

Abstract
Background
Muscle wasting is a common complication of chronic kidney disease (CKD) that is associated with higher mortality. Although the mechanisms of myofibre loss in CKD has been widely studied, the contribution of muscle precursor cell (MPC) senescence remains poorly understood. Senescent MPCs no longer proliferate and can produce proinflammatory factors or cytokines. In this study, we tested the hypothesis that the senescence associated secretory phenotype (SASP) of MPCs contributes to CKD‐induced muscle atrophy and weakness.
Methods
CKD was induced in mice by 5/6th nephrectomy. Kidney function, muscle size, and function were measured, and markers of atrophy, inflammation, and senescence were evaluated using immunohistochemistry, immunoblots, or qPCR. To study the impact of senescence, a senolytics cocktail of dasatinib + quercetin (D&Q) was given orally to mice for 8 weeks. To investigate CKD‐induced senescence at the cellular level, primary MPCs were incubated with serum from CKD or control subjects. The roles of specific proteins in MPC senescence were studied using adenoviral transduction, siRNA, and plasmid transfection.
Results
In the hindlimb muscles of CKD mice, (i) the senescence biomarker SA‐β‐gal was sharply increased (~30‐fold); (ii) the DNA damage response marker γ‐H2AX was increased 1.9‐fold; and (iii) the senescence pathway markers p21 and p16INK4a were increased 1.99‐fold and 2.82‐fold, respectively (all values, P < 0.05), whereas p53 was unchanged. γ‐H2AX, p21, and p16INK4A were negatively correlated at P < 0.05 with gastrocnemius weight, suggesting a causal relationship with muscle atrophy. Administration of the senolytics cocktail to CKD mice for 8 weeks eliminated the disease‐related elevation of p21, p16INK4a, and γ‐H2AX, abolished positive SA‐β‐gal, and depressed the high levels of the SASP cytokines, TNF‐α, IL‐6, IL‐1β, and IFN (all values, P < 0.05). Skeletal muscle weight, myofibre cross‐sectional area, and grip function were improved in CKD mice receiving D&Q. Markers of protein degradation, inflammation, and MPCs dysfunction were also attenuated by D&Q treatment compared with the vehicle treatment in 5/6th nephrectomy mice (all values, P < 0.05). Uraemic serum induced senescence in cultured MPCs. Overexpression of FoxO1a in MPCs increased the number of p21+ senescent cells, and p21 siRNA prevented uraemic serum‐induced senescence (P < 0.05).
Conclusions
Senescent MPCs are likely to contribute to the development of muscle wasting during CKD by producing inflammatory cytokines. Limiting senescence with senolytics ameliorated muscle wasting and improved muscle strength in vivo and restored cultured MPC functions. These results suggest potential new therapeutic targets to improve muscle health and function in CKD.
Keywords: Cachexia, CKD, CDK (cyclin‐dependent kinase), DNA damage, Muscle precursor cells, Senescence
Introduction
Chronic kidney disease (CKD) is a global health challenge that impacts more than 700 million patients. 1 The ensuing uraemia and other pathological responses frequently lead to muscle wasting or atrophy. Loss of muscle mass and reduced function negatively impact the quality of life of CKD patients, leading to higher risks of frailty, co‐morbidities and mortality. 2
Muscle loss in CKD results from abnormalities in physiologic signals that collectively produce an imbalance between protein synthesis and degradation. Metabolic acidosis, insulin resistance, inflammation, and other factors can decrease protein synthesis and consistently upregulate protein degradation via the ubiquitin–proteasome system (UPS), lysosomal proteolysis and caspase‐3. 2 , 3 Downregulation of IGF‐PI3K‐AKT signalling in myofibres and muscle precursor cells (MPCs), which are also known as satellite cells or muscle stem cells, has been linked to CKD‐associated muscle atrophy. 2 In patients and animals with CKD, persistent systemic inflammation was positively correlated with all‐cause mortality and muscle wasting. 4 , 5 , 6 Levels of inflammatory cytokines, specifically TNF and IL‐6, were increased in muscle biopsies of patients and mice with CKD. 7 , 8 The causes and sources of inflammation in muscle during CKD remain poorly understood.
Senescence is a cellular state that is characterized by irreversible cell‐cycle arrest and associated with many diseases including CKD. 9 , 10 Physiological processes and pathologic stresses, such as aging, DNA damage, and mitochondrial or metabolic dysfunction can activate the p16INK4a/Rb and p53/p21 senescence pathways which arrest the cell cycle. 9 , 11 Although senescent cells no longer divide and proliferate, they remain capable of producing and secreting various proinflammatory factors, a state known as senescence associated secretory phenotype (SASP). 12 Secreted factors such as cytokines, chemokines, growth factors, and other inflammatory molecules are associated with chronic inflammation and tissue damage. 9 , 10 For example, p16INK4a+ cells have been observed in kidney in different types of kidney diseases, including membranous nephropathy, focal segmental glomerulosclerosis, IgA nephropathy, and in transplanted kidneys. 13 , 14 SA‐β‐gal and p16INK4a were also increased in kidney epithelial cells of patients with diabetic nephropathy. 13
Low‐grade chronic systemic inflammation is a hallmark feature of CKD that is associated with pathological features of the disease such as muscle wasting. 4 Senescence has been linked to the progression of muscle injury and age‐related sarcopenia. Chiche et al. 15 reported that cardiotoxin‐injured tibialis anterior (TA) muscle contained SA‐β‐gal+ cells, and expression of p16INK4a, p21, and the SASP‐related molecules IL‐6, MMP3, and MMP13 was increased. Oxidative stress also has been associated with increased p21 expression and SASP factors, including IL‐6, and prevented activation and regeneration of MPCs. 16 Although cellular senescence and muscle atrophy are both linked to inflammation and chronic diseases, the causal relationship between the two conditions is unclear.
When considered together, these findings led us to hypothesize that cellular senescence contributes to CKD‐induced muscle atrophy and weakness. To test this possibility, hindlimb muscles of mice with CKD were evaluated for markers of cellular senescence by SA‐β‐gal staining and measuring the abundance of DNA damage response marker γ‐H2AX, p16INK4a, p53, p21, and SASP‐associated proteins. Mice with CKD were fed a cocktail of senolytics to eliminate senescent cells, and the treatment impact on muscle size, function, and markers of muscle atrophy was evaluated. We also conducted experiments with MPCs incubated with uraemic serum from CKD patients. Our results indicate that cellular senescence has a contributory role in the inflammatory environment and muscle wasting associated with CKD.
Methods
Human serum collection
The human subject protocol was reviewed and approved by the Emory University Institutional Review Board. Human sera were obtained from non‐CKD volunteers and CKD stage 4 patients of different ages, ranging from 55 to 70 years old. Details of those patients are provided in Table S1.
Mouse models and drug treatments
All animal experiments protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at Emory University. C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbour, ME, USA). CKD was induced in mice by two‐step 5/6th nephrectomy protocol. 17 Mice were randomly distributed to four cohorts: control, CKD, CKD with glycerol vehicle, and CKD with senolytics. The senolytics dasatinib (D) (5 mg/kg body weight) and quercetin (Q) (50 mg/kg body weight) were given to CKD mice by oral gavage in 100–150 μL glycerol twice per week. 18 Food and body weights were measured three times per week. Grip strength was measured with a grip strength meter equipped with dual computerized sensors (Columbus Instruments, Columbus, OH) 8 weeks after surgery just prior to the harvest of muscles and serum. CKD was confirmed by measuring blood urea nitrogen (BUN) and creatinine with a BUN Kinetic Procedure Kit (Thermo Electron, Louisville, CO) and Creatinine (Serum) Colorimetric Assay Kit (Caymen Chemical Co), respectively.
Muscle progenitor cell (MPC) isolation and culture
MPCs were isolated from mouse skeletal muscles using a Mouse Satellite Cell Isolation Kit (MACS Miltenyi Biotec Inc. 130‐104‐268). 17 Briefly, single‐cell suspensions were obtained from mouse skeletal muscle use the Skeletal Muscle Dissociation Kit (MACS 130‐098‐305). Non‐target cells were depleted using a MACS column and separator column. MPCs in the effluent were cultured on a Cell Attachment Matrix Gel (Millipore/Sigma, Burlington MA #08‐110) coated dish in Ham's F‐12 Nutrient Mixture medium (Invitrogen) supplemented with 20% fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin and 5 ng/mL human β‐FGF (fibroblast growth factor) (Atlanta Biologicals, Atlanta, GA). For experiments, MPCs were seeded in collagen‐coated plates on Day 1 using medium without β‐FGF. When cells reached 60% confluence on Day 2, differentiation of MPCs was induced by switching medium to Ham's F‐12 medium containing 2% horse serum. On Day 3, MPCs were treated with F‐12 medium with 2% pooled control or uraemic human serum with or without 12.5 nM dasatinib and 25 nM quercetin (dissolved in DMSO). When applicable, adenovirus transduction or plasmid/siRNA transfection were also performed on Day 3. MPCs were harvested for assessment on Day 5.
