

Abstract
Single-cell RNA-sequencing (scRNA-seq) provides unique insights into the pathology and cellular origin of disease. We introduce scDRS, an approach that links scRNA-seq with polygenic disease risk at single-cell resolution, independent of annotated cell-types. scDRS identifies cells exhibiting excess expression across disease-associated genes implicated by genome-wide association studies (GWAS). We applied scDRS 74 diseases/traits and 1.3M single-cell gene-expression profiles across 31 tissues/organs. Cell-type-level results broadly recapitulated known cell-type-disease associations. Individual-cell-level results identified subpopulations of disease-associated cells not captured by existing cell-type labels, including T cell subpopulations associated with inflammatory bowel disease, partially characterized by their effector-like states; neuron subpopulations associated with schizophrenia, partially characterized by their spatial locations; hepatocyte subpopulations associated with triglyceride levels, partially characterized by their higher ploidy levels. Genes whose expression was correlated with the scDRS score across cells (reflecting co-expression with GWAS disease-associated genes) were strongly enriched for gold-standard drug target and Mendelian disease genes.
Editor summary:
scDRS associates individual cells in scRNA-seq with disease by scoring single-cell transcriptomes using GWAS gene signatures. Applied to 74 GWAS and 1.3 million single-cell profiles, scDRS identifies specific cellular subpopulations associated with these diseases.
Introduction
The mechanisms through which risk variants identified by genome-wide association studies (GWASs) impact critical tissues and cell types are largely unknown1; identifying these tissues and cell types is central to our understanding of disease etiologies and will inform efforts to develop therapeutic treatments2. Single-cell RNA sequencing (scRNA-seq) has emerged as the tool of choice for measuring gene expression abundance at single-cell resolution3, providing an increasingly clear picture of which genes are active in which cell types and also being able to identify novel cell populations within classically defined cell types. Integrating scRNA-seq with GWAS data offers the potential to identify critical tissues, cell types, and cell populations through which GWAS risk variants impact disease4–6, thus providing finer resolution than studies using bulk transcriptomic data7–10.
Previous studies integrating scRNA-seq with GWAS have largely focused on predefined cell type annotations (e.g., classical cell types defined using known marker genes), aggregating cells from the same cell type followed by evaluating overlap of the cell type-level information with GWAS4–6. However, this approach overlooks the considerable heterogeneity of individual cells within cell types that has been reported in studies of scRNA-seq data alone11–16; the underlying methods (e.g., Seurat cell-scoring function13, Vision14, and VAM16) have sought to explain transcriptional heterogeneity in scRNA-seq data by scoring cells based on predefined gene sets such as cellular pathways, but do not consider polygenic disease risk from GWAS and generally do not provide individual cell-level association p-values. Integrating information from scRNA-seq data at fine-grained resolution (e.g., individual cells both within and across cell types) with polygenic signals from disease GWAS has considerable potential to produce new biological insights.
Here, we introduce single-cell Disease Relevance Score (scDRS), a method to evaluate polygenic disease enrichment of individual cells in scRNA-seq data. scDRS assesses whether a given cell has excess expression levels across a set of putative disease genes derived from GWAS, using an appropriately matched empirical null distribution to estimate well-calibrated p-values. We performed extensive simulations to assess the calibration and power of scDRS. We applied scDRS to 74 diseases/traits (average GWAS N=346K) and 16 scRNA-seq data sets (including the Tabula Muris Senis (TMS) mouse cell atlas17), assessing cell type-disease associations and within-cell type association heterogeneity, including heterogeneity of T cells in association with autoimmune diseases, neurons in association with brain-related diseases/traits, and hepatocytes in association with metabolic traits; we analyzed a broader set of scRNA-seq data sets to provide validation across species (human vs. mouse) and across sequencing platforms, and to include scRNA-seq data sets with experimentally determined cell types and cell states.
Results
Overview of methods
scDRS integrates gene expression profiles from scRNA-seq with polygenic disease information from GWAS to associate individual cells to disease, by assessing the excess expression of GWAS putative disease genes in a given cell relative to other genes with similar expression across all cells. scDRS consists of three steps (Figure 1). First, scDRS constructs a set of putative disease genes from GWAS summary statistics using MAGMA18, an existing gene scoring method (top 1,000 MAGMA genes). Second, scDRS quantifies the aggregate expression of the putative disease genes in each cell to generate cell-specific raw disease scores; to maximize power, each putative disease gene is weighted by its GWAS MAGMA z-score and inversely weighted by its gene-specific technical noise level in the single-cell data, estimated via modeling the mean-variance relationship across genes16,19. To determine statistical significance, scDRS also generates 1,000 sets of cell-specific raw control scores at Monte Carlo (MC) samples of matched control gene sets (matching gene set size, mean expression, and expression variance of the putative disease genes). Third, scDRS normalizes the raw disease score and raw control scores for each cell (producing the normalized disease score and normalized control scores), and then computes cell-level p-values based on the empirical distribution of the pooled normalized control scores across all control gene sets and all cells. Further details are provided in Methods, Supplementary Note, and Supplementary Figures 1–3.
Figure 1. Overview of scDRS method.
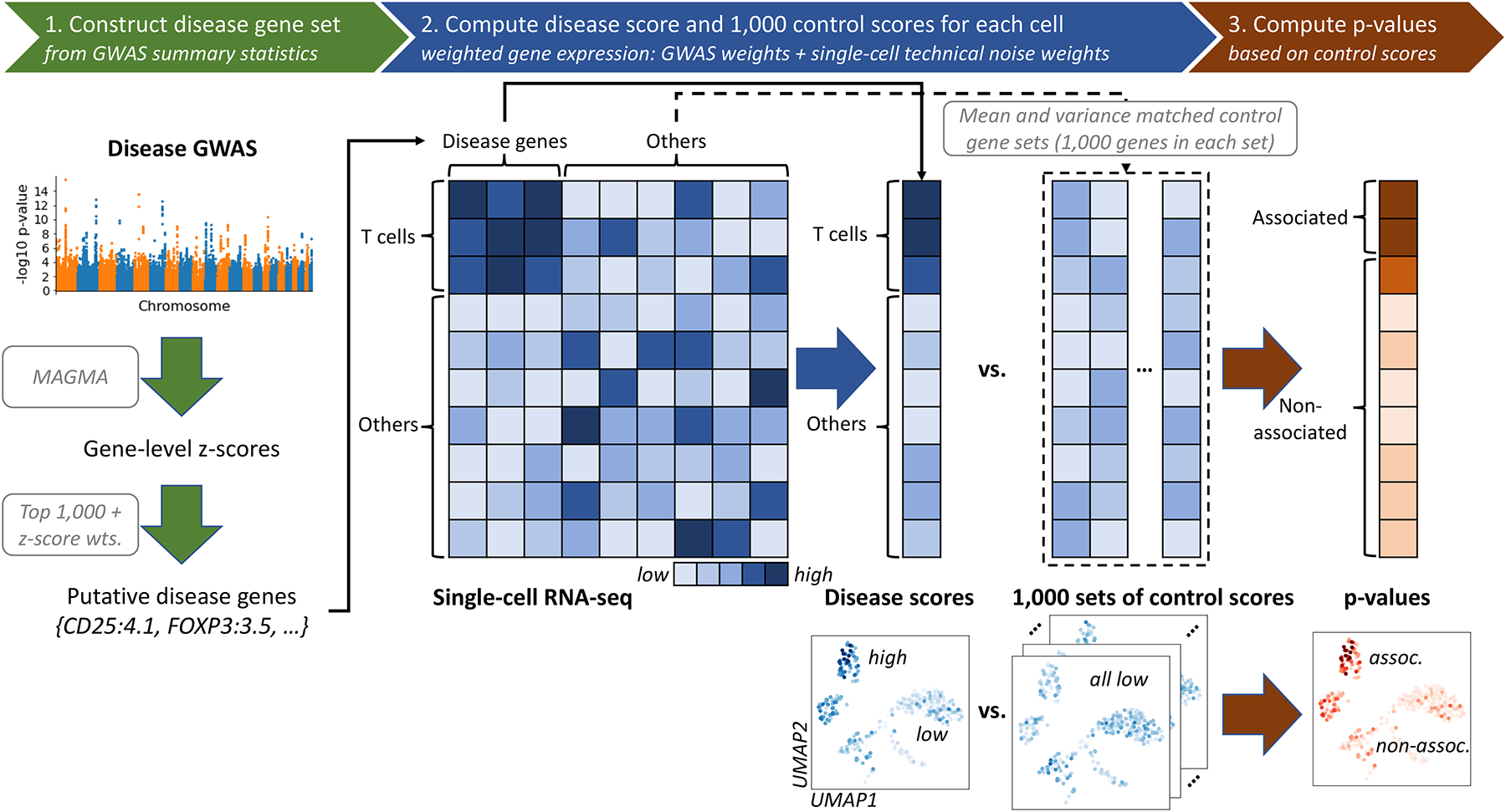
scDRS takes a disease GWAS and an scRNA-seq data set as input and outputs individual cell-level p-values for association with the disease. (1) scDRS constructs a set of putative disease genes from GWAS summary statistics by selecting the top 1,000 MAGMA genes; these putative disease genes are expected to have higher expression levels in the relevant cell population. (2) scDRS computes a raw disease score for each cell, quantifying the aggregate expression of the putative disease genes in that cell; to maximize power, each putative disease gene is weighted by its GWAS MAGMA z-score and inversely weighted by its gene-specific technical noise level in scRNA-seq. scDRS also computes a set of 1,000 Monte Carlo raw control scores for each cell, in each case using a random set of control genes matching the gene set size, mean expression, and expression variance of the putative disease genes. (3) scDRS normalizes the raw disease score and raw control scores across gene sets and across cells, and then computes a p-value for each cell based on the empirical distribution of the pooled normalized control scores across all control gene sets and all cells. The choice of 1,000 for the number of putative disease genes and the choice of 1,000 for the number of control scores are independent.
scDRS outputs individual cell-level disease-association p-values, normalized disease scores, and 1,000 sets of normalized control scores (referred to as “disease scores” and “control scores” in the rest of the paper) that can be used for a wide range of downstream applications (Methods). Here, we focus on three downstream analyses. First, we perform cell type-level analyses to associate predefined cell types to disease and assess heterogeneity in association to disease across cells within a predefined cell type. Second, we perform individual cell-level analyses to associate individual cells to disease and correlate individual cell-level variables to the scDRS disease score. Third, we perform gene-level analyses to prioritize disease-relevant genes whose expression is correlated with the scDRS disease score, reflecting co-expression with genes implicated by disease GWAS.
We analyzed publicly available GWAS summary statistics of 74 diseases/traits (average N=346K; Supplementary Table 1) in conjunction with 16 scRNA-seq or single-nucleus RNA-seq (snRNA-seq) data sets spanning 1.3 million cells from 31 tissues/organs from mouse (mus musculus) and human (homo sapiens) (Supplementary Tables 2–7, Supplementary Figure 4). The single-cell data sets include two data sets from the Tabula Muris Senis (TMS) mouse cell atlases17 collected using different technologies, the Tabula Sapiens (TS) human cell atlas20, and other data sets focusing on specific tissues containing well-annotated cell types/states. We focused on the TMS FACS data in our primary analyses due to its comprehensive coverage of 23 tissues and 120 cell types and more accurate quantification of gene expression levels (via Smart-seq221); we used the other 15 data sets to validate our results. We note the extensive use of mouse gene expression data to study human diseases and complex traits4–7,10,22 (Supplementary Note).
Simulations assessing calibration and power
We performed null simulations and causal simulations to assess the calibration and power of scDRS, comparing scDRS to three state-of-art methods for scoring individual cells with respect to a specific gene set: Seurat (cell-scoring function)13, Vision14, and VAM16 (Methods).
First, we evaluated each method in null simulations where no cells have systematically higher expression across the putative disease genes analyzed. We subsampled 10,000 cells from the TMS FACS data and randomly selected 1,000 putative disease genes. We simulated GWAS gene weights for scDRS matching the MAGMA z-score distributions in real traits and used binary disease gene sets for the other 3 methods. scDRS and Seurat produced well-calibrated p-values, Vision suffered slightly inflated type I error, and VAM suffered severely inflated type I error (Figure 2a and Supplementary Table 8).
Figure 2. Results for null and causal simulations.
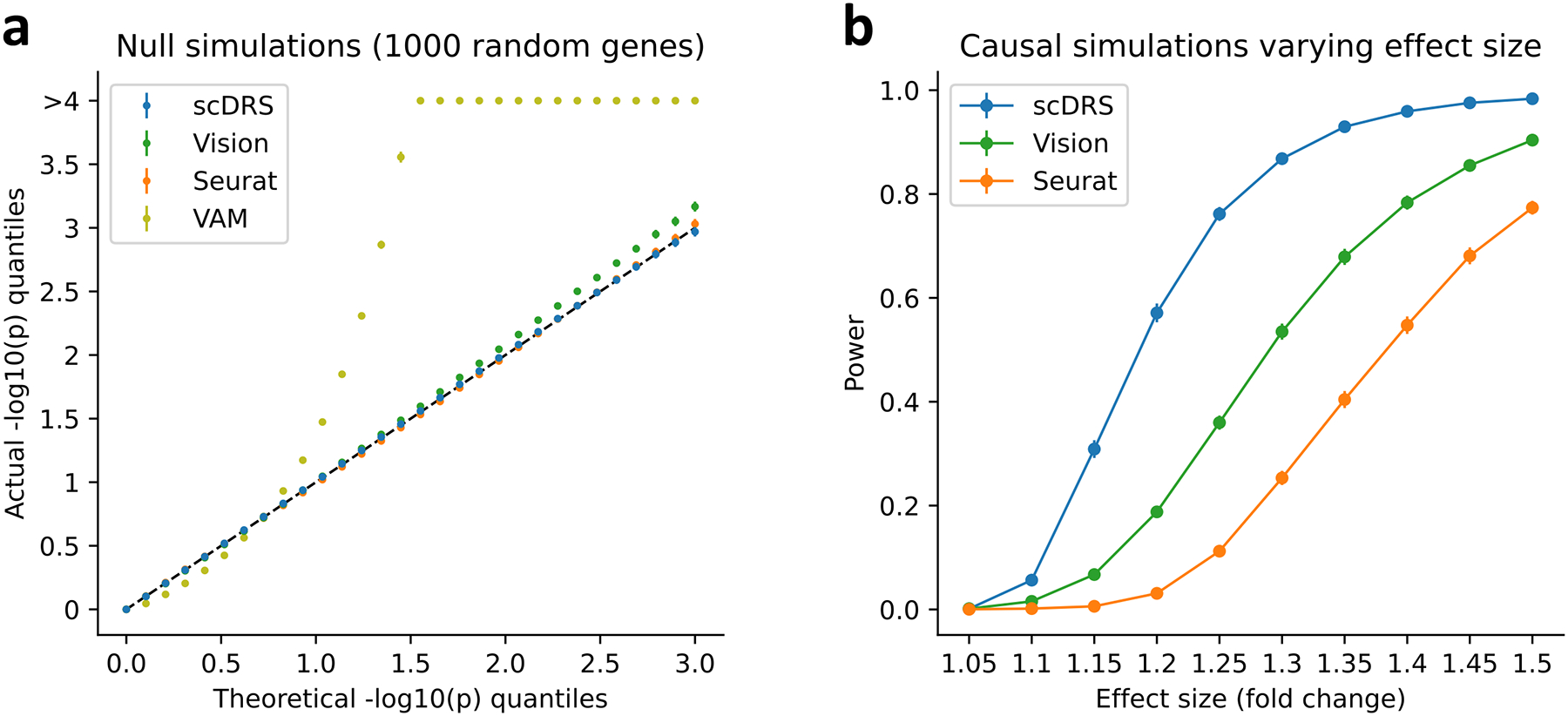
(a) Q-Q plot for null simulations using 1,000 randomly selected genes as the putative disease genes. Random GWAS gene weights were used for scDRS matching the MAGMA z-score distributions in real traits while binary gene sets were used for the other 3 methods. X-axis denotes theoretical −log10 p-value quantiles and y-axis denotes actual −log10 p-value quantiles for different methods. Error bars denote 95% confidence intervals around the mean of 100 simulation replicates (with 10,000 cells per simulation replicate); all error bars are <0.05 from the point estimate. Numerical results are reported in Supplementary Table 8 and additional results are reported in Extended Data Figure 1 and Supplementary Figure 5. (b) Power for casual simulations with perturbed expression of causal genes in causal cells. We report the power at FDR=0.1 for different methods and different effect sizes. Error bars denote 95% confidence intervals around the mean of 100 simulation replicates (with 10,000 cells per simulation replicate); all error bars are <0.02 from the point estimate. Numerical results are reported in Supplementary Table 9 and additional results are reported in Extended Data Figure 2.
Next, we evaluated scDRS, Seurat and Vision in causal simulations where a subset of causal cells has systematically higher expression across putative disease genes (we did not include VAM, which was not well-calibrated in null simulations). We used the same 10,000 cells subsampled from the TMS FACS data, randomly selected 1,000 causal disease genes, randomly selected 500 of the 10,000 cells as causal cells and artificially perturbed their expression levels to be higher (1.05–1.50X for different simulations) across the 1,000 causal disease genes, and randomly selected 1,000 putative disease genes (provided as input to each method) with 25% overlap with the 1,000 causal disease genes. We used the binary gene set for all 3 methods because there were no GWAS weights involved in data generation. We determined that scDRS attained higher power than Seurat and Vision to detect individual cell-disease associations at FDR<0.1 (Figure 2b and Supplementary Table 9); the improved power of scDRS may be due to its incorporation of gene-specific weights that downweight genes with higher levels of technical noise.
