

Abstract
Rationale
Knowledge on biomarkers of interstitial lung disease is incomplete. Interstitial lung abnormalities (ILAs) are radiologic changes that may present in its early stages.
Objectives
To uncover blood proteins associated with ILAs using large-scale proteomics methods.
Methods
Data from two prospective cohort studies, the AGES-Reykjavik (Age, Gene/Environment Susceptibility–Reykjavik) study (N = 5,259) for biomarker discovery and the COPDGene (Genetic Epidemiology of COPD) study (N = 4,899) for replication, were used. Blood proteins were measured using DNA aptamers, targeting more than 4,700 protein analytes. The association of proteins with ILAs and ILA progression was assessed with regression modeling, as were associations with genetic risk factors. Adaptive Least Absolute Shrinkage and Selection Operator models were applied to bootstrap data samples to discover sets of proteins predictive of ILAs and their progression.
Measurements and Main Results
Of 287 associations, SFTPB (surfactant protein B) (odds ratio [OR], 3.71 [95% confidence interval (CI), 3.20–4.30]; P = 4.28 × 10−67), SCGB3A1 (Secretoglobin family 3A member 1) (OR, 2.43 [95% CI, 2.13–2.77]; P = 8.01 × 10−40), and WFDC2 (WAP four-disulfide core domain protein 2) (OR, 2.42 [95% CI, 2.11–2.78]; P = 4.01 × 10−36) were most significantly associated with ILA in AGES-Reykjavik and were replicated in COPDGene. In AGES-Reykjavik, concentrations of SFTPB were associated with the rs35705950 MUC5B (mucin 5B) promoter polymorphism, and SFTPB and WFDC2 had the strongest associations with ILA progression. Multivariate models of ILAs in AGES-Reykjavik, ILAs in COPDGene, and ILA progression in AGES-Reykjavik had validated areas under the receiver operating characteristic curve of 0.880, 0.826, and 0.824, respectively.
Conclusions
Novel, replicated associations of ILA, its progression, and genetic risk factors with numerous blood proteins are demonstrated as well as machine-learning–based models with favorable predictive potential. Several proteins are revealed as potential markers of early fibrotic lung disease.
Keywords: interstitial lung abnormalities, interstitial lung disease, idiopathic pulmonary fibrosis, proteomics, biomarkers
At a Glance Commentary
Scientific Knowledge on the Subject
Knowledge of protein biomarkers of interstitial lung disease is insufficient, especially in its early forms.
What This Study Adds to the Field
We present a large-scale proteomic study, the first such study of interstitial lung abnormalities, that suggests several potential biomarkers of changes suggestive of early pulmonary fibrosis.
Many biologically active proteins have been proposed as potential biomarkers for advanced interstitial lung diseases (ILDs) that can result in pulmonary fibrosis, such as idiopathic pulmonary fibrosis (IPF). These are indicators of alveolar epithelial cell damage, proteins involved in extracellular remodeling, proteins involved in immune response, adhesion molecules, and growth factors (1–5). Still, novel ILD biomarkers are needed (1).
Interstitial lung abnormalities (ILAs) are chest computed tomography (CT) abnormalities resembling the radiologic appearance of ILD (6, 7). They are associated with risk factors common in patients with IPF, such as age, smoking, restrictive lung deficits, and certain genetic polymorphisms, most notably the rs35705950 promoter polymorphism of the MUC5B gene (8). In addition, some patients with ILA have histopathological evidence of pulmonary fibrosis (6, 8–10), progression of ILAs has been reported (11), and ILAs are associated with increased mortality (12). Patterns of ILA have been classified, among which associations with progression and mortality vary (11, 13). Interest in ILAs stems from an ambition to detect an early stage of pulmonary fibrosis before advanced architectural remodeling develops (7). Blood biomarkers of ILA could aid with identifying those at greatest risk of progression to pulmonary fibrosis and improving understanding of the pathogenesis of early disease stages (7, 14).
Blood-based proteomics methods are emerging as an effective way of uncovering accessible biomarkers of human disease (15). Although such methods have been applied in small cohorts of patients with IPF and their relatives (2–5), no study exists in which methods of proteomics are applied to early stages of fibrotic lung disease.
Therefore, the objective of this study was to apply large-scale proteomics methods to identify biomarkers of ILA and their progression.
Methods
Study Design
The AGES-Reykjavik (Age, Gene/Environment Susceptibility–Reykjavik) study was designed to explore risk factors of disease among the elderly with a multidisciplinary approach (16). The 5,764 participants underwent a range of examinations, including CT imaging of the thorax. Five years later, 3,167 participants had a follow-up examination and CT.
The COPDGene (Genetic Epidemiology of COPD) study is a multicenter cohort study of non-Hispanic White and African American individuals, 99% of whom were smokers, designed to investigate the genetics and epidemiology of chronic obstructive pulmonary disease (COPD) (6). Subjects with significant interstitial lung disease at enrollment were ineligible for COPDGene. Data were used from 5,339 participants who participated in the 5-year follow-up (phase 2) visit, who had chest CT imaging and fresh-frozen plasma samples available (17).
Definitions of ILA, ILA Subtypes, and ILA Progression
Images from initial and follow-up examinations were visually assessed for the presence of ILAs in AGES-Reykjavik (6, 11), as were images from the COPDGene 5-year follow-up visit. ILAs were defined per recent Fleischner Society guidelines (14). Changes present in <5% of any lung zone were deemed indeterminate (6). Images from participants with ILAs were classified with regard to the presence of definite fibrosis and the usual interstitial pneumonia (UIP) pattern (6, 11). For participants who had ILA present at initial examination or at follow-up, CT scans of AGES-Reykjavik participants were simultaneously compared for the development and progression (heretofore referred to as “progression”) of ILA (11).
Protein Profiling
Proteomic measurements were performed coincident with the initial chest CT in AGES-Reykjavik and with the 5-year follow-up visit chest CT in COPDGene. Single-stranded DNA aptamers designed to recognize target proteins, termed Slow-Off rate Modified Aptamers (SOMAmers), were used for protein detection and measurements. Serum samples from 5,457 participants were incubated with a mixture of 5,034 SOMAmers, creating SOMAmer–protein complexes. After washout of unbound proteins and SOMAmers, these enriched SOMAmers were quantified using a hybridization array. Median intraassay and interassay coefficients of variation were found to be <5%. SOMAmer specificity was validated by cross-platform validation, and the specificity of 779 SOMAmers was directly confirmed using mass spectrometry techniques. Details of this process are previously described (18). Human proteins were targeted by 4,782 SOMAmers. Because some proteins were targeted by multiple SOMAmers and some were annotated to multiple genes, these SOMAmers targeted 4,137 human proteins with 4,115 unique genetic targets. For statistical analyses, one SOMAmer per genetic target was used, selecting the SOMAmer with the stronger association with ILA at baseline for proteins targeted by multiple SOMAmers.