Virus reagents
All experiments involving recombinant adenovirus were approved by the Emory University Environmental Health and Safety Office. Ad‐CDK1 and Ad‐CDK4 were provided by Dr Mohamed (University of Louisville) 19 ; the constitutively active FoxO3a virus (CA‐FOXO3a,) was purchased from Vector Biolabs (#1025; Malvern, PA).
Plasmid and siRNA transfection
A FOXO1a plasmid was a gift from Domenico Accili (Addgene plasmid #17551; Watertown, MA) 20 ; a p16INK4a plasmid (SR403922) was from OriGene Technologies (Rockville, MD); p21Waf1/Cip1(sc‐29427) siRNA was from Santa Cruz, (Dallas, TX). Effectene transfection reagent (Qiagen, Germantown, MD) was used for plasmid transfection whereas Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific; Waltham, MA) was used for siRNA transfection.
Proliferation assay
The Cell Proliferation Assay (Chemicon/Fisher, Waltham, MA) was used for quantification of MPCs proliferation. MPCs were seeded in collagen coated 96‐well plates and differentiated for 24 h. Experimental reagents were added to cells for 48 h. To evaluate proliferation, WST‐1/ECS solution was added to each well, and cells were incubated for 1 h before measuring absorbance at 450 nm using a TECAN microtiter plate reader.
Apoptosis assay
Apoptosis was measured using the Click‐iT™ Plus TUNEL Assay (Invitrogen/Fisher, Waltham, MA). Cell images were captured with a Olympus 1 × 51 inverted microscope equipped with a DP73‐1‐51‐17MP colour camera; 10–20 fields/plate were randomly acquired and quantified.
Histology
Gastrocnemius muscles were frozen and cut into 10 μm sections. For immunochemical analyses, muscle sections or MPC cells were fixed in 4% paraformaldehyde and washed with PBS. After permeabilization in 0.05% Triton X‐100 and quench‐fixation in 50‐mM NH4Cl, tissues were blocked in 3% bovine serum albumin for 0.5 h and incubated with primary antibody overnight at 4°C. The sections were washed in TBST and incubated with FITC‐labelled secondary antibody for 1 h at RT. Nuclei were stained by DAPI. Cell images were captured with an Olympus 1 × 51 inverted microscope equipped with a DP73‐1‐51‐17MP colour camera; 10–20 fields/plate were randomly acquired and quantified. To evaluate the senescent state of MPCs in vivo, frozen sections were double stained for p21, a senescent marker, and pax 7, a MPC marker. Skeletal muscle fibre cross‐sectional areas (700–900 fibres in each cohort) were measured after staining with an anti‐laminin antibody (Sigma‐Aldrich). Images were analysed using NIH ImageJ.
SA‐β‐gal staining
Senescence associate β‐gal was evaluated with a β‐galactosidase staining kit from Cell Biolabs (San Diego, CA) for in vitro experiments; an X‐Gal staining kit from Genlantis (San Diego, CA) was used for in vivo experiments. For whole tissue staining, skeletal muscles were fixed in 4% PFA for 1 h at RT, washed with PBS 3 times and then incubated in 1× X‐Gal solution overnight at 37°C in a humidifying container. The next day, the tissues were sectioned to 10 μm slides, and the images were captures and analysed as described above.
Protein immunoblotting and antibodies
Gastrocnemius muscle and MPC cell lysates were homogenized in RIPA buffer containing protease/phosphatase inhibitors. Equal amounts of protein were separated by SDS‐PAGE, proteins were transferred to PVDF membranes, and the blots were blocked and processed with primary and secondary antibodies. Processed blots were quantified using a LI‐COR Odyssey infrared scanning system (Li‐COR Biosciences, Lincoln, NE). Antibodies used for experiments are listed in Table S2.
Quantitative real‐time PCR
RNA was extracted from frozen muscle samples using Tri‐Reagent (Molecular Research Inc., Cincinnati, OH) and reverse‐transcribed to cDNA using a Thermoscript RT‐PCR kit (Invitrogen Carlsbad, CA). Real‐time quantitative PCR was performed using SYBR Green PCR reagent (Bio‐Rad, Hercules, CA) using the following PCR cycle parameters: 94°C for 2 min followed by 40 cycles of 94°C for 15 s, 55°C for 30 s, and 72°C for 30 s with final extension at 72°C for 10 min. 21 The 18S rRNA was used as control for comparison. Primers are listed in Table S3.
Statistical analysis
Data were expressed as mean ± SE. A two‐tailed Student's t‐test was performed to identify statistically significant differences between two cohorts. A one‐way analysis of variance (ANOVA) with a post hoc analysis using Tukey's multiple comparisons test was used to compare more than two cohorts. Differences were considered significant when P ≤ 0.05.
Results
CKD induces cellular senescence markers in the muscles of mice
To investigate the relationship between muscle atrophy and senescence in CKD, a mouse partial nephrectomy model was used. Two‐month‐old C57BL/6J mice were randomly assigned to sham‐operated, or 5/6th nephrectomy (CKD) cohorts, and skeletal muscles were harvested 8 weeks after the partial nephrectomy. Renal dysfunction in the CKD mice was confirmed by measuring blood urea nitrogen (BUN) and serum creatinine (Figure 1A). CKD mice had lower body weights, muscle grip strength (Figure 1B), and individual wet muscle weights (Figure 1C). Abundance of the DNA damage marker γ‐H2AX and the cell cycle arrest markers p21 and p16INK4a were increased in the skeletal muscle of the CKD cohort compared to the sham operated cohort (Figure 1D). Notably, another senescence marker, p53, was not significantly changed by CKD. Senescence‐associated beta‐galactosidase (SA‐β‐gal) staining was higher in skeletal muscle of the CKD mice (Figure 1E). All three senescence markers γ‐H2AX, p21, and p16INK4a were negatively correlated with the weights of the gastrocnemius muscles (Figure 1F). These results documented the presence of a senescent phenotype in hindlimb muscles of mice with CKD.
Figure 1.
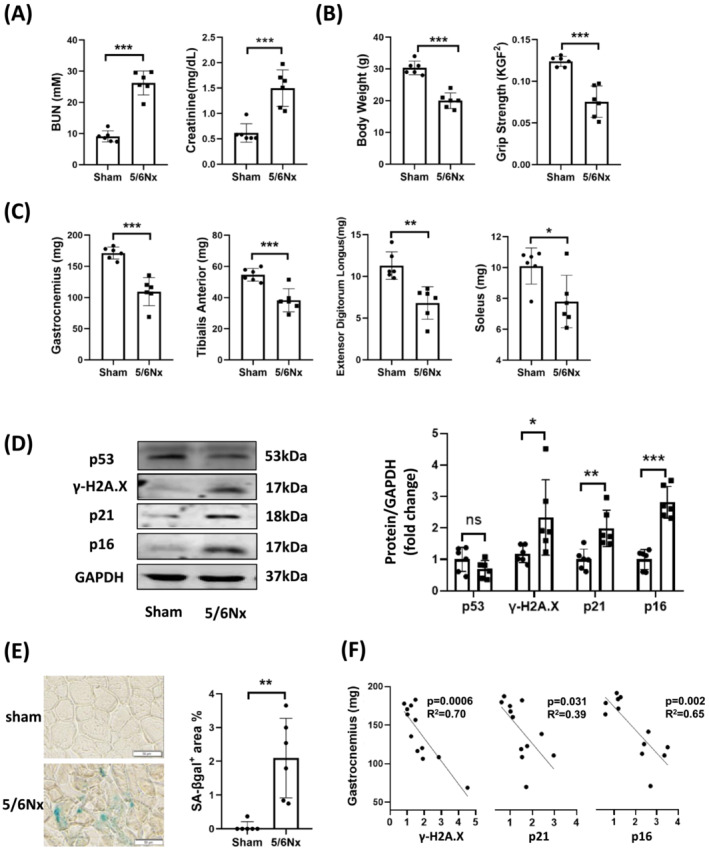
Cellular senescence appeared in the muscle of chronic kidney disease mice. A partial nephrectomy (5/6Nx) or sham operation (S) was performed in two‐month‐old C57BL6 mice; sham and CKD mice were harvested 2‐month later. (A) Blood urea nitrogen (BUN) serum and creatinine and (B) body weight and hindlimb muscle grip strength were measured. (C) Changes of muscle weight in gastrocnemius, tibialis anterior (TA), extensor digitorum longus (EDL) and soleus were evaluated in sham and CKD group. (D) the amount of p53, γ‐H2A.X, p21, and 16INK4a were measured by immunoblot analysis in gastrocnemius muscle. The results are reported in the bar graph as the fold change of each protein, normalized to the GAPDH (mean ± SE; n = 6/cohort; *P < 0.05; **P < 0.01; ***P < 0.001 vs. sham). (E) Muscles were stained with SA‐β‐gal (scale bar 50 μm), and the staining was quantified using ImageJ software. The results are reported in the bar/point graph as percentage of positive area (mean ± SE; n = 6/group; *P < 0.05; **P < 0.01; ***P < 0.001 vs. sham). (F) The gastrocnemius muscle weight was plotted versus the quantified senescence markers γ‐H2A.X, p21, and p16 and the data were analysed by linear regression. Each point represents one animal (n = 12).
