

Abstract
G protein–coupled receptors (GPCRs) are the largest family of cell surface receptors and signal through the proximal effectors, G proteins and β-arrestins, to influence nearly every biological process. The G protein and β-arrestin signaling pathways have largely been considered separable; however, direct interactions between Gα proteins and β-arrestins have been described that appear to be part of a distinct GPCR signaling pathway. Within these complexes, Gαi/o, but not other Gα protein subtypes, directly interacts with β-arrestin, regardless of the canonical Gα protein that is coupled to the GPCR. Here, we report that the endogenous biased chemokine agonists of CXCR3 (CXCL9, CXCL10, and CXCL11), together with two small-molecule biased agonists, differentially formed Gαi:β-arrestin complexes. Formation of the Gαi:β-arrestin complexes did not correlate well with either G protein activation or β-arrestin recruitment. β-arrestin biosensors demonstrated that ligands that promoted Gαi:β-arrestin complex formation generated similar β-arrestin conformations. We also found that Gαi:β-arrestin complexes did not couple to the mitogen-activated protein kinase ERK, as is observed with other receptors such as the V2 vasopressin receptor, but did couple with the clathrin adaptor protein AP-2, which suggests context-dependent signaling by these complexes. These findings reinforce the notion that Gαi:β-arrestin complex formation is a distinct GPCR signaling pathway and enhance our understanding of the spectrum of biased agonism.
A complex to complicate chemokine signaling
G protein–coupled receptors (GPCRs) represent about 30% of the targets of FDA-approved drugs. GPCRs signal through two pathways that are considered separable: one dependent on direct activation of heterotrimeric G proteins and the other on the recruitment of #-arrestin proteins. Building on their previous work, Zheng et al. showed that some, but not all, of the endogenous agonists of the chemokine receptor CXCR3 induced the formation of a G#i:#-arrestin complex, which was independent of the relative extent of activation of either G protein or #-arrestin signaling pathways. Signaling downstream of the G#i:#-arrestin complex was different from that stimulated by #-arrestin alone. Together, these data add to the evidence of crosstalk between G proteins and #-arrestins and may have implications for the development of GPCR-targeting drugs.
INTRODUCTION
G protein–coupled receptors (GPCRs) enable cells to sense and respond appropriately to hormonal and environmental signals. Targeting GPCR signaling has proven to be of therapeutic benefit because about 30% of all Food and Drug Administration–approved medications target GPCRs (1). Classically, each GPCR couples to distinct Gα protein families, such as Gαs, Gαi, Gαq, or Gα12/13, and β-arrestins (2, 3). These transducer proteins are used by nearly every GPCR to translate and integrate extracellular stimuli into intracellular signals. It is thought that G proteins and β-arrestins constitute separable GPCR signaling pathways, with β-arrestins acting both as independent signaling scaffolds for downstream effectors while also serving as critical negative regulators of G protein signaling (4–9). GPCRs can also form “megaplex” signaling complexes in which a GPCR simultaneously binds to both G protein and β-arrestin (10, 11).
We previously reported that agonist binding to GPCRs, regardless of which Gα protein the GPCR canonically couples to, can promote the formation of noncanonical Gαi:β-arrestin signaling complexes to regulate pathways, including signaling by the mitogen-activated protein kinase (MAPK) extracellular signal–regulated kinase (ERK) (12). Gαi appears to play a distinct role in influencing β-arrestin signaling relative to the other G protein isoforms. Such Gαi:β-arrestin complexes can form complexes with effectors such as ERK downstream of the V2 vasopressin receptor (V2R). These signaling complexes potentially have widespread physiological and therapeutic implications. However, it remains unclear whether different agonists for the same GPCR form Gαi:β-arrestin complexes in a conserved fashion, whether Gαi:β-arrestin complex formation correlates with other established arms of GPCR activation, and whether Gαi:β-arrestin complexes interact with receptors or effectors, such as ERK, in a conserved manner.
One set of tools that can be helpful to address such questions is biased agonists. Biased agonists bind to the same receptor and differentially activate distinct downstream signaling pathways. Typically, G proteins and β-arrestins promote signaling through relatively independent pathways (10, 11). Synthetic G protein– and β-arrestin–biased agonists have been identified for many GPCRs, although only a few GPCRs have multiple endogenous ligands that act as biased agonists. One of the best systems demonstrating endogenous biased agonism is the chemokine system. Most chemokine receptors bind to more than one chemokine, and many chemokines act as biased agonists of their receptors (13). The receptor C-X-C motif chemokine receptor 3 (CXCR3) has three endogenous ligands: CXCL9, CXCL10, and CXCL11. We previously demonstrated that CXCL11 is β-arrestin–biased relative to CXCL9 and CXCL10 (13, 14). In addition, CXCR3 has small-molecule biased agonists, such as VUF10661 and VUF11418 (15, 16), which we previously validated as β-arrestin– and G protein–biased agonists, respectively (17). CXCR3 is also an attractive drug target because it is implicated in multiple diseases including cancer, infection, atherosclerosis, hypersensitivity reactions, and autoimmune disorders (18–22).
To investigate the potential linkage between canonical and noncanonical GPCR signaling, we used multiple agonists to study signaling pathways downstream of CXCR3, including G protein signaling, β-arrestin recruitment, and the formation of Gαi:β-arrestin complexes. We found that biased agonists had distinct G protein recruitment, G protein signaling, and β-arrestin recruitment profiles. However, none of these profiles corresponded with the ability to form Gαi:β-arrestin complexes. Through a panel of β-arrestin biosensors, we found that ligands that promoted the formation of Gαi:β-arrestin complexes generated similar β-arrestin conformations to one another. Last, we identified the ability of CXCL11 to form Gαi:β-arrestin complexes that coupled with the clathrin adapter protein AP-2 but not ERK. This study provides insights into GPCR activation events that determine Gαi:β-arrestin complex formation and expands our concept of biased agonism.
RESULTS
Assessment of canonical CXCR3 signaling
We first compared the activities of five CXCR3 agonists—CXCL9, CXCL10, CXCL11, VUF10661, and VUF11418—in multiple assays: Gαi/o protein recruitment, G protein signaling [by measuring the inhibition of cyclic adenosine 3′,5′-monophosphate (cAMP) synthesis], and β-arrestin2 recruitment. Note that G protein recruitment and G protein signaling (such as the inhibition of cAMP synthesis in the case of Gαi) may not correlate with one another because of differences in G protein precoupling or other mechanisms (23). Furthermore, because CXCR3 is a canonically Gαi-coupled receptor, either G protein recruitment or G protein signaling may more directly correlate with Gαi:β-arrestin2 complex formation. First, we tested the ability of the panel of CXCR3 agonists to recruit Gαi subunits to CXCR3 in human embryonic kidney (HEK) 293T cells using a previously described nanoBiT complementation system (Fig. 1A) (18, 24). The Gαi family members Gαi and Gαo, but not Gαs, were recruited to CXCR3 after treatment with CXCL11, the β-arrestin–biased agonist VUF10661, and the G protein–biased agonist VUF11418 (Fig. 1, B and C, and fig. S1, A and B). CXCL9, CXCL10, and VUF11418 did not induce Gαi/o protein recruitment to CXCR3 as robustly as CXCL11 and VUF10661 did (Fig. 1, B and C, and fig. S1A). The interaction of Gαi/o with CXCR3 was sensitive to pretreatment with pertussis toxin (PTX) (fig. S1, C and D), which promotes the adenosine 5′-diphosphate ribosylation of cysteine-352 in helix 5 of Gαi, and substantially attenuated the observed interaction between CXCR3 and Gαi/o. Treatment with PTX had no effect on the inability of Gαs to be recruited to the receptor (fig. S1E). Overexpression of β-arrestin2, which sterically hinders G proteins from interacting with GPCRs, also significantly attenuated the interaction between Gαi and CXCR3 (fig. S2A). Combining PTX pretreatment and overexpression of β-arrestin2 completely blocked G protein recruitment to CXCR3 (fig. S2B). Overexpression of β-arrestin2, pretreatment with PTX, or a combination of both had no significant effect on the lack of recruitment of Gαs to CXCR3 (fig. S2, C and D).
Fig. 1. Gαi protein recruitment, inhibition of cAMP synthesis, and β-arrestin recruitment by biased agonists of CXCR3.
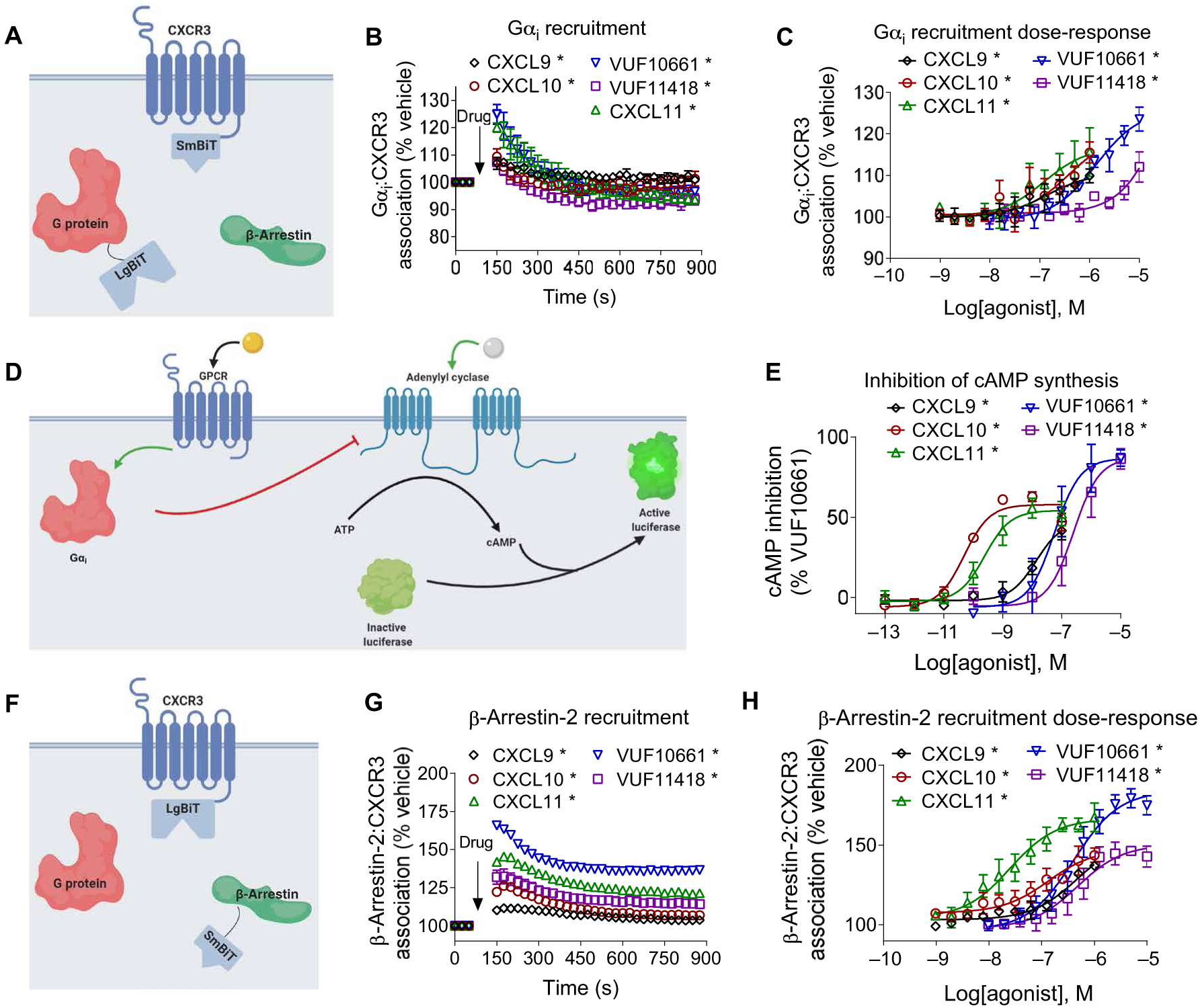
(A) Arrangement of the nanoBiT luciferase fragments used to assess G protein recruitment. (B) HEK293T transiently expressing CXCR3-smBiT and Gαi-LgBiT were treated with 100 nM CXCL9, 100 nM CXCL10, 100 nM CXCL11, 1 μM VUF10661, 1 μM VUF11418, or vehicle, and the resulting luminescence was analyzed for 750 s after an initial preread. (C) Gαi recruitment to CXCR3 after treatment with the indicated concentrations of agonists 1 min after treatment. (D) Scheme illustrating the Gαi-regulated cAMP assay. Before the cells were treated with biased agonists of CXCR3, the cellular cAMP concentration was increased by treatment with 10 μM forskolin. (E) HEK293T cells transiently expressing cAMP-activated modified firefly luciferase and CXCR3 were treated with vehicle, 100 nM CXCL9, 100 nM CXCL10, 100 nM CXCL11, 1 μM VUF10661, or 1 μM VUF11418. The inhibition of cAMP synthesis is displayed as a percentage of that in VUF10661-treated cells. (F) Arrangement of the luciferase fragments CXCR3-LgBiT and smBiT–β-arrestin2 to assess β-arrestin2 recruitment to CXCR3. (G) HEK293T transiently expressing CXCR3-LgBiT and smBiT–β-arrestin2 were treated with 100 nM CXCL9, 100 nM CXCL10, 100 nM CXCL11, 1 μM VUF10661, 1 μM VUF11418, or vehicle and then were analyzed for 750 s after an initial preread. (H) β-arrestin2 recruitment after treatment with the indicated concentrations of agonist 6 min after treatment. Data are means ± SEM of three or four independent experiments. *P < 0.05 by two-way ANOVA; Dunnett’s post hoc analysis showed a significant difference relative to pretreatment.
