

Abstract
In the lungs, defective CFTR associated with cystic fibrosis (CF) represents the nidus for abnormal mucus clearance in the airways and consequently a progressive lung disease. Defective CFTR-mediated Cl− secretion results in altered mucus properties, including concentration, viscoelasticity, and the ratio of the two mucins, MUC5B and MUC5AC. In the past decades, therapies targeting the CF mucus defect, directly or indirectly, have been developed; nevertheless, better treatments to prevent the disease progression are still needed. This review summarizes the existing knowledge on the defective mucus in CF disease and highlights it as a barrier to the development of future inhaled genetic therapies. The use of new mucus-targeting treatments is also discussed, focusing on their potential role to halt the progress of CF lung disease.
1. Mucus transport in normal airways vs. CF airways
Cystic Fibrosis (CF) is characterized by impaired transport of chloride and bicarbonate across the cystic fibrosis transmembrane regulator (CFTR) channel, which results, in the airways, in a critical disease-initiating step: a reduction of mucus transport and clearance [1–3].
In normal airways, an effective mucus transport and clearance requires a well-hydrated airway surface layer (ASL), composed of a mucus layer and a periciliary layer (PCL), which acts as a lubricating layer in which the cilia beat. The water flux from the cells to the ASL is tightly regulated by an interplay of coordinated chloride secretion and sodium absorption across the low-resistance epithelium [4]. Chloride is secreted via CFTR, and the calcium-activated chloride channels (CaCCs) and sodium is absorbed via the epithelial sodium channel (ENaC), while the water is transported passively with the resulting osmotic gradient. Together, the balance of water and ions across the epithelium ensures a properly hydrated ASL and an efficient mucus clearance [5–8].
Mucus production and its clearance are part of our innate immune system designed to protect the pulmonary surfaces from the persistent onslaught of inhaled infectious and noxious substances. Airways mucus can bind to inhaled foreign microorganisms, and the ciliated epithelial cells lining the airways sweep the mucus material until it is either swallowed or coughed out of the airways. This efficient system deters the establishment of lung infections [9–11]. (Figure 1A).
Figure 1: Mucus transport in healthy airways vs. CF airways.
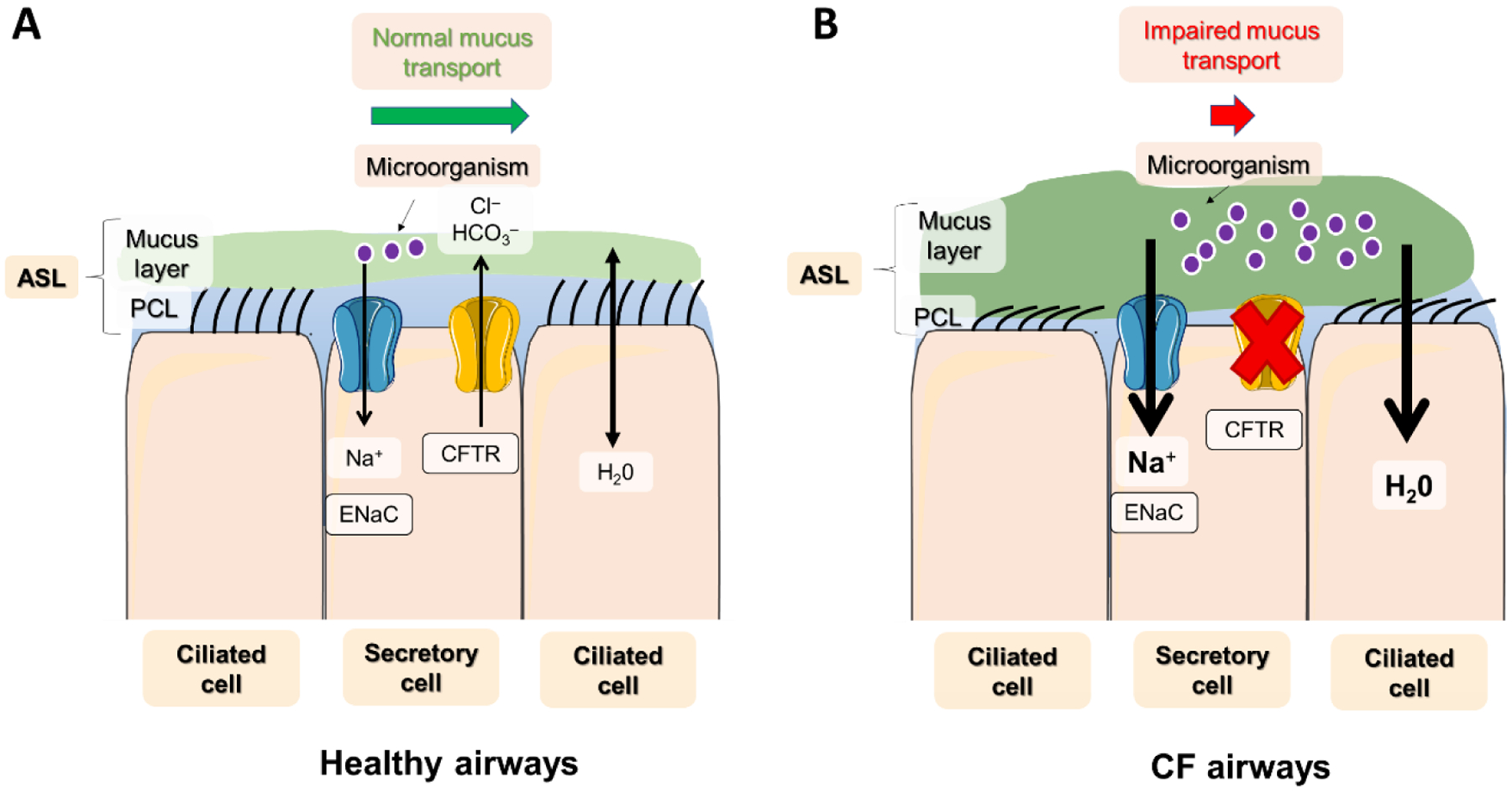
A) In healthy airways, a well-balanced activity of ions (Na+ and Cl−), water, and mucus, across the lung epithelium results in an efficient mucus transport: Here, a well-hydrated PCL layer ensures that cilia can efficiently sweep away mucus and noxious inhaled microorganisms, preventing their accumulation in the airways. B) In CF airways, defective CFTR (mainly expressed in secretory cells) leads to impaired fluid secretion activity in mucin-producing goblet cells, leading to ASL dehydration and a PCL and cilia collapse that fails to transport mucus and infectious particles out of the lungs. The result is mucus obstruction and chronic bacterial infection in the airways and progressive lung disease.