Limiting senescence with the senolytics ameliorated muscle wasting in CKD mice
In earlier reports, a senolytic cocktail consisting of dasatinib plus quercetin (D&Q) selectively eliminated senescent cells in vivo and in vitro. 18 , 22 To investigate the relationship between CKD‐induced cellular senescence and muscle atrophy, CKD mice were administered D&Q twice a week for 8 weeks and muscles were evaluated for muscle loss and markers of senescence. The amounts of histologically detectable γ‐H2AX, retinoblastoma protein (Rb: inhibitor of cell cycle progression) and SA‐β‐gal were increased in the gastrocnemius muscles of the CKD cohort vs shams (Figure 2A) and administration of D&Q reduced these markers of senescence. The intervention also prevented the CKD‐induced increase in p21 and p16INK4a and increased the amounts of two cyclin‐dependent kinases (CDKs) family members, CDK1/2 and CDK4, in the muscle of CKD mice. CDKs support progression of the cell cycle by inhibiting Rb.
Figure 2.
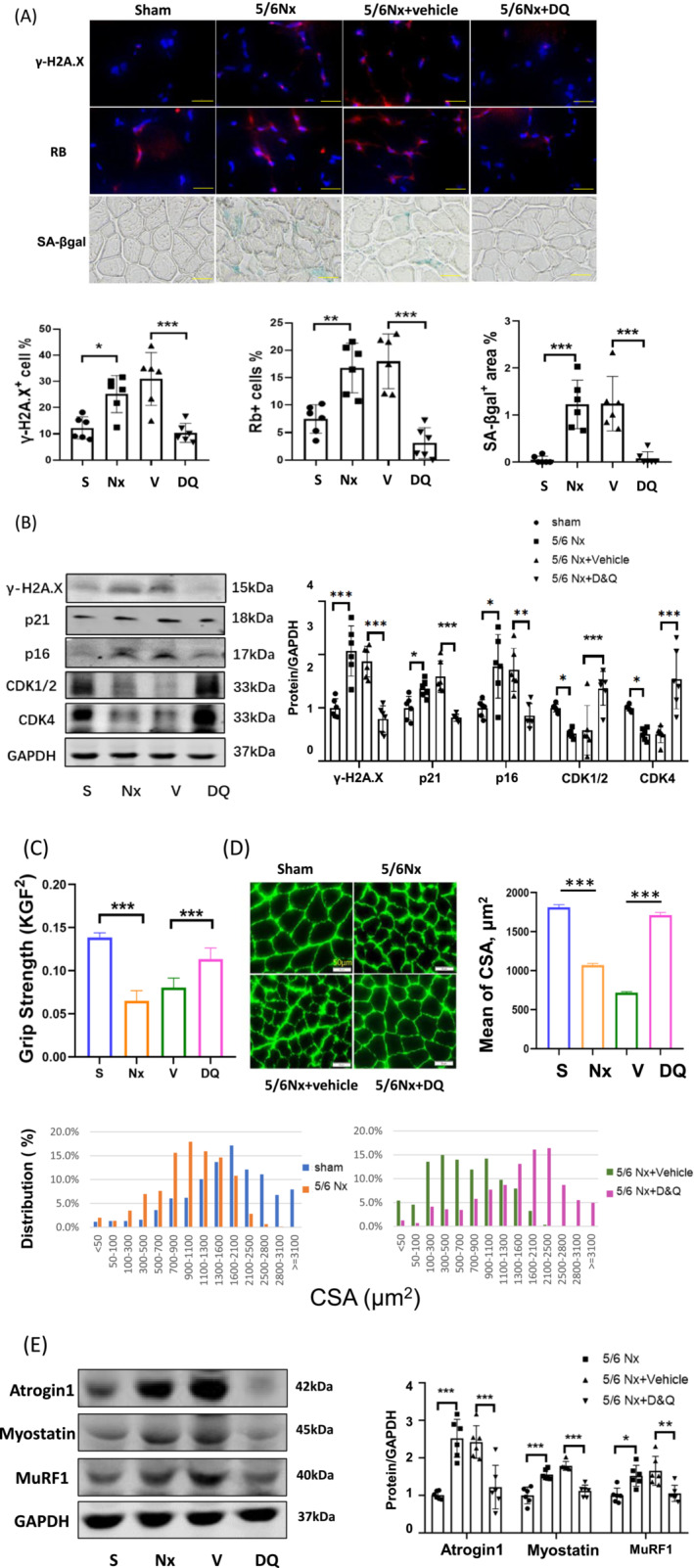
Limiting senescence with the senolytics ameliorated muscle wasting in CKD mice. CKD mice were fed a cocktail of dasatinib and quercetin (D&Q) two times per week and skeletal muscles were harvested 8 weeks later. (A) γ‐H2A.X+, Rb+, and SA‐β‐gal+ were stained in gastrocnemius (scale bar 50 μm). The positive cells were counted using ImageJ software. The results are reported in the bar/point graphs as the percentage positive cells or area for sham (S), CKD (Nx) CKD plus vehicle (V), and CKD plus D&Q (DQ) (n = 6/group; mean ± SE; *P < 0.05; **P < 0.01; ***P < 0.001). (B) The protein level of senescence markers, γ‐H2A.X, p21, 16INK4a, cyclin‐dependent kinases (CDK)1/2, and CDK4 were measured by immunoblot analysis. The graphs show the normalized fold changes of the indicated proteins compared with the levels of sham group (presented as one‐fold) (n = 6/group; mean ± SE; *P < 0.05; **P < 0.01; ***P < 0.001). (C) Muscle function was measured using a mouse grip strength meter with dual computerized sensors to detect and record the grip force in each group of mice. Data are presented as mean ± SE (n = 6/group; KGF = kilogram force, ***P < 0.001). (D) Representative images of muscle cross‐sections immunostained with anti‐laminin antibody are presented. Cross‐sectional areas of 700–900 fibres per cohort were measured and the frequency distribution of fibre CSA are reported in the bar graph: Sham (blue), 5/6Nx (red), 5/6Nx + vehicle (green), and 5/6Nx + D&Q (purple) (mean ± SE; n = 6/group; scale bars = 50 μm). (E) The muscle atrophy markers atrogin‐1, MuRF1, and myostatin in the gastrocnemius muscle were measured by immunoblot analysis and normalized data are reported in arbitrary units in the bar/point graph (n = 6/group; mean ± SE; *P < 0.05; **P < 0.01; ***P < 0.001).
Curiously, administration of D&Q did not significantly improve the CKD‐induced overall loss of body weight. However, the treatment did cause an increase in the wet weights of the gastrocnemius and TA muscles over the same muscles of vehicle‐treated CKD mice; the EDL and soleus muscles also tended to be bigger but the differences from vehicle were not significant. This improvement in muscle mass in CKD mice translated into muscle function, indicated by an increased in muscle grip strength (Figure 2(C)). The average cross‐sectional areas (CSA) of the myofibres were also larger in the CKD plus D&Q cohort (means ± SE: 7845 ± 329) than in the vehicle‐treated CKD cohort (4907 ± 240, n = 500, P < 0.001 vs. CKD/D&Q). The frequency distribution of fibre cross‐sectional area in the CKD plus D&Q cohort has a significant right shift (toward bigger cross‐sectional areas) compared to the CKD/vehicle cohort (Figure 2(D)). Markers of muscle atrophy, FBXO32/atrogin‐1, myostatin, and TRIM63/MuRF1, were increased in the gastrocnemius of CKD mice, versus controls and were reduced following treatment with D&Q (Figure 2E). These data suggest that a rise in the population of senescent cells in muscle contributes to muscle loss and dysfunction.
Senolytics reduce inflammation in muscles of CKD mice
SASP is a key feature of senescent cells that if persistent, can lead to chronic inflammation and tissue damage. 11 Inflammation has been linked to CKD‐induced muscle atrophy by through activation of multiple proteolytic pathways. 2 , 3 To evaluate the contribution of senescent cells to muscle inflammation in CKD mice, inflammatory cytokines in the gastrocnemius were evaluated by immunoblot analyses and real time quantitative PCR. IL‐1β and TNF‐α were increased in the muscles of the CKD cohort and when the senolytics cocktail was administered, these proteins were reduced in muscle to the levels in the control cohort (Figure 3A). Similarly, the expression of IL‐1β, IL‐6, TNF‐α and IFN‐γ mRNAs were increased in muscles of CKD mice and provision of the senolytics eliminated these responses (Figure 3B). These results indicate senescent cells contribute to deleterious CKD‐related inflammation in muscle.