Next, we tested the ability of CXCR3 to promote canonical Gαi signaling through the inhibition of cAMP synthesis (Fig. 1D). Consistent with the predicted signaling properties of Gαi-coupled CXCR3, all five ligands reduced the concentration of intracellular cAMP in a concentration-dependent fashion, with potencies that mirrored the affinities of each agonist for CXCR3. Both small-molecule agonists had a higher maximal response (Emax) than those of the endogenous chemokines (Fig. 1E). This reduction in cAMP synthesis in response to these ligands was dependent on CXCR3, because cells transiently transfected with empty vector instead of CXCR3 expression vector did not inhibit cAMP synthesis when treated with agonist (fig. S3, A to E). Despite the inability of CXCL9 and CXCL10 to robustly recruit Gαi/o, both chemokines inhibited cAMP synthesis to similar degrees relative to the other three agonists. This indicates that CXCR3-regulated G protein recruitment and G protein signaling may not always correlate well with one another, especially in assays with different levels of amplification.
We then assessed the recruitment of β-arrestin2 to CXCR3 using a nanoLuc complementation assay (Fig. 1F). Consistent with previous observations, all five agonists led to β-arrestin2 recruitment, albeit with different median effective concentration (EC50) and Emax values. VUF10661 had the highest Emax for β-arrestin2 recruitment, which was followed by CXCL11, VUF11418, CXCL10, and CXCL9 (Fig. 1G). Furthermore, VUF10661 and CXCL11 promoted substantially more β-arrestin2 recruitment than the other three agonists (Fig. 1H). Pretreatment with PTX did not affect agonist-induced β-arrestin2 recruitment to CXCR3, consistent with previous observations for other GPCRs (fig. S4, A to E) (12).
Biased agonists of CXCR3 differentially promotethe formation of Gαi:β-arrestin complexes
We then examined whether CXCR3 biased agonists could promote Gαi:β-arrestin complex formation. We previously demonstrated that an association between Gαi family proteins and β-arrestins could be stimulated by ligand treatment of various GPCRs, even those that do not classically couple to Gαi, such as the V2R, the β2-adrenergic receptor (β2AR), and the neurotensin receptor type 1 (NTSR1) (12). However, it is unknown whether biased agonists can differentially promote the formation of Gαi:β-arrestin complexes. To address this, we treated HEK293 cells overexpressing CXCR3, G protein–Large Binary Technology (LgBiT), and Small Binary Technology (SmBiT)–β-arrestin2 with our CXCR3 agonist panel and tested for an association between Gαi and β-arrestin2 (Fig. 2A). Treatment of CXCR3 with CXCL11, the β-arrestin–biased agonist VUF10661, or the G protein–biased agonist VUF11418 resulted in the association of Gαi with β-arrestin2 (Fig. 2, B and C). Similarly, treatment of CXCR3 with CXCL11 or VUF10661 resulted in an association between Gαo and β-arrestin2 (fig. S5A). Note that neither CXCL9 nor CXCL10 stimulated any appreciable association of Gαi or Gαo with β-arrestin2 (Fig. 2B and fig. S5A).
Fig. 2. Biased agonists of CXCR3 differentially promote the formation of Gαi:β-arrestin complexes.
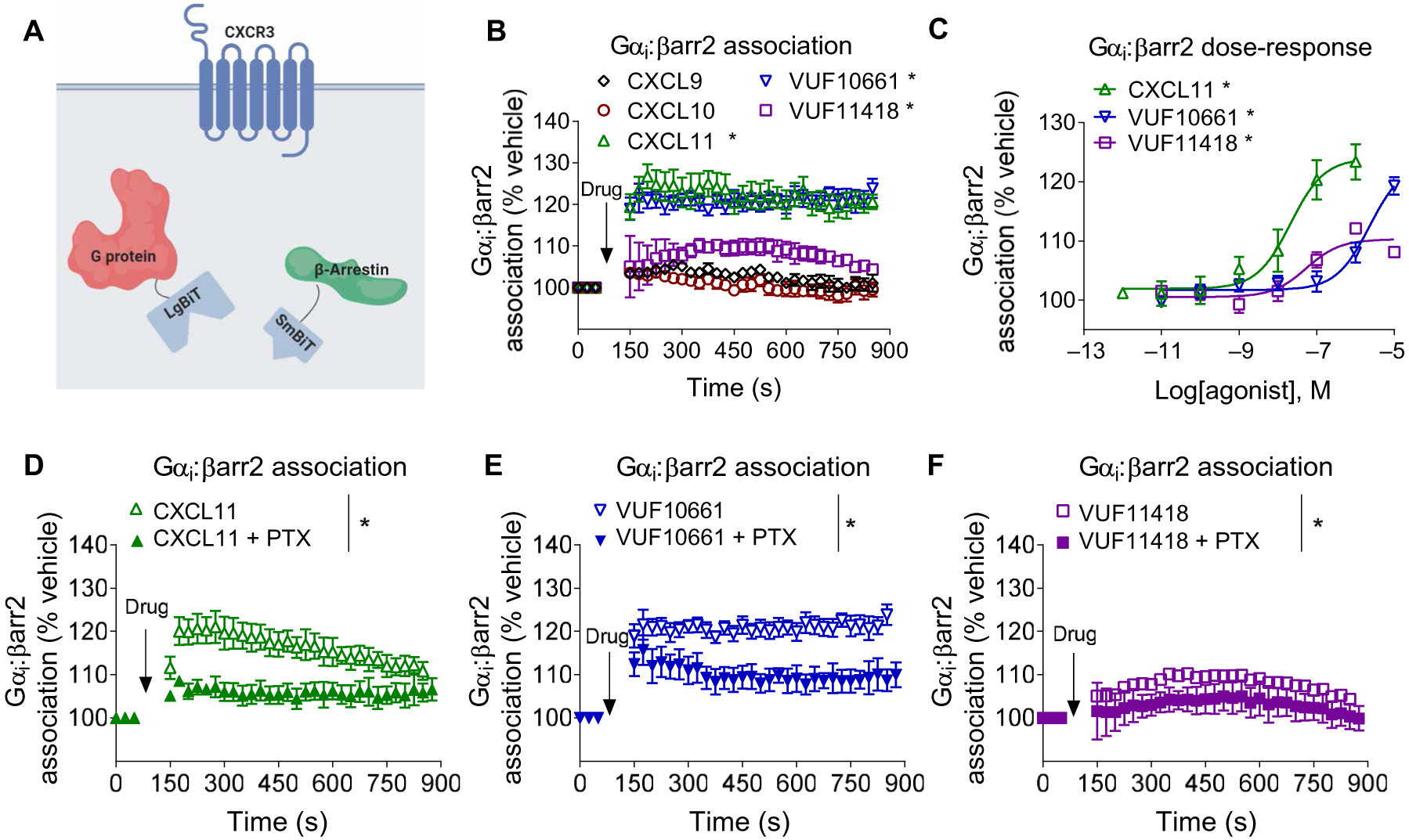
(A) Scheme of the assay used to assess Gαi:β-arrestin complex formation. (B) HEK293T cells transiently expressing CXCR3, smBiT–β-arrestin2 (βarr2), and Gαi-LgBiT were treated with 100 nM CXCL9, 100 nM CXCL10, 100 nM CXCL11, 1 μM VUF10661, 1 μM VUF11418, or vehicle, and luminescence was monitored for 15 min. (C) Transfected HEK293T cells expressing CXCR3, smBiT–β-arrestin2, and Gαi-LgBiT were treated with the indicated concentrations of CXCL11, VUF10661, VUF11418, or vehicle and were assayed 6 min after treatment. (D to F) HEK293T cells transiently expressing CXCR3, smBiT–β-arrestin2, and Gαi-LgBiT were pretreated with PTX (200 ng/m) and subsequently treated with (D) CXCL11, (E) VUF10661, or (F) VUF11418 before luminescence was measured to determine the formation of Gαi:β-arrestin complexes. Data are means ± SEM of three to five independent experiments. *P < 0.05 by two-way ANOVA. Data in (B) and (C) included Dunnett’s post hoc analysis to show significant differences between treatments.