However, in CF airways, defective CFTR function leads to unregulated absorption of sodium and airway fluid resulting in dehydration of both the mucus and PCL layers and abnormal mucus clearance [12]. The dehydration of these layers leads to compressed cilia, impeding their ability to efficiently sweep the mucus and trapped infectious agents out of the airways, thus favoring airway infections. The result is the accumulation of mucus and pathogenic microorganisms, and the development of chronic airway inflammation and progressive lung function deterioration in CF patients [13–15] (Figure 1B).
Importantly, in 2012, our group’s research introduced a significant revision to our understanding of the airway surface organization by describing the gel-on-brush model, depicting a high molecular weight secreted mucous layer juxtaposed to airway cell surface-tethered mucins. This work demonstrated that the secreted mucins in the mucus layer, in health, cannot penetrate the PCL as a result of the osmotic forces established by the densely expressed tethered mucins lining the airway surface. However, in CF, the dehydrated airway surface results in an increased mucus osmotic pressure, higher than that of the PCL. The result is water drawn out of the PCL, compression of the cilia, and mucostasis, as mentioned above [5].
In addition, recent studies have indicated that airway mucus obstruction associated with inflammation already occurs during the early stages of CF pulmonary disease. For instance, air trapping has been documented in newborn CFTR–/– piglets devoid of bacterial infection [16], and similar findings were reported in CF infants and pre-school children [17].
2. CF mucus defect
The healthy mucus layer that covers the human airways is a complex mixture of many components, such as mucins, globular proteins, DNA, ions, cells, antimicrobial peptides, salts, and water [18,19]. The interdependency of these components is most apparent in CF airways, where the ion-mediated changes in mucus concentration have a tremendously adverse effect on its overall biophysical properties [20,21]. Here, we explored mucus properties primarily affected by CF: concentration, viscoelasticity, and MUC5B/MUC5AC ratio.
2.1. Mucus concentration
The healthy mucus layer is composed of mostly water (roughly 98%) and only 2% solid materials such as mucins (~0.5%), globular proteins (~0.5%), and salt (~0.9%) [21,22]. In CF, however, as described above, the defective CFTR Cl− secretion combined with upregulated Na+ absorption leads to ASL dehydration, which directly results in a higher concentration of mucus solids to values of ~4–5% for expectorated mucus and 12% for mucus that is collected from freshly excised CF lungs. This higher mucus concentration leads to two main deleterious consequences: 1) increased mucus viscoelasticity and 2) the mucus layer osmotic compression of the PCL. Combined, they reduce mucus clearance which fuels the CF pathogenic loop. Thus, mucus hyper-concentration has a central role in CF disease [2,5,6,13].
2.2. Viscoelasticity
The flowability of mucus is typically described by its rheological properties: viscosity (resistance to flow) and elasticity (stiffness or gel-like properties). In addition, mucus is a non-Newtonian gel that, under high shear stress, behaves as a viscous liquid; thus, the shear properties are essential to maintain its flowable properties [18,19].
As a unique organ, the lungs are constantly subjected to various shear forces during tidal breathing and coughing [23] (PMID: 19166889). For example, during respiration, airflow and transepithelial pressure gradients are two fundamental shear stress forces that act within the surface of airway cells [24,25]. However, more extreme stress can happen on the airways during cough due to mechanical forces, such as transpulmonary pressure. The glottis closure creates this high pressure when combined with the rapid expiratory muscle activity [26].
Changes in mucus rheological properties in the airways can profoundly affect its functioning as a first-line defense against harmful infections. For example, due to the mucus hyper-concentration in CF, the mucus layer’s viscoelasticity is higher than normal non-CF values [20]. In addition, the viscoelasticity threshold for which the cilia beating is still effective (100 mPa·s) is surpassed by an airway mucus with concentrations over 2.5% solids [27]. Also, PCL height was shown to decrease when the mucus concentration is around 5% solids, and it completely collapses at concentration above 8% (similar values are found in CF mucus) [5,27]. Therefore, the reduced cilia beat efficiency associated with the osmotic compression of the cilia, momentum transfer from the cilia to the viscoelastic mucus (i.e., motive force) is significantly reduced, resulting in mucostasis and disease progression [27–29].
In addition, the accumulation of static mucus combined with chronic infections in CF airways is associated with inflammation and necrosing neutrophils that release DNA (i.e., Netosis), which further increases mucus viscoelasticity [30,31]. Furthermore, it has also been suggested that the high oxidative stress from neutrophilic inflammation is associated with the formation of additional mucin disulfide bonds, thereby forming a highly cross-linked, and viscoelastic, mucus gel [32].
2.3. MUC5B and MUC5AC mucins in the airways
Mucins are large, multimerized, gel-forming glycoproteins (up to 100 megadaltons in size). MUC5B and MU5AC are the prominent secreted mucin isoforms in human airways produced by submucosal glands (MUC5B) and superficial goblet and club cells (MUC5B and MUC5AC). In health, MUC5B is the dominant mucin isoform which is expressed approximately 75 fold higher than MUC5AC [6]. However, up-regulation of MUC5AC has been demonstrated to be more prominent during pulmonary exacerbations in CF [33], worsening COPD (Gold) status [34], and bacterial [35] and Trichuris muri’s (enteric nematodes) infections [36,37]. Increased MUC5AC has also been associated with allergic inflammation in asthma [38].
It has been suggested that MUC5AC is a “stickier” and stiffer mucin that can be beneficial during pulmonary infections to help bind and clear the inhaled pathogens out of the airways [39,40]. However, numerous data suggest that prolonged and elevated expression of MUC5AC can have detrimental effects on mucus clearance [40,41]. For example, MUC5B knockout mice were shown to have reduced mucus clearance and develop chronic infections that failed to resolve spontaneously [41]. In addition, patients homozygous for a novel splicing variant in the MUC5B gene were found to have impaired mucociliary clearance [42]. Therefore, the role that chronically increased MUC5AC expression plays in disease pathogenesis should be further investigated.
