

Abstract
Structural variations (SVs) are large genomic rearrangements that can be challenging to identify with current short read sequencing technology due to various confounding factors such as existence of genomic repeats and complex SV structures. Hi-C breakfinder is the first computational tool that utilizes the technology of high-throughput chromatin conformation capture assay (Hi-C) to systematically identify SVs, without being interfered by regular confounding factors. SVs change the spatial distance of genomic regions and cause discontinuous signals in Hi-C, which are difficult to analyze by routine informatics practice. Here we provide step-by-step guidance for how to identify SVs using Hi-C data and how to reconstruct Hi-C maps in the presence of SVs.
Keywords: Structural variation, Hi-C, Chromatin conformation, 3D genome organization, Cancer genomics
1. Introduction
Structural variations (SVs) are large genomic rearrangements including inversions, deletions, duplications, aneuploidy, translocations, and chromoplexy. Every normal individual genome can harbor thousands of germline SVs. While the majority of them have neutral impact to human health, a small proportion of germline SVs that disrupt know tumor suppressor genes like BRCA2 and ATM can confer predisposition to certain cancers [1]. Meanwhile, the presence of a large number of somatic SVs can be found in most cancer genomes [2]. Oncogene activation events have been identified as the product of recurrent SVs, such as translocation-induced gene fusion events involving ABL1 and MLL1 that drive the development of leukemia [3]. SVs have provided clear diagnostic and prognostic information in the clinic [4], as well as successful targets for drug therapies [5–9].
SVs can be detected by various technologies. G-band karyotyping, fluorescence in situ hybridization, and microarray have been historically widely used [10, 11]. Due to their limited throughput and resolution, genome-wide-throughput sequencing-based technologies like whole genome sequencing (WGS) have emerged as attractive alternatives for SV detection in recent years [12, 13]. Despite its success, WGS is limited by its reliance on short sequence reads with much of the structural continuity of the genome being lost, calling for novel method that can effectively identify SVs on repetitive sequences in the genome and resolve complex SVs at affordable cost. In 2018, high-throughput chromosome conformation capture (Hi-C), a technology that had been initially invented for profiling chromatin spatial structure [14], was for the first time adopted for SV detection [15].
Hi-C is capable for SV detection due to its unique experiment design. It starts with fixation of the cells to preserve the spatial structure of chromatin, then the DNA is cleaved by restriction enzyme. DNA ends are marked with biotin treated with DNA ligase, such that the DNA that is spatially close to each other will have a higher chance to be religated and can be further enriched by biotin-selection. Then, reverse cross-linking is performed to free the reconnected DNA from proteins. Sequencing libraries from such DNA are finally constructed and sequenced to generate hundreds of millions of sequencing reads. Religated reads originated from two loci of the genome are mapped back as pairs, and numerous such pairs eventually form a square matrix to describe the frequency of chromatin contacts between any two given regions [14].
Hi-C showed the potential to be a good fit for SV detection, because SVs can alter the spatial distance of two genomic regions that are originally far apart, which greatly increases chance of DNA from those regions to be religated and such reads can be aberrantly enriched as in comparison to the background with similar genomic distance. Indeed, with more and more Hi-C data generated since 2009, it has been visually noticed that some cancer cell lines show specific patterns in Hi-C matrix and they are speculated to be caused by SVs [16]. Hi-C breakfinder is the first method that can systematically identify SVs genome-wide, including SVs’ type, loci, orientation, and confidence score. SVs are called when the increased number of Hi-C contacts significantly deviates from expectation by taking into account the genomic distance, A/B compartment, TADs, and interactions between small chromosomes and between sub-telomeric regions [15]. As an important advantage over WGS, Hi-C breakfinder can robustly identify largescale SVs utilizing reads mapped to anywhere of the joined SVs arms, without being interfered by complex genomic sequences like repeats around the SV breakpoints. The long insert reads also preserve the genetic contiguity for resolving and reconstructing clusters of complex SVs.
Recent studies showed that SVs induce aberrant gene expression by juxtaposing distal enhancers to important cancer genes, a phenomenon known as enhancer hijacking, and reorganizing local chromatin conformation [17]. Therefore, elucidating the SV-induced chromatin conformation change could shed light on novel mechanisms in gene regulation. However, difficulties remain to systematically identify novel chromatin conformation changes such as formation of novel TADs and loop domains. This is further confounded by the heterozygosity of SVs and copy number variations (CNV) at SV loci, and heterogeneity of tumor samples. More sophisticated computational methods are needed to address these questions in future work.
2. Materials
Materials used for the analysis of Hi-C data include computers running Unix or Mac OS operating systems and various software to perform preprocessing of raw data and identification and visualization of SVs. All the necessary softwares are listed in Sect. 2.1, along with the installation instructions on Sect. 2.2.
2.1. Computation Resource Requirement
Unix, Linux, or Mac OS X.