Figure 3.
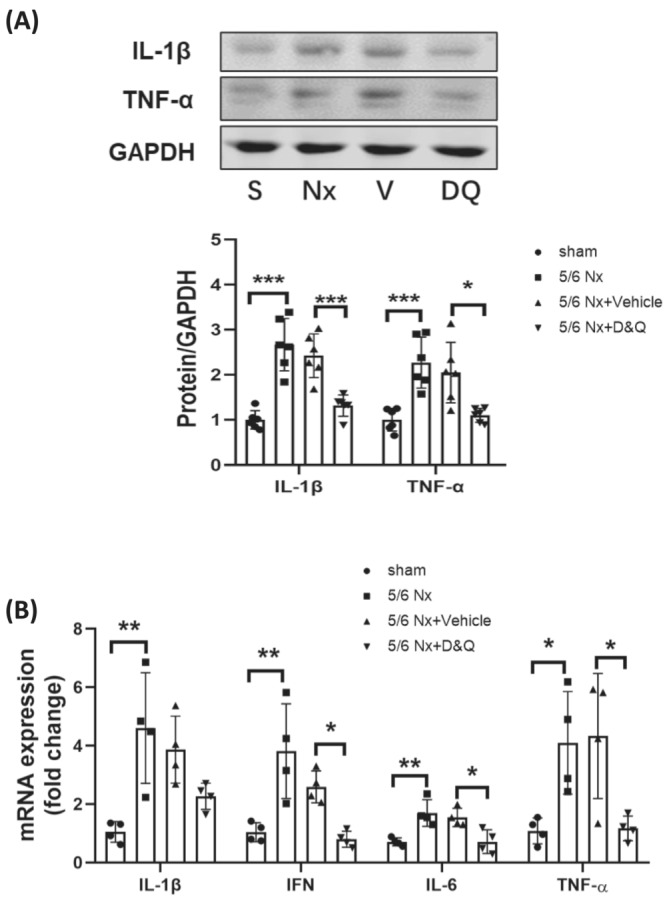
Senolytics reduce inflammation and senescent responses in muscle. CKD mice were treated with D&Q twice per week and gastrocnemius muscles were harvested 8 weeks after surgery. (A) The abundance of IL‐1β and TNF‐α, proteins in gastrocnemius muscles, were measured by immunoblot analyses for sham (S), CKD (Nx) CKD plus vehicle (V), and CKD plus D&Q (DQ) cohorts. Data are reported in the bar/point graphs as the fold change of normalized proteins of interest (n = 6/group; mean ± SE; *P < 0.05; **P < 0.01; ***P < 0.001). (B) Total RNA was isolated from gastrocnemius muscles of sham, CKD, CKD plus vehicle, and CKD plus D&Q mice. The relative abundances of mRNAs encoding IL‐1β, IL‐6, interferon gamma (IFN) and TNF‐α were measured by real time qPCR. Results are reported in the bar/point graph as the fold changes of individual mRNAs, normalized with 18S, for each treatment cohort (n = 4/group; mean ± SE; *P < 0.05; **P < 0.01; ***P < 0.001).
Senolytics suppress senescent markers in muscles of CKD mice
Healthy skeletal muscle is a post‐mitotic tissue. In response to injury or other physiologic stresses such as exercise, muscle regenerates via a process that is involves activation, proliferation, differentiation, and fusion of MPCs to myofibres. When MPCs become senescent, muscle repair is impaired. To evaluate the regenerative capacity of muscle in CKD mice, we evaluated the proliferation marker ki67, the MPC marker Pax7+, and the senescent marker p21 by immunostaining muscle cross‐sections. The percentage of cells that were positive for ki67 (Figure 4A) and Pax7+ (Figure 4B1) were similarly reduced in CKD mice whereas the percentage of cells that were positive for p21 was increased; the ratio of Pax7+p21+/Pax7+ cells, which indicates the proportion of senescent MPC in the total MPC population, was also increased (Figure 4B2). Treatment of CKD mice with the senolytic cocktail prevented these changes in proliferative and senescence markers (Figure 4A,B). To further explore the impact of senolytics on myogenesis, we performed immunoblot analysis of pax7 and myogenin proteins in muscle. Consistent with our other data, CKD suppressed both myogenesis markers and provision of the senolytics cocktail reversed the effect in muscle (Figure 4C). These results suggest that senolytics improve MPC proliferation (increasing ki67 and pax7 positive cells and protein) and decrease the population of resident senescent cells (p21 positive cells) in muscle of CKD mice.
Figure 4.
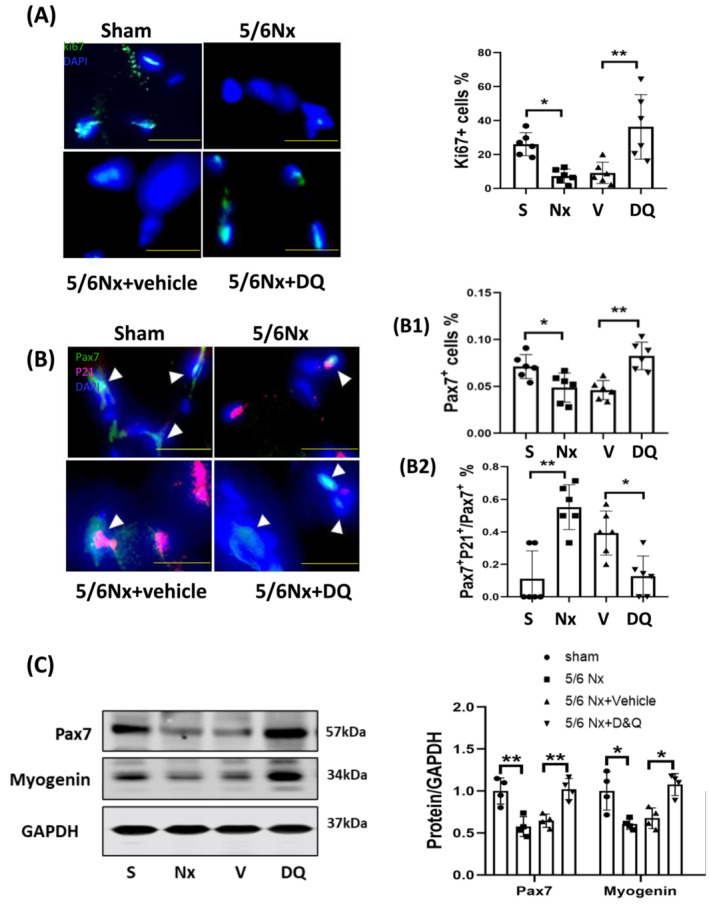
Senolytics suppressed senescent markers in muscles of CKD mice. (A) Immunostaining of ki67 was performed in frozen sections of gastrocnemius muscles harvested from sham (S), CKD (Nx), CKD + vehicle (V), and CKD + D&Q (DQ) mice. Results are reported in the Bar/point graphs as the percentage of ki67 positive cells in uraemic muscles compared to shams by the arbitrary unit (bar: mean ± SE; scale bar = 10 μm; n = 6/group; *P < 0.05; **P < 0.01; ***P < 0.001). (B) Sections of gastrocnemius muscles were double immunostained for p21 and Pax7. White arrows indicate Pax7+ cells (muscle progenitors) and pink staining represents p21+ cells (senescent cells). DAPI staining was used to determine the number of nuclei (total cell population). The percentages of pax7+ cells in the total cell population (B1) and p21+ cells in total pax7+ cells population (B2) were analysed using image‐J software and results are summarized in the bar/point graphs (bars: Mean ± s.e.; scale bar = 10 μm; n = 6/group; *P < 0.05; **P < 0.01; ***P < 0.001). (C) Proteins involved in muscle regeneration (Pax7 and myogenin) were evaluated by immunoblot analysis in samples from the gastrocnemius muscle of sham, CKD (Nx), CKD + vehicle (V), and CKD + D&Q (DQ) mice. Results are reported in the bar/point graph as the fold change of each protein, normalized to GAPDH (n = 4/group; bar: mean ± SE; *P < 0.05; **P < 0.01; ***P < 0.001).
Senolytics suppress uraemic serum‐induced senescence in MPCs
To determine if CKD‐related serum factors are responsible for inducing the MPC senescent phenotype, cultured mouse MPCs were incubated with serum from CKD or healthy patients. Uraemic serum reduced the percentage of proliferating cells expressing ki67 (Figure 5A) and increased the DNA damage indicator γ‐H2AX, as well as the senescence markers p21 and p16INK4a (Figure 5A). To test whether the reduction in MPC proliferation was due to a transition to senescence, p21 or p16INK4a were silenced using siRNA and the rate of cell proliferation was measured. Uraemic serum significantly decreased the proliferation rate of culture MPCs and silencing p16INK4a or p21 restored their ability to proliferate (Figure 5B). The silencing of p21 or p16INK4a by siRNA was confirmed by immunohistological analysis for these proteins in MPCs (Figure S3).