In the COPDGene study, proteomic measurements were conducted on ethylenediaminetetraacetic acid plasma samples collected during the phase 2 clinical visit (n = 6,018). Samples were stored at −80°C until the time of assaying by SomaLogic on their SomaScan version 4.0 (5K) assay for human plasma. This version of SomaScan contained 5,285 SOMAmers, of which 4,979 target human proteins, representing 4,776 unique proteins with 4,720 unique Uniprot numbers. SomaLogic standardized the SomaScan data per their protocol. It consisted of within-plate hybridization to control for variability across array signals, median signal normalization to control for technical variability of replicates within a run, plate scaling and calibration of SOMAmers to control for interassay variation between analytes, and batch differences between plates. Finally, median normalization to a reference using adaptive normalization by maximum likelihood is applied within dilution group to quality control (QC) replicates and individual samples to remove edge effects and technical variance. After data cleaning (n = 101), removing those with lung reduction or transplant surgery (n = 19), and samples that failed QC (n = 228), 5,670 results were available for analysis. The QC coefficient of variation at the 10%, 50%, and 90% percentiles were 3.1%, 5.0%, and 9.8%, respectively. SomaScan results were reported in relative fluorescence units.
Statistical Analysis
A flow chart of study design is shown in Figure E1 in the online supplement. Because of variability in measurement values, protein data were transformed using a variant of the Box-Cox transformation, providing results per SD (19). Extreme outliers (0.2% of values) were removed because of likelihood of measurement error and imputed before analysis using K-nearest neighbor imputing. All logistic regression models were adjusted for age, sex, pack-years, and smoking status at the beginning of the study. Bonferroni-corrected P values < 0.05 were considered significant for single-point analyses. Actual P values are shown throughout the paper. Statistical analyses were performed using R.
Analyses of ILA in AGES-Reykjavik
Of 5,764 AGES-Reykjavik participants, 5,259 (91%) had data on ILA status at baseline and protein measurements. Logistic regression models of the associations of single proteins with ILA were fitted for all proteins. For comparison, associations of single proteins with indeterminate ILA were also assessed with logistic regression; otherwise, participants with indeterminate ILA status were excluded from all analyses. The 1,609 proteins with suggestive associations (P < 0.05) with ILA in single-protein models were explored using adaptive LASSO (Least Absolute Shrinkage and Selection Operator) modeling of 200 bootstrap data samples (20). The associations of the eight proteins that occurred in all 200 LASSO models were analyzed using multivariate logistic regression models. The areas under the receiver operating characteristic curves (AUROCs) were calculated for these models and validated using resampling methods (21). The variance inflation factor was calculated for the multivariate regression model. Further methodological details, comparative analyses of proteins occurring in fewer LASSO models, tissue expression analyses, and functional enrichment analyses based on the GTEx, KEGG, WikiPathways, TRANSFAC, CORUM, and HPO databases are described in online supplemental methods.
Replication of ILA Analyses in COPDGene
In the COPDGene study, SOMAmer data were available for 4,899 participants (92% of participants with CT data available). After exclusion of those indeterminate for ILA and with missing covariate data, data were available for 2,974 participants. Single-protein logistic regression models using the three proteins with the strongest associations with ILA in AGES-Reykjavik (SFTPB [surfactant protein B], SCGB3A1 [Secretoglobin family 3A member 1], and WFDC2 [WAP four-disulfide core domain protein 2]) were fitted, as well as the eight-protein multivariate logistic regression model based on adaptive LASSO modeling in AGES-Reykjavik. The AUROCs of these models were calculated and validated, with methods identical to those in AGES-Reykjavik. Because of their association with variable standardization, models in COPDGene were additionally adjusted for white blood cell and platelet count and study center.
Analyses of Pulmonary Fibrosis–associated SNPs
Genotyping was done for 5,656 AGES-Reykjavik participants, as previously described (22). For the 5,368 participants with both genotyping and protein measurements available, linear regression analyses adjusted for age, sex, pack-years of smoking, and smoking at study entry were performed to evaluate the associations of previously reported pulmonary fibrosis–related SNPs (SNPs previously associated with either IPF or ILA [9]) with all 4,782 human SOMAmers.
Analyses of ILA Imaging Patterns
The 287 proteins that were significantly associated with ILA in AGES-Reykjavik were assessed by logistic regression models for associations with ILA imaging patterns (i.e., the definite fibrosis pattern and the UIP pattern). Logistic regression models of the associations of these proteins with each pattern were constructed. Comparisons were made with participants without ILA, excluding participants with other imaging patterns. For analyses involving the UIP pattern, participants with probable or definite UIP were regarded as having UIP, and participants with no or indeterminate UIP were not.
Analyses of ILA Progression
Included in analyses of ILA progression in AGES-Reykjavik were the 223 participants who had ILA progression at follow-up examination and the 1,425 who did not have ILA at either baseline or follow-up. Participants with definite or probable progression on follow-up examinations were compared with participants with no ILA in both examinations (11). ILA progression was not assessed in COPDGene, as data collection is not yet complete.
The associations of single proteins with the progression of ILA were tested with logistic regression. As in analyses of ILA at baseline, the proteins suggestively associated with progression (P < 0.05) in single-protein logistic regression models (1,562 proteins) were used to create 200 bootstrap data samples. An adaptive LASSO regression model was created in each sample. A multivariate regression model was constructed, using proteins in all 200 LASSO models with the AUROC calculated and validated. Comparative analyses and further details are found in the online supplement.
Data Availability
The custom-design Novartis SOMAscan is available through a collaboration agreement with the Novartis Institutes for BioMedical Research (lori.jennings@novartis.com). Data from the AGES Reykjavik study are available through collaboration (AGES_data_request@hjarta.is) under a data usage agreement with the IHA in accordance with participants’ informed consent.
Results
Participant characteristics at baseline in AGES-Reykjavik and in phase 2 of COPDGene are shown in Table 1. Representative ILA cases are shown in Figure E2.
Table 1.
Overview of the Study Participants with Available Protein Measurements and Characterization of Interstitial Lung Abnormalities
| Participants | AGES-Reykjavik |
COPDGene |
||||
|---|---|---|---|---|---|---|
| No ILAs (n = 3,187) | Indeterminate for ILAs (n = 1,703) | ILAs (n = 329) | No ILAs (n = 2,484) | Indeterminate for ILAs (n = 1,891) | ILAs (n = 524) | |
| Age, mean (SD) | 75.9 (5.4) | 77.3 (5.6) | 78.1 (5.6) | 64.0 (8.3) | 66.4 (8.7) | 69.0 (8.8) |
| Women, n (%) | 1,889 (59) | 950 (56) | 149 (45) | 1,221 (49) | 955 (50) | 249 (48) |
| BMI, mean (SD) | 27.2 (4.4) | 26.8 (4.4) | 27.2 (4.7) | 29.0 (6.4) | 28.8 (6.4) | 29.6 (6.1) |
| History of smoking, n (%) | 1,742 (55) | 1,015 (60) | 237 (72) | 2,448 (99) | 1,872 (99) | 518 (99) |
| Pack-years, median (IQR) | 0 (0–17) | 2.9 (0–23) | 11 (0–28) | 38.0 (25.1–51.0) | 41.4 (28.2–57.0) | 43.9 (28.4–59.1) |
| Current smoker, n (%) | 371 (12) | 203 (12) | 54 (16) | 911 (37) | 762 (40) | 199 (38) |
| Definite fibrosis pattern, n (%) | ||||||
| Without fibrosis | — | — | 206 (63) | — | — | 469 (90) |
| Definite fibrosis | — | — | 123 (37) | — | — | 55 (10) |
| UIP pattern, n (%) | ||||||
| No UIP | — | — | 104 (32) | — | — | 129 (25) |
| Indeterminate for UIP | — | — | 131 (40) | — | — | 346 (66) |
| Probable UIP | — | — | 77 (23) | — | — | 46 (9) |
| Definite UIP | — | — | 17 (5) | — | — | 3 (0.6) |
Definition of abbreviations: AGES-Reykjavik = Age, Gene/Environment Susceptibility–Reykjavik; BMI = body mass index; COPDGene = Genetic Epidemiology of COPD; ILA = interstitial lung abnormality; IQR = interquartile range; UIP = usual interstitial pneumonia.
