

Abstract
NRF2 is a transcription factor that controls the cellular response to various stressors, such as reactive oxygen and nitrogen species. As such, it plays a key role in the suppression of carcinogenesis, but constitutive NRF2 expression in cancer cells leads to resistance to chemotherapeutics and promotes metastasis. As a result, inhibition of the NRF2 pathway is a target for new drugs, especially for use in conjunction with established chemotherapeutic agents like carboplatin and 5-fluorouracil. A new class of NRF2 inhibitors has been discovered with substituted nicotinonitriles, such as MSU38225. In this work, the effects on NRF2 inhibition with structural changes were explored. Through these studies, we identified a few compounds with as good or better activity than the initial hit but with greatly improved solubility. The syntheses involved a variety of metal-catalyzed reactions, including titanium multicomponent coupling reactions and various Pd and Cu coupling reactions. In addition to inhibiting NRF2 activity, these new compounds inhibited the proliferation and migration of lung cancer cells in which the NRF2 pathway is constitutively activated.
SAR on NRF2 inhibitor MSU38225 has led to a compound that inhibits expression of NRF2 target genes, reduces proliferation of lung cancer cells through G2/M arrest, and lowers cell migration.
Introduction
NFE2-related factor 2 (NRF2) is a transcription factor that is activated by cellular stress to direct multiple responses to restore homeostasis. NRF2, and its negative regulator Kelch-like ECH-associated protein 1 (KEAP1), play a key role in the regulation of reactive species, such as hydrogen peroxide, reactive nitrogen species (e.g., nitric oxide), and alkylating agents, in the cell.
Extensive evidence demonstrates that NRF2 activation suppresses carcinogenesis.1,2 Further, genetic deletion of NRF2 leads to a greater susceptibility to tumor formation.3 In cancer prevention studies, numerous compounds that activate the pathway are potential preventative drugs, many of which are found naturally in foods, such as isothiocyanate-containing sulforaphane found in broccoli and Brussels sprouts.4–7
However, constitutive NRF2 pathway activation in cancer cells reduces their sensitivity to chemo- and radiotherapy.2 Loss-of-function mutations of KEAP1 and gain-of-function mutations of NFE2L2 (the gene that codes for NRF2) are found in many cancers. Mutations affecting this pathway (NFE2L2, KEAP1, CUL3, BTRC, and MAFG) are particularly prevalent in lung cancer cells with ∼30% of all lung cancers containing such mutations. Consistent with these data, recent studies suggest that constitutive activation of the NRF2 pathway promotes tumor cell survival, proliferation, and metastasis.8–11
Brusatol was the first NRF2 inhibitor to be identified and is still the most potent known;12 however, profiling studies suggest that brusatol, rather than being a specific inhibitor, acts to inhibit global protein synthesis.13 Brusatol is also expensive to isolate or synthesize, which limits its potential clinical use. A few other small molecule inhibitors of NRF2 have been identified from screens, e.g., AEM1,14 ML385,15 and IM3829.16 In addition, a few natural compounds,17,18 such as stigmasterol, have been shown to inhibit the pathway. In all cases, however, these inhibitors have lacked efficacy, selectivity or drug-like physical properties, and a review in 2019 declared NRF2 inhibitors a “completely open field for pharmacology”.19
In a recent publication, the Liby and Odom laboratories reported the discovery of a novel NRF2 pathway inhibitor, the small molecule MSU38225, which was effective in vivo and in vitro.20 The compound showed promising selectivity for the NRF2 pathway and did not alter the concentrations of common off-target cellular proteins. Cells with constitutive NRF2-activity (KEAP1 mutant) were more sensitive than wild-type cells to the compound. For example, MSU38225 inhibited proliferation of lung cancer cells with KEAP1 mutations but did not affect normal epithelial cell growth. Importantly, the compound was found to act synergistically with chemotherapeutics like carboplatin (CI = 0.82) and 5-fluorouracil (CI = 0.54).
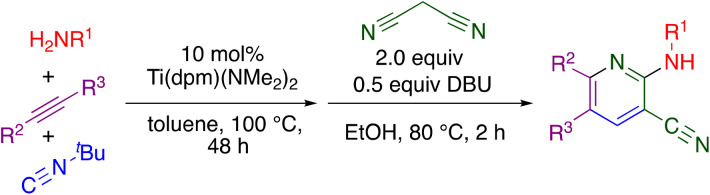
The “hit” compound MSU38225 was prepared using a novel titanium-catalyzed21–23 multicomponent coupling strategy in a one-pot procedure as shown in Scheme 1.24 The Ti(dpm)(NMe2)2 (ref. 25)-catalyzed reaction, where dpm = deprotonated 5,5-dipyrrolylmethane, involves coupling of a primary amine, alkyne, and isonitrile, which forms both C–C and C–N bonds in the same catalytic cycle, alkyne iminoamination.23 The product of the multicomponent coupling was not isolated but instead was treated with malononitrile and DBU to give a 2-aminonicotinonitrile.24
Scheme 1. Synthesis of MSU38225 using titanium-catalyzed multicomponent coupling in a one-pot procedure.

In this work, NRF2-pathway inhibitory activity as a function of structure for this class of compounds was explored. The activity is quite sensitive to the structure of the inhibitor, and some compounds in the class can be activators rather than inhibitors with seemingly small chemical changes. In these, the first SAR studies on the class, changes were made around the core pyridine ring as well as a few changes to the central core. One major drawback of MSU38225 was its poor solubility, and, in addition to probing the effect of structure on activity, we were seeking a more soluble derivative with as good or better activity.
Results and discussion
The structural investigations were undertaken with a variety of goals, and the structure of MSU38225 was modified to determine (1) the functionality necessary for NFR2 inhibition, (2) where groups may be added to the structure without greatly affecting activity, (3) where heterocycles are tolerated, (4) what functional groups can be added to increase water solubility while retaining biological activity. We sought to increase or retain activity while improving water solubility to give a clog P ≤ 3. To monitor NRF2 inhibition activities, a standardized luciferase assay with standard errors calculated based on multiple runs was employed.20
To achieve these goals, a variety of different catalytic methods were brought to bear including titanium multicomponent, palladium, and copper coupling reactions. In some cases, further modifications were made, including protection/deprotection and hydrogenation. A few different core structures were investigated, but the focus was largely on changes to the periphery. Once the effects of substituents are better understood, the core may be further explored.
Synthesis of compounds and NRF2 inhibition assay results
The titanium-catalyzed chemistry shown in Scheme 1 was designed to allow rapid access to 2-aminonicotonitriles in a one-pot procedure.22 As a result, it was further utilized here to produce derivatives of MSU38225 (1a), which was reprepared and tested for activity under identical conditions to the other compounds in this study. The initial hit 1a and the other compounds prepared using this procedure are shown in Table 1.