Consistent with our previously reported results with other receptors, none of the agonists promoted the association of β-arrestin2 with Gαs, Gαq, or Gα12 (fig. S5, B to D) (12). Because many of these chemokines have alternative receptors, we also tested the effects of these ligands on cells transfected with plasmids to express solely the β-arrestin2 and Gαi components without CXCR3. We did not observe a significant increase in the association between Gαi and β-arrestin2 for any ligand apart from CXCL9 (fig. S5E), which is likely through off-target activity at high concentrations and is consistent with the ability of CXCL9 to reduce cAMP synthesis in cells that did not express CXCR3 (fig. S3A). Consistent with our previously reported observations, pretreatment with PTX significantly attenuated the association between Gαi and β-arrestin2 after treatment with agonists that promoted Gαi:β-arrestin complex formation (CXCL11, VUF10661, and VUF11418) (Fig. 2, D to F). Pretreatment with PTX attenuated Gαi:β-arrestin formation and Gαi recruitment but had no measurable effect on β-arrestin2 recruitment (fig. S4, A to E).
Assessment of CXCR3 biased signaling through G proteins, β-arrestins, and Gαi:β-arrestin complexes
We first qualitatively compared Gαi:β-arrestin complex formation, β-arrestin2 recruitment, G protein recruitment, and the inhibition of cAMP synthesis by constructing bias plots as previously described (Fig. 3, A to G) (19, 20). Bias plots enable an assessment of assay amplification effects, which can confound efforts to identify biased ligands (7). For example, because of second messenger amplification, cAMP inhibition assays often demonstrate left-shifted potencies and higher Emax values relative to those of G protein recruitment assays. If a ligand does not appear to be biased with a bias plot, then it is unlikely to be biased, because calculated bias factors can frequently have large errors (19). This is primarily because bias plots are not prone to errors introduced from different fitting approaches. Whereas the relationship among β-arrestin2 recruitment and Gαi:β-arrestin complex formation was relatively similar between CXCL11, VUF10661, and VUF11418 (Fig. 3F), there was considerably more variance in the relationship between G protein signaling (Gαi recruitment or inhibition of cAMP synthesis) and Gαi:β-arrestin complex formation (Fig. 3, E and G), with VUF10661 and VUF11418 displaying relative bias toward G protein signaling and CXCL11 toward β-arrestin2 recruitment and Gαi:β-arrestin complex formation (Fig. 3, C and D). Note that, whereas both CXCL9 and CXCL10 promoted G protein signaling and β-arrestin2 recruitment to CXCR3 (Fig. 1, B, E, and G), neither CXCL9 nor CXCL10 promoted Gαi:β-arrestin2 complex formation. Thus, although CXCL9 and CXCL10 were not included in this analysis (Fig. 3, E to G), their exclusion is evidence of biased signaling toward G protein signaling and β-arrestin2 recruitment relative to Gαi:β-arrestin complex formation.
Fig. 3. Bias plots of biased ligands of CXCR3 in G protein recruitment, G protein signaling, β-arrestin2 recruitment, and Gαi:β-arrestin2 complex formation.
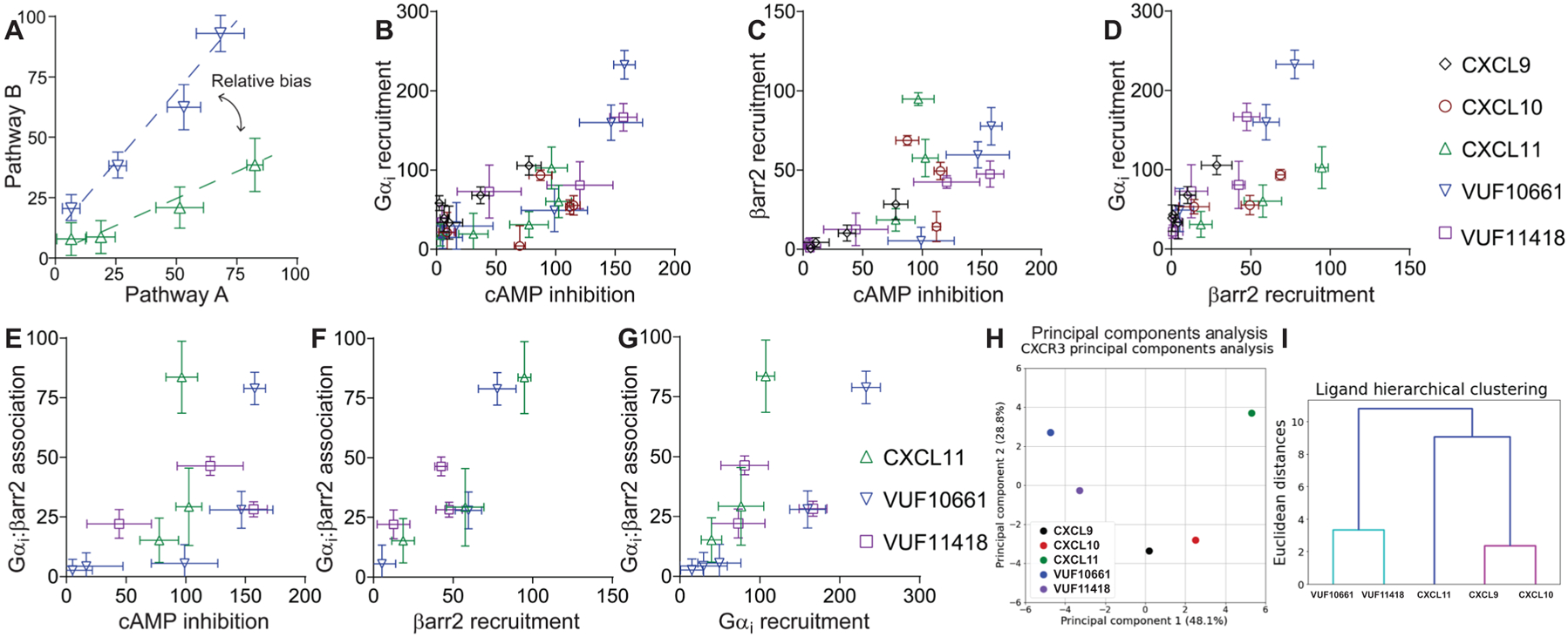
(A) Example of the interpretation of a bias plot. A difference in the two response-response curves (at the same agonist concentration) is indicative of a relative bias between agonists. (B to G) Bias plots of cAMP inhibition and Gαi recruitment (B); cAMP inhibition and β-arrestin2 recruitment (C); β-arrestin2 recruitment and Gαi recruitment (D); cAMP inhibition and Gαi:β-arrestin2 association, β-arrestin2 recruitment, and Gαi:β-arrestin association (F); and Gαi recruitment and Gαi:β-arrestin association (G). Because CXCL11 is the only full endogenous agonist for both the G protein and β-arrestin pathways, it was used as the reference ligand. (H) PCA was used to assess CXCR3 agonist similarity based on signaling assays; computed principal components are visualized with the top two principal components. Principal component 1 contributes to 48% of the observed variation, and principal component 2 contributes to 29% of observed variation. Points denote the composite response of a single ligand at varying concentrations. (I) Dendrogram of hierarchical clustering to determine the number of clusters and the relationship between the ligand signaling profiles. PCA and dendrogram analyses were of means from three to five independent experiments for each signaling assay.
To quantify these relationships further, we then calculated fitted curve parameters (EC50 and Emax) based on the dose-response data used for the bias plots (Table 1). These parameters were then used to calculate the logarithm of ratios of relative intrinsic activities for each pair of pathways to determine bias factors (Table 2). Consistent with the bias plots, the relationships between Gαi:β-arrestin complex formation and β-arrestin recruitment showed smaller bias factors than those between Gαi:β-arrestin complex formation and either Gαi recruitment or cAMP inhibition. We subsequently performed linear regression of each ligand per bias plot to calculate an R2 (coefficient of determination) value, describing the linear similarity between two signaling pathways per agonist (table S1). We then performed principal components analyses (PCAs), with the activity of each ligand at each pathway serving as input components. This analysis enabled a two-dimensional reduction of each ligand’s assessed signaling characteristics to evaluate agonist similarity (Fig. 3H). Subsequent hierarchical clustering revealed that the signaling of these five agonists fell into three clusters: one centered around CXCL9 and CXCL10, another around VUF10661 and VUF11418, and the last around CXCL11 (Fig. 3H). These relationships were further detailed in a clustering dendrogram that is consistent with the visual observation from the PCA plot, with VUF10661 and VUF11418 forming one cluster, CXCL9 and CXCL10 another cluster, and CXCL11 by itself (Fig. 3I). Together, these analyses demonstrate that these ligands display substantial bias between their signaling pathways, suggesting that both ligands and pathways, including the Gαi:β-arrestin pathway, are functionally distinct.
Table 1. Pharmacological parameters for canonical and noncanonical signaling by CXCR3.
Summary of the parameters obtained from fits of signaling assays (Figs. 1 and 2) from three to five independent experiments. EC50 and Emax (expressed as a percentage of the CXCL11 signal) measurements for agonists were calculated from a three-parameter fit [y = Min + (Max − Min)/(1 + 10LogEC50−X)]. For Gαi:β-arrestin complex formation, values were only calculated with CXCL11, VUF10661, and VUF11418, because the other agonists did not induce complex formation. If the three-parameter fit of ligand-receptor interaction produced a poor fit, then the data were omitted from the table. N/A, not applicable.
| Gαi:β-arrestin2 | CXCR3:β-arrestin2 | CXCR3:Gαi | cAMP inhibition | ||
|---|---|---|---|---|---|
| CXCL9 | LogEC50 | N/A | −7.7 ± 0.40 | −7.6 ± 0.57 | −7.9 ± 0.19 |
| E max | N/A | 35 ± 9.0 | 79 ± 30 | 87 ± 10 | |
| CXCL10 | LogEC50 | N/A | −8.5 ± 0.17 | −9.1 ± 0.55 | −10 ± 0.15 |
| E max | N/A | 76 ± 6.0 | 76 ± 19 | 110 ± 7.3 | |
| CXCL11 | LogEC50 | −7.6 ± 0.29 | −8.2 ± 0.16 | −8.8 ± 0.44 | −9.6 ± 0.21 |
| E max | 100 ± 20 | 100 ± 8.8 | 100 ± 21 | 100 ± 8.9 | |
| VUF10661 | LogEC50 | −6.0 ± 0.18 | −6.4 ± 0.17 | −6.3 ± 0.21 | −7.2 ± 0.26 |
| E max | 94 ± 10 | 92 ± 7.8 | 230 ± 25 | 160 ± 19 | |
| VUF11418 | LogEC50 | −5.9 ± 0.25 | −6.7 ± 0.28 | −5.7 ± 0.43 | −6.6 ± 0.23 |
| E max | 85 ± 13 | 52 ± 6.8 | 160 ± 43 | 160 ± 18 |
Table 2. Bias factors comparing signaling through different pathways.
