

Abstract
Systemic sclerosis, also known as scleroderma, is a rare and complex autoimmune connective-tissue disease. Once considered an untreatable and unpredictable condition, research advancements have improved our understanding of its disease pathogenesis and clinical phenotypes and expanded our treatment armamentarium. Early and accurate diagnosis is essential, while ongoing efforts to risk stratify patients have a central role in predicting both organ involvement and disease progression. A holistic approach is required when choosing the optimal therapeutic strategy, balancing the side-effect profile with efficacy and tailoring the treatment according to the goals of care of the patient. This Seminar reviews the multiple clinical dimensions of systemic sclerosis, beginning at a precursor very early stage of disease, with a focus on timely early detection of organ involvement. This Seminar also summarises management considerations according to the pathological hallmarks of systemic sclerosis (eg, inflammation, fibrosis, and vasculopathy) and highlights unmet needs and opportunities for future research and discovery.
Introduction
Systemic sclerosis is an orphan disease characterised by autoimmunity, fibrosis of the skin and internal organs, and vasculopathy. Although the prevalence of systemic sclerosis is relatively low, the burden of disease is substantial.1,2 Systemic sclerosis has the highest mortality among all rheumatic diseases.3,4 The heterogeneous expression of this rare disease poses challenges to both the patient and clinician, particularly with regard to predicting the development of serious internal organ involvement. Clinicians often struggle to diagnose systemic sclerosis early in the disease course. Late referrals to specialty centres, scarcity of optimal multidisciplinary care, and a paucity of policy concerning patient pathways can hinder proper patient care. Furthermore, evidence-based clinical practice guidelines concerning the treatment of certain organ complications, such as gastrointestinal tract and cardiac involvement, do not exist.5
Patients with systemic sclerosis experience substantial uncertainty regarding systemic sclerosis-related taxo nomy and management, and many have limited access to knowledgeable health-care providers. In addition, patients with systemic sclerosis will invariably incur considerable personal health-care expenses (eg, non-reim burse ment of certain therapies) over the course of their disease, and it is not uncommon for patients to have to stop working.6,7 In essence, the experience of having systemic sclerosis can drastically alter quality of life and life expectancy.
Promisingly, the European Commission has recently launched European reference networks, virtual networks involving health-care providers across Europe, to address some of the aforementioned challenges.8
Clinical presentation
The earliest signs of systemic sclerosis include Raynaud phenomenon and fatigue, which are non-specific for systemic sclerosis and can have several alternative causes.9 Efforts have been made to identify patients with Raynaud phenomenon who are at greatest risk of the development of systemic sclerosis. Two validated sets of criteria exist to detect this early stage of systemic sclerosis. In 2001, LeRoy and Medsger10 proposed criteria for the early diagnosis of systemic sclerosis, which included Raynaud phenomenon, systemic sclerosis-speci fic autoimmune antibodies, and scleroderma-type changes on nailfold capillaroscopy. A recent prospective study demonstrated that a definite diagnosis of systemic sclerosis occurred in 65·9% of patients at 5 years and 72·7% of patients at 10 years in those with Raynaud phenomenon who had both a systemic sclerosis-specific antibody and a scleroderma pattern on capillaroscopy (figure 1).12,13,15 In 2011, very early diagnosis of systemic sclerosis criteria were proposed and consisted of similar criteria to the LeRoy and Medsger10 criteria for early systemic sclerosis, with the addition of puffy fingers.14,16
Figure 1: Clinical features of (very) early systemic sclerosis.