Compounds prepared using titanium multicomponent coupling catalysisa.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Clog Pb (solub.) | % yield (% activity) | Compound | Clog Pb (solub.) | % yield (% activity) | Compound | Clog Pb (solub.) | % yield (% activity) |
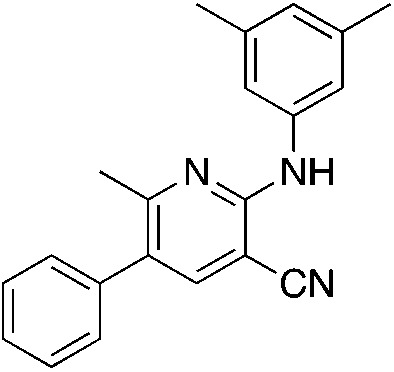 1a MSU38225 1a MSU38225 |
4.6 (3 μM) | 34 (54.6 ± 2.1) |
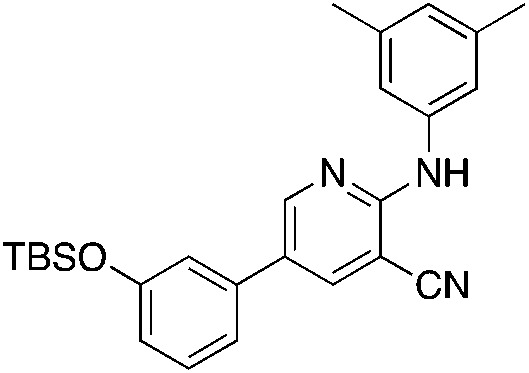 1i 1i |
6.9 | 19 (12.6 ± 5.5) |
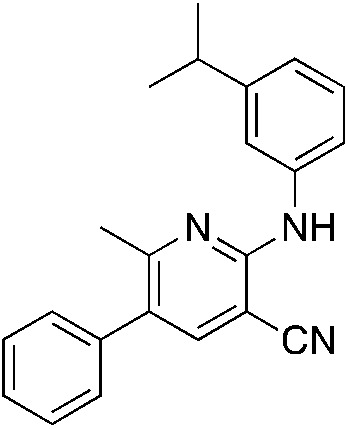 1q 1q |
5.1 (0.9 μM) | 18 (55.2 ± 2.3) |
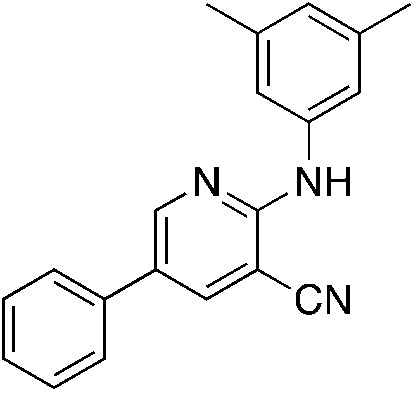 1b 1b |
4.4 (4 μM) | 19 (38.4 ± 1.5) |
 1j 1j |
4.8 | 14 (−32.3 ± 7.8) |
 1r 1r |
5.1 | 28 (15.7 ± 1.8) |
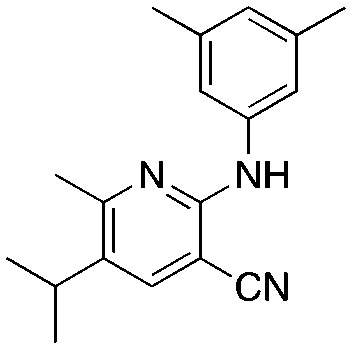 1c 1c |
4.4 | 17, 35c (−6.3 ± 13.6) |
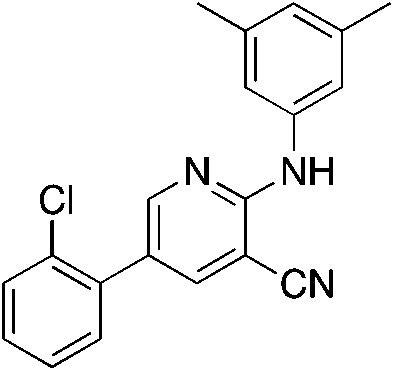 1k 1k |
4.9 | 22 (32.9 ± 1.3) |
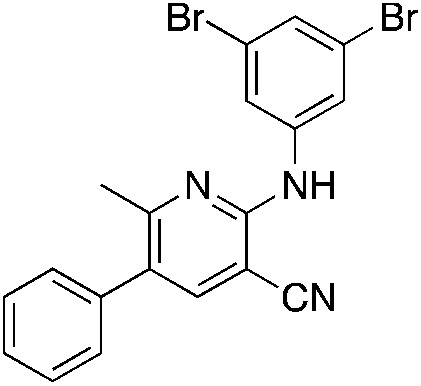 1s 1s |
5.4 | 19 (7.7 ± 4.7) |
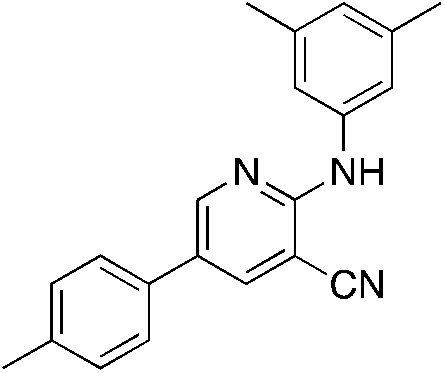 1d 1d |
4.9 | 10 (−29.6 ± 4.1) |
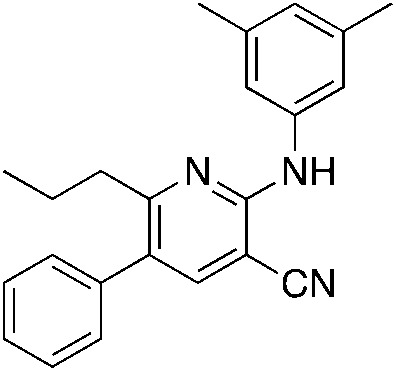 1l 1l |
5.7 | 34 (−11.8 ± 18.1) |
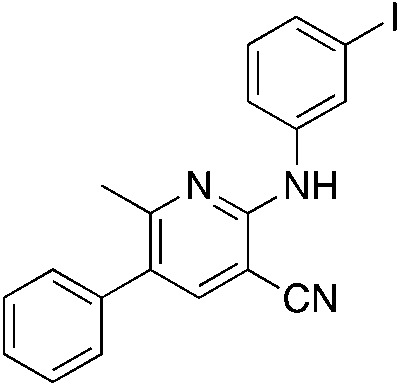 1t 1t |
4.8 | 38 (51.8 ± 2.3) |
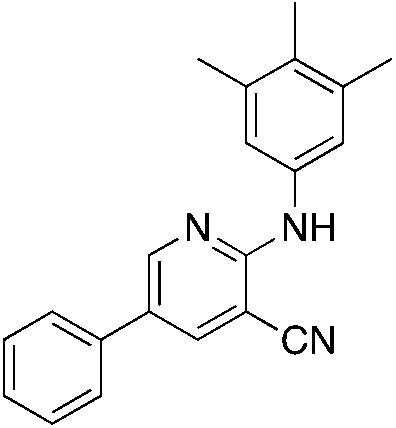
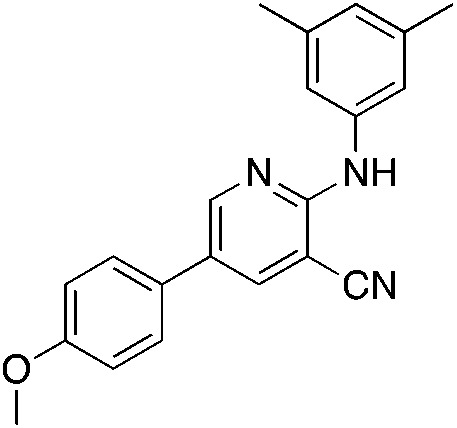 1e 1e |
4.4 | 12 (−1.5 ± 12.1) |
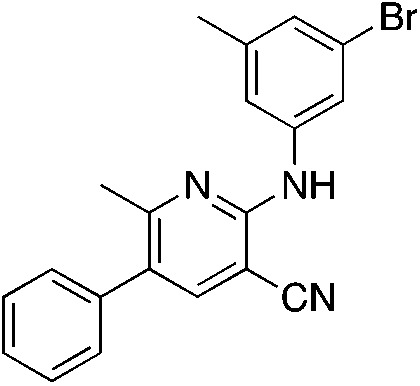 1m 1m |
5.0 | 19 (58.4 ± 6.2) |
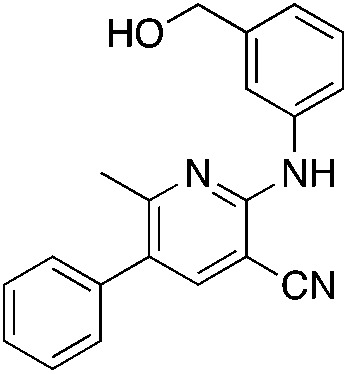 1u 1u |
2.6 | 45, 56d (16.7 ± 12.4) |
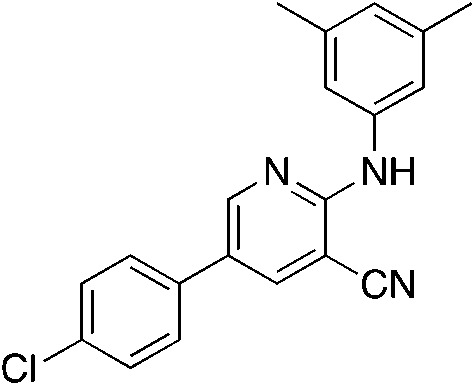 1f 1f |
5.2 | 17 (−4.9 ± 21.1) |
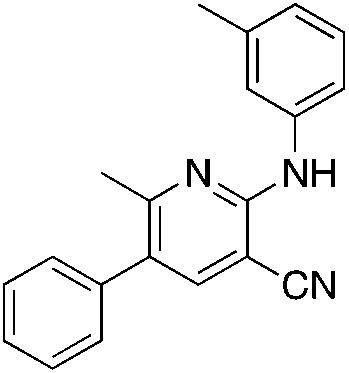 1n 1n |
4.1 | 29 (40.9 ± 5.2) |
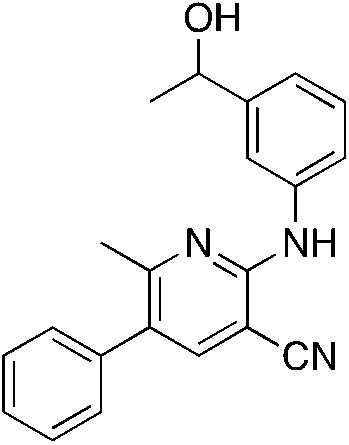 1v 1v |
2.9 | 33, 54d (26.0 ± 6)e (5.2 ± 7.5) |
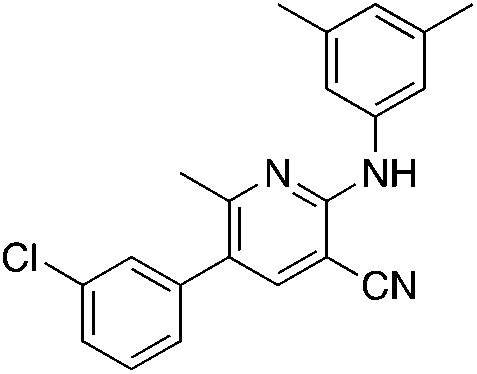 1g 1g |
5.4 | 19 (49.2 ± 4.2) |
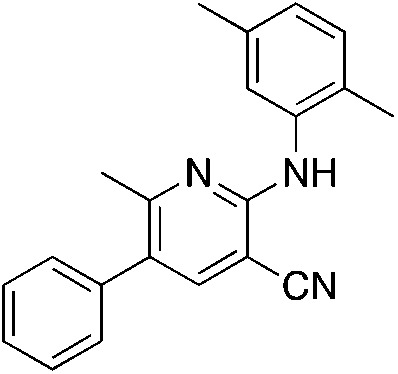 1o 1o |
4.6 | 15 (51 ± 2.6) | |||
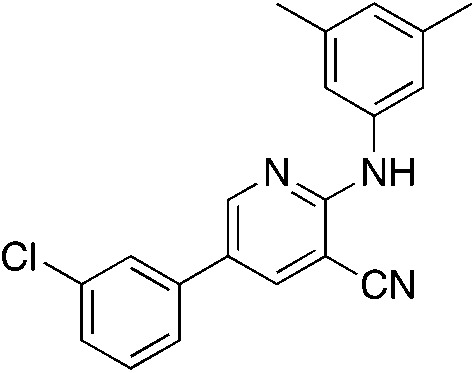 1h 1h |
5.2 | 23 (18.2 ± 12.9) |
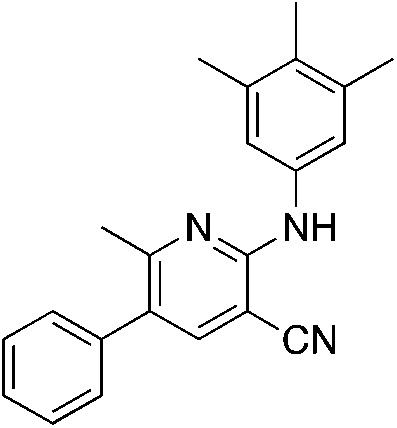 1p 1p |
5.0 | 15 (37.2 ± 0.5) | |||
All compounds were purified to >98% purity by GC-FID. Yields are of purified compound after two synthetic steps, except as specified. Cellular activity where positive values indicate inhibition and negative values indicate activation.