Associations between Serum Proteins and ILA in AGES-Reykjavik
Of the 4,137 proteins tested, 287 were associated with ILA after Bonferroni adjustment (P < 1.22 × 10−5) (Figure 1A and Tables E1 and E2). The association of SFTPB was the most significant (P = 4.28 × 10−67) and was associated with the greatest odds increase of ILA (odds ratio [OR], 3.71 [95% confidence interval [CI], 3.20–4.30]). Among other significant associations of ILA at baseline were associations with SCGB3A1 (OR, 2.43 [95% CI, 2.13–2.77]; P = 8.01 × 10−40) and WFDC2 (OR, 2.42 [95% CI, 2.11–2.78]; P = 4.01 × 10−36). The distributions of these three proteins grouped by ILA status are shown in Figure E3. Proteins associated with ILA were enriched for lung-tissue specificity when compared with all proteins in the genome but not when compared with all proteins with available SOMAmer measurements in AGES-Reykjavik (Figure E4). Full results of analyses of indeterminate ILAs, tissue expression, and functional enrichment, and direct comparisons with previously published ILA biomarkers are shown in online supplement results, Figures E4 and E5, and Tables E2–E4.
Figure 1.

The associations of single proteins with interstitial lung abnormalities (ILAs) at baseline and progression of ILAs. Shown in red circles are results from the AGES-Reykjavik (Age, Gene/Environment Susceptibility–Reykjavik) study. Results from COPDGene (Genetic Epidemiology of COPD) are shown in blue triangles. Models are (A) logistic regression models of a single protein with ILAs at baseline, and (B) progression of ILAs, adjusted for age, sex, pack-years, and smoking at study entry. Models in COPDGene are additionally adjusted for white blood cell count, platelet count, and study center. BPIFB1 = BPI fold containing family B member 1; CTSH = cathepsin H; GDF-15 = growth differentiation factor 15; SCGB3A1 = secretoglobin family 3A member 1; SFTPB = surfactant protein B; UBE2E1 = ubiquitin conjugating enzyme E2 E1; WFDC2 = WAP four-disulfide core domain protein 2.
Eight proteins featured in all 200 adaptive LASSO models of ILAs (Table E5). In a multivariate logistic regression model exploring the association of these proteins with ILA, WFDC2 (OR, 3.15 [95% CI, 2.55–3.89]; P = 2.81 × 10−26) and SFTPB (OR, 3.14 [95% CI, 2.66–3.71]; P = 1.78 × 10−41) had the largest effect on ILAs. SCGB3A1 (OR, 1.62 [95% CI, 1.37–1.91]; P = 1.84 × 10−8) and CBLN4 (Cerebellin 4 Precursor; OR, 1.26 [95% CI, 1.09–1.45]; P = 0.0019) were also positively associated with ILAs, whereas WFIKKN2 (WAP, Kazal, immunoglobulin, Kunitz and NTR domain-containing protein 2; OR, 0.42 [95% CI, 0.35–0.51]; P = 3.83 × 10−19), ADAM Metallopeptidase Domain 9 (ADAM9) (OR, 0.59 [95% CI, 0.50–0.70]; P = 6.93 × 10−10), and Annexin A9 (ANXA9) (OR, 0.69 [95% CI, 0.59–0.81]; P = 6.39 × 10−6) were negatively associated (Figure 2A and Table E6). The validated AUROC of this model, based on 200-fold resampling, was 0.880, compared with 0.670 for a model with only age, sex, pack-years, and smoking at beginning of the study and 0.749, 0.760, and 0.826 for single-protein models with SCGB3A1, WFDC2, and SFTPB, respectively, added to these demographic factors (Figure 3A). The variance inflation factor was less than two for all components of the eight-protein multivariate model. Results of comparative models based on proteins used in a lower number of LASSO models are seen in Figure E6.
Figure 2.
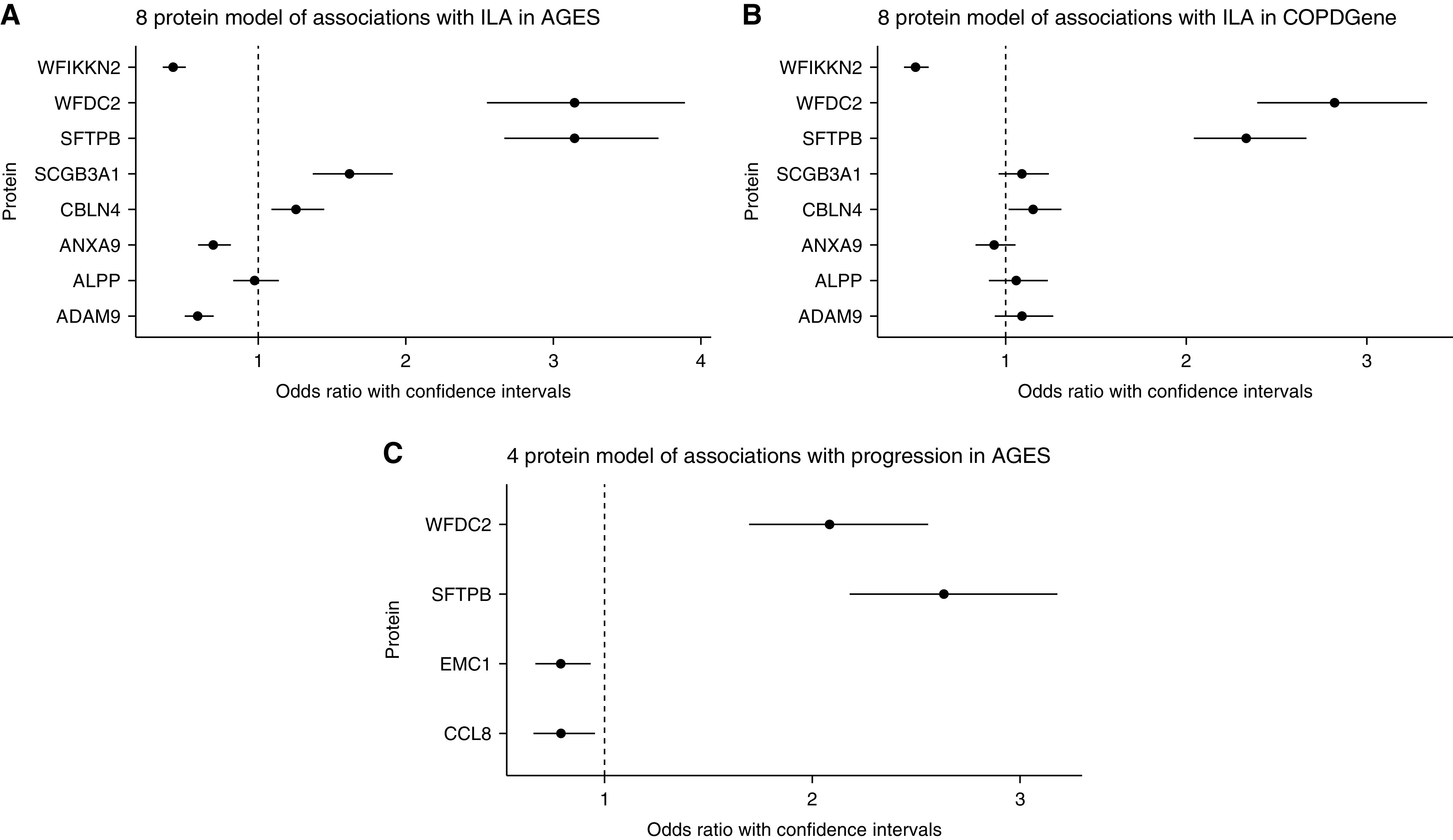