Results of additional null and causal simulations are reported in the Supplementary Note, Extended Data Figures 1,2, Supplementary Figure 5, and Supplementary Table 10.
Results across 120 TMS cell types for 74 diseases and traits
We analyzed GWAS data from 74 diseases/traits (average N=346K; Supplementary Tables 1,11) in conjunction with the TMS FACS data with 120 cell types (cells from different tissues were combined for a given cell type; Supplementary Table 5). We first report scDRS cell type-level results, aggregated for each cell type from the scDRS individual cell-level results; the individual cell-level results are discussed in subsequent sections. Results for a representative subset of 19 cell types and 22 diseases/traits are reported in Figure 3. Within this subset, scDRS identified 80 associated cell type-disease pairs (FDR<0.05; squares in Figure 3) and detected significant within-cell type disease-association heterogeneity for 43 of these 80 associated cell type-disease pairs (FDR<0.05; cross symbols in Figure 3) (273 of 597 across all pairs of the 120 cell types and 74 diseases/traits; Extended Data Figure 3 and Supplementary Table 12).
Figure 3. Disease associations at the cell type-level.
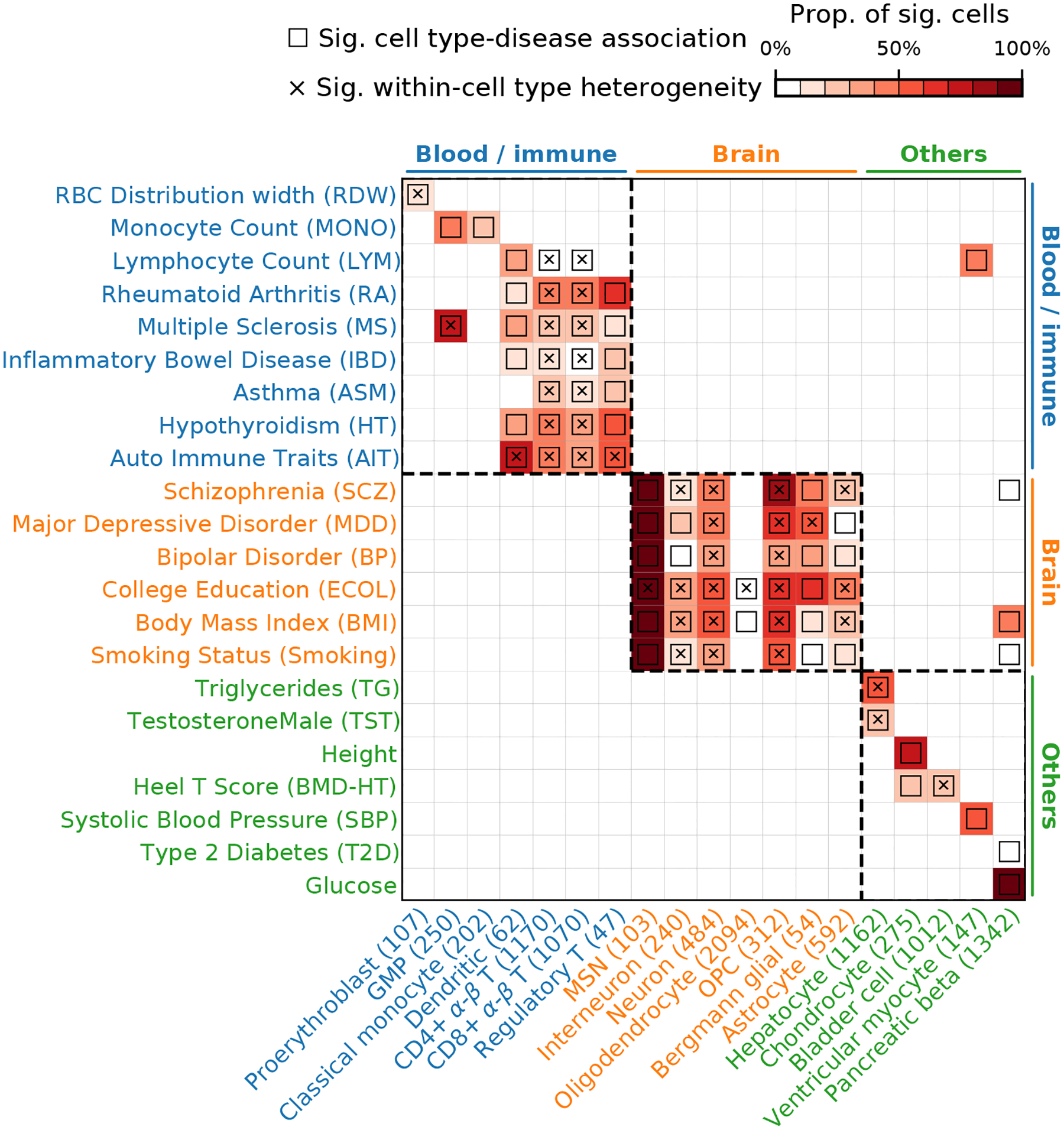
We report scDRS results for individual cells aggregated at the cell type-level for a subset of 19 cell types and 22 diseases/traits in the TMS FACS data. Each row represents a disease/trait and each column represents a cell type (with number of cells indicated in parentheses). Heatmap colors for each cell type-disease pair denote the proportion of significantly associated cells (FDR<0.1 across all cells for a given disease). Squares denote significant cell type-disease associations (FDR<0.05 across all pairs of the 120 cell types and 74 diseases/traits; p-values via MC test; Methods). Cross symbols denote significant heterogeneity in association with disease across individual cells within a given cell type (FDR<0.05 across all pairs; p-values via MC test; Methods). Heatmap colors (>10% of cells associated) and cross symbols are omitted for cell type-disease pairs with non-significant cell type-disease associations via MC test (heatmap colors omitted for 1 pair (Dendritic-ASM) and cross symbols omitted for 6 pairs (CD4+ α-β T-MONO, CD8+ α-β T-MONO, bladder cell-RA, bladder cell-ASM, oligodendrocyte-BP, and dendritic-BMD-HT)). Auto Immune Traits (AIT) represents a collection of diseases in the UK Biobank that characterize autoimmune physiopathogenic etiology62(Supplementary Table 1). Abbreviated cell type names include red blood cell (RBC), granulocyte monocyte progenitor (GMP), medium spiny neuron (MSN), and oligodendrocyte precursor cell (OPC). Neuron refers to neuronal cells with undetermined subtypes (whereas MSN and interneuron (non-overlapping with neuron) refer to neuronal cells with those inferred subtypes). Complete results for 120 cell types and 74 diseases/traits are reported in Extended Data Figure 3 and Supplementary Table 12.
For cell type-disease associations, as expected, scDRS broadly associated blood/immune cell types with blood/immune-related diseases/traits, brain cell types with brain-related diseases/traits, and other cell types with other diseases/traits (block-diagonal pattern in Figure 3). Most scDRS discoveries recapitulated well-established cell type-disease associations, including blood/immune cell types with blood cell traits, immune cell types with immune diseases, neuronal cell types with brain-related traits/diseases6,22,23, and hepatocytes with metabolic traits24. In addition, chondrocytes, bladder cells, ventricular myocytes and pancreatic beta cells were associated with their corresponding expected diseases/traits25–28. Exceptions to the block-diagonal pattern and further details are discussed in the Supplementary Note.
We also discuss several less well-established results. First, granulocyte monocyte progenitors (GMP) were strongly associated with multiple sclerosis (MS), highlighting the role of myeloid cells in MS29. Second, oligodendrocytes, oligodendrocyte precursor cells (OPCs) were associated with multiple brain-related diseases/traits; these associations are less clear in existing genetic studies4,6,22,30, but are biologically plausible, consistent with the increasingly discussed role of oligodendrocyte lineage cells in brain diseases/traits: the differentiation and myelination activity of oligodendrocyte lineage cells are important to maintain the functionality of neuronal cells31. We detected significant heterogeneity across OPCs in their association with many brain-related diseases/traits, consistent with recent evidence of functionally diverse states of OPCs32, traditionally considered to be a homogeneous population (Supplementary Figure 6). Third, we detected significant heterogeneity across cells for the association between proerythroblasts and red cell distribution width (RDW), which may correspond to erythrocytes at different differentiation stages33 (Supplementary Figure 7). We also detected other instances of significant within-cell type association heterogeneity, including T cells with immune diseases, neurons with brain-related diseases/traits, and hepatocytes with metabolic traits, discussed in subsequent sections.
Additional secondary analyses assessing robustness of the results and alternative versions of scDRS are reported in Methods, Supplementary Note, Extended Data Figures 1,4,5, Supplementary Figures 8–11, and Supplementary Tables 13–16.
Heterogeneous T cells subpopulations in autoimmune disease
We investigated the heterogeneity across TMS FACS T cells in association with autoimmune diseases (Figure 3). We jointly analyzed all TMS FACS T cells (3,769 cells, spanning 15 tissues). Since the original study clustered cells from different tissues separately17, we reclustered these T cells, resulting in 11 clusters (Figure 4a; Methods); we verified that batch effects were not observed for tissue, age, or sex (Supplementary Figure 12). We considered 10 autoimmune diseases: inflammatory bowel disease (IBD), Crohn’s disease (CD), ulcerative colitis (UC), rheumatoid arthritis (RA), MS, autoimmune traits (AIT; a general term for autoimmune diseases), hypothyroidism (HT), eczema, asthma (ASM), and respiratory and ear-nose-throat diseases (RR-ENT) (Supplementary Table 1); we considered height as a negative control trait.
Figure 4. Associations of T cells with autoimmune diseases.
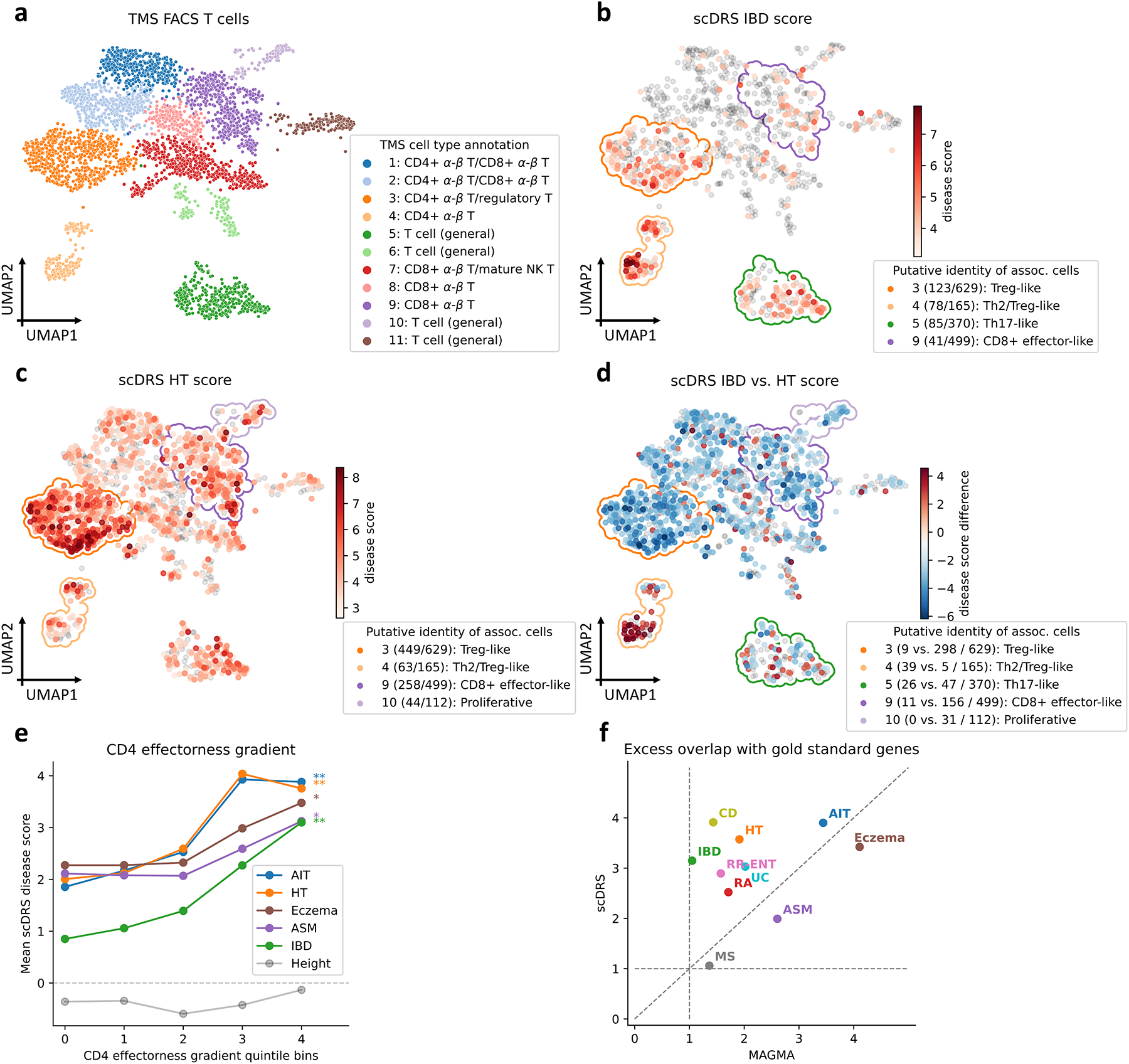
(a) UMAP visualization of T cells in TMS FACS. Cluster labels are based on annotated TMS cell types in the cluster. (b-c) Subpopulations of T cells associated with IBD and HT, respectively. Significantly associated cells (FDR<0.1) are denoted in red, with shades of red denoting scDRS disease scores; other cells are denoted in grey. Cluster boundaries indicate the corresponding T cell clusters from panel a. Clusters are annotated based on the putative identities of associated cells in the cluster, for the top 4 clusters (out of 11) with the strongest level of association (highest average disease score for associated cells in the cluster); number of disease-associated cells and number of all cells in the cluster are provided in parentheses. (d) Differences in individual cell-level associations between IBD and HT. Differentially associated cells (absolute scDRS disease score difference>2) are denoted in red and blue, with shades of colors denoting scDRS disease score differences; other cells are denoted in grey. Cluster boundaries indicate the corresponding T cell clusters from panel a. Clusters are annotated the same as in panels b,c; number of IBD-enriched cells, HT-enriched cells, and all cells in the cluster are provided in parentheses. For panels b-d, results for the other 8 autoimmune diseases are reported in Supplementary Figures 13,18. (e) Association between scDRS disease score and CD4 effectorness gradient across CD4+ T cells for 5 representative autoimmune diseases and height, a negative control trait. X-axis denotes CD4 effectorness gradient quintile bins and y-axis denotes the average scDRS disease score in each bin for each disease. * denotes P<0.05 and ** denotes P<0.005 (one-sided MC test). Numerical results for all 10 autoimmune diseases are reported in Supplementary Table 20. (f) Excess overlap with gold standard gene sets. X-axis denotes excess overlap of genes prioritized by MAGMA and y-axis denotes excess overlap of genes prioritized by scDRS, for each of the 10 autoimmune diseases. Median ratio of (excess overlap − 1) for scDRS vs. MAGMA was 2.07. Numerical results are reported in Supplementary Table 22.
We focused on individual cells associated with IBD, a representative autoimmune disease (Figure 4b). The 387 IBD-associated cells (FDR<0.1) formed subpopulations of 4 of the 11 T cell clusters. The subpopulation of 123 IBD-associated cells in cluster 3 (labeled as “Treg”) had high expression of regulatory T cell (Treg) marker genes (FOXP3+, CTLA4+, LAG3+; Supplementary Figure 17a), and their specifically expressed genes significantly overlapped with Treg signatures (P=6.0×10−8 for MSigDB signatures and P=4.0×10−68 for an effector-like Treg program34, two-sided Fisher’s exact test; Supplementary Figure 17c,d), suggesting these cells had Treg immunosuppressive functions. Interestingly, these 123 IBD-associated cells were non-randomly distributed in cluster 3 on the UMAP plot (P<0.001, MC test; Methods). Genes specifically expressed in these IBD-associated cells were preferentially enriched (compared to the 506 non-IBD-associated cells in the same cluster) in pathways defined by NF-κB signaling, T helper cell differentiation, and tumor necrosis factor-mediated signaling (Supplementary Figure 17e); these pathways are closely related to inflammation, a distinguishing feature of IBD35. In addition, the subpopulations of IBD-associated cells in clusters 4,5,9 were labeled as “Th2/Treg-like”, “Th17-like”, and “CD8+ effector-like”, respectively, consistent with previous studies associating subpopulations of effector T cells to IBD, particularly Tregs and Th17 cells35. Results for other autoimmune diseases, details for annotating disease-associated T cell subpopulations, and replication analyses on 2 human scRNA-seq data sets36,37 are reported in Figure 4c, Methods, Supplementary Note, Supplementary Figures 13–17, and Supplementary Tables 17,18.
We further compared the individual T cell associations of IBD to HT, another representative autoimmune disease (Figure 4c,d). The top 4 HT-associated subpopulations included 3 IBD-associated subpopulations (cells in clusters 3,4,9; Figure 4c), but also a unique subpopulation of cells in cluster 10 (labeled as “Proliferative”). Despite the stronger associations to HT overall (possibly due to higher GWAS power), IBD was more strongly associated with cells in cluster 4 (labeled as “Th2/Treg-like”; Figure 4d). Additional results are reported in the Supplementary Note, Extended Data Figure 6, Supplementary Figure 18, and Supplementary Table 19.