3. Current mucus-targeting treatments in use for CF
3.1. DNAse
As mentioned above, increased DNA released from neutrophils in CF is associated with a higher viscoelasticity. Therefore, an inhaled form of a recombinant human DNAse enzyme that selectively hydrolyzes DNA, named Dornase Alfa, was developed for inhaled delivery. This drug was one of the first approved to treat CF in 1993 and is still commonly used by most patients in their CF management [43,44].
As expected, hydrolyzing DNA with Dornase Alfa is capable at decreasing the viscoelasticity of CF sputum [45]. Clinical studies with Dornase Alfa showed that this treatment significantly decreases pulmonary exacerbations and slightly increases lung function [46]. [44,46]. These positive clinical trial results made Dornase Alfa a standard CF therapy that is widely used today. However, while Dornase Alfa reduces the frequency of pulmonary exacerbations, it does not entirely prevent them from happening. Thus, the development of new mucus-targeting agents continues to be essential to treat CF.
3.2. Mucus hydrators
-. Hypertonic Saline
Given the role of mucus dehydration to the pathogenesis of CF lung disease, rehydration can be achieved by osmotic agents, such as hypertonic saline (HS). The beneficial effects HS on mucociliary clearance were first identified in 1997 [47]. However, it was only in 2006 that HS was evaluated for its long-term efficacy by the National Hypertonic Saline Study Group [47]. Subsequently, many studies showed that this therapy significantly reduces exacerbation and mildly increases lung function [48–50].
In addition, in vitro studies suggested that HS increases the mucus clearance rate in patients due to mucus dilution and osmotically active ASL expansion. Interestingly, these effects were found to be prolonged on hyper-concentrated mucus samples, likely due to increased osmotic forces of the concentrated mucus [51]. Based on these findings, HS is consolidated as an additional therapy shown to be effective and inexpensive for patients. Nevertheless, studies have shown that HS has a short lifetime in the airways due to sodium absorption via ENaC [48,51,52]. Therefore, the development of other therapies with a prolonged effect would be of interest; for instance, combining HS with ENaC inhibitors could extend the hydration benefit as a result of reduced ENaC-mediated ASL fluid reabsorption elicited by the inhalation of HS [51]. The net effect would be an increase in the duration of mucus hydration compared to HS alone [51]. Nevertheless, future clinical studies are needed to clarify if airway mucosal hydration is enhanced by the combined therapies [53].
-. Mannitol
Several studies suggested that HS may inactivate important cationic antimicrobial defensins and contribute to infections and inflammation due to its ionic constitution [54–56]. Therefore, as an alternative non-ionic osmotic agent, mannitol was tested and approved in several countries as a dry inhalation powder named Bronchitol, which has been subsequently approved in the US in 2020 [57,58].
In comparison to HS, Branchitol’s use reduces treatment burden and microbial contamination of nebulizers, as it is administrated via a small and portable dry-powder inhaler. Bronchitol safety and efficacy were evaluated in various phase 3 studies over the course of 26 weeks. The studies demonstrated that it improved the patient’s forced expiratory volume in one second (FEV1) when compared to the placebo group (NCT00630812, NCT00446680, NCT02134353). Although the drug was found to be safe, patients must do a Bronchitol tolerance test before starting treatment, as some can develop an allergic reaction [58–61].
3.3. CFTR Modulators
Small-molecule CFTR modulator drugs either correct and/or potentiate the CFTR protein by modulating its functioning. Correctors fix the defective folding of the protein and rescue its trafficking to the plasmatic membrane, while potentiators allow enhanced anion transport via CFTR by increasing this channel open probability [62–64]. The net result is a restoration of CFTR-mediated fluid secretion, reduction in mucus concentration, and reduced mucus viscoelastic properties. In the past few years, the ability to correct/modulate CFTR by different compounds has shown great promise to partially restore defective chloride and bicarbonate transport in CF cells and significantly improve CF patients’ lung function [65].
The most recently approved modulator was Trikafta (Ivacaftor, Tezacaftor, and Elexacaftor) in 2019, which made CFTR modulators approved for 90% of the CF population with specific CFTR mutations [66–69]. A recent in vitro study pointed to a reduction in mucus concentration in response to Trikafta [70], but additional studies are necessary to address the long-term effects of CFTR modulators on mucus and mucociliary clearance restoration. The possible combination of modulators and mucus-targeting therapies can be even more relevant for CF adult patients with established lung disease and bronchiectasis. In addition, because ~ 10% of the CF population is still not eligible for current CFTR modulator therapy, there is an unmet need for new therapies for these patients [71].
4. Future perspectives
4.1. Reducing agents/mucolytics
As mentioned above, CF sputum has higher oxidative stress due to airway inflammation which generates mucin cross-link via multiple intermolecular disulfide bonds. Therefore, researchers have pursued the development of disulfide bond reducing agents as a mucolytic therapy for many decades [28,49].
In the 1960s, Mucomyst (N-acetylcysteine [NAC]) started being used to treat CF; however, later clinical research demonstrated a limited effect of this drug in addition to adverse effects, such as bronchospasm and off-targeting irritation effect. Different studies highlighted that the NAC failure as a drug was due to its rapid transepithelial absorption and low intrinsic activity at neutral pH [72–74].
To compensate for the adverse effects, a more recent paper tested a novel reducing agent named “P3001” (Parion Sciences) that has a longer residence time and activity on the airway surfaces. The authors in vitro studies revealed that this compound is more effective than NAC and DNAse in reducing CF sputum viscoelasticity [73].
4.2. Inhibition of mucin production/secretion
In healthy airways, mucin production and secretion are essential to maintaining the ciliary clearance of harmful microorganisms. However, in CF, it is documented that the constant presence of airways insults leads to a detrimental upregulated synthesis and secretion of mucins [75,76]. Jaramillo et al., 2018, highlighted that, despite functionally interacting, the mucin synthesis and secretion processes involve different and complex pathways that have been studied as potential therapeutical targets [77].