Clog P calculated using ChemDraw 19.1 for the various structures. The experimental kinetic solubilities in μM were measured for a few derivatives.
Hydrogenation Yield.
Deprotection Yield.
Activity before deprotection.
The titanium chemistry tolerates many functional groups such as ethers, tertiary amines, secondary amines, halides, olefins, and others; however, the catalyst is sensitive to nitro groups, esters, amides, carboxylic acids, and alcohols. Alcohols, for example, may be protected using the typical procedures, such as silylation, which was employed for the synthesis of 1i, 1u, and 1v. The yields were generally low for the derivatives described here, but the compounds were rapidly prepared using the one-pot procedure on scales more than adequate for biological testing and characterization, and the compounds were readily purified from the reaction mixtures.
Titanium iminoamination was used to explore the effects of various aromatic groups on the 2-NH position (R1, Table 1) by simply changing anilines, of which many are commercially available. In addition, the groups on the alkyne (R2 and R3, Table 1) could be readily varied to explore the effects of these positions on activity. There is a strong tendency for sp2-carbons on the alkyne (vinyl or aromatic) to be placed in the 5-position of the pyridine during the synthesis; in fact, the other regioisomer is typically not observed in crude reaction mixtures by GC-MS. This was used to advantage in the synthesis of 1c, where an enyne was used to direct the vinyl group to the 5-position followed by hydrogenation to the isopropyl group.
Several different methods of modifying MSU38225 (1a) were also investigated, and the activities of the resulting compounds were tested. These are shown in Table 2.
Derivatization of MSU38225 (1a)a.
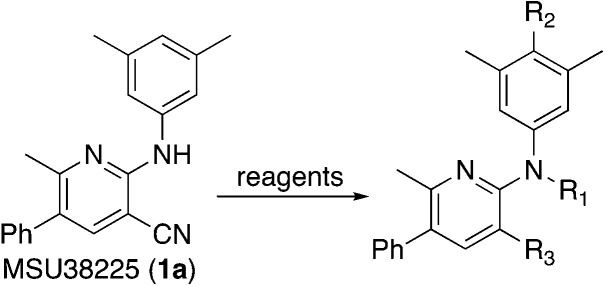
| |||
|---|---|---|---|
| Compound | Reagents | Clog Pb | % yield (% activity) |
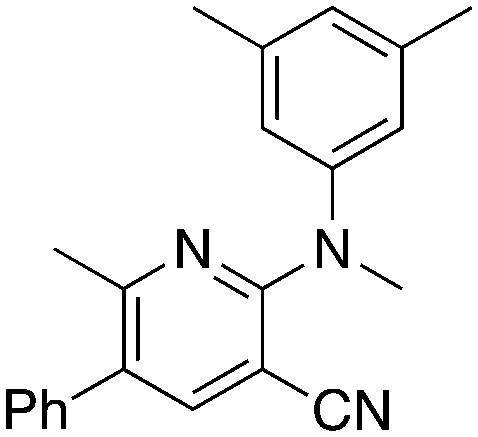 2a 2a |
KOtBu, MeI | 5.1 | 55 (21.9 ± 2.9) |
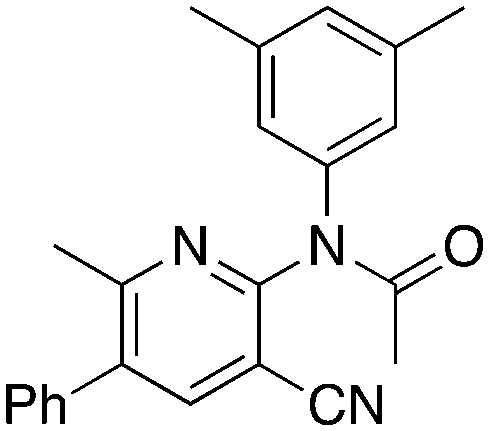 2b 2b |
Ac2O | 3.1 | 69 (12.5 ± 8.7) |
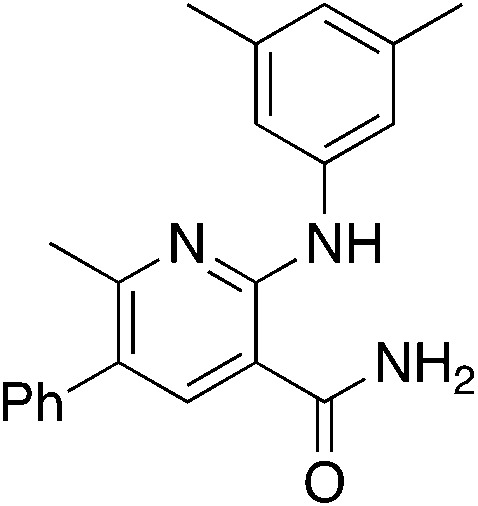 2c 2c |
{NBu4}+ OH−, EtOH | 4.1 | 23 (−18.1 ± 8.9) |
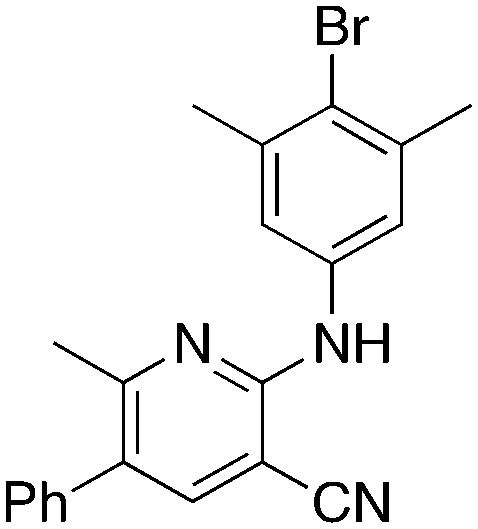 2d 2d |
NBS, (PhCO2)2 | 5.5 | 86 (25.7 ± 2.1) |
All compounds were purified to >98% purity by GC-FID. Yields are of purified compound after two synthetic steps, except as specified. Cellular activity where positive values indicate inhibition and negative values indicate activation.
Clog P calculated using ChemDraw 19.1 for the various structures.
Methylation (2a) or acylation (2b) of the amine nitrogen substantially reduced activity. Base-catalyzed hydration of the nitrile to an amide resulted in a compound (2c) that was an NRF2 pathway activator (negative % activity), which could be an indication that hydrogen bonding donors/acceptors are important for the inhibition. Even simple bromination of the 4-position of the aromatic group to give 2d lowered the biological activity compared to 1a.