Multivariate logistic regression models of the association of proteins with interstitial lung abnormalities (ILAs) and ILA progression. (A) A model of the associations of the eight proteins selected for 200 adaptive Least Absolute Shrinkage and Selection Operator (LASSO) models with ILAs in the AGES-Reykjavik (Age, Gene/Environment Susceptibility–Reykjavik) cohort. (B) A model of the associations of the eight proteins selected for 200 adaptive LASSO models with ILA in the COPDGene (Genetic Epidemiology of COPD) cohort. (C) A model of the associations of the four proteins selected for 200 adaptive LASSO models with progression of ILA in the AGES-Reykjavik cohort. Models in AGES-Reykjavik are logistic regression models, adjusted for age, sex, pack-years, and smoking at study entry. The model in COPDGene is additionally adjusted for white blood cell count, platelet count, and study center. ADAM9 = ADAM metallopeptidase domain 9; ALPP = alkaline phosphatase, placental; ANXA9 = annexin A9; CBLN4 = cerebellin 4 precursor; CCL8 = C-C motif chemokine ligand 8; EMC1 = ER membrane protein complex subunit 1; SCGB3A1 = secretoglobin family 3A member 1; SFTPB = surfactant protein B; WFDC2 = WAP four-disulfide core domain protein 2; WFIKKN2 = WAP, Kazal, immunoglobulin, Kunitz and NTR domain-containing protein 2.
Figure 3.

Receiver operating characteristic (ROC) curves of models of the associations of proteins with interstitial lung abnormalities (ILAs) at baseline and ILA progression. ROC curves for the specified logistic regression models. (A) Curves for models in the AGES-Reykjavik (Age, Gene/Environment Susceptibility–Reykjavik) cohort with ILAs at baseline as the outcome. (B) Curves for models in the COPDGene (Genetic Epidemiology of COPD) cohort with ILAs at baseline as the outcome. (C) Curves for models in the AGES-Reykjavik cohort with progression of ILAs as the outcome. Baseline: A model with age, sex, pack-years, and smoking at study entry, for which all other models are adjusted. Names of proteins refer to single-protein models of that protein with ILAs at baseline or at progression. Eight proteins (A and B): A model with proteins used in 200 adaptive Least Absolute Shrinkage and Selection Operator (LASSO) models of ILAs at baseline. Four proteins (C): A model with proteins used in 200 adaptive LASSO models of ILA progression. AUC = area under curve; SCGB3A1 = secretoglobin family 3A member 1; SFTPB = surfactant protein B; WFDC2 = WAP four-disulfide core domain protein 2; vAUC = area under curve, validated with 200-fold resampling.
Replication Analyses of ILAs in COPDGene
In models of single proteins, SFTPB (OR, 2.70 [95% CI, 2.39–3.05]; P = 1.13 × 10−57), WFDC2 (OR, 2.61 [95% CI, 2.27–2.99]; P = 5.89 × 10−43), and SCGB3A1 (OR, 1.49 [95% CI, 1.33–1.67]; P = 1.77 × 10−12) were all associated with ILA in the COPDGene cohort (Figure 1A and Table E7). The validated AUROCs of these models were 0.787, 0.763, and 0.709, respectively, compared with 0.692 for demographic factors only (Figure 3B). In a multivariate model with the eight proteins selected using LASSO modeling, SFTPB (OR, 2.33 [95% CI 2.04–2.66]; P = 9.33 × 10−36), WFDC2 (OR, 2.82 [95% CI, 2.39–3.33]; P = 2.33 × 10−34), WFIKKN2 (OR, 0.50 [95% CI, 0.43–0.57]; P = 1.64 × 10−22), and CBLN4 (OR, 1.15 [95% CI, 1.02–1.31]; P = 0.03) were associated with ILAs (Figure 2B and Table E7), and the validated AUROC of this model was 0.826 (Figure 3B).
Associations with Pulmonary Fibrosis–associated SNPs in AGES-Reykjavik
Results of linear regression analyses modeling the relationship of pulmonary fibrosis–related SNPs with human proteins are shown in Table 2, for proteins with associations reaching genome-wide significance (P < 5 × 10−8). Also shown are the associations of these proteins with ILA and the previously reported associations of the SNPs with ILAs (9). Four proteins were found to be associated with these SNPs, and three of these were themselves associated with ILAs. These SNPs were the rs35705950 MUC5B promoter polymorphism associated with SFTPB, the rs2736100 TERT (Telomerase reverse transcriptase) polymorphism associated with thrombopoietin, and the rs4727443 polymorphism associated with Paired Immunoglobin Like Type 2 Receptor Alpha (PILRA). Of those, only the MUC5B promoter polymorphism was significantly associated with ILAs in the cohort. This association was consistent when conducted with stratification based on ILA status (Table E8). The associations of the MUC5B promoter polymorphism with proteins are shown graphically in Figure E7 and numerically in Table E9.
Table 2.