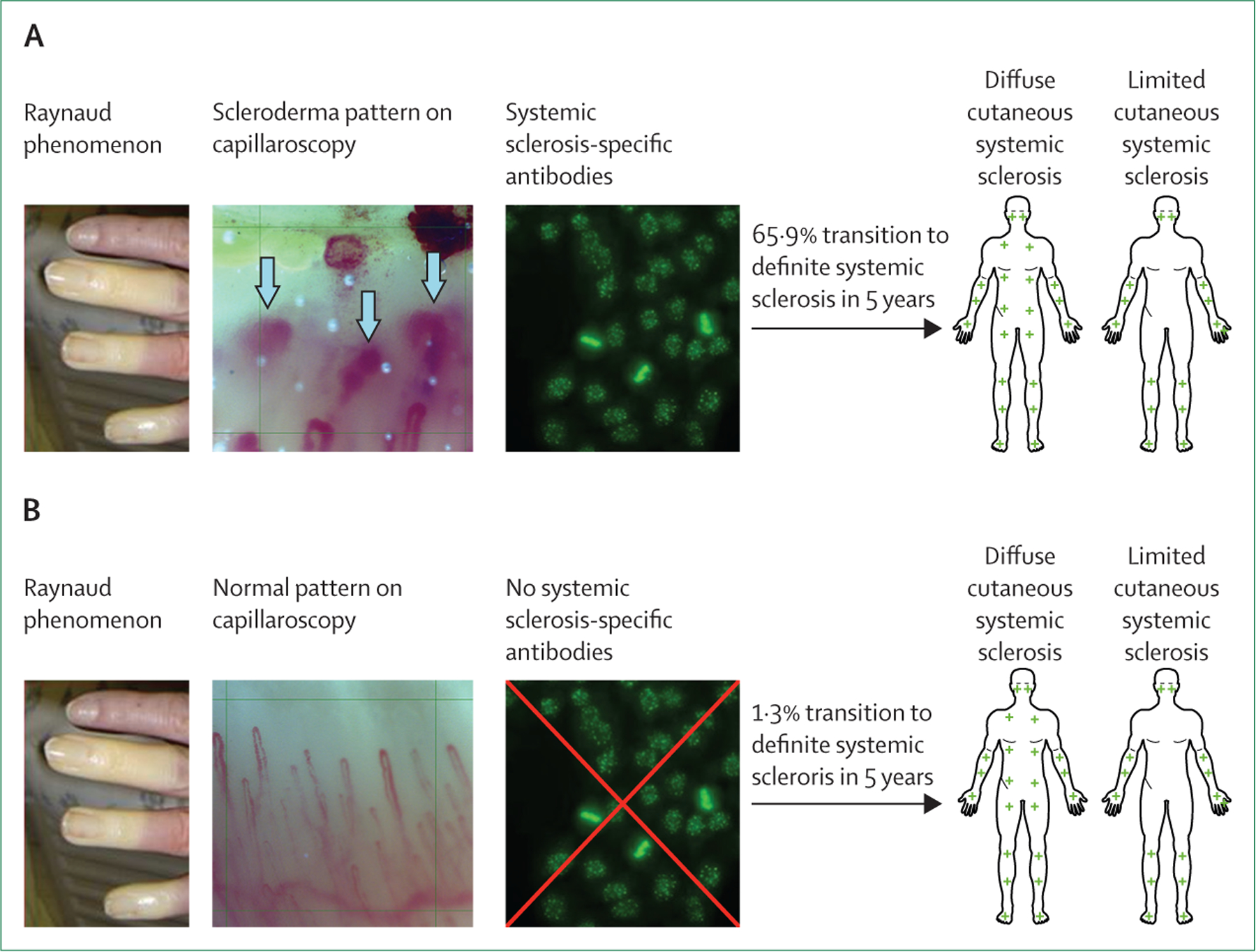
(A) The proportion of patients fulfilling the 2013 American College of Rheumatology and European Alliance of Associations for Rheumatology Classification Criteria for systemic sclerosis11 at 5 years was as follows for the following combinations of criteria: in patients with a scleroderma pattern on capillaroscopy plus systemic sclerosis-specific antibodies, 65·9% developed definite systemic sclerosis at 5 years;12,13 in patients with positive antinuclear antibodies and puffy fingers, 79·0% developed definite systemic sclerosis at 5 years; in patients with systemic sclerosis-specific autoantibodies and puffy fingers, 94·1% developed definite systemic sclerosis at 5 years; and in patients with capillaroscopic abnormalities (both systemic sclerosis specific as well as non-specific abnormalities) and systemic sclerosis-specific antibodies, 82·2% developed systemic sclerosis at 5 years.14 (B) 1·3% of patients with Raynaud phenomenon in the absence of scleroderma patterns on capillaroscopy and systemic sclerosis-specific antibodies developed systemic sclerosis at 5 years follow-up.11
Rate of progression to definite systemic sclerosis varies on the basis of systemic sclerosis subtype. Typically, patients with diffuse cutaneous systemic sclerosis develop their first non-Raynaud phenomenon symptom of systemic sclerosis within 1–2 years of the onset of Raynaud phenomenon; however, some patients with diffuse cutaneous systemic sclerosis can develop non-Raynaud phenomenon symptoms in parallel to or within weeks of the onset of Raynaud phenomenon. By contrast, patients with limited cutaneous systemic sclerosis develop their first non-Raynaud-phenomenon symptom of systemic sclerosis between 5 years and 10 years after onset of Raynaud phenomenon.17 This temporal sequence of events, although not universal, can help to establish disease subset early and improve risk stratification. Each cutaneous subtype is associated with a different natural history of the disease, severity of organ involvement, and mortality rate.4,18 Although no diagnostic criteria exist, clinicians sometimes incorrectly use research classification criteria (figure 2) to make a diagnosis of systemic sclerosis.11 However, fulfilment of research classification criteria is not necessary for a systemic sclerosis diagnosis.