Our group has focused on the proinflammatory IL-1 cytokines pathway as it has been observed that such cytokines are highly expressed in the supernatant of mucopurulent material (SMM) from the airways of excised CF lungs [78,79] and have been linked with CF airway inflammatory responses and mucus obstruction [80]. In agreement with these findings, Chen et al. demonstrated that IL-1β increases the production and secretion of MUC5B and MUC5AC in normal and CF HBE, with the predominant effect being MUC5B up-regulation [80]. Furthermore, the studies by Chen et al. demonstrated that mucin production was blunted in mice lacking either IL-1R1 or SAM Pointed Domain Containing ETS Transcription Factor (SPDEF), a mediator of Muc5b expression. Another study showed that the ER stress transducer IRE1b, a key mediator of airway mucin production and whose expression is up-regulated in CF, was decreased in Spdef–/– mice treated with IL-1β [81]. These findings indicate that IL-1β up-regulates MUC5B production via SPDEF-IRE1β signaling, suggesting that this pathway can be a therapeutic target for the hyper-concentrated CF mucus.
Recent research by our group indicated that the small molecule KIRA6 (kinase inhibiting RNase attenuator 6) decreases SMM-up-regulated IRE1α-dependent generation of the spliced form of the transcription factor XBP-1, which promotes cytokine production, including IL-1β, in CF airways [82]. Together, these findings suggest that the IRE1α/XBP-1 pathway may also be a therapeutic target for the hyper-concentrated CF mucus due to its functional role in IL-1β production. Decreasing the burden on the CF lung resulting from inflammatory mediator-increased mucus production is expected to be therapeutically beneficial, especially if combined with other therapies such as mucolytic agents.
4.3. Genetic therapies
Genetic therapies for cystic fibrosis have been in the spotlight since the CFTR gene identification in 1989. Like CFTR modulator therapy, gene therapies could treat the underlying cause of CF and, therefore, indirectly have a positive effect on CF mucus properties. Since the initial trials, the main challenges have been the delivery of the corrected CFTR gene and its persistence in the airways [83,84]. Nevertheless, the scientific knowledge about CF disease and the use of new models and technologies have improved drastically over the years. For example, in vitro models, such as human epithelial organoids and air-liquid interface lung cells, and animal models, such as CF ferrets and pigs, are essential to access the physiological effects of genetic therapies [85–87]. Furthermore, to improve the delivery of the corrected CFTR gene, other technologies developed were new viral and non-viral approaches such as reprogrammed AAV and lipid nanoparticles. CRISPR gene-editing, mRNA and genetically corrected airway stem cells are also growing prospects that are rapidly changing the field and broadening possibilities for a successful CF gene therapy on the horizon [88–90].
It is also important to note that many of the gene therapies in development focus on an inhaled delivery form, which can directly benefit the main organ affected by CF, the lungs. However, to successfully deliver gene therapy vectors to the airways, these inhaled genetic therapies have the challenge of overcoming a dense glycocalyx (tethered mucins) in the PCL, in addition to a viscoelastic mucus layer exacerbated in CF [91,92]. To this end, new nanoparticles (NPs) and viruses capable of penetrating the mucus barrier are being tested [93–95]. For example, Wan et al. showed that polymeric NPs combined with highly integrated lipid shells could significantly increase NPs penetration within the mucus layer [96]. In addition, it has been suggested that pretreatment with agents that can help hydrate mucus (i.e, mannitol, or HS) and/or cleave mucins (i.e., NAC) could enhance inhaled genetic therapies delivery efficiency [97]. Hence, further studies with mucus-targeting agents utilizing translational in-vitro models that mimic the CF airways mucus properties will be necessary to provide a proof-of-principle for these therapeutics approaches.
5. Conclusion
This review highlights that the abnormal mucus in CF airways is a key pathological factor, which is already observed in the early stages of CF lung disease. Therefore, studies on mucus cell biology, production, and biophysical properties in normal and CF airways are essential. Furthermore, this knowledge is promising for developing novel approaches in genetic therapy, small molecule inhibition of signaling pathways, CFTR modulation, and mucolytics. Therefore, tackling the mucus defects in CF is critical for successfully developing better therapies beneficial to all CF patients.
Acknowledgments
This work was supported by National Institutes of Health grants R01HL125280 and P30DK065988; and by Cystic Fibrosis Foundation grants FIGUEI18F0; BUTTON19G0; RIBEIR18G0; RIBEIR20P0 and BOUCHE19R0.
References
- 1.Livraghi A, Randell SH: Cystic Fibrosis and Other Respiratory Diseases of Impaired Mucus Clearance. Toxicol Pathol 2007, 35:116–129. [DOI] [PubMed] [Google Scholar]
- 2.Hill DB, Long RF, Kissner WJ, Atieh E, Garbarine IC, Markovetz MR, Fontana NC, Christy M, Habibpour M, Tarran R, et al. : Pathological mucus and impaired mucus clearance in cystic fibrosis patients result from increased concentration, not altered pH. Eur Respir J 2018, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sellers ZM, Illek B, Figueira MF, Hari G, Joo NS, Sibley E, Souza-Menezes J, Morales MM, Fischer H, Wine JJ: Impaired PGE2-stimulated Cl- and HCO3- secretion contributes to cystic fibrosis airway disease. PLoS One 2017, 12:e0189894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boucher RC: Cystic fibrosis: a disease of vulnerability to airway surface dehydration. Trends Mol Med 2007, 13:231–240. [DOI] [PubMed] [Google Scholar]
- 5.Button B, Cai LH, Ehre C, Kesimer M, Hill DB, Sheehan JK, Boucher RC, Rubinstein M: A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science (80- ) 2012, 337:937–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henderson AG, Ehre C, Button B, Abdullah LH, Cai LH, Leigh MW, DeMaria GC, Matsui H, Donaldson SH, Davis CW, et al. : Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J Clin Invest 2014, 124:3047–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Figueira MF, Webster MJ, Tarran R: CrossTalk proposal: mucosal acidification drives early progressive lung disease in cystic fibrosis. J Physiol 2018, 596:3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennett WD, Laube BL, Corcoran T, Zeman K, Sharpless G, Thomas K, Wu J, Mogayzel PJ, Pilewski J, Donaldson S: Multisite comparison of mucociliary and cough clearance measures using standardized methods. J Aerosol Med Pulm Drug Deliv 2013, 26:157–164. [DOI] [PubMed] [Google Scholar]
- 9.Butler R, Hunt T, Smith NJ: ENaC inhibitors for the treatment of cystic fibrosis. http://dx.doi.org/104155/ppa1451 2015, 4:17–27. [DOI] [PubMed] [Google Scholar]
- 10.Althaus M: ENaC inhibitors and airway re-hydration in cystic fibrosis: state of the art. Curr Mol Pharmacol 2013, 6:3–12. [DOI] [PubMed] [Google Scholar]
- 11.Hill DB, Button B, Rubinstein M, Boucher RC: Physiology and Pathophysiology of Human Airway Mucus. https://doi.org/101152/physrev000042021 2022, doi:10.1152/PHYSREV.00004.2021. [DOI] [PMC free article] [PubMed]
- 12.Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC: Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 1998, 95:1005–1015. [DOI] [PubMed] [Google Scholar]
- 13.Anderson WH, Coakley RD, Button B, Henderson AG, Zeman KL, Alexis NE, Peden DB, Lazarowski ER, Davis CW, Bailey S, et al. : The Relationship of Mucus Concentration (Hydration) to Mucus Osmotic Pressure and Transport in Chronic Bronchitis. Am J Respir Crit Care Med 2015, doi: 10.1164/rccm.201412-2230OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boucher RC: An overview of the pathogenesis of cystic fibrosis lung disease. Adv Drug Deliv Rev 2002, 54:1359–1371. [DOI] [PubMed] [Google Scholar]
- 15.Murphy SV, Ribeiro CMP: Cystic Fibrosis Inflammation: Hyperinflammatory, Hypoinflammatory, or Both? Am J Respir Cell Mol Biol 2019, 61:273–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adam RJ, Michalski AS, Bauer C, Alaiwa MHA, Gross TJ, Awadalla MS, Bouzek DC, Gansemer ND, Taft PJ, Hoegger MJ, et al. : Air trapping and airflow obstruction in newborn cystic fibrosis piglets. Am J Respir Crit Care Med 2013, 188:1434–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stick SM, Brennan S, Murray C, Douglas T, von Ungern-Sternberg BS, Garratt LW, Gangell CL, De Klerk N, Linnane B, Ranganathan S, et al. : Bronchiectasis in infants and preschool children diagnosed with cystic fibrosis after newborn screening. J Pediatr 2009, 155. [DOI] [PubMed] [Google Scholar]
- 18.Lai SK, Wang YY, Wirtz D, Hanes J: Micro- and macrorheology of mucus. Adv Drug Deliv Rev 2009, 61:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thornton DJ, Sheehan JK: From mucins to mucus: toward a more coherent understanding of this essential barrier. Proc Am Thorac Soc 2004, 1:54–61. [DOI] [PubMed] [Google Scholar]
- 20.Henderson AG, Ehre C, Button B, Abdullah LH, Cai LH, Leigh MW, DeMaria GC, Matsui H, Donaldson SH, Davis CW, et al. : Cystic fibrosis airway secretions exhibit mucin hyperconcentration andincreased osmotic pressure. J Clin Invest 2014, 124:3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ehre C, Ridley C, Thornton DJ: Cystic fibrosis: an inherited disease affecting mucin-producing organs. Int J Biochem Cell Biol 2014, 52:136–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheehan JK, Kesimer M, Pickles R: Innate immunity and mucus structure and function. In Innate Immunity to Pulmonary Infection. . 2008:155–169. [PubMed] [Google Scholar]
- 23.Lai SK, Wang YY, Wirtz D, Hanes J: Micro- and macrorheology of mucus. Adv Drug Deliv Rev 2009, 61:86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Button B, Picher M, Boucher RC: Differential effects of cyclic and constant stress on ATP release and mucociliary transport by human airway epithelia. J Physiol 2007, 580:577–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tarran R, Button B, Picher M, Paradiso AM, Ribeiro CM, Lazarowski ER, Zhang L, Collins PL, Pickles RJ, Fredberg JJ, et al. : Normal and cystic fibrosis airway surface liquid homeostasis. The effects of phasic shear stress and viral infections. J Biol Chem 2005, 280:35751–35759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Button B, Boucher RC: Role of mechanical stress in regulating airway surface hydration and mucus clearance rates. Respir Physiol Neurobiol 2008, 163:189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hill DB, Vasquez PA, Mellnik J, McKinley SA, Vose A, Mu F, Henderson AG, Donaldson SH, Alexis NE, Boucher RC, et al. : A Biophysical Basis for Mucus Solids Concentration as a Candidate Biomarker for Airways Disease. PLoS One 2014, 9:e87681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Puchelle E, Zahm JM: Influence of rheological properties of human bronchial secretions on the ciliary beat frequency. Biorheology 1984, 21:265–272. [DOI] [PubMed] [Google Scholar]
- 29.Galabert C, Jacquot J, Zahm JM, Puchelle E: Relationships between the lipid content and the rheological properties of airway secretions in cystic fibrosis. Clin Chim Acta 1987, 164:139–149. [DOI] [PubMed] [Google Scholar]
- 30.Linssen RS, Chai G, Ma J, Kummarapurugu AB, van Woensel JBM, Bem RA, Kaler L, Duncan GA, Zhou L, Rubin BK, et al. : Neutrophil extracellular traps increase airway mucus viscoelasticity and slow mucus particle transit. Am J Respir Cell Mol Biol 2021, 64:69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kreda SM, Davis CW, Rose MC: CFTR, Mucins, and Mucus Obstruction in Cystic Fibrosis. Cold Spring Harb Perspect Med 2012, 2:a009589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan S, Hollinger M, Lachowicz-Scroggins ME, Kerr SC, Dunican EM, Daniel BM, Ghosh S, Erzurum SC, Willard B, Hazen SL, et al. : Oxidation increases mucin polymer cross-links to stiffen airway mucus gels. Sci Transl Med 2015, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henke MO, John G, Germann M, Lindemann H, Rubin BK: MUC5AC and MUC5B mucins increase in cystic fibrosis airway secretions during pulmonary exacerbation. Am J Respir Crit Care Med 2007, 175:816–821. [DOI] [PubMed] [Google Scholar]
- 34.S A, F J, K BT, M M, C MT, F LJ, TT M, C M, Z J, A J, et al. : MUC5AC drives COPD exacerbation severity through amplification of virus-induced airway inflammation. 2019, doi: 10.1101/706804. [DOI] [Google Scholar]
- 35.Ha UH, Jae HL, Kim HJ, Wu W, Jin S, Xu H, Li JD: MKP1 regulates the induction of MUC5AC mucin by Streptococcus pneumoniae pneumolysin by inhibiting the PAK4-JNK signaling pathway. J Biol Chem 2008, 283:30624–30631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hasnain SZ, Evans CM, Roy M, Gallagher AL, Kindrachuk KN, Barron L, Dickey BF, Wilson MS, Wynn TA, Grencis RK, et al. : Muc5ac: a critical component mediating the rejection of enteric nematodes. J Exp Med 2011, 208:893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campbell L, Hepworth MR, Whittingham-Dowd J, Thompson S, Bancroft AJ, Hayes KS, Shaw TN, Dickey BF, Flamar AL, Artis D, et al. : ILC2s mediate systemic innate protection by priming mucus production at distal mucosal sites. J Exp Med 2019, 216:2714–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans CM, Raclawska DS, Ttofali F, Liptzin DR, Fletcher AA, Harper DN, McGing MA, McElwee MM, Williams OW, Sanchez E, et al. : The polymeric mucin Muc5ac is required for allergic airway hyperreactivity. Nat Commun 2015, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carpenter J, Wang Y, Gupta R, Li Y, Haridass P, Subramani DB, Reidel B, Morton L, Ridley C, O’Neal WK, et al. : Assembly and organization of the N-terminal region of mucin MUC5AC: Indications for structural and functional distinction from MUC5B. Proc Natl Acad Sci U S A 2021, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]; **This paper brings insights into mucin protein structure, which can open pathways for developing targeted therapies. The main finding was a distinct N- terminal domain in the MUC5AC protein that may explain why this mucin forms a more static and “tethered” mucus layer when compared to MUC5B.
- 40.Livraghi-Butrico A, Grubb BR, Wilkinson KJ, Volmer AS, Burns KA, Evans CM, O’Neal WK, Boucher RC: Contribution of mucus concentration and secreted mucins Muc5ac and Muc5b to the pathogenesis of muco-obstructive lung disease. Mucosal Immunol 2017, 10:395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roy MG, Livraghi-Butrico A, Fletcher AA, McElwee MM, Evans SE, Boerner RM, Alexander SN, Bellinghausen LK, Song AS, Petrova YM, et al. : Muc5b is required for airway defence. Nature 2014, 505:412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Costain G, Liu Z, Mennella V, Radicioni G, Goczi AN, Albulescu A, Walker S, Ngan B, Manson D, Vali R, et al. : Hereditary Mucin Deficiency Caused by Biallelic Loss-of-Function of MUC5B. Am J Respir Crit Care Med 2022, doi: 10.1164/RCCM.202106-1456OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Costello CM, O’Connor CM, Finlay GA, Shiels P, FitzGerald MX, Hayes JP: Effect of nebulised recombinant DNase on neutrophil elastase load in cystic fibrosis. Thorax 1996, 51:619–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, Rosenstein BJ, Smith AL, Wohl ME: Effect of Aerosolized Recombinant Human DNase on Exacerbations of Respiratory Symptoms and on Pulmonary Function in Patients with Cystic Fibrosis. http://dx.doi.org/101056/NEJM199409083311003 2010, 331:637–642. [DOI] [PubMed] [Google Scholar]
- 45.Dawson M, Wirtz D, Hanes J: Enhanced viscoelasticity of human cystic fibrotic sputum correlates with increasing microheterogeneity in particle transport. J Biol Chem 2003, 278:50393–50401. [DOI] [PubMed] [Google Scholar]
- 46.Quan JM, Tiddens HAWM, Sy JP, McKenzie SG, Montgomery MD, Robinson PJ, Wohl MEB, Konstan MW: A two-year randomized, placebo-controlled trial of dornase alfa in young patients with cystic fibrosis with mild lung function abnormalities. J Pediatr 2001, 139:813–820. [DOI] [PubMed] [Google Scholar]
- 47.Elkins MR, Robinson M, Rose BR, Harbour C, Moriarty CP, Marks GB, Belousova EG, Xuan W, Bye PTP: A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med 2006, 354:229–240. [DOI] [PubMed] [Google Scholar]
- 48.Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC: Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med 2006, 354:241–250. [DOI] [PubMed] [Google Scholar]
- 49.Reeves EP, McCarthy C, McElvaney OJ, Vijayan MSN, White MM, Dunlea DM, Pohl K, Lacey N, McElvaney NG: Inhaled hypertonic saline for cystic fibrosis: Reviewing the potential evidence for modulation of neutrophil signalling and function. World J Crit Care Med 2015, 4:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robinson M, Hemming AL, Regnis JA, Wong AG, Bailey DL, Bautovich GJ, King M, Bye PTP: Effect of increasing doses of hypertonic saline on mucociliary clearance in patients with cystic fibrosis. Thorax 1997, 52:900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goralski JL, Wu D, Thelin WR, Boucher RC, Button B: The in vitro effect of nebulised hypertonic saline on human bronchial epithelium. Eur Respir J 2018, 51:1702652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nicolson CHH, Stirling RG, Borg BM, Button BM, Wilson JW, Holland AE: The long term effect of inhaled hypertonic saline 6% in non-cystic fibrosis bronchiectasis. Respir Med 2012, 106:661–667. [DOI] [PubMed] [Google Scholar]
- 53.Blé FX, Cannet C, Collingwood S, Danahay H, Beckmann N: ENaC-mediated effects assessed by MRI in a rat model of hypertonic saline-induced lung hydration. Br J Pharmacol 2010, 160:1008–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robinson M, Daviskas E, Eberl S, Baker J, Chan HK, Anderson SD, Bye PTP: The effect of inhaled mannitol on bronchial mucus clearance in cystic fibrosis patients: a pilot study. Eur Respir J 1999, 14:678–685. [DOI] [PubMed] [Google Scholar]
- 55.Smith JJ, Travis SM, Greenberg EP, Welsh MJ: Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell 1996, 85:229–236. [DOI] [PubMed] [Google Scholar]
- 56.Goldman MJ, Anderson GM, Stolzenberg ED, Kari UP, Zasloff M, Wilson JM: Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 1997, 88:553–560. [DOI] [PubMed] [Google Scholar]
- 57.Daviskas E, Anderson SD, Jaques A, Charlton B: Inhaled mannitol improves the hydration and surface properties of sputum in patients with cystic fibrosis. Chest 2010, 137:861–868. [DOI] [PubMed] [Google Scholar]
- 58.Duman I, Ünal G, Yilmaz AI, Güney AY, Durduran Y, Pekcan S: Inhaled Dry Powder Mannitol Treatment in Pediatric Patients with Cystic Fibrosis: Evaluation of Clinical Data in a Real-World Setting. Pediatr Allergy Immunol Pulmonol 2022, 35:19–26. [DOI] [PubMed] [Google Scholar]
- 59.Bilton D, Robinson P, Cooper P, Gallagher CG, Kolbe J, Fox H, Jaques A, Charlton B: Inhaled dry powder mannitol in cystic fibrosis: An efficacy and safety study. Eur Respir J 2011, 38:1071–1080. [DOI] [PubMed] [Google Scholar]
- 60.Bilton D, Bellon G, Charlton B, Cooper P, De Boeck K, Flume PA, Fox HG, Gallagher CG, Geller DE, Haarman EG, et al. : Pooled analysis of two large randomised phase III inhaled mannitol studies in cystic fibrosis. J Cyst Fibros 2013, 12:367–376. [DOI] [PubMed] [Google Scholar]
- 61.Aitken ML, Bellon G, De Boeck K, Flume PA, Fox HG, Geller DE, Haarman EG, Hebestreit HU, Lapey A, Schou IM, et al. : Long-term inhaled dry powder mannitol in cystic fibrosis: an international randomized study. Am J Respir Crit Care Med 2012, 185:645–652. [DOI] [PubMed] [Google Scholar]
- 62.Lopes-Pacheco M: CFTR Modulators: Shedding Light on Precision Medicine for Cystic Fibrosis. Front Pharmacol 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pinto MC, Silva IAL, Figueira MF, Amaral MD, Lopes-Pacheco M: Pharmacological Modulation of Ion Channels for the Treatment of Cystic Fibrosis. J Exp Pharmacol 2021, 13:693–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, Griese M, McKone EF, Wainwright CE, Konstan MW, et al. : A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011, 365:1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jih KY, Lin WY, Sohma Y, Hwang TC: CFTR potentiators: From Bench to Bedside. Curr Opin Pharmacol 2017, 34:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Taylor-Cousar JL, Mall MA, Ramsey BW, McKone EF, Tullis E, Marigowda G, McKee CM, Waltz D, Moskowitz SM, Savage J, et al. : Clinical development of triple-combination CFTR modulators for cystic fibrosis patients with one or two F508del alleles. ERJ open Res 2019, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hoy SM: Elexacaftor/Ivacaftor/Tezacaftor: First Approval. Drugs 2019, 79:2001–2007. [DOI] [PubMed] [Google Scholar]
- 68.Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, Ramsey BW, Taylor-Cousar JL, Tullis E, Vermeulen F, et al. : Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med 2019, 381:1809–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heijerman HGM, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, Mall MA, Welter JJ, Ramsey BW, McKee CM, et al. : Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet (London, England) 2019, 394:1940–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morrison CB, Shaffer KM, Araba KC, Markovetz MR, Wykoff JA, Quinney NL, Hao S, Delion MF, Flen AL, Morton LC, et al. : Treatment of cystic fibrosis airway cells with CFTR modulators reverses aberrant mucus properties via hydration. Eur Respir J 2021, doi: 10.1183/13993003.00185-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]; *For the first time, the authors of this study showed the in vitro impact of the CFTR modulators on mucus concentration.
- 71.Pinto MC, Silva IAL, Figueira MF, Amaral MD, Lopes-Pacheco M: Pharmacological Modulation of Ion Channels for the Treatment of Cystic Fibrosis. J Exp Pharmacol 2021, 13:693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Duijvestijn YCM, Brand PLP: Systematic review of N-acetylcysteine in cystic fibrosis. Acta Paediatr 1999, 88:38–41. [DOI] [PubMed] [Google Scholar]
- 73.Ehre C, Rushton ZL, Wang B, Hothem LN, Morrison CB, Fontana NC, Markovetz MR, Delion MF, Kato T, Villalon D, et al. : An Improved Inhaled Mucolytic to Treat Airway Muco-obstructive Diseases. Am J Respir Crit Care Med 2019, 199:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rochat T, Lacroix JS, Jornot L: N-acetylcysteine inhibits Na+ absorption across human nasal epithelial cells. J Cell Physiol 2004, 201:106–116. [DOI] [PubMed] [Google Scholar]
- 75.Jones LC, Moussa L, Fulcher ML, Zhu Y, Hudson EJ, O’Neal WK, Randell SH, Lazarowski ER, Boucher RC, Kreda SM: VAMP8 is a vesicle SNARE that regulates mucin secretion in airway goblet cells. J Physiol 2012, 590:545–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Williams OW, Sharafkhaneh A, Kim V, Dickey BF, Evans CM: Airway mucus: From production to secretion. Am J Respir Cell Mol Biol 2006, 34:527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jaramillo AM, Azzegagh Z, Tuvim MJ, Dickey BF: Airway Mucin Secretion. Ann Am Thorac Soc 2018, 15:S164–S170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Montgomery ST, Dittrich AS, Garratt LW, Turkovic L, Frey DL, Stick SM, Mall MA, Kicic A: Interleukin-1 is associated with inflammation and structural lung disease in young children with cystic fibrosis. J Cyst Fibros 2018, 17:715–722. [DOI] [PubMed] [Google Scholar]
- 79.Fritzsching B, Zhou-Suckow Z, Trojanek JB, Schubert SC, Schatterny J, Hirtz S, Agrawal R, Muley T, Kahn N, Sticht C, et al. : Hypoxic epithelial necrosis triggers neutrophilic inflammation via IL-1 receptor signaling in cystic fibrosis lung disease. Am J Respir Crit Care Med 2015, 191:902–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen G, Sun L, Kato T, Okuda K, Martino MB, Abzhanova A, Lin JM, Gilmore RC, Batson BD, O’Neal YK, et al. : IL-1β dominates the promucin secretory cytokine profile in cystic fibrosis. J Clin Invest 2019, 129:4433–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martino MB, Jones L, Brighton B, Ehre C, Abdulah L, Davis CW, Ron D, O’Neal WK, Ribeiro CMP: The ER stress transducer IRE1β is required for airway epithelial mucin production. Mucosal Immunol 2013, 6:639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hull-Ryde EA, Minges JT, Martino MEB, Kato T, Norris-Drouin JL, Ribeiro CMP: IRE1α Is a Therapeutic Target for Cystic Fibrosis Airway Inflammation. Int J Mol Sci 2021, 22:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This paper showed for the first time that the small molecule RE1αkinase and RNase inhibitor KIRA6 is a potential therapy for treating the increased inflammation in CF disease.
- 83.Christopher Boyd A, Guo S, Huang L, Kerem B, Oren YS, Walker AJ, Hart SL: New approaches to genetic therapies for cystic fibrosis. J Cyst Fibros 2020, 19:S54–S59. [DOI] [PubMed] [Google Scholar]
- 84.Hart SL, Harrison PT: Genetic therapies for cystic fibrosis lung disease. Curr Opin Pharmacol 2017, 34:119–124. [DOI] [PubMed] [Google Scholar]
- 85.Sun X, Sui H, Fisher JT, Yan Z, Liu X, Cho HJ, Joo NS, Zhang Y, Zhou W, Yi Y, et al. : Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J Clin Invest 2010, 120:3149–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Choi KYG, Wu BC, Lee AHY, Baquir B, Hancock REW: Utilizing Organoid and Air-Liquid Interface Models as a Screening Method in the Development of New Host Defense Peptides. Front Cell Infect Microbiol 2020, 10:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bouzek DC, Abou Alaiwa MH, Adam RJ, Pezzulo AA, Reznikov LR, Cook DP, Aguilar Pescozo MI, Eyck P Ten, Wu C, Gross TJ, et al. : Early Lung Disease Exhibits Bacteria-Dependent and -Independent Abnormalities in Cystic Fibrosis Pigs. Am J Respir Crit Care Med 2021, 204:692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This paper has a crucial role in describing the CF pig as an animal model to understand CF pathophysiology. The study demonstrated that CF lung disease in pigs starts right after birth and that early antibiotics intervention only partially prevents lung abnormities.
- 88.Vaidyanathan S, Salahudeen AA, Sellers ZM, Bravo DT, Choi SS, Batish A, Le W, Baik R, de la O S, Kaushik MP, et al. : High-Efficiency, Selection-free Gene Repair in Airway Stem Cells from Cystic Fibrosis Patients Rescues CFTR Function in Differentiated Epithelia. Cell Stem Cell 2020, 26:161–171.e4. 26:161–171.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]; *In this paper the authors provided a new important strategy for gene therapy. They showed in vitro that different gene correction techniques can be used on autologous CF patient’s airway stem cells and achieve >30% CFTR function relative to non-CF levels in DF508 alleles.
- 89.Chen OG, Mather SE, Brommel CM, Hamilton BA, Ehler A, Villacreses R, Girgis RE, Alaiwa MA, Stoltz DA, Zabner J, et al. : Transduction of Pig Small Airway Epithelial Cells and Distal Lung Progenitor Cells by AAV4. Cells 2021, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]; *The authors developed a novel preclinical gene therapy study using Adeno-associated virus 4 (AAV4) and CF pig cells from the small airway epithelia. In addition, they showed that AAV4 can be a safe vector and successfully infect pig lungs in vivo.
- 90.Lee JA, Cho A, Huang EN, Xu Y, Quach H, Hu J, Wong AP: Gene therapy for cystic fibrosis: new tools for precision medicine. J Transl Med 2021 191 2021, 19:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hida K, Lai SK, Suk JS, Won SY, Boyle MP, Hanes J: Common gene therapy viral vectors do not efficiently penetrate sputum from cystic fibrosis patients. PLoS One 2011, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kesimer M, Ehre C, Burns KA, Davis CW, Sheehan JK, Pickles RJ: Molecular organization of the mucins and glycocalyx underlying mucus transport over mucosal surfaces of the airways. Mucosal Immunol 2013, 6:379–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pei Y, Bao Y, Sacchetti C, Brady J, Gillard K, Yu H, Roberts S, Rajappan K, Tanis SP, Perez-Garcia CG, et al. : Synthesis and bioactivity of readily hydrolysable novel cationic lipids for potential lung delivery application of mRNAs. Chem Phys Lipids 2022, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Duncan GA, Kim N, Colon-Cortes Y, Rodriguez J, Mazur M, Birket SE, Rowe SM, West NE, Livraghi-Butrico A, Boucher RC, et al. : An Adeno-Associated Viral Vector Capable of Penetrating the Mucus Barrier to Inhaled Gene Therapy. Mol Ther Methods Clin Dev 2018, 9:296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schneider CS, Xu Q, Boylan NJ, Chisholm J, Tang BC, Schuster BS, Henning A, Ensign LM, Lee E, Adstamongkonkul P, et al. : Nanoparticles that do not adhere to mucus provide uniform and long-lasting drug delivery to airways following inhalation. Sci Adv 2017, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wan F, Nylander T, Klodzinska SN, Foged C, Yang M, Baldursdottir SG, Nielsen HM: Lipid Shell-Enveloped Polymeric Nanoparticles with High Integrity of Lipid Shells Improve Mucus Penetration and Interaction with Cystic Fibrosis-Related Bacterial Biofilms. ACS Appl Mater Interfaces 2018, 10:10678–10687. [DOI] [PubMed] [Google Scholar]
- 97.Da Silva Sanchez A, Paunovska K, Cristian A, Dahlman JE: Treating Cystic Fibrosis with mRNA and CRISPR. Hum Gene Ther 2020, 31:940–955. [DOI] [PMC free article] [PubMed] [Google Scholar]