

Abstract
Background:
Crosstalk between sterol metabolism and inflammatory pathways has been demonstrated to significantly impact the development of atherosclerosis. Cholesterol biosynthetic intermediates and derivatives are increasingly recognized as key immune regulators of macrophages in response to innate immune activation and lipid overloading. 25-hydroxycholesterol (25-HC) is produced as an oxidation product of cholesterol by the enzyme cholesterol 25-hydroxylase (CH25H) and belongs to a family of bioactive cholesterol derivatives produced by cells in response to fluctuating cholesterol levels and immune activation. Despite the major role of 25-HC as a mediator of innate and adaptive immune responses, its contribution during the progression of atherosclerosis remains unclear.
Methods:
The levels of 25-HC were analyzed by liquid chromatography-mass spectrometry, and the expression of CH25H in different macrophage populations of human or mouse atherosclerotic plaques, respectively. The effect of CH25H on atherosclerosis progression was analyzed by bone marrow (BM) adoptive transfer of cells from WT or Ch25h−/− mice to lethally irradiated Ldlr−/− mice, followed by a Western diet (WD) feeding for 12 weeks. Lipidomic, transcriptomic analysis and effects on macrophage function and signaling were analyzed in vitro from lipid-loaded macrophage isolated from Ldlr−/− or Ch25h−/−;Ldlr−/− mice. The contribution of secreted 25-HC to fibrous cap formation was analyzed using a smooth muscle cell (SMC) lineage tracing mouse model, Myh11ERT2CREmT/mG;Ldlr −/−, adoptively transferred with WT or Ch25h−/− mice BM followed by 12 weeks of WD feeding.
Results:
We found that 25-HC accumulated in human coronary atherosclerotic lesions and that macrophage-derived 25-HC accelerated atherosclerosis progression promoting plaque instability via autocrine and paracrine actions. 25-HC amplified the inflammatory response of lipid-loaded macrophages and inhibited the migration of SMCs within the plaque. 25-HC intensified inflammatory responses of lipid-laden macrophages by modifying the pool of accessible cholesterol in the plasma membrane, which altered Toll-like receptor 4 (TLR4) signaling, promoted nuclear factor-κB (NFκB)-mediated pro-inflammatory gene expression, and increased apoptosis susceptibility. Interestingly, these effects were independent of 25-HC-mediated modulation of liver X receptor (LXR) or sterol regulatory-element binding protein (SREBP) transcriptional activity.
Conclusions:
Production of 25-HC by activated macrophages amplifies their inflammatory phenotype, thus promoting atherogenesis.
Keywords: Atherosclerosis, macrophages, inflammation, 25-hydroxycholesterol
INTRODUCTION
Atherosclerosis is a metabolic and inflammatory disease, characterized by the accumulation of cholesterol-rich lipoproteins and inflammatory cells in the artery wall. Retention of native low-density lipoproteins (LDL) in the intima, favors the formation of oxidized LDLs (Ox-LDL) and other modified lipoproteins, rich in bioactive lipids, that trigger inflammation, leading to subendothelial leukocyte recruitment1–3. Recruited monocytes differentiate into macrophages that proliferate4 and acquire an inflammatory phenotype5,6. The inflammatory mediators released by macrophages further activate vascular endothelium, to further promote monocyte recruitment, thus perpetuating a chronic inflammatory state within the artery7. Once within the vessel wall, macrophages start scavenging Ox-LDL via surface scavenger receptors8,9. Accumulation of cholesterol and other LDL-derived lipids gives rise to lipid-loaded macrophages or foam cells if they are completely filled with lipid droplets10. Lipid accumulation may have a beneficial impact11; however, in later stages foamy macrophages can be more susceptible to death, promoting a proinflammatory microenvironment. In advanced atherosclerosis, certain lesions evolve into a vulnerable/unstable plaque characterized by necrotic areas rich in dying foam cells, low collagen content, a thin fibrous cap, and non-resolving inflammation12 that can lead to plaque disruption and acute thrombotic events.
Intracellular cholesterol content is tightly regulated by feedback mechanisms that involve two families of transcription factors, the sterol regulatory element binding proteins (SREBPs) and the liver X receptors (LXRs)13,14. SREBP2 drives the transcription of genes involved in cholesterol synthesis and uptake, e.g., hydroxy methyl glutaryl-coenzyme A (HMG-CoA) reductase (HMGCR) and LDL receptor (LDLR), while LXR upregulates cholesterol efflux genes, including ATP-binding cassette A1 (ABCA1)13,14. Besides controlling cholesterol homeostasis, SREBPs and LXRs regulate inflammation to establish crosstalk with cholesterol metabolism15–21. In addition to cholesterol, other sterol intermediates of cholesterol biosynthesis22 and cholesterol derivatives (oxysterols), such as 25-hydroxycholesterol (25-HC)23,24 accumulate within the plaque. 25-HC is an oxidation product of cholesterol generated by 25-hydroxylase (CH25H)25, and reduces cholesterol biosynthesis25 via inactivation of SREBP226,27, stimulates cholesterol efflux through agonism of LXR13,28–30, and enhances cholesterol esterification by activating Acyl-CoA:cholesterol acyltransferase31. However, mice deficient in Ch25h have intact cholesterol metabolism32, challenging the physiological role of 25-HC as a regulator of cholesterol homeostasis33–35. On the other hand, Ch25h expression is transcriptionally-induced, in macrophages, in response to various inflammatory mediators32,35–38, thus increasing 25-HC synthesis and participating in immune-related functions. Ch25h is an interferon (IFN)-stimulated gene that shows antiviral activities against a range of enveloped viruses37–39. These antiviral effects have been linked to the ability of 25-HC to inhibit SREBP237,38, to activate LXRs40, and to reduce cholesterol biosynthesis37,38. Other investigations have determined that 25-HC regulates immunoglobulin A production32, augments the production of some inflammatory cytokines41,42, and mediates feedback inhibition of interleukin-1 (IL-1) family cytokine production43 by mediating a type I IFN inhibitory effect on inflammasome activation through repression of SREBP2 activation and concomitant decrease of the cholesterol biosynthetic pathway44.
Although numerous studies have highlighted the role of 25-HC as a mediator of inflammation-metabolism crosstalk in macrophages, the role of 25-HC in atherosclerosis remains unclear45,46. While one study showed that decreased 25-HC via ATF3-mediated repression of Ch25h expression, protected against foam cell formation and atherosclerosis46, another concluded that global Ch25h deficiency accelerated atherosclerosis45. Here we report that 25-HC accumulates in human coronary atherosclerotic lesions and that CH25H is expressed in pro-inflammatory macrophages that populate the plaque. We further show that hematopoietic deficiency of Ch25h attenuates the progression of atherosclerosis and promotes lesion stability characterized by reduced plaque necrosis and thicker fibrous cap. Notably, RNA-sequencing (RNA-seq) analysis of lipid-loaded elicited macrophages revealed that Ch25h levels, and hence 25-HC, amplifies the inflammatory response of macrophages, increasing the expression of pro-inflammatory genes independently of SREBP2 or LXR-mediated gene transcription. Instead, our findings indicate that these effects were mediated by changes in the pool of accessible cholesterol in the plasma membrane (PM). Furthermore, our data demonstrate that 25-HC did not restrain cholesterol biosynthesis in activated lipid-laden macrophages and did not alter inflammasome activation. Additionally, we demonstrate that activated lipid-laden macrophages release 25-HC into the extracellular media mediating a paracrine effect on SMCs, reducing their migratory phenotype. Altogether, our findings indicate that CH25H-mediated production of 25-HC by activated macrophages amplifies their inflammatory phenotype, thus promoting atherogenesis.
METHODS
Detailed methods are provided in the Supplemental Material. The authors declare that all supporting data are available within the article [and its online supplementary files]. Bulk RNA-seq data are accessible in NCBI Gene Expression Omnibus and through GEO Series accession number GSE189079. Data sets from previously published work analyzed in the present study are described in the Supplemental Methods. All animal studies have been approved by the Institutional Animal Care Use Committee of Yale University School of Medicine and conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Human samples used in this study are fully described in Supplemental Methods. Briefly, human left main coronary arteries were obtained from the explanted hearts of transplant recipients or cadaver organ donors as described previously47, and approved by the Institutional Review Boards of Yale University and the New England Organ Bank and were used for liquid chromatography–mass spectrometry (LC/MS-MS) and immunohistochemistry analysis.
Statistical analysis
The number of animals used in each study is listed in the figure legends. In vitro experiments were routinely repeated at least three times unless otherwise noted. Analysts were blinded to experimental groups. Data are expressed as average ± SD or ± SEM. Statistical differences were measured using an unpaired two-sided Student’s t-test, one-way ANOVA and two-way ANOVA with Bonferroni correction for multiple comparisons. Normality was checked using the Kolmogorov-Smirnov test. A nonparametric test (Mann-Whitney) was used when data did not pass the normality test or sample size was small. A value of P ≤ 0.05 was considered statistically significant. Data analysis was performed using GraphPad Prism Software Version 7 (GraphPad, San Diego, CA). For Bulk RNA-seq, the Differential Gene Expression-GSA algorithm was implemented. A default P-value < 0.05 was considered statistically significant with a fold-change > 1.5 for up-regulated transcripts or < −1.5 for down-regulated transcripts.
RESULTS
Human and mouse pro-inflammatory atherosclerotic plaque macrophages exhibit increased CH25H mRNA levels
We have previously described that 25-HC is present in atherosclerotic mouse aortas24. To understand the relevance of 25-HC in atherosclerosis, we analyzed the levels of 25-HC in human coronary arteries with mild, moderate, and severe atherosclerotic plaques (Figure 1A) by LC/MS-MS (Figure 1B). We found that plaques with more severe atherosclerosis presented higher levels of 25-HC (Figure 1B). Interestingly, this finding is in line with higher macrophage content (CD68+ cells) in these plaques (Figure 1A), suggesting that 25-HC levels are related to the accumulation of macrophages within the plaque.
Figure 1. 25-hydroxycholesterol accumulates in human coronary atherosclerotic plaque and CH25H is highly express in human and mouse inflammatory plaque macrophages.
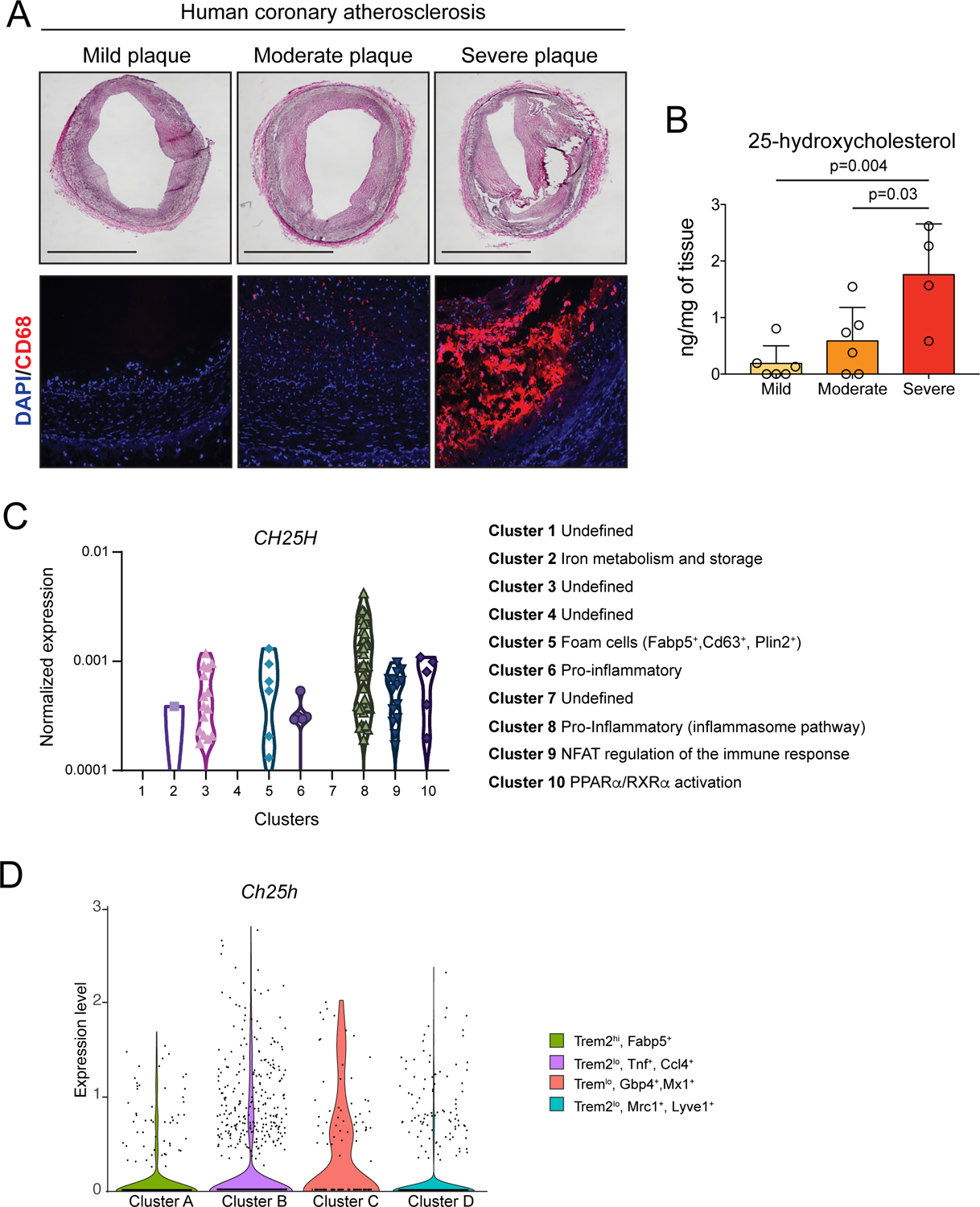
A, Representative hematoxylin, and eosin (H&E) staining (upper panels) and CD68 (macrophage marker) immunofluorescence (lower panels) of atherosclerotic plaques isolated from human coronary arteries. Scale bar: 1 mm. B, 25-hydroxycholesterol quantification in atherosclerotic plaques isolated from human coronary arteries. Data were analyzed by one-way ANOVA, post-hoc Bonferroni’s multiple comparison test (n= 6 mild plaques, 6 moderate plaques and 4 severe plaques). C, Violin plots showing CH25H expression among different macrophages clusters presents in human plaque tissue48. D, Violin plot showing the expression of Ch25h withing the indicated macrophage clusters from leukocyte isolated from Ldlr−/− aortic tissue10 of mice fed a WD for 12 weeks.