Figure 5.
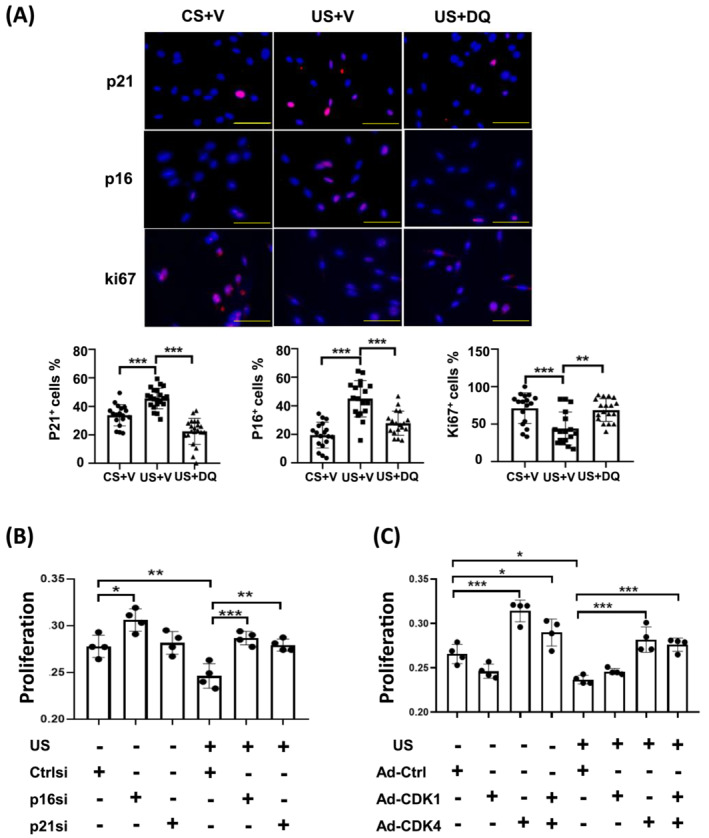
Senolytics suppress uraemic serum‐induced senescence in MPCs. (A) MPCs were isolated from the skeletal muscle of normal C57BL6 mice and cultured in 2% control (CS) or uraemic serum (US) for 48 h and the amount of γ‐H2A.X, p21, 16INK4a, and ki67 immunofluorescence was measured. DAPI staining was used to determine the number of nuclei (total cell population). The percentages of positive cells for each marker are reported in the bar/point graphs (scale bar = 50 μm). (B) MPCs were treated in 2% control or uraemic serum for 48 h. siRNAs corresponding to control (ctrlsi), p16 (p16si), or p21 (p21si) were transfected into the cells and cell proliferation was measured 48 h later. (C) MPCs were treated with 2% control or uraemic serum for 48 h, followed by transduction with ad‐ctrl, ad‐CDK1 or ad‐CDK4. After 48 h, cell proliferation was measured. Results are reported in the bar/point graph; data represent the absorbance in arbitrary units for each group. N = 6/group; bar: mean ± SE; *P < 0.05; **P < 0.01; ***P < 0.001.
In our in vivo studies, CDK1/2 and CDK4 were decreased in the muscle of uraemic mice (Figure 2B), and the senolytic cocktail reversed these changes. We next examined the role of cyclin‐dependent kinases on the CKD changes in MPC proliferation by overexpressing CKD1 or CKD4 in MPCs using adenovirus mediated gene transfer (Figure S1A,B). Overexpression of CDK1 did not significantly change the proliferation rate of MPCs; however, augmenting CDK4 sharply increased the proliferation rate of MPCs incubated with either control or uraemic serum (Figure 5C). The results indicate that the CKD induced reduction in MPC proliferation is mediated by serum factors that upregulate p21 and p16INK4a while simultaneously downregulating CDK4.
FOXO1 inhibits MPC proliferation via upregulating p21
The FOXO transcription factors have been linked to induction of p21 and senescence in non‐muscle cell types. 23 , 24 Consistent with earlier studies, FOXO1 and FOXO3a were increased in muscles of mice with CKD 3 and administering senolytics to CKD‐mice prevented induction of the FOXOs (Figure 6A). To explore the relationship between FOXO1/3a and cell proliferation, constitutively active FOXO1 or FOXO3a were overexpressed in MPCs. The number of p21+ cells was increased following overexpression of activated FoxO1 but not activated FOXO3a (Figures 6B and S2). As seen in earlier experiments, treating MPCs with uraemic serum increased the number of p21+ cells and co‐treatment with senolytics prevented the increase by CKD serum (Figure 6C). Inclusion of D&Q with uraemic serum also increased the number of ki67+ cells over MPCs treated with uraemic serum alone, suggesting the senolytic cocktail increased cell proliferation (Figure 6C2). Importantly, overexpression of activated FoxO1 blocked the responses to senolytics in MPCs treated with uraemic serum (Figure 6C) Moreover, knockdown of p21 by siRNA prevented the senescence‐inducing effect of FOXO1 as evidenced by maintenance of the number of ki67+ MPCs (Figure 6C). These findings suggest that CKD induces FoxO1, which then inhibits proliferation of MPCs by upregulating p21.
Figure 6.
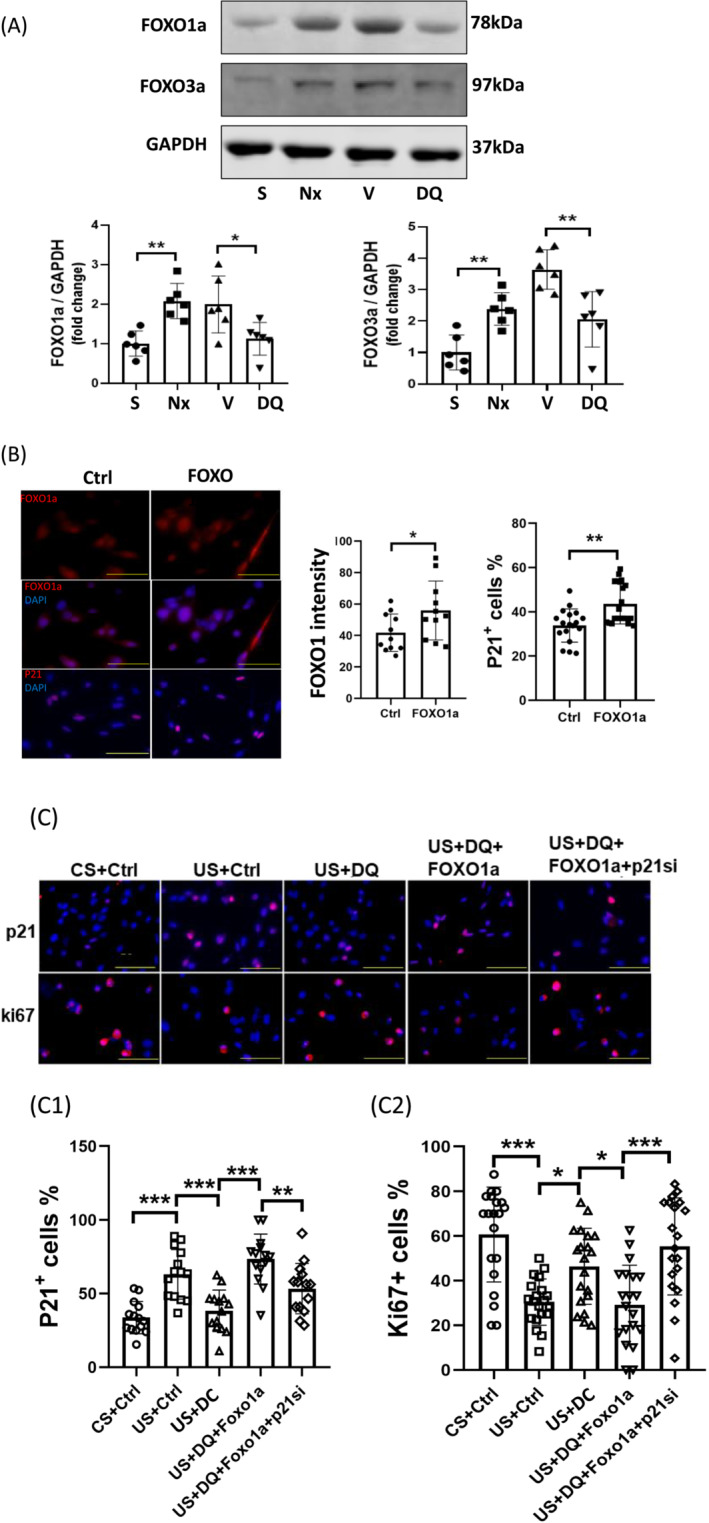
FOXO1 inhibited MPC proliferation via p21. (A) FOXO proteins in gastrocnemius muscle of sham, CKD (Nx), CKD + vehicle (V), and CKD + D&Q (DQ) mice were evaluated by immunoblot analysis. Results are reported in the bar/point graph as the fold change of FOXO proteins, normalized to GAPDH (n = 6/group; bar: mean ± SE; *P < 0.05; **P < 0.01; ***P < 0.001). (B) MPCs were transfected with a plasmid encoding FOXO1a and the levels of p21 immunofluorescence were measured. DAPI staining was used to measure the number of nuclei. FoxO1 intensity, the percentages of p21+ cells were counted, and the results are presented in the bar/point graph (bars: mean ± SE; scale bar = 50 μm; *P < 0.05; **P < 0.01; ***P < 0.001). (C) MPCs were cultured in 2% control or uraemic serum and transfected with FOXO1a plasmid and/or p21 siRNA. Some cells were also treated with D&Q. the number of p21+ or Ki67+ cells were measured. DAPI staining was used to determine the number of cell nuclei. The percentages of p21+ cells or ki67+ cells in the cell population were calculated and summarized in the bar/point graphs (bars: mean ± SE; scale bar = 50 μm; *P < 0.05; **P < 0.01; ***P < 0.001).