Due to the slow turnover frequency with heterocycle substrate in titanium-catalyzed iminoamination reactions, a different synthetic route was developed to expand the types of aromatic groups that would be added to the pyridine framework. We prepared 2-bromo-5-phenylnicotinonitrile 3-III (Scheme 2) on multigram scales in a few steps. The first step was a condensation involving 3-dimethylamino-2-phenylacrylaldehyde 3-I, itself prepared by Vilsmeier–Haack formylation of phenylacetaldehyde dimethyl acetal. The aldehyde reacts with cyanoacetamide in the presence of base to give the 2-hydroxypyridine 3-II. The conversion of the hydroxyl to bromo occurs in high yield with NBu4+Br− and P2O5.
Scheme 2. Synthesis of 2-bromo-5-phenylnicotinonitrile (3-III).

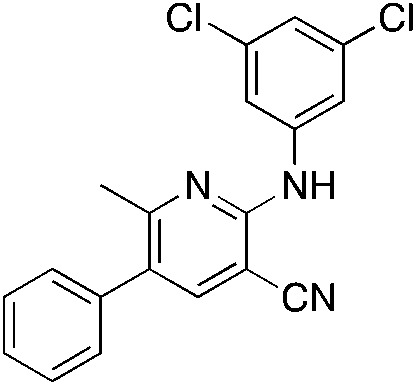
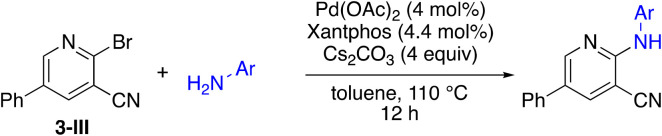
The 2-bromo-5-phenylnicotinonitrile 3-III in Scheme 2 was found to be a good substrate for Buchwald–Hartwig coupling with various aniline derivatives, as shown in Table 3. Of these compounds, of specific note is 3i.§ All the compounds in Table 3 are closely related, e.g., 3f–3i are isomers; as such, they have a clog P of ∼2.5. However, 3i is substantially more biologically active and shows good kinetic solubility. MSU38225 has a solubility of 3 μM in pH = 7.4 phosphate-buffered saline. The solubility of 3i is 70 times greater at 209 μM. Once purified, it was obvious that 3i exhibits two isomers in solution, and, to clarify, an X-ray diffraction study was undertaken. As shown in Fig. 1, the compound undergoes an intramolecular cyclization involving nucleophilic attack of one of the pyridine rings on the nitrile. This intramolecular cyclization was not observed on 3d or 3f due to methyl substitution adjacent to the pyridine nitrogen.
MSU38225 derivatives prepared by Buchwald–Hartwig couplinga.
| ||||
|---|---|---|---|---|
| Compound | H2NAr | Clog Pb (solub.) | % yield | % activity |
| 3a |
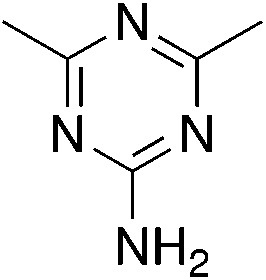
|
−0.05 | 12 | 9.6 ± 0.2 |
| 3b |
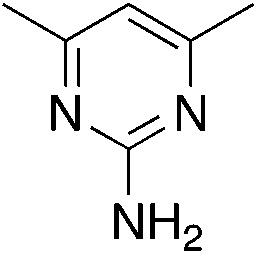
|
1.4 | 69 | 11.2 ± 5.8 |
| 3c |
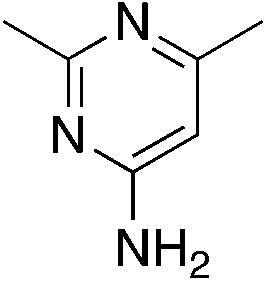
|
1.4 | 21 | −33.9 ± 13.6 |
| 3d |
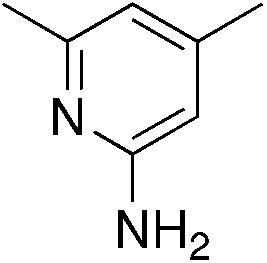
|
2.9 | 50 | 20.0 ± 7.6 |
| 3e |
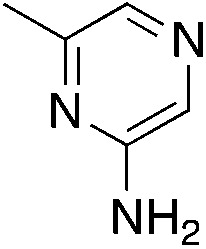
|
0.95 | 35 | 13.2 ± 7.2 |
| 3f |
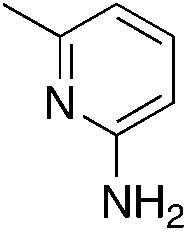
|
2.4 | 63 | −13.8 ± 5.5 |
| 3g |
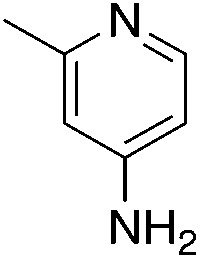
|
2.4 | 68 | −20.3 ± 9.2 |
| 3h |
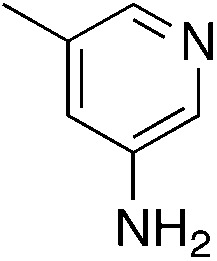
|
2.4 | 68 | 25.3 ± 5.2 |
| 3i |
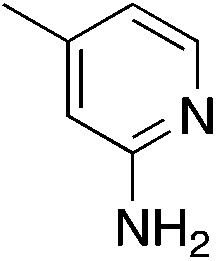
|
2.4 | 42 | 46.7 ± 4.0 |
| 1.5c (209 μM) | ||||
| 3j |
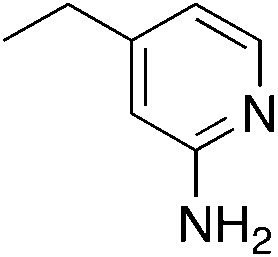
|
2.9 | 30 | 42.3 ± 3.9 |
| 2.1c | ||||
| 3k |
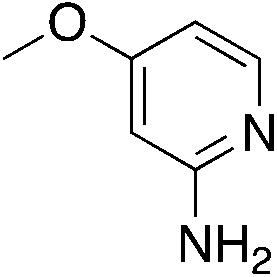
|
1.9 | 62 | 39.7 ± 4.2 |
| 0.9c | ||||
| 3l |
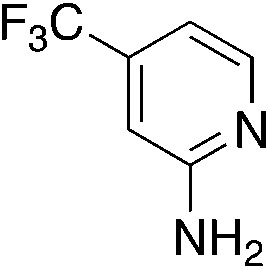
|
2.8 | 77 | 55.3 ± 6.8 |
| 1.9c | ||||
| 3m |
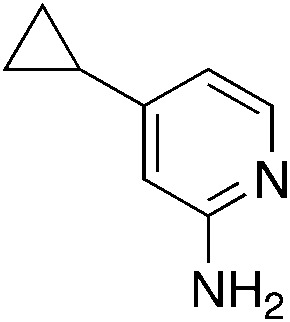
|
2.9 | 22 | −23.6 ± 7.8 |
| 2.0c | ||||
| 3n |
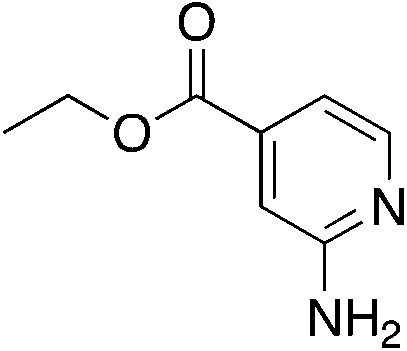
|
2.4 | 15 | −0.1 ± 4.3 |
| 1.5c | ||||
| 3o |
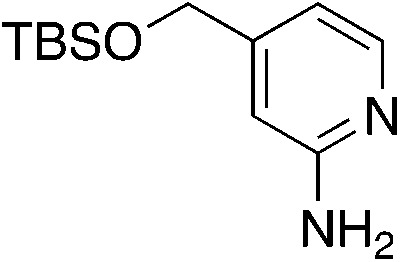
|
3.7 | 82 | 43.3 ± 6.8 |
| 2.8c | ||||
| 3p |
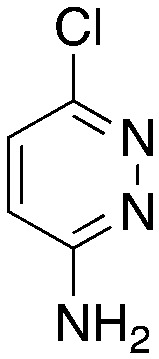
|
1.4 | 23 | −130.6 ± 33.4 |
| 0.6c | ||||
| 3q |
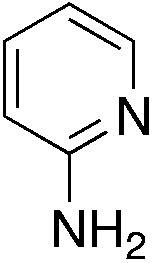
|
1.9 | 19 | 25.5 ± 11.8 |
| 1.0c | ||||
| 3r |
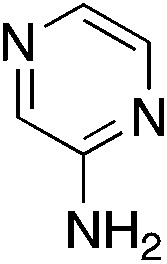
|
0.5 | 40 | 28.3 ± 12.6 |
| −0.5c | ||||
All compounds were purified to >98% purity by GC-FID. Yields are of purified compound after two synthetic steps, except as specified. Cellular activity where positive values indicate inhibition and negative values indicate activation.
Clog P calculated using ChemDraw 19.1 for the various structures. The experimental kinetic solubilities in μM were measured for a few derivatives.
Clog P value of cyclized form.