Next, we analyzed the expression of CH25H in macrophages from human48 (Figure 1C) and mouse (Figure 1D and Figures S1) atherosclerotic plaques10,49. Single cell RNA-seq (scRNA-seq) revealed that macrophages from human atherosclerotic plaques with a pro-inflammatory gene signature exhibited more cells with high CH25H mRNA levels (Figure 1C). ScRNA-seq analysis from total leukocytes obtained from Ldlr−/− aortas10 showed that inflammatory clusters of aortic macrophages, Clusters B (Trem2lo, Tnf+) and C (Trem2lo, Gbp4+,Mx1+), exhibited more cells with high levels of Ch25h, whereas in non-inflammatory clusters, Clusters A (Trem2hi, Fabp5+) or D (Trem2lo, Mrc1+, Lyve1+) exhibited fewer cells expressing high levels of Ch25h (Figure 1D and Figure S1A–C). Similar results were obtained by analyzing a different scRNA-seq data set from cells obtained from Ldlr−/− aortas49 with equivalent macrophage clusters (Figure S1G–J). Furthermore, bulk RNA-seq analysis of intimal macrophages isolated from Apolipoprotein E (Apoe) deficient murine aortas10 (Figure S1D), manifested that foamy (BODIPYhi, Side Scatter/SSChi) non-inflammatory macrophages exhibited diminished Ch25h expression when compared to inflammatory macrophages with a broad range of lipid-loading but with no evident signs of lipid droplet accumulation (BODIPYlo,SSClo).
Collectively, these results indicate that 25-HC accumulates in the artery wall during atherogenesis and that CH25H is expressed at higher levels in both human and mouse pro-inflammatory plaque macrophages, suggesting that 25-HC is associated with inflammation and atherosclerosis.
Hematopoietic Ch25h deficiency diminishes the progression of atherosclerosis
To determine the in vivo function of Ch25h and, hence 25-HC, during the progression of atherosclerosis, we transplanted bone marrow (BM) cells from WT or Ch25h−/− mice to lethally irradiated Ldlr−/− mice (hence forth referred to as WT→Ldlr−/− and Ch25h−/−→Ldlr−/− mice), followed by a WD feeding for 12 weeks. We determined the levels of 25-HC in lipid extracts from Ch25h−/−→Ldlr−/− mouse aortas (Figure S2A). 25-HC was undetectable, whereas other oxysterols (e.g., 27-HC or 24-HC) and sterols such as desmosterol were detectable. Interestingly, 27-HC, an oxysterol produced from cholesterol by cytochrome P450 family 27 subfamily A (Cyp27a), which is almost specifically expressed in macrophages (Figure S1E and F, see also S1B and H), was efficiently detected in the aortas of Ch25h−/−→Ldlr−/− mice (Figure S2A). Thus, in atherosclerotic murine aortas, 25-HC is mostly produced by hematopoietic cells and the contribution of other cell types from the recipient Ldlr−/− mice to the production of 25-HC is negligible. These results are in line with almost exclusive expression of Ch25h in macrophages when compared to other cell types present in mouse aortas (Figure S1F).
Histological analysis of the aortic root revealed that Ch25h−/−→Ldlr−/− mice developed smaller lesions than WT→Ldlr−/− mice (Figure 2A). Although oil red O (ORO) staining in the aortic root did not show differences in the accumulation of neutral lipids (Figure S2B), en face analysis of the whole aorta showed reduced lipid accumulation in Ch25h−/−→Ldlr−/− mice (Figure 2B). Additional morphological analysis of the aortic roots revealed that Ch25h−/−→Ldlr−/− mice had smaller necrotic cores (Figure 2C) and increased collagen content (Figure 2D), indicating more stable atherosclerotic lesions. All these effects were observed with no statistical differences in body weight, plasma lipid levels, or circulating leukocytes (Figure S3A–H).
Figure 2. Ch25h deficiency in hematopoietic cells protects against atherosclerosis.
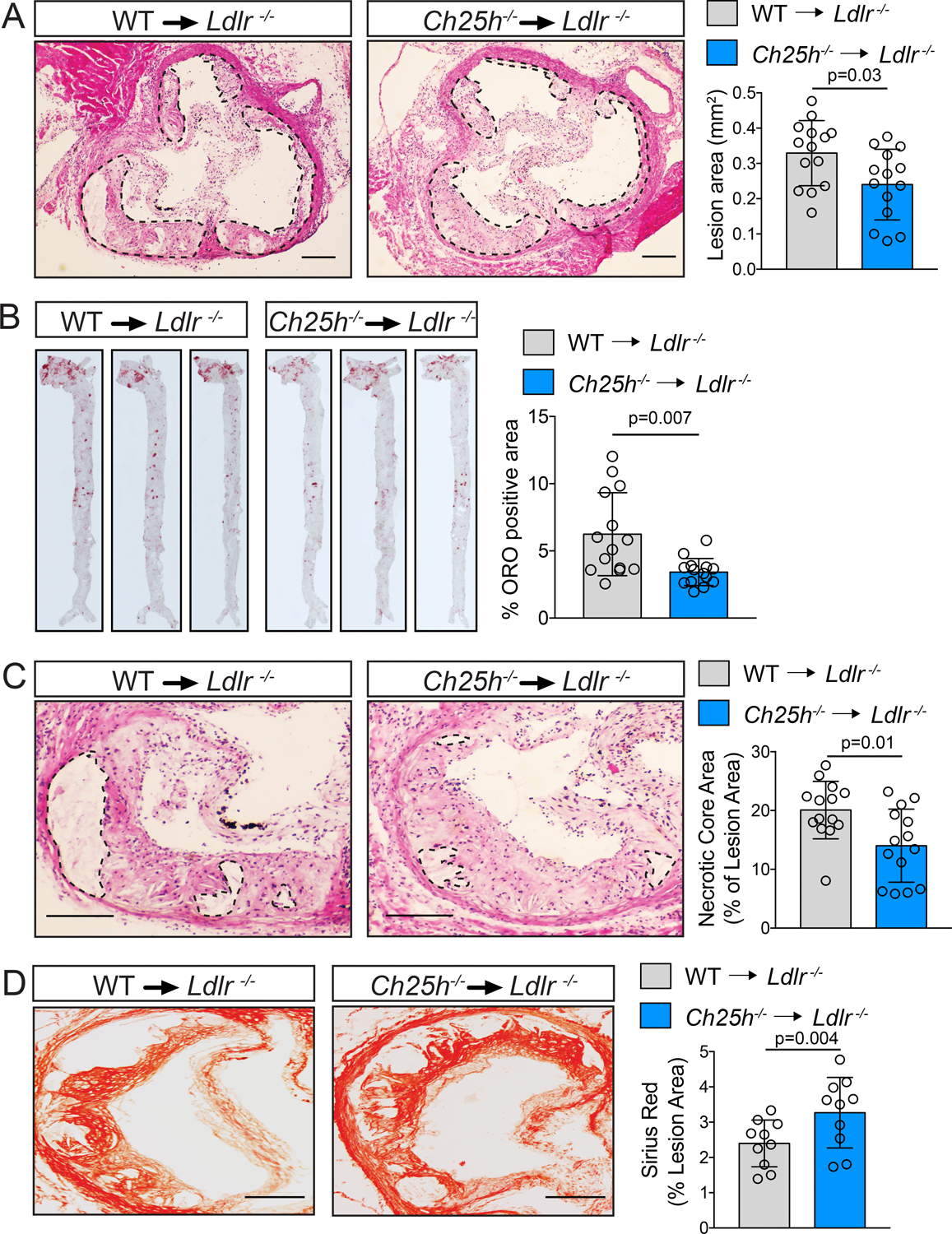
Analysis of Ldlr−/− mice transplanted with WT or Ch25h−/− bone marrow and fed for 12 weeks on a WD. A, C, Representative histological analysis of cross sections of the aortic sinus stained with hematoxylin and eosin (H&E). Dashed lines delimit the plaque area (A) and show the edge of the developing necrotic core (C). Quantification of plaque size (A) and necrotic core (C) are shown on the right panels (mean ±SD, n=14 mice per group). B, Representative en face ORO staining of aortas. Quantification of the ORO positive area is shown in the right panel and represents the mean ±SD (n=14 mice per group). D, Representative histological analysis of cross sections of the aortic sinus stained with Picrosirius Red. Quantification of the Picrosirius Red positive area is shown at the right panel and represents the mean ±SD (n=10 mice per group). All data were analyzed by Mann-Whitney non-parametric test. Scale bar: 100 μm.
Complex advanced lesions are characterized by the accumulation of macrophages, SMCs, and dead cells that in turn promote chronic inflammation within the artery wall12. Immunodetection in the aortic root for CD68 positive macrophages or smooth alpha actin (SMA) positive SMCs showed that Ch25h−/−→Ldlr−/− mice exhibited reduced CD68-positive plaque area and increased SMA-positive area when compared to WT→Ldlr−/− mice (Figure 3A). The decrease in macrophage content within the plaques of Ch25h−/−→Ldlr−/− mice was accompanied by reduced inflammation, as indicated by reduced plasma levels of circulating pro-inflammatory mediators, including chemokine (C-X-X) motif ligand 1 (CXCL1), 9 (CXCL9) and the monocyte chemoattractant protein-1 (MCP-1) or CCL2 (Figure S2C), as well as reduced immunodetection of vascular cell adhesion molecule-1 (VCAM-1) (Figure 3B). In agreement with the reduced necrotic core area (Figure 2B), we found that Ch25h−/−→Ldlr−/− mice had reduced numbers of TUNEL-positive CD68-positive cells within plaques than control mice (Figure 3C). These findings indicated that loss of Ch25h in hematopoietic cells reduced the progression of atherosclerosis and promoted plaque stability by decreasing vascular inflammation and reducing macrophage accumulation and death within the lesions.
Figure 3. Ch25h deficiency in hematopoietic cells reduces vascular inflammation and promotes plaque stability.
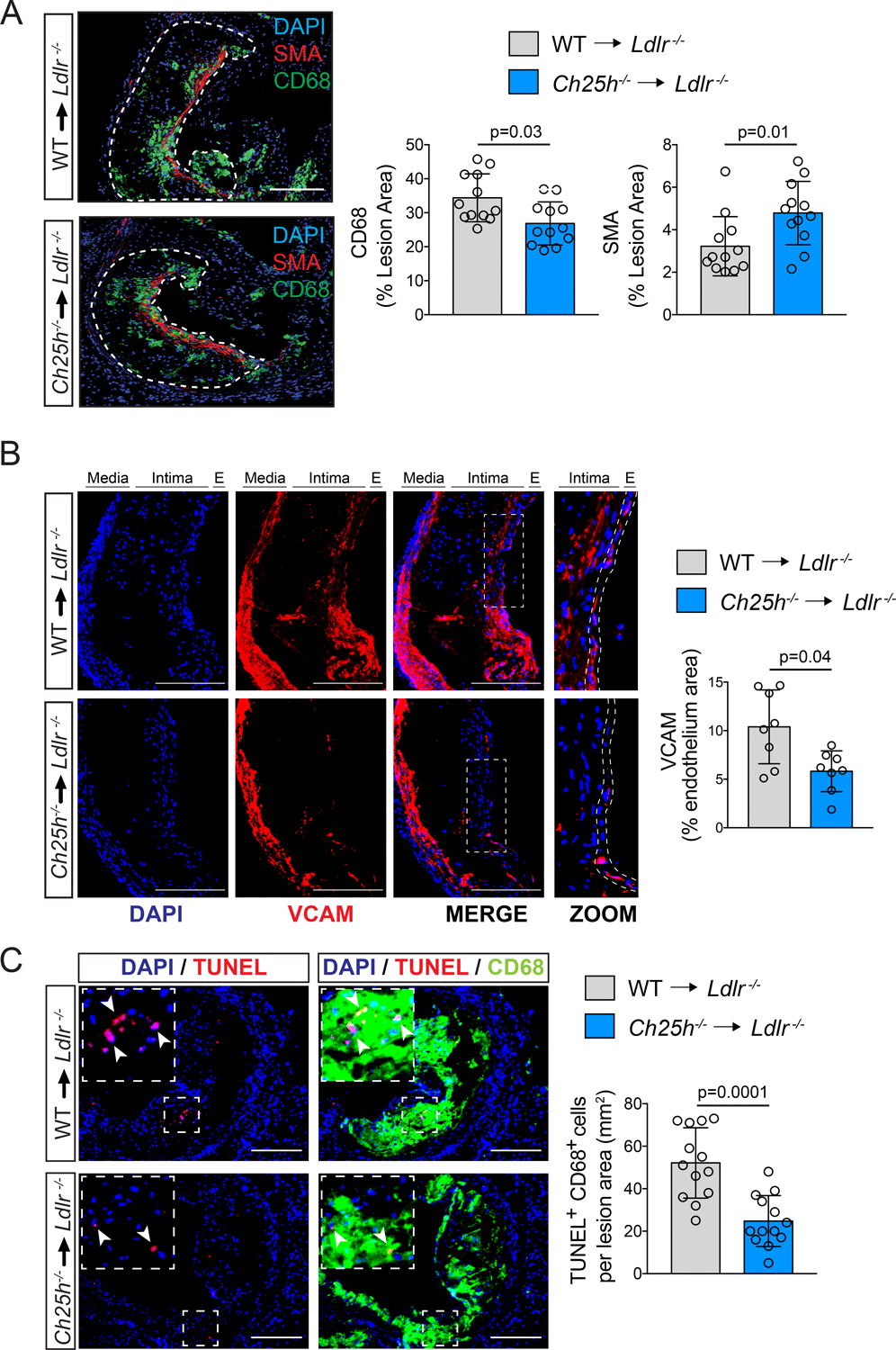
Analysis of Ldlr−/− mice transplanted with WT or Ch25h−/− bone marrow and fed for 12 weeks on a WD. A, Representative histological analysis of cross sections of the aortic sinus stained with CD68 or smooth muscle actin (SMA). Dashed lines delimit the plaque area (A) Quantification of the CD68 or SMA positive area is shown on the right panels and represents the mean ±SD (n=12 mice per group). B, Representative histological analysis of cross sections of the aortic sinus stained with VCAM and DAPI. Quantification of the VCAM positive endothelium area is shown in the right panel and represents the mean ±SD (n=8 mice per group). In the enlarged images the dashed lines show how the endothelium area was delimited for quantification. C, Representative histological analysis of cross sections of the aortic sinus stained with CD68 and TUNEL. Quantification of the CD68-positive cells with TUNEL-positive nuclei is shown in the right panel and represents the mean ±SD (n=13 mice per group). DAPI was used to stain the nuclei. All data were analyzed by Mann-Whitney non-parametric test. Scale bar: 100 μm.