Senolytics induce apoptosis in senescent MPCs
An important characteristic of senescent cells is they are resistant to apoptosis. 25 To test whether senolytics treatment makes senescent cells more susceptible to apoptosis, MPCs were incubated with uraemic serum with or without D&Q and then cellular senescence markers (p16INK4a and p21) were measured and apoptosis assessed using the TUNEL method (Figure 7A,B). Uraemic serum alone slightly increased apoptosis as evidenced by the elevated the number of TUNEL+ cells in MPCs; the change was not statistically significant. However, senolytics significantly increased apoptosis in MPCs that expressed p16INK4a (Figure 7A) and p21 (Figure 7B). These results indicate that senolytics only increase apoptosis in senescent MPCs.
Figure 7.
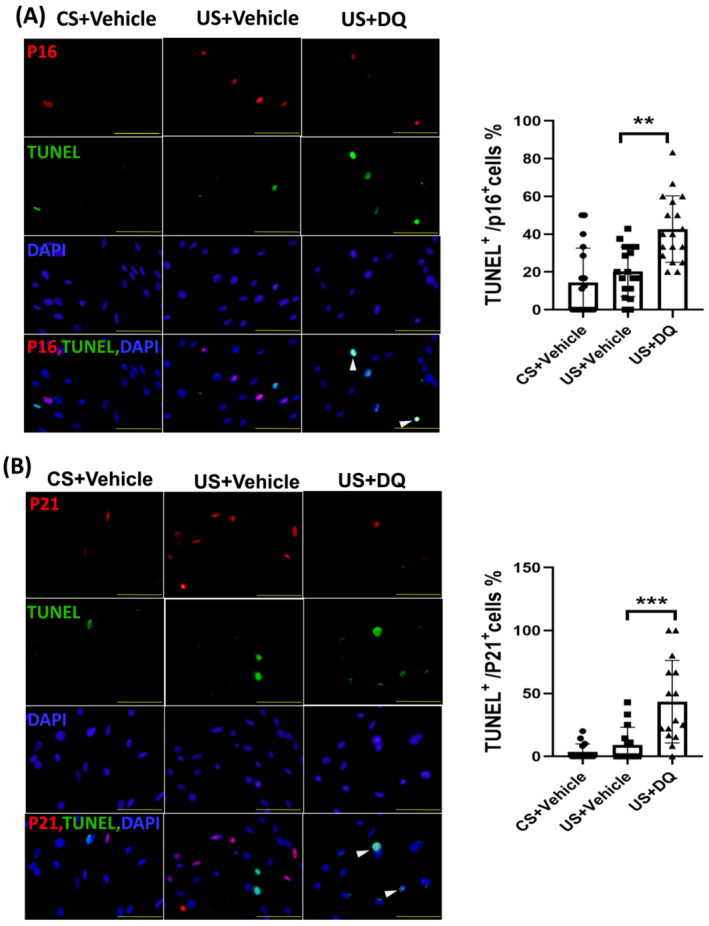
Senolytics induced apoptosis in senescent MPCs. MPCs were cultured in 2% control or uraemic serum and treated with vehicle or D&Q for 48 h. (A) MPCs were evaluated for apoptosis by TUNEL and for p16INK4a by immunostaining; DAPI staining was used to determine the number of nuclei. TUNEL and p16INK4a positive cells are indicated by white arrows. The percentages of TUNEL positive cells that were also p16INK4a positive cells were calculated and summarized in the bar/point graph (bars: mean ± SE; scale bar = 50 μm.; *P < 0.05; **P < 0.01; ***P < 0.001). (B) MPCs were evaluated for apoptosis by TUNEL and for p21 by immunostaining; DAPI staining was used to determine the number of nuclei. Both TUNEL and p21+ cells were indicated by white arrows. The percentages of TUNEL+ cells that were also p21+ cells were calculated and summarized in the bar/point graph (bars: mean ± SE; scale bar = 50 μm.; *P < 0.05; **P < 0.01; ***P < 0.001).
Discussion
Our investigation provides evidence that experimental CKD leads to MPC senescence in the muscles of mice. In mice with CKD for 8 weeks, elevated levels of the SA‐β‐gal, γ‐H2AX which is a marker for DNA damage, and p16INK4a and p21 which are senescence signalling proteins, as well as cytokine production are present in muscle; all are associated with a senescence‐associated secretory phenotype. Furthermore, we found that a cocktail of senolytics reduced the levels of senescent MPCs, improved MPC proliferation and markers of myogenesis, and attenuated muscle wasting induced by CKD. These positive actions of the senolytics were linked to a suppression of FOXO1, p21, and p16INK4a (Figure 8).
Figure 8.
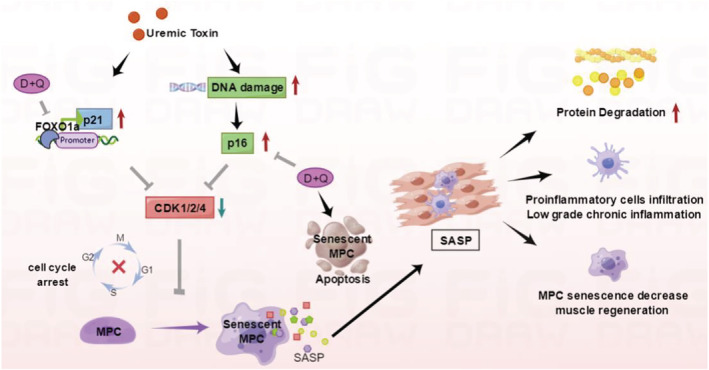
Mechanisms responsible for MPC senescence in CKD. CKD induces DNA damage in MPCs and activates the FOXO1 transcription factor. DNA damage increases p16INK4a whereas the activation of FOXO1 transcription factor increases p21. Both p16INK4a and p21 are cyclin‐dependent kinase (CDKs) inhibitors, which prevent cell cycle progression and lead to cell senescence. MPC senescence prevents muscle progenitor proliferation and induces the SASP in MPCs which leads to their production of inflammatory mediators and subsequent muscle wasting. Administration of dasatinib plus quercetin (D&Q) reduces CKD‐induced muscle atrophy by downregulation of FoxO1 and P16INK4a as well as inducing apoptosis of senescent cells. This reduction of senescent cells limits inflammation and attenuates muscle wasting and dysfunction.
In general, there are two causes of cellular senescence. Telomerase dysfunction leads to telomere erosion that is linked to proliferative exhaustion. 26 This process is called replicative senescence and is associated with the aging process. In contrast, stress‐induced premature senescence is caused by stress stimuli in the absence of telomere shortening and the process is reversible. Chronic exposure to oxidative stress, hyperglycaemia, or radiation are examples of signals that can cause stress‐induced premature senescence. 27 Stressors that induce DNA damage can cause cell cycle arrest and loss of its proliferative capacity, 28 leading to replicative or stress‐induced cellular senescence. 28 , 29 The fact that knockdown of the p21 or p16INK4a in uraemic serum‐treated MPCs led to increased expression of the proliferation marker ki67 indicates that CKD causes stress‐induced premature senescence.