Fig. 1. Equilibrium between cyclized and uncyclized isomers for 3i. The compound crystallizes as the cyclized species, which was characterized by single crystal X-ray diffraction and an ORTEP diagram (thermal ellipsoids = 50%) is shown. Hydrogens in calculated positions, a second chemically equivalent molecule in the asymmetric unit, and CH2Cl2 of crystallization are omitted. The rate constants in DMSO-d6 were found by fitting the concentrations of both isomers versus an internal standard of ferrocene as functions of time by 1H NMR spectroscopy (see the ESI†).

For 3i, the closed form has a clog P of ∼1.5, which suggests that the cyclized form is more water soluble compared to uncyclized form. When the compound is placed in solution, an equilibrium begins that takes ∼36 h to reach completion in DMSO-d6. The cyclized compound is favored in this polar medium with Keq = 0.49 measured by 1H NMR. The forward (k1 = 1.5 × 10−5 s−1) and back (k−1 = 3.0 × 10−5 s−1) rate constants were also measured by fitting the 1H NMR-derived concentrations as a function of time (see the ESI†).
In CDCl3, the cyclized form is also favored with a Keq = 0.77 (k1 = 3.9 × 10−4 s−1, k−1 = 5.1 × 10−4 s−1); however, in less polar toluene the open form becomes favored with a Keq = 1.48 (k1 = 4.8 × 10−5 s−1, k−1 = 3.3 × 10−5 s−1). DFT calculations (B3PW91/cc-PVTZ) suggest that 3i should favor the uncyclized species in a nonpolar environment as well (see the ESI†).
To explore the effect of moving the 5-phenyl group to the 4-position of the core pyridine, an alternative substrate for C–N bond coupling, commercially available 2-chloro-4-phenylnicotinonitrile, was utilized (Table 4). Testing of the 4-phenyl isomers of MSU38225 (4a) demonstrated that these compounds also display reasonable inhibition activity, although slightly less than their 5-phenyl analogs, cf., 4a (36.4%) vs.1a (51%) and 4c (27.9%) vs.3i (46.7%). No intramolecular cyclization products were observed for 4d–f, presumably due steric inhibition by phenyl adjacent to the nitrile.
Buchwald–Hartwig coupling to generate 4-phenylpyridine derivativesa.
| ||||
|---|---|---|---|---|
| Compound | H2NAr | Clog Pb | % yield | % activity |
| 4a |
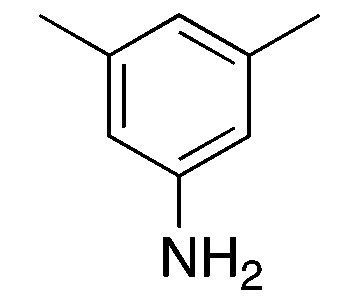
|
4.4 | 68 | 39.0 ± 4.4 |
| 4b |
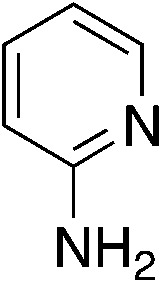
|
1.9 | 94 | −78.1 ± 11.4 |
| 4c |
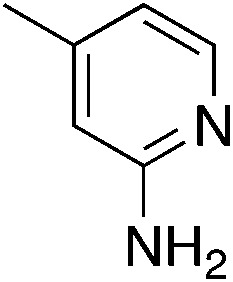
|
2.4 | 73 | 35.4 ± 7.4 |
| 4d |
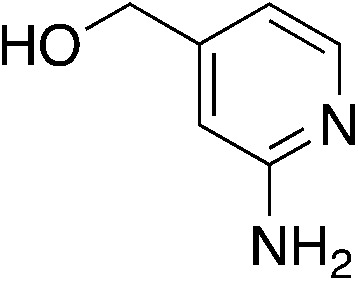
|
1.4 | 74 | −4.65 ± 5.5d |
| 75c | −31.77 ± 4.0 | |||
| 4e |
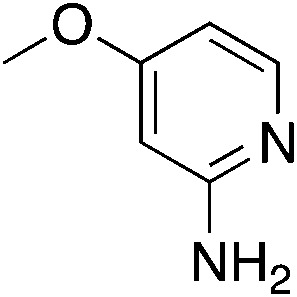
|
1.9 | 87 | −4.5 ± 1.8 |
| 4f |
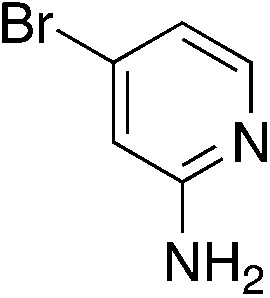
|
2.8 | 37 | 7.5 ± 5.1 |
All compounds were purified to >98% purity by GC-FID. Yields are of purified compound after two synthetic steps, except as specified. Cellular activity where positive values indicate inhibition and negative values indicate activation.
Clog P calculated using ChemDraw 19.1 for the various structures.
Deprotection yield.
Activity before deprotection.
Due to the strong tendency for sp2-carbons on the alkyne to be placed in the 5-position of the pyridine during the synthesis, Buchwald–Hartwig coupling was used to prepare 5b (Table 5), the 6-phenyl derivative of active compound 1b. For good measure, the 6-methyl derivative with no substituent on the 5-position was also prepared, which is MSU38225 (1a) sans 5-phenyl. These derivatizations extinguished the NRF2 inhibition activity observed in 1a and 1b.
Buchwald–Hartwig coupling to generate 6-substituted pyridine derivativesa.
| ||||
|---|---|---|---|---|
| Compound | Product | Clog Pb | % yield | % activity |
| 5a |
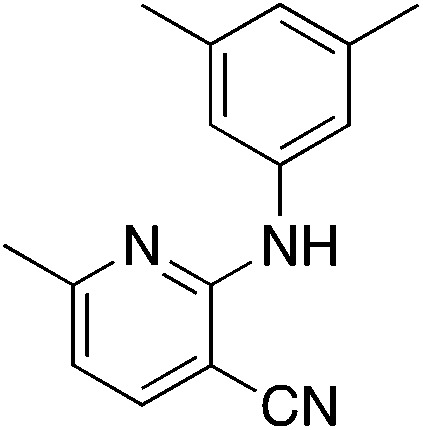
|
3.1 | 56 | 6.1 ± 12.5 |
| 5b |
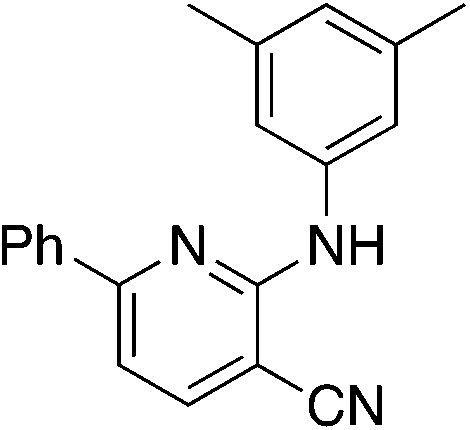
|
4.7 | 95 | 3.7 ± 5.9 |
All compounds were purified to >98% purity by GC-FID. Yields are of purified compound after two synthetic steps, except as specified. Cellular activity where positive values indicate inhibition and negative values indicate activation.
Clog P calculated using ChemDraw 19.1 for the various structures.
A complementary synthetic route to titanium-catalyzed pyridine synthesis to incorporate heterocycles at the 5-position was developed using a selective chloride replacement at the 2-position of a pyridine in the presence of a 5-bromo group via Buchwald–Hartwig coupling using Pd2(dba)3 with DMAPF ((eqn 1)).26 This left the 5-bromo-2-((3,5-dimethylphenyl)amino)nicotinonitrile (6-I), which is amenable to functionalization. Suzuki coupling utilized palladium(0) tetrakis(triphenylphosphine) with aqueous potassium carbonate in dioxane.27 The reactions, generally, were high yielding using this synthetic protocol (Table 6); however, the substituted phenyl and pyridine compounds prepared in this class thus far have not exhibited NRF2 activity on par with 1a or 3i.
Suzuki coupling reactions with 5-bromopyridinesa.
| ||||
|---|---|---|---|---|
| Compound | ArB(OH)2 | Clog Pb | % yield | % activity |
| 6a |
|
5.4 | 74 | 25.1 ± 7.1 |
| 6b |
|
4.3 | 84 | −23.3 ± 2.7 |
| 6c |
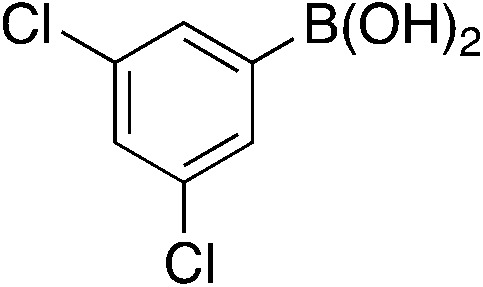
|
5.9 | 20 | 2.9 ± 2.4 |
| 6d |
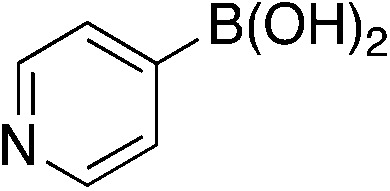
|
2.9 | 81 | −42.8 ± 1.4 |
| 6e |
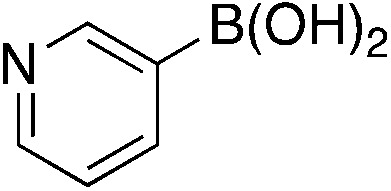
|
2.9 | 79 | −8.7 ± 4.4 |
All compounds were purified to >98% purity by GC-FID. Yields are of purified compound after two synthetic steps, except as specified. Cellular activity where positive values indicate inhibition and negative values indicate activation.