Figure 2: The 2013 American College of Rheumatology and European Alliance of Associations for Rheumatology Classification Criteria for systemic sclerosis.
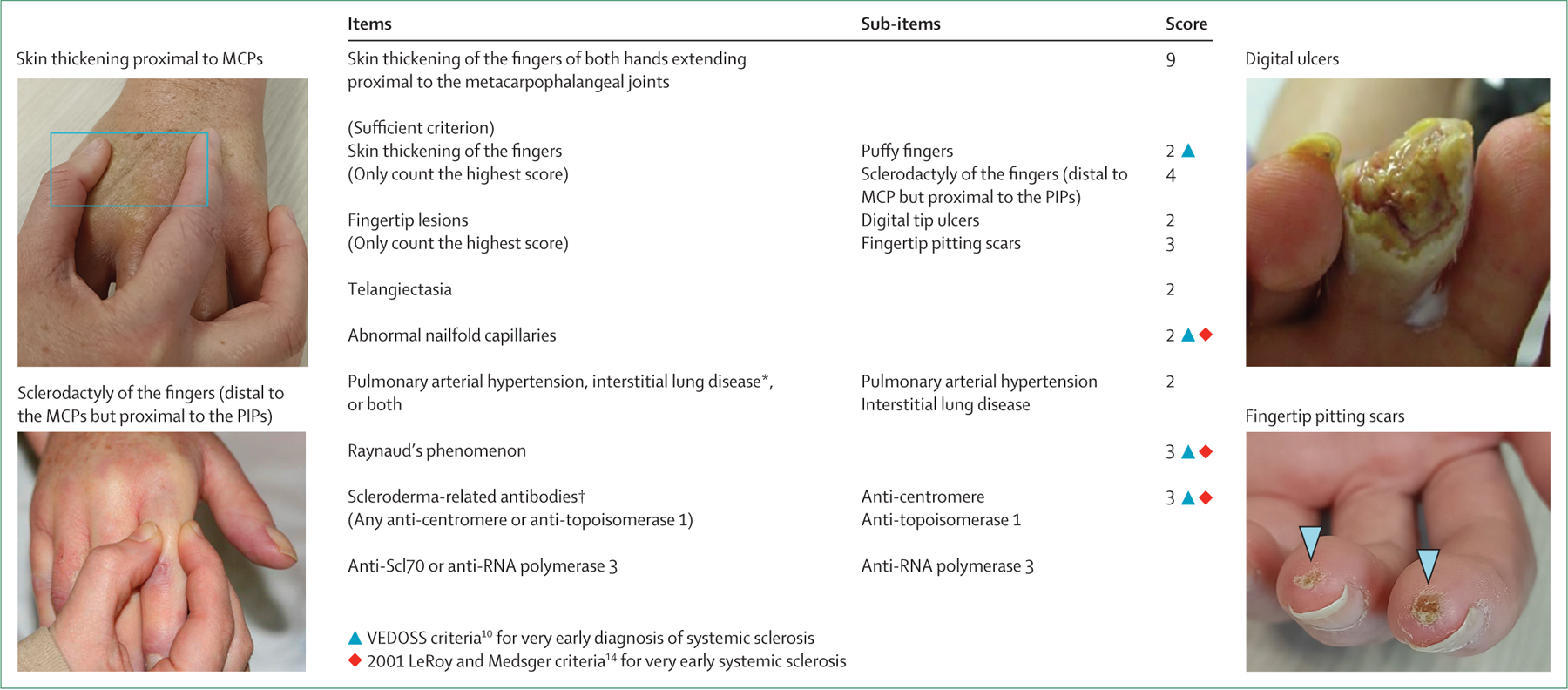
A total score of 9 or higher is sufficient to classify a patient as having definite systemic sclerosis, in the absence of other causes for the individual criterion.18 These criteria were developed for research purposes and do not represent diagnostic criteria. *Maximum score of 2. †Maximum score of 3. MCP=metacarpophalangeal joint. PIP=proximal interphalangeal joint. VEDOSS=very early diagnosis of systemic sclerosis.