The data from this study support a model whereby CKD activates two senescence‐inducing pathways (Figure 7). The first pathway involves DNA damage and increased amounts of γ‐H2A.X and p16INK4a. 28 The second pathway involves a FoxO1/p21 axis. In general, p53 is a major transcriptional regulator of p21 but in our study, p53 was not increased in the muscles of CKD mice. p53‐independent p21 expression has been described during cell‐cycle arrest 30 and others have reported that expression of p21 is regulated by non‐coding RNA, 31 the GSK3β/β‐catenin axis, 32 the p62‐Skp2 axis 33 and PI3K‐FOXO1/3a signalling. 34 Both P21 and p16INK4a are cyclin‐dependent kinase inhibitors which can block the G1/S and/or G2/M transition and impede cell cycle progression. 35
The regulation of p21 by FOXOs was notable because we and others have reported that FOXO1/3 in muscle are upregulated by CKD and are linked to myoblast regeneration. 17 , 24 , 36 In the current study, overexpression of FOXO1 in MPCs resulted in increased p21 expression and reduced proliferation. Furthermore, administration of senolytics to mice with CKD resulted in downregulation of FOXO1 and improved MPCs proliferation plus the levels of Pax7 and myogenin. An interesting finding in this study was that overexpression of FoxO1, but not FoxO3, induced p21 in MPCs. This selectivity of FOXO1 is consistent with our earlier reports that FOXO1 is the dominant mediator of muscle wasting in CKD. 17 , 37
Senescent MPCs have two properties that likely contribute to frequent characterization of CKD as a chronic inflammatory state. First, senescent cells are resistant to apoptosis and therefore persist is the senescence state. 38 Second, they develop a SASP that features persistent cytokine production. 12 Initially, cytokine production is beneficial to promote tissue repair but because the cells remain frozen in their cell cycle, they keep generating high levels of cytokines which produce a local and systemic chronic inflammatory state that causes tissue damage, can lead to muscle wasting, and increase the risks of all‐cause mortality. 4 , 35 In this study, we took advantage of senolytic agents that selectively target senescent cells. Dasatinib is an inhibitor of multiple tyrosine kinases 39 and quercetin is a plant pigment (flavonoid); together, they induce apoptosis in senescent cells, but not in proliferating or quiescent cells. 35 , 40 Treatment of CKD mice and isolated MPCs with D&Q reduced the expression of SA‐β‐gal, p16INK4a, and the inflammatory cytokines—TNF‐α, IL‐6, and IL‐1β—while restoring the proliferative capability of MPCs. These actions coincided with improvements in the muscle fibre size, grip strength and a reduction in cachexia‐related atrogenes. Others have similarly reported that D&Q administration to irradiated mice reduced p16INK4a expression and SA‐β‐gal positive cells plus increased exercise endurance and total work capacity. 41
Another interesting observation in our study was the improvement in renal function seen in CKD mice treated with senolytics. D&Q lowered BUN and serum creatinine while increasing the size of some muscles and overall muscle function. Inflammation is a key event in the progression of renal dysfunction and the anti‐inflammatory effects of senolytic agents may help improve kidney function. 3 Recent evidence suggests that cellular senescence plays an important role in the pathogenesis of different forms of renal damage, including acute and chronic kidney disease, and renal transplantation. 42 The systemic nature of D&Q treatment suggests that senescent cells will be eliminated in multiple tissues including kidney. Others recently reported that reductions in kidney function were associated with increased renal production and release of activin A and other pro‐cachectic factors which acted on skeletal muscle to induce proteolytic pathways and muscle wasting. 43 Given the latter findings, any improvement in renal function due to a reduction in renal cell senescence could contribute to the improvements in muscle size and function.
In summary, our study links cellular senescence to uraemia‐induced muscle atrophy and persistence of low‐grade systemic inflammation (Figure 8). The induction of MPC senescence in CKD results from activation of FOXO1/p21 and DNA damage induced upregulation of p16INK4a. These actions inhibit cyclin‐dependent kinases and cell cycle progression (1–4). MPC senescence limits muscle progenitor proliferation and increases inflammation via the SASP, leading to muscle wasting. Administration of a senolytic cocktail attenuates CKD‐induced muscle loss by reducing the production of inflammatory mediators, and by improving the proliferative capability of MPCs. The treatment also reduces activation of FOXO1 in myofibres which mediates other atrophy‐related changes in metabolism such as increased muscle protein degradation. The findings suggest that senolytics might be effective agents in the treatment of muscle atrophy and weakness due to CKD.
Conflicts of interest
The authors declare no conflicts of interest.
Supporting information
Figure S1. Uremic serum (US) decreased the amount of cyclindependent kinase (CDK). MPCs were transduced with Ad‐ctrl, Ad‐CDK1 (A) or Ad‐CDK4 (B) and treated with either control (CS) or uremic serum (US) for 48 hours. Immunofluorescence staining showed that CDK1/2 + and CDK4 + cells were increased after adenovirus transduction. DAPI staining was used to determine the number of nuclei (total cell population). The percent of CDK1 + (A) or CDK4 + (B) cells in total cell population were calculated and summarized in bar/point graph. (Bars: mean ± s.e.; scale bar = 50 μm; n = 6/group; * = p < 0.05; ** = p < 0.01; *** = p < 0.001).
Figure S2. p21 was not regulated by FOXO3a in MPCs. MPCs were transduced with constitutive activation Ad‐(CA)‐FOXO3a‐GFP or Ad‐GFP adenovirus for control. (A) The shape of adenovirus transduced MPCs were shown (scale bar = 100 μm). (B) p21 positive cells were measured by immunofluorescence. The percent of p21 positive cells (red) in total cell population were calculated and summarized in bar/point graph. (Bars: mean ± s.e.; scale bar = 50 μm; n = 6/group).
Figure S3. confirmation of knockdown by the siRNAs in uremic serum treated MPCs
MPCs were treated in 2% control or uremic serum for 48 hours. The siRNA of control (NC), p16 (p16si) or p21 (p21si) were transfected into the cells. Immunofluorescence staining showed that p16 and p21 positive cells were decreased in uremic treated group. DAPI staining was used to determine the number of nuclei (total cell population).
Table S1. Supporting Information
Table S2. Antibody list
Table S3. Primer list
Table S4. The effect of D&Q treatments on body or muscle weights, BUN, creatinine, and grip strength
Acknowledgements
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) of the National Institutes of Health under Award Number R01 AR060268 to XHW; by the Department of Veteran Affairs Merit Award 5I01BX000994 to HC; by the NIH National Heart, Lung, and Blood Institute (R01HL135183 and R61AT010457) and the Department of Veteran Affairs Merit Award (I01CX001065) to JP; by the National Natural Science Foundation of China (81973385 and 81773801) to XZ. This work was supported by grants from Shanghai Science and Technology Innovation (No. 20410713300). The content is solely the responsibility of the authors and does not necessarily reflect the official views of the NIH, the Department of Veterans Affairs, or the US Government. The authors gratefully acknowledge Dr Mohamed in Institute of Molecular Cardiology of University of Louisville for providing adenovirus Ad‐CDK1/4, CCNB, and CCND. The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. 44
Huang Y., Wang B., Hassounah F., Price S. R., Klein J., Mohamed T. M. A., Wang Y., Park J., Cai H., Zhang X., and Wang X. H. (2023) The impact of senescence on muscle wasting in chronic kidney disease, Journal of Cachexia, Sarcopenia and Muscle, 14, 126–141, 10.1002/jcsm.13112
Ying Huang and Bin Wang contributed equally to this work.
Contributor Information
Xuemei Zhang, Email: xuemzhang@fudan.edu.cn.
Xiaonan H. Wang, Email: xwang03@emory.edu.