Bias factors were calculated by the logarithm of ratios of relative intrinsic activities method (see Materials and Methods). Listed are the bias factors ± SEM from the fit parameters obtained from three to five independent experiments.
| CXCR3:β-arrestin2 | CXCR3:Gαi | cAMP inhibition | CXCR3:Gαi | cAMP inhibition | cAMP inhibition | |
|---|---|---|---|---|---|---|
| CXCL9 | N/A | N/A | N/A | 0.39 ± 0.87 | 0.89 ± 0.54 | 0.50 ± 0.80 |
| CXCL10 | N/A | N/A | N/A | 0.07 ± 0.76 | −0.52 ± 0.36 | −0.59 ± 0.77 |
| CXCL11 | 0 | 0 | 0 | 0 | 0 | 0 |
| VUF10661 | 0.22 ± 0.44 | 0.62 ± 0.61 | 0.55 ± 0.50 | 0.40 ± 0.55 | 0.33 ± 0.42 | −0.07 ± 0.60 |
| VUF11418 | 0.05 ± 0.52 | 1.2 ± 0.75 | 1.1 ± 0.51 | 1.2 ± 0.71 | 1.0 ± 0.46 | −0.15 ± 0.71 |
Biased agonists of CXCR3 induce differential conformational signatures in β-arrestin
To better characterize the effects of CXCR3 activation by biased agonists on β-arrestin2 conformations, we used a panel of previously described fluorescent arsenical hairpin (FlAsH) bioluminescence resonance energy transfer (BRET) conformational biosensors of β-arrestin2 (21). Each FlAsH BRET biosensor has a full-length Renilla luciferase (RLuc) at the N terminus of β-arrestin2, as well as an arsenical hairpin with a six–amino acid motif, CCPGCC, that binds arsenic at different positions (Fig. 4, A and B). The biosensors perform intramolecular BRET between the RLuc donor and the FlAsH acceptor to report on β-arrestin2 conformations. We modeled the positions of each FlAsH acceptor on the structures of inactive (22) and active (25) β-arrestin1 (Fig. 4, B and C; colored tags represent each FlAsH probe location).
Fig. 4. Biased agonists that induce Gαi:β-arrestin complex formation induce similar conformations of β-arrestin.
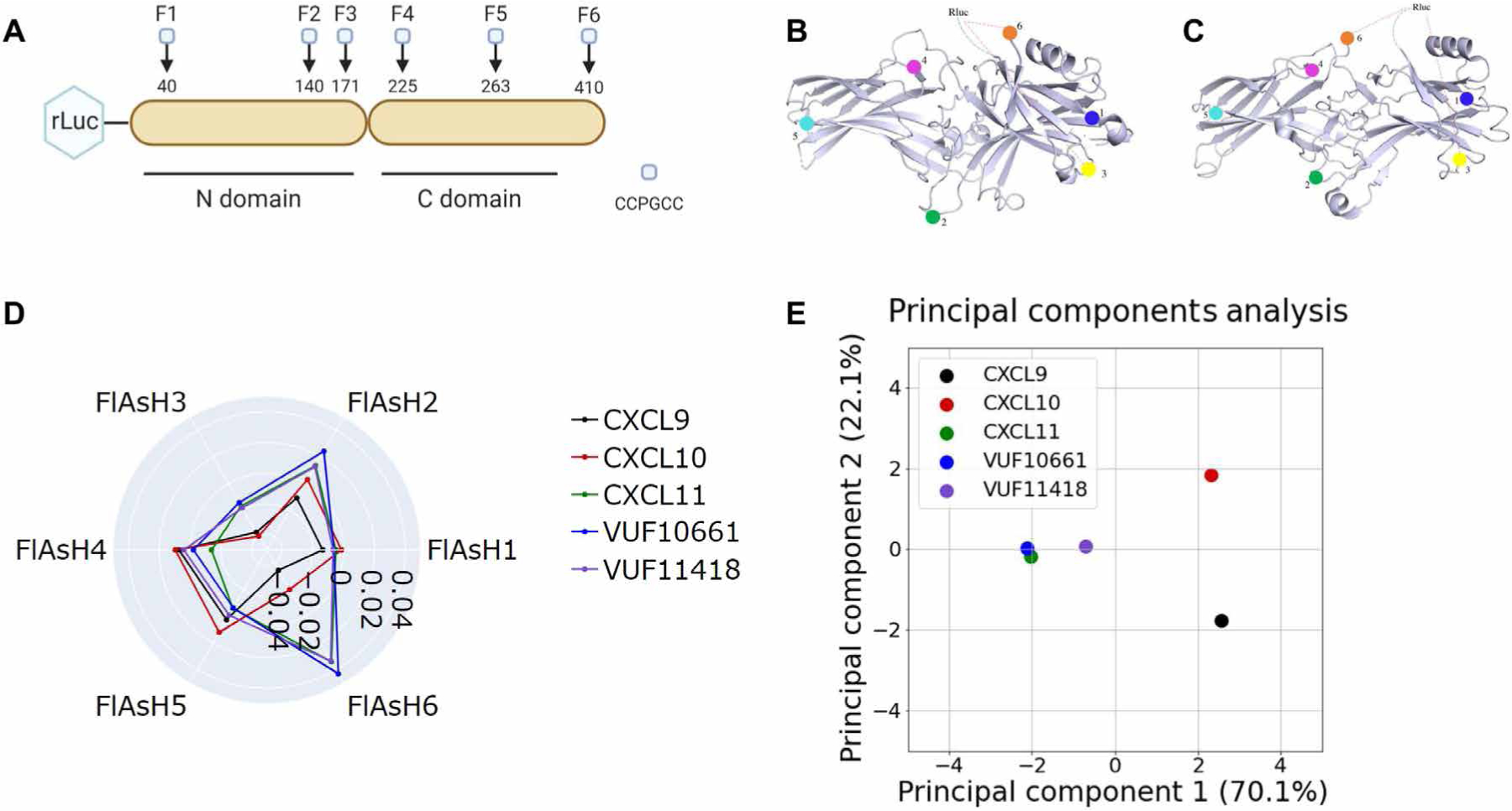
(A) The RLuc–β-arrestin2–FlAsH1 to RLuc–β-arrestin2–FlAsH6 reporters (F1 to F6) have the amino acid motif CCPGCC inserted after residues 40, 140, 171, 225, 263, and 410 of β-arrestin2. This motif acts as a dipole acceptor to assess conformational changes in β-arrestin2. (B and C) The positions of the FlAsH binding motifs are highlighted in the inactive (B) and active (C) structures of β-arrestin1. (D) HEK293N cells were transfected with plasmids encoding CXCR3 and one of the RLuc–β-arrestin2–FlAsH reporters (FlAsH1 to FlAsH6). The cells were then treated with 100 nM CXCL9, 100 nM CXCL10, 100 nM CXCL11, 1 μM VUF10661, 1 μM VUF11418, or vehicle. The radar plot depicts the intramolecular net BRET ratio calculated from subtracting the vehicle from treatment groups. (E) PCA was used to assess biased agonist–induced β-arrestin conformation similarities; computed principal components are visualized with the top two principal components. Principal component 1 contributes to 70% of the observed variation, and principal component 2 contributes to 22% of the observed variation. Points denote the composite response of a single ligand at varying doses. Data are means of three to five independent experiments.
For all FlAsH probes other than FlAsH2 and FlAsH3, statistically significant differences in net BRET were observed between ligands (Fig. 4D, and fig. S6, A to F). For FlAsH6, significant differences were observed between CXCL9 versus CXCL11, VUF10661, and VUF11418 (fig. S6F). For FlAsH4, significant differences were observed only between CXCL9 and CXCL10 versus CXCL11 (fig. S6D). For all FlAsH probes, no statistically significant differences were observed between CXCL11, VUF10661, and VUF11418 (fig. S6, A to F). The overall signal that we observed was more consistent with an active β-arrestin2 structure based on the change in FlAsH4 and FlAsH5 signal, although the FlAsH6 signal was more difficult to interpret because of the substantial flexibility of the N and C termini of β-arrestin2.
We then performed a PCA, with the intramolecular BRET signal of each ligand for all FlAsH probes as input components (Fig. 4E). PCA captured 92% of the total observed variance of the observed β-arrestin2 conformational signatures. The conformational changes of β-arrestin2 induced by CXCL11, VUF10661, and VUF11418 were extremely similar, as indicated by similar PC1 and PC2 values. In contrast, both CXCL9 and CXCL10 are grouped much further from the other three ligands. In short, ligands that promoted Gαi:β-arrestin complex formation also generated similar β-arrestin2 conformations with features that are associated with β-arrestin2 activation.
CXCL11, VUF10661, and VUF11418 promote the formation of Gαi:β-arrestin:CXCR3 complexes
We previously demonstrated that Gαi:β-arrestin scaffolds form selective complexes with the canonically Gαs-coupled receptors V2R and β2AR after stimulation with an endogenous agonist (12). However, it is unknown whether canonically Gαi-coupled receptors can form ternary complexes with Gαi:β-arrestin scaffolds. To address this, we used the three CXCR3 agonists that were observed to form the Gαi:β-arrestin complex, CXCL11, VUF10661, and VUF11418, in a “complex BRET” assay. Complex BRET is similar to other BRET-based strategies to assess complex formation (26, 27) and requires complementation of a low-affinity nanoBiT split luciferase system followed by energy transfer to a third protein tagged with a fluorescent protein acceptor, monomeric Kusabira Orange (mKO), generating a BRET response (Fig. 5A). Thus, this technique enables real-time assessment of interactions between a two-protein complex and a third protein in cells. Treatment with all three ligands that we showed can form Gαi:β-arrestin complexes (CXCL11, VUF10661, and VUF11418) promoted the formation of Gαi:β-arrestin:CXCR3 ternary complexes (Fig. 5B). Although VUF10661 and CXCL11 produced the strongest Gαi:β-arrestin signal, the rank order of Gαi:β-arrestin:CXCR3 Emax values changed such that VUF10661 and VUF11418 induced a greater net BRET ratio than that induced by CXCL11. Because the complex BRET signal depends both on distance and orientation, these results could indicate that these ligands generate distinct conformations of Gαi:β-arrestin complexes with CXCR3.
Fig. 5. Formation of a Gαi:β-arrestin:CXCR3 ternary complex.