Clinical evaluation
Systemic sclerosis can affect multiple organ systems with varying degrees of severity. Although some systemic sclerosis manifestations are clinically silent or insidious and progress slowly over time, others can have an acute onset and progress rapidly. Comprehensive clinical, radiographic, and laboratory analyses are necessary to phenotype patients at initial presentation.5,19
Serological profile
Although a positive antinuclear antibody as determined by indirect immunofluorescence is observed in up to 95% of patients with systemic sclerosis, more specific systemic sclerosis-related autoantibodies are found in distinct groups of patients with the disease, and their presence helps to predict organ involvement and severity (table 1).20,21 A subgroup of patients can exhibit positive antinuclear autoantibodies without the presence of any systemic sclerosis-specific autoantibodies. These patients can develop rapidly progressive disease and have an associated high mortality rate.22
Table 1:
Autoantibodies associated with systemic sclerosis and their clinical correlates
| Cutaneous subtype | Clinical correlates | |
|---|---|---|
| Anti-centromere | Limited cutaneous systemic sclerosis | Pulmonary arterial hypertension |
| Anti-topoisomerase 1 (anti-ScI 70)* | Diffuse cutaneous systemic sclerosis | Progressive interstitial lung disease |
| Anti-RNA polymerase 3 | Diffuse cutaneous systemic sclerosis | Renal crisis and malignancy |
| Anti-U1 ribonucleoprotein (anti-ribonucleoprotein) | Limited cutaneous systemic sclerosis | Mixed connective tissue disease |
| Anti-U3 ribonucleoprotein (anti-fibrillarin) | Diffuse cutaneous systemic sclerosis | Pulmonary arterial hypertension and myositis |
| Anti-Pm-Scl | Limited cutaneous systemic sclerosis | Myositis |
| Anti-Th/To | Limited cutaneous systemic sclerosis | Interstitial lung disease and pulmonary arterial hypertension |
Testing for systemic sclerosis-associated auto-antibodies is recommended early in the evaluation for systemic sclerosis because it can help establish a systemic sclerosis diagnosis and inform risk-stratification strategies. Patients will seldomly produce more than one systemic sclerosis-related auto-antibody concurrently.
Immunodiffusion is the preferred method for detecting anti-topoisomerase 1 antibodies, given that multibead assays are associated with high false-positive rates.
Patients with systemic sclerosis may have coexistent overlapping symptoms of other autoimmune diseases (eg, rheumatoid arthritis, Sjögrens syndrome, myositis, and primary biliary cholangitis). Although overlap syndromes remain poorly defined from a clinical standpoint, broadening immunological assessment beyond systemic sclerosis autoantibodies is prudent if symptoms of an overlap syndrome are present.
Biomarkers
Several candidate biomarkers have been studied in systemic sclerosis, of which a few are available and feasible to measure in clinical practice. Among the biomarkers that are readily available for measurement in most clinical practices, C-reactive protein (CRP) elevation is present in a subset of patients with systemic sclerosis and is associated with increased risk of progressive fibrosis, pulmonary arterial hypertension, and mortality.23–26 CRP elevation is mediated by interleukin (IL)-6, and serum concentrations of IL-6 are associated with poorer disease course in patients with interstitial lung disease.27 CRP and IL-6 have been used to stratify patients in clinical studies and are therefore of importance when applying these studies in clinical practice.28 The serum biomarkers Krebs von den Lungen glycoprotein-6 and chemokine ligand 18 have also been found to predict interstitial lung disease severity and progression.29,30 Baseline elevations in these biomarkers, and persistent elevations in these biomarkers despite treatment, might help guide therapeutic decision making in the near future.