References
- 1. Cockwell P, Fisher LA. The global burden of chronic kidney disease. Lancet 2020;395:662–664. [DOI] [PubMed] [Google Scholar]
- 2. Wang XH, Mitch WE. Mechanisms of muscle wasting in chronic kidney disease. Nat Rev Nephrol 2014;10:504–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang XH, Mitch WE, Price SR. Pathophysiological mechanisms leading to muscle loss in chronic kidney disease. Nature reviews. Nephrol Ther 2021;18:138–152. [DOI] [PubMed] [Google Scholar]
- 4. Mihai S, Codrici E, Popescu ID, Enciu AM, Albulescu L, Necula LG, et al. Inflammation‐Related Mechanisms in Chronic Kidney Disease Prediction, Progression, and Outcome. J Immunol Res 2018;–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang C, Li Y, Wu Y, Wang L, Wang X, Du J. Interleukin‐6/signal transducer and activator of transcription 3 (STAT3) pathway is essential for macrophage infiltration and myoblast proliferation during muscle regeneration. J Biol Chem 2013;288:1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang L, Wang XH, Wang H, Du J, Mitch WE. Satellite cell dysfunction and impaired IGF‐1 signaling cause CKD‐induced muscle atrophy. J Am Soc Nephrol 2010;21:419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deger SM, Hung AM, Gamboa JL, Siew ED, Ellis CD, Booker C, et al. Systemic inflammation is associated with exaggerated skeletal muscle protein catabolism in maintenance hemodialysis patients. JCI. Insight 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang L, Du J, Hu Z, Han G, Delafontaine P, Garcia G, et al. IL‐6 and serum amyloid A synergy mediates angiotensin II‐induced muscle wasting. J Am Soc Nephrol 2009;20:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular Senescence: Defining a Path Forward. Cell 2019;179:813–827. [DOI] [PubMed] [Google Scholar]
- 10. Dai L, Qureshi AR, Witasp A, Lindholm B, Stenvinkel P. Early Vascular Ageing and Cellular Senescence in Chronic Kidney Disease. Comput Struct Biotechnol J 2019;17:721–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest 2018;128:1238–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lopes‐Paciencia S, Saint‐Germain E, Rowell MC, Ruiz AF, Kalegari P, Ferbeyre G. The senescence‐associated secretory phenotype and its regulation. Cytokine 2019;117:15–22. [DOI] [PubMed] [Google Scholar]
- 13. Docherty MH, O'Sullivan ED, Bonventre JV, Ferenbach DA. Cellular Senescence in the Kidney. J Am Soc Nephrol 2019;30:726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schmitt R, Melk A. Molecular mechanisms of renal aging. Kidney Int 2017;92:569–579. [DOI] [PubMed] [Google Scholar]
- 15. Chiche A, Le Roux I, von Joest M, Sakai H, Aguin SB, Cazin C, et al. Injury‐Induced Senescence Enables In Vivo Reprogramming in Skeletal Muscle. Cell Stem Cell 2017;20:407–14.e4. [DOI] [PubMed] [Google Scholar]
- 16. Sugihara H, Teramoto N, Yamanouchi K, Matsuwaki T, Nishihara M. Oxidative stress‐mediated senescence in mesenchymal progenitor cells causes the loss of their fibro/adipogenic potential and abrogates myoblast fusion. Aging (Albany NY) 2018;10:747–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang B, Zhang C, Zhang A, Cai H, Price SR, Wang XH. MicroRNA‐23a and MicroRNA‐27a Mimic Exercise by Ameliorating CKD‐Induced Muscle Atrophy. J Am Soc Nephrol 2017;28:2631–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med 2018;24:1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mohamed TMA, Ang YS, Radzinsky E, Zhou P, Huang Y, Elfenbein A, et al. Regulation of Cell Cycle to Stimulate Adult Cardiomyocyte Proliferation and Cardiac Regeneration. Cell 2018;173:104–16.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Frescas D, Valenti L, Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt‐dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem 2005;280:20589–20595. [DOI] [PubMed] [Google Scholar]
- 21. Zhang A, Li M, Wang B, Klein JD, Price SR, Wang XH. miRNA‐23a/27a attenuates muscle atrophy and renal fibrosis through muscle‐kidney crosstalk. J Cachexia Sarcopenia Muscle 2018;9:755–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 2017;8:14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Furukawa‐Hibi Y, Kobayashi Y, Chen C, Motoyama N. FOXO transcription factors in cell‐cycle regulation and the response to oxidative stress. Antioxid Redox Signal 2005;7:752–760. [DOI] [PubMed] [Google Scholar]
- 24. Shi X, Wallis AM, Gerard RD, Voelker KA, Grange RW, DePinho RA, et al. Foxk1 promotes cell proliferation and represses myogenic differentiation by regulating Foxo4 and Mef2. J Cell Sci 2012;125:5329–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017;169:132–47 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Masutomi K, Yu EY, Khurts S, Ben‐Porath I, Currier JL, Metz GB, et al. Telomerase maintains telomere structure in normal human cells. Cell 2003;114:241–253. [DOI] [PubMed] [Google Scholar]
- 27. Shakeri H, Lemmens K, Gevaert AB, De Meyer GRY, Segers VFM. Cellular senescence links aging and diabetes in cardiovascular disease. Am J Physiol Heart Circ Physiol 2018;315:H448–H462. [DOI] [PubMed] [Google Scholar]
- 28. Robles SJ, Adami GR. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene 1998;16:1113–1123. [DOI] [PubMed] [Google Scholar]
- 29. d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA‐damage response. Nat Rev Cancer 2008;8:512–522. [DOI] [PubMed] [Google Scholar]
- 30. Sato T, Koseki T, Yamato K, Saiki K, Konishi K, Yoshikawa M, et al. p53‐independent expression of p21(CIP1/WAF1) in plasmacytic cells during G(2) cell cycle arrest induced by Actinobacillus actinomycetemcomitans cytolethal distending toxin. Infect Immun 2002;70:528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xu CL, Sang B, Liu GZ, Li JM, Zhang XD, Liu LX, et al. SENEBLOC, a long non‐coding RNA suppresses senescence via p53‐dependent and independent mechanisms. Nucleic Acids Res 2020;48:3089–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ruan B, Liu W, Chen P, Cui R, Li Y, Ji M, et al. NVP‐BEZ235 inhibits thyroid cancer growth by p53‐ dependent/independent p21 upregulation. Int J Biol Sci 2020;16:682–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jung D, Khurana A, Roy D, Kalogera E, Bakkum‐Gamez J, Chien J, et al. Quinacrine upregulates p21/p27 independent of p53 through autophagy‐mediated downregulation of p62‐Skp2 axis in ovarian cancer. Sci Rep 2018;8:2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Muñoz‐Espín D, Cañamero M, Maraver A, Gómez‐López G, Contreras J, Murillo‐Cuesta S, et al. Programmed cell senescence during mammalian embryonic development. Cell 2013;155:1104–1118. [DOI] [PubMed] [Google Scholar]
- 35. Kirkland JL, Tchkonia T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017;21:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luo J, Liang A, Liang M, Xia R, Rizvi Y, Wang Y, et al. Serum Glucocorticoid‐Regulated Kinase 1 Blocks CKD‐Induced Muscle Wasting Via Inactivation of FoxO3a and Smad2/3. J Am Soc Nephrol 2016;27:2797–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu J, Li R, Workeneh B, Dong Y, Wang X, Hu Z. Transcription factor FoxO1, the dominant mediator of muscle wasting in chronic kidney disease, is inhibited by microRNA‐486. Kidney Int 2012;82:401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res 1995;55:2284–2292. [PubMed] [Google Scholar]
- 39. Montero JC, Seoane S, Ocana A, Pandiella A. Inhibition of SRC family kinases and receptor tyrosine kinases by dasatinib: possible combinations in solid tumors. Clin Cancer Res 2011;17:5546–5552. [DOI] [PubMed] [Google Scholar]
- 40. Kirkland JL, Tchkonia T. Senolytic drugs: from discovery to translation. J Intern Med 2020;288:518–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 2015;14:644–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li Y, Lerman LO. Cellular Senescence: A New Player in Kidney Injury. Hypertension 2020;76:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Solagna F, Tezze C, Lindenmeyer MT, Lu S, Wu G, Liu S, et al. Pro‐cachectic factors link experimental and human chronic kidney disease to skeletal muscle wasting programs. J Clin Invest 2021;131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2021. J Cachexia Sarcopenia Muscle 2021;12:2259–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Uremic serum (US) decreased the amount of cyclindependent kinase (CDK). MPCs were transduced with Ad‐ctrl, Ad‐CDK1 (A) or Ad‐CDK4 (B) and treated with either control (CS) or uremic serum (US) for 48 hours. Immunofluorescence staining showed that CDK1/2 + and CDK4 + cells were increased after adenovirus transduction. DAPI staining was used to determine the number of nuclei (total cell population). The percent of CDK1 + (A) or CDK4 + (B) cells in total cell population were calculated and summarized in bar/point graph. (Bars: mean ± s.e.; scale bar = 50 μm; n = 6/group; * = p < 0.05; ** = p < 0.01; *** = p < 0.001).
Figure S2. p21 was not regulated by FOXO3a in MPCs. MPCs were transduced with constitutive activation Ad‐(CA)‐FOXO3a‐GFP or Ad‐GFP adenovirus for control. (A) The shape of adenovirus transduced MPCs were shown (scale bar = 100 μm). (B) p21 positive cells were measured by immunofluorescence. The percent of p21 positive cells (red) in total cell population were calculated and summarized in bar/point graph. (Bars: mean ± s.e.; scale bar = 50 μm; n = 6/group).
Figure S3. confirmation of knockdown by the siRNAs in uremic serum treated MPCs
MPCs were treated in 2% control or uremic serum for 48 hours. The siRNA of control (NC), p16 (p16si) or p21 (p21si) were transfected into the cells. Immunofluorescence staining showed that p16 and p21 positive cells were decreased in uremic treated group. DAPI staining was used to determine the number of nuclei (total cell population).
Table S1. Supporting Information
Table S2. Antibody list
Table S3. Primer list
Table S4. The effect of D&Q treatments on body or muscle weights, BUN, creatinine, and grip strength