Associations between Previously Identified Pulmonary Fibrosis–associated Genetic Loci, Single-Protein Measurements, and Interstitial Lung Abnormalities
| SNPs and ILAs (Previously Reported) |
SNPs and Proteins |
Proteins and ILAs |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| rsID | Chromosomal Location | Nearest Gene | OR | P Value | Protein | β (SE) | P Value | OR | P Value |
| rs73199442 | 3q13 | FCF1P3 | 1.68 (1.39–2.02) | 5 × 10−8 | – | – | – | – | – |
| rs6886640 | 5q12 | IPO11 | 1.28 (1.18–1.41) | 4 × 10−8 | – | – | – | – | – |
| rs7744971 | 6q15 | HTR1E | 1.26 (1.16–1.37) | 1 × 10−7 | – | – | – | – | – |
| rs35705950 | 11p15 | MUC5B | 1.97 (1.74–2.22) | 3 × 10−27 | SFTPB | 0.26 (0.030) | 8 × 10−18 | 3.71 | 4.28 × 10−67 |
| rs2609255 | 4q22 | FAM13A | 1.18 (1.07–1.29) | 5 × 10−4 | – | – | – | – | – |
| rs2076295 | 6p24 | DSP | 1.14 (1.05–1.2) | 0.001 | – | – | – | – | – |
| rs2034650 | 15q15 | IVD | 1.08 (0.99–1.17) | 0.07 | – | – | – | – | – |
| rs12610495 | 19p13 | DPP9 | 1.14 (1.03–1.26) | 0.01 | N/A | N/A | N/A | N/A | N/A |
| rs6793295 | 3q26 | LRRC34 | 1.06 (0.97–1.15) | 0.2 | N/A | N/A | N/A | N/A | N/A |
| rs1981997 | 17q21 | MAPT | 1.16 (1.03–1.30) | 0.01 | – | – | – | – | – |
| rs2736100 | 5p15 | TERT | 1.03 (0.95–1.12) | 0.44 | THPO | 0.11 (0.019) | 4 × 10−9 | 0.77* | 1.39 × 10−4 |
| 1.19* | 3.93 × 10−3 | ||||||||
| rs11191865 | 10q24 | OBFC1 | 1.03 (0.95–1.12) | 0.46 | – | – | – | – | – |
| rs1278769 | 13q34 | ATP11A | 1.04 (0.95–1.15) | 0.37 | F7 | −0.13 (0.020) | 6 × 10−11 | 0.91 | 0.17 |
| rs62025270 | 15q25 | AKAP13 | 1.09 (0.99–1.20) | 0.08 | – | – | – | – | – |
| rs4727443 | 7q22 | LOC1001 28334/LOC1053 75423 | 0.95 (0.87–1.03) | 0.19 | PILRA | −0.50 (0.019) | 2 × 10−151 | 0.97* | 0.66 |
| 0.97* | 0.56 | ||||||||
| 1.30* | 1.20 × 10−5 | ||||||||
| 1.21* | 1.69 × 10−3 | ||||||||
| 1.07* | 0.26 | ||||||||
Definition of abbreviations: AGES-Reykjavik = Age, Gene/Environment Susceptibility–Reykjavik; CI = confidence interval; ILA = interstitial lung abnormality; OR = odds ratio; PILRA = Paired Immunoglobin Like Type 2 Receptor Alpha; SFTPB = surfactant protein B; SOMAmers = Slow-Off rate Modified Aptamers; THPO = thrombopoietin.
SNPs and ILAs: the associations between the listed SNPs and ILAs as previously reported (8). SNPs and proteins: the associations between the listed SNPs and proteins in the AGES-Reykjavik cohort, adjusted for age, sex, pack-years of smoking, and smoking at study entry. Proteins with an association with a P value < 5 × 10−8 are shown. Proteins and ILAs: the associations between shown proteins and ILAs, calculated with logistic regression adjusted for age, sex, pack-years of smoking, and smoking at study entry. N/A indicates data for SNP not available in AGES-Reykjavik.
Data shown for multiple SOMAmers binding to the same protein.
Associations with ILA Imaging Patterns
Results of models of the associations of ILA-associated proteins with imaging patterns are shown in Tables E10 and E11. The 20 proteins most significantly associated with ILAs had significant associations regardless of the presence or absence of specific imaging patterns. All twenty proteins were associated with greater odds of ILA with definite fibrosis than ILA without definite fibrosis. Similar results were seen for the UIP pattern; all proteins were associated with ILAs with and without UIP, but ORs were higher for ILA with UIP (Table E10).
Analyses of ILA Progression in AGES-Reykjavik
In single-protein models of ILA progression, 121 proteins were significant after Bonferroni correction (P < 1.22 × 10−5) (Figure 1B and Tables E12 and E13). The protein associated with the greatest odds of ILA progression was SFTPB (OR, 3.08 [95% CI, 2.56–3.69]; P = 1.59 × 10−39). Other strongly associated proteins were WFDC2 (OR, 2.72 [95% CI, 2.25–3.29]; P = 4.90 × 10−25), GDF-15 (Growth Differentiation Factor 15; OR, 2.14 [95% CI, 1.79–2.55]; P = 3.01 × 10−17), and CTSH (Cathepsin H; OR, 2.02 [95% CI, 1.70–2.40]; P = 1.72 × 10−15).
The proteins used in all 200 adaptive LASSO models of ILA progression (Table E14) were all associated with ILA progression in multiprotein models. The most significant associations were for SFTPB (OR, 2.63 [95% CI, 2.17–3.18]; P = 1.96 × 10−23) and WFDC2 (OR, 2.08 [95% CI, 1.69–2.55]; P = 3.05 × 10−12), whereas ER Membrane Protein Complex Subunit 1 (EMC1) (OR, 0.79 [95% CI, 0.67–0.94]; P = 0.0063) and C-C Motif Chemokine Ligand 8 (CCL8) (OR, 0.79 [95% CI, 0.66–0.95]; P = 0.015) were also associated with progression (Figure 2C and Table E15). The validated AUROC of this model was 0.824, compared with 0.669 for a model with only demographic factors and 0.760 and 0.798 for single-protein models with WFDC2 and SFTPB (Figure 3C).
Discussion
This study represents the first proteomic assessment of ILAs and is the largest blood proteomic assessment of ILD or pulmonary fibrosis to date (5, 23). In addition to a comprehensive assessment of thousands of proteins, uncovering hundreds of associations with ILAs and ILA progression, these results provide replicable evidence for the association between protein measures, alone (e.g., SFTPB, WFDC2, and SCGB3A1) or in machine-learning–based models, and ILAs across independent populations. The magnitudes of the associations of single proteins are strong compared with known risk factors (6, 8), and multiprotein models demonstrate replicable ability to improve risk prediction of ILA and its progression over demographic risk factors. The proteins most strongly associated with ILA consistently had larger associations with imaging patterns that are correlated with unfavorable outcomes, such as progression and mortality (11).
These findings greatly expand on the small but growing number of independent studies demonstrating that peripheral blood protein measures can help detect ILAs (24–27). These proteins were all suggestively associated with ILAs based on an unadjusted P value, and most were associated with ILAs and/or ILA progression after multiple testing (Tables E1, E2, E4, E12, and E13). Although magnitudes of associations are hard to directly compare owing to differences in the units in which they are provided, the directionality of associations was consistent for all previously published protein biomarkers of ILA (Table E4). In addition to providing an independent replication of these prior findings, this reproducibility across different platforms provides some assurance that other associations are not platform specific, as protein measurements for these previously published studies were mainly done using ELISA (24–27).