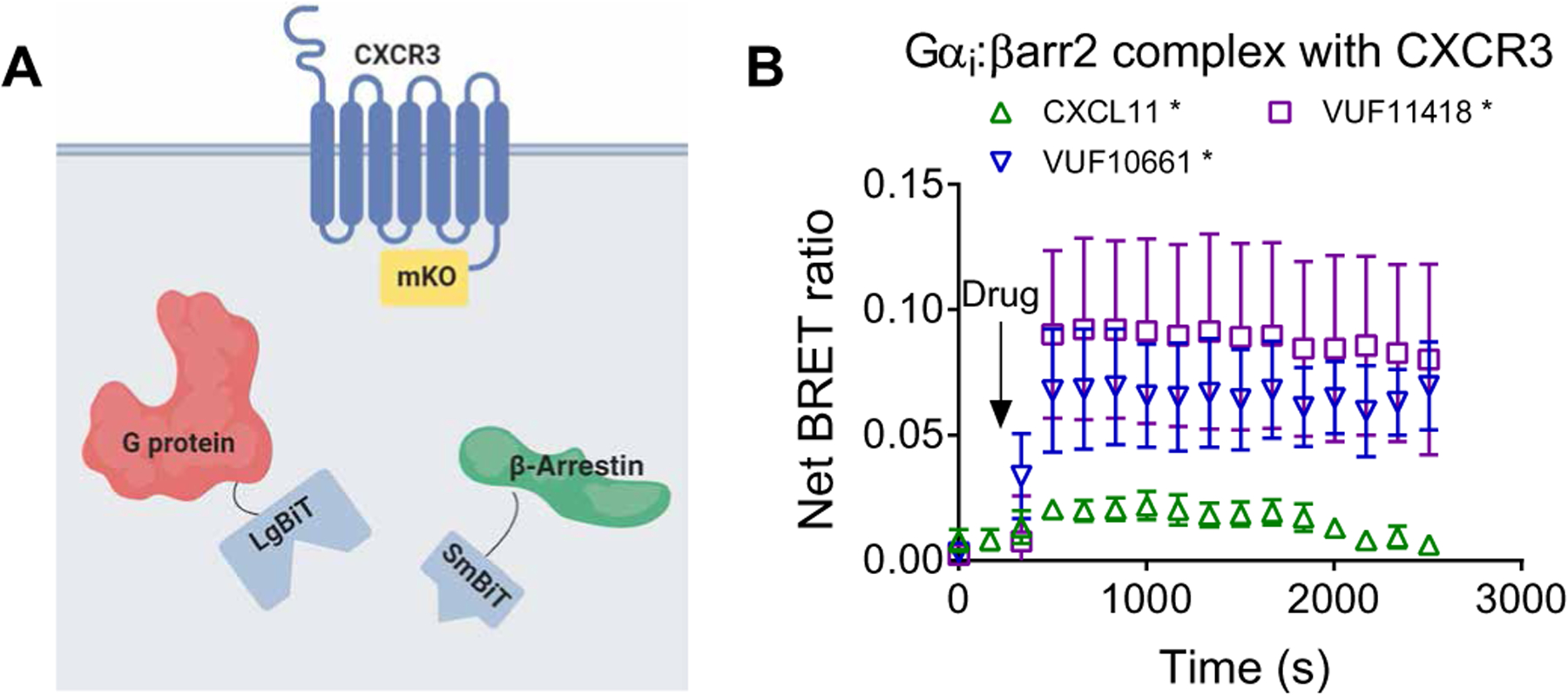
(A) Arrangement of the luciferase fragments and the monomeric Kusabira Orange (mKO) acceptor fluorophore for complex BRET on Gαi (LgBiT), β-arrestin2 (SmBiT), and CXCR3 (mKO). (B) HEK293T cells transiently transfected with plasmids encoding CXCR3-mKO, Gαi-LgBiT, and SmBiT–β-arrestin2 were stimulated with 100 nM CXCL11, 1 μM VUF10661, 1 μM VUF11418, or vehicle. Complex BRET ratios were calculated for the Gαi:β-arrestin:CXCR3 ternary complex after the indicated treatments. Data are means ± SEM of three to five independent experiments. *P < 0.05 by two-way ANOVA, with Dunnett’s post hoc analysis showing a significant difference relative to vehicle control.
CXCR3 does not promote β-arrestin–dependent ERK phosphorylation or the formation of Gαi:β-arrestin:ERK complexes
Different GPCRs stimulate ERK through different mechanisms involving G proteins and β-arrestins (6, 28, 29). We previously showed that agonist treatment of the V2R promotes the formation of Gαi:β-arrestin:ERK complexes that were associated with ERK phosphorylation (12). CXCR3 activation also increases ERK1/2 phosphorylation, with CXCL9, CXCL10, and CXCL11 known to activate ERK1/2 (14, 30). Given that all three ligands activate ERK, but only CXCL11 induced the formation of Gαi:β-arrestin complexes, this suggested that Gαi:β-arrestin complex formation was not necessary for the activation of ERK1/2 downstream of CXCR3.
We proceeded to test the ability of CXCL11, as well as VUF10661, to form Gαi:β-arrestin:ERK complexes (Fig. 6A). Unlike the V2R, agonist treatment of CXCR3 did not induce the formation of Gαi:β-arrestin:ERK complexes (Fig. 6B). We then investigated the requirement of β-arrestin2 for CXCL11-induced activation of ERK in experiments with two different HEK293 cell lines in which both β-arrestin1 and β-arrestin2 were knocked out by CRISPR-Cas9 (31, 32), which we refer to as βarr1/2-CRISPR knockout (KO)-“A” and KO-“B” cells. In addition, we performed small interfering RNA (siRNA)–mediated knockdown of β-arrestin2 in the two different parental cell lines from which the CRISPR KO cells were derived, which we refer to as HEK293-A and HEK293-B cells. Unlike the V2R, for which either CRISPR-Cas9–mediated knockout or siRNA-mediated knockdown of β-arrestin significantly reduced ERK phosphorylation (6), knockout or knockdown of β-arrestin did not reduce CXCR3-dependent ERK phosphorylation (Fig. 6, C to J). Rather, knockdown or knockout of β-arrestin resulted in inconsistent effects on ERK phosphorylation, with significantly increased phosphorylation observed in two of the four conditions tested (Fig. 6, D and J). In addition, we confirmed siRNA-mediated β-arrestin2 knockdown, CRISPR-Cas9–mediated β-arrestin1/2 knockout, and CXCR3 surface expression on the tested cell lines (fig. S7, A to E). Together, these findings further support findings that ERK activation is a complex process that is regulated by both G proteins and β-arrestins but does not universally depend on Gαi:β-arrestin:ERK complex formation.
Fig. 6. β-Arrestin is not necessary for CXCR3-dependent ERK activation, and no Gαi:β-arrestin:ERK complex is observed.
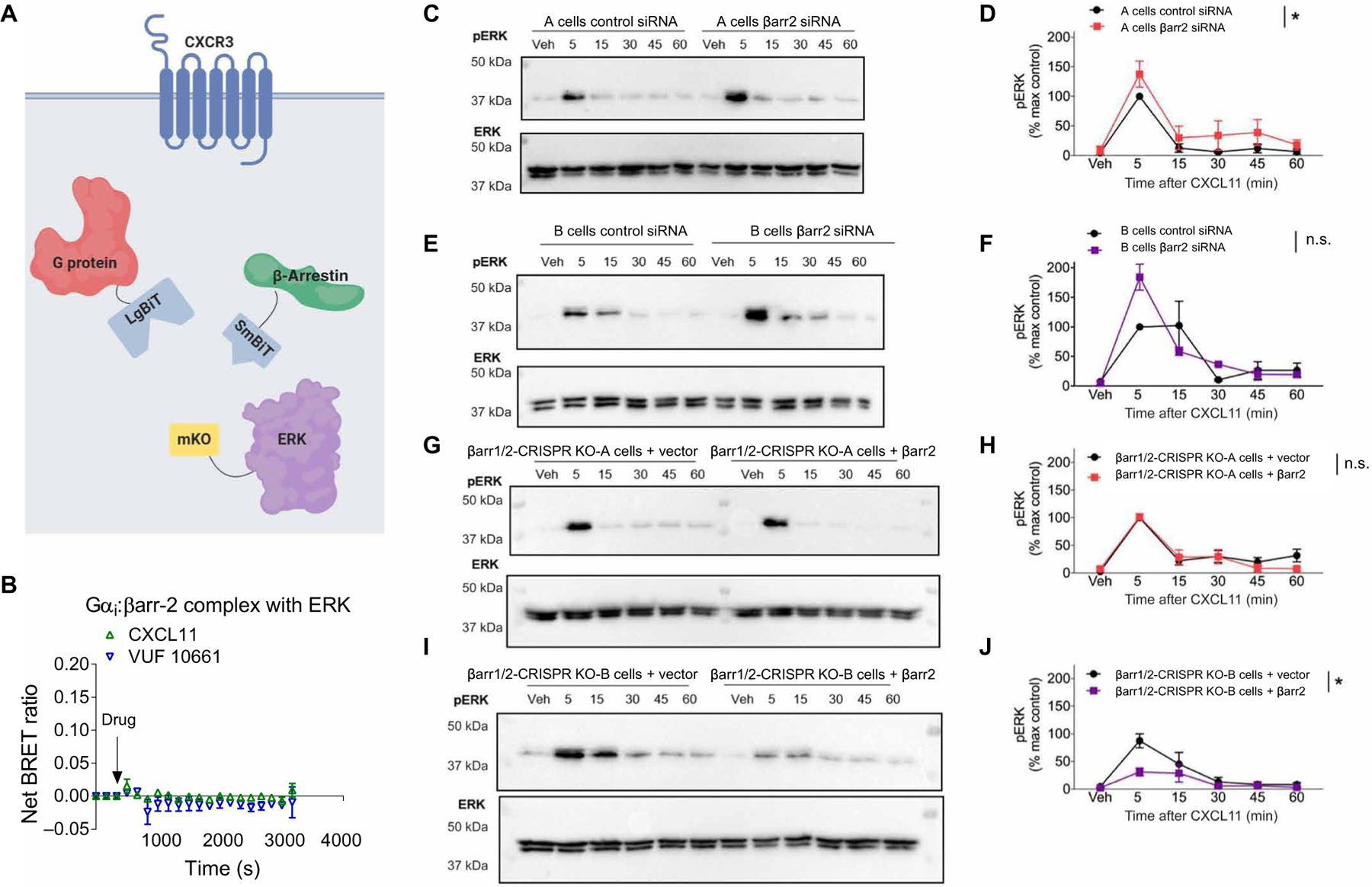
(A) Arrangement of the luciferase fragments and mKO acceptor fluorophore for complex BRET on Gαi (LgBiT), β-arrestin2 (SmBiT), and ERK2 (mKO). (B) Complex BRET ratio for Gαi:β-arrestin2:ERK after treatment with 100 nM CXCL11, 1 μM VUF10661, or vehicle. Data were normalized to both vehicle treatment and cytosolic mKO. (C to G) Western blotting analysis of the time course of ERK phosphorylation in A and B parental and β-arrestin1/2 CRISPR KO HEK293 cells stimulated with 100 nM CXCL11. (C) Western blotting analysis of phospho-ERK (pERK) and (D) its quantification in A parental cells transfected with control siRNA or β-arrestin2–specific siRNA. (E) Western blotting analysis of phospho-ERK and (F) its quantification in B parental cells transfected with control siRNA or β-arrestin2–specific siRNA. (G) Western blotting analysis of phospho-ERK and (H) its quantification in A β-arrestin1/2 CRISPR KO cells transfected with the control or β-arrestin2 rescue plasmid. (I) Western blotting analysis of phospho-ERK and (J) its quantification in B β-arrestin1/2 CRISPR KO cells transfected with the control or β-arrestin2 rescue plasmid. *P < 0.05 by two-way ANOVA to determine the main effect of either the siRNA or rescue. Data are from three experiments per condition. n.s., not significant.