Clog P calculated using ChemDraw 19.1 for the various structures.
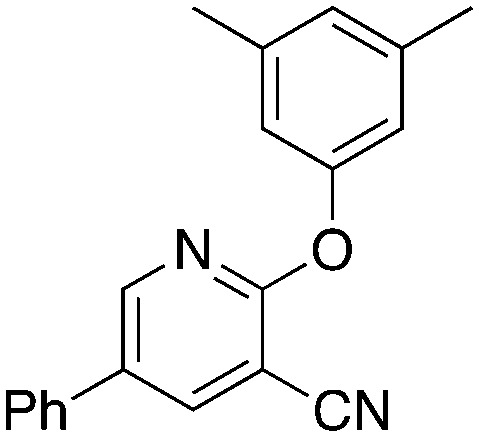
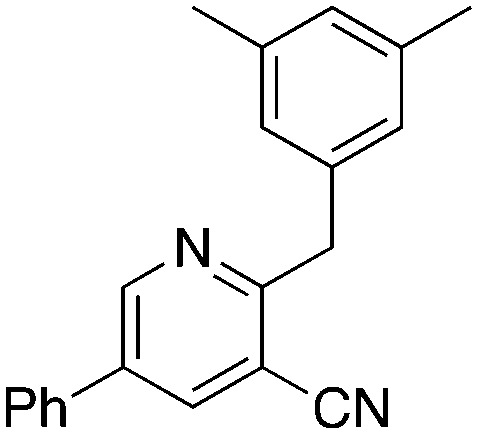
To further investigate the effect of the 2-position on activity, 1b derivatives with bridging O and CH2 linkers in place of NH (Table 7) were prepared. Replacement of the bromide in 2-bromo-5-phenylnicotinonitrile (3-III) occurs with simple nucleophiles. The O-linked compound was prepared by reaction with the sodium arylate, and the CH2-bridged compound was accessed using BrZnCH2(3,5-Me2C6H3) prepared in situ. Both of these substitutions of the NH linker resulted in compounds that acted as NRF2 activators in the assay.
 |
1 |
Replacements on 2-bromo-5-phenylnicotinonitrile to give O and CH2 linkersa.
| ||||
|---|---|---|---|---|
| Compound | Clog Pb | Reagents | % yield | % activity |
 7a 7a |
5.1 | 3,5-Dimethylphenol, NaH | 58 | −45.4 ± 10.4 |
 7b 7b |
5.2 | 3,5-Dimethylbenzyl bromide, Zn dust | 88 | −33.9 ± 13.6 |
All compounds were purified to >98% purity by GC-FID. Yields are of purified compound after two synthetic steps, except as specified. Cellular activity where positive values indicate inhibition and negative values indicate activation.
Clog P calculated using ChemDraw 19.1 for the various structures.
Finally, a few different cores in place of pyridine were explored. To increase solubility while maintaining a structure very similar to MSU38225, we explored the use of pyrazines in place of the pyridine. A nearly ideal starting material, 2-amino-3-cyano-5-phenylpyrazine 8-I, for making derivatives like 1b can be synthesized with a 2-step synthesis (Scheme 3).28 Chan–Lam coupling was used to install aromatic groups onto the NH2-group in this starting material to make a compound that only differs from 1b by having a nitrogen in the 4-position of the core in place of a CH (8a); interestingly, this compound showed no NRF2 inhibition activity. Placing a 3,5-(OMe)2C6H3 group on the 2-nitrogen gave a compound that activated the NRF2 pathway. The 2-NH2 starting material (8-I) was also tested and found to be an NRF2 pathway activator, −25 ± 20% (Table 8).
Scheme 3. Synthesis of 3-amino-6-phenylpyrazine-2-carbonitrile (8-I).

Chan–Lam coupling to generate pyrazine derivativesa.
| ||||
|---|---|---|---|---|
| Compound | Ar | Clog Pb | % yield | % activity |
| 8a | 3,5-Me2C6H3 | 3.4 | 9 | −7.8 ± 5.2 |
| 8b | 3,5-(OMe)2C6H3 | 2.2 | 11 | −66.0 ± 21.4 |
All compounds were purified to >98% purity by GC-FID. Yields are of purified compound after two synthetic steps, except as specified. Cellular activity where positive values indicate inhibition and negative values indicate activation.
Clog P calculated using ChemDraw 19.1 for the various structures.
In place of pyridine, quinoline and 1,8-napthyridine were also briefly explored as core structures as shown in Table 9. The 2-amino starting materials were readily prepared using a Friedlander reaction between the 2-aminoarylaldehydes and malononitrile. Substituted aromatics were then installed onto the 2-amino group using Buchwald–Hartwig coupling catalyzed by Pd(OAc)2 and XantPhos. However, these alternative cores were not inhibitors, and, in the case of 9b and 9c, are NRF2 activators.
Synthesis of quinoline and 1,8-napthyridine derivativesa.
| |||
|---|---|---|---|
| Compound | Clog Pb | % yield | % activity |
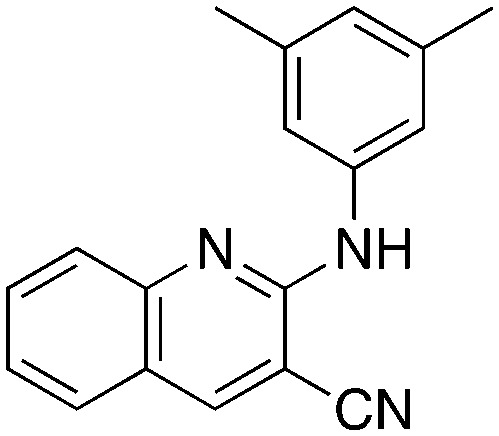 9a 9a |
3.9 | 93 | 21.2 ± 13.8 |
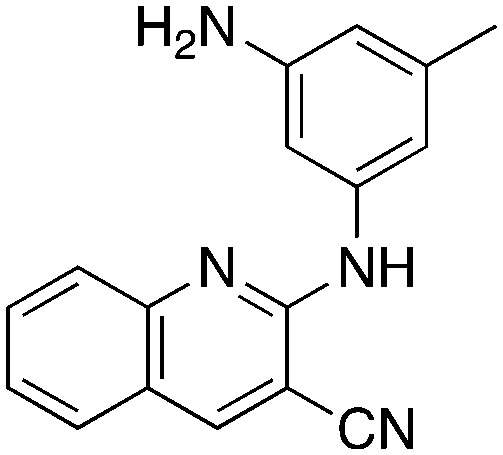 9b 9b |
2.2 | 62 | −9.5 ± 2.4 |
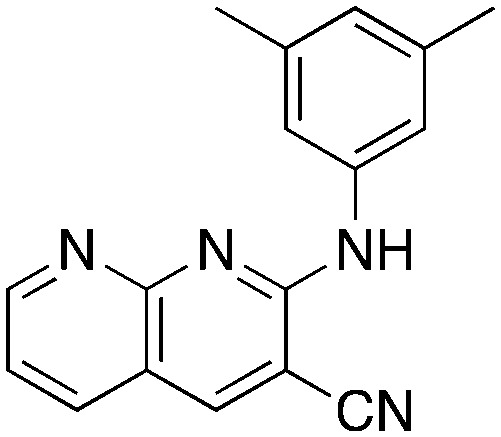 9c 9c |
2.4 | 30 | −42.7 ± 16.6 |
All compounds were purified to >98% purity by GC-FID. Yields are of purified compound after two synthetic steps, except as specified. Cellular activity where positive values indicate inhibition and negative values indicate activation.
Clog P calculated using ChemDraw 19.1 for the various structures.
Proliferation assay
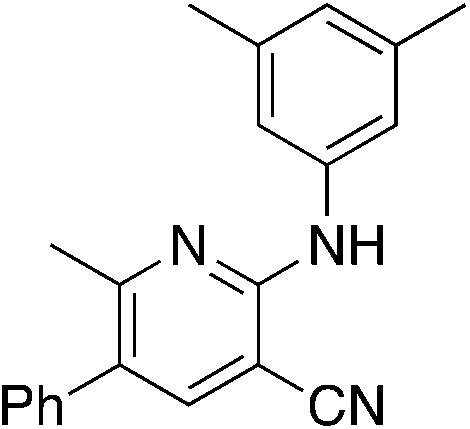
Three of the more active compounds for inhibiting NRF2 pathway activation in the luciferase reporter assay were tested for their ability to inhibit proliferation of A549 human lung cancer cells. Cells were treated with 5 μmol L−1 of compound for 72 h and viability was evaluated with an MTT assay. As shown in Table 10, all of these compounds inhibit proliferation of lung cancer cells, and 3i was the most potent tested.