2.1.1. System Requirement
2.1.2. Software Requirement
Samtools: https://github.com/samtools/samtools/releases
Python version 3.x: https://www.python.org/download/releases/3.0/
Conda package: https://docs.conda.io/en/latest/miniconda.html
Pairtools: https://github.com/mirnylab/pairtools
Cooler package: https://github.com/mirnylab/cooler
HiGlass: https://github.com/higlass/higlass-manage
Hi-C breakfinder: https://github.com/dixonlab/hic_breakfinder
2.2. Tool Installation
You will need Unix-based system to install the following tools.
2.2.1. Install BWA
BWA is a fast short-read aligner. We will use it for Hi-C reads mapping.
git clone https://github.com/lh3/bwa.git cd bwa; make
2.2.2. Install Samtools
Samtools is a tool for interacting with high-throughput sequencing data.
wget https://github.com/samtools/samtools/releases/download/ 1.10/samtools-1.10.tar.bz2 tar xvfj samtools-1.10.tar.bz2 cd samtools-1.10 ./configure --prefix=$PWD make && make install
2.2.3. Install Conda
Conda is an open source package management system. We recommend installing conda through Miniconda. Miniconda is a minimal installer for conda which includes conda, Python, and a small number of useful packages. Miniconda installers for Python 3 can be found at: https://docs.conda.io/en/latest/miniconda.html
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86.sh bash Miniconda3-latest-Linux-x86.sh
2.2.4. Install Pairtools
Pairtools is fast command-line tool to process aligned Hi-C reads and perform various operations on Hi-C pairs and matrix.
conda install -c conda-forge -c bioconda pairtools
2.2.5. Install Cooler
Cooler is a Python library for manipulating .cool/.mcool files for Hi-C maps.
conda install -c conda-forge -c bioconda cooler
2.2.6. Install HiGlass
HiGlass is a visualization tool for Hi-C maps in .cool/.mcool format. Here we install the higlass-manage Python package to visualize Hi-C maps on a local machine.
pip install higlass-manage
2.2.7. Install Hi-C Breakfinder
Hi-C breakfinder is a command line tools for identifying structural variants using Hi-C data. Hi-C breakfinder requires Eigen C++ library and the BamTools library.
First, we need to download and compile Eigen C++ library.
wget https://gitlab.com/libeigen/eigen/-/archive/3.3.7/eigen-3.3.7.tar.bz2 tar xvjf eigen-3.3.7.tar.bz2 cd eigen-3.3.7 mdkir build_dir cd build_dir cmake .. -DCMAKE_INSTALL_PREFIX=$PWD make install
Next, we need to do the same thing for BamTools library. CMake (version >= 3.0) is required to build BamTools. Check CMake version before building BamTools. If CMake is missing or an older version, make sure to install the CMake version >= 3.0.
cmake --version git clone git://github.com/pezmaster31/bamtools.git cd bamtools mdkir build_dir cd build_dir cmake -DCMAKE_INSTALL_PREFIX=$PWD .. make
Last, we install Hi-C breakfinder by linking to the Eigen and Bamtools library.
git clone https://github.com/dixonlab/hic_breakfinder.git cd hic_breakfinder ./configure CPPFLAGS=“-I /path/to/bamtools/include -I /path/ to/eigen” LDFLAGS=“-L/path/to/bamtools/lib/” make make install
2.3. Example Data Sets
In this chapter, we will use the Hi-C data for Caki2 cell line to illustrate the process of discovering SVs using Hi-C data. Caki2 is a human renal cancer cell line. We will download the publicly available Hi-C dataset for Caki2 from ENCODE (https://www.encodeproject.org/, accession number: ENCSR401TBQ). First, we will set up a working directory and then download the data into the directory, for example, creating a folder named “project” under your home directory.
mkdir ~/project cd ~/project wget -O Caki2.R1.fastq.gz https://www.encodeproject.org/ files/ENCFF190NLS/@@download/ENCFF190NLS.fastq.gz wget -O Caki2.R2.fastq.gz https://www.encodeproject.org/ files/ENCFF914EQR/@@download/ENCFF914EQR.fastq.gz
3. Methods
Several computational methods have been proposed to detect structural variation and copy number variations (CNVs) using Hi-C as a tool (Table 1). Among these methods, Hi-C breakfinder developed by our lab detects the most types of SVs with a resolution as high as 1 kb. In this chapter, we will focus on discovering SVs using Hi-C breakfinder.
Table 1.