CXCR3 selectively promotes the formation of Gαi:β-arrestin:AP-2 complexes
Because we did not observe regulation of ERK by CXCR3 through Gαi:β-arrestin complexes, this raised the question as to whether these complexes could interact with other potential effectors in the context of CXCR3 stimulation. We tested whether Gαi:β-arrestin formed a complex with the clathrin adapter AP-2, a known binding partner of β-arrestins (33). We found that CXCL11 promoted the formation of a ternary complex between Gαi:β-arrestin and AP-2 after CXCR3 stimulation (Fig. 7A). These results were consistent when controlled for by either a cytosolic monomeric Kusabira orange (mKO) control (Fig. 7, A and B) or an mKO-CAAX control (fig. S8). In addition, we performed confocal microscopy to visualize the localization of Gαi, β-arrestin, and AP-2. Colocalization of all three proteins occurred after stimulation of the cells with CXCL11 (Fig. 7C). These data suggest that, similar to the role of β-arrestins in regulating GPCR signaling, Gαi:β-arrestin complexes can differentially form ternary complexes with signaling partners such as ERK and AP-2 in a context-dependent manner (Fig. 7D).
Fig. 7. Formation of a Gαi:β-arrestin:AP-2 ternary complex.
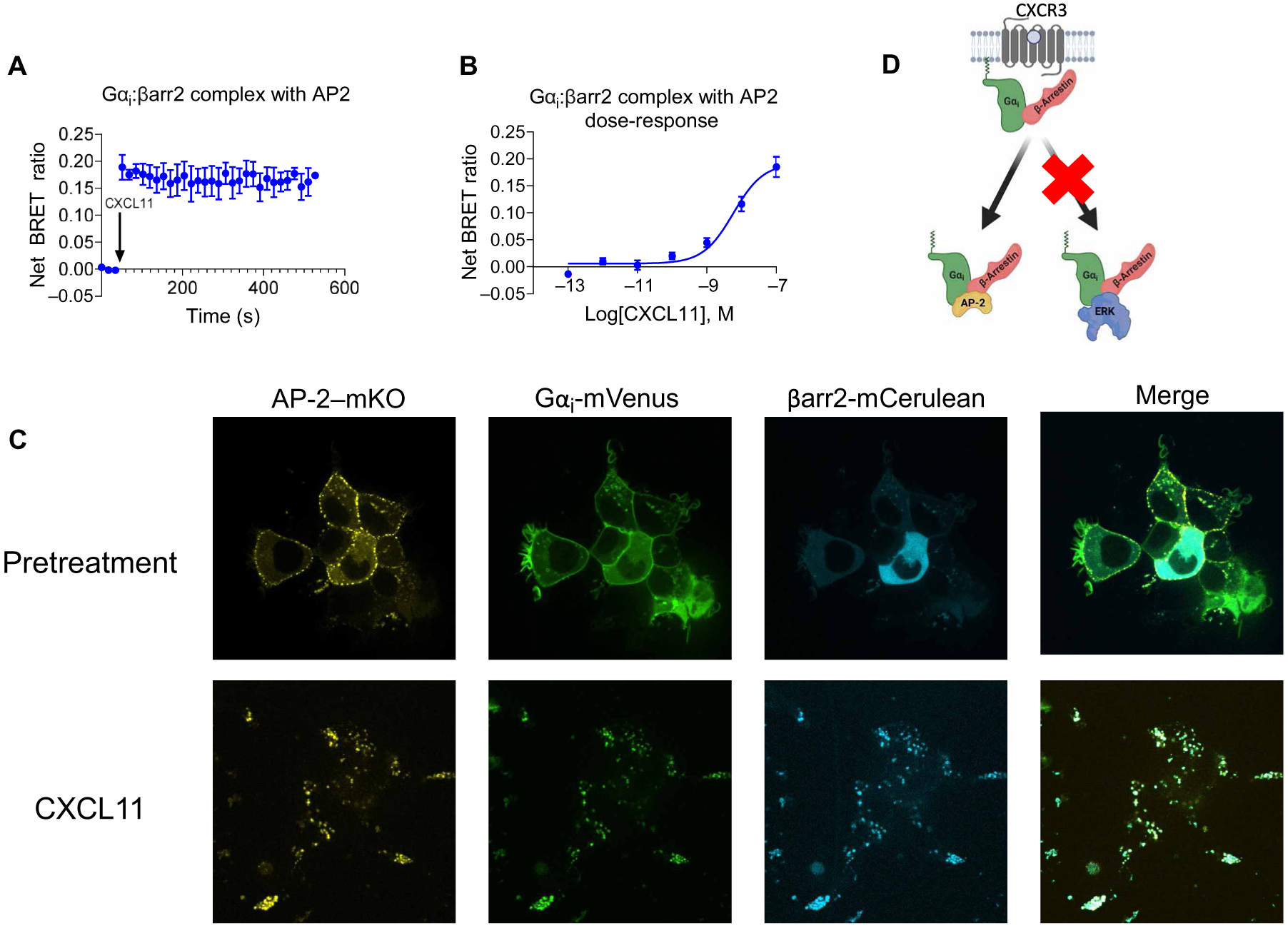
(A to D) Transfected HEK293T cells expressing CXCR3, smBiT–β-arrestin2, Gαi-LgBiT, and AP-2-mKO or cytosolic mKO were treated with vehicle or the indicated concentrations of CXCL11. (A) Complex BRET ratio for Gαi:β-arrestin2:AP-2 after treatment with 100 nM CXCL11 or vehicle. (B) Transfected HEK293T cells were treated with vehicle or the indicated concentrations of CXCL11. Net BRET was measured 6 min after treatment. (C) Confocal microscopy analysis of CXCL11-induced complexes of Gαi:β-arrestin:AP-2 in HEK293T cells transfected with mVenus-tagged Gαi, mKO-tagged AP-2, and mCerulean-tagged β-arrestin2. Cells were imaged before (basal) and 20 min after treatment. Data are representative of three experiments per condition. For BRET experiments, data were normalized to both vehicle treatment and cytosolic mKO transfection conditions. (D) Scheme demonstrating the selective formation of Gαi:β-arrestin:AP-2 rather than Gαi:β-arrestin:ERK in response to the activation of CXCR3 by CXCL11.
DISCUSSION
Our results demonstrate that biased agonists of CXCR3 differentially promote the formation of Gαi:β-arrestin complexes. We also showed that CXCR3 agonists have pluridimensional efficacy through canonical G protein and β-arrestin pathways, as well as Gαi:β-arrestin complex formation. All five CXCR3 agonists inhibited cAMP synthesis and recruited Gαi and β-arrestin2, albeit to different degrees. However, only three of the five agonists tested (CXCL11, VUF10661, and VUF11418) stimulated Gαi:β-arrestin complex formation, consistent with Gαi:β-arrestin complex formation being a distinct GPCR signaling pathway.
Using bias plots together with traditional Emax and EC50 calculations, we showed that five CXCR3 agonists have distinct signaling profiles and that activity at one signaling pathway does not necessarily predict activity at another pathway. However, we observed some pathways that had a high degree of correlation, for example, a strong positive correlation between the inhibition of cAMP synthesis and G protein recruitment, most easily visualized within bias plots (Fig. 3). When comparing these two processes, we observed (at nonsaturating concentrations of agonist) more inhibition of cAMP synthesis relative to G protein recruitment. This is likely due to differences in amplification between these assays, consistent with decades of work demonstrating hormone amplification, for example, GPCRs catalyze guanine nucleotide exchange at G proteins, which can, in turn, catalyze second messenger signaling, such as cAMP (34, 35).
In comparing the inhibition of cAMP synthesis to β-arrestin recruitment, we found that CXCL11 demonstrated bias toward β-arrestin recruitment relative to the two synthetic compounds, VUF10661 and VUF11418. VUF11418 appeared to be G protein biased relative to VUF10661, consistent with previous findings with different methods (17), but with additional nuances of signaling pathway potency and efficacy now appreciated. CXCL11 appeared to be biased toward Gαi:β-arrestin complex formation relative to inhibition of cAMP synthesis when compared to the two small-molecule CXCR3 agonists. As might be expected with a positive correlation between Gαi recruitment and inhibition of cAMP synthesis, CXCL11 was also biased toward Gαi:β-arrestin complex formation relative to G protein recruitment. In comparison to VUF10661, CXCL11 generated Gαi:β-arrestin complex formation to a similar extent but exhibited much less G protein recruitment. To reduce the complexity of these signaling profiles for each ligand, we dissected these results by PCA, which enabled us to demonstrate that the signaling of CXCL9 and CXCL10 were similar to one another and that VUF10661 and VUF11418 were similar to one other, whereas CXCL11 had its own signaling profile. Note that VUF10661 and VUF11418 differentially regulate chemotaxis and inflammation, suggesting that there is substantial granularity of signaling within these clusters (17). Together, these findings illuminate signaling granularity within the activity of CXCR3 agonists. Our findings suggest that detailed signaling analyses of multiple pathways are likely necessary to design a drug capable of targeting CXCR3 with a desired signaling profile.
Through a panel of established β-arrestin biosensors (21), we correlated signaling activity with conformational changes in β-arrestin. Consistent with bias calculations and pathway activity, CXCL9 and CXCL10 displayed different β-arrestin conformational patterns relative to those of CXCL11, VUF10661, and VUF11418. These data further support the idea that distinct conformational signatures are adopted by β-arrestin when CXCR3 is stimulated by biased agonists, consistent with previous reports about other GPCRs (21, 36–38). These agonist-induced conformational changes in β-arrestin2 that were reported on by the FlAsH biosensors may reflect the specific β-arrestin2 conformations needed to form Gαi:β-arrestin complexes. Conformational differences in β-arrestin may thus explain, in part, the inability of CXCL9 and CXCL10 to induce the formation of Gαi:β-arrestin complexes; however, delineating finer structural details will be necessary to test this hypothesis. Such conformational changes in β-arrestin2 likely represent a receptor-unbound conformation (39, 40) and may also correlate with spatially distinct pools of GPCR signaling (41, 42).
PTX inactivates canonical Gαi signaling, enabling the dissection of certain G protein and β-arrestin contributions to signaling events. Whereas PTX had no effect on CXCR3-mediated β-arrestin2 recruitment, it reduced Gαi recruitment and altered the Gαi:β-arrestin association. This suggests that both G protein recruitment and Gαi:β-arrestin association may be partially governed by biochemical mechanisms that are disrupted by PTX. Such mechanisms could include the disruption of a critical interaction between the α5 helix of Gαi and CXCR3. Interactions between the G protein α5 helix and the receptor core are thought to be conserved across GPCRs and are critical for disrupting key G protein:GDP contacts and initiating canonical G protein signaling (43–45). However, attenuation of signal and lack of elimination of Gαi:β-arrestin complex formation by pretreatment with PTX suggests an alternative interaction site beyond the α5 helix, because PTX might be altering the conformational state of Gαi.
Unlike with the V2R (12), we did not observe the formation of Gαi:β-arrestin:ERK ternary complexes with CXCR3. However, our observations are consistent with previously described CXCR3 signaling, whereby CXCL9, CXCL10, and CXCL11 activate ERK1/2 (14, 30). Given that the binding of CXCL9 and CXCL10 to CXCR3 do not result in the formation of Gαi:β-arrestin complexes but still resulted in ERK phosphorylation, it is likely that CXCR3 signals through ERK through a different mechanism than that used by the V2R. Rather, we found that Gαi:β-arrestin formed a ternary complex with AP-2, consistent with a role in promoting receptor endocytosis. This is consistent with context-dependent Gαi:β-arrestin signaling. Further work with a panel of receptors will be necessary to establish whether ligand-generated Gαi:β-arrestin complexes with ERK are a common mechanism of ERK activation, and whether established receptor properties correlate with the ability to form Gαi:β-arrestin:ERK complexes.