Motivated by the effector-like T cell subpopulations associated to IBD, we investigated whether the heterogeneity of T cells in association with autoimmune diseases was correlated with T cell effectorness gradient, a continuous classification from naive to effector T cells. We separately computed the effectorness gradients for CD4+ T cells (1,686 cells) and CD8+ T cells (2,197 cells) using pseudotime analysis36,38, and assessed whether the CD4 (resp., CD8) effectorness gradient was correlated with scDRS disease scores for the 10 autoimmune diseases, across CD4+ T cells (resp., CD8+ T cells). Results are reported in Figure 4e and Supplementary Table 20. The CD4 effectorness gradient had strong associations with IBD, CD, UC, AIT, and HT (P<0.005, MC test), weak associations with Eczema and ASM (P<0.05), but non-significant associations with RA, MS, or RR-ENT, implying these autoimmune diseases are associated with more effector-like CD4+ T cells. The CD8 effectorness gradient had weaker associations (P<0.05 for IBD,CD,AIT, non-significant for others), suggesting that CD4+ effector T cells may be more important than CD8+ effector T cells for these diseases. The association of T cell effectorness gradients with autoimmune diseases has not been formally evaluated previously, but is consistent with previous studies linking T cell effector functions to autoimmune disease39 or characterizing similarities among effector T cell subtypes36,40. Additional results on T cell effectorness gradient and comparison to cluster-level LDSC-SEG are reported in the Supplementary Note, Supplementary Figures 19,20, and Supplementary Tables 17,20.
Finally, we prioritized disease-relevant genes by computing the correlation (across all TMS FACS cells) between the expression of a given gene and the scDRS score for a given disease; this approach identifies genes co-expressed with genes implicated by disease GWAS. We compared the top 1,000 genes prioritized using this approach with putative drug targets41 (for 8 autoimmune diseases except RR-ENT and HT) or genes known to cause a Mendelian form of the disease42 (for RR-ENT and HT whose drug targets are not available; Supplementary Table 21). Results are reported in Figure 4f and Supplementary Table 22. We determined that scDRS attained a more accurate prioritization of disease-relevant genes compared to the top 1,000 MAGMA genes (2.07 for median ratio of (excess overlap − 1); Methods), likely by capturing disease-relevant genes with weak GWAS signal43. For example, scDRS prioritized ITGB7 for IBD (rank 11; drug target for IBD using vedolizumab44) and JAK1 for RA (rank 358; drug target for RA using baricitinib45), both missed by MAGMA (ranks 10565,5228, P=0.54,0.26, respectively). Additional results are reported in the Supplementary Note, Extended Data Figure 7, and Supplementary Table 21.
Heterogeneous neuron subpopulations in brain traits
We investigated the heterogeneity across neurons in association with brain-related diseases/traits (Figure 3). We considered 7 brain-related diseases/traits: schizophrenia (SCZ), major depressive disorder (MDD), bipolar disorder (BP), neuroticism (NRT), college education (ECOL), body mass index (BMI), Smoking (Supplementary Table 1); we considered height as a negative control trait. Since the TMS FACS data has limited coverage of neuronal subtypes, we focused on a separate mouse brain scRNA-seq data (Zeisel & Muñoz-Manchado et al.46; 3,005 cells), which has better coverage of neuronal subtypes and has been analyzed at cell type level in previous genetic studies6,22. Results for TMS FACS neurons are reported in the Supplementary Note, Extended Data Figure 6, Supplementary Figures 21,22, and Supplementary Table 19.
scDRS associated several neuronal subtypes (CA1 pyramidal neurons, SS pyramidal neurons, and interneurons) with the 7 brain-related diseases/traits (Supplementary Figure 23a, Supplementary Table 23), consistent with previous genetic studies6,22,47. We focused on CA1 pyramidal neurons from the hippocampus (827 cells), which exhibited the strongest within-cell type heterogeneity (FDR<0.005 for all 7 brain-related traits, MC test; Supplementary Table 23). Individual cell-trait associations for SCZ are reported in Figure 5a (results for all 7 brain-related traits in Supplementary Figure 23b). We observed a continuous gradient of CA1 pyramidal neuron associations to the 7 brain-related traits.
Figure 5. Associations of neurons with brain-related disease/traits and hepatocytes with metabolic traits.
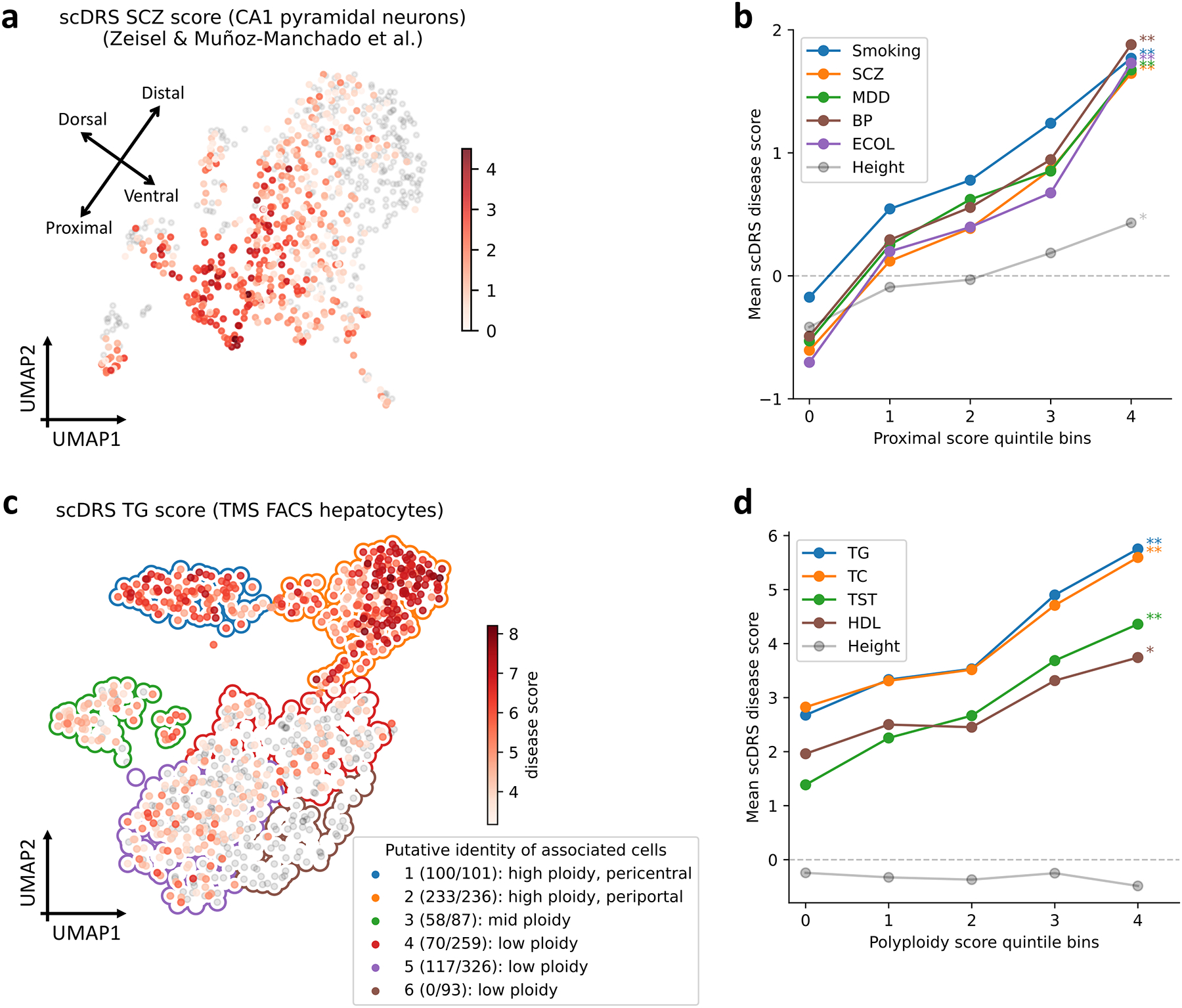
(a) Subpopulations of CA1 pyramidal neurons associated with SCZ in the Zeisel & Muñoz-Manchado et al. data. Colors of cells denote scDRS disease scores (negative disease scores are denoted in grey). We include a visualization of putative dorsal-ventral and proximal-distal axes (see text). Results for all 7 brain-related diseases/traits and height are reported in Supplementary Figure 23b. (b) Association between scDRS disease score and proximal score across CA1 pyramidal neurons for 5 representative brain-related disease/traits and height, a negative control trait. The x-axis denotes proximal score quintile bins and the y-axis denotes average scDRS disease score in each bin for each disease. * denotes P<0.05 and ** denotes P<0.005 (one-sided MC test). Results for all 6 spatial scores and all 7 brain traits (and height) are reported in Extended Data Figure 8 and Supplementary Table 25. (c) Subpopulations of hepatocytes associated with TG in the TMS FACS data. Significantly associated cells (FDR<0.1) are denoted in red, with shades of red denoting scDRS disease scores; non-significant cells are denoted in grey. Cluster boundaries indicate the corresponding hepatocyte clusters. In the legend, numbers in parentheses denote the number of TG-associated cells vs. the total number of cells. Cluster labels are based on the putative identity of cells in the cluster. Results for the other 8 metabolic traits and height are reported in Supplementary Figure 25. (d) Association between scDRS disease score and polyploidy score for 4 representative metabolic traits and height, a negative control trait. The x-axis denotes polyploidy score quintile bins and the y-axis denotes average scDRS disease score in each bin for each disease. * denotes P<0.05 and ** denotes P<0.005 (one-sided MC test). Results for all 3 scores (polyploidy score, pericentral score, periportal score) and all 9 metabolic traits (and height) are reported in Extended Data Figure 9 and Supplementary Table 26.
Motivated by known location-specific functions of hippocampal neurons15, we investigated whether the heterogeneity observed in Figure 5a was correlated with spatial location. We considered the natural CA1 spatial axes48 (dorsal-ventral long axis, proximal-distal transverse axis, and superficial-deep radial axis) and inferred spatial coordinates for each cell in terms of 6 continuous individual cell-level scores for these spatial regions (Supplementary Figures 23c,24, Supplementary Table 24; Methods). The inferred spatial scores for the dorsal-ventral and proximal-distal axes varied along the top two UMAP axes, providing visual evidence of stronger neuron-SCZ associations in dorsal and proximal regions (Figure 5a, Supplementary Figure 23).
We used the results of scDRS for individual cells to assess whether the inferred spatial scores for each of the 6 spatial regions (dorsal/ventral/proximal/distal/superficial/deep) were correlated to the scDRS disease scores for each of the 7 brain-related traits across CA1 pyramidal neurons (Methods). Results are reported in Figure 5b (for the proximal region, which had the strongest associations), Extended Data Figure 8, and Supplementary Table 25. The proximal score was strongly associated with all 7 brain-related traits (all P<0.002, MC test), suggesting proximal CA1 pyramidal neurons may be more relevant to these brain-related traits (instead of distal CA1 pyramidal neurons). The association between the proximal region and brain-related traits is consistent with the fact that the proximal region of the hippocampus receives synaptic inputs in the perforant pathway, which is the main input source of the hippocampus49. Validations of the spatial scores using independent data and results on other spatial scores and 6 additional mouse and human data sets50–55 are reported in the Supplementary Note and Extended Data Figure 8.
Heterogeneous hepatocyte subpopulations in metabolic traits
We investigated the heterogeneity across TMS FACS hepatocytes (in the liver) in their association with metabolic traits (Figure 3). We considered 9 metabolic traits: triglyceride levels (TG), high-density lipoprotein (HDL), low-density lipoprotein (LDL), total cholesterol (TC), testosterone (TST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), sex hormone-binding globulin (SHBG), and total bilirubin (TBIL) (Supplementary Table 1); we considered height as a negative control trait.
We focused on individual cells associated with TG, a representative metabolic trait (Figure 5c; results for other traits in Supplementary Figure 25). The 530 TG-associated cells (FDR<0.1) formed subpopulations of 5 of the 6 hepatocyte clusters; we characterized these subpopulations based on ploidy level (number of sets of chromosomes in a cell) and zonation (pericentral/mid-lobule/periportal spatial location in the liver lobule), which have been extensively investigated in previous studies of hepatocyte heterogeneity56,57. We inferred the ploidy level and zonation for each individual cell in terms of continuous individual cell-level polyploidy, pericentral, and periportal scores (Supplementary Figure 26; Methods). The inferred ploidy level and zonation varied across clusters, providing visual evidence of stronger cell-TG associations in high-ploidy clusters (clusters 1,2), particularly the periportal high-ploidy cluster (cluster 2; Figure 5c).
We used the results of scDRS for individual cells to assess whether the inferred polyploidy, pericenteral and periportal scores were correlated to the scDRS disease score for each of the 9 metabolic traits across hepatocytes; we jointly regressed the scDRS disease score for each trait on the polyploidy score, pericentral score, and periportal score (because the polyploidy score was positively correlated with the other 2 scores; Methods). Results are reported in Figure 5d (for the polyploidy score which had the strongest associations), Extended Data Figure 9, and Supplementary Table 26. The polyploidy score was strongly associated with all 9 metabolic traits (all P<0.007, MC test), suggesting that high-ploidy hepatocytes may be more relevant to these metabolic traits. The association between ploidy level and metabolic traits is consistent with previous findings that ploidy levels are associated with changes in the expression level of genes for metabolic processes such as de novo lipid biosynthesis and glycolysis57,58, and supports the hypothesis that liver functions are enhanced in polyploid hepatocytes57. In addition, the periportal score was associated with the 9 metabolic traits (all P<0.005 except P=0.04,0.02,0.24 for HDL,SHBG,TBIL, MC test). While the pericentral score was not significantly associated with these traits in TMS FACS, we detected significant associations across multiple other data sets (Supplementary Note). These results suggest that these metabolic traits are impacted by complex processes involving both pericentral and periportal hepatocytes. Validations of the polyploidy and zonation scores using independent data and results on 5 additional mouse and human data sets17,59–61 (the unpublished Taychameekiatchai et al. data was provided by co-authors A. Taychameekiatchai, P. Rao, and B. Wang) are reported in the Supplementary Note, Extended Data Figure 9, Supplementary Figure 27, and Supplementary Tables 24,27.
Discussion
We have introduced scDRS, a method that leverages polygenic GWAS signals to associate individual cells in scRNA-seq data with diseases and complex traits; we showed via extensive simulations that scDRS is well-calibrated and powerful in realistic scenarios. We applied scDRS to 74 diseases/traits in conjunction with 16 scRNA-seq data sets and detected extensive heterogeneity in disease associations of individual cells within classical cell types. These findings may prove useful for targeting the relevant cell populations for in vitro experiments to elucidate the molecular mechanisms through which GWAS risk variants impact disease.
We have demonstrated the value in associating individual cells to disease; assessing the heterogeneity across individual cells within predefined cell types in their association to disease; identifying cell-level variables partially characterizing the individual cells that are associated to disease; and broadly associating predefined cell types to disease. Analyses of larger scRNA-seq/snRNA-seq and GWAS data sets using approaches such as scDRS will continue to further these goals.
We note several limitations and future directions of our work. First, identifying a statistical correlation between individual cells (or cell types) and disease does not imply causality, but may instead reflect indirect tagging of causal cells/cell types, analogous to previous works4,5,10,18. However, even in such cases, the implicated cells/cell types are likely to be closely biologically related to the causal cells/cell types, based on their similar expression patterns. Second, the fact that scDRS assesses the statistical significance of an individual cell’s association to disease by implicitly comparing it to other cells via matched control genes may reduce power if most cells in the data are truly causal. For example, association with IBD in a data set containing only Tregs (one of the causal cell types for IBD) will likely yield largely non-significant results. This limitation did not impact our main analyses, because the TMS data includes a broad set of cell types; in more specialized data sets (which may be preferred in some settings due to the more comprehensive profiling of the focal cell population), this limitation can potentially be addressed by selecting matched control genes based on a broad cell atlas (e.g., the TMS or TS data). Third, while we have primarily focused on the associations involving a single disease/trait, further investigation of differences between diseases/traits within the same category is an important future direction. Additional limitations are discussed in the Supplementary Note. Despite all these limitations, scDRS is a powerful method for distinguishing disease associations of individual cells in single-cell RNA-seq data.
Methods
Ethical statement
This study analyzed publicly available data sets and hence did not require ethical approval.
scDRS method
We consider an scRNA-seq data set with ncell cells (not cell types) and ngene genes. We denote the cell-gene matrix as , where Xcg represents the expression level of cell c and gene g. We assume that X is size-factor-normalized (e.g., 10,000 counts per cell) and log-transformed (log(x + 1)) from the original raw count matrix19. We regress the covariates out from the normalized data19 (with a constant term in the regressors to center the data), before adding the original log mean expression of each gene back to the residual data. Such a procedure preserves the mean-variance relationship in the covariate-corrected data, necessary for estimating the gene-specific technical noise levels (Supplementary Note). Gene-level statistics for the scRNA-seq data are reported in Supplementary Figure 4 and Supplementary Tables 3,4; estimated technical noise levels are moderately correlated across genes between the 16 data sets (avg. cor. 0.34) and are highly correlated between data sets with similar cell type compositions (e.g., 0.74 between TMS FACS and TS FACS).