25-HC promotes the expression of inflammatory genes in lipid-laden macrophages
To elucidate the contribution of Ch25h in regulating atherogenic phenotypes, we isolated thioglycolate-elicited peritoneal macrophages (TG-EPM) from hypercholesterolemic mice, which accumulate and store broad amounts of lipids in the cytosol, mimicking the lipid-loaded macrophages present in atherosclerotic lesions50–52, and profiled their transcriptomes by RNA-seq (Figure S4 and Figures 4 and 5). Macrophages from both Ldlr−/− or Ch25h−/−;Ldlr−/− mice exhibited increased neutral lipid accumulation when compared to mice (WT or Ch25h−/−) that were not on the Ldlr−/− background (Figure S5A). However, the absence of Ch25h did not affect the accumulation of neutral lipids (Figure S5B). TG-EPM from Ldlr−/− or Ch25h−/−;Ldlr−/− mice were stimulated for 4 or 12 h with lipopolysaccharide (LPS) to increase basal expression levels of Ch25h36,37 ,38,41,53,54. As expected, LPS stimulation of control Ldlr−/− macrophages increased Ch25h mRNA levels (Figure 4A), which was accompanied with an increase in intracellular 25-HC36 (Figure 4B). This effect was not observed in Ch25h−/−;Ldlr−/− elicited macrophages (Figure 4A and B). In mock-treatment conditions, ~ 200 genes were differentially and significantly expressed (FC>1.5, P ≤ 0.05) in Ch25h−/−;Ldlr−/− macrophages when compared to control Ldlr−/− (Figure 4C). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis showed that extracellular matrix remodeling factors were over-represented in Ch25h−/−;Ldlr−/− macrophages (Figure 4D, upper panel). However, after 4 h of LPS stimulation there were more than 500 differentially expressed genes in Ch25h−/−;Ldlr−/− vs. Ldlr−/− macrophages (Figure 4C). The vast majority of these genes were downregulated, and KEGG pathway enrichment analysis revealed that pathways related to macrophage inflammatory response were over-represented in Ch25h−/−;Ldlr−/− mice (Figure 4D, lower-left panel). Interestingly, transcripts of the TLR4-induced transcriptional program, including NFκB, activator protein-1 (AP-1), and interferon regulatory factors (IRFs) (e.g., Rela, Nfkb2, Irf5, Jund), as well as genes that encode cytokines and chemokines (e.g., Tnf, Cxcl10, Il12a, Ccl5) were significantly downregulated in Ch25h−/−;Ldlr−/− macrophages (Figure 4E). Remarkably, within the upregulated transcripts in Ch25h−/−;Ldlr−/− macrophages, we found numerous anti-inflammatory genes (e.g., Il10, Arg1, Mrc1) (Figure 4E). After 12 h of LPS stimulation, Ch25h−/−;Ldlr−/− macrophages presented 552 differentially regulated transcripts. In this case, most of the genes were up-regulated (Figure 4C) and pathways related to macrophage phagocytic activity were over-represented (Figure 4D, lower-right panel). Specifically, genes involved in phagocytosis (e.g., Mrc1, Cd36, Fcgr1, Cyba), lysosomal activity (e.g., Ctsl, Atp6v0b, Lgmn, Psap) and intracellular cholesterol transport (e.g., Npc2, Stard3, Tspo) were significantly upregulated (Figure 4F). Interestingly, the genes that were downregulated in Ch25h−/−;Ldlr−/− macrophages were also pro-inflammatory (e.g., Il6, Il12b, Cxcl9) (Figure 4F).
Figure 4. 25-hydroxycholesterol in lipid-laden macrophages promotes an inflammatory gene expression profile.
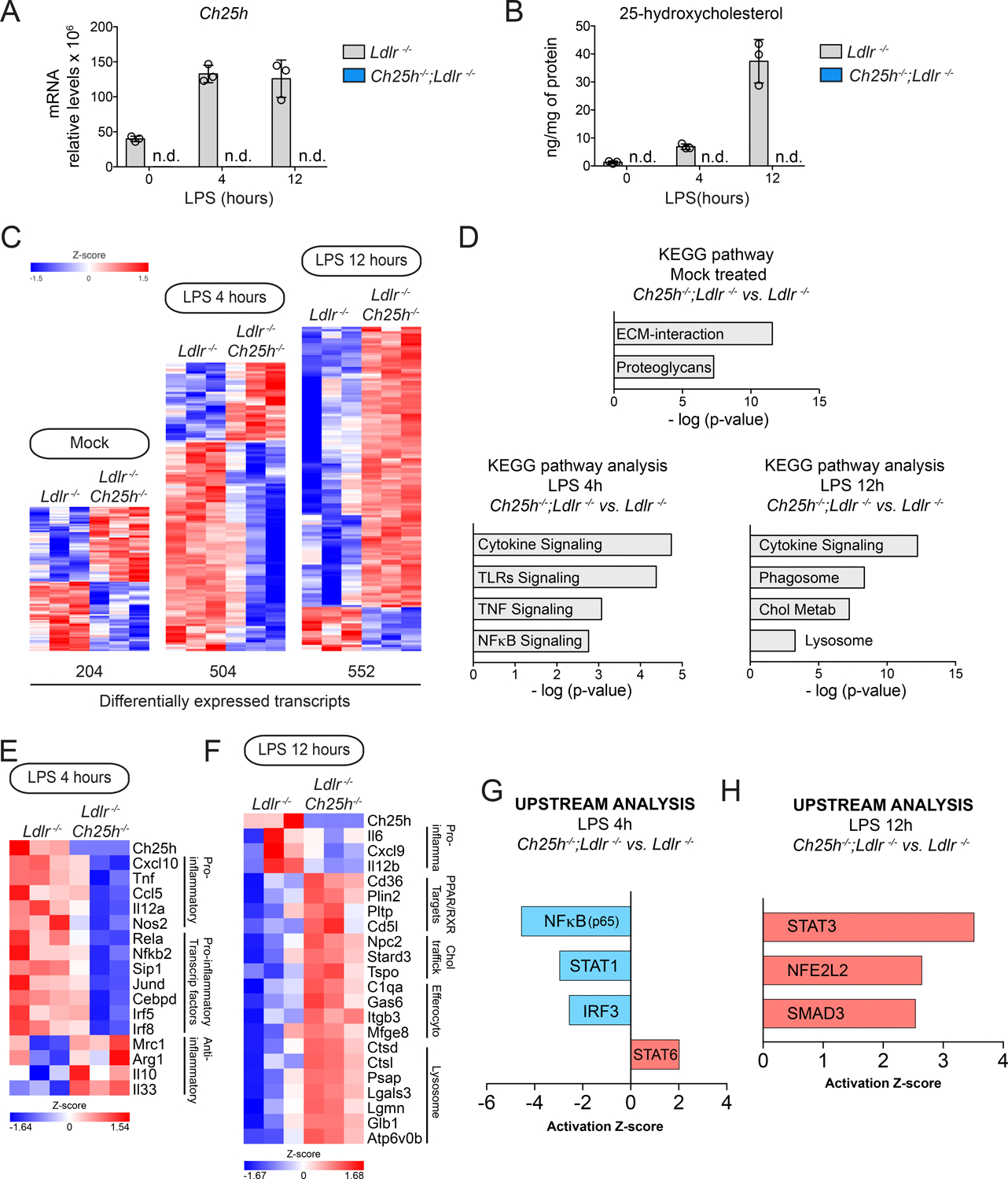
Analysis of lipid-laden TG-EPM isolated from Ldlr−/− or Ch25h−/−;Ldlr−/− mice (n=3 mice per genotype), treated with LPS (100 ng/ml) for 4 or 12 hours. A, Ch25h relative mRNA expression levels in where n.d. means not detected B, LC/MS-MS quantification of 25-hydroxycholesterol, in where n.d. means not detected. C, Heatmaps of differentially expressed genes. A default P-value ≤ 0.05 was considered statistically significant with a fold-change ≥ 1.5 for up-regulated transcripts or ≤ −1.5 for down-regulated transcripts. D, Kyoto Encyclopedia of Gene and Genomes (KEGG) pathway enrichment analysis of differentially expressed genes at different time points. E & F, Heatmaps of representative genes differentially expressed involved in inflammation and resolution of inflammation. A default P-value ≤ 0.05 was considered statistically significant with a fold-change ≥ 1.5 for up-regulated transcripts or ≤ −1.5 for down-regulated transcripts. G & H, Upstream analysis using Ingenuity Pathway Analysis Software of Transcription factors in the above indicated macrophages treated with 100ng/ml LPS for 4 hours (G) or 12 hours (H). Transcription factors predicted to be activated (z-score > 0) appear in red and those that were predicted to be inactive (z-score < 0) appear in blue.
Figure 5. 25-hydroxycholesterol in lipid-laden macrophages is not necessary for LXR or SREBP transcriptional regulation.
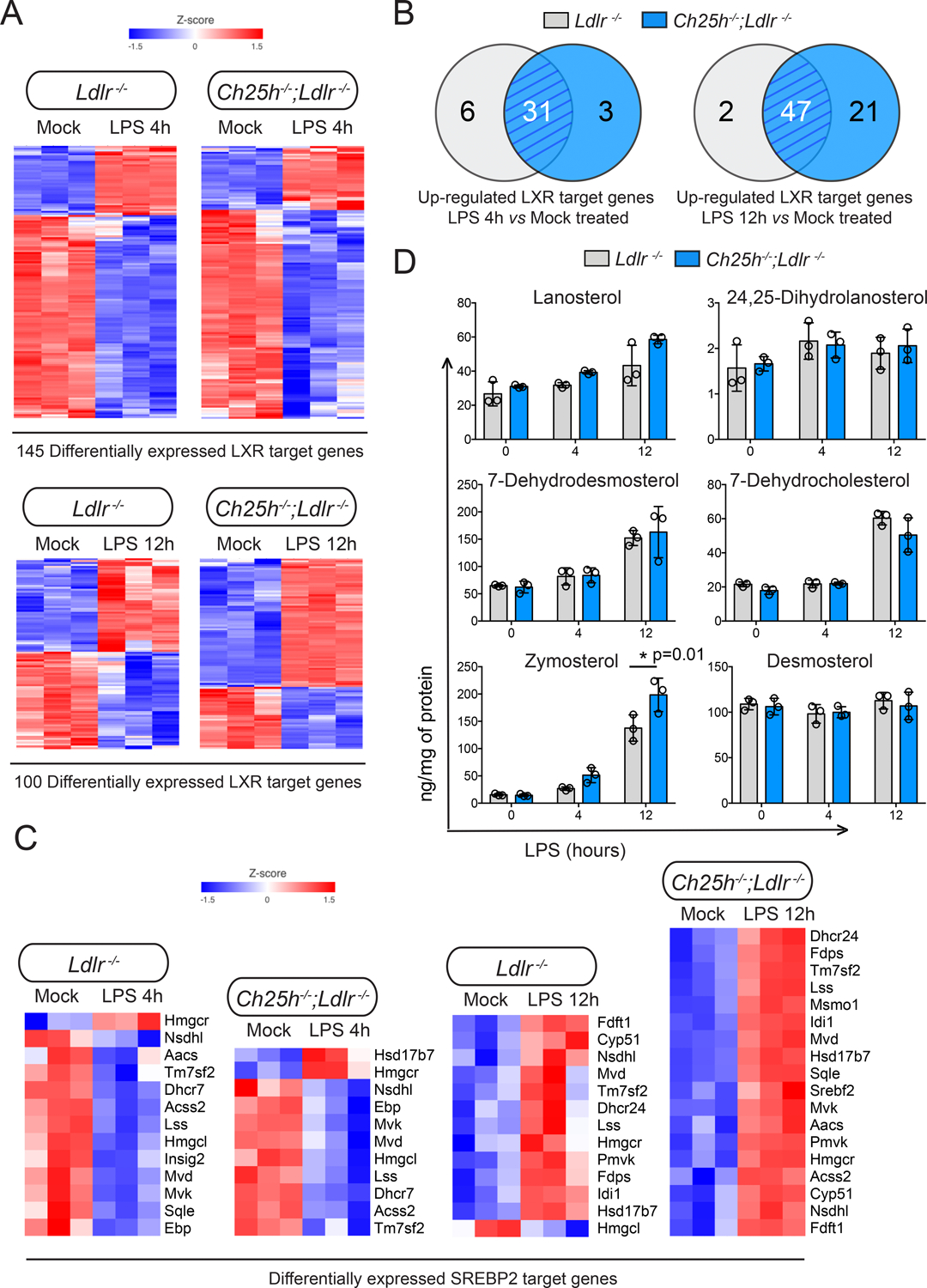
Analysis of lipid-laden TG-EPM isolated from Ldlr−/− or Ch25h−/−;Ldlr−/− mice (n=3 mice per genotype), treated with LPS (100 ng/ml) for 4 or 12 hours. A, Heatmaps showing LXR target genes differentially expressed in response to LPS treatment for 4 or 12 hours respectively. A default P-value ≤ 0.05 was considered statistically significant with a fold-change ≥ 1.5 for up-regulated transcripts or ≤ −1.5 for down-regulated transcripts. B, Venn diagrams depicting the overlap of upregulated LXR target genes. C, Heatmaps illustrating the effect of LPS treatment for 4 or 12 hours on SREBP2. A default P-value ≤ 0.05 was considered statistically significant with a fold-change ≥ 1.5 for up-regulated transcripts or ≤ −1.5 for down-regulated transcripts. D, Lipidomic analysis of sterol biosynthetic intermediates in macrophages isolated and treated as above. Data were analyzed by two-way ANOVA, post-hoc Bonferroni’s multiple comparison test (n=3 mice per genotype).
To identify molecules that potentially participate in the observed changes in gene expression, we used Ingenuity Pathway Analysis (IPA®) to identify potential upstream regulators and focused the analysis on transcription factors (TFs). Upstream regulator analysis after 4 h of LPS stimulation predicted inactivation of the pro-inflammatory TFs NFκB, STAT1 and IRF3 in Ch25h−/−;Ldlr−/− vs. Ldlr−/− macrophages (Figure 4G). This aligns with the decreased levels of Tnf, Cxcl10, Il12a, and Ccl5 in Ch25h−/−;Ldlr−/− macrophages (Figure 4E). On the other hand, the anti-inflammatory TF STAT6 was predicted to be activated (Figure 4G), in line with increased levels of some of its targets genes, such as Arg1 and Mrc155, in Ch25h−/−;Ldlr−/− macrophages (Figure 4E). After 12 h of TLR4 stimulation, upstream analysis predicted that the TFs STAT3, NFE2L2, and SMAD3 were activated in Ch25h−/−;Ldlr−/− macrophages (Figure 4H). STAT3 is crucial for the suppressive effect of IL-10 on LPS-induced genes56. NFE2L2-NFκB interplay regulates inflammatory responses57. NFE2L2 negatively regulates the transcription of Il658, which is also a pro-inflammatory cytokine gene downregulated in Ch25h−/−;Ldlr−/− macrophages (Figure 4F). SMAD3 has been shown to promote a phagocytic phenotype in macrophages59. Activation of SMAD3 could account for the increase in some phagocytic genes, including Mfge8, that are observed in Ch25h−/−;Ldlr−/− macrophages (Figure 4F).
These results indicate that hypercholesterolemic Ch25h−/− macrophages, that do not synthesize 25-HC upon TLR4-stimulation, exhibit a reduced pro-inflammatory gene signature and a pro-resolving transcriptional profile. This suggests that 25-HC synthesis by macrophages during atherosclerosis amplifies inflammation.