Our approach has several limitations. In particular, our approach uses modified proteins in the setting of heterologous overexpression, which may not fully represent physiological signaling patterns. In addition, our inclusion of bias factors is highly limited by the inability of several of our tested compounds of reaching a saturating concentration due to the risk of nonspecific effects and cellular toxicity at higher doses. Bias factor calculation is highly reliant on fit parameters with propagation of errors, which are particularly sensitive to reaching a saturating response for accurate calculation. This limitation does not apply to bias plots, which demonstrated bias between agonists across multiple signaling axes.
In summary, our findings further demonstrate that Gαi:β-arrestin complexes are a distinct GPCR signaling pathway. We showed that biased agonists differentially formed Gαi:β-arrestin complexes and that established biased ligands have greater signaling granularity than was previously appreciated. Gαi:β-arrestin complexes scaffolded with CXCR3 and AP-2 but not with ERK. Further efforts to understand and incorporate Gαi:β-arrestin signaling complexes into biased agonist development may aid in the discovery of novel pharmacologic therapies capable of differentially targeting these pathways.
MATERIALS AND METHODS
Cell culture and transfection
HEK293T cells were maintained in minimum essential medium (MEM) supplemented with 1% penicillin/streptomycin and 10% fetal bovine serum (FBS). Cells were grown at 37°C within a humidified atmosphere of 5% CO2. For BRET and luminescence studies, HEK293T cells were transiently transfected through an optimized calcium phosphate protocol as previously described (14).
Split luciferase and complex BRET assays
HEK293T cells seeded in six-well plates were cotransfected with plasmids encoding smBiT-, LgBiT-, and mKO-tagged components as previously described (12). Twenty-four hours later, the cells were plated onto clear bottom, white-walled 96-well plates at 50,000 to 100,000 cells per well in BRET medium [clear MEM (Gibco) supplemented with 2% FBS, 10 mM Hepes, 1× GlutaMAX, and 1% penicillin/streptomycin/amphotericin B (Gibco)]. Select cells were then treated overnight with PTX at a final concentration of 200 ng/ml. For luminescence split luciferase studies, plates were read with a BioTek Synergy Neo2 plate reader set at 37°C with a 485-nm emission filter. Cells were stimulated with either vehicle [Hanks’ balanced salt solution (HBSS) with 20 mM Hepes] or the appropriate concentration of agonist. For split luciferase luminescence experiments, plates were read both before and after ligand treatment to calculate the Δnet change in luminescence and were subsequently normalized to vehicle treatment. For complex BRET experiments, plates were read on a Berthold Mithras LB 940 with prewarmed medium at 37°C with a standard RLuc emissions filter (480 nm) with a custom mKO 542-nm long-pass emission filter (Chroma Technology Co.) as previously described (12).
FlAsH Intramolecular BRET
Intramolecular BRET at β-arrestin2 conformational biosensors was measured on the basis of a modified version of the steps and procedures previously described (21). Briefly, HEK293N cells were seeded in six-well plates at a cell density of 5 × 105 cells per well and transfected with plasmids encoding CXCR3 (2000 ng) and one of the six FlAsH constructs (200 ng). Clear bottom, white-walled 96-well plates were prepared by coating them with rat collagen. Twenty-four hours after transfection, cells were plated onto these coated 96-well plates at 50,000 to 100,000 cells per well in MEM supplemented with 1% antibiotic-antimycotic and 10% FBS. In preparation of being read, cells were treated with the biarsenical labeling reagent FlAsH-EDT2 for 45 min, washed with British anti-Lewisite (BAL) wash buffer, and suspended in HBSS with 20 mM Hepes. Cells were treated with either vehicle (HBSS with 20 mM Hepes) or the appropriate concentration of agonist. Immediately before reading, cells were treated with coelenterazine and read on a Berthold Mithras LB 940 using a prewarmed instrument at 37°C with a standard RLuc emissions filter (480 nm) with a custom enhanced yellow fluorescent protein filter (530 nm).
Inhibition of cAMP synthesis
GloSensor cAMP inhibition was conducted similar to that previously described (14). GloSensor biosensor (Promega) uses a modified form of firefly luciferase containing a cAMP-binding motif. Upon cAMP binding, a conformational change leads to enzyme complementation, and incubation with a luciferase substrate results in a luminescence readout. Analysis of cAMP accumulation was performed in HEK293 cells transiently transfected with the GloSensor construct and plasmid encoding human CXCR3. Cells were seeded in 96-well white, clear-bottomed plates at 80,000 cells per well in MEM supplemented with 1% penicillin/streptomycin and 10% FBS. The next day, the GloSensor reagent [4% (v/v); Promega] was incubated at room temperature for 2 hours. Cells were then stimulated with a range of CXCR3 agonists for 5 min, and increases in luminescence were read on a BioTek Synergy Neo2 plate reader set at 37°C with a 485-nm emission filter.
Bias factor calculations
Concentration-response data were fit with a three-parameter dose-response model with Emax, EC50, and baseline in Prism 9.1 (GraphPad). Bias factors were calculated from the Emax and EC50 values as previously performed (46). Briefly, a bias factor, which quantifies the relative stabilization of one signaling state over another compared to the reference agonist (CXCL11), was calculated as
SEM values for these bias factors were calculated by error propagation of the errors from the fits for the Emax and EC50.
Structural protein representations
Schematic representations of inactive [Protein Data Bank (PDB) code: 1G4M] and NTSR1-bound (PDB code: 6PWC) β-arrestin1 were derived from crystal structures of inactive and active β-arrestin. A sequence alignment of β-arrestin1 and β-arrestin2 was used to transpose FlAsH reporter sequence location from β-arrestin2 to β-arrestin1.
Western blotting analysis of ERK phosphorylation
We used HEK293 cell lines in which β-arrestin1 and β-arrestin2 had knocked out by CRISPR-Cas9 in the laboratories of A.I. (31) and Laporte (32), which we refer to as βarr1/2-CRISPR KO-A and βarr1/2-CRISPR KO-B cells, respectively. In addition, we performed siRNA-mediated knockdown of β-arrestin2 in the parental cells from which the CRISPR KO cells were derived, which we refer to as HEK293-A and HEK293-B cells. Parental and β-arrestin1/2 knockout HEK293 cell lines were previously generated and validated (6). Reconstitution of β-arrestin1/2 in CRISPR-Cas9 HEK293 cells by transient transfection was conducted as previously described (6, 14). β-arrestin1/2 knockdown in parental HEK293 cells by siRNA was performed with Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s specifications, similar to that previously described (6). HEK293-A parental and CRISPR KO cells were maintained in MEM supplemented with 10% FBS and 1% penicillin/streptomycin or gentamicin (20 μg/ml). HEK293-B parental and CRISPR KO cells were maintained in Dulbecco’s modified Eagle’s medium without pyruvate supplemented with 10% FBS and 1% penicillin/streptomycin or gentamicin (20 μg/ml). Antibodies specific for β-arrestin1, β-arrestin2, phosphorylated ERK1/2 (Cell Signaling Technology), and total ERK (Millipore) were used to detect β-arrestin1 and analyze ERK activation (phosphorylation) as previously described (14).
Confocal microscopy
HEK293T cells plated in rat tail collagen-coated, 35-mm, glass-bottomed dishes (MatTek Corp., no. P35G-0–14-C) were transiently transfected with polyethylenimine with plasmids encoding AP-2–mKO, Gαi-mVenus, and β-arrestin2–mCerulean. Forty-eight hours later, the cells were serum-starved for 2 hours before being stimulated with 100 nM CXCL11 for 20 min. Samples were imaged with a Zeiss CSU-X1 spinning disk confocal microscope.
Drugs
CXCL9, CXCL10, and CXCL11 were prepared and stored according to the manufacturer’s specifications (PeproTech) with 0.1% bovine serum albumin used as a carrier protein. VUF10661 (Sigma-Aldrich) and VUF11418 (Aobious) were dissolved in dimethyl sulfoxide to make stock solutions and stored in a desiccator cabinet. All drug dilutions were performed with BRET medium or cell culture medium. PTX was obtained from List Biological Laboratories. All compound stocks were stored at −20°C until use.
Statistical analysis
Dose-response curves were fitted to a log agonist versus stimulus with three parameters (span, baseline, and EC50) after baseline correction with Prism 9.1 (GraphPad). Statistical tests were performed using a one- or two-way analysis of variance (ANOVA). Further details of statistical analysis and replicates are included in the figure legends. Lines represent the mean, and error bars signify the SEM, unless otherwise noted.
Supplementary Material
Acknowledgments:
We thank N. Nazo for administrative assistance and C. Lee for helpful discussion and thoughtful feedback. We thank S. Laporte for the β-arrestin CRISPR KO cells. Diagrams were created with Biorender.com.
Funding:
This work was supported by T32GM7171 (to J.S.S.); the Duke Medical Scientist Training Program (to J.S.S. and D.S.E.); F31DA041160 (to T.F.P.); the Japan Agency for Medical Research and Development 19gm5910013 (to A.I.), JP19gm0010004 (to A.I.), and JP20am0101095 (to A.I.); the Japan Society for the Promotion of Science 21H04791 (to A.I.); Japan Science and Technology Agency JPMJMS2023 (to A.I.); The Uehara Memorial Foundation (to A.I.), 17K08264R37MH073853 (to M.G.C.), 1R01GM122798-01A1 (to S.R.), K08HL114643-01A1 (to S.R.); and the Burroughs Wellcome Career Award for Medical Scientists (to S.R.).
Footnotes
Competing interests: J.S.S., T.P., and S.R. are inventors on patent application no. 16/876,934 submitted by Duke University that covers Complex BRET Technique for Measuring Biological Interactions. All other authors declare that they have no competing interests.
View the article online:
Data and materials availability:
All data associated with this study are available in the main text or the Supplementary Materials. Constructs for CXCR3, CXCR3-LgBiT, smBiT–β-arrestin2, β-arrestin2–mKO, ERK-mKO, AP-2–mKO, and β-arrestin2–mCerulean are available from S.R. under a materials transfer agreement with Duke University.