Inhibition of A549 cell proliferation by novel NRF2 inhibitors.
| Compound | Structure | % inhibitiona |
|---|---|---|
| 1a (MSU38225) |
|
9.8 ± 12.9 |
| 3i |
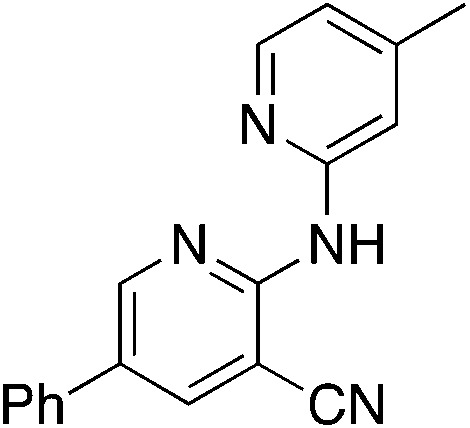
|
59.8 ± 1.8 |
| 1m |
|
35.3 ± 8.7 |
| 1t |
|
26.6 ± 0.9 |
% inhibition of proliferation relative to standard. See ESI† for more details.
Expression of NRF2 and downstream targets results
To evaluate the impact of 3i on NRF2 pathway signalling, NRF2 protein levels were assessed in non-small cell lung cancer cell lines with KEAP1-inactivating mutations that promote constitutive NRF2 pathway activity. Consistent with previous results using MSU38225,203i decreased NRF2 protein levels (Fig. 2A) to a similar extent as MSU38225 in A549 and H460 epithelial-like lung cancer cells (ATCC in Manassas, Virginia). Brusatol, a protein synthesis inhibitor and canonical inhibitor of the NRF2 pathway, was included as a positive control.29 To demonstrate functional significance of 3i-mediated NRF2 pathway downregulation, changes in NRF2 transcriptional activity in A549 cells were evaluated by measuring NRF2 target gene expression with RT-PCR. The mRNA levels of NQO1, GCLC, AKR1C2, and GCLM were significantly (p < 0.001) reduced by treatment with MSU38225 and 3i (Fig. 2B).
Fig. 2. Compound 3i inhibits expression of NRF2 target genes, reduces viability in lung cancer cells, and blocks migration. A549 or H460 cells were incubated for 24 h and (A) NRF2 protein levels or (B) mRNA expression in A549 cells were evaluated by western blotting or RT-PCR. NRF2 target genes were normalized to GAPDH and the DMSO vehicle control. Data are representative of 3 independent experiments. (C) Cells were treated for 72 h with various concentrations of 3i. Cell viability was measured using an MTT assay and values normalized as a percentage of vehicle (DMSO) control. n = 3 replicates. (D) Cells were synchronized in low serum media for 12 h, then treated for 24 h with inhibitors. Cell stage was analyzed by propidium iodide staining and flow cytometry, specifically univariate cell cycle analysis using a Watson pragmatic model with G0/1 and G2/M peak constraints (n = 2–3 independent experiments). Data are plotted as mean ± SE percentage of total cells. A549 cells were seeded in serum-free medium containing (E) DMSO vehicle, 10 μM of NRF2 pathway inhibitors MSU38225 or 3i; or (F) DMSO vehicle, and the indicated doses of 3i in the top chamber of transwells. Media containing compounds and 5% FBS as a chemoattractant was used in the bottom chamber and cells were allowed to migrate for 24 h, at which point transwell filters were fixed and unmigrated cells were removed. The remaining cells were stained with DAPI, and imaged (9 fields per transwell insert, representative image shown). Two transwell inserts were used for each condition, n = 2–3 experimental replicates. Data were analyzed by one-way or two-way ANOVA. * = p < 0.05; ** = p < 0.01; *** = p < 0.001; **** = p < 0.0001 vs. vehicle control. # = p < 0.05; ### = p < 0.001 vs. brusatol.

Viability and cell cycle assays
Given the cytoprotective effects of NRF2 pathway activation, changes in viability of the lung cancer cells with constitutive NRF2 activity were analyzed using MTT assays. Compound 3i decreased viability of A549 and H358 lung cancer cells in a dose-dependent manner with IC50 values of 0.61 and 0.58 μmol L−1, respectively, following 72 h of treatment (Fig. 2C). As determined by univariate cell cycle analysis, both MSU38225 and 3i, but not brusatol, arrested A549 and H460 cells in the G2/M phase (Fig. 2D). These results highlight differences in the mode of action between 3i and brusatol. Brusatol has been shown to globally inhibit protein synthesis.29 In contrast, MSU38225 and 3i have not been found to inhibit proteins unrelated to the NRF2 pathway.20
Migration assay
Recently, an important role for NRF2 in lung cancer metastasis has emerged.10,30 To investigate the ability of NRF2 pathway inhibitors to block cancer cell migration, we used a transwell migration assay with 5% FBS as a chemoattractant. When A549 cells were treated with vehicle or 10 μmol L−1 of MSU38225 and 3i, migration was significantly (p < 0.0001) reduced by 65% and 80%, respectively (Fig. 2E). To determine the dose-dependence of the 3i results, the transwell assay was repeated with vehicle or 1, 5, and 10 μmol L−1 of 3i. While 1 μmol L−1 of 3i was insufficient to alter A549 migration, 5 μmol L−13i reduced migration by 72% and 10 μmol L−13i reduced migration by 79% (Fig. 2F).
Conclusions
Our initial hit, MSU38225 (1a), is of low solubility and has modest (IC50 ∼ 5 μmol L−1) activity, but it is a first-in-class NRF2 inhibitor with a structure amenable to synthetic investigation. Here, ∼50 compounds with related structures were prepared to increase the activity and improve solubility. As noted, the activity is extremely sensitive to the structure (Fig. 3), which varies widely with small changes to some positions of the hit compound. Surprisingly, some of the newly synthesized compounds act as activators instead of inhibitors. In a series of experiments (Table 2), many simple modifications of 1a did not lead to improved activity, but the experiments give some insight into the features necessary for NRF2 inhibition by the class. In particular, hydration of the nitrile to an amide removed the activity (2c).
Fig. 3. Graphical summary of selected results.

Modifications of the 2-NH group were surveyed by replacing the hydrogen with a variety of groups such as Me (2a) and Ac (2b), which removed the inhibition activity. Similarly, replacing the NH with O (7a) or CH2 (7b) gave NRF2 activators.
While 1b shows activity for NRF2 inhibition, in general, removal of the 6-methyl group from structures like 1a did lower the activity, cf.1g and 1h. However, lengthening the chain in this position to an n-Pr gave a compound with no activity (1l).
There is a 5-phenyl group in 1b and moving the phenyl group to the unoccupied 4-position gave a compound with some activity (4a). This was seen in other compounds as well (3ivs.4c), where the 4-Ph was somewhat less active than if this group is in the 5-position. Placing the phenyl group in the 6-position extinguished the activity entirely, consistent with the activity of other compounds larger than methyl (e.g., 1l).
Replacement of 5-phenyl with aromatics containing donor atoms and heterocycles can give potent activators. In fact, para-substitution does not seem to be well tolerated, but chloro and methyl can be added to meta-positions to get inhibitors.
Most core changes led to no activity or compounds that were activators. One exception is the switch from pyridine to quinoline (1i), which is promising.
Similarly promising results were found by replacing the groups on the 2-NH with various heterocycles. In particular, 3i shows activity slightly higher than our initial hit (1a, MSU38225). The compound also demonstrates a reversible cyclization reaction (Fig. 1). Considering the nitrile seems to be important for activity (vide supra), it seems likely that the “open” form is what leads to the activity. This cyclization seems to be blocked by addition of a methyl group adjacent to the pyridine nitrogen (3d); however, this compound is inactive, muddling conclusions regarding the utility of the cyclization in the desired biological activity.
A very promising lead compound was found in 3i, which reduces NRF2 protein levels and mRNA of downstream target genes, inhibits proliferation of lung cancer cells by inducing G2/M arrest, and suppresses invasion. Compound 3i has markedly better anti-proliferative and anti-migratory activity (Fig. 2E) than MSU38225 (1a) and works through a different mechanism than brusatol (Fig. 2D). In addition, 3i has a kinetic solubility ∼70 higher (209 μM) than 1a. Further studies on the activities of 3i and related compounds and their anticancer activity in vivo are underway.
Abbreviations
- DMAPF
1,1′-Bis(bis(dimethylamino)phosphino)ferrocene
- Xantphos
4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene
- BINAP
(2,2′-Bis(diphenylphosphino)-1,1′-binaphthyl)
- NBS
N-Bromosuccinimide
- mCPBA
meta-Chloroperoxybenzoic acid
- H2dpm
5,5-Dimethyldipyrrolylmethane
- tBHQ
tert-Butylhydroquinone
- MTT
3-(4,5-Demethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
Conflicts of interest
A patent application has been submitted on the compounds in this article.