25-HC in lipid-laden macrophages does not promote LXR activation or the repression of SREBP2 activation.
SREBP and LXR transcription factor families are well known as master regulators of cholesterol homeostasis and are also implicated in macrophage inflammatory response13,20,21,29,44. Previous studies have established that 25-HC acts as a ligand for LXRs11,13 and as a repressor for SREBPs23. However, there is only limited in vivo evidence that 25-HC functions via these pathways in the context of lipid-laden macrophages. We analyzed how the levels of LXR60 and SREBP61 target genes were altered in our RNA-seq data set. We found that the activation and repression of LXR target genes after TLR4 stimulation was similar regardless of Ch25h expression (Figure 5A). Ch25h−/−;Ldlr−/− macrophages exhibited almost the same number of upregulated LXR target genes as control Ldlr−/− macrophages after 4 h of LPS stimulation. However, after 12 h of TLR4 stimulation, more LXR target genes were upregulated in Ch25h deficient macrophages than in Ldlr−/− macrophages (Figure 5B), despite the latter accumulating substantial amounts of 25-HC (Figure 4B), suggesting that 25-HC was not required for LXR activation. Moreover, comparing the normalized counts of well-established LXR target genes (e.g., Abca1, Abcg1, Lpl, Mylip, Pltp, Plin2) at different time points after LPS treatment, did not reveal significant differences between the genotypes (Figure S5A).
Next, we analyzed mRNA levels of classical SREBP2 target genes in LPS treated TG-EPM from Ldlr−/− or Ch25h−/−;Ldlr−/− mice. We found an overall reduction in the expression of SREBP2 target genes after 4 hours of LPS stimulation in control Ldlr−/− macrophages (Figure 5C). However, after 12 hours of LPS stimulation, the expression of SREBP2 target genes were elevated compared to the mock-treated macrophages (Figure 5C). Interestingly, Ch25h deficiency did not alter this pattern of expression (Figure 5C); indeed, the mRNA levels of several well-known SREBP2 target genes (e.g., Hmgcs1, Hmgcr, Hmgcl, Cyp51, Ebp, Scd5, Dhcr7, Dhcr24) were similar in Ch25h−/−;Ldlr−/− vs. Ldlr−/− macrophages (Figure S6B). To further evaluate the contribution of 25-HC to the regulation of cholesterol biosynthesis in lipid-laden macrophages, we employed LC/MS-MS to quantify alterations in sterol intermediates because of 25-HC-mediated regulation of SREBP2 signaling. As described above, control Ldlr−/− macrophages but not Ch25h−/−;Ldlr−/− accumulated 25-HC (Figure 4B). However, the absence or presence of 25-HC in macrophages did not alter the amount of key sterol intermediates such as lanosterol, 24,25-dehydrolanosterol, 7-dehydrodesmosterol, 7-dehydrocholesterol or desmosterol. Only increased levels of zymosterol were found in Ch25h−/−;Ldlr−/− macrophages (Figure 5D).
Altogether, our results indicate that in activated lipid-loaded macrophages, 25-HC does not significantly alter SREBP or LXR-mediated gene expression and, that the accumulation of sterol intermediates in cholesterol biosynthesis after inflammatory stimulation is independent of the presence of 25-HC.
25-HC in activated lipid-laden macrophages reduces cell survival and efferocytosis capacity
To explore the effect of 25-HC on cell death, we first analyzed how the absence of Ch25h affects susceptibility to LPS-induced apoptosis62. As shown in Figure 6A, after LPS stimulation, lipid-laden Ch25h−/−;Ldlr−/− macrophages presented reduced active/cleaved caspase 3 levels when compared to Ldlr−/− counterparts, in agreement with the reduced TUNEL-positive CD68-positive cells found in plaques from Ch25h−/−→Ldlr−/− mice (Figure 3C). This suggests that a lack of 25-HC synthesis in activated lipid-laden macrophages reduced apoptosis susceptibility. Additionally, we studied caspase-1 activation, as a read-out of inflammasome activation, given that pyroptosis is an inflammasome-dependent form of cell death that occurs within atherosclerotic plaques63, and that 25-HC regulates inflammasome activation43,44. As shown in Figure 6B, Ch25h−/−;Ldlr−/− macrophages exhibited a significant increase in pro-IL-1β protein despite Il1b mRNA levels being unchanged. Regardless, we did not find differences in inflammasome activation or IL-1β secretion between Ldlr−/− and Ch25h−/−;Ldlr−/− foamy macrophages (Figure 6B). This is in contrast with previous work that has shown that Ch25h−/− macrophages exhibit increased inflammasome activation and IL-1β secretion43,44. However, in non-lipid-laden BM-derived macrophages, we observed increased levels of active caspase-1 and secreted IL-1β, in line with previous work43 (Figure S7A).
Figure 6. Ch25h deficiency in macrophage reduces LPS-induced apoptosis independently of inflammasome activation but increases macrophages efferocytotic activity.
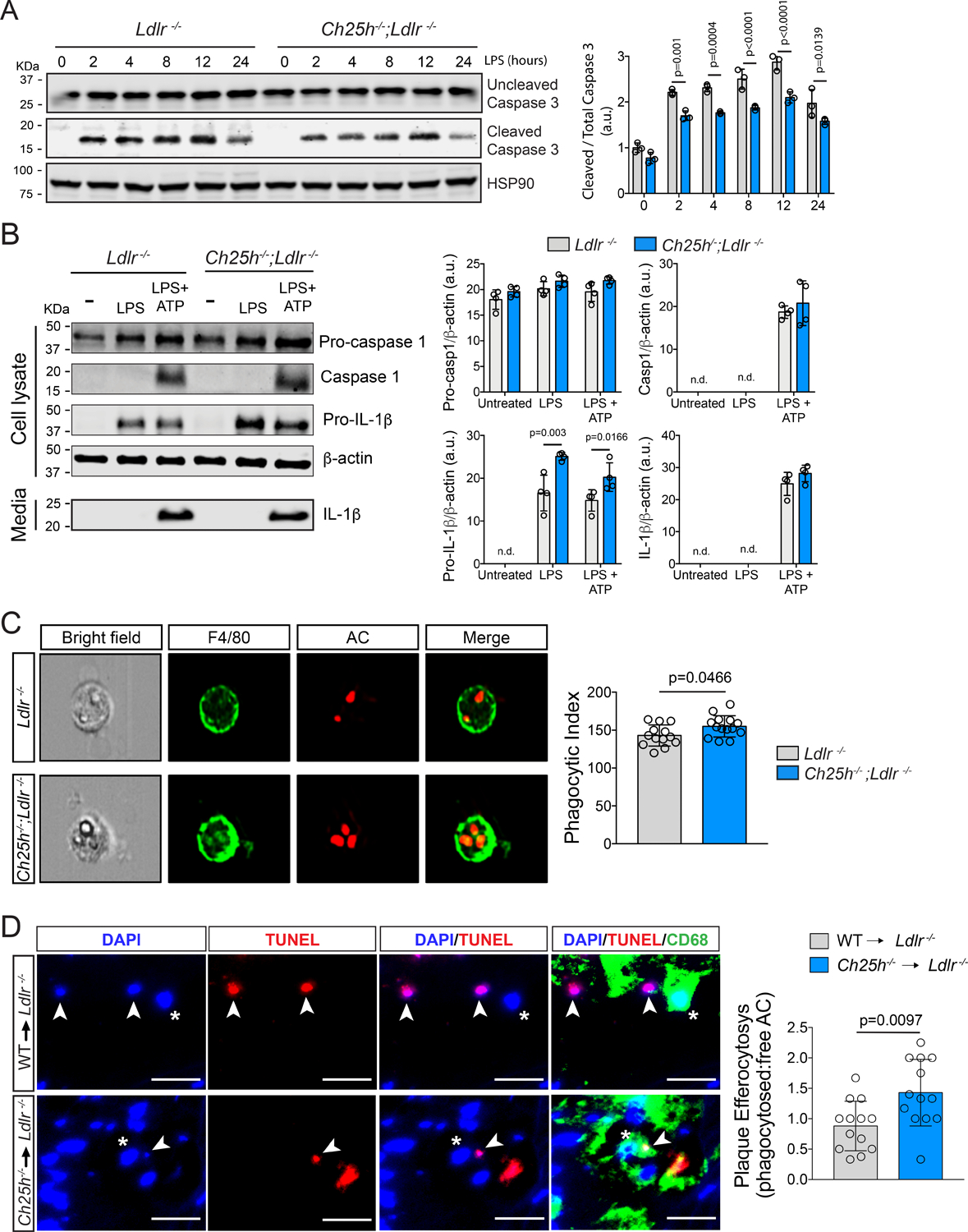
A, Representative Western blot analysis of cleaved and total caspase-3 in Ldlr−/− or Ch25h−/−;Ldlr−/− lipid-laden TG-EPM treated with LPS (100 ng/ml) at the indicated times. HSP90 was used as a loading control. Relative protein quantification by band densitometry is shown in the right panel. Data were analyzed by two-way ANOVA, post-hoc Bonferroni’s multiple comparison test (n=3). n.d, not detectable. B, Representative Western blot analysis of caspase-1 and IL-1β as a read out of inflammasome activation of Ldlr−/− or Ch25h−/−;Ldlr−/− lipid-laden TG-EPM with LPS (100 ng/ml) for 8 hours. ATP (5 mM) was added the last 30 minutes and prior to samples collection. Relative protein quantification by band densitometry is shown in the right panels. β-Actin was used as a loading control. Data were analyzed by two-way ANOVA, post-hoc Bonferroni’s multiple comparison test (n=3). n.d. not detectable. C, Representative images of the in vitro engulfment of CellTracker Deep Red labeled apoptotic Jurkat cells by lipid-laden TG-EPM from Ldlr−/− or Ch25h−/−;Ldlr−/− mice (n=14 mice per group). Data were acquired by Amnis Imagestream-X MarkII Imaging Flow Cytometer. Right panel shows efferocytosis quantification as phagocytic index, which is the number of apoptotic cells (red) ingested in 1 h per F4/80-positive macrophage (green) × 100. Significance was determined by Mann-Whitney non-parametric test. D, Representative images of the in situ efferocytosis assay. Apoptotic bodies were identified as TUNEL-positive nuclei (white arrow heads) and macrophages as CD68 positive cells (asteriscs). Free apoptotic bodies were those TUNEL-positive nuclei that do not overlap with CD68 (upper panels), whereas TUNEL-positive nuclei that do overlap with CD68 (lower panels) are considered phagocytosed apoptotic bodies. Right panel shows the quantification of the phagocytosed versus free apoptotic bodies ratio (n= 13 mice per group). Data were analyzed by Mann-Whitney non-parametric test. Scale bar: 10 μm.
Since previous studies have correlated 25-HC synthesis to mitochondrial reactive oxygen species (ROS) production44,64 and because disbalance in mitochondrial ROS can lead to apoptosis, we also measured mitochondrial mass and mitochondrial ROS production by flow cytometry in LPS-stimulated TG-EPM form Ldlr−/− and Ch25h−/−;Ldlr−/− mice. As shown in Figure S7 B and C, there were no statistical differences in mitochondrial mass or ROS production. These results suggest that in lipid-laden macrophages, the reduced susceptibility to LPS-induced apoptosis observed in Ch25h−/−;Ldlr−/− macrophages (Figure 6A) could be unrelated to inflammasome activation or mitochondrial ROS production. RNA-seq analysis revealed that activated Ch25h−/−;Ldlr−/− lipid-laden macrophages showed reduced mRNA levels of Tnf (Figure 4E), and autocrine secretion of TNF has been involved in LPS-induced apoptosis62. Alternatively, activated Ch25h−/−;Ldlr−/− lipid-laden macrophages showed increased levels of Cd5l (Figure S7D), which is highly expressed in foamy macrophages within atherosclerotic lesions (Figure S1B,C, H and I) and protective65–67, promoting their survival68. These findings are in line with the diminished inflammatory and apoptotic phenotype exhibited by Ch25h−/−;Ldlr−/− lipid-laden macrophages.
We also found that Ch25h−/−;Ldlr−/− macrophages displayed increased efferocytotic capacity compared to Ldlr−/− macrophages (Figure 6C). This correlates with increased mRNA levels of several efferocytosis genes such as Gas6, C1q, Mfge8 and Itgb3 found in Ch25h−/−;Ldlr−/− macrophages (Figure 4F). Furthermore, Mertk expression was found in macrophage clusters with low or no detectable Ch25h expression (Figure S1B and H). To evaluate the biological relevance of this finding, we analyzed in situ efferocytosis by counting the number of macrophage-associated apoptotic cells vs. free apoptotic cells in individual aortic root sections69–72. As shown in Figure 6D, Ch25h−/−→Ldlr−/− mice exhibit a higher ratio of macrophage-associated to free apoptotic cells, consistent with the improved efferocytotic activity observed in the absence of Ch25h. These results agree with the smaller necrotic core found in Ch25h−/−→Ldlr−/− mice (Figure 2B) and reveal that in the absence of 25-HC production, lipid-laden activated macrophages have improved efferocytosis and are more resistant to inflammation-induced apoptosis.