SV detection tools for Hi-C data
| SV type | Link | Publication | |
|---|---|---|---|
| Hi-C breakfinder | Deletion, inversion, duplication, translocation | https://github.com/dixonlab/hic_breakfinder | Dixon et al. Nature Genetics, 2018 [15] |
| HiCtrans, HiCnv | Translocation, CNVs | https://github.com/ay-lab/HiCtrans https://github.com/ay-lab/HiCnv | Chakraborty et al. Bioinformatics, 2018 [29] |
| HiNT | Translocation, CNVs | https://github.com/parklab/HiNT | Wang et al. Genome Biology, 2020 [30] |
The Hi-C data analysis generally involves the following procedures: (1) mapping Hi-C reads onto a reference genome; (2) generating and normalizing Hi-C matrices; and (3) detection of hierarchies of chromatin structures including A/B compartments [14], Topologically Associating Domains (TADs) [18], sub-TADs [19], or loop domains [20]. Many computational methods have been developed to streamline above procedures [21–24]. The comparison of different Hi-C analysis methods has been described elsewhere [25].
The only user-provided input for Hi-C breakfinder is the read alignment file. Therefore, one can utilize the alignment file (usually, the “bam” file) generated from various Hi-C analysis pipeline. Based on our observations, the differences of alignment files from different pipelines have no impact on the results of Hi-C breakfinder. In the following sections, we will show an in-house pipeline starting from Hi-C read alignment to SV discovery using Hi-C breakfinder and finally visualization of SVs on a Hi-C map.
3.1. Overview of the SV Discovery Pipeline Using Hi-C Breakfinder
We first process the raw Hi-C reads using a pipeline adapted from the 4D Nucleome Hi-C processing pipeline (https://data.4dnucleome.org/resources/data-analysis/hi_c-processing-pipeline). This step will generate an alignment file (.bam) which will be the input for the next step of SV detection, and a matrix file in cooler format from which we can plot Hi-C heatmaps at the SV loci (Fig. 1).
Fig. 1.

An overview of SV discovery with Hi-C data
3.2. Hi-C Read Alignment
We align the Caki2 raw reads to human genome reference GRCh38 using BWA aligner. Users can choose their reference version based on their needs. First, we need to download the reference data and index the reference.
mkdir -p project/ref; cd project/ref wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/001/405/GCA_000001405.15_GRCh38/seqs_for_alignment_pipelines.ucsc_ids/GCA_000001405.15_GRCh38_no_alt_plus_hs38d1_analysis_set.fna.gz gunzip GCA_000001405.15_GRCh38_no_alt_plus_hs38d1_analysis_set.fna.gz bwa index -a bwtsw GCA_000001405.15_GRCh38_no_alt_plus_hs38d1_analysis_set.fna
Next, we align the Caki2 paired-end reads to the indexed reference genome. Note that older pipelines may require the user to align each pair of reads separately due to problematic estimation of insert size during the alignment. This is not necessary in newer versions (> 0.7.15) of BWA in which it is possible to align in one command two mates of a paired-end reads without forcing them to be from one single genomic segment.
cd project REF= ref/GCA_000001405.15_GRCh38_no_alt_plus_hs38d1_analysis_set.fna bwa mem -SP5M -t 8 $REF Caki2.R1.fastq.gz Caki2.R2.fastq.gz | samtools view -bS > Caki2.bam
Next, we want to filter out unmapped/single-sided/duplicated/multi-mapped reads using Pairtools. The minimum mapping quality is set by --min-mapq. Two common values are 1 and 30 as used in Juicer. Here we will set --min-mapq to 1. Pairtools convert alignments to contact pairs. Pair type are denoted as “U” or “R” if the read is uniquely mapped (defined by –min-mapq) or “rescued” which are those derived from chimeric alignment but are informative Hi-C contacts. We will retain pairs marked as “UU,” “UR,” or “RU.”
CHROM_SIZES=/path/to/GRCh38.chrom.sizes samtools view Caki2.bam | \ pairtools parse -c $CHROM_SIZES --min-mapq 1 --assembly hg38 | pairtools sort | \ pairtools select ‘(pair_type == “UU” or pair_type == “UR” or pair_type == “RU”)’ | \ pairtools dedup --max-mismatch 3 --output \ >( pairtools split --output-pairs Caki2.nodups.pairs.gz -- output-sam Caki2.nodups.bam )
The “Caki2.nodups.bam” from Pairtools will serve as input for Hi-C breakfinder. If the restriction enzyme information is available, one may want to filter out reads that fall in the same restriction fragment or self-circles. We will need to generate a restriction site file and then filter the pairs against this restriction site file. Here, we use the “MboI” restriction enzyme as an example.
