tubbs A, Chan EM, Callen E, Wu W, Belinky F, Tripathi V, Wong N et al (2020) Repeat expansions confer WRN dependence in microsatellite‐unstable cancers. Nature 586: 292–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wietmarschen N, Nathan WJ, Nussenzweig A (2021) The WRN helicase: resolving a new target in microsatellite unstable cancers. Curr Opin Genet Dev 71: 34–38 [DOI] [PubMed] [Google Scholar]
- Young SJ, Sebald M, Shah Punatar R, Larin M, Masino L, Rodrigo‐Brenni MC, Liang CC, West SC (2020) MutSbeta stimulates Holliday junction resolution by the SMX complex. Cell Rep 33: 108289 [DOI] [PubMed] [Google Scholar]