Absence of 25-HC alters cell membrane composition, resulting in a reduction in the TLR4/p38/NFkB signaling cascade
As shown above, Ch25h−/−;Ldlr−/− lipid-laden macrophages had reduced expression of inflammatory genes after LPS stimulation when compared to control Ldlr−/− macrophages. Interestingly, Tnf, Il12a and Il6 were among the significantly downregulated genes (Figure 4E), that are indeed hallmarks of both NF-κB and AP-1 activation55. Thus, we wondered whether TLR4 signaling was affected in Ch25h−/−;Ldlr−/− lipid-laden macrophages to account for the reduced expression of inflammatory genes. LPS/TLR4 signaling can be divided into MyD88-dependent and MyD88-independent pathways, which mediate the activation of pro-inflammatory cytokines and type I IFN induced genes, respectively. Hence, we first analyzed the activation of three key proteins in the MyD88-dependent and independent pathways, p65, MAP-kinase p38, and IRF3. As shown in Figure 7A, after long exposure to LPS, Ch25h−/−;Ldlr−/− macrophages exhibited significantly reduced activation (phosphorylation) of p38 and IRF3 than Ldlr−/− macrophages. Similar results were also observed under acute TLR4 stimulation (Figure 7B), in this case with reduced phosphorylation of p65. Interestingly, we also found a delay in the p38 activation peak, from 15 minutes in Ldlr−/− macrophages to 30 min in Ch25h−/−;Ldlr−/− macrophages. The IRF3 activation peak went from 30 min in Ldlr−/− macrophages to 60 min in Ch25h−/−;Ldlr−/− macrophages (Figure 7B). Altogether, these results indicate that absence of 25-HC in Ch25h−/−;Ldlr−/− macrophages downregulates both TLR4 signaling branches. Since the accumulation of free cholesterol in the PM can hypersensitize macrophages to LPS73–77, we used the fluorescently-labeled, non-lytic cholesterol-binding protein, Anthrolysin O domain 4 (ALOD4), to measure the amount of accessible cholesterol in the PM of Ch25h−/−;Ldlr−/− and Ldlr−/− lipid-laden macrophages by flow cytometry. As shown in Figure 7C, we found a diminished accumulation of accessible cholesterol in the PM of Ch25h−/−;Ldlr−/− macrophages in comparison to control Ldlr−/−. Although mRNA levels of membrane cholesterol transporters Abca1 and Abcg1 were similar in the presence or absence of Ch25h (Figure S5), Ch25h−/−;Ldlr−/− macrophages showed higher protein levels of both ABCA1 and ABCG1 (Figure S7E). Of interest, Abca1- and Abcg1-deficient peritoneal macrophages have enhanced inflammatory responses which is associated with increased membrane free cholesterol76. These results suggests that the reduction in TLR4-mediated signaling that we observed in hypercholesterolemic Ch25h−/−;Ldlr−/− macrophages could be due to diminished free cholesterol in the PM. Thus, we pre-treated the cells with sphingomyelinase (SMase), to release cholesterol sequestered by sphingomyelin to increase the pool of accessible cholesterol in the PM78,79, prior to the stimulation with LPS and assessed p65 activation. Cells were also incubated with anthrolysin O peptide (ALOD4) to concomitantly measure accessible free cholesterol. SMase pre-treatment increased ALOD4 binding in both groups of macrophages (Figure 7D). Interestingly, the SMase-mediated increase of free cholesterol in the PM of Ch25h−/−;Ldlr−/− macrophages rescued the reduced activation of p65 observed in Ch25h−/−;Ldlr−/− in the absence of SMAse (Figure 7B), resulting in comparable levels of p65 activation to that present in Ldlr−/− macrophages in the absence of SMase (Figure 7D). These results indicate that in activated lipid-laden macrophages, lack of 25-HC production decreases TLR4-mediated signaling, at least in part by altering the accumulation of free cholesterol in the PM.
Figure 7. 25-hydroxycholesterol alters TLR4 downstream signaling by reducing the accessible cholesterol in the plasma membrane.
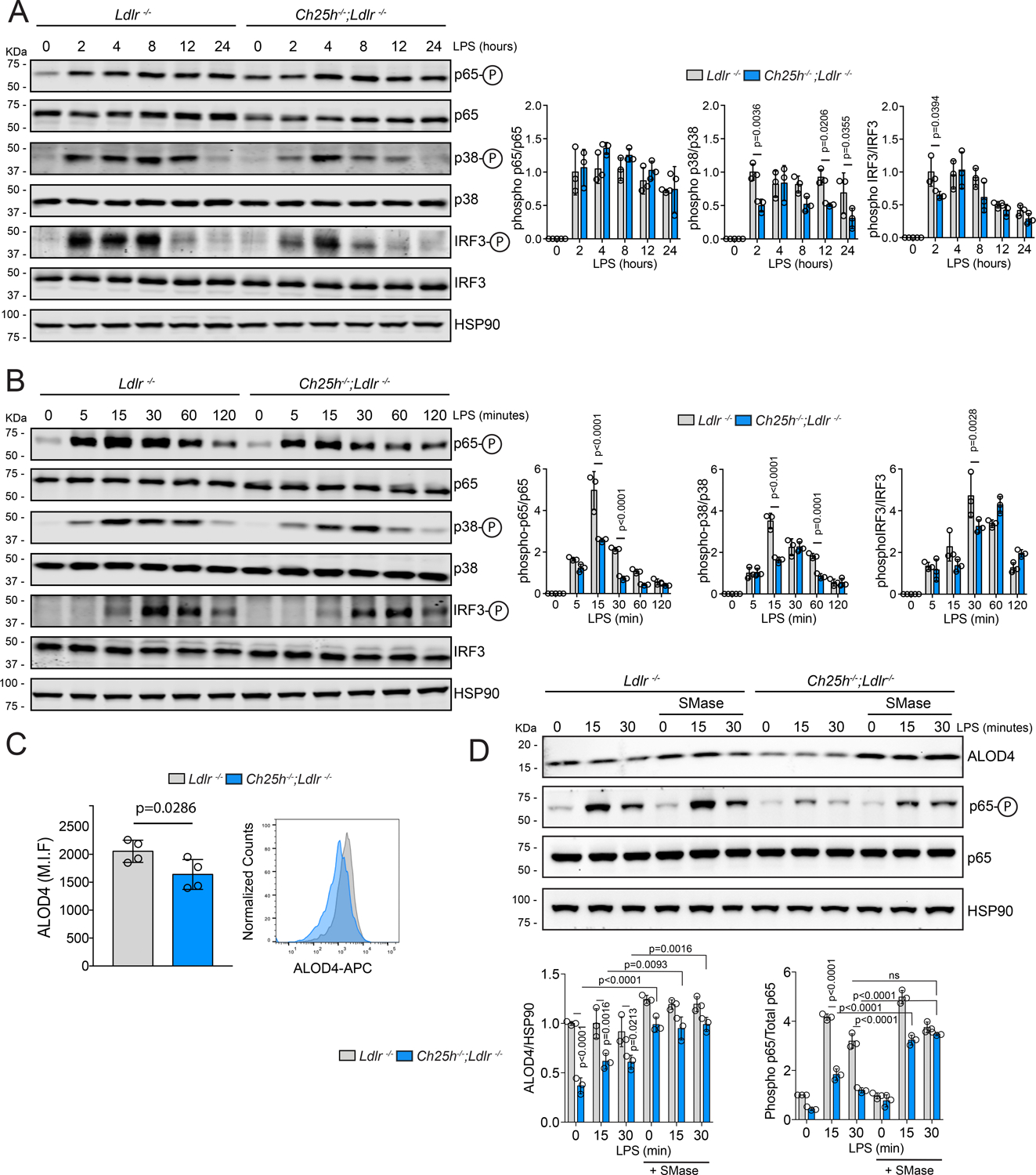
A-B, Representative Western blot of phospho-p65, p65, phospho-p38, p38, phospho-IRF3, IRF3 or HSP90 in Ldlr−/− or Ch25h−/−;Ldlr−/− lipid-laden TG-EPM treated with LPS (100 ng/ml) at the indicated time points. HSP90 was used as loading control. Quantification of the relative phosphorylation is showing at the right panels. Data were analyzed by two-way ANOVA, post-hoc Bonferroni’s multiple comparison test (n=3). C, Quantification of ALOD4 PM binding, in lipid-laden TG-EPM from Ldlr−/− or Ch25h−/−;Ldlr−/− mice, by flow cytometry. Representative histogram is showing in the bottom panel. Data are average of the mean of Median Intensity Fluorescence (M.I.F.) in arbitrary units (a.u.) and analyzed by Mann-Whitney non-parametric test (n=4). D, Representative Western blot of phospho-p65, p65, ALOD4 or HSP90 in lipid-laden TG-EPM from Ldlr−/− or Ch25h−/−;Ldlr−/− treated with LPS (100 ng/ml) in the presence or absence of Sphingomyelinase (200 mU/ml) at the indicated time points. Quantification of ALOD4 and the relative p65 phosphorylation is showed in the right panel. HSP90 was used as loading control. Data were analyzed by two-way ANOVA, post-hoc Bonferroni’s multiple comparison test (n=3).
25-HC is released by activated macrophages to inhibit SMC migration
Activated macrophages release 25-HC into the extracellular space with a paracrine effect on cells in the immediate vicinity32,38,41. In consonance, we found that LPS-stimulated lipid-laden macrophages release 25-HC to the media in a time-dependent manner, while macrophages deficient in Ch25h did not (Figure 8A). In both cases, the secretion of other oxysterols (e.g., 24-HC or 27-HC) was unaltered (Figure 8A). Since migration of SMCs from the tunica media into the intima is a key event for fibrous cap formation, collagen deposition, and plaque stability, and we found an increase in SMC content within the plaques of Ch25h−/−→Ldlr−/− mice, we sought to investigate whether 25-HC influences cellular responses of SMCs during atherogenesis. We analyzed the effect of 25-HC on SMC migration in response to platelet-derived growth factor (PDGF) in a trans-well assay. As shown in Figure 8B pretreatment of VSMCs with 25-HC reduced PDGF-induced migration. These findings suggest that macrophage-derived 25-HC reduces SMC migration to populate the fibrous cap. Since PDGF receptor β (PDGFRβ) is relocated into cholesterol-rich domains within the PM for proper intracellular signaling transduction80 and the pool of free cholesterol in the PM is altered when cells are treated with 25-HC23, we investigated whether 25-HC alters PDGFRβ signaling in SMC. As expected, addition of 25-HC into the culture media reduced the amount of free cholesterol in the PM of SMC (Figure 8C) without affecting cell viability, since total cell number was not affected (Figure S8A). In line with this, SMC pretreated with 25-HC showed decreased phosphorylated protein kinase B (aka AKT) and extracellular signal-regulated kinase (ERK1/2) after stimulation with PDGF, suggesting that 25-HC diminishes PDGFRβ signaling, in part by altering the pool of free cholesterol in the PM. Furthermore, dorsal ruffle formation after PDGF stimulation81, an early event in cell migration, was reduced when SMC were pretreated with 25-HC (Figure S8B) These findings suggest that macrophage-derived 25-HC may alter PDGF signaling in SMC and thus affecting their migration.
Figure 8. 25-hydroxycholesterol secreted by macrophages inhibits vascular smooth muscle cells migration.
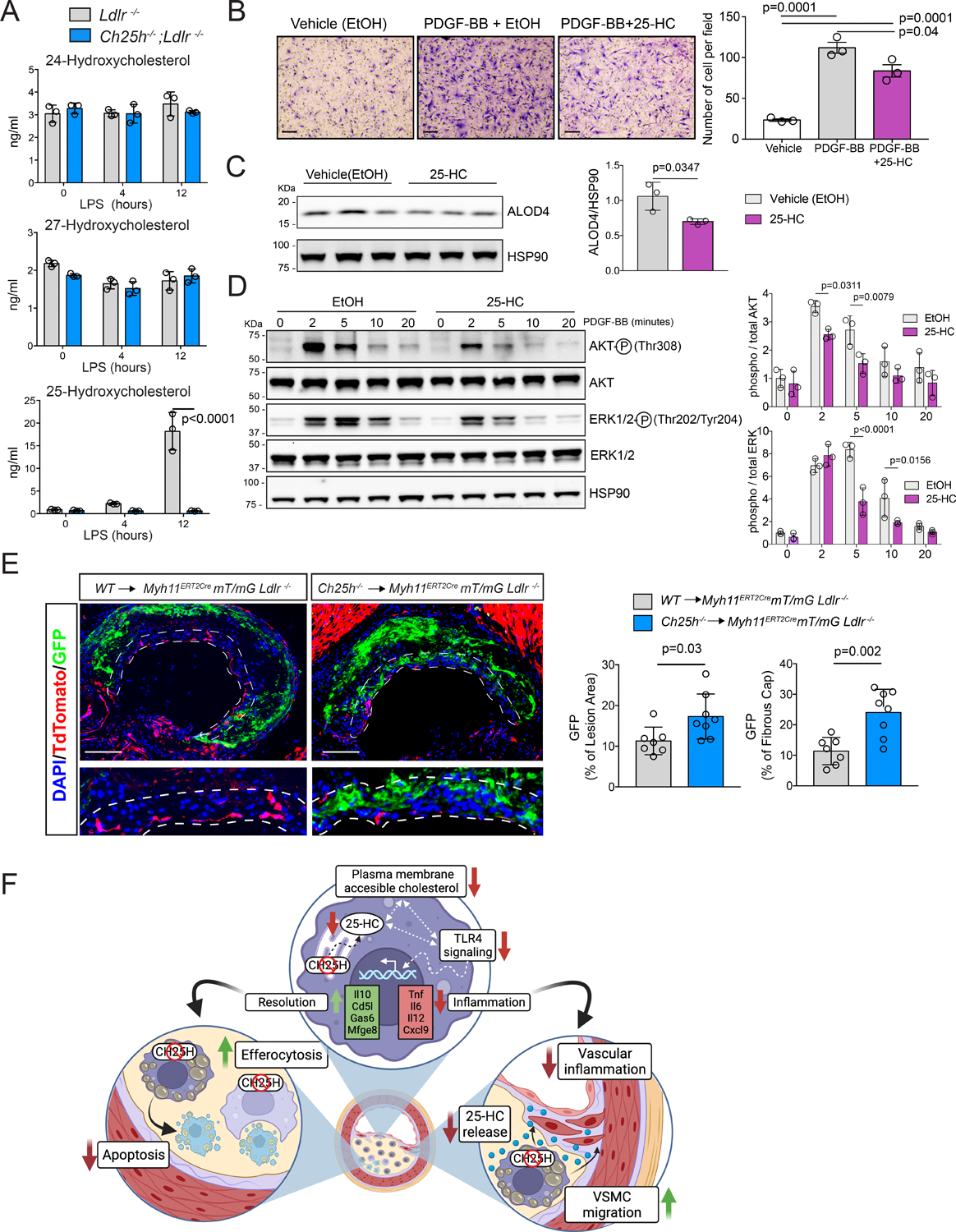
A, LC/MS-MS quantification of several oxysterols presents in the media of cultured lipid-laden TG-EPM from Ldlr−/− or Ch25h−/−;Ldlr−/− (n=3 mice per genotype) treated with LPS (100 ng/ml) for 4 or 12 hours. Data were analyzed by two-way ANOVA, post-hoc Bonferroni’s multiple comparison test. B, SMC migration in the presence of PDGF-BB (10 ng/ml) or PDGF-BB plus 25-HC (5μM). 5 images were taken randomly per trans-well. Quantification is showing in the right panel and represent the mean ±SEM of 3 independent experiments. Data were analyzed by one-way ANOVA, post-hoc Bonferroni’s multiple comparison test. Scale bar: 100 μm. C, Western blot analysis of ALOD4 binding into the PM in SMC treated with ethanol (EtOH) or 25-hydroxycholesterol (25-HC) at a concentration of 5μM for 2 hours. Quantification of ALOD4 is showed in the right panel. Data were analyzed by Mann-Whitney non-parametric test. D, Representative Western blot of phospho-AKT, AKT, phospho-ERK1/2, ERK1/2 or HSP90 in SMC pre-treated with EtOH or 25-HC (5μM) for 2 hours and then stimulated with PDGF-BB (10 ng/ml) for the indicated times. Quantification of the relative AKT and ERK1/2 phosphorylation is showed in the right panel. Data were analyzed by two-way ANOVA, post-hoc Bonferroni’s multiple comparison test (n=3). E, Representative histological analysis of cross sections of the aortic sinus stained with eGFP and Tdtomato of Myh11CRE;mT/mG; Ldlr−/− mice transplanted with bone marrow from WT or Ch25h−/− donor mice and fed for 12 weeks a WD. Dashed lines define the fibrous cap area used for quantification. Scale bar: 100 μm. Quantification of the eGFP positive area within the whole plaque or the fibrous cap are shown at the right panels and represents the mean ±SD (n=8 mice per group) DAPI was used to stain the cell nucleus. All data were analyzed by Mann-Whitney non-parametric test. F, Proposed working model of 25-HC in macrophages in the context of atherosclerosis. Macrophages-derived 25-HC accelerates atherosclerosis progression and promotes plaque instability. Mechanistically we found that lack of 25-HC synthesis favors a reduction of accessible cholesterol in the PM, what diminishes pro-inflammatory response initiated by pattern recognition receptors in lipid-laden macrophages and promotes a reprogramming into a more pro-resolving phenotype (up). This pro-resolving phenotype is characterized by a better efferocytotic capacity and a lower susceptibility to stress-associated apoptosis in hypercholesterolemic macrophages Ch25h deficient (right). Additionally, the lack of macrophage-derived 25-HC released allows the migration of tunica media smooth muscle cells into the intima to form the fibrous cap (left).