cooler digest -o restrict_sites.MboI.hg38.bed $CHROM_SIZE $REF
MboI
pairtools select ‘len(chrom1) < 6 and len(chrom2) < 6 and
chrom1 != “chrM” and chrom2 != “chrM”‘ Caki2.nodups.pairs.gz |
\
paritools restrict -f restrict_sites.MboI.hg38.bed | \
awk ‘$0 ~ /^#/{print} $0 !~ /^#/ && $9 != $12{print}’ | bgzip
-c > Caki2.nodups.valid.pairs.gz3.3. SV Discovery with Hi-C Breakfinder
Besides the alignment file, Hi-C breakfinder requires two expectation files as input. The expectation file is generated by averaging the expected chromatin contacts from a number of normal cell lines. These expected contacts will be used in a negative binomial model for detecting SV-induced chromatin contacts. We can download the expectation file from https://salkinstitute.app.box.com/s/m8oyv2ypf8o3kcdsybzcmrpg032xnrgx
hic_breakfinder --bam-file Caki2.nodups.bam --exp-file-inter inter_expect_1Mb.hg38.txt --exp-file-intra intra_expect_100kb.hg38.txt --name Caki2
Hi-C breakfinder can detect SVs at 1 kb final resolution by setting the option --min-1 kb. This requires frequent cutting enzyme such as MboI and DnpII. One caveat is that if the Hi-C sequencing depth is high, adding this option can significantly increase the run time.
The final SV predictions are in the file named “Caki2.breaks. txt”. The file contains 10 columns (Table 2, the actual result file does not contain the header row), where the “score” column describes the confidence of the SV call. The columns 2–4 denote the possible range of coordinates for the first SV breakpoint, and similarly column 6–8 for the second SV breakpoint. The strand indicates whether the predicted breakpoints are closer to the end (“+” strand) or the start (“−” strand) coordinates. Column 10 is the resolution at which the SV were detected.
Table 2.
Example SV predictions from Hi-C breakfinder
| Score | chrl | startl | endl | strandl | chr2 | start2 | end2 | strand2 | Resolution |
|---|---|---|---|---|---|---|---|---|---|
| 2145.07 | chrl2 | 116800000 | 117920000 | − | chrl 1 | 124590000 | 125170000 | + | 10 lcb |
| 1953.23 | chr4 | 53550000 | 55660000 | − | chr6 | 55380000 | 56530000 | + | 10 kb |
| 805.469 | chr4 | 64330000 | 64630000 | + | chrl 2 | 64980000 | 66570000 | + | 10 kb |
| 146.297 | chr5 | 69370000 | 72050000 | + | chrl | 197010000 | 197310000 | − | 10 kb |
| 599.733 | chr5 | 71800000 | 72070000 | − | chrl | 198420000 | 198540000 | − | 10 kb |
SVs are often categorized into different types such as deletion, inversion, and translocation. The categorization of an SV can be derived from its coordinates and strands of two breakpoints. Here we provide a set of rules for classifying an SV (Table 3). Note that an inversion is called only if two SVs (four breakpoints) come from the same event. Translocations can be interchromosomal or intrachromosomal.
Table 3.
SV classification
| Two breakpoints on the same chromosome? | Strand1/Strand2 | SV type |
|---|---|---|
| Yes | +/− | Deletion |
| Yes | −/+ | Duplication |
| Yes | +/+ and −/− | Inversion |
| Yes | +/+ or −/− | Translocation |
| No | Any | Translocation |
3.4. Visualization of SVs on Normal Hi-C Maps
Now we have the SV predictions, we can visualize the SVs on a normal Hi-C map or a reconstructed Hi-C map. There are many visualization tools available for viewing Hi-C map including standalone software (e.g., Juicerbox, HiGlass) or web-based tools (e.g., 3D Genome Browser [26], WashU Epigenome Browser [27]). Each tool may require as input specific file format of Hi-C matrices. For simplicity, we choose HiGlass and in-house Python scripts for visualization of SVs on Hi-C maps.
First, we will need to generate Hi-C matrix in cooler format, and then normalize the matrix using ICE-normalization [28].
cooler cload pairs -c1 2 -p1 3 -c2 4 -p2 5 --assembly hg38 $CHROM_SIZE:10000 Caki2.nodups.valid.pairs.gz Caki2.10kb.cool cooler balance Caki2.10kb.cool cooler zoomify -p 8 --balance -o Caki2.10kb.mcool –r 20000,40000,100000,250000,500000,1000000 Caki2.10kb.cool
To visualize the Hi-C matrix with HiGlass, we need to set up a local HiGlass instance. Note that Docker needs to be installed on the computer before running HiGlass instances. To add the Hi-C matrix to a HiGlass instance, we run the command “higlass-manage ingest.”
higlass-manage ingest Caki2.10kb.mcool higlass-manage start
With a HiGlass instance running, we can access this tool by visiting localhost:8989 in a web browser. Click the “+” sign on the right upper corner and select Caki2.10kb.mcool we just added. It is easy to spot block-like signals in the interchromosomal regions which are different from a normal Hi-C map. Below is example of such signals between chr11 and chr12 (Fig. 2), which corresponds to the chr11-chr12 translocation in the Table 2.
Fig. 2.