Although some of the proteins most significantly associated with ILAs in the present findings have previously been associated with IPF (1), many associations are novel. SFTPB is a small hydrophobic protein that is an essential component of the regulation and function of pulmonary surfactant (28). Its concentration in plasma is normally low, and its elevation is believed to represent a breakdown of the alveolar–capillary membrane, which likely contributes to its elevation in other conditions (29, 30). Rare genetic variants in multiple surfactant proteins (31) have been implicated in IPF. Common variants in SFTPB have been previously reported in association with IPF (32), although these findings have not been confirmed in larger studies (33), and elevated concentrations have been reported in patients with pulmonary fibrosis in some (34), but not all (35), studies. This reflects the protein’s different isoforms and the challenges of reliably measuring it in plasma (29). Although WFDC2 (also referred to as HE-4 [human epididymis protein 4]) is highly expressed in epithelial cells and submucosal glands in the human respiratory tract, its function remains incompletely characterized (36). Elevation of WFDC2 in plasma has been associated with IPF (37). SCGB3A1 (also referred to as UGRP2 [uteroglobin-related protein 2] or HIN-1 [high in normal 1]) is a tumor-suppressor gene known to be secreted throughout the conducting airways (38, 39). To the best of our knowledge, it has not been previously studied in IPF. WFIKKN2 has a replicable negative association with ILAs. This protein is an antagonist of GDF-8 and GDF-11, implicated in various aging-related processes and diseases, and interacts with other members of the Transforming Growth Factor-β (TGF-β) superfamily (40, 41). The TGF-β family of proteins, a member of which is the previously mentioned GDF-15, is central to the tissue-injury response and plays a pivotal role in the pathogenesis of IPF as it is currently understood (42). The proposed role of cytokine signaling in ILD pathogenesis extends well beyond that of TGF-β, possibly explaining the enrichment of gene ontology terms related to such signaling among ILA-associated proteins (42).
The potential of SFTPB as a biomarker of ILAs and ILA progression is supported by the association of the rs35705950 promoter polymorphism of MUC5B, the best-established genetic risk factor for pulmonary fibrosis (33). The mechanism of this association is unknown but of great interest. Although it is possible that this finding is a result of two separate strong associations with ILAs, no other proteins had associations with MUC5B that reached genome-wide significance. Therefore, it is conceivable that this association represents an unknown biological mechanism, considering that the distal airways are a major site of MUC5B expression, that MUC5B is known to be coexpressed with other surfactant proteins in distal airways (43, 44), and the suggestion that an appropriate ratio of mucins relative to surfactant may be necessary for normal physiology of certain airway zones (44). Another notable finding was the association of TERT, an established genetic risk factor of IPF (42), with THPO (thrombopoietin), concentrations of which were associated with ILAs. Although data on the role of thrombopoietin in pulmonary fibrosis are scarce, an increase in its amount in lungs of mice with medication-induced lung fibrosis has been documented (45). Still, the meaning of this finding is unclear, because TERT is not associated with ILAs. This could be because THPO is more strongly associated with ILAs than TERT or because the association of THPO with ILAs is independent of its association with TERT.
This study has several important limitations. First, although this is the largest proteomic analysis of ILAs, and any form of ILD, to date, it is possible that larger sample sizes of those with ILAs will be needed to uncover additional important proteins and pathways associated with the early stages of pulmonary fibrosis. Second, although we demonstrate replicable evidence for associations of proteins and multiprotein models with ILAs across independent populations, it is likely that associations of some of these proteins are not specific to ILAs alone. Third, not all the proteins selected for replication from the AGES-Reykjavik cohort were validated in COPDGene. It is possible that demographic differences between these two cohorts (e.g., age, smoking history, and racial background) could explain some of these discrepancies. Fourth, the validation of results is based on SOMAmer technology, a novel measurement platform. The specificity of this method compared with other methods has been extensively studied, and a minority of proteins have potential cross-reactivity with protein isoforms or related proteins with high amino acid homology (46). Although validation data from recent studies based on both mass spectrometry and antibody-based technology, shown for the proteins highlighted in the results (Table E16), were reassuring, not all proteins had such data available (18, 47). Therefore, as the SOMAmer technology is novel and the validation of SOMAmer specificity is ongoing, future replication of the findings with standard platforms would be useful. Fifth, the preferred method of selecting multiprotein models for biomarker discovery has not been established in proteomics studies to date. The method of machine learning by adaptive LASSO regression of bootstrap data samples provides sets of proteins that jointly predict ILAs. This selection method is based on statistical correlations but not biological mechanisms, possibly explaining why not all proteins selected had significant associations when externally validated. The decision to use only proteins that featured in all 200 adaptive LASSO models can be questioned. However, as shown in Figure E6, the gain in prediction by using more inclusive models is slim. Finally, effects of residual confounding (e.g., with chronic diseases that could coincide with ILAs) cannot be excluded for the multitude of associations presented.
In this first proteomic assessment of ILAs and ILA progression, replicable associations of several proteins, notably SFTPB, WFDC2, SCGB3A1, and WFIKKN2, are presented. In conjunction with prior studies of IPF, these findings demonstrate the utility of proteomics in generating reproducible models of ILA that may help to detect patients at risk for pulmonary fibrosis.
Acknowledgments
Acknowledgment
The authors thank other investigators, staff, and particularly the participants of the AGES-Reykjavik and the COPDGene studies.
Footnotes
Supported by National Institutes of Health grants K08 HL140087 (R.K.P.); K08 HL136928 (B.D.H.); R01 HL135142 and R01 HL137927 (M.H.C.); U01 HL089856, R01 HL113264, R01 137927, R01 133135, and P01 HL114501 (E.K.S.); R01 HL137995 and R01 HL152735 (R.P.B.); R01 HL111024, R01 HL130974, and R01 135142 (G.M.H., H.H.); and R01CA203636 and 5U01CA209414-03 (H.H.). Supported by National Institute on Aging grant 27120120022C (V. Gudnason). Supported by the Icelandic Centre for Research, project grants 141513-051 (G.G., V. Gudnason, and G.M.H.); 195761-051 (V.E.); and 184845-051 and 206692-051 (V. Gudmundsdottir). Supported by Landspítali Háskólasjúkrahús grants A-2019-029, A-2019-030, A-2020-018, and A-2020-017 (G.G.), University of Iceland Research Fund 2021, and the Eimskip University Fund (G.T.A.). The Age, Gene/Environment Susceptibility-Reykjavik Study was supported by National Institutes of Health contracts N01-AG-1-2100 and HHSN27120120022C, the National Institute on Aging Intramural Research Program, Hjartavernd (the Icelandic Heart Association), and the Althingi (the Icelandic Parliament). COPDGene: The COPDGene study (NCT00608764) is also supported by the COPD Foundation through contributions made to an Industry Advisory Committee that has included AstraZeneca, Bayer Pharmaceuticals, Boehringer-Ingelheim, Genentech, GlaxoSmithKline, Novartis, Pfizer, and Sunovion. The project described was supported by National Heart, Lung, and Blood Institute grants U01 HL089897, U01 HL089856, R01 HL137995, and R01 HL129937. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health. SCCOR: This project was supported by National Heart, Lung, and Blood Institute grants P50HL084948 and R21HL129917 and Pennsylvania Department of Health CURE SAP 4100062224. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute. Funding sources of the study had no role in the collection, analysis, or interpretation of the data, writing of the manuscript, or the decision to submit for publication.