The scDRS algorithm is described below. Given a disease GWAS and an scRNA-seq data set, scDRS computes a p-value for each individual cell for association with the disease. scDRS also outputs cell-level normalized disease scores and B sets of normalized control scores (default B= 1,000) that can be used for data visualization and Monte Carlo-based statistical inference (see below). scDRS consists of three steps. First, scDRS constructs a set of putative disease genes from the GWAS summary statistics. Second, scDRS computes a raw disease score and B MC samples of raw control scores for each cell. Third, after gene set-wise and cell-wise normalization, scDRS computes an association p-value for each cell by comparing its normalized disease score to the empirical distribution of the pooled normalized control scores across all control gene sets and all cells.
We discuss guidelines for using scDRS. First, scDRS relies on assumptions which require the disease gene set to have a moderate size (e.g., >50 genes and <20% of all genes). Second, to ensure a reasonable number of scDRS discoveries, we recommend using GWAS data with a heritability z-score greater than 5 or a sample size greater than 100K. We also recommend using single-cell RNA-seq data with a diverse set of cells potentially relevant to disease, although a smaller number of cells should not affect the scDRS power. However, scDRS will not produce false positives for less ideal GWAS or single-cell data sets. Third, scDRS is computationally efficient and scales linearly with the number of cells and number of control gene sets for both computation and memory use; it takes around 3 hours and 60GB for a single-cell data set with a million cells). Fourth, scDRS can be used in conjunction with any gene sets (instead of the MAGMA GWAS gene sets) to evaluate the enrichment of aggregate expression for cells in a single-cell data set. Fifth, we provide an option to adjust for cell-type proportions (or any cell group annotations), recommended only for extremely unbalanced data sets. Further details and evaluations of alternative versions of scDRS are provided in the Supplementary Note.
Algorithm description
We describe the scDRS algorithm.
Input: Disease GWAS summary statistics (or putative disease gene set G with GWAS gene weights {wg}g∈G), scRNA-seq data .
Parameter: Number of MC samples of control gene sets B (default 1,000).
- Construct putative disease gene set
- Construct putative disease gene set G ⊂ {1, 2, … , ngene} with GWAS gene weights {wg}g∈G from GWAS summary statistics using MAGMA.
- Compute disease scores and control scores
- Sample B sets of control genes matching mean expression and expression variance of disease genes.
- Estimate gene-specific technical noise level .
- Compute raw disease score and B raw control scores for each cell c = 1, … ,ncell, ,
(1)
- Compute disease association p-values
- First gene set alignment by mean and variance. Let be the expression variance of gene g. For each cell c,
(2) - Cell-wise standardization for each cell c by the mean and SD of control scores of that cell,
(3) - Second gene set alignment by mean. For each cell c,
(4) - Compute cell-level p-values based on the empirical distribution of the pooled normalized control scores for each cell c,
(5)
Output: cell-level p-values pc, normalized disease scores sc, and normalized control scores .
Downstream applications and MC test
We use a unified MC test for the scDRS downstream analyses based on the (normalized) disease and control scores. Specifically, let t be a test statistic computed from the disease score of the given set of cells (different downstream analyses differ by the test statistics) and let be the same test statistics computed from the B sets of control scores of the same set of cells. The MC p-value can be written as
| (6) |
The MC test avoids the assumption that the cells are independent—a strong assumption in scRNA-seq analyses, e.g., when analyzing cells in the same cluster that are dependent due to the clustering process. However, the MC p-value pMC cannot be smaller than 1/(1 + B) by Equation (6), limiting its ability in distinguishing highly-significant associations. We can also compute an MC z-score as ; this MC z-score is not restricted by the MC limit of 1/(1 + B) but relies the assumption that the control test statistics approximately follow a normal distribution. We recommend using MC p-values to determine statistical significance and using MC z-scores to further prioritize associations whose MC p-values have reached the MC limit. Below, we describe the test statistics used by the 3 analyses listed above. Besides the 3 analyses below, the MC test can in principle be extended to any analysis that computes a test statistic from the disease scores of a set of cells.
Associating cell type to disease.
We use the top 5% quantile of the disease scores of cells from the given cell type as the test statistic. This test statistic is robust to annotation outliers, e.g., a few misannotated but highly significant cells. One can also use other test statistics such as the top 1% quantile or the maximum.
Assessing within-cell type heterogeneity in association with disease.
We use Geary’s C14,65 as the test statistic. Geary’s C measures the spatial autocorrelation of the disease score across a set of cells (e.g., cells from the same cell type or cell cluster) with respect to a cell-cell similarity matrix. Given a set of n cells, the corresponding disease scores s1, … ,sn, and the cell-cell similarity matrix , Geary’s C is calculated as
| (7) |
where . A value significantly lower than 1 suggests a high level of disease-association heterogeneity across the given set of cells. Details are provided in the Supplementary Note.
Correlating a cell-level variable to disease across a given set of cells.
For associating a single cell-level variable with disease, we use the Pearson’s correlation between the cell-level variable and the disease score across the given set of cells as the test statistic. For jointly associating multiple cell-level variables with disease, we use the regression t-statistic as the test statistic, obtained from jointly regressing the disease score against the cell-level variables.
Simulations
We considered 3 comparison methods: Seurat13 (“score_genes” as implemented in Scanpy66 v1.6.0), Vision14 (implemented in scDRS v1.0.1), and VAM16 (v0.5.1). To our knowledge, VAM is the only published cell-scoring method that provides cell-level association p-values. We chose to include Seurat due to its wide use. We standardized its output cell-level scores (mean 0 and SD 1) before computing the cell-level p-values based on the standard normal distribution. We chose to include Vision because its outputs are nominal cell-level z-scores that can be easily converted to p-values; we again added the standardization step because otherwise the Vision results were highly unstable. We did not include other methods like PAGODA11 or AUCell12 because it is not straightforward to convert their outputs to cell-level association p-values and also because the z-scoring methods (e.g., Vision) outperformed other methods in a recent evaluation16.
We performed simulations on a data set with 10,000 cells subsampled from the TMS FACS data. In null simulations, we randomly selected putative disease genes from a set of non-informative genes. We considered four numbers of putative disease genes (100, 500, 1,000, or 2,000) and four types of genes to sample from: (1) the set of all genes, (2) the set of top 25% genes with high mean expression, (3) the set of top 25% genes with high expression variance, (4) the set of top 25% overdispersed genes, where the level of overdispersion is calculated as the difference between the actual variance and the estimated technical variance in the log scale data. For the default version of scDRS, we simulated GWAS gene weights by first randomly selecting a disease (out of the 74 diseases/traits) and then randomly permuting the top MAGMA z-scores from the selected disease. We did not simulate gene-specific technical noise-based single-cell weights because these weights were inherent to the single-cell data. For the MC test for cell type-disease association, we used the top 5% quantile as the test statistic (see above).
In causal simulations, we randomly selected 1,000 causal disease genes, randomly selected 500 of the 10,000 cells as causal cells and artificially perturbed their expression levels to be higher (at various effect sizes) across the 1,000 causal disease genes, and randomly selected 1,000 putative disease genes (provided as input to scDRS and other methods) with various levels of overlap with the 1,000 causal disease genes. Here, the effect size corresponds to the fold change of expression of the causal genes in the causal cells (multiplicative in the original count space and additive in the log space). We performed three sets of causal simulations: (1) varying effect size from 5% to 50% while fixing 25% overlap, (2) varying level of overlap from 5% to 50% while fixing 25% effect size, (3) assigning the 528 B cells in the subsampled data to be causal (instead of the 500 randomly selected cells; varying effect size while fixing 25% overlap). The FDR and power are based on applying the B-H procedure67 to all cells at nominal FDR=0.1.
GWAS and single-cell data sets
We analyzed GWAS summary statistics of 74 diseases and complex traits from the UK Biobank68 (47 of the 74 diseases/traits with average N=415K) and other publicly available sources (27 of the 74 diseases/traits with average N=225K); average N=346K for all 74 diseases/traits; Supplementary Table 1). All diseases/traits were well-powered (heritability z-score>5), except celiac disease (Celiac), systemic lupus erythematosus (SLE), multiple sclerosis (MS), subject well being (SWB), and type 1 diabetes (T1D), which were included due to their clinical importance. The major histocompatibility complex (MHC) region was removed from all analyses because of its unusual LD and genetic architecture69.
We analyzed 16 scRNA-seq or snRNA-seq data sets. The 3 atlas-level data sets (TMS FACS, TMS droplet, and TS FACS) allow us to broadly associate diverse cell types/populations to disease and to compare results between species (mouse/human) and between technologies (FACS/droplet). The other 13 data sets focus on a single tissue and contain finer-grained annotations of cell types/states and/or experimentally determined annotations, which allow for better validation (Supplementary Table 2).
Analysis of T cells and autoimmune diseases
We collectively analyzed all TMS FACS T cells (4,125 cells labeled as “CD4+ α-β T cell”, “CD8+ α-β T cell”, “regulatory T cell”, “mature NK T cell”, “mature α-β T cell”, or “T cell” in the TMS data; Supplementary Table 5); the more general terms like “T cell” and “mature α-β T cell” were used for cells whose more specific identities were not clear. We processed the T cells following the same procedure as described in the original paper17,66. First, we performed size factor normalization (10,000 counts per cell) and log transformation. Second, we selected highly variable genes and computed the batch-corrected PCA embedding using Harmony70, treating each mouse as a batch. Finally, we constructed KNN graphs and clustered the cells using the Leiden algorithm71 (resolution=0.7), followed by computing the UMAP embedding. We removed 376 cells either from small clusters (less than 100 cells) or whose identities are ambiguous, resulting in 3,769 cells. We annotated the clusters based on the major TMS cell types in the cluster; the label “mature α-β T cell” was omitted because a more specific TMS cell type label (e.g., “CD8+ α-β T”) was available in the corresponding cluster. We further characterized disease-associated T cell subpopulations based on marker gene expression, automatic T cell subtype annotation72, and overlap of specifically expressed genes in each subpopulation with T cell signature gene sets (Supplementary Figures 14–17; Supplementary Note). We considered cells from clusters 1–4 as clear CD4+ T cells (1,686 cells) and cells from clusters 1,2,7–9 as clear CD8+ T cells (2,197 cells; the shared clusters 1 and 2 contain a mix of naive CD4+ and CD8+ T cells). We used diffusion pseudotime (DPT)38 to assign effectorness gradient for CD4+ and CD8+ T cells separately, where we used the leftmost cell in cluster 2 on the UMAP as the root cell (clearly naive T cell).
We used MSigDB73 (v7.1) to curate T cell signature gene sets, including naive CD4, memory CD4, effector CD4, naive CD8, memory CD8, effector CD8, Treg, Th1 (T helper 1), Th2 (T helper 2), and Th17 (T helper 17) signatures. For each T cell signature gene set, we identified a set of relevant MSigDB gene sets (22–34 gene sets, Supplementary Table 17), followed by selecting the top 100 most frequent genes in these MSigDB gene sets as the T cell signature genes; a gene was required to appear at least twice and genes appearing the same number of times were all included, resulting in 62 to 513 genes for the 10 T cell signature gene sets (Supplementary Table 24). For gold-standard gene sets used in the analysis of disease gene prioritization, we curated 27 putative drug target gene sets from Open Targets41 (mapped to 27 of the 74 diseases/traits); for a given disease, we selected all genes with drug score >0 (clinical trial phase 1 and above) and only considered diseases with at least 10 putative drug target genes. We curated 16 Mendelian diseases gene sets from Freund et al.42 (mapped to 45 of the 74 diseases/traits) (Supplementary Table 21). For comparison of two gene sets, the p-value is based on two-sided Fisher’s exact test and excess overlap is defined as the ratio between the observed overlap of the two gene sets and the expected overlap (by chance). Of note, for a given query gene set with a fixed size and a fixed level of excess overlap with the reference gene set, the −log10 p-value increases with the size of the reference gene set; we report both excess overlap and −log10 p-value while using the former as our primary metric, which is more interpretable.
Analysis of neurons and brain-related diseases/traits
For the TMS FACS data, we focused on the 484 neurons (TMS label “neuron”, excluding cells with TMS label “medium spiny neuron” or “interneuron”). For the Zeisel & Muñoz-Manchado et al. data, we applied scDRS to all 3,005 cells and then focused on the 827 CA1 pyramidal neurons (“level1class” label “pyramidal CA1”). For inferring spatial coordinates, we curated differentially expressed genes for each of the 6 spatial regions (dorsal vs. ventral, ventral vs. dorsal, proximal vs. distal, distal vs. proximal, deep vs. superficial, and superficial vs. deep) using the gene expression data from Cembrowski et al.48 (GEO GSE67403; gene sets in Supplementary Table 24). For each differential gene expression analysis, we selected genes based on FPKM>10 for the average expression in the enriched region (e.g., dorsal for the dorsal vs. ventral comparison), q-value<0.05, and log2(fold change) >2. We used scDRS and these signature gene sets to assign 6 spatial scores for each cell. For the regression analysis, we separately regressed the scDRS disease scores for each of the 7 brain-related traits (and height, a negative control trait) on each of the 6 spatial scores. We performed marginal regression instead of joint regression for these spatial scores because the inferred spatial scores for opposite regions on the same axis (e.g., dorsal vs. ventral) were highly collinear (strongly negatively correlated), and the inferred spatial scores for dorsal, proximal, and deep regions (which had strong marginal associations to diseases) had very low pairwise correlations (average |r|=0.10; Supplementary Figure 23d), suggesting these associations were independent.
Analysis of hepatocytes and metabolic traits
We considered all hepatocytes in the TMS FACS data (1,162 cells). Since the original study clustered all cells from the liver together17 (limiting the resolution for distinguishing cell states within hepatocytes), we reprocessed and reclustered these cells following the same procedure as we did for the T cells. We further filtered out low-quality cells (proportion of mitochondrial gene counts ≥0.3; likely apoptotic or lysing), resulting in 1,102 hepatocytes. We computed the polyploidy, pericentral, and periportal scores by applying scDRS to published polyploidy/zonation signature gene sets (instead of MAGMA putative disease gene sets). We curated signature gene sets for ploidy level, zonation (pericentral/periportal), and putative zonated pathways. We curated 4 sets of polyploidy signatures, including differentially expressed genes (DEGs) for partial hepatectomy (PH) vs. pre-PH58 (used for the polyploidy score), Cdk1 knockout (case) vs. control58, 4n vs. 2n hepatocytes60, large vs. small hepatocytes58. We curated 3 sets of diploidy signatures, including DEGs for pre-PH vs. PH58, control vs. Cdk1 knockout58, and 2n vs. 4n hepatocytes60. We curated signature gene sets for pericentral (CV) and periportal (PN) hepatocytes from Halpern et al.59. We curated gene sets for putative zonated pathways from MSigDB73 (v7.1), including glycolysis (pericentral), bile acid production (pericentral), lipogenesis (pericentral), xenobiotic metabolism (pericentral), beta-oxidation (periportal), cholesterol biosynthesis (periportal), protein secretion (periportal), and gluconeogenesis (periportal) (Supplementary Table 24). For the joint regression analysis of scDRS disease score on ploidy and zonation scores, we regressed the polyploidy score out of both the pericentral and periportal score before the joint regression because the ploidy level confounded both zonation scores. We performed joint regression instead of marginal regression here (unlike the regression analysis in the neuron section) because the polyploidy score was positively correlated with the pericentral and periportal scores (unlike the analysis in the neuron section where the 3 sets of scores had low correlations).
Statistics & Reproducibility
We analyzed only existing data sets. No statistical method was used to predetermine sample size. No data were excluded from the analyses. We did not use any study design that required randomization or blinding. We replicated our results by performing the same analyses on additional independent data sets; all attempts at replication were successful.
Extended Data
Extended Data Fig. 1. Additional null simulations.