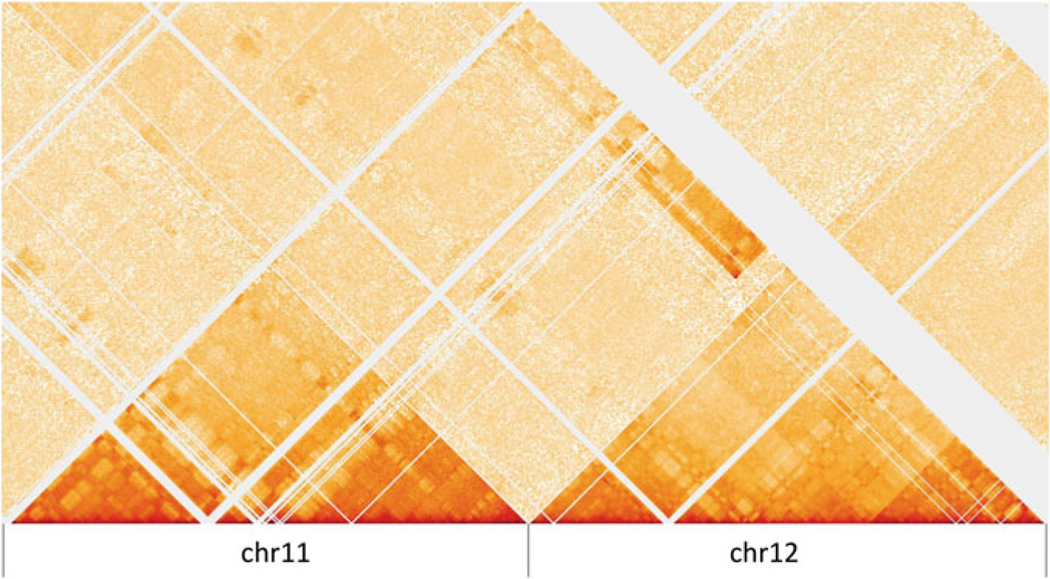
Visualization of the chr11-chr12 translocation in HiGlass
The 3D Genome Browser (3dgenome.org) provides a more convenient way to visualize the SV on a Hi-C map. We can type in the region of interest in the “Inter-chrom” module of the website, e.g., the whole chr11 and chr12. We will see a similar Hi-C map as above (Fig. 3).
Fig. 3.

Visualization of the chr11-chr12 translocation in the 3D Genome Browser
Based on Dixon et al. 2018, the SV calls from Hi-C breakfinder is highly accurate and supported by orthogonal techniques such as BioNano optical mapping and whole genome sequencing. In the example in Fig. 2, the unusual signals in the Hi-C heatmap provide a partial proof that the SV detected by Hi-C breakfinder is correct. This SV is further validated by whole genome sequencing using data from Dixon et al. 2018. We observed WGS reads spanning the breakpoints by mapping WGS reads to the concatenated reference genome of chr11 and chr12 (Fig. 4).
Fig. 4.

WGS reads spanning the SV breakpoints. Reads spanning the breakpoint (Colored), Forward read (purple), reverse read (red); Reads not spanning the breakpoint (gray). (Figure generated by svviz)
3.5. Visualization of SVs on Reconstructed Hi-C Maps
The block-like signal in the interchromosomal regions is originated from the strong contact of two distal regions that was brought into proximity by the SV events. When we map the reads onto the reference genome, these contacts would look like “interchromosomal” contacts. We will need to reconstruct the Hi-C map to better understand the change of local chromosomal organization at the SV loci. However, reconstruction of Hi-C map is challenging due to the complexity of SV orientation combinations. An SV is typically composed of two breakpoint each having one of two possible orientations (5′ to 3′, or 3′ to 5′). Based on different combinations of breakpoint orientation, Hi-C map needs to flip or rotate to reflect the altered linear genome.
Here, we provide a Python script for reconstructing and plotting Hi-C map at SV loci (see Appendix). We rebuilt the local Hi-C map of the above chr11-chr12 translocation in Caki2 (Fig. 5). The fine continuity of Hi-C signals and TAD-like structure across the translocation breakpoint are indicative of chromatin reorganization and formation of novel interactions at this region.
Fig. 5.

Reconstructed Hi-C map at chr11-chr12 translocation
clr_path = ‘Caki2.10kb.mcool’ plotHiC_SV(clr_path, (‘chr11’, 125170000, ‘+’), (‘chr12’, 116800000, ‘-’), w=1500000, res=40000)
In the above code, the plotHiC_SV() function is loaded from the script in Appendix. The first parameter is the path to the cooler file, second and third parameters are just the SV breakpoints and orientation from Hi-C breakfinder. We also require the Hi-C map to be plotted at a region of 1.5 Mb up-or-downstream of the breakpoints with resolution of 40 kb.
Lastly, the reconstructed Hi-C map provides a paradigm for studying novel mechanisms of gene regulation in cancer genomics such as enhancer hijacking where a distal enhancer is brought to the proximity of its target gene by SVs. Therefore, we believe the method we described here will shed light on the process of tumorigenesis.