N-terminal pro-B-type natriuretic peptide is a valid biomarker of early pulmonary arterial hypertension. It is often used in conjunction with other disease characteristics to select patients for right-heart catheterisation.31
RNA sequencing represents a promising tool in systemic sclerosis research, including biomarker discovery, characterisation of systemic sclerosis phenotypes, and precision medicine. For example, a study demonstrated that specific baseline transcript-module scores predicted response to treatment with mycophenolate in patients with systemic sclerosis associated with interstitial lung disease.32
Screening for organ involvement
Screening for specific organ involvement is warranted both at disease onset and repeatedly during disease course. Screening for disease manifestations over time can be modified on the basis of dynamic patient-related factors, including disease duration and progression of organ involvement.
Skin involvement
Wide heterogeneity in the extent of skin involvement exists between patients and within the individual patient over time.16 The skin fibrosis in systemic sclerosis starts in the distal fingers and toes and progresses proximally. In the initial phases, the fingers of the hands become puffy as a result of microvascular changes and inflammation. Over time, excessive collagen deposition leads to skin thickening and limited movement over joints resulting in contractures of both large and small joints in some patients. After years of disease, the skin can become tight, hide bound, and sometimes atrophic.
The modified Rodnan Skin Score (mRSS) is a physician-performed assessment measure used to examine progression of cutaneous fibrosis over time. Although this tool has acceptable reliability and interobserver variability in patients with diffuse cutaneous systemic sclerosis, it is not able to distinguish early fibrotic skin from the hide-bound skin in later disease.33
Several alternative tools for assessment of skin fibrosis have emerged, notably ultrasound and durometry.34–37 Optical coherence elastography together with optical coherence tomography is another promising tool which needs further exploration in systemic sclerosis.38 Finally, self-assessment of skin thickness has received increased interest during the COVID-19 pandemic.39 This tool could empower patients to self-monitor their disease in a manner that helps patients receive earlier medical follow-up if they detect disease progression between clinic visits.
In addition to cutaneous fibrosis, approximately 25% of patients with systemic sclerosis will develop cutaneous calcinosis.40 This manifestation is often painful as it can protrude both inwards into deeper tissues and outwards to cause ulceration. Of note, a small subset (<5%) of patients with established systemic sclerosis have no clinically detectable cutaneous fibrosis; these patients are classified as having systemic sclerosis sine scleroderma.41
Lung involvement
Studies estimate that approximately 50–65% of patients with systemic sclerosis will exhibit interstitial lung abnormalities on high-resolution computed tomography (HRCT).42,43 Although some patients with systemic sclerosis and interstitial lung disease will have a stable course of interstitial lung disease in the absence of treatment, others will have progression of interstitial lung disease with some demonstrating rapid progression and others slow progression over time.44 Interstitial lung disease is now the leading cause of death in systemic sclerosis.44–46 Observational studies have shown that certain systemic sclerosis patient subgroups (eg, male individuals, African American people, and patients who possess the Scl70 autoantibody) are more likely to have a progressive interstitial lung disease phenotype.17,47,48 However, a study demonstrated that African American and non-African American participants of two systemic sclerosis and interstitial lung disease trials had similar short-term and long-term outcomes, suggesting that confounding factors (eg, access to care) should be considered when interpreting the results of observational studies.49
Non-specific interstitial pneumonia is the most common histological pattern observed in systemic sclerosis; usual interstitial pneumonia is present in 10–25% of cases.50–52 HRCT is the most sensitive way to detect early systemic sclerosis associated with interstitial lung disease and can also identify other features of systemic sclerosis, including a patulous oesophagus, dilated pulmonary artery, and comorbidities, such as pulmonary malignancy.53
Pulmonary function testing (PFT) is also important in the initial assessment of a patient with systemic sclerosis and should include measurement of the diffusing capacity for carbon monoxide (DLCO) and forced vital capacity (FVC). Although PFTs have limited sensitivity in diagnosing interstitial lung disease, they are central to monitoring interstitial lung disease progression.54 PFTs are associated with intraindividual variation during repeated