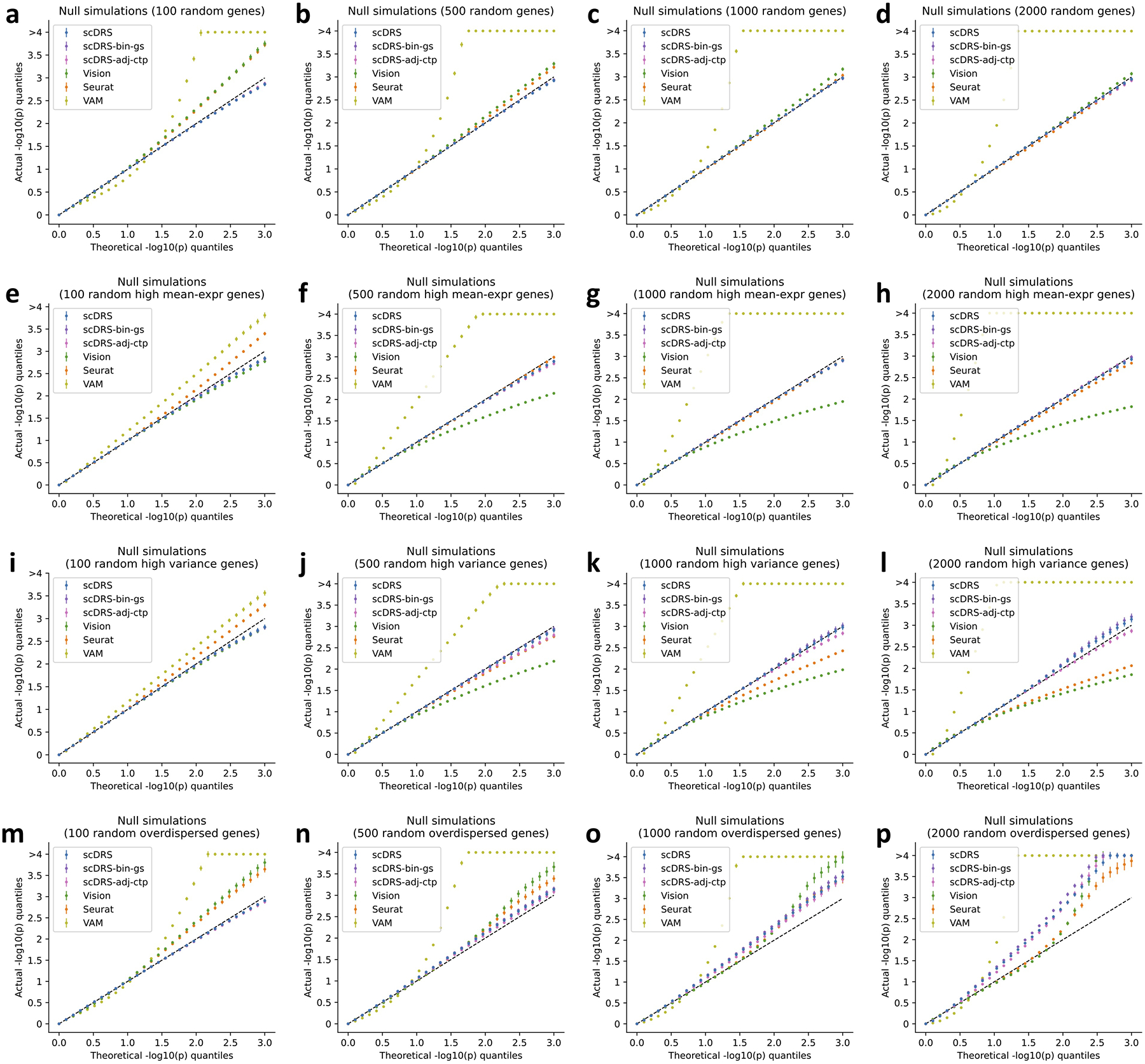
We performed null simulations for various numbers of putative disease genes (100, 500, 1,000, and 2,000 for the four columns respectively) and various types of genes to randomly sample from: all genes (first row), and top 25% genes with high expression (second row), top 25% genes with high expression variance (third row), top 25% overdispersed genes (fourth row). We considered two additional versions of scDRS: scDRS-bin-gs (binary gene sets instead of MAGMA z-score gene weights) and scDRS-adj-ctp (adjusting for cell type proportion). For scDRS-adj-ctp, we simulated random biased gene sets (high-mean/high-variance/overdispersed) based on the balanced data (inversely weighting cells by cell type proportion) to better match the model assumption, namely testing for excess expression relative to cells in the balanced data. In each panel, the x-axis denotes theoretical −log10 p-value quantiles and the y-axis denotes actual −log10 p-value quantiles for different methods. The 3 versions of scDRS produced well-calibrated p-values in most settings and suffered slightly inflated type I error in panels o,p, possibly because it is hard to match a large number of overdispersed putative disease genes using the remaining set of genes. In comparison, all other methods are less well-calibrated and are particularly problematic when the numbers of putative disease genes are small. Error bars denote 95% confidence intervals around the mean of 100 simulation replicates.
Extended Data Fig. 2. Additional causal simulations.
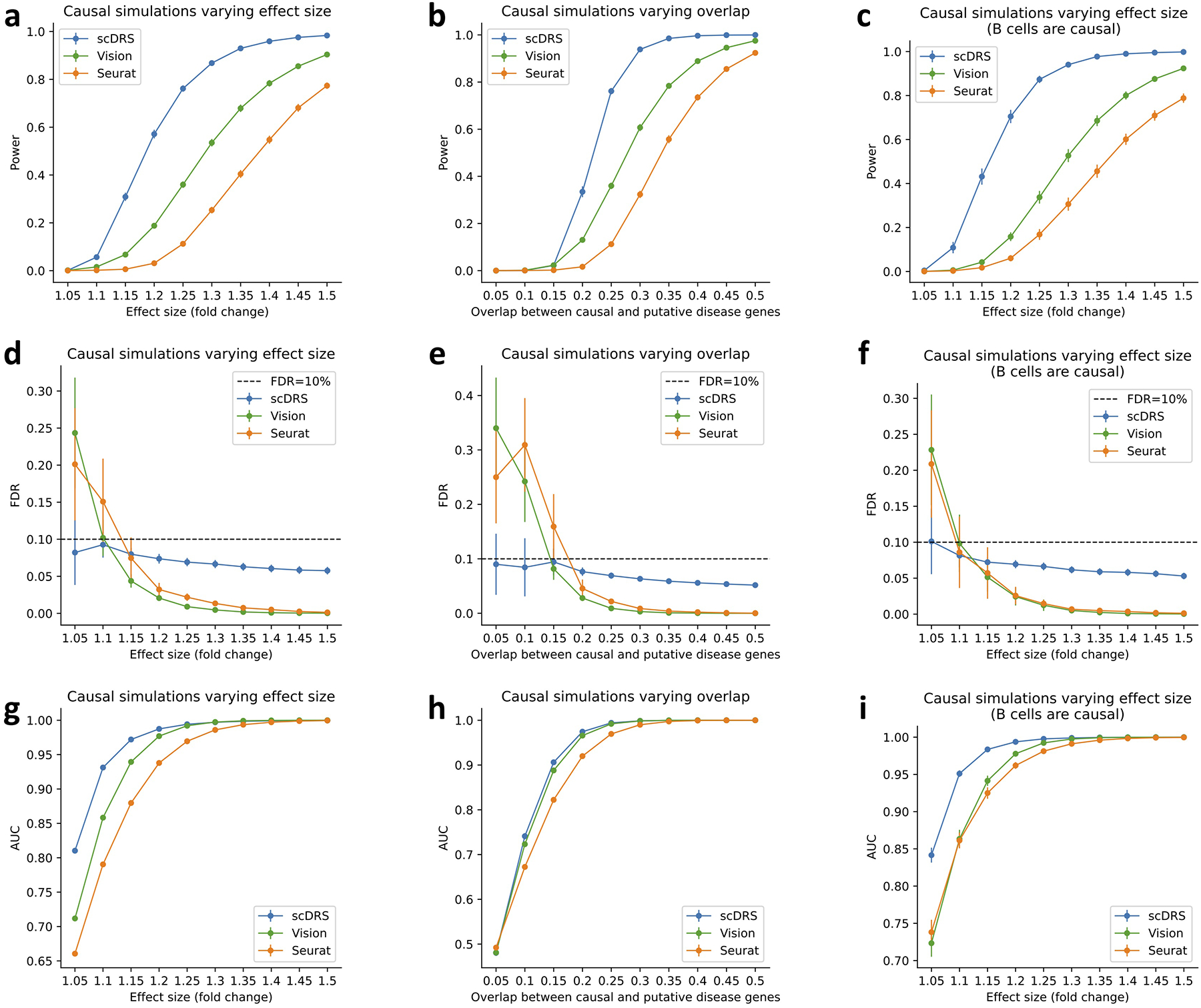
We performed three sets of causal simulations: (1) varying effect size from 5% to 50% while fixing 25% overlap (first column), (2) varying level of overlap from 5% to 50% while fixing 25% effect size (second column), (3) assigning the 528 B cells in the subsampled data to be causal (instead of the 500 randomly selected cells; varying effect size while fixing 25% overlap; third column). We report the power (first row), FDR (second row), and AUC for classifying causal from non-causal cells based on the p-values (third row). scDRS outperformed other methods under all metrics. Error bars denote 95% confidence intervals around the mean of 100 simulation replicates.
Extended Data Fig. 3. Complete results for cell type-level disease associations for 74 diseases/traits and TMS FACS 120 cell types.
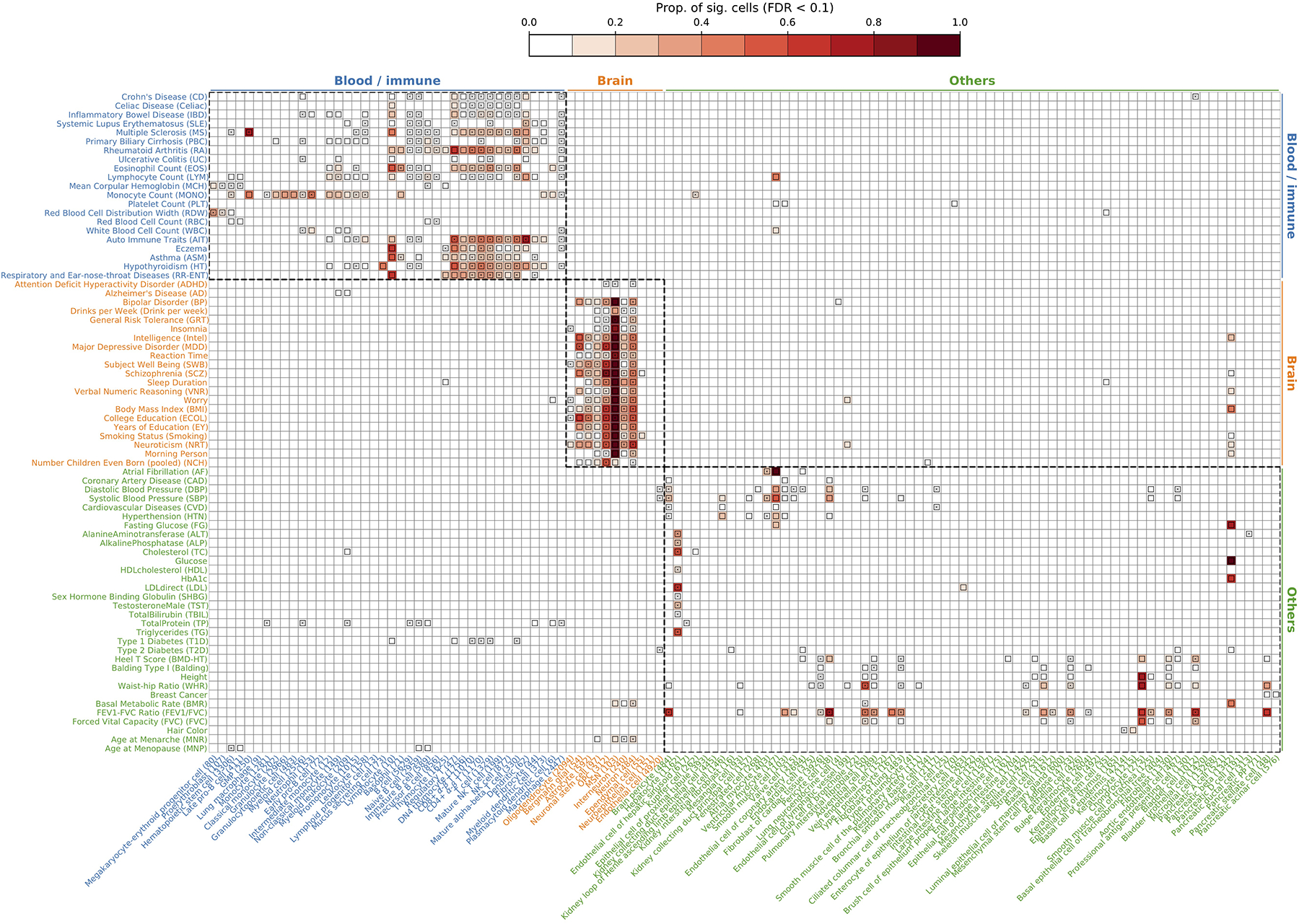
Each row represents a disease/trait and each column represents a cell type (number of cells in parentheses). Heatmap colors denote the proportion of significantly associated cells (FDR<0.1 across all cells for a given disease). Squares denote significant cell type-disease associations (FDR<0.05 across all pairs of the 120 cell types and 74 diseases/traits; 597 significant pairs; MC test; Methods). Cross symbols denote significant heterogeneity in association with disease across individual cells within a given cell type (FDR<0.05 across all pairs; 273 significant pairs; MC test; Methods). Heatmap colors and cross symbols are omitted for cell type-disease pairs with non-significant cell type-disease associations. Within the blood/immune block (40 cell types and 21 diseases/traits), 136 of 264 cell type-disease pairs with significant association also had significant heterogeneity. Within the brain block (11 cell types and 21 diseases/traits), 64 of 133 cell type-disease pairs with significant association also had significant heterogeneity. Within the other block (69 cell types and 32 diseases/traits), 54 of 146 cell type-disease pairs with significant association also had significant heterogeneity. We discuss the results for FEV1/FVC. We identified 20 cell types associated with FEV1/FVC (FDR<0.05), including 5 lung cell types and 15 cell types from other tissues. They can be categorized into 5 sets of associations: (1) type II pneumocyte (2) skin-related cells (3) smooth muscle cells (4) fibroblast-and-MSC-like cells (5) pericyte-like cells. The first 4 sets of associations are consistent with a previous work63. The 5th set of pericyte associations is also plausible because pericytes are known to regulate lung morphogenesis64. We note that the cell type associations from the lung are more likely to be causal and those from the other tissues are more likely tagging the causal cell types due to shared expression. Numerical results are reported in Supplementary Table 12.
Extended Data Fig. 4. Comparison of cell type-level disease association results between TMS FACS and TMS droplet (different technologies), TS FACS (different species).
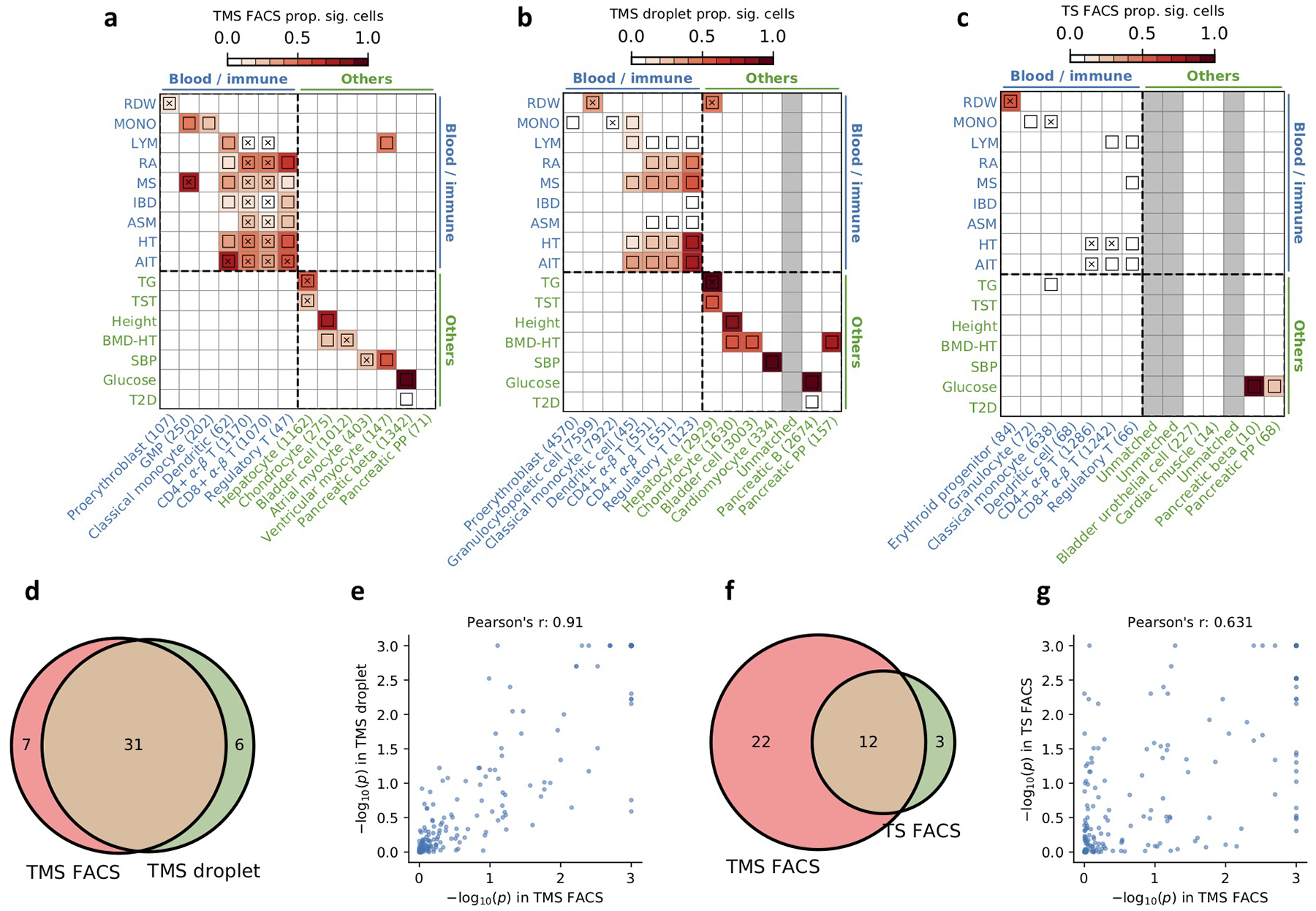
(a-c) Results for disease association at the cell type-level for TMS FACS, TMS droplet, and TS FACS for diseases and cell types in the blood/immune block (upper left) and the other cell types/diseases block (lower right) in Figure 3 (TMS droplet and TS FACS do not contain brain data; Supplementary Tables 6,7). The plotting style is the same as Figure 3. Heatmap colors for each cell type-disease pair denote the proportion of significantly associated cells (FDR<0.1); squares denote significant cell type-disease associations (FDR<0.05); and cross symbols denote significant heterogeneity in association with disease across individual cells within a given cell type (FDR<0.05). Heatmap colors (>10% of cells associated) and cross symbols are omitted for cell type-disease pairs with non-significant cell type-disease associations via MC test. We matched each TMS FACS cell type using the closest cell type in the TMS droplet and TS FACS data; unmatched cell types were colored in grey. (d) Overlap of significant cell type-disease associations between TMS FACS and TMS droplet (P=2.8×10−24, two-sided Fisher’s exact test). (e) Pearson’s correlation of −log10 p-values for cell type-disease associations between TMS FACS and TMS droplet. (f) Overlap of significant cell type-disease associations between TMS FACS and TS FACS (P=1.3×10−7, two-sided Fisher’s exact test). (g) Pearson’s correlation of −log10 p-values for cell type-disease associations between TMS FACS and TS FACS. We determined that the results are highly consistent between TMS FACS and TMS droplet, and are reasonably consistent between TMS FACS and TS FACS. Our method is underpowered in the TS FACS data, possibly due to the smaller sample size (27K cells in TS FACS vs. 110K cells in TMS FACS). The current TS FACS data corresponds to the initial data release and there will likely be more cells in future releases20.