Acknowledgments
F. Y. is supported by NIH grants R35GM124820, R01HG009906, and U01CA200060 (F.Y.), R24DK106766 (R.C.H. and F.Y.). J.D. is supported by DP5 OD023071.
Appendix Python script for making reconstructed Hi-C map at SV loci
import cooler
import numpy as np
import matplotlib.pyplot as plt
import matplotlib.colors as mcol
from itertools import product
cmap = mcol.LinearSegmentedColormap.from_list(‘custom red’,
[(0, ‘white’), (0.15, ‘white’), (1, ‘#e60000’)], N=256)
def matrix_reader(cool_path, chrom, res, iced, chrom2=None):
cool_handle = cooler.Cooler(cool_path + “::/resolutions/” +
str(res))
cool_matrix = cool_handle.matrix(balance=iced).fetch
(chrom)[:]
if chrom2 is not None:
cool_matrix = cool_handle.matrix(balance=iced).fetch
(chrom, chrom2)[:]
else:
np.fill_diagonal(cool_matrix, 0)
np.nan_to_num(cool_matrix, copy=False)
return cool_matrix
def concat_matrix(cool_path, c1, o1, c2, o2, res, iced):
m1 = matrix_reader(cool_path, c1, res, iced)
m2 = matrix_reader(cool_path, c2, res, iced)
mi = matrix_reader(cool_path, c1, res, iced, chrom2=c2)
nrow_1 = m1.shape[0]
nrow_2 = m2.shape[0]
nrow_cm = nrow_1 + nrow_2
cm = np.zeros((nrow_cm, nrow_cm))
if o1 + o2 == “+-”:
pass
elif o1 + o2 == “++”:
m2 = np.flip(np.flip(m2, 0), 1)
mi = np.flip(mi, 1)
elif o1 + o2 == “--”:
m1 = np.flip(np.flip(m1, 0), 1)
mi = np.flip(mi, 0)
elif o1 + o2 == “-+”:
m1 = np.flip(np.flip(m1, 0), 1)
m2 = np.flip(np.flip(m2, 0), 1)
mi = np.flip(np.flip(mi, 0), 1)
cm[0:nrow_1, 0:nrow_1] = m1
cm[nrow_1:nrow_cm, nrow_1:nrow_cm] = m2
cm[0:nrow_1, nrow_1:nrow_cm] = mi * 2
cm[nrow_1:nrow_cm, 0:nrow_1] = np.rot90(np.flip(mi, 1), 1) *
2
return cm
def concat_tri(fig, ax, mat, c1, o1, c2, o2, res, cmap,
title=None, xlabel=None, ylabel=None):
n = mat.shape[0]
t = np.array([[1, 1], [−1, 1]])
A = np.dot(np.array([(i[1], i[0]) for i in product(range(n
+1), range(n+1))]), t)
vmax = np.percentile(mat, 92)
if vmax == 0:
while vmax == 0 and (perc - 5) > 0:
perc = perc - 5
vmax = np.percentile(mat, perc)
vmin = np.min(mat)
img = ax.pcolormesh(A[:, 1].reshape(n+1, n+1), A[:, 0].
reshape(n+1, n+1), mat, cmap=cmap, vmax=vmax, vmin=vmin)
img.set_rasterized(True)
ax.set_frame_on(False)
ax.set_ylim([0, n])
ax.set_aspect(1)
ax.yaxis.set_visible(False)
# set ticks
merge_point = 2 * (c1[2] - c1[1] ) // res
ticks = [0, merge_point // 2, merge_point, merge_point//2+n,
2*n]
ax.set_xticks(ticks)
# set labels
ax.set_xticklabels([“, c1[0], “, c2[0], “], size=16)
# ax title
title = f”{c1[0]}:{c1[1]}-{c1[2]}({o1})\n{c2[0]}:{c2[1]}-{c2
[2]}({o2})”
ax.set_title(title, pad=20, size=15)
if xlabel != None:
ax.set_xlabel(xlabel, labelpad=10, fontsize=15)
if ylabel != None:
ax.set_ylabel(ylabel, labelpad=10, fontsize=15)
# set color bar
pos0 = ax.get_position()
cax = fig.add_axes([pos0.x0+pos0.width*0.9, pos0.y0+pos0.