REFERENCES AND NOTES
- 1.Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al-Lazikani B, Hersey A, Oprea TI, Overington JP, A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov 16, 19–34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hamm HE, The many faces of G protein signaling. J. Biol. Chem 273, 669–672 (1998). [DOI] [PubMed] [Google Scholar]
- 3.Smith JS, Rajagopal S, The β-arrestins: Multifunctional regulators of G protein-coupled receptors. J. Biol. Chem 291, 8969–8977 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hübner H, Huang X-P, Sassano MF, Giguère PM, Löber S, Duan D, Scherrer G, Kobilka BK, Gmeiner P, Roth BL, Shoichet BK, Structure-based discovery of opioid analgesics with reduced side effects. Nature 537, 185–190 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cahill TJ, Thomsen ARB, Tarrasch JT, Plouffe B, Nguyen AH, Yang F, Huang L-Y, Kahsai AW, Bassoni DL, Gavino BJ, Lamerdin JE, Triest S, Shukla AK, Berger B, Little J, Antar A, Blanc A, Qu C-X, Chen X, Kawakami K, Inoue A, Aoki J, Steyaert J, Sun J-P, Bouvier M, Skiniotis G, Lefkowitz RJ, Distinct conformations of GPCR-β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc. Natl. Acad. Sci. U.S.A 114, 2562–2567 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luttrell LM, Wang J, Plouffe B, Smith JS, Yamani L, Kaur S, Jean-Charles P-Y, Gauthier C, Lee M-H, Pani B, Kim J, Ahn S, Rajagopal S, Reiter E, Bouvier M, Shenoy SK, Laporte SA, Rockman HA, Lefkowitz RJ, Manifold roles of β-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci. Signal 11, eaat7650 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith JS, Lefkowitz RJ, Rajagopal S, Biased signalling: From simple switches to allosteric microprocessors. Nat. Rev. Drug Discov 17, 243–260 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Picard L-P, Schonegge A-M, Bouvier M, Structural insight into G protein-coupled receptor signaling efficacy and bias between Gs and β-arrestin. ACS Pharmacol. Transl. Sci 2, 148–154 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zidar DA, Violin JD, Whalen EJ, Lefkowitz RJ, Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc. Natl. Acad. Sci. U.S.A 106, 9649–9654 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB, Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther 320, 1–13 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Kenakin T, Christopoulos A, Signalling bias in new drug discovery: Detection, quantification and therapeutic impact. Nat. Rev. Drug Discov 12, 205–216 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Smith JS, Pack TF, Inoue A, Lee C, Zheng K, Choi I, Eiger DS, Warman A, Xiong X, Ma Z, Viswanathan G, Levitan IM, Rochelle LK, Staus DP, Snyder JC, Kahsai AW, Caron MG, Rajagopal S, Noncanonical scaffolding of Gαi and β-arrestin by G protein–coupled receptors. Science 371, eaay1833 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rajagopal S, Bassoni DL, Campbell JJ, Gerard NP, Gerard C, Wehrman TS, Biased agonism as a mechanism for differential signaling by chemokine receptors. J. Biol. Chem 288, 35039–35048 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith JS, Alagesan P, Desai NK, Pack TF, Wu J-H, Inoue A, Freedman NJ, Rajagopal S, C-X-C motif chemokine receptor 3 splice variants differentially activate beta-arrestins to regulate downstream signaling pathways. Mol. Pharmacol 92, 136–150 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scholten DJ, Canals M, Wijtmans M, de Munnik S, Nguyen P, Verzijl D, de Esch IJP, Vischer HF, Smit MJ, Leurs R, Pharmacological characterization of a small-molecule agonist for the chemokine receptor CXCR3. Br. J. Pharmacol 166, 898–911 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wijtmans M, Scholten DJ, Roumen L, Canals M, Custers H, Glas M, Vreeker MCA, de Kanter FJJ, de Graaf C, Smit MJ, de Esch IJP, Leurs R, Chemical subtleties in small-molecule modulation of peptide receptor function: The case of CXCR3 biaryl-type ligands. J. Med. Chem 55, 10572–10583 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Smith JS, Nicholson LT, Suwanpradid J, Glenn RA, Knape NM, Alagesan P, Gundry JN, Wehrman TS, Atwater AR, Gunn MD, MacLeod AS, Rajagopal S, Biased agonists of the chemokine receptor CXCR3 differentially control chemotaxis and inflammation. Sci. Signal 11, eaaq1075 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inoue A, Raimondi F, Kadji FMN, Singh G, Kishi T, Uwamizu A, Ono Y, Shinjo Y, Ishida S, Arang N, Kawakami K, Gutkind JS, Aoki J, Russell RB, Illuminating G-protein-coupling selectivity of GPCRs. Cell 177, 1933–1947.e25 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Onaran HO, Rajagopal S, Costa T, What is biased efficacy? Defining the relationship between intrinsic efficacy and free energy coupling. Trends Pharmacol. Sci 35, 639–647 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Onaran HO, Ambrosio C, Uğur Ö, Madaras Koncz E, Grò MC, Vezzi V, Rajagopal S, Costa T, Systematic errors in detecting biased agonism: Analysis of current methods and development of a new model-free approach. Sci. Rep 7, 44247 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee M-H, Appleton KM, Strungs EG, Kwon JY, Morinelli TA, Peterson YK, Laporte SA, Luttrell LM, The conformational signature of β-arrestin2 predicts its trafficking and signalling functions. Nature 531, 665–668 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C, Crystal structure of beta-arrestin at 1.9 A: Possible mechanism of receptor binding and membrane translocation. Structure 9, 869–880 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Chung KY, Rasmussen SGF, Liu T, Li S, DeVree BT, Chae PS, Calinski D, Kobilka BK, Woods VL, Sunahara RK, Conformational changes in the G protein Gs induced by the β2 adrenergic receptor. Nature 477, 611–615 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wan Q, Okashah N, Inoue A, Nehmé R, Carpenter B, Tate CG, Lambert NA, Mini G protein probes for active G protein-coupled receptors (GPCRs) in live cells. J. Biol. Chem 293, 7466–7473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yin W, Li Z, Jin M, Yin Y-L, de Waal PW, Pal K, Yin Y, Gao X, He Y, Gao J, Wang X, Zhang Y, Zhou H, Melcher K, Jiang Y, Cong Y, Edward Zhou X, Yu X, Eric Xu H, A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res. 29, 971–983 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Urizar E, Yano H, Kolster R, Galés C, Lambert N, Javitch JA, CODA-RET reveals functional selectivity as a result of GPCR heteromerization. Nat. Chem. Biol 7, 624–630 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cotnoir-White D, El Ezzy M, Boulay P-L, Rozendaal M, Bouvier M, Gagnon E, Mader S, Monitoring ligand-dependent assembly of receptor ternary complexes in live cells by BRETFect. Proc. Natl. Acad. Sci. U.S.A 115, E2653–E2662 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hawes BE, van Biesen T, Koch WJ, Luttrell LM, Lefkowitz RJ, Distinct pathways of Gi- and Gq-mediated mitogen-activated protein kinase activation. J. Biol. Chem 270, 17148–17153 (1995). [DOI] [PubMed] [Google Scholar]
- 29.Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, Eckhardt AE, Cowan CL, Spurney RF, Luttrell LM, Lefkowitz RJ, Distinct β-Arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J. Biol. Chem 281, 10856–10864 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Berchiche YA, Sakmar TP, CXC chemokine receptor 3 alternative splice variants selectively activate different signaling pathways. Mol. Pharmacol 90, 483–495 (2016). [DOI] [PubMed] [Google Scholar]
- 31.O’Hayre M, Eichel K, Avino S, Zhao X, Steffen DJ, Feng X, Kawakami K, Aoki J, Messer K, Sunahara R, Inoue A, von Zastrow M, Gutkind JS, Genetic evidence that β-arrestins are dispensable for the initiation of β2-adrenergic receptor signaling to ERK. Sci. Signal 10, eaal3395 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Namkung Y, Le Gouill C, Lukashova V, Kobayashi H, Hogue M, Khoury E, Song M, Bouvier M, Laporte SA, Monitoring G protein-coupled receptor and β-arrestin trafficking in live cells using enhanced bystander BRET. Nat. Commun 7, 12178 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SS, Caron MG, Barak LS, The β2-adrenergic receptor/βarrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc. Natl. Acad. Sci. U.S.A 96, 3712–3717 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pedersen SE, Ross EM, Functional reconstitution of beta-adrenergic receptors and the stimulatory GTP-binding protein of adenylate cyclase. Proc. Natl. Acad. Sci. U.S.A 79, 7228–7232 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gilman AG, The Albert Lasker Medical Awards. G proteins and regulation of adenylyl cyclase. JAMA 262, 1819–1825 (1989). [PubMed] [Google Scholar]
- 36.Shukla AK, Violin JD, Whalen EJ, Gesty-Palmer D, Shenoy SK, Lefkowitz RJ, Distinct conformational changes in beta-arrestin report biased agonism at seven-transmembrane receptors. Proc. Natl. Acad. Sci. U.S.A 105, 9988–9993 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zimmerman B, Beautrait A, Aguila B, Charles R, Escher E, Claing A, Bouvier M, Laporte SA, Differential β-arrestin-dependent conformational signaling and cellular responses revealed by angiotensin analogs. Sci. Signal 5, ra33 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Nuber S, Zabel U, Lorenz K, Nuber A, Milligan G, Tobin AB, Lohse MJ, Hoffmann C, β-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature 531, 661–664 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eichel K, Jullié D, Barsi-Rhyne B, Latorraca NR, Masureel M, Sibarita J-B, Dror RO, von Zastrow M, Catalytic activation of β-arrestin by GPCRs. Nature 557, 381–386 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Latorraca NR, Wang JK, Bauer B, Townshend RJL, Hollingsworth SA, Olivieri JE, Xu HE, Sommer ME, Dror RO, Molecular mechanism of GPCR-mediated arrestin activation. Nature 557, 452–456 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SGF, Sunahara RK, El-Samad H, Huang B, von Zastrow M, Conformational biosensors reveal GPCR signalling from endosomes. Nature 495, 534–538 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsvetanova NG, von Zastrow M, Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat. Chem. Biol 10, 1061–1065 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dror RO, Mildorf TJ, Hilger D, Manglik A, Borhani DW, Arlow DH, Philippsen A, Villanueva N, Yang Z, Lerch MT, Hubbell WL, Kobilka BK, Sunahara RK, Shaw DE, Structural basis for nucleotide exchange in heterotrimeric G proteins. Science 348, 1361–1365 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koehl A, Hu H, Maeda S, Zhang Y, Qu Q, Paggi JM, Latorraca NR, Hilger D, Dawson R, Matile H, Schertler GFX, Granier S, Weis WI, Dror RO, Manglik A, Skiniotis G, Kobilka BK, Structure of the μ-opioid receptor-Gi protein complex. Nature 558, 547–552 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kato HE, Zhang Y, Hu H, Suomivuori C-M, Kadji FMN, Aoki J, Krishna Kumar K, Fonseca R, Hilger D, Huang W, Latorraca NR, Inoue A, Dror RO, Kobilka BK, Skiniotis G, Conformational transitions of a neurotensin receptor 1-Gi1 complex. Nature 572, 80–85 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rajagopal S, Ahn S, Rominger DH, Gowen-MacDonald W, Lam CM, Dewire SM, Violin JD, Lefkowitz RJ, Quantifying ligand bias at seven-transmembrane receptors. Mol. Pharmacol 80, 367–377 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are available in the main text or the Supplementary Materials. Constructs for CXCR3, CXCR3-LgBiT, smBiT–β-arrestin2, β-arrestin2–mKO, ERK-mKO, AP-2–mKO, and β-arrestin2–mCerulean are available from S.R. under a materials transfer agreement with Duke University.