To validate these in vitro results and to understand the contribution of macrophage-derived 25-HC to fibrous cap formation, we used a SMC lineage tracing mouse model (Myh11ERT2CREmT/mG;Ldlr−/−) since SMCs within the atherosclerotic plaque cannot solely be identified by SMA staining82, and performed BM adoptive transfer using WT or Ch25h−/− mice as BM donors to study the progression of atherosclerosis. After 12 weeks on WD, reporter mice that received Ch25h−/− BM showed a significant increase in enhanced green fluorescence protein (eGFP)+ SMCs within the plaque (Figure 8E) primarily due to accumulation within the fibrous cap (Figure 8E). These findings suggest that expression of Ch25h, and consequent production of 25-HC by macrophages reduces the migration of SMCs that populate the fibrous cap, thus promoting plaque instability.
DISCUSSION
In this study we determined that macrophage-derived 25-HC accelerates atherosclerosis progression and promotes plaque instability. We found that, in lipid-laden macrophages, lack of 25-HC production favors lower levels of accessible cholesterol in the PM, which alters TLR4 signaling, decreases NFκB-mediated pro-inflammatory gene expression, and reduces apoptosis susceptibility. These effects are not a result of 25-HC-mediated modulation of LXR or SREBP transcription. Interestingly, unlike other oxysterols, 25-HC is released by activated macrophages as a secondary messenger that inhibits SMC migration, contributing to plaque instability.
Our in vivo studies demonstrate that hematopoietic Ch25h deficiency significantly reduces atherosclerosis and enhances plaque stability. Our results align with a previous study that showed that ATF3-mediated suppression of Ch25h reduces atherosclerosis46, while another study by Li et al. showed that Ch25h deficiency promoted atherosclerosis45. The discrepancies could be due to the differences in the animal model approach used. While we used a BM adoptive transfer approach in Ldlr−/− mice to determine the role of Ch25h in hematopoietic cells, and hence macrophages, Li et al. used a global Ch25h knockout in the atherogenic Apoe−/− background. With this approach, the effect of Ch25h deficiency in other tissues (e.g., liver and adipose tissue) and its participation in atherosclerotic progression could not be ruled out. On the other hand, in a model of diet-induced obesity, global deficiency of Ch25h reduces adipose tissue inflammation83. Additionally, it has been shown that Ch25h−/− mice are protected against inflammatory-induced pathology in a model of influenza infection41. These results, together with the findings of our present study, support the idea that 25-HC promotes inflammation in vivo.
To understand the molecular mechanisms that underlie the proatherogenic effects of macrophage-derived 25-HC, we performed RNA-seq in lipid-laden macrophages. We stimulated macrophages with LPS for 4 or 12 h to mimic an activation phase and resolution phase of inflammation19. In the activation phase, Ch25h deficiency reduced the expression of inflammatory genes, which agrees with previous studies showing that BM derived macrophages (BMDMs) isolated from Ch25h−/− mice have reduced levels of Nos2, Il12b, Il6 and Tnf after LPS-mediated TLR4 stimulation41,83. In line with this, we also found that aortic macrophages from atherosclerotic mice with reduced expression of inflammatory genes also had reduced Ch25h levels. Mechanistically, we found that the absence of 25-HC synthesis decreased the accumulation of accessible cholesterol in the PM and increased ABCA1 and ABCG1 protein levels, altogether resulting in reduced TLR4 mediated signaling and diminished NFκB-mediated transcription of inflammatory genes. This aligns with previous studies that have demonstrated that macrophages deficient in cholesterol Abc transporters accumulate free cholesterol in the PM, increasing TLR4-dependent signaling73,74, 75–77. Interestingly, the accumulation of sterols is quite different. While Abcg1−/− macrophages accumulate 7-ketocholesterol and desmosterol, as well as 27-OH, the absence of Ch25h promotes the accumulation of zymosterol. This indicates that the underlying effect on inflammation could not be attributed to a specific sterol and that all may contribute to the observed effect in PM cholesterol composition, or that other factors could be responsible for the phenotype. Indeed, diminished p38 activation upon the absence of 25-HC synthesis in Ch25h−/−;Ldlr−/− macrophages could influence ABCG1 phosphorylation and degradation84 and, therefore increase ABCG1 protein levels. Altogether, our results indicate that deficiency in 25-HC production alters free cholesterol PM composition and macrophage responses to inflammatory stimulus.
In the resolution phase, lipid-laden Ch25h deficient macrophages were transcriptionally reprogrammed into a pro-phagocytic phenotype, which was supported by the in vitro and in situ efferocytosis assays. This is in contrast with recent work showing that 25-HC production promotes efferocytosis in alveolar and peritoneal macrophages85. However, in our efferocytosis experiments, macrophages were stimulated with LPS before co-culture with apoptotic cells to increase the expression of Ch25h and hence the amount of 25-HC in Ldlr−/− control macrophages. In the presence of LPS, the efferocytosis mediator MERTK is cleaved from the macrophage membrane surface by the metalloprotease ADAM17 in a p38-dependent manner, thus reducing macrophage efferocytosis capacity86. Since in the absence of Ch25h, TLR4 stimulated signaling and p38 activation are reduced, it is possible that Ch25h−/−;Ldlr−/− macrophages conserve more MERTK on the surface after LPS treatment, which could explain their increased efferocytotic activity. This is in line with the increased expression of Mertk in aortic macrophage clusters with low or nondetectable levels of Ch25h. Nevertheless, our in vivo data showed that absence of hematopoietic Ch25h reduces necrotic core area and the accumulation of dead macrophages in the plaque, due to more efficient efferocytosis. A second factor that influences necrosis in atherosclerotic plaques is extensive apoptosis that occurs in lesions87. In this regard, Ch25h−/−;Ldlr−/− macrophages have increased levels of the pro-survival gene Cd5l, which has been reported to protect lipid-laden macrophages from apoptosis68, and make them less susceptible to LPS-induced apoptosis65–67. Furthermore, Cd5l expression was restricted to foamy and non-inflammatory macrophage clusters with barely detectable levels of Ch25h expression. These results demonstrate that 25-HC increases cell apoptosis, reduces efferocytosis and promotes inflammation, three hallmarks of defective inflammatory resolution.
For many years, 25-HC has been proposed to regulate cholesterol metabolism26, and it has been demonstrated that 25-HC added into culture media suppresses SREBP proteolytic processing88, 26,89 and activates LXR-mediated gene expression30,89. On the other hand, animals deficient in Ch25h do not exhibit any alteration in cholesterol metabolism32 and there is no evidence that Ch25h knockout animals have altered SREBP or LXR transcriptional activity. However, a recent study showed that 2h after LPS inhalation, WT animals had increased 25-HC in the lungs, and several LXR target genes (e.g., Abca1, Abcg1, Srebf1, Nr1h3 and Nr1h2) were upregulated, while this response was not present in Ch25h deficient mice85. Our data obtained in TG-EPM from hypercholesterolemic Ch25h−/−;Ldlr−/− mice indicate unaltered LXR transcriptional activity in basal and stimulated conditions, i.e., no significant differences in mRNA levels of more than 300 validated LXR target genes60 when compared to Ldlr−/− macrophages. However, it is possible that the effect described by Madenspatcher et al. in some specific LXR target genes85 can be buffered in our model, since LXR activity is presumably higher in lipid-loaded macrophages. In line with this, accumulation of desmosterol, an endogenous LXR ligand24,50, was not altered in the absence or presence of 25-HC. Similarly, secreted levels of the LXR ligand, 27-HC, that normally correlate with its cellular levels, were unaltered in Ldlr−/− and Ch25h−/−;Ldlr−/− macrophages. Furthermore, while TLR4-stimulated normolipidemic BMDMs downregulate Cyp51 expression and accumulate lanosterol to diminish innate immune responses36, lipid-laden peritoneal macrophages did not exhibit this effect regardless of the absence or presence of 25-HC. This also provides relevance of our results to hypercholesterolemic proatherogenic conditions.
In normolipidemia, increased 25-HC production after pathogen recognition reduces SREBP2 activity and cholesterol biosynthesis to prevent cholesterol-dependent mitochondrial dysfunction and inflammasome activation43,44. In lipid-laden macrophages, we did not find any alteration in mitochondrial ROS production or inflammasome activation in Ch25h−/−;Ldlr−/− macrophages compared to Ldlr−/− controls. This is consistent with unchanged mRNA levels of SREBP2 target genes, cholesterol, or desmosterol levels, which have also been associated with mito-ROS production and NLRP3-dependent inflammasome activation24, further indicating that 25-HC does not restrain SREBP2 activation and cholesterol biosynthesis in this setting. On the other hand, they suggested that Ch25h is not necessary for the initial downregulation of cholesterol biosynthesis but required to maintain this effect over time in a mTORC1-dependent manner44. Lysosomes are key for mTORC1 signaling90 and disturbances in lysosome biology have a clear impact on mTORC1 activation90. Interestingly, lipid-laden TG-EPM present significant differences in lysosome biology compared to BMDMs91. Therefore, some of the discrepancies could be additionally explained by a different mTORC1 response in peritoneal macrophages vs. the BMDMs used in their studies.
Atherosclerosis progression and plaque remodeling are determined by the crosstalk between vascular cells and macrophages in advanced plaques. In this regard, several chemokines and cytokines, among other bioactive molecules secreted by macrophages, influence the response of different cell types within the artery wall. Activated macrophages release 25-HC into the extracellular media32,38,41,92,93, this in turn promotes paracrine effects in nearby cells, including alterations in membrane composition38, migratory stimulation93, or NFκB activation92. Our present study shows that in response to LPS, lipid-laden macrophages secrete 25-HC, without inducing the secretion of other oxysterols. This suggests that 25-HC has a predominant role under inflammatory conditions. Furthermore, 25-HC blunts SMC migration in vitro and reduces SMC content within the fibrous cap of WT→Ldlr−/− mice, suggesting that 25-HC could participate as a lipid mediator in the crosstalk among these two cell types. Mechanistically, we found that 25-HC alters PDGFRβ signaling in SMC in vitro by affecting the pool of PM free cholesterol which alters PDGFRβ location within high-cholesterol PM microdomains and subsequent signal events80, which is in line with recent work that shows that SMCs that populate the fibrous cap in a PDGFRβ-dependent manner94.
Our work emphasizes the pro-inflammatory role of macrophage-derived 25-HC and demonstrates that 25-HC promotes inflammation and atherogenesis. Mechanistically, our experiments showed that 25-HC produced by activated lipid-loaded macrophages boosts the inflammatory response, increases apoptosis sensitivity, reduces efferocytotic capacity and promotes paracrine effects in neighbor cells. Additionally, present results show that 25-HC is not a major regulator of LXR or SREBP2 transcriptional activity in lipid-laden activated macrophages. This work opens the door for future studies further exploring the role of 25-HC as a biomarker of inflammation and plaque stability in human atherosclerosis.
Supplementary Material
CLINICAL PERSPECTIVE.
What is new?
25-hydroxycholesterol accumulates in human coronary atherosclerosis.
25-hydroxycholesterol produced by macrophages accelerates atherosclerosis progression and promotes plaque instability by promoting the inflammatory response in macrophages, and via paracrine actions on smooth muscle cells migratory response.
25-hydroxycholesterol in lipid-loaded macrophages amplifies their inflammatory response independently of the modulation of LXR or SREBP transcriptional activity.
What are the clinical implications?
Inhibition of 25-hydroxycholesterol production might delay atherosclerosis progression and promotes plaque stability.
Our study opens the door to explore the role of 25-hydroxycholesterol as a target to control inflammation and plaque stability in human atherosclerosis.
Acknowledgements
We thank Christopher Castaldi at Yale Center for Genome Analysis for RNA expression studies by bulk RNA-Seq and Rolando Garcia-Milian at Medical library for RNA-seq analysis advice. We also thank Dr. Jordan Pober at Yale School of Medicine for helpful suggestions. Working model graph was created with BioRender.com.
Source of Funding
The work at Suarez and Fernandez-Hernando laboratories is supported by grants from National Heart and Lung Institute (NHLBI), NIH (R35HL155988 to YS and R35HL135820 to CF-H) and the American Heart Association (AHA), (20TPA35490202 to YS and 20TPA35490416 to CFH). The collection of human coronary arteries was supported by R01 HL146723 to GT. Additional support is from R01 HL153712-01, NIH-NCATS UH3TR002067, AHA (20SFRN35210252) and CZI (NFL-2020-218415) to CG and 5F32DK10348902 to NLP. ACD and NR were recipient of grants from AHA (19POST34430108 to ACD and 17SDG33110002 to NR) and DG-C from Grant RTI2018-098113-B-I00 (Plan Estatal de Investigación Científica y Técnica y de Innovación 2017-2020, Ministerio de Ciencia, Innovación y Universidades, Spain, and the European Development Regional Fund (ERDF).