height*0.8, pos0.width*0.01, pos0.height*0.2])
cbar = fig.colorbar(img, cax=cax, ticks=[vmin, vmax])
cbar.ax.set_yticklabels([f”{vmin:.1f}”, f”{vmax:.1f}”],
fontsize=12)
# add an arrow/dashed line at the merge_point
xmin, xmax, ymin, _ = ax.axis()
x1, y1 = xmin + merge_point, ymin
x2, y2 = xmin + merge_point/2, ymin + merge_point/2
x3, y3 = x1 + (xmax-xmin-merge_point)/2, ymin + (xmax-xmin-
merge_point)/2
ax.plot((x1, x2), (y1, x2), linestyle=‘:’, linewidth=2,
color=“black”)
ax.plot((x1, x3), (y1, y3), linestyle=‘:’, linewidth=2,
color=“black”)
return None
def plotHiC_SV(clr, c1, c2, w, res, iced=False):
o1 = c1[2]
o2 = c2[2]
if o1 == “+”:
start = 0 if (c1[1] - w) < 0 else (c1[1] - w)
end = c1[1]
else:
start = c1[1]
end = c1[1] + w
c1 = (c1[0], start, end)
if o2 == “+”:
start = 0 if (c2[1] - w) < 0 else (c2[1] - w)
end = c2[1]
else:
start = c2[1]
end = c2[1] + w
c2 = (c2[0], start, end)
mat = concat_matrix(clr, c1, o1, c2, o2, res, iced)
fig, ax = plt.subplots(1, 1, figsize=(8, 4.3), constraine-
d_layout=True)
concat_tri(fig, ax, mat, c1, o1, c2, o2, res, cmap=cmap)
plt.show()4 Notes
It is recommended to install the Python packages in Sect. 2.1.2 in a new conda environment to avoid package incompatibility and dependency issues. If doing so, make sure to activate this conda environment before running the scripts in Sect. 3.2
If Hi-C breakfinder is successfully installed but complains about missing bamtools library when running, make sure to add the bamtools library path to system LD_LIBRARY_PATH. For example, add this line “export LD_LIBRARY_PATH=/path/to/bamtools/lib:$LD_LIBRARY_PATH” to your ~/.bashrc.
Hi-C breakfinder performs well even for low sequencing depth Hi-C data. However, it is recommended to have at least 50 ~ 100 million usable reads to confidently detect structural variants with Hi-C breakfinder
When reconstructing Hi-C maps in Sect. 3.5, one may need to set different window sizes (the “w” parameter) up to several Mb to examine change of chromatin conformation at different scales.
The script for visualizing reconstructed Hi-C map in the Appendix can take as input coordinates of structural variants from not just Hi-C, but other data source as well such as those obtained by whole genome sequencing in base-pair resolution.
References
- 1.Consortium ITP-CAoWG (2020) Pan-canceranalysis of whole genomes. Nature 578 (7793):82–93. 10.1038/s41586-020-1969-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144 (5):646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 3.Fielding AK (2010) How I treat Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood 116(18):3409–3417. 10.1182/blood-2010-01-242750 [DOI] [PubMed] [Google Scholar]
- 4.Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127(20):2391–2405. 10.1182/blood-2016-03-643544 [DOI] [PubMed] [Google Scholar]
- 5.Futreal PA, Coin L, Marshall M, Down T,Hubbard T, Wooster R, Rahman N, Stratton MR (2004) A census of human cancer genes. Nat Rev Cancer 4(3):177–183. 10.1038/nrc1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soda M, Choi YL, Enomoto M, Takada S,Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, Aburatani H, Niki T, Sohara Y, Sugiyama Y, Mano H (2007) Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448(7153):561–566. 10.1038/nature05945 [DOI] [PubMed] [Google Scholar]
- 7.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Janne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, Fidias P, Stubbs H, Engelman JA, Sequist LV, Tan W, Gandhi L, Mino-Kenudson M, Wei GC, Shreeve SM, Ratain MJ, Settleman J, Christensen JG, Haber DA, Wilner K, Salgia R, Shapiro GI, Clark JW, Iafrate AJ (2010) Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 363(18):1693–1703. 10.1056/NEJMoa1006448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rowley JD (1973) Letter: a new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 243 (5405):290–293 [DOI] [PubMed] [Google Scholar]
- 9.Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C, Niederwieser D, Resta D, Capdeville R, Zoellner U, Talpaz M, Druker B, Goldman J, O’Brien SG, Russell N, Fischer T, Ottmann O, Cony-Makhoul P, Facon T, Stone R, Miller C, Tallman M, Brown R, Schuster M, Loughran T, Gratwohl A, Mandelli F, Saglio G, Lazzarino M, Russo D, Baccarani M, Morra E, International STICMLSG (2002) Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med 346(9):645–652. 10.1056/NEJMoa011573 [DOI] [PubMed] [Google Scholar]