Author Contributions: Conceptualization, methodology, and design of the study: G.T.A., G.G., J.L.S., B.D.H., M.H.C., E.K.S., R.P.B., L.J.L., L.L.J., G.M.H., V.E., and V. Gudnason. Data acquisition: R.K.P., E.F.G., H.H., V. Gudmundsdottir, A.G., T. Hino, T. Hida, B.D.H., E.K.S., R.P.B., G.M.H., V.E., and V. Gudnason. Analysis of the data: G.T.A., G.G., K.A.P., T.A., V. Gudmundsdottir, and A.G. Administration and supervision: E.K.S., R.P.B., L.J.L., L.L.J., G.M.H., V.E., and V. Gudnason. Drafting of the initial manuscript: G.T.A., G.G., G.M.H., and V. Gudnason. All authors edited the manuscript for scientific content. All the authors agree to be accountable for the work with regard to accuracy and integrity.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202110-2296OC on April 19, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Guiot J, Moermans C, Henket M, Corhay J-L, Louis R. Blood biomarkers in idiopathic pulmonary fibrosis. Lung . 2017;195:273–280. doi: 10.1007/s00408-017-9993-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mathai SK, Cardwell J, Metzger F, Powers J, Walts AD, Kropski JA, et al. Preclinical pulmonary fibrosis circulating protein biomarkers. Am J Respir Crit Care Med . 2020;202:1720–1724. doi: 10.1164/rccm.202003-0724LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. O’Dwyer DN, Norman KC, Xia M, Huang Y, Gurczynski SJ, Ashley SL, et al. The peripheral blood proteome signature of idiopathic pulmonary fibrosis is distinct from normal and is associated with novel immunological processes. Sci Rep . 2017;7:46560. doi: 10.1038/srep46560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Niu R, Liu Y, Zhang Y, Zhang Y, Wang H, Wang Y, et al. iTRAQ-based proteomics reveals novel biomarkers for idiopathic pulmonary fibrosis. PLoS One . 2017;12:e0170741. doi: 10.1371/journal.pone.0170741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Todd JL, Neely ML, Overton R, Durham K, Gulati M, Huang H, et al. IPF-PRO Registry investigators Peripheral blood proteomic profiling of idiopathic pulmonary fibrosis biomarkers in the multicentre IPF-PRO Registry. Respir Res . 2019;20:227. doi: 10.1186/s12931-019-1190-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Washko GR, Hunninghake GM, Fernandez IE, Nishino M, Okajima Y, Yamashiro T, et al. COPDGene Investigators Lung volumes and emphysema in smokers with interstitial lung abnormalities. N Engl J Med . 2011;364:897–906. doi: 10.1056/NEJMoa1007285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hunninghake GM. Interstitial lung abnormalities: erecting fences in the path towards advanced pulmonary fibrosis. Thorax . 2019;74:506–511. doi: 10.1136/thoraxjnl-2018-212446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hunninghake GM, Hatabu H, Okajima Y, Gao W, Dupuis J, Latourelle JC, et al. MUC5B promoter polymorphism and interstitial lung abnormalities. N Engl J Med . 2013;368:2192–2200. doi: 10.1056/NEJMoa1216076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hobbs BD, Putman RK, Araki T, Nishino M, Gudmundsson G, Gudnason V, et al. Overlap of genetic risk between interstitial lung abnormalities and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med . 2019;200:1402–1413. doi: 10.1164/rccm.201903-0511OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miller ER, Putman RK, Vivero M, Hung Y, Araki T, Nishino M, et al. Histopathology of interstitial lung abnormalities in the context of lung nodule resections. Am J Respir Crit Care Med . 2018;197:955–958. doi: 10.1164/rccm.201708-1679LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Putman RK, Gudmundsson G, Axelsson GT, Hida T, Honda O, Araki T, et al. Imaging patterns are associated with interstitial lung abnormality progression and mortality. Am J Respir Crit Care Med . 2019;200:175–183. doi: 10.1164/rccm.201809-1652OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Putman RK, Hatabu H, Araki T, Gudmundsson G, Gao W, Nishino M, et al. Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators; COPDGene Investigators Association between interstitial lung abnormalities and all-cause mortality. JAMA . 2016;315:672–681. doi: 10.1001/jama.2016.0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Putman RK, Gudmundsson G, Araki T, Nishino M, Sigurdsson S, Gudmundsson EF, et al. The MUC5B promoter polymorphism is associated with specific interstitial lung abnormality subtypes. Eur Respir J . 2017;50:1700537. doi: 10.1183/13993003.00537-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hatabu H, Hunninghake GM, Richeldi L, Brown KK, Wells AU, Remy-Jardin M, et al. Interstitial lung abnormalities detected incidentally on CT: a position paper from the Fleischner Society. Lancet Respir Med . 2020;8:726–737. doi: 10.1016/S2213-2600(20)30168-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Emilsson V, Gudnason V, Jennings LL. Predicting health and life span with the deep plasma proteome. Nat Med . 2019;25:1815–1816. doi: 10.1038/s41591-019-0677-y. [DOI] [PubMed] [Google Scholar]
- 16. Harris TB, Launer LJ, Eiriksdottir G, Kjartansson O, Jonsson PV, Sigurdsson G, et al. Age, Gene/Environment Susceptibility-Reykjavik Study: multidisciplinary applied phenomics. Am J Epidemiol . 2007;165:1076–1087. doi: 10.1093/aje/kwk115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wan ES, Fortis S, Regan EA, Hokanson J, Han MK, Casaburi R, et al. COPDGene Investigators Longitudinal phenotypes and mortality in preserved ratio impaired spirometry in the COPDGene study. Am J Respir Crit Care Med . 2018;198:1397–1405. doi: 10.1164/rccm.201804-0663OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Emilsson V, Ilkov M, Lamb JR, Finkel N, Gudmundsson EF, Pitts R, et al. Co-regulatory networks of human serum proteins link genetics to disease. Science . 2018;361:769–773. doi: 10.1126/science.aaq1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yeo IK, Johnson RA. A new family of power transformations to improve normality or symmetry. Biometrika . 2000;87:954–959. [Google Scholar]
- 20. Zou H. The adaptive lasso and its oracle properties. J Am Stat Assoc . 2006;101:1418–1429. [Google Scholar]
- 21.Efron B, Tibshirani RJ. An introduction to the bootstrap. New York: Chapman & Hall; 1993. [Google Scholar]