Non-standard Abbreviations and Acronyms
- 25-HC
25-hydroxycholesterol
- CH25H
25-hydroxylase
- BM
Bone marrow
- WD
Western diet
- SMC
Smooth muscle cell
- TLR4
Toll-like receptor 4
- NF-κB
Nuclear factor-κB
- LXR
Liver X receptor
- SREBP
Sterol regulatory-element binding protein
- LDL
Low-density lipoproteins
- Ox-LDL
Oxidized LDLs
- HMG-CoA
Hydroxy methyl glutaryl-coenzyme A
- LDLR
LDL receptor
- ABC
ATP-binding cassette
- IFN
Interferon
- IL
Interleukin
- RNA-seq
RNA-sequencing
- PM
Plasma membrane
- LC/MS-MS
Liquid chromatography–mass spectrometry
- scRNA-seq
Single cell RNA-seq
- Apo E
Apolipoprotein E
- SSC
Side Scatter
- Cyp
Cytochrome P450 family
- ORO
Oil red O
- SMA
Smooth alpha actin
- TG-EPM
Thioglycolate-elicited peritoneal macrophages
- TFs
Transcription factors
- ROS
Reactive oxygen species
- LPS
Lipopolysaccharide
- SMase
Sphingomyelinase
- ALOD4
Anthrolysin O peptide
- eGFP
Enhanced green fluorescence protein
- BMDMs
BM derived macrophages
Footnotes
Disclosures
None
Supplemental Materials
Online-only Supplemental Figures S1–8
Online-only Supplemental Figure legends for Figures S1–8
References
- 1.Cushing SD, Berliner JA, Valente AJ, Territo MC, Navab M, Parhami F, Gerrity R, Schwartz CJ, Fogelman AM. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc Natl Acad Sci U S A. 1990;87:5134–5138. doi: 10.1073/pnas.87.13.5134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hurt-Camejo E, Camejo G, Rosengren B, Lopez F, Ahlstrom C, Fager G, Bondjers G. Effect of arterial proteoglycans and glycosaminoglycans on low density lipoprotein oxidation and its uptake by human macrophages and arterial smooth muscle cells. Arterioscler Thromb. 1992;12:569–583. doi: 10.1161/01.atv.12.5.569 [DOI] [PubMed] [Google Scholar]
- 3.Skalen K, Gustafsson M, Rydberg EK, Hulten LM, Wiklund O, Innerarity TL, Boren J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002;417:750–754. doi: 10.1038/nature00804 [DOI] [PubMed] [Google Scholar]
- 4.Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, Gorbatov R, Sukhova GK, Gerhardt LM, Smyth D, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19:1166–1172. doi: 10.1038/nm.3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–161. doi: 10.1038/ni.1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, Becker CE, Ediriweera HN, Mullick AE, Golenbock DT, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. 2013;14:812–820. doi: 10.1038/ni.2639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tabas I, Garcia-Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209:13–22. doi: 10.1083/jcb.201412052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moore KJ, Freeman MW. Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler Thromb Vasc Biol. 2006;26:1702–1711. doi: 10.1161/01.ATV.0000229218.97976.43 [DOI] [PubMed] [Google Scholar]
- 9.Medzhitov R, Janeway C Jr., Innate immune recognition: mechanisms and pathways. Immunol Rev. 2000;173:89–97. doi: 10.1034/j.1600-065x.2000.917309.x [DOI] [PubMed] [Google Scholar]
- 10.Kim K, Shim D, Lee JS, Zaitsev K, Williams JW, Kim KW, Jang MY, Seok Jang H, Yun TJ, Lee SH, et al. Transcriptome Analysis Reveals Nonfoamy Rather Than Foamy Plaque Macrophages Are Proinflammatory in Atherosclerotic Murine Models. Circ Res. 2018;123:1127–1142. doi: 10.1161/CIRCRESAHA.118.312804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spann NJ, Glass CK. Sterols and oxysterols in immune cell function. Nat Immunol. 2013;14:893–900. doi: 10.1038/ni.2681 [DOI] [PubMed] [Google Scholar]
- 12.Back M, Yurdagul A Jr., Tabas I, Oorni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol. 2019;16:389–406. doi: 10.1038/s41569-019-0169-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383:728–731. doi: 10.1038/383728a0 [DOI] [PubMed] [Google Scholar]
- 14.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. 2003;9:213–219. doi: 10.1038/nm820 [DOI] [PubMed] [Google Scholar]
- 16.Castrillo A, Joseph SB, Marathe C, Mangelsdorf DJ, Tontonoz P. Liver X receptor-dependent repression of matrix metalloproteinase-9 expression in macrophages. J Biol Chem. 2003;278:10443–10449. doi: 10.1074/jbc.M213071200 [DOI] [PubMed] [Google Scholar]
- 17.Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, Rosenfeld MG, Glass CK. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol Cell. 2007;25:57–70. doi: 10.1016/j.molcel.2006.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghisletti S, Huang W, Jepsen K, Benner C, Hardiman G, Rosenfeld MG, Glass CK. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes Dev. 2009;23:681–693. doi: 10.1101/gad.1773109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oishi Y, Spann NJ, Link VM, Muse ED, Strid T, Edillor C, Kolar MJ, Matsuzaka T, Hayakawa S, Tao J, et al. SREBP1 Contributes to Resolution of Pro-inflammatory TLR4 Signaling by Reprogramming Fatty Acid Metabolism. Cell Metab. 2017;25:412–427. doi: 10.1016/j.cmet.2016.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo C, Chi Z, Jiang D, Xu T, Yu W, Wang Z, Chen S, Zhang L, Liu Q, Guo X, et al. Cholesterol Homeostatic Regulator SCAP-SREBP2 Integrates NLRP3 Inflammasome Activation and Cholesterol Biosynthetic Signaling in Macrophages. Immunity. 2018;49:842–856 e847. doi: 10.1016/j.immuni.2018.08.021 [DOI] [PubMed] [Google Scholar]
- 21.Kusnadi A, Park SH, Yuan R, Pannellini T, Giannopoulou E, Oliver D, Lu T, Park-Min KH, Ivashkiv LB. The Cytokine TNF Promotes Transcription Factor SREBP Activity and Binding to Inflammatory Genes to Activate Macrophages and Limit Tissue Repair. Immunity. 2019;51:241–257 e249. doi: 10.1016/j.immuni.2019.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muse ED, Yu S, Edillor CR, Tao J, Spann NJ, Troutman TD, Seidman JS, Henke A, Roland JT, Ozeki KA, et al. Cell-specific discrimination of desmosterol and desmosterol mimetics confers selective regulation of LXR and SREBP in macrophages. Proc Natl Acad Sci U S A. 2018;115:E4680–E4689. doi: 10.1073/pnas.1714518115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adams CM, Reitz J, De Brabander JK, Feramisco JD, Li L, Brown MS, Goldstein JL. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J Biol Chem. 2004;279:52772–52780. doi: 10.1074/jbc.M410302200 [DOI] [PubMed] [Google Scholar]
- 24.Zhang X, McDonald JG, Aryal B, Canfran-Duque A, Goldberg EL, Araldi E, Ding W, Fan Y, Thompson BM, Singh AK, et al. Desmosterol suppresses macrophage inflammasome activation and protects against vascular inflammation and atherosclerosis. Proc Natl Acad Sci U S A. 2021;118. doi: 10.1073/pnas.2107682118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lund EG, Kerr TA, Sakai J, Li WP, Russell DW. cDNA cloning of mouse and human cholesterol 25-hydroxylases, polytopic membrane proteins that synthesize a potent oxysterol regulator of lipid metabolism. J Biol Chem. 1998;273:34316–34327. doi: 10.1074/jbc.273.51.34316 [DOI] [PubMed] [Google Scholar]
- 26.Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, Goldstein JL. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc Natl Acad Sci U S A. 2007;104:6511–6518. doi: 10.1073/pnas.0700899104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022 [DOI] [PubMed] [Google Scholar]
- 28.Yang C, McDonald JG, Patel A, Zhang Y, Umetani M, Xu F, Westover EJ, Covey DF, Mangelsdorf DJ, Cohen JC, et al. Sterol intermediates from cholesterol biosynthetic pathway as liver X receptor ligands. J Biol Chem. 2006;281:27816–27826. doi: 10.1074/jbc.M603781200 [DOI] [PubMed] [Google Scholar]
- 29.Chen W, Chen G, Head DL, Mangelsdorf DJ, Russell DW. Enzymatic reduction of oxysterols impairs LXR signaling in cultured cells and the livers of mice. Cell Metab. 2007;5:73–79. doi: 10.1016/j.cmet.2006.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA, Tontonoz P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc Natl Acad Sci U S A. 2000;97:12097–12102. doi: 10.1073/pnas.200367697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du X, Pham YH, Brown AJ. Effects of 25-hydroxycholesterol on cholesterol esterification and sterol regulatory element-binding protein processing are dissociable: implications for cholesterol movement to the regulatory pool in the endoplasmic reticulum. J Biol Chem. 2004;279:47010–47016. doi: 10.1074/jbc.M408690200 [DOI] [PubMed] [Google Scholar]
- 32.Bauman DR, Bitmansour AD, McDonald JG, Thompson BM, Liang G, Russell DW. 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc Natl Acad Sci U S A. 2009;106:16764–16769. doi: 10.1073/pnas.0909142106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712 [DOI] [PubMed] [Google Scholar]
- 34.Bjorkhem I Are side-chain oxidized oxysterols regulators also in vivo? J Lipid Res. 2009;50 Suppl:S213–218. doi: 10.1194/jlr.R800025-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diczfalusy U On the formation and possible biological role of 25-hydroxycholesterol. Biochimie. 2013;95:455–460. doi: 10.1016/j.biochi.2012.06.016 [DOI] [PubMed] [Google Scholar]
- 36.Araldi E, Fernandez-Fuertes M, Canfran-Duque A, Tang W, Cline GW, Madrigal-Matute J, Pober JS, Lasuncion MA, Wu D, Fernandez-Hernando C, et al. Lanosterol Modulates TLR4-Mediated Innate Immune Responses in Macrophages. Cell Rep. 2017;19:2743–2755. doi: 10.1016/j.celrep.2017.05.093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, Lacaze P, Watterson S, Griffiths SJ, Spann NJ, et al. The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity. 2013;38:106–118. doi: 10.1016/j.immuni.2012.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, Pernet O, Guo H, Nusbaum R, Zack JA, et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity. 2013;38:92–105. doi: 10.1016/j.immuni.2012.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li C, Deng YQ, Wang S, Ma F, Aliyari R, Huang XY, Zhang NN, Watanabe M, Dong HL, Liu P, et al. 25-Hydroxycholesterol Protects Host against Zika Virus Infection and Its Associated Microcephaly in a Mouse Model. Immunity. 2017;46:446–456. doi: 10.1016/j.immuni.2017.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y, Wei Z, Zhang Y, Ma X, Chen Y, Yu M, Ma C, Li X, Cao Y, Liu J, et al. Activation of liver X receptor plays a central role in antiviral actions of 25-hydroxycholesterol. J Lipid Res. 2018;59:2287–2296. doi: 10.1194/jlr.M084558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gold ES, Diercks AH, Podolsky I, Podyminogin RL, Askovich PS, Treuting PM, Aderem A. 25-Hydroxycholesterol acts as an amplifier of inflammatory signaling. Proc Natl Acad Sci U S A. 2014;111:10666–10671. doi: 10.1073/pnas.1404271111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koarai A, Yanagisawa S, Sugiura H, Ichikawa T, Kikuchi T, Furukawa K, Akamatsu K, Hirano T, Nakanishi M, Matsunaga K, et al. 25-Hydroxycholesterol enhances cytokine release and Toll-like receptor 3 response in airway epithelial cells. Respir Res. 2012;13:63. doi: 10.1186/1465-9921-13-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reboldi A, Dang EV, McDonald JG, Liang G, Russell DW, Cyster JG. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science. 2014;345:679–684. doi: 10.1126/science.1254790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dang EV, McDonald JG, Russell DW, Cyster JG. Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell. 2017;171:1057–1071 e1011. doi: 10.1016/j.cell.2017.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Z, Martin M, Zhang J, Huang HY, Bai L, Zhang J, Kang J, He M, Li J, Maurya MR, et al. Kruppel-Like Factor 4 Regulation of Cholesterol-25-Hydroxylase and Liver X Receptor Mitigates Atherosclerosis Susceptibility. Circulation. 2017;136:1315–1330. doi: 10.1161/CIRCULATIONAHA.117.027462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gold ES, Ramsey SA, Sartain MJ, Selinummi J, Podolsky I, Rodriguez DJ, Moritz RL, Aderem A. ATF3 protects against atherosclerosis by suppressing 25-hydroxycholesterol-induced lipid body formation. J Exp Med. 2012;209:807–817. doi: 10.1084/jem.20111202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen PY, Qin L, Li G, Tellides G, Simons M. Smooth muscle FGF/TGFbeta cross talk regulates atherosclerosis progression. EMBO Mol Med. 2016;8:712–728. doi: 10.15252/emmm.201506181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, Khan NS, Wong CK, Shamailova R, Hill CA, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. 2019;25:1576–1588. doi: 10.1038/s41591-019-0590-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pan H, Xue C, Auerbach BJ, Fan J, Bashore AC, Cui J, Yang DY, Trignano SB, Liu W, Shi J, et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation. 2020;142:2060–2075. doi: 10.1161/CIRCULATIONAHA.120.048378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, Reichart D, Fox JN, Shaked I, Heudobler D, et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell. 2012;151:138–152. doi: 10.1016/j.cell.2012.06.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Price NL, Rotllan N, Canfran-Duque A, Zhang X, Pati P, Arias N, Moen J, Mayr M, Ford DA, Baldan A, et al. Genetic Dissection of the Impact of miR-33a and miR-33b during the Progression of Atherosclerosis. Cell Rep. 2017;21:1317–1330. doi: 10.1016/j.celrep.2017.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li AC, Binder CJ, Gutierrez A, Brown KK, Plotkin CR, Pattison JW, Valledor AF, Davis RA, Willson TM, Witztum JL, et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J Clin Invest. 2004;114:1564–1576. doi: 10.1172/JCI18730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diczfalusy U, Olofsson KE, Carlsson AM, Gong M, Golenbock DT, Rooyackers O, Flaring U, Bjorkbacka H. Marked upregulation of cholesterol 25-hydroxylase expression by lipopolysaccharide. J Lipid Res. 2009;50:2258–2264. doi: 10.1194/jlr.M900107-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park K, Scott AL. Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J Leukoc Biol. 2010;88:1081–1087. doi: 10.1189/jlb.0610318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tugal D, Liao X, Jain MK. Transcriptional control of macrophage polarization. Arterioscler Thromb Vasc Biol. 2013;33:1135–1144. doi: 10.1161/ATVBAHA.113.301453 [DOI] [PubMed] [Google Scholar]
- 56.Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169:2253–2263. doi: 10.4049/jimmunol.169.5.2253 [DOI] [PubMed] [Google Scholar]
- 57.Cuadrado A, Manda G, Hassan A, Alcaraz MJ, Barbas C, Daiber A, Ghezzi P, Leon R, Lopez MG, Oliva B, et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol Rev. 2018;70:348–383. doi: 10.1124/pr.117.014753 [DOI] [PubMed] [Google Scholar]
- 58.Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, Tanaka N, Moriguchi T, Motohashi H, Nakayama K, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. 2016;7:11624. doi: 10.1038/ncomms11624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen B, Huang S, Su Y, Wu YJ, Hanna A, Brickshawana A, Graff J, Frangogiannis NG. Macrophage Smad3 Protects the Infarcted Heart, Stimulating Phagocytosis and Regulating Inflammation. Circ Res. 2019;125:55–70. doi: 10.1161/CIRCRESAHA.119.315069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramon-Vazquez A, de la Rosa JV, Tabraue C, Lopez F, Diaz-Chico BN, Bosca L, Tontonoz P, Alemany S, Castrillo A. Common and Differential Transcriptional Actions of Nuclear Receptors Liver X Receptors alpha and beta in Macrophages. Mol Cell Biol. 2019;39. doi: 10.1128/MCB.00376-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goedeke L, Canfran-Duque A, Rotllan N, Chaube B, Thompson BM, Lee RG, Cline GW, McDonald JG, Shulman GI, Lasuncion MA, et al. MMAB promotes negative feedback control of cholesterol homeostasis. Nat Commun. 2021;12:6448. doi: 10.1038/s41467-021-26787-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xaus J, Comalada M, Valledor AF, Lloberas J, Lopez-Soriano F, Argiles JM, Bogdan C, Celada A. LPS induces apoptosis in macrophages mostly through the autocrine production of TNF-alpha. Blood. 2000;95:3823–3831. [PubMed] [Google Scholar]
- 63.Rayner KJ. Cell Death in the Vessel Wall: The Good, the Bad, the Ugly. Arterioscler Thromb Vasc Biol. 2017;37:e75–e81. doi: 10.1161/ATVBAHA.117.309229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Viaud M, Ivanov S, Vujic N, Duta-Mare M, Aira LE, Barouillet T, Garcia E, Orange F, Dugail I, Hainault I, et al. Lysosomal Cholesterol Hydrolysis Couples Efferocytosis to Anti-Inflammatory Oxysterol Production. Circ Res. 2018;122:1369–1384. doi: 10.1161/CIRCRESAHA.117.312333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Valledor AF, Hsu LC, Ogawa S, Sawka-Verhelle D, Karin M, Glass CK. Activation of liver X receptors and retinoid X receptors prevents bacterial-induced macrophage apoptosis. Proc Natl Acad Sci U S A. 2004;101:17813–17818. doi: 10.1073/pnas.0407749101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, Hogenesch J, O’Connell RM, Cheng G, Saez E, et al. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell. 2004;119:299–309. doi: 10.1016/j.cell.2004.09.032 [DOI] [PubMed] [Google Scholar]
- 67.Im SS, Osborne TF. Protection from bacterial-toxin-induced apoptosis in macrophages requires the lipogenic transcription factor sterol regulatory element binding protein 1a. Mol Cell Biol. 2012;32:2196–2202. doi: 10.1128/MCB.06294-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arai S, Shelton JM, Chen M, Bradley MN, Castrillo A, Bookout AL, Mak PA, Edwards PA, Mangelsdorf DJ, Tontonoz P, et al. A role for the apoptosis inhibitory factor AIM/Spalpha/Api6 in atherosclerosis development. Cell Metab. 2005;1:201–213. doi: 10.1016/j.cmet.2005.02.002 [DOI] [PubMed] [Google Scholar]
- 69.Schlegel M, Sharma M, Brown EJ, Newman AAC, Cyr Y, Afonso MS, Corr EM, Koelwyn GJ, van Solingen C, Guzman J, et al. Silencing Myeloid Netrin-1 Induces Inflammation Resolution and Plaque Regression. Circ Res. 2021;129:530–546. doi: 10.1161/CIRCRESAHA.121.319313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aprahamian T, Rifkin I, Bonegio R, Hugel B, Freyssinet JM, Sato K, Castellot JJ Jr., Walsh K. Impaired clearance of apoptotic cells promotes synergy between atherogenesis and autoimmune disease. J Exp Med. 2004;199:1121–1131. doi: 10.1084/jem.20031557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:1256–1261. doi: 10.1161/01.ATV.0000166517.18801.a7 [DOI] [PubMed] [Google Scholar]
- 72.Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe−/− mice. Arterioscler Thromb Vasc Biol. 2008;28:1421–1428. doi: 10.1161/ATVBAHA.108.167197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Koseki M, Hirano K, Masuda D, Ikegami C, Tanaka M, Ota A, Sandoval JC, Nakagawa-Toyama Y, Sato SB, Kobayashi T, et al. Increased lipid rafts and accelerated lipopolysaccharide-induced tumor necrosis factor-alpha secretion in Abca1-deficient macrophages. J Lipid Res. 2007;48:299–306. doi: 10.1194/jlr.M600428-JLR200 [DOI] [PubMed] [Google Scholar]
- 74.Zhu X, Lee JY, Timmins JM, Brown JM, Boudyguina E, Mulya A, Gebre AK, Willingham MC, Hiltbold EM, Mishra N, et al. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J Biol Chem. 2008;283:22930–22941. doi: 10.1074/jbc.M801408200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhu X, Owen JS, Wilson MD, Li H, Griffiths GL, Thomas MJ, Hiltbold EM, Fessler MB, Parks JS. Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J Lipid Res. 2010;51:3196–3206. doi: 10.1194/jlr.M006486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N, Tall AR. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837–1847. doi: 10.1161/CIRCULATIONAHA.108.793869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ito A, Hong C, Rong X, Zhu X, Tarling EJ, Hedde PN, Gratton E, Parks J, Tontonoz P. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. Elife. 2015;4:e08009. doi: 10.7554/eLife.08009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhou QD, Chi X, Lee MS, Hsieh WY, Mkrtchyan JJ, Feng AC, He C, York AG, Bui VL, Kronenberger EB, et al. Interferon-mediated reprogramming of membrane cholesterol to evade bacterial toxins. Nat Immunol. 2020;21:746–755. doi: 10.1038/s41590-020-0695-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Abrams ME, Johnson KA, Perelman SS, Zhang LS, Endapally S, Mar KB, Thompson BM, McDonald JG, Schoggins JW, Radhakrishnan A, et al. Oxysterols provide innate immunity to bacterial infection by mobilizing cell surface accessible cholesterol. Nat Microbiol. 2020;5:929–942. doi: 10.1038/s41564-020-0701-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Seong J, Huang M, Sim KM, Kim H, Wang Y. FRET-based Visualization of PDGF Receptor Activation at Membrane Microdomains. Sci Rep. 2017;7:1593. doi: 10.1038/s41598-017-01789-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fernandez-Hernando C, Jozsef L, Jenkins D, Di Lorenzo A, Sessa WC. Absence of Akt1 reduces vascular smooth muscle cell migration and survival and induces features of plaque vulnerability and cardiac dysfunction during atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:2033–2040. doi: 10.1161/ATVBAHA.109.196394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–637. doi: 10.1038/nm.3866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Russo L, Muir L, Geletka L, Delproposto J, Baker N, Flesher C, O’Rourke R, Lumeng CN. Cholesterol 25-hydroxylase (CH25H) as a promoter of adipose tissue inflammation in obesity and diabetes. Mol Metab. 2020;39:100983. doi: 10.1016/j.molmet.2020.100983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nagelin MH, Srinivasan S, Nadler JL, Hedrick CC. Murine 12/15-lipoxygenase regulates ATP-binding cassette transporter G1 protein degradation through p38- and JNK2-dependent pathways. J Biol Chem. 2009;284:31303–31314. doi: 10.1074/jbc.M109.028910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Madenspacher JH, Morrell ED, Gowdy KM, McDonald JG, Thompson BM, Muse G, Martinez J, Thomas S, Mikacenic C, Nick JA, et al. Cholesterol 25-hydroxylase promotes efferocytosis and resolution of lung inflammation. JCI Insight. 2020;5. doi: 10.1172/jci.insight.137189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thorp E, Vaisar T, Subramanian M, Mautner L, Blobel C, Tabas I. Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cdelta, and p38 mitogen-activated protein kinase (MAPK). J Biol Chem. 2011;286:33335–33344. doi: 10.1074/jbc.M111.263020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10:36–46. doi: 10.1038/nri2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sudhof TC, Van der Westhuyzen DR, Goldstein JL, Brown MS, Russell DW. Three direct repeats and a TATA-like sequence are required for regulated expression of the human low density lipoprotein receptor gene. J Biol Chem. 1987;262:10773–10779. [PubMed] [Google Scholar]
- 89.Shibata N, Carlin AF, Spann NJ, Saijo K, Morello CS, McDonald JG, Romanoski CE, Maurya MR, Kaikkonen MU, Lam MT, et al. 25-Hydroxycholesterol activates the integrated stress response to reprogram transcription and translation in macrophages. J Biol Chem. 2013;288:35812–35823. doi: 10.1074/jbc.M113.519637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014;24:400–406. doi: 10.1016/j.tcb.2014.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Weber K, Schilling JD. Distinct lysosome phenotypes influence inflammatory function in peritoneal and bone marrow-derived macrophages. Int J Inflam. 2014;2014:154936. doi: 10.1155/2014/154936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pokharel SM, Shil NK, Gc JB, Colburn ZT, Tsai SY, Segovia JA, Chang TH, Bandyopadhyay S, Natesan S, Jones JCR, et al. Integrin activation by the lipid molecule 25-hydroxycholesterol induces a proinflammatory response. Nat Commun. 2019;10:1482. doi: 10.1038/s41467-019-09453-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yi T, Wang X, Kelly LM, An J, Xu Y, Sailer AW, Gustafsson JA, Russell DW, Cyster JG. Oxysterol gradient generation by lymphoid stromal cells guides activated B cell movement during humoral responses. Immunity. 2012;37:535–548. doi: 10.1016/j.immuni.2012.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Newman AAC, Serbulea V, Baylis RA, Shankman LS, Bradley X, Alencar GF, Owsiany K, Deaton RA, Karnewar S, Shamsuzzaman S, et al. Multiple cell types contribute to the atherosclerotic lesion fibrous cap by PDGFRbeta and bioenergetic mechanisms. Nat Metab. 2021;3:166–181. doi: 10.1038/s42255-020-00338-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rotllan N, Chamorro-Jorganes A, Araldi E, Wanschel AC, Aryal B, Aranda JF, Goedeke L, Salerno AG, Ramirez CM, Sessa WC, et al. Hematopoietic Akt2 deficiency attenuates the progression of atherosclerosis. FASEB J. 2015;29:597–610. doi: 10.1096/fj.14-262097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Canfran-Duque A, Rotllan N, Zhang X, Fernandez-Fuertes M, Ramirez-Hidalgo C, Araldi E, Daimiel L, Busto R, Fernandez-Hernando C, Suarez Y. Macrophage deficiency of miR-21 promotes apoptosis, plaque necrosis, and vascular inflammation during atherogenesis. EMBO Mol Med. 2017;9:1244–1262. doi: 10.15252/emmm.201607492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang X, Sun J, Canfran-Duque A, Aryal B, Tellides G, Chang YJ, Suarez Y, Osborne TF, Fernandez-Hernando C. Deficiency of histone lysine methyltransferase SETDB2 in hematopoietic cells promotes vascular inflammation and accelerates atherosclerosis. JCI Insight. 2021;6. doi: 10.1172/jci.insight.147984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Seimon TA, Wang Y, Han S, Senokuchi T, Schrijvers DM, Kuriakose G, Tall AR, Tabas IA. Macrophage deficiency of p38alpha MAPK promotes apoptosis and plaque necrosis in advanced atherosclerotic lesions in mice. J Clin Invest. 2009;119:886–898. doi: 10.1172/JCI37262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099 [DOI] [PubMed] [Google Scholar]
- 100.McDonald JG, Smith DD, Stiles AR, Russell DW. A comprehensive method for extraction and quantitative analysis of sterols and secosteroids from human plasma. J Lipid Res. 2012;53:1399–1409. doi: 10.1194/jlr.D022285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mitsche MA, McDonald JG, Hobbs HH, Cohen JC. Flux analysis of cholesterol biosynthesis in vivo reveals multiple tissue and cell-type specific pathways. Elife. 2015;4:e07999. doi: 10.7554/eLife.07999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Endapally S, Infante RE, Radhakrishnan A. Monitoring and Modulating Intracellular Cholesterol Trafficking Using ALOD4, a Cholesterol-Binding Protein. Methods Mol Biol. 2019;1949:153–163. doi: 10.1007/978-1-4939-9136-5_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba AE, Zernecke A. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ Res. 2018;122:1661–1674. doi: 10.1161/CIRCRESAHA.117.312509 [DOI] [PubMed] [Google Scholar]
- 104.Willemsen L, de Winther MP. Macrophage subsets in atherosclerosis as defined by single-cell technologies. J Pathol. 2020;250:705–714. doi: 10.1002/path.5392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Do TH, Ma F, Andrade PR, Teles R, de Andrade Silva BJ, Hu C, Espinoza A, Hsu JE, Cho CS, Kim M, et al. TREM2 macrophages induced by human lipids drive inflammation in acne lesions. Sci Immunol. 2022;7:eabo2787. doi: 10.1126/sciimmunol.abo2787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.El Kharbili M, Aviszus K, Sasse SK, Zhao X, Serban KA, Majka SM, Gerber AN, Gally F. Macrophage programming is regulated by a cooperative interaction between fatty acid binding protein 5 and peroxisome proliferator-activated receptor gamma. FASEB J. 2022;36:e22300. doi: 10.1096/fj.202200128R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Guo Y, Liu Y, Zhao S, Xu W, Li Y, Zhao P, Wang D, Cheng H, Ke Y, Zhang X. Oxidative stress-induced FABP5 S-glutathionylation protects against acute lung injury by suppressing inflammation in macrophages. Nat Commun. 2021;12:7094. doi: 10.1038/s41467-021-27428-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Seo J, Jeong DW, Park JW, Lee KW, Fukuda J, Chun YS. Fatty-acid-induced FABP5/HIF-1 reprograms lipid metabolism and enhances the proliferation of liver cancer cells. Commun Biol. 2020;3:638. doi: 10.1038/s42003-020-01367-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Umbarawan Y, Enoura A, Ogura H, Sato T, Horikawa M, Ishii T, Sunaga H, Matsui H, Yokoyama T, Kawakami R, et al. FABP5 Is a Sensitive Marker for Lipid-Rich Macrophages in the Luminal Side of Atherosclerotic Lesions. Int Heart J. 2021;62:666–676. doi: 10.1536/ihj.20-676 [DOI] [PubMed] [Google Scholar]
- 110.Di Gregoli K, Somerville M, Bianco R, Thomas AC, Frankow A, Newby AC, George SJ, Jackson CL, Johnson JL. Galectin-3 Identifies a Subset of Macrophages With a Potential Beneficial Role in Atherosclerosis. Arterioscler Thromb Vasc Biol. 2020;40:1491–1509. doi: 10.1161/ATVBAHA.120.314252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu BC, Sarhan J, Panda A, Muendlein HI, Ilyukha V, Coers J, Yamamoto M, Isberg RR, Poltorak A. Constitutive Interferon Maintains GBP Expression Required for Release of Bacterial Components Upstream of Pyroptosis and Anti-DNA Responses. Cell Rep. 2018;24:155–168 e155. doi: 10.1016/j.celrep.2018.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Goo YH, Son SH, Yechoor VK, Paul A. Transcriptional Profiling of Foam Cells Reveals Induction of Guanylate-Binding Proteins Following Western Diet Acceleration of Atherosclerosis in the Absence of Global Changes in Inflammation. J Am Heart Assoc. 2016;5:e002663. doi: 10.1161/JAHA.115.002663 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.