Supplementary Material
Acknowledgments
This work was supported by the National Science Foundation CHE-1953254 and MRI-1919565, MTRAC for Life Sciences Innovation Hub-Mi-Kickstart Award, a Molecular Discovery Group Pilot Grant from MSU, Discovery Funding Initiative (DFI) funding from MSU's Office of Research and Innovation, ACS-Petroleum Research Fund 65702-ND3, and NIH R01 grant (RC226690). Di Zhang was supported by the Integrative Pharmacology Sciences Training Program (IPSTP) training fellowship 5T32GM092715-07, a fellowship from the Aitch Foundation, a Penner Fund Fellowship, and a NIH NCI F99/K00 fellowship (CA245473). Kelly Aldrich was supported by a grant from the American Association of Women. Zhilin Hou was supported by a fellowship from Michigan State University. The authors thank Edmund Ellsworth for helpful discussion.
Electronic supplementary information (ESI) available: Supplementary schemes for intermediates, synthetic details, characterization data for new compounds, data for kinetic studies related to ring closure, details for DFT studies, details for biological assays, representative purity analysis traces by GC/FID or HPLC, and 1H/13C NMR spectra for new compounds. CCDC data for the X-ray diffraction study is available from the Cambridge Crystallographic Database under reference number 2091559. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2md00211f
Footnotes
Closely related, fluorinated 3l has activity and solubility comparable to 3i but hasn't seen as extensive study.
Notes and references
- Menegon S. Columbano A. Giordano S. Trends Mol. Med. 2016;22:578–593. doi: 10.1016/j.molmed.2016.05.002. [DOI] [PubMed] [Google Scholar]
- Sporn M. B. Liby K. T. Nat. Rev. Cancer. 2012;12:564–571. doi: 10.1038/nrc3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor T. O. Huang M. T. Prawan A. Liu Y. Hao X. P. Yu S. W. Cheung W. K. L. Chan J. Y. Reddy B. S. Yang C. S. Kong A. N. Cancer Prev. Res. 2008;1:187–191. doi: 10.1158/1940-6207.CAPR-08-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkova-Kostova A. T. Ann. Nutr. Metab. 2015;67:14. [Google Scholar]
- Dinkova-Kostova A. T. Fahey J. W. Kostov R. V. Kensler T. W. Trends Food Sci. Technol. 2017;69:257–269. doi: 10.1016/j.tifs.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagishita Y. Fahey J. W. Dinkova-Kostova A. T. Kensler T. W. Molecules. 2019;24:3593. doi: 10.3390/molecules24193593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson H. Dinkova-Kostova A. T. Hayes J. D. Cancers. 2020;12:3609. doi: 10.3390/cancers12123609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collisson E. A. Campbell J. D. Brooks A. N. Berger A. H. Lee W. Chmielecki J. Beer D. G. Cope L. Creighton C. J. Danilova L. et al. . Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerman P. S. Lawrence M. S. Voet D. Jing R. Cibulskis K. Sivachenko A. Stojanov P. McKenna A. Lander E. S. Gabriel S. et al. . Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lignitto L. LeBoeuf S. E. Homer H. Jiang S. W. Askenazi M. Karakousi T. R. Pass H. I. Bhutkar A. J. Tsirigos A. Ueberheide B. Sayin V. I. Papagiannakopoulos T. Pagano M. Cell. 2019;178:316. doi: 10.1016/j.cell.2019.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Lang F. C. Yang C. Z. Pharmacol. Ther. 2021;217:107664. doi: 10.1016/j.pharmthera.2020.107664. [DOI] [PubMed] [Google Scholar]
- Ren D. M. Villeneuve N. F. Jiang T. Wu T. D. Lau A. Toppin H. A. Zhang D. D. Proc. Natl. Acad. Sci. U. S. A. 2011;108:1433–1438. doi: 10.1073/pnas.1014275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian S. Ma T. P. Lee J. Haverty P. M. Kirkpatrick D. S. Yu K. B. Stokoe D. Mol. Cell. Proteomics. 2016;15:1220–1231. doi: 10.1074/mcp.M115.055509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollong M. J. Yun H. Sherwood L. Woods A. K. Lairson L. L. Schultz P. G. ACS Chem. Biol. 2015;10:2193–2198. doi: 10.1021/acschembio.5b00448. [DOI] [PubMed] [Google Scholar]
- Singh A. Venkannagari S. Oh K. H. Zhang Y. Q. Rohde J. M. Liu L. Nimmagadda S. Sudini K. Brimacombe K. R. Gajghate S. Ma J. F. Wang A. Xu X. Shahane S. A. Xia M. G. Woo J. Y. Mensah G. A. Wang Z. B. Ferrer M. Gabrielson E. Li Z. Y. Rastinejad F. Shen M. Boxer M. B. Biswal S. ACS Chem. Biol. 2016;11:3214–3225. doi: 10.1021/acschembio.6b00651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. Lim M. J. Kim M. H. Yu C. H. Yun Y. S. Ahn J. Song J. Y. Free Radical Biol. Med. 2012;53:807–816. doi: 10.1016/j.freeradbiomed.2012.05.038. [DOI] [PubMed] [Google Scholar]
- Liao H. Zhu D. Bai M. Z. Chen H. F. Yan S. H. Yu J. Zhu H. T. Zheng W. X. Fan G. R. Cancer Cell Int. 2020;20:480. doi: 10.1186/s12935-020-01470-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D. Liu Y. Zhou Y. Q. Ruiz-Rodado V. Larion M. Xu G. W. Yang C. Z. Proc. Natl. Acad. Sci. U. S. A. 2020;117:9964–9972. doi: 10.1073/pnas.1913633117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado A. Rojo A. I. Wells G. Hayes J. D. Cousin S. P. Rumsey W. L. Attucks O. C. Franklin S. Levonen A. L. Kensler T. W. Dinkova-Kostova A. T. Nat. Rev. Drug Discovery. 2019;18:295–317. doi: 10.1038/s41573-018-0008-x. [DOI] [PubMed] [Google Scholar]
- Zhang D. Hou Z. Aldrich K. E. Lockwood L. Odom A. L. Liby K. T. Mol. Cancer Ther. 2021;20:1692–1701. doi: 10.1158/1535-7163.MCT-21-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom A. L. Dalton Trans. 2005:225–233. doi: 10.1039/b415701j. doi: 10.1039/b415701j. [DOI] [PubMed] [Google Scholar]
- Odom A. L. McDaniel T. J. Acc. Chem. Res. 2015;48:2822–2833. doi: 10.1021/acs.accounts.5b00280. [DOI] [PubMed] [Google Scholar]
- Cao C. S. Shi Y. H. Odom A. L. J. Am. Chem. Soc. 2003;125:2880–2881. doi: 10.1021/ja0284714. [DOI] [PubMed] [Google Scholar]
- Dissanayake A. A. Staples R. J. Odom A. L. Adv. Synth. Catal. 2014;356:1811–1822. doi: 10.1002/adsc.201301046. [DOI] [Google Scholar]
- Shi Y. H. Hall C. Ciszewski J. T. Cao C. S. Odom A. L. Chem. Commun. 2003:586–587. doi: 10.1039/b212423h. doi: 10.1039/b212423h. [DOI] [PubMed] [Google Scholar]
- Keylor M. H. Niemeyer Z. L. Sigman M. S. Tan K. L. J. Am. Chem. Soc. 2017;139(31):10613–10616. doi: 10.1021/jacs.7b05409. doi: 10.1021/jacs.7b05409. [DOI] [PubMed] [Google Scholar]
- Cuny G. D. Yu P. B. Laha J. K. Xing X. C. Liu J. F. Lai C. S. Deng D. Y. Sachidanandan C. Bloch K. D. Peterson R. T. Bioorg. Med. Chem. Lett. 2008;18:4388–4392. doi: 10.1016/j.bmcl.2008.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haight A. R. Bailey A. E. Baker W. S. Cain M. H. Copp R. R. DeMattei J. A. Ford K. L. Henry R. F. Hsu M. C. Keyes R. F. King S. A. McLaughlin M. A. Melcher L. M. Nadler W. R. Oliver P. A. Parekh S. I. Patel H. H. Seif L. S. Staeger M. A. Wayne G. S. Wittenberger S. J. Zhang W. J. Org. Process Res. Dev. 2004;8:897–902. doi: 10.1021/op049889k. [DOI] [Google Scholar]
- Harder B. Tian W. La Clair J. J. Tan A. C. Ooi A. Chapman E. Zhang D. D. Mol. Carcinog. 2017;56:1493–1500. doi: 10.1002/mc.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiel C. Le Gal K. Ibrahim M. X. Jahangir C. A. Kashif M. Yao H. D. Ziegler D. V. Xu X. F. Ghosh T. Mondal T. Kanduri C. Lindahl P. Sayin V. I. Bergo M. O. Cell. 2019;178:330. doi: 10.1016/j.cell.2019.06.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.