- 22. Gudjonsson A, Gudmundsdottir V, Axelsson GT, Gudmundsson EF, Jonsson BG, Launer LJ, et al. A genome-wide association study of serum proteins reveals shared loci with common diseases. Nat Commun . 2022;13:480. doi: 10.1038/s41467-021-27850-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alqalyoobi S, Adegunsoye A, Linderholm A, Hrusch C, Cutting C, Ma S-F, et al. Circulating plasma biomarkers of progressive interstitial lung disease. Am J Respir Crit Care Med . 2020;201:250–253. doi: 10.1164/rccm.201907-1343LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ho JE, Gao W, Levy D, Santhanakrishnan R, Araki T, Rosas IO, et al. Galectin-3 is associated with restrictive lung disease and interstitial lung abnormalities. Am J Respir Crit Care Med . 2016;194:77–83. doi: 10.1164/rccm.201509-1753OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Armstrong HF, Podolanczuk AJ, Barr RG, Oelsner EC, Kawut SM, Hoffman EA, et al. MESA (Multi-Ethnic Study of Atherosclerosis) Serum matrix metalloproteinase-7, respiratory symptoms, and mortality in community-dwelling adults. Am J Respir Crit Care Med . 2017;196:1311–1317. doi: 10.1164/rccm.201701-0254OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McGroder CF, Aaron CP, Bielinski SJ, Kawut SM, Tracy RP, Raghu G, et al. Circulating adhesion molecules and subclinical interstitial lung disease: the Multi-Ethnic Study of Atherosclerosis. Eur Respir J . 2019;54:19900295. doi: 10.1183/13993003.00295-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sanders JL, Putman RK, Dupuis J, Xu H, Murabito JM, Araki T, et al. The association of aging biomarkers, interstitial lung abnormalities, and mortality. Am J Respir Crit Care Med . 2021;203:1149–1157. doi: 10.1164/rccm.202007-2993OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martínez-Calle M, Olmeda B, Dietl P, Frick M, Pérez-Gil J. Pulmonary surfactant protein SP-B promotes exocytosis of lamellar bodies in alveolar type II cells. FASEB J . 2018;32:4600–4611. doi: 10.1096/fj.201701462RR. [DOI] [PubMed] [Google Scholar]
- 29. Leung JM, Mayo J, Tan W, Tammemagi CM, Liu G, Peacock S, et al. Plasma pro-surfactant protein B and lung function decline in smokers. Eur Respir J . 2015;45:1037–1045. doi: 10.1183/09031936.00184214. [DOI] [PubMed] [Google Scholar]
- 30. Sin DD, Tammemagi CM, Lam S, Barnett MJ, Duan X, Tam A, et al. Pro-surfactant protein B as a biomarker for lung cancer prediction. J Clin Oncol . 2013;31:4536–4543. doi: 10.1200/JCO.2013.50.6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ley B, Brown KK, Collard HR. Molecular biomarkers in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol . 2014;307:L681–L691. doi: 10.1152/ajplung.00014.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Selman M, Lin H-M, Montaño M, Jenkins AL, Estrada A, Lin Z, et al. Surfactant protein A and B genetic variants predispose to idiopathic pulmonary fibrosis. Hum Genet . 2003;113:542–550. doi: 10.1007/s00439-003-1015-4. [DOI] [PubMed] [Google Scholar]
- 33. Allen RJ, Guillen-Guio B, Oldham JM, Ma SF, Dressen A, Paynton ML, et al. Genome-wide association study of susceptibility to idiopathic pulmonary fibrosis. Am J Respir Crit Care Med . 2020;201:564–574. doi: 10.1164/rccm.201905-1017OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kahn N, Rossler A-K, Hornemann K, Muley T, Grünig E, Schmidt W, et al. C-proSP-B: a possible biomarker for pulmonary diseases? Respiration . 2018;96:117–126. doi: 10.1159/000488245. [DOI] [PubMed] [Google Scholar]
- 35. Papaioannou AI, Kostikas K, Manali ED, Papadaki G, Roussou A, Spathis A, et al. Serum levels of surfactant proteins in patients with combined pulmonary fibrosis and emphysema (CPFE) PLoS One . 2016;11:e0157789. doi: 10.1371/journal.pone.0157789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bingle L, Armes H, Bingle C. Expression and function of murine WFDC2 in the respiratory tract. Eur Respir J . 2020;56:2775. [Google Scholar]
- 37. Raghu G, Richeldi L, Jagerschmidt A, Martin V, Subramaniam A, Ozoux M-L, et al. Idiopathic pulmonary fibrosis: prospective, case-controlled study of natural history and circulating biomarkers. Chest . 2018;154:1359–1370. doi: 10.1016/j.chest.2018.08.1083. [DOI] [PubMed] [Google Scholar]
- 38. Reynolds SD, Reynolds PR, Pryhuber GS, Finder JD, Stripp BR. Secretoglobins SCGB3A1 and SCGB3A2 define secretory cell subsets in mouse and human airways. Am J Respir Crit Care Med . 2002;166:1498–1509. doi: 10.1164/rccm.200204-285OC. [DOI] [PubMed] [Google Scholar]
- 39. Mazumdar J, Hickey MM, Pant DK, Durham AC, Sweet-Cordero A, Vachani A, et al. HIF-2alpha deletion promotes Kras-driven lung tumor development. Proc Natl Acad Sci USA . 2010;107:14182–14187. doi: 10.1073/pnas.1001296107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Szláma G, Kondás K, Trexler M, Patthy L. WFIKKN1 and WFIKKN2 bind growth factors TGFβ1, BMP2 and BMP4 but do not inhibit their signalling activity. FEBS J . 2010;277:5040–5050. doi: 10.1111/j.1742-4658.2010.07909.x. [DOI] [PubMed] [Google Scholar]
- 41. Frohlich J, Vinciguerra M. Candidate rejuvenating factor GDF11 and tissue fibrosis: friend or foe? Geroscience . 2020;42:1475–1498. doi: 10.1007/s11357-020-00279-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol . 2014;9:157–179. doi: 10.1146/annurev-pathol-012513-104706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hancock LA, Hennessy CE, Solomon GM, Dobrinskikh E, Estrella A, Hara N, et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat Commun . 2018;9:5363. doi: 10.1038/s41467-018-07768-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Okuda K, Chen G, Subramani DB, Wolf M, Gilmore RC, Kato T, et al. Localization of secretory mucins MUC5AC and MUC5B in normal/healthy human airways. Am J Respir Crit Care Med . 2019;199:715–727. doi: 10.1164/rccm.201804-0734OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhou Y, Zhang B, Li C, Huang X, Cheng H, Bao X, et al. Megakaryocytes participate in the occurrence of bleomycin-induced pulmonary fibrosis. Cell Death Dis . 2019;10:648. doi: 10.1038/s41419-019-1903-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, et al. Genomic atlas of the human plasma proteome. Nature . 2018;558:73–79. doi: 10.1038/s41586-018-0175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pietzner M, Wheeler E, Carrasco-Zanini J, Kerrison ND, Oerton E, Koprulu M, et al. Synergistic insights into human health from aptamer- and antibody-based proteomic profiling. Nat Commun . 2021;12:6822. doi: 10.1038/s41467-021-27164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The custom-design Novartis SOMAscan is available through a collaboration agreement with the Novartis Institutes for BioMedical Research (lori.jennings@novartis.com). Data from the AGES Reykjavik study are available through collaboration (AGES_data_request@hjarta.is) under a data usage agreement with the IHA in accordance with participants’ informed consent.