Extended Data Fig. 5. Optimizing parameters of scDRS based on expected and unexpected control cell types across 20 traits.
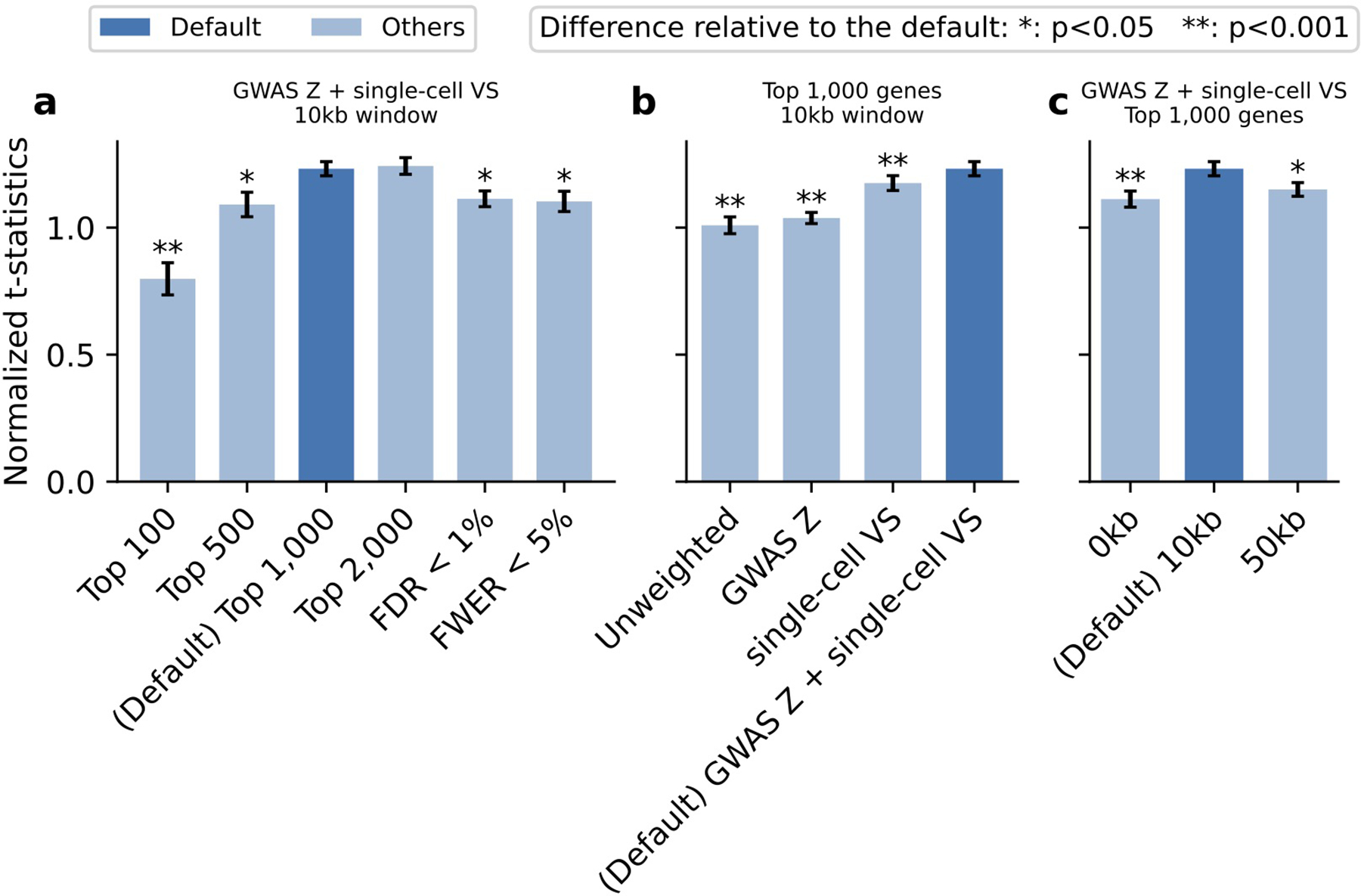
We considered different versions of scDRS by varying methods for selecting (1) putative disease genes (2) weights for the disease genes (3) MAGMA window size. We considered 6 methods for selecting putative disease genes, 4 methods for selecting gene weights, and 3 MAGMA gene window sizes (Supplementary Note). We applied each version of scDRS to the subsampled TMS FACS data (20 repetitions with 10K cells each) and a curated set of 20 traits with expected and unexpected disease-critical cell types (Supplementary Table 15). For a given scDRS version and a given trait, we computed the t-statistic between cells from the expected and unexpected cell types, and divided it by the average t-statistics of results of the given trait from all data sets and all scDRS versions to correct for trait-specific baseline. We evaluated each version by first computing the mean and SE of the normalized t-statistics for a given trait across the 20 repetitions and then combining the estimates across the 20 traits via random-effect meta-analysis. We compared the performance of a pair of scDRS versions by applying the same procedure to the difference of the normalized t-statistics between the two versions. (a) Varying gene selection methods while fixing other parameters as the default. (b) Varying gene weighting methods while fixing other parameters as the default. (c) Varying MAGMA gene window size while fixing other parameters as the default. The default version was denoted in dark blue. Error bars denote 95% confidence intervals around the mean based on meta-analysis across 20 sub-sampled data sets and 20 traits, using procedures as described above. * denotes P<0.05 and ** denotes P<0.001 for significant differences relative to the default configuration; one-sided tests based on the estimated mean and CIs. Numerical results are reported in Supplementary Table 16.
Extended Data Fig. 6. Numbers of overlapping genes (upper triangle) and correlations of the scDRS disease scores across all TMS FACS cells (lower triangle) between the 26 autoimmune, brain, and metabolic traits analyzed in the main paper.
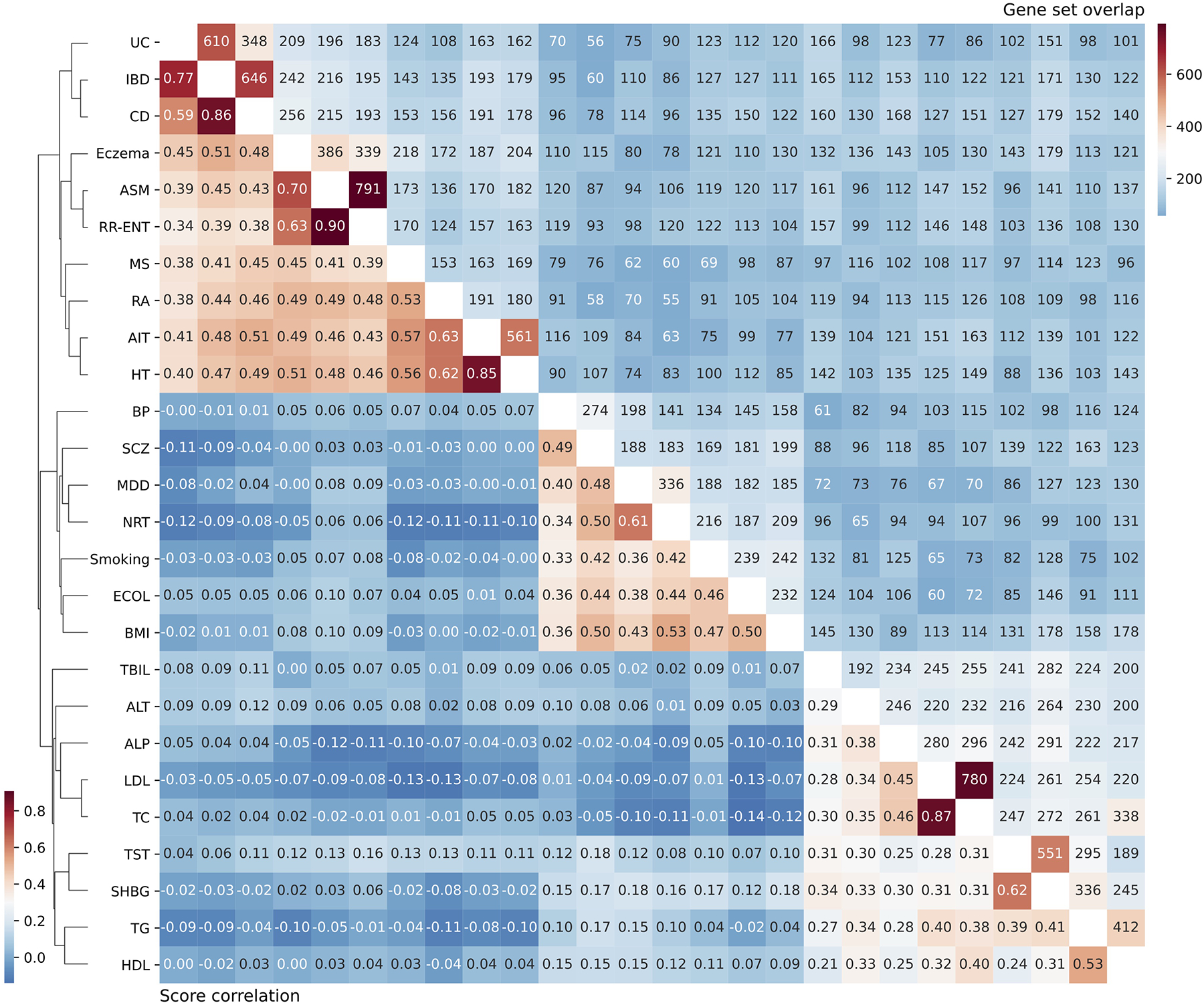
Traits are ordered via hierarchical clustering of the scDRS score correlation and the clustering dendrogram was provided. The level of gene set overlap is moderate. scDRS disease score correlations distinguish diseases/traits from the 3 categories as well as subgroups of diseases/traits in the same category.
Extended Data Fig. 7. Additional results on disease gene prioritization.
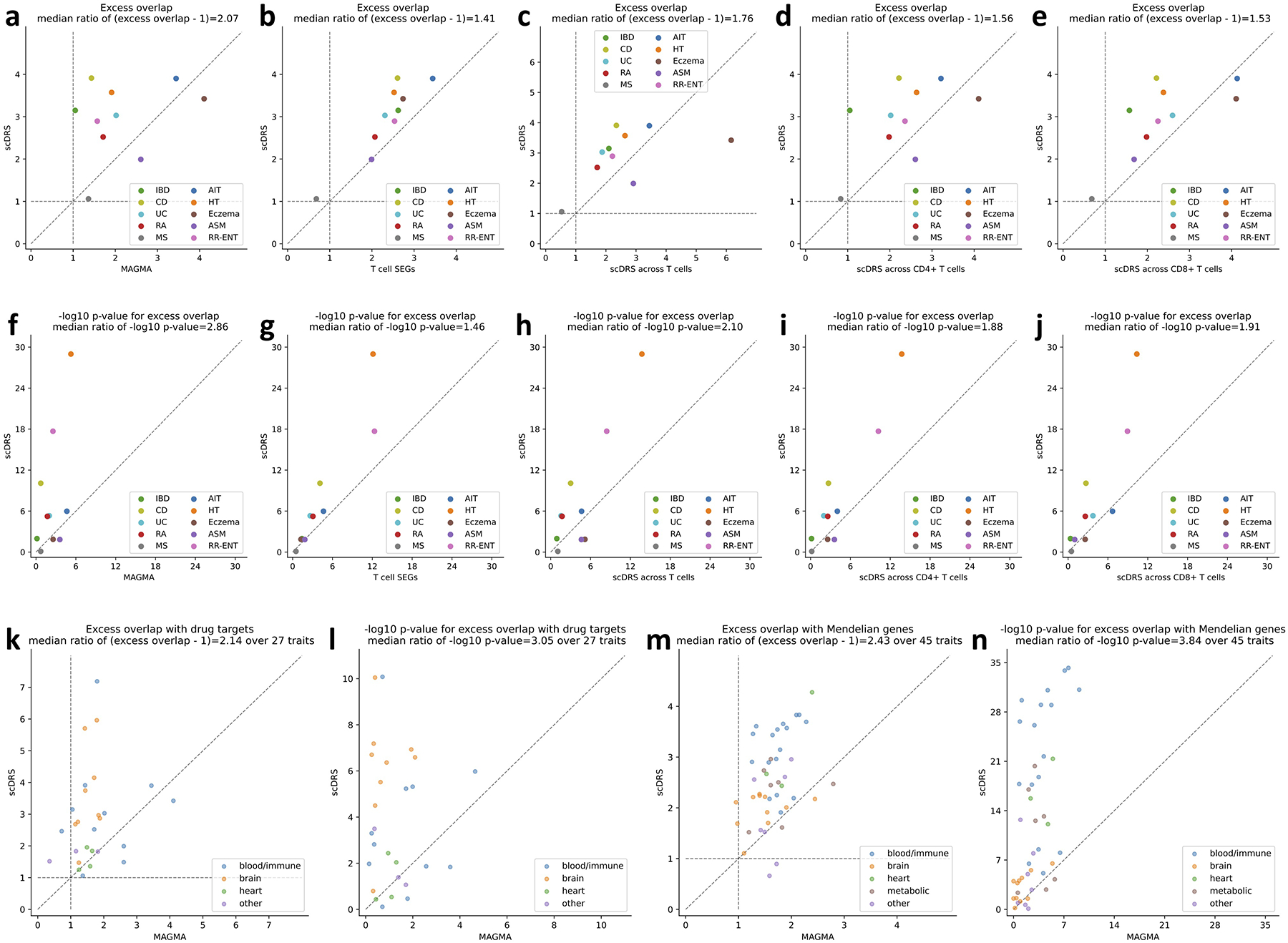
(a-j) Comparison to alternative disease gene prioritization methods for the 10 autoimmune diseases. The first row shows levels of excess overlap between the prioritized disease genes and the gold standard gene sets while the second row shows the corresponding −log10 p-values for excess overlap. Each dot corresponds to a disease, the y-axis shows results for the proposed prioritization method (correlating gene expression levels with the scDRS disease score across all TMS FACS cells), and the x-axis shows results from comparison methods, including (from left to right) top 1,000 MAGMA genes, top 1,000 genes specifically expressed in T cells (vs. the rest of cells in TMS FACS), prioritization based on correlation across T cells (instead of all TMS FACS cells), prioritization based on correlation across CD4+ T cells (instead of all TMS FACS cells), and prioritization based on correlation across CD8+ T cells (instead of all TMS FACS cells). (k-l) Overlap with drug target genes for 27 diseases. (m-n) Overlap with Mendelian disease genes for 45 diseases. The median ratio of −log10 p-values and (excess overlap − 1) between the y- and x-values (median of ratios) was provided in the figure title. P-values are based on two-sided Fisher’s exact tests.
Extended Data Fig. 8. Complete results of correlations between scDRS disease scores and inferred spatial coordinates across CA1 pyramidal neurons in 7 single-cell data sets (extending results in Figure 5b).
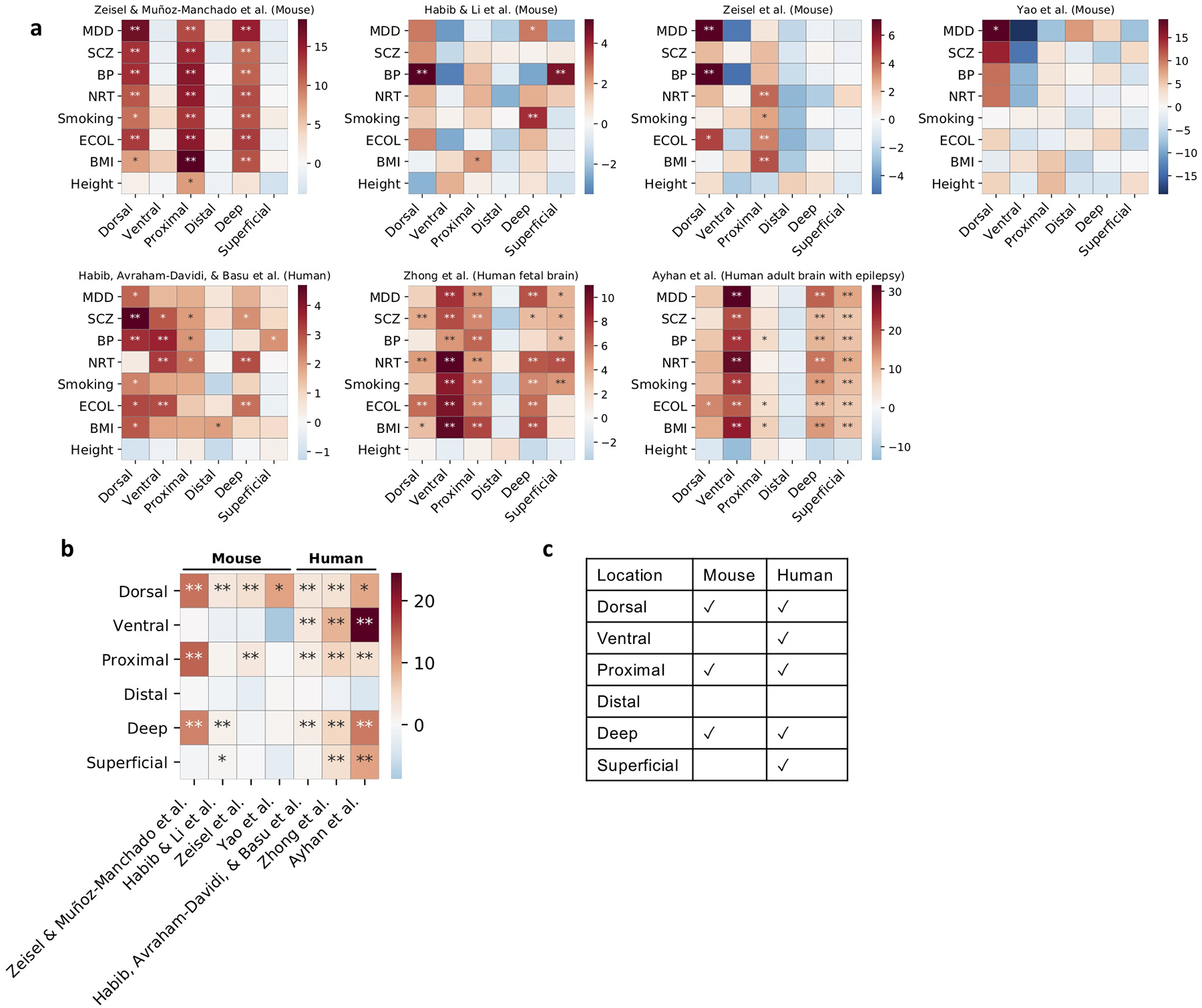
(a) Results for regressing the scDRS disease scores against the inferred spatial coordinates for each disease/trait and each inferred spatial coordinate. Color represents the t-statistics and stars represent significant associations (* denotes P<0.05 and ** denotes P<0.005, one-sided MC test; Methods). For clarification, Zeisel & Muñoz-Manchado et al. refers to the data from Zeisel & Muñoz-Manchado et al. 2015 Science46 and Zeisel et al. refers to the data from Zeisel et al. 2018 Cell51. (b) Summary of results in panel a. Heatmap color represent the average t-statistics across the 7 brain-related diseases/traits (excluding height) for each data set and stars represent significant associations by combining p-values across datasets using Fisher’s combined probability test. (c) Summary of the association between brain-related diseases and the inferred spatial coordinates for the mouse and human data sets in panel b.
Extended Data Fig. 9. Complete results of joint regression analysis for GWAS metabolic traits and putative zonated metablic processes across the 6 data sets (extending results in Figure 5d).
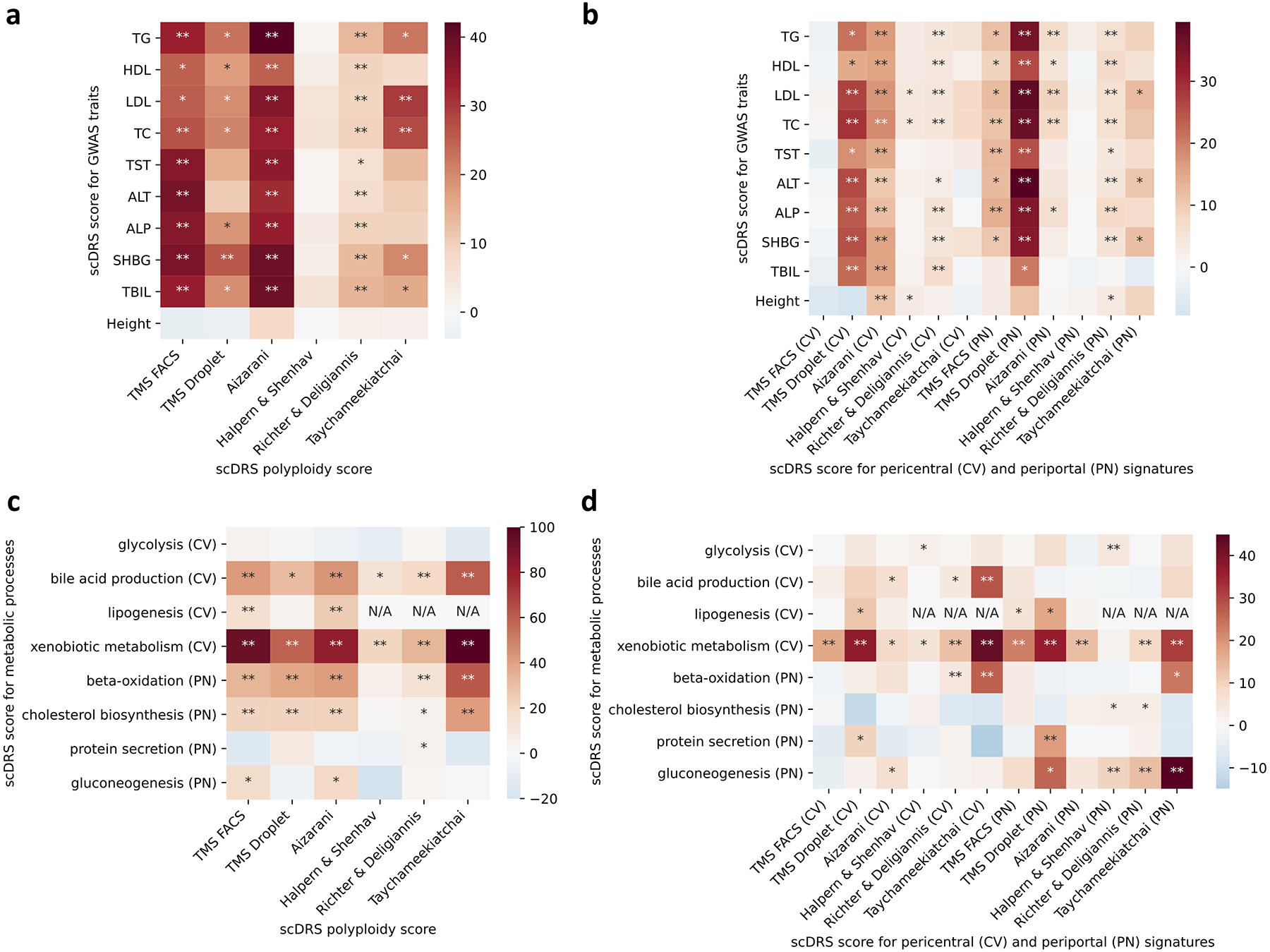
(a-b) Results for the 9 metabolic traits and height, a negative control trait. The polyploidy score (panel a) and both the pericentral and periportal score (panel b) were consistently associated with the 9 metabolic traits across the data sets. The strong association (P<0.005) between the pericentral score and height in the Aizarani et al. data may be because that we inferred the pericentral score using mouse gene signatures, which are less conserved in human (as also mentioned in the original paper61). (c-d) Results for the 8 metabolic pathways. Overall, as shown in panel d, the pericentral score was associated with pericentral-specific pathways (first 4 rows) while the periportal score was associated with periportal-specific pathways (last 4 rows). * denotes P<0.05 and ** denotes P<0.005 based on one-sided MC tests.
Supplementary Material
Acknowledgements
We thank Huwenbo Shi, Katherine Siewert-Rocks, Tiffany Amariuta, Xiaoyu Xu, Benjamin J Strober, and Alexander Gusev for helpful suggestions. This research was conducted using the UK Biobank Resource (application number: 16549). This research was funded by NIH grants U01 HG009379 (A.L.P. and S.R.), R01 MH101244 (A.L.P.), R37 MH107649 (A.L.P.), R01 MH115676 (A.L.P. and B.P.), U01 HG012009 (A.L.P. and S.R.), and R01 HG006399 (A.L.P.). The authors were also supported by NIH grants P01-AI148102 (S.R.), UC2-AR081023 (S.R.), R01-AR063759 (S.R.), UH2-AR067677 (S.R.), R01MH121521 (M.G.), and R01MH123922 (M.G.), and NIH NHGRI grants K99HG012203 (K.K.D.) and F32 HG011434-03 (K.A.J.). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Competing Interests Statement
The authors declare no competing interests.
Code availability
Software implementing scDRS and its downstream applications and a web interface for interactively exploring results of scDRS are available at https://github.com/martinjzhang/scDRS75. Code for generating all results of the paper is available at https://github.com/martinjzhang/scDRS_paper76.
Data availability
We release our data at https://figshare.com/projects/Single-cell_Disease_Relevance_Score_scDRS_/11890274 (instructions at https://github.com/martinjzhang/scDRS), including GWAS summary statistics of the 74 diseases/traits, TMS FACS scRNA-seq data, reprocessed TMS FACS data (for T cells and hepatocytes), MAGMA and gold standard gene sets, and scDRS results for TMS FACS (disease scores and control scores for the 74 diseases/traits). The 16 scRNA-seq data sets were obtained as follows (15 out of 16 publicly available). The TMS FACS data and TMS droplet data17 was downloaded from the official release https://figshare.com/articles/dataset/Processed_files_to_use_with_scanpy_/8273102. The TS FACS data20 was downloaded from the official release https://figshare.com/articles/dataset/Tabula_Sapiens_release_1_0/14267219. The Cano-Gamez & Soskic et al. data36 was downloaded from https://www.opentargets.org/projects/effectorness. The Nathan et al. data37 was downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158769. The Zeisel & Muñoz-Manchado et al. data46 was downloaded from http://linnarssonlab.org/cortex/. The Zeisel et al. data51 was downloaded from http://mousebrain.org/adolescent/downloads.html. The Habib & Li et al. data50 and Habib, Avraham-Davidi, & Basu et al. data53 were downloaded from https://singlecell.broadinstitute.org/single_cell. The Ayhan et al. data55 was downloaded from https://cells.ucsc.edu/human-hippo-axis/. The Yao et al. data52 was downloaded from https://assets.nemoarchive.org/dat-jb2f34y. The Zhong et al. data54 was downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE119212. The Aizarani et al. data61 was downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE124395. The Halpern & Shenhav et al. data59 was downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE84498. The Richter & Deligiannis et al. data60 (annotated count matrix) was obtained via communication with the authors (raw data publicly available via links in the paper). The Taychameekiatchai et al. data is not publicly available and was provided by co-authors A. Taychameekiatchai, P. Rao, and B. Wang. The MSigDB73 (v7.1) was downloaded from the official website http://www.gsea-msigdb.org/gsea/index.jsp. The Open Targets41 data was downloaded from the official website https://www.opentargets.org/.
References
- 1.Visscher Peter M, Wray Naomi R, Zhang Qian, Sklar Pamela, McCarthy Mark I, Brown Matthew A, and Yang Jian. 10 years of gwas discovery: biology, function, and translation. The American Journal of Human Genetics, 101(1):5–22, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hekselman Idan and Yeger-Lotem Esti. Mechanisms of tissue and cell-type specificity in heritable traits and diseases. Nature Reviews Genetics, 21(3):137–150, 2020. [DOI] [PubMed] [Google Scholar]
- 3.Regev Aviv, Teichmann Sarah A, Lander Eric S, Amit Ido, Benoist Christophe, Birney Ewan, Bodenmiller Bernd, Campbell Peter, Carninci Piero, Clatworthy Menna, et al. Science forum: the human cell atlas. elife, 6:e27041, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calderon Diego, Bhaskar Anand, Knowles David A, Golan David, Raj Towfique, Fu Audrey Q, and Pritchard Jonathan K. Inferring relevant cell types for complex traits by using single-cell gene expression. The American Journal of Human Genetics, 101(5):686–699, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watanabe Kyoko, Mirkov Maša Umicevic, de Leeuw Christiaan A, van den Heuvel Martijn P, and Posthuma Danielle. Genetic mapping of cell type specificity for complex traits. Nature communications, 10(1):1–13, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bryois Julien, Skene Nathan G, Hansen Thomas Folkmann, Kogelman Lisette JA, Watson Hunna J, Liu Zijing, Brueggeman Leo, Breen Gerome, Bulik Cynthia M, Arenas Ernest, et al. Genetic identification of cell types underlying brain complex traits yields insights into the etiology of parkinson’s disease. Nature genetics, 52(5):482–493, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu Xinli, Kim Hyun, Stahl Eli, Plenge Robert, Daly Mark, and Raychaudhuri Soumya. Integrating autoimmune risk loci with gene-expression data identifies specific pathogenic immune cell subsets. The American Journal of Human Genetics, 89(4):496–506, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gormley Padhraig, Anttila Verneri, Winsvold Bendik S, Palta Priit, Esko Tonu, Pers Tune H, Farh Kai-How, Cuenca-Leon Ester, Muona Mikko, Furlotte Nicholas A, et al. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nature genetics, 48(8):856–866, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ongen Halit, Brown Andrew A, Delaneau Olivier, Panousis Nikolaos I, Nica Alexandra C, and Dermitzakis Emmanouil T. Estimating the causal tissues for complex traits and diseases. Nature genetics, 49(12):1676–1683, 2017. [DOI] [PubMed] [Google Scholar]
- 10.Finucane Hilary K, Reshef Yakir A, Anttila Verneri, Slowikowski Kamil, Gusev Alexander, Byrnes Andrea, Gazal Steven, Loh Po-Ru, Lareau Caleb, Shoresh Noam, et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nature genetics, 50(4):621–629, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan Jean, Salathia Neeraj, Liu Rui, Kaeser Gwendolyn E, Yung Yun C, Herman Joseph L, Kaper Fiona, Fan Jian-Bing, Zhang Kun, Chun Jerold, et al. Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nature methods, 13(3):241–244, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aibar Sara, González-Blas Carmen Bravo, Moerman Thomas, Imrichova Hana, Hulselmans Gert, Rambow Florian, Marine Jean-Christophe, Geurts Pierre, Aerts Jan, van den Oord Joost, et al. Scenic: single-cell regulatory network inference and clustering. Nature methods, 14(11):1083–1086, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butler Andrew, Hoffman Paul, Smibert Peter, Papalexi Efthymia, and Satija Rahul. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature biotechnology, 36(5):411–420, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeTomaso David, Jones Matthew G, Subramaniam Meena, Ashuach Tal, Chun J Ye, and Yosef Nir. Functional interpretation of single cell similarity maps. Nature communications, 10(1):1–11, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cembrowski Mark S and Spruston Nelson. Heterogeneity within classical cell types is the rule: lessons from hippocampal pyramidal neurons. Nature Reviews Neuroscience, 20(4):193–204, 2019. [DOI] [PubMed] [Google Scholar]
- 16.Frost Hildreth Robert. Variance-adjusted mahalanobis (vam): a fast and accurate method for cell-specific gene set scoring. Nucleic acids research, 48(16):e94–e94, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.The Tabula Muris Consortium. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature, 583(7817):590–595, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Leeuw Christiaan A, Mooij Joris M, Heskes Tom, and Posthuma Danielle. Magma: generalized gene-set analysis of gwas data. PLoS Comput Biol, 11(4):e1004219, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stuart Tim, Butler Andrew, Hoffman Paul, Hafemeister Christoph, Papalexi Efthymia, Mauck William M III, Hao Yuhan, Stoeckius Marlon, Smibert Peter, and Satija Rahul. Comprehensive integration of single-cell data. Cell, 177(7):1888–1902, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tabula Sapiens Consortium*, Jones Robert C, Karkanias Jim, Krasnow Mark A, Pisco Angela Oliveira, Quake Stephen R, Salzman Julia, Yosef Nir, Bulthaup Bryan, Brown Phillip, et al. The tabula sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science, 376(6594):eabl4896, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Picelli Simone, Faridani Omid R, Björklund Åsa K, Winberg Gösta, Sagasser Sven, and Sandberg Rickard. Full-length rna-seq from single cells using smart-seq2. Nature protocols, 9(1):171, 2014. [DOI] [PubMed] [Google Scholar]
- 22.Skene Nathan G, Bryois Julien, Bakken Trygve E, Breen Gerome, Crowley James J, Gaspar Héléna A, Giusti-Rodriguez Paola, Hodge Rebecca D, Miller Jeremy A, Muñoz-Manchado Ana B, et al. Genetic identification of brain cell types underlying schizophrenia. Nature genetics, 50(6):825–833, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coleman Jonathan RI, Gaspar Héléna A, Bryois Julien, Byrne Enda M, Forstner Andreas J, Holmans Peter A, de Leeuw Christiaan A, Mattheisen Manuel, McQuillin Andrew, Pavlides Jennifer M Whitehead, et al. The genetics of the mood disorder spectrum: genome-wide association analyses of more than 185,000 cases and 439,000 controls. Biological psychiatry, 88(2):169–184, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alves-Bezerra Michele and Cohen David E. Triglyceride metabolism in the liver. Comprehensive Physiology, 8(1):1, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo Michael, Liu Zun, Willen Jessie, Shaw Cameron P, Richard Daniel, Jagoda Evelyn, Doxey Andrew C, Hirschhorn Joel, and Capellini Terence D. Epigenetic profiling of growth plate chondrocytes sheds insight into regulatory genetic variation influencing height. elife, 6:e29329, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kemp John P, Morris John A, Medina-Gomez Carolina, Forgetta Vincenzo, Warrington Nicole M, Youlten Scott E, Zheng Jie, Gregson Celia L, Grundberg Elin, Trajanoska Katerina, et al. Identification of 153 new loci associated with heel bone mineral density and functional involvement of gpc6 in osteoporosis. Nature genetics, 49(10):1468–1475, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warren Helen R, Evangelou Evangelos, Cabrera Claudia P, Gao He, Ren Meixia, Mifsud Borbala, Ntalla Ioanna, Surendran Praveen, Liu Chunyu, Cook James P, et al. Genome-wide association analysis identifies novel blood pressure loci and offers biological insights into cardiovascular risk. Nature genetics, 49(3):403–415, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiou Joshua, Zeng Chun, Cheng Zhang, Han Jee Yun, Schlichting Michael, Miller Michael, Mendez Robert, Huang Serina, Wang Jinzhao, Sui Yinghui, et al. Single-cell chromatin accessibility identifies pancreatic islet cell type–and state-specific regulatory programs of diabetes risk. Nature Genetics, 53(4):455–466, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Bondt Mirre, Hellings Niels, Opdenakker Ghislain, and Struyf Sofie. Neutrophils: Underestimated players in the pathogenesis of multiple sclerosis (ms). International Journal of Molecular Sciences, 21(12):4558, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Agarwal Devika, Sandor Cynthia, Volpato Viola, Caffrey Tara M, Monzón-Sandoval Jimena, Bowden Rory, Alegre-Abarrategui Javier, Wade-Martins Richard, and Webber Caleb. A single-cell atlas of the human substantia nigra reveals cell-specific pathways associated with neurological disorders. Nature communications, 11(1):1–11, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ettle Benjamin, Schlachetzki Johannes CM, and Winkler Jürgen. Oligodendroglia and myelin in neurodegenerative diseases: more than just bystanders? Molecular neurobiology, 53(5):3046–3062, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spitzer Sonia Olivia, Sitnikov Sergey, Kamen Yasmine, Evans Kimberley Anne, Kronenberg-Versteeg Deborah, Dietmann Sabine, de Faria Omar Jr, Agathou Sylvia, and Káradóttir Ragnhildur Thóra. Oligodendrocyte progenitor cells become regionally diverse and heterogeneous with age. Neuron, 101(3):459–471, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang Peng, Zhao Yongzhong, Zhong Jianmei, Zhang Xinhua, Liu Qifa, Qiu Xiaoxia, Chen Shaoke, Yan Hongxia, Hillyer Christopher, Mohandas Narla, et al. Putative regulators for the continuum of erythroid differentiation revealed by single-cell transcriptome of human bm and ucb cells. Proceedings of the National Academy of Sciences, 117(23):12868–12876, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Amy, Herbst Rebecca H, Canner David, Schenkel Jason M, Smith Olivia C, Kim Jonathan Y, Hillman Michelle, Bhutkar Arjun, Cuoco Michael S, Rappazzo C Garrett, et al. Il-33 signaling alters regulatory t cell diversity in support of tumor development. Cell reports, 29(10):2998–3008, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abraham Clara and Cho Judy H.. Inflammatory bowel disease. New England Journal of Medicine, 361(21):2066–2078, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cano-Gamez Eddie, Soskic Blagoje, Roumeliotis Theodoros I, So Ernest, Smyth Deborah J, Baldrighi Marta, Willé David, Nakic Nikolina, Esparza-Gordillo Jorge, Larminie Christopher GC, et al. Single-cell transcriptomics identifies an effectorness gradient shaping the response of cd4+ t cells to cytokines. Nature communications, 11(1):1–15, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nathan Aparna, Beynor Jessica I, Baglaenko Yuriy, Suliman Sara, Ishigaki Kazuyoshi, Asgari Samira, Huang Chuan-Chin, Luo Yang, Zhang Zibiao, Lopez Kattya, et al. Multimodally profiling memory t cells from a tuberculosis cohort identifies cell state associations with demographics, environment and disease. Nature Immunology, 22(6):781–793, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haghverdi Laleh, Büttner Maren, Wolf F Alexander, Buettner Florian, and Theis Fabian J. Diffusion pseudotime robustly reconstructs lineage branching. Nature methods, 13(10):845–848, 2016. [DOI] [PubMed] [Google Scholar]
- 39.Leung Stewart, Liu Xuebin, Fang Lei, Chen Xi, Guo Taylor, and Zhang Jingwu. The cytokine milieu in the interplay of pathogenic th1/th17 cells and regulatory t cells in autoimmune disease. Cellular & molecular immunology, 7(3):182–189, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gutierrez-Arcelus Maria, Teslovich Nikola, Mola Alex R, Polidoro Rafael B, Nathan Aparna, Kim Hyun, Hannes Susan, Slowikowski Kamil, Watts Gerald FM, Korsunsky Ilya, et al. Lymphocyte innateness defined by transcriptional states reflects a balance between proliferation and effector functions. Nature communications, 10(1):1–15, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koscielny Gautier, An Peter, Carvalho-Silva Denise, Cham Jennifer A, Fumis Luca, Gasparyan Rippa, Hasan Samiul, Karamanis Nikiforos, Maguire Michael, Papa Eliseo, et al. Open targets: a platform for therapeutic target identification and validation. Nucleic acids research, 45(D1):D985–D994, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freund Malika Kumar, Burch Kathryn S, Shi Huwenbo, Mancuso Nicholas, Kichaev Gleb, Garske Kristina M, Pan David Z, Miao Zong, Mohlke Karen L, Laakso Markku, et al. Phenotype-specific enrichment of mendelian disorder genes near gwas regions across 62 complex traits. The American Journal of Human Genetics, 103(4):535–552, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Connor Luke J, Schoech Armin P, Hormozdiari Farhad, Gazal Steven, Patterson Nick, and Price Alkes L. Extreme polygenicity of complex traits is explained by negative selection. The American Journal of Human Genetics, 105(3):456–476, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Hailong, Zheng Yajuan, Pan Youdong, Lin Changdong, Wang Shihui, Yan Zhanjun, Lu Ling, Ge Gaoxiang, Li Jinsong, Zeng Yi Arial, et al. A mutation that blocks integrin α 4 β 7 activation prevents adaptive immune-mediated colitis without increasing susceptibility to innate colitis. BMC biology, 18(1):1–15, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choy Ernest HS, Miceli-Richard Corinne, González-Gay Miguel A, Sinigaglia Luigi, Schlichting Douglas E, Meszaros Gabriella, de la Torre Inmaculada, and Schulze-Koops Hendrik. The effect of jak1/jak2 inhibition in rheumatoid arthritis: efficacy and safety of baricitinib. Clin Exp Rheumatol, 37(4):694–704, 2019. [PubMed] [Google Scholar]
- 46.Zeisel Amit, Muñoz-Manchado Ana B, Codeluppi Simone, Lönnerberg Peter, La Manno Gioele, Juréus Anna, Marques Sueli, Munguba Hermany, He Liqun, Betsholtz Christer, et al. Cell types in the mouse cortex and hippocampus revealed by single-cell rna-seq. Science, 347(6226):1138–1142, 2015. [DOI] [PubMed] [Google Scholar]
- 47.Skene Nathan G and Grant Seth GN. Identification of vulnerable cell types in major brain disorders using single cell transcriptomes and expression weighted cell type enrichment. Frontiers in neuroscience, 10:16, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cembrowski Mark S, Bachman Julia L, Wang Lihua, Sugino Ken, Shields Brenda C, and Spruston Nelson. Spatial gene-expression gradients underlie prominent heterogeneity of ca1 pyramidal neurons. Neuron, 89(2):351–368, 2016. [DOI] [PubMed] [Google Scholar]
- 49.Henriksen Espen J, Colgin Laura L, Barnes Carol A, Witter Menno P, Moser May-Britt, and Moser Edvard I. Spatial representation along the proximodistal axis of ca1. Neuron, 68(1):127–137, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Habib Naomi, Li Yinqing, Heidenreich Matthias, Swiech Lukasz, Avraham-Davidi Inbal, Trombetta John J, Hession Cynthia, Zhang Feng, and Regev Aviv. Div-seq: Single-nucleus rna-seq reveals dynamics of rare adult newborn neurons. Science, 353(6302):925–928, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zeisel Amit, Hochgerner Hannah, Lönnerberg Peter, Johnsson Anna, Memic Fatima, Van Der Zwan Job, Häring Martin, Braun Emelie, Borm Lars E, La Manno Gioele, et al. Molecular architecture of the mouse nervous system. Cell, 174(4):999–1014, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yao Zizhen, van Velthoven Cindy TJ, Nguyen Thuc Nghi, Goldy Jeff, Sedeno-Cortes Adriana E, Baftizadeh Fahimeh, Bertagnolli Darren, Casper Tamara, Chiang Megan, Crichton Kirsten, et al. A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell, 184(12):3222–3241, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Habib Naomi, Inbal Avraham-Davidi Anindita Basu, Burks Tyler, Shekhar Karthik, Hofree Matan, Choudhury Sourav R, Aguet François, Gelfand Ellen, Ardlie Kristin, et al. Massively parallel single-nucleus rna-seq with dronc-seq. Nature methods, 14(10):955–958, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhong Suijuan, Ding Wenyu, Sun Le, Lu Yufeng, Dong Hao, Fan Xiaoying, Liu Zeyuan, Chen Ruiguo, Zhang Shu, Ma Qiang, et al. Decoding the development of the human hippocampus. Nature, 577(7791):531–536, 2020. [DOI] [PubMed] [Google Scholar]
- 55.Ayhan Fatma, Kulkarni Ashwinikumar, Berto Stefano, Sivaprakasam Karthigayini, Douglas Connor, Lega Bradley C, and Konopka Genevieve. Resolving cellular and molecular diversity along the hippocampal anterior-to-posterior axis in humans. Neuron, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ben-Moshe Shani and Itzkovitz Shalev. Spatial heterogeneity in the mammalian liver. Nature Reviews Gastroenterology & Hepatology, 16(7):395–410, 2019. [DOI] [PubMed] [Google Scholar]
- 57.Donne Romain, Saroul-Aïnama Maëva, Cordier Pierre, Celton-Morizur Séverine, and Desdouets Chantal. Polyploidy in liver development, homeostasis and disease. Nature Reviews Gastroenterology & Hepatology, 17(7):391–405, 2020. [DOI] [PubMed] [Google Scholar]
- 58.Miettinen Teemu P, Pessa Heli KJ, Caldez Matias J, Fuhrer Tobias, Diril M Kasim, Sauer Uwe, Kaldis Philipp, and Björklund Mikael. Identification of transcriptional and metabolic programs related to mammalian cell size. Current Biology, 24(6):598–608, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Halpern Keren Bahar, Shenhav Rom, Matcovitch-Natan Orit, Tóth Beáta, Lemze Doron, Golan Matan, Massasa Efi E, Baydatch Shaked, Landen Shanie, Moor Andreas E, et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature, 542(7641):352–356, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Richter ML, Deligiannis IK, Yin K, Danese A, Lleshi E, Coupland P, Vallejos Catalina A, Matchett KP, Henderson NC, Colome-Tatche M, et al. Single-nucleus rna-seq2 reveals functional crosstalk between liver zonation and ploidy. Nature communications, 12(1):1–16, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aizarani Nadim, Saviano Antonio, Mailly Laurent, Durand Sarah, Herman Josip S, Pessaux Patrick, Baumert Thomas F, Grün Dominic, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature, 572(7768):199–204, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gazal Steven, Loh Po-Ru, Finucane Hilary K, Ganna Andrea, Schoech Armin, Sunyaev Shamil, and Price Alkes L. Functional architecture of low-frequency variants highlights strength of negative selection across coding and non-coding annotations. Nature genetics, 50(11):1600–1607, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Method-only references
- 63.Sakornsakolpat Phuwanat, Prokopenko Dmitry, Lamontagne Maxime, Reeve Nicola F, Guyatt Anna L, Jackson Victoria E, Shrine Nick, Qiao Dandi, Bartz Traci M, Kim Deog Kyeom, et al. Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell-type and phenotype associations. Nature genetics, 51(3):494–505, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kato Katsuhiro, Diéguez-Hurtado Rodrigo, Park Do Young, Hong Seon Pyo, Kato-Azuma Sakiko, Adams Susanne, Stehling Martin, Trappmann Britta, Wrana Jeffrey L, Koh Gou Young, et al. Pulmonary pericytes regulate lung morphogenesis. Nature communications, 9(1):1–14, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Geary Robert C. The contiguity ratio and statistical mapping. The incorporated statistician, 5(3):115–146, 1954. [Google Scholar]
- 66.Wolf F Alexander, Angerer Philipp, and Theis Fabian J. Scanpy: large-scale single-cell gene expression data analysis. Genome biology, 19(1):1–5, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Benjamini Yoav and Hochberg Yosef. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal statistical society: series B (Methodological), 57(1):289–300, 1995. [Google Scholar]
- 68.Bycroft Clare, Freeman Colin, Petkova Desislava, Band Gavin, Elliott Lloyd T, Sharp Kevin, Motyer Allan, Vukcevic Damjan, Delaneau Olivier, O’Connell Jared, et al. The uk biobank resource with deep phenotyping and genomic data. Nature, 562(7726):203–209, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Finucane Hilary K, Bulik-Sullivan Brendan, Gusev Alexander, Trynka Gosia, Reshef Yakir, Loh Po-Ru, Anttila Verneri, Xu Han, Zang Chongzhi, Farh Kyle, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nature genetics, 47(11):1228–1235, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Korsunsky Ilya, Millard Nghia, Fan Jean, Slowikowski Kamil, Zhang Fan, Wei Kevin, Baglaenko Yuriy, Brenner Michael, Loh Po-ru, and Raychaudhuri Soumya. Fast, sensitive and accurate integration of single-cell data with harmony. Nature methods, 16(12):1289–1296, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Traag Vincent A, Waltman Ludo, and Jan Van Eck Nees. From louvain to leiden: guaranteeing well-connected communities. Scientific reports, 9(1):1–12, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Andreatta Massimo, Corria-Osorio Jesus, Müller Sören, Cubas Rafael, Coukos George, and Carmona Santiago J. Interpretation of t cell states from single-cell transcriptomics data using reference atlases. Nature communications, 12(1):1–19, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liberzon Arthur, Subramanian Aravind, Pinchback Reid, Thorvaldsdóttir Helga, Tamayo Pablo, and Mesirov Jill P. Molecular signatures database (msigdb) 3.0. Bioinformatics, 27(12):1739–1740, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang Martin Jinye and Hou Kangcheng. scdrs data release 030122. Figshare, 10.6084/m9.figshare.19312583.v1, 2022. [DOI]
- 75.Zhang Martin Jinye and Hou Kangcheng. scdrs software v1.0.1. Zenodo, 10.5281/zenodo.6615722, 2022. [DOI]
- 76.Zhang Martin Jinye and Hou Kangcheng. scdrs data analysis code v1.0.1. Zenodo, 10.5281/zenodo.6615791, 2022. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We release our data at https://figshare.com/projects/Single-cell_Disease_Relevance_Score_scDRS_/11890274 (instructions at https://github.com/martinjzhang/scDRS), including GWAS summary statistics of the 74 diseases/traits, TMS FACS scRNA-seq data, reprocessed TMS FACS data (for T cells and hepatocytes), MAGMA and gold standard gene sets, and scDRS results for TMS FACS (disease scores and control scores for the 74 diseases/traits). The 16 scRNA-seq data sets were obtained as follows (15 out of 16 publicly available). The TMS FACS data and TMS droplet data17 was downloaded from the official release https://figshare.com/articles/dataset/Processed_files_to_use_with_scanpy_/8273102. The TS FACS data20 was downloaded from the official release https://figshare.com/articles/dataset/Tabula_Sapiens_release_1_0/14267219. The Cano-Gamez & Soskic et al. data36 was downloaded from https://www.opentargets.org/projects/effectorness. The Nathan et al. data37 was downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158769. The Zeisel & Muñoz-Manchado et al. data46 was downloaded from http://linnarssonlab.org/cortex/. The Zeisel et al. data51 was downloaded from http://mousebrain.org/adolescent/downloads.html. The Habib & Li et al. data50 and Habib, Avraham-Davidi, & Basu et al. data53 were downloaded from https://singlecell.broadinstitute.org/single_cell. The Ayhan et al. data55 was downloaded from https://cells.ucsc.edu/human-hippo-axis/. The Yao et al. data52 was downloaded from https://assets.nemoarchive.org/dat-jb2f34y. The Zhong et al. data54 was downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE119212. The Aizarani et al. data61 was downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE124395. The Halpern & Shenhav et al. data59 was downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE84498. The Richter & Deligiannis et al. data60 (annotated count matrix) was obtained via communication with the authors (raw data publicly available via links in the paper). The Taychameekiatchai et al. data is not publicly available and was provided by co-authors A. Taychameekiatchai, P. Rao, and B. Wang. The MSigDB73 (v7.1) was downloaded from the official website http://www.gsea-msigdb.org/gsea/index.jsp. The Open Targets41 data was downloaded from the official website https://www.opentargets.org/.