- 10.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhsng CZ, Wala J, Mermel CH, Sougnez C, Gabriel SB, Hernandez B, Shen H, Laird PW, Getz G, Meyerson M, Beroukhim R (2013) Pan-cancer patterns of somatic copy number alteration. Nat Genet 45(10):1134–1140. 10.1038/ng.2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wan TS (2014) Cancer cytogenetics: methodology revisited. Ann Lab Med 34(6):413–425. 10.3343/alm.2014.34.6.413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mardis ER, Wilson RK (2009) Cancer genome sequencing: a review. Hum Mol Genet 18(R2): R163–R168. 10.1093/hmg/ddp396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inaki K, Hillmer AM, Ukil L, Yao F, Woo XY, Vardy LA, Zawack KF, Lee CW, Ariyaratne PN, Chan YS, Desai KV, Bergh J, Hall P, Putti TC, Ong WL, Shahab A, Cacheux-Rataboul V, Karuturi RK, Sung WK, Ruan X, Bourque G, Ruan Y, Liu ET (2011) Transcriptional consequences of genomic structural aberrations in breast cancer. Genome Res 21(5):676–687. 10.1101/gr.113225.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326 (5950):289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dixon JR, Xu J, Dileep V, Zhan Y, Song F, Le VT, Yardimci GG, Chakraborty A, Bann DV, Wang Y, Clark R, Zhang L, Yang H, Liu T, Iyyanki S, An L, Pool C, Sasaki T, Rivera-Mulia JC, Ozadam H, Lajoie BR, Kaul R, Buckley M, Lee K, Diegel M, Pezic D, Ernst C, Hadjur S, Odom DT, Stamatoyannopoulos JA, Broach JR, Hardison RC, Ay F, Noble WS, Dekker J, Gilbert DM, Yue F (2018) Integrative detection and analysis of structural variation in cancer genomes. Nat Genet 50(10):1388–1398. 10.1038/s41588-018-0195-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harewood L, Kishore K, Eldridge MD, Wingett S, Pearson D, Schoenfelder S, Collins VP, Fraser P (2017) Hi-C as a tool for precise detection and characterisation of chromosomal rearrangements and copy number variation in human tumours. Genome Biol 18(1):125. 10.1186/s13059-017-1253-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weischenfeldt J, Dubash T, Drainas AP, Mardin BR, Chen Y, Stutz AM, Waszak SM, Bosco G, Halvorsen AR, Raeder B, Efthymiopoulos T, Erkek S, Siegl C, Brenner H, Brustugun OT, Dieter SM, Northcott PA, Petersen I, Pfister SM, Schneider M, Solberg SK, Thunissen E, Weichert W, Zichner T, Thomas R, Peifer M, Helland A, Ball CR, Jechlinger M, Sotillo R, Glimm H, Korbel JO (2017) Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat Genet 49(1):65–74. 10.1038/ng.3722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y,Shen Y, Hu M, Liu JS, Ren B (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485(7398):376–380. 10.1038/nature11082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wijchers PJ, Krijger PHL, Geeven G, Zhu Y,Denker A, Verstegen M, Valdes-Quezada C, Vermeulen C, Janssen M, Teunissen H, Anink-Groenen LCM, Verschure PJ, de Laat W (2016) Cause and consequence of tethering a SubTAD to different nuclear compartments. Mol Cell 61(3):461–473. 10.1016/j.molcel.2016.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159(7):1665–1680. 10.1016/j.cell.2014.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Servant N, Varoquaux N, Lajoie BR, Viara E,Chen CJ, Vert JP, Heard E, Dekker J, Barillot E (2015) HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16:259. 10.1186/s13059-015-0831-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, Aiden EL (2016) Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst 3(1):99–101. 10.1016/j.cels.2015.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hwang YC, Lin CF, Valladares O, Malamon J, Kuksa PP, Zheng Q, Gregory BD, Wang LS (2015) HIPPIE: a high-throughput identification pipeline for promoter interacting enhancer elements. Bioinformatics 31(8):1290–1292. 10.1093/bioinformatics/btu801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abdennur N, Mirny LA (2020) Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics 36 (1):311–316. 10.1093/bioinformatics/btz540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forcato M, Nicoletti C, Pal K, Livi CM, Ferrari F, Bicciato S (2017) Comparison of computational methods for Hi-C data analysis. Nat Methods 14(7):679–685. 10.1038/nmeth.4325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Song F, Zhang B, Zhang L, Xu J, Kuang D, Li D, Choudhary MNK, Li Y, Hu M, Hardison R, Wang T, Yue F (2018) The 3D genome browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol 19(1):151. 10.1186/s13059-018-1519-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li D, Hsu S, Purushotham D, Sears RL, Wang T (2019) WashU epigenome browser update 2019. Nucleic Acids Res 47(W1): W158–W165. 10.1093/nar/gkz348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, Mirny LA (2012) Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods 9(10):999–1003. 10.1038/nmeth.2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chakraborty A, Ay F (2018) Identification of copy number variations and translocations in cancer cells from Hi-C data. Bioinformatics 34 (2):338–345. 10.1093/bioinformatics/btx664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang S, Lee S, Chu C, Jain D, Kerpedjiev P, Nelson GM, Walsh JM, Alver BH, Park PJ (2020) HiNT: a computational method for detecting copy number variations and translocations from Hi-C data. Genome Biol 21 (1):73. 10.1186/s13059020-01986-5 [DOI] [PMC free article] [PubMed] [Google Scholar]