

Abstract
Motivation
Amplicon sequencing is widely applied to explore heterogeneity and rare variants in genetic populations. Resolving true biological variants and quantifying their abundance is crucial for downstream analyses, but measured abundances are distorted by stochasticity and bias in amplification, plus errors during polymerase chain reaction (PCR) and sequencing. One solution attaches unique molecular identifiers (UMIs) to sample sequences before amplification. Counting UMIs instead of sequences provides unbiased estimates of abundance. While modern methods improve over naïve counting by UMI identity, most do not account for UMI reuse or collision, and they do not adequately model PCR and sequencing errors in the UMIs and sample sequences.
Results
We introduce Deduplication and Abundance estimation with UMIs (DAUMI), a probabilistic framework to detect true biological amplicon sequences and accurately estimate their deduplicated abundance. DAUMI recognizes UMI collision, even on highly similar sequences, and detects and corrects most PCR and sequencing errors in the UMI and sampled sequences. DAUMI performs better on simulated and real data compared to other UMI-aware clustering methods.
Availability and implementation
Source code is available at https://github.com/DormanLab/AmpliCI.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Amplicon sequencing has been widely applied to explore complex genetic populations, such as viral quasispecies (Bhiman et al., 2015; Caskey et al., 2017; Jabara et al., 2011; Seifert et al., 2016; Zhou et al., 2015), microbial communities (Callahan et al., 2016; Fields et al., 2021; Karst et al., 2021), cancer tumors (Newman et al., 2016; Shu et al., 2017), immune receptor repertoires (Rosati et al., 2017; Shugay et al., 2014; Vander Heiden et al., 2014) and engineered barcodes (Blundell and Levy, 2014; Kebschull and Zador, 2018; McKenna and Gagnon, 2019). These experiments generate high-coverage sequence data for targeted genomic regions, enabling the detection of variants with single-nucleotide differences or low abundance (Callahan et al., 2017; Hathaway et al., 2018). However, to attach sequencing adapters (Rohland and Reich, 2012) and achieve deep coverage, the targets are amplified by polymerase chain reaction (PCR) and sequenced using error-prone high-throughput technologies. Stochasticity, bias in amplification, along with PCR and sequencing errors obscure the true variants and distort abundances (Kebschull and Zador, 2015; Varghese et al., 2010). Bias is especially problematic in low-biomass samples, like single cells, where few template molecules are intensely amplified (Kivioja et al., 2011; Sze and Schloss, 2019). Errors, chimeras and amplification bias accelerate with more PCR cycles (Sze and Schloss, 2019; Varghese et al., 2010), burying real sequences in increasing noise.
PCR bias is solved by removing PCR-duplicated molecules, but duplicate detection is difficult for amplicon data. Unique molecular identifiers (UMIs), attached to sample molecules before amplification (Hug and Schuler, 2003; König et al., 2010) can directly mark duplicates. UMIs can detect ultra-low frequency variants (Kinde et al., 2011; Xu et al., 2019), eliminate amplification bias (Jabara et al., 2011; Kivioja et al., 2011) and provide cleaner data for downstream analysis (Kim et al., 2020; Svensson, 2020; Townes et al., 2019).
The first UMI processing pipelines (Chen et al., 2019; Shugay et al., 2017; Stoler et al., 2016; Vander Heiden et al., 2014) clustered reads by UMI identity and generated a consensus sample sequence per cluster. Unfortunately, UMIs can collide, identically marking two or more sampled molecules (Clement et al., 2018) and leading to undercount, loss or misestimation of sampled variants. And UMIs experience both PCR and sequencing errors, leading to the discovery of false (error) variants and inflated counts of sampled molecules (Smith et al., 2017). Modern deduplication tools account for errors in UMIs, and sometimes, UMI collisions (Supplementary Table S1). Calib (Orabi et al., 2019) clusters whole reads, the UMI and sample sequence, by similarity. Use of whole reads can resolve UMI collisions attached to distinct sample sequences, but clustering without accounting for sequence abundance is difficult to calibrate, likely to over-merge similar reads or under-merge multi-error misreads of abundant haplotypes (Amir et al., 2017; Callahan et al., 2016; Peng and Dorman, 2021). Starcode-umi (Zorita et al., 2015) clusters UMIs and sample sequences separately, using distance and cluster abundance thresholds that need careful tuning (Orabi et al., 2019) then reconstructs error-free reads from denoised UMIs and sample sequences. UMI-tools (Smith et al., 2017) clusters UMIs on a network, using abundance and similarity, but it only avoids UMI collision by partitioning UMIs on reference mapping position.
We introduce DAUMI, Deduplication and Abundance estimation with Unique Molecular Identifiers, a novel probabilistic framework to detect true biological amplicon sequences and accurately estimate their deduplicated abundance (Fig. 1). DAUMI better detects and corrects PCR, sequencing and collision errors on simulation and real data than traditional UMI-aware consensus methods. DAUMI is incorporated in AmpliCI, our amplicon deduplication software, previously only capable of denoising amplicons without UMIs (Peng and Dorman, 2021).
Fig. 1.
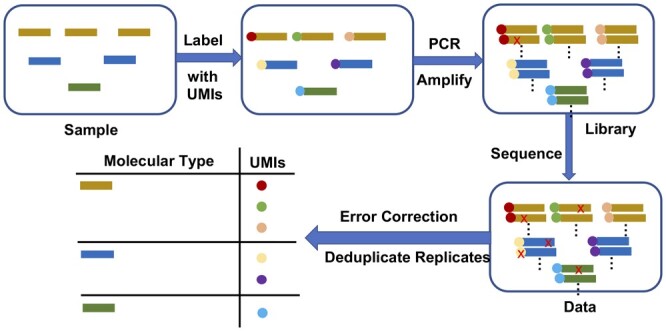
Deduplication and error correction with UMIs. Red crosses are PCR or sequencing errors. Our goal is to resolve the true biological sequences in the sample, as well as their sampled absolute abundance
2 Materials and methods
We start with a dataset of n single-end reads, where the ith read contains an observed UMI and sample sequence . A pair of hidden variables for read i encodes the unknown source of UMI sequence , drawn from a set of N true UMIs, , and the unknown source of sample sequence , drawn from a set of K true sample sequences (haplotypes), .
2.1 Model
Each read is assumed to be an independent realization of a one-step Hidden markov model (Fig. 2). True UMI is chosen with mixing proportion ηs, a value determined by stochastic amplification, UMI collision and accidental inclusion of low-frequency error UMIs in . Next, haplotype is attached to with probability γsk. Finally, the observed barcode and sample read are sequenced from and with possible errors. The set of all observed barcodes and sample reads has observed log likelihood
| (1) |
where , with and , are model parameters.
Fig. 2.
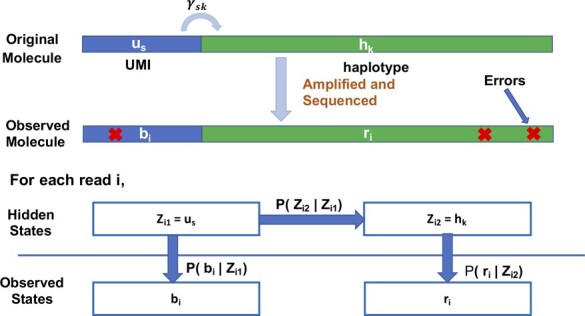
One-step hidden Markov model. A read has two hidden states, true UMI attached to haplotype by transition matrix , and two observed noisy emission states, UMI and sample sequence
We expect , i.e. only one haplotype is marked by UMI , but recombination during PCR (e.g. chimeras) (Kebschull and Zador, 2015; Potapov and Ong, 2017) and UMI collision may create more than one positive γsk per UMI . To encourage the sparsity in we expect in a well-executed experiment, we maximize the observed penalized log likelihood, where penalty (Armagan et al., 2013; Candès et al., 2008),
| (2) |
for constants , is subtracted from (1). As ω approaches 0, the penalty becomes -like, such that estimated transitions when the expected amplified count of molecule drops below threshold ρ. UMI collision is implied when expected counts of and exceed ρ. Because expected counts consider sequence similarity, error rates and abundance, DAUMI can identify UMI collision even on highly similar sampled molecules (Supplementary Fig. S1).
For emission probabilities and , we use an existing sequencing error model (Peng and Dorman, 2021). First, we align the read and the candidate UMI and haplotype . The emission of observed read X ( or with indels) from true sequence Y ( or with indels) is
| (3) |
where l is the alignment length, d is the number of observed insertion/deletion (indel) events, and is the probability (1 for indels) of generating Xj from Yj with quality score qj at aligned position j. Indels follow a truncated with indel error rate δ per position per read, but we assume d = 0 in UMIs since indel errors are rare in these short, typically high quality parts of the reads.
The goal of DAUMI is to estimate true haplotypes and abundances. We choose candidate haplotypes , UMIs and parameters of (3) (see Section 2.2.1). Then, we maximize the penalized log likelihood to obtain estimates and using an expectation–maximization (EM) algorithm (details in Supplementary Section S1; demonstrated accuracy in Supplementary Fig. S8). and are fixed, but we can discard UMIs with expected abundance and the penalty may eliminate false haplotypes. Finally, the deduplicated abundance of haplotype is .
2.2 Implementation
DAUMI is implemented in C. We describe EM algorithm initialization, approximations used for speed, and selection of penalty parameters.
2.2.1 Initialization
To include all true UMIs and haplotypes in candidate pools and , while excluding most error UMIs and haplotypes, we extend UMI-unaware AmpliCI (Peng and Dorman, 2021). We set to the ‘denoised’ UMIs (option ––umi) assuming quality scores provide PHRED error probabilities (Ewing and Green, 1998). Quality scores are generally accurate (Zanini et al., 2017), but they slightly underestimate error rates (Schirmer et al., 2016; Shugay et al., 2017), suggesting this approach will liberally identify UMIs. To populate , AmpliCI is applied to full reads (UMI length set via –-trim_umi_length), using an error profile estimated from reads clustered by the above-denoised UMIs, further split by a UMI-tools-like algorithm (Smith et al., 2017). Specifically for error profile estimation, if any sample sequence in a UMI cluster has over half the abundance of the most abundant sample sequence, we partition the cluster as a possible UMI collision. The two sequences are promoted to new cluster centers, and each read in the original cluster is assigned to the closer center, by Hamming distance, or the more abundant center if equidistant. To ensure cluster centers are error free, we drop clusters where the center sequence is not sufficiently replicated (default, observed abundance < 2), then assume all differences between cluster members and cluster centers are errors. The fitted error profile is plugged into (3) and the unique, ‘denoised’ sequences, without UMI tags, form . Finally with and given, we initialize mixing proportions to relative observed abundances of UMIs and transition matrix to observed transition counts, normalizing row sums to one.
2.2.2 Approximations
For large datasets, we reduce computation and memory expended on unlikely UMI-haplotype combinations. Since Illumina errors are <0.1% per nucleotide (Stoler and Nekrutenko, 2021), we assume each true molecule has been observed without error at least once and otherwise force . Most surviving combinations have exceptionally small posterior probabilities eisk (defined in Supplementary Section S1). In the E step, we only keep the top T (default 10) most likely candidates for each read i, resetting
where is the Tth largest value among NK entries. With these approximations, estimate is possible, in which case we assume is an error UMI and exclude the sth row of . Increasing T to 30 had no effect on estimation in simulation (data not shown). DAUMI computational complexity depends on the number of UMIs , haplotypes and EM iterations (see Supplementary Table S3). Additional improvements, especially in the E step, are likely possible.
2.2.3 Selection of the penalty parameters
Fixing for an penalty (Yin, 2016), ρ is chosen to separate true from error UMIs. To automate ρ selection, we model the observed UMI abundance distribution. Specifically, we modify a Galton–Watson branching process (Stolovitzky and Cecchi, 1996) to account for amplification and sequencing error. We fit the model by minimizing the Kolmogorov–Smirnov statistic (Stephens, 1974) applied to the observed UMI abundances right-truncated for long tails caused by UMI collision or other artifacts. Selecting ρ as the fifth percentile of the fitted distribution worked well for all datasets, but we manually selected ρ = 20 for HIV V3 based on visual assessment. Details and limitations of this procedure are provided in Supplementary Material Section S2. Supplementary Table S4 shows fitted models and selected ρ for all datasets.
2.3 Data simulation
We conducted a brief simulation (pipeline in Supplementary Fig. S2 and settings, including PCR efficiencies and cycle numbers, in Supplementary Table S2). The relative abundance of K = 25 haplotypes were obtained from power law distribution . Then, N = 400 molecules were sampled from a Multinomial distribution and randomly attached to N 9 bp UMIs. For datasets with UMI collision, we resampled the N distinct UMIs with replacement before attachment. The 400 true UMIs and 25 true haplotypes were randomly selected from UMIs and haplotypes found in an HIV dataset (Bhiman et al., 2015, SRR2241783) after AmpliCI denoising (Peng and Dorman, 2021). Next, molecules were PCR amplified with error probability per base, per cycle from Quince et al. (2011) (–, depending on substitution). ART Illumina (Huang et al., 2012) simulated single-end reads with fixed length 250 bp, using error profile MSv1 to mimic Miseq reads. Indel rates were set to per position.
2.4 Running competing methods
We ran Calib (Orabi et al., 2019), UMI-tools (Smith et al., 2017), Starcode-umi (Zorita et al., 2015) and a ‘Naïve method’ that clusters reads by UMI identity (Clement et al., 2018; Vander Heiden et al., 2014) with default parameter settings, except as discussed here. Since Calib (v0.3.4) requires paired-end reads, we passed in UMI-tagged reads as forward reads and UMI-detagged reads as reverse reads (a hack suggested by the author). We disabled sequence trimming (–-seq-trim 0) in Starcode-umi (v1.3) to ensure a fair comparison with other algorithms, and set maximum edit distance one (–-umi-d 1), a common setting for UMI clustering. For UMI-tools, we used the core algorithm from its Application Programming Interface, which is stable across versions, to cluster UMIs. For the Naïve method, sequences were clustered by UMI identity, discarding singleton UMIs and attached sample sequences as likely errors. For Calib, UMI-tools and the Naïve method, we took the majority rule consensus sequence for the true UMI and haplotype (first seen of {A, T, C, G} when tied) per cluster. For Starcode-umi, we used the canonical UMIs and sample sequences estimated during clustering. Supplementary Table S6 verifies chosen parameters were optimal or near-optimal for all methods (details in Supplementary Section S3).
3 Results
We compared DAUMI to four UMI clustering methods, Calib (Orabi et al., 2019), UMI-tools (Smith et al., 2017), Starcode-umi (Zorita et al., 2015) and the Naïve method, on both simulated and real datasets. For comparison to UMI-unaware AmpliCI, see Supplementary Figure S7.
3.1 Benchmarking on simulated data
To compare methods, we simulated four datasets (Supplementary Table S2). Of 25 true haplotypes, only 23 were sampled in Simulation 2 and 22 in the others. Calib ran on subsets of Simulation 2 (50%), 3 (45%) and 4 (50%), since it failed on the whole dataset (true abundances adjusted accordingly).
DAUMI is superior in all four simulations (Fig. 3, estimates and goodness-of-fit in Supplementary Table S5). Without UMI collision, DAUMI, Calib, UMI-tools and Naïve identify all true haplotypes, but DAUMI introduces fewer false positives (Fig. 3, Simulations 1 and 2). Starcode-umi finds less true haplotypes and is the only method to overestimate abundance; its estimates also contain more noise. DAUMI excels on data with UMI collision (Fig. 3, Simulations 3 and 4). UMI-tools and Naïve, which cannot detect collision, underestimate haplotype abundance, but Calib also underestimates abundance, possibly by merging UMI clusters with similar tagged reads.
Fig. 3.

Abundance estimation on simulated data without (Simulations 1 and 2) or with (Simulations 3 and 4) UMI collision. Blue is fitted (y = bx) and red is expected (y = x) line. Number of estimated (true) haplotypes at upper left of each scatter plot
3.2 Application to HIV amplicon data
We analyzed reverse reads with attached UMIs from two HIV env amplicon datasets, V1V2 (Bhiman et al., 2015, SRR2241783) and V3 (Caskey et al., 2017, SRR5105420). For V1V2, UMIs were extracted from the reads using a custom method (Supplementary Section S5). For V3, UMIs were extracted from the read starts, dropping the following primer sequences. Reads with ambiguous nucleotides (1%) were discarded, and remaining reads were trimmed to the same length (details in Supplementary Table S8).
To assess performance when true haplotypes and abundances are unknown, we evaluated method consistency across a random bipartition (repeated five times). We report for each subset (Table 1) average number of haplotypes found and two similarity metrics: Jaccard Index measures agreement in found haplotypes (Jaccard, 1912); Ruzicka Similarity (Deza and Deza, 2006) is further weighted by estimated abundances. Methods are more consistent across subsets for V1V2 (Supplementary Fig. S11) than V3 (Supplementary Fig. S9).
Table 1.
Agreement across random partition of HIV V1V2 and V3 datasets
| Deduplicated abundance 1 |
Deduplicated abundance 2 |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dataset | Methods | Hap1 | Hap2 | SD | Jaccard | Ruzicka | Hap1 | Hap2 | SD | Jaccard | Ruzicka | ||||
| V1V2 | Calib | 808 | 797 | (21) | 0.08 | (0.00) | 0.23 | (0.00) | 43 | 43 | (4) | 0.67 | (0.03) | 0.82 | (0.02) |
| DAUMI | 90 | 90 | (5) | 0.72 | (0.03) | 0.84 | (0.02) | 37 | 37 | (2) | 0.77 | (0.05) | 0.87 | (0.02) | |
| Naïve | 293 | 303 | (9) | 0.29 | (0.01) | 0.57 | (0.01) | 43 | 43 | (3) | 0.75 | (0.03) | 0.88 | (0.03) | |
| Starcode-umi | 2304 | 2311 | (48) | 0.00 | (0.00) | 0.19 | (0.00) | 15 | 13 | (1) | 0.73 | (0.05) | 0.80 | (0.02) | |
| UMI-tools | 542 | 538 | (16) | 0.14 | (0.00) | 0.36 | (0.00) | 42 | 42 | (3) | 0.77 | (0.04) | 0.87 | (0.01) | |
| V3 | Calib | 5048 | 5021 | (57) | 0.06 | (0.00) | 0.11 | (0.00) | 64 | 66 | (4) | 0.55 | (0.04) | 0.71 | (0.02) |
| DAUMI | 180 | 171 | (7) | 0.39 | (0.01) | 0.72 | (0.01) | 68 | 72 | (4) | 0.72 | (0.03) | 0.83 | (0.01) | |
| Naïve | 1561 | 1543 | (10) | 0.06 | (0.00) | 0.22 | (0.00) | 91 | 84 | (4) | 0.64 | (0.04) | 0.74 | (0.02) | |
| Starcode-umi | 15077 | 15097 | (79) | 0.02 | (0.00) | 0.09 | (0.00) | 96 | 103 | (5) | 0.08 | (0.01) | 0.77 | (0.02) | |
| UMI-tools | 3718 | 3724 | (36) | 0.06 | (0.00) | 0.14 | (0.00) | 72 | 74 | (3) | 0.64 | (0.03) | 0.75 | (0.01) | |
Note: The mean and standard deviation (SD in parentheses) of five replicates of: Hap1 and Hap2: no. inferred haplotypes in each subset (rounded to integer); Jaccard: Jaccard Index of inferred haplotype sets; Ruzicka: Ruzicka Similarity, abundance-weighted form of Jaccard Index. For DAUMI, ρ = 10 for V3, ρ = 7 for V1V2, both subsets. Best performance is bolded per dataset.
Overall, DAUMI achieves the highest agreement in recovered haplotypes and estimated abundances on both HIV datasets (Table 1). Not surprisingly, abundant haplotypes are easier to detect, but DAUMI can also reliably find low-abundance haplotypes. Only UMI-tools and Naïve are competitive and only on the subset of haplotypes attached to at least two UMIs in V1V2. Though UMI collision is rare in these data, DAUMI and Starcode-umi detect a collision on two haplotypes with nine mismatches (Fig. 4). Within this UMI cluster (126 sequences), DAUMI infers two haplotype sequences with high observed abundance (15 and 7) that also dominate other UMI clusters. Starcode-umi finds one of these haplotypes but chooses for the other a sequence never seen in the reads. DAUMI substantially outperforms all competing methods on V3, probably because of an elevated collision rate. Supplementary Figures S5 and S6 show a long right tail despite low PCR amplification, and Supplementary Figure S10 finds excess nucleotide T, suggesting degeneracy in the random UMIs.
Fig. 4.

UMI collision in V1V2 data resolved by DAUMI. (a) Distribution of pairwise distances between unique sequences labeled with UMI GTGTCGGTA, indicating at least two clusters linked this UMI. (b) DAUMI finds two clusters (green, left, and black, right). Network nodes are unique sample sequences; edges link sequences within edit distance six
3.3 Application to single-cell data
We further compared methods on a SMART-Seq3 scRNA-seq benchmark dataset (Ziegenhain et al., 2022, E-MTAB-10372). These data include a molecular spike carrying an 18-bp internal UMI. Molecular counting by the 8 bp SMART-Seq3 UMI can be calibrated by comparing to counts of the 18-bp spike UMIs. DAUMI is not intended for general scRNA-seq data, but it can be applied to this molecular spike data and targeted single-cell data (Pokhilko et al., 2021; Woyke and Jarett, 2015). We demultiplexed using pheniqs (Galanti et al., 2021) and retained only 5′ reads of the molecular spike. These reads contain a 57-bp molecular spike sequence, including the 18-bp spike UMI and surrounding fixed sequence, which we treat as the sampled amplicon. Only reads containing the spike at the expected location and no ambiguous nucleotides are kept.
For 41 cells, we denoised the 18-bp spike UMIs using DADA2 (Callahan et al., 2016) with singletons (DETECT_SINGLETONS = TRUE) to produce gold-standard haplotypes. When comparing resolved haplotypes to the gold standard, DAUMI had the highest precision but lower recall than Naïve and Starcode-umi (Fig. 5), probably because the initialization of the UMI set is conservative on this very clean data (Supplementary Table S11). When evaluating clusters, Naïve produced slightly more homogeneous clusters than DAUMI, but DAUMI had far superior completeness (Dorman et al., 2021). Naïve oversplits clusters, spreading spike UMIs across multiple clusters, leading to overestimated UMI counts (and low completeness).
Fig. 5.

Single-cell molecular spike data. Jaccard index, precision and recall were computed by comparing the resolved haplotypes with the DADA2-estimated gold standard for each cell. Cluster homogeneity and completeness were computed by treating the 18 bp raw spike UMIs as the true class label (Starcode-umi does not provide read assignments). For DAUMI, ρ = 9 was auto-selected for all 41 cells
4 Discussion
DAUMI is a novel probabilistic framework to correct errors and deduplicate reads for accurate molecular counts of amplicon sequence data with UMIs. DAUMI correctly resolves the origin of more molecules than competing methods even with UMI collisions. Here, we discuss advantages, limitations and directions for future research.
UMI collision happens, especially when UMIs are short (Clement et al., 2018). Low nucleotide diversity in high-frequency UMIs was reported for single-cell RNA-seq (scRNA-seq) (Petukhov et al., 2018), and we observed similar patterns in the V3 data. Most UMI-based quantification methods for scRNA-seq, e.g. UMI-tools derivative Alevin (Srivastava et al., 2019), map reads to avoid UMI collision, but mapping cannot resolve amplicons. Even in scRNA-seq, mapping may under-collapse clusters (Srivastava et al., 2019), since reads of the same molecule can map to slightly different genome positions (Sena et al., 2018). Calib and Starcode-umi can detect UMI collision but performed overall worse than UMI-tools and DAUMI.
DAUMI and all methods compared here cannot detect errors introduced before UMI tagging, e.g. during cDNA synthesis. Downstream methods may detect such errors, like UMI-based variant callers (Shugay et al., 2017; Xu et al., 2019), which compare variation to background error rates after UMI clustering. Methods also struggle to detect early-cycle PCR errors, which are highly amplified and difficult to distinguish from true variation. If such errors occur in the UMI, methods may overestimate abundance by treating both UMIs as valid. Post hoc detection is possible by checking for similar UMIs attached to identical haplotypes. More likely, errors strike the sampled molecule. Other methods vary in their resolution of such cases, but DAUMI will treat the result as a UMI collision and overestimate the abundance of a false haplotype. If good experimental design has eliminated UMI collision, DAUMI abundance estimation can assign each UMI to one and only one haplotype, namely (with option –-ncollision), but when collision and early-cycle PCR error coexist, explicit modeling of the PCR process may be necessary.
DAUMI is moderately affected by penalty parameter ρ, a molecular count distinguishing error and true molecules. With UMI collisions, high ρ may merge similar haplotypes, and regardless of UMI collision, low ρ may over-split (overcount in UMI; false positive in haplotype) PCR errors. Automated selection of ρ worked well for simulated, V1V2, and single-cell data. For V3, with low UMI replication, we manually overrode the auto-selected ρ = 1 with ρ = 20, but both choices worked well (Supplementary Table S9). We also demonstrated that DAUMI has superior performance for a range of reasonable ρ (Supplementary Table S6). It may be possible to use cross-validation in large datasets or control variants to calibrate ρ or adaptively select distinct ρsk for each UMI/haplotype combination, perhaps as a simple function of abundance or the proximity and abundance of other observed molecules. Such attempts are increasingly complex approximations of an explicit PCR model.
DAUMI is affected by the candidate UMI and haplotype sets. If candidates are known a priori, DAUMI works very well (Supplementary Section S4), but prior knowledge of is rare. We used UMI-unaware AmpliCI (Peng and Dorman, 2021) to populate and . This method assumes each true sample sequence is observed with the same UMI at least twice. Singleton discard is a blunt way to increase agreement across subsets, as shown for Calib, UMI-tools and Starcode-umi, which do not discard singletons by default (Table 1 versus Supplementary Table S10). The resulting UMI counts are biased downward, which can be corrected (Pflug and von Haeseler, 2018), but the discarded haplotypes cannot be recovered. In low-complexity samples with low amplification or high downsampling, multiple singleton or low-frequency UMIs may be linked to the same sampled molecule. DAUMI’s poorer performance on V1V2 when restricting to haplotypes with deduplicated abundance (Supplementary Table S10) is due to the discard of barely amplified UMI-tagged haplotypes. AmpliCI sensitivity can be increased (see options –-diagnostic, –-useAIC, –-abundance), but the defaults used here are set to work on varied datasets and without posthoc filtering.
DAUMI is easily extended. Overpopulation of UMI set could be ameliorated by penalizing the relative abundances ηs of . A hierarchical model on ηs could detect early PCR errors or other sources of variation in UMI abundance, such as biased nucleotide usage. Transition probabilities in that link UMI to haplotype could model transcript selection followed by random fragmentation, as utilized in some scRNA-seq protocols (Zilionis et al., 2017). Finally, the DAUMI framework is not limited to Illumina sequence data and could, with appropriate error model and good initialization, be applied to long-read technology.
Supplementary Material
Contributor Information
Xiyu Peng, Department of Epidemiology and Biostatistics, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA.
Karin S Dorman, Department of Statistics, Iowa State University, Ames, IA 50011, USA; Department of Genetics, Development and Cell Biology, Iowa State University, Ames, IA 50011, USA; Bioinformatics and Computational Biology Program, Iowa State University, Ames, IA 50011, USA.
Funding
This work was supported in part by the United States Department of Agriculture (USDA) National Institute of Food and Agriculture (NIFA) Hatch project [IOW03717]. The findings and conclusions in this publication are those of the author(s) and should not be construed to represent any official USDA or U.S. Government determination or policy.
Conflict of Interest: none declared.
Data availability
Code and links to existing data resources used to in this manuscript can be found in the public repository for the software: https://github.com/DormanLab/AmpliCI.
References
- Amir A. et al. (2017) Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems, 2, e00191-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armagan A. et al. (2013) Generalized double Pareto shrinkage. Stat. Sin., 23, 119–143. [PMC free article] [PubMed] [Google Scholar]
- Bhiman J.N. et al. (2015) Viral variants that initiate and drive maturation of V1V2-directed HIV-1 broadly neutralizing antibodies. Nat. Med., 21, 1332–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell J.R., Levy S.F. (2014) Beyond genome sequencing: lineage tracking with barcodes to study the dynamics of evolution, infection, and cancer. Genomics, 104, 417–430. [DOI] [PubMed] [Google Scholar]
- Callahan B.J. et al. (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods, 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan B.J. et al. (2017) Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J., 11, 2639–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candès E.J. et al. (2008) Enhancing sparsity by reweighted minimization. J. Fourier Anal. Appl., 14, 877–905. [Google Scholar]
- Caskey M. et al. (2017) Antibody 10-1074 suppresses viremia in HIV-1-infected individuals. Nat. Med., 23, 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. et al. (2019) Gencore: an efficient tool to generate consensus reads for error suppressing and duplicate removing of NGS data. BMC Bioinformatics, 20, 606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement K. et al. (2018) AmpUMI: design and analysis of unique molecular identifiers for deep amplicon sequencing. Bioinformatics, 34, i202–i210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deza E., Deza M.-M. (2006) Dictionary of Distances. Elsevier Science, Amsterdam, The Netherlands. [Google Scholar]
- Dorman K.S. et al. (2021) Denoising methods for inferring microbiome community content and abundance. In: Datta S., Guha S. (eds) Statistical Analysis of Microbiome Data. Springer Nature, Cham, Switzerland, pp. 3–25. [Google Scholar]
- Ewing B., Green P. (1998) Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res., 8, 186–194. [PubMed] [Google Scholar]
- Fields B. et al. (2021) MAUI-seq: metabarcoding using amplicons with unique molecular identifiers to improve error correction. Mol. Ecol. Resour., 21, 703–720. [DOI] [PubMed] [Google Scholar]
- Galanti L. et al. (2021) Pheniqs 2.0: accurate, high-performance Bayesian decoding and confidence estimation for combinatorial barcode indexing. BMC Bioinformatics, 22, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hathaway N.J. et al. (2018) SeekDeep: single-base resolution de novo clustering for amplicon deep sequencing. Nucleic Acids Res., 46, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W. et al. (2012) ART: a next-generation sequencing read simulator. Bioinformatics, 28, 593–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug H., Schuler R. (2003) Measurement of the number of molecules of a single mRNA species in a complex mRNA preparation. J. Theor. Biol., 221, 615–624. [DOI] [PubMed] [Google Scholar]
- Jabara C.B. et al. (2011) Accurate sampling and deep sequencing of the HIV-1 protease gene using a primer ID. Proc. Natl. Acad. Sci. USA, 108, 20166–20171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaccard P. (1912) The distribution of the flora in the alpine zone. New Phytol., 11, 37–50. [Google Scholar]
- Karst S.M. et al. (2021) High-accuracy long-read amplicon sequences using unique molecular identifiers with Nanopore or PacBio sequencing. Nat. Methods, 18, 165–169. [DOI] [PubMed] [Google Scholar]
- Kebschull J.M., Zador A.M. (2015) Sources of PCR-induced distortions in high-throughput sequencing data sets. Nucleic Acids Res., 43, e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebschull J.M., Zador A.M. (2018) Cellular barcoding: lineage tracing, screening and beyond. Nat. Methods, 15, 871–879. [DOI] [PubMed] [Google Scholar]
- Kim T.H. et al. (2020) Demystifying “drop-outs” in single-cell UMI data. Genome Biol., 21, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinde I. et al. (2011) Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA, 108, 9530–9535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivioja T. et al. (2011) Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods, 9, 72–74. [DOI] [PubMed] [Google Scholar]
- König J. et al. (2010) iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol., 17, 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A., Gagnon J.A. (2019) Recording development with single cell dynamic lineage tracing. Development, 146, dev169730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman A.M. et al. (2016) Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol., 34, 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orabi B. et al. (2019) Alignment-free clustering of UMI tagged DNA molecules. Bioinformatics, 35, 1829–1836. [DOI] [PubMed] [Google Scholar]
- Peng X., Dorman K. (2021) AmpliCI: a high-resolution model-based approach for denoising Illumina amplicon data. Bioinformatics, 36, 5151–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petukhov V. et al. (2018) dropEst: pipeline for accurate estimation of molecular counts in droplet-based single-cell RNA-seq experiments. Genome Biol., 19, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflug F.G., von Haeseler A. (2018) TRUmiCount: correctly counting absolute numbers of molecules using unique molecular identifiers. Bioinformatics, 34, 3137–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokhilko A. et al. (2021) Targeted single-cell RNA sequencing of transcription factors enhances the identification of cell types and trajectories. Genome Res., 31, 1069–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapov V., Ong J.L. (2017) Examining sources of error in PCR by single-molecule sequencing. PLoS One, 12, e0169774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quince C. et al. (2011) Removing noise from pyrosequenced amplicons. BMC Bioinformatics, 12, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohland N., Reich D. (2012) Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res., 22, 939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosati E. et al. (2017) Overview of methodologies for T-cell receptor repertoire analysis. BMC Biotechnol., 17, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer M. et al. (2016) Illumina error profiles: resolving fine-scale variation in metagenomic sequencing data. BMC Bioinformatics, 17, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert D. et al. (2016) A comprehensive analysis of primer IDs to study heterogeneous HIV-1 populations. J. Mol. Biol., 428, 238–250. [DOI] [PubMed] [Google Scholar]
- Sena J.A. et al. (2018) Unique molecular identifiers reveal a novel sequencing artefact with implications for RNA-seq based gene expression analysis. Sci. Rep., 8, 13121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y. et al. (2017) Circulating tumor DNA mutation profiling by targeted next generation sequencing provides guidance for personalized treatments in multiple cancer types. Sci. Rep., 7, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shugay M. et al. (2014) Towards error-free profiling of immune repertoires. Nat. Methods, 11, 653–655. [DOI] [PubMed] [Google Scholar]
- Shugay M. et al. (2017) MAGERI: computational pipeline for molecular-barcoded targeted resequencing. PLoS Comput. Biol., 13, e1005480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T. et al. (2017) UMI-tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res., 27, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A. et al. (2019) Alevin efficiently estimates accurate gene abundances from dscRNA-seq data. Genome Biol., 20, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M.A. (1974) EDF statistics for goodness of fit and some comparisons. J. Am. Stat. Assoc., 69, 730–737. [Google Scholar]
- Stoler N., Nekrutenko A. (2021) Sequencing error profiles of Illumina sequencing instruments. NAR Genom. Bioinform., 3, lqab019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoler N. et al. (2016) Streamlined analysis of duplex sequencing data with Du Novo. Genome Biol., 17, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolovitzky G., Cecchi G. (1996) Efficiency of DNA replication in the polymerase chain reaction. Proc. Natl. Acad. Sci. USA, 93, 12947–12952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson V. (2020) Droplet scRNA-seq is not zero-inflated. Nat. Biotechnol., 38, 147–150. [DOI] [PubMed] [Google Scholar]
- Sze M.A., Schloss P.D. (2019) The impact of DNA polymerase and number of rounds of amplification in PCR on 16S rRNA gene sequence data. mSphere, 4, e00163–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townes F.W. et al. (2019) Feature selection and dimension reduction for single-cell RNA-seq based on a multinomial model. Genome Biol., 20, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden J.A. et al. (2014) pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics, 30, 1930–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varghese V. et al. (2010) Nucleic acid template and the risk of a PCR-induced HIV-1 drug resistance mutation. PLoS One, 5, e10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woyke T., Jarett J. (2015) Function-driven single-cell genomics. Microb. Biotechnol., 8, 38–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C. et al. (2019) smCounter2: an accurate low-frequency variant caller for targeted sequencing data with unique molecular identifiers. Bioinformatics, 35, 1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X. (2016) Probabilistic methods for quality improvement in high-throughput sequencing data. PhD Thesis, Iowa State University.
- Zanini F. et al. (2017) Error rates, PCR recombination, and sampling depth in HIV-1 whole genome deep sequencing. Virus Res., 239, 106–114. [DOI] [PubMed] [Google Scholar]
- Zhou S. et al. (2015) Primer ID validates template sampling depth and greatly reduces the error rate of next-generation sequencing of HIV-1 genomic RNA populations. J. Virol., 89, 8540–8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegenhain C. et al. (2022) Molecular spikes: a gold standard for single-cell RNA counting. Nat. Methods, 19, 560–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilionis R. et al. (2017) Single-cell barcoding and sequencing using droplet microfluidics. Nat. Protoc., 12, 44–73. [DOI] [PubMed] [Google Scholar]
- Zorita E. et al. (2015) Starcode: sequence clustering based on all-pairs search. Bioinformatics, 31, 1913–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Code and links to existing data resources used to in this manuscript can be found in the public repository for the software: https://github.com/DormanLab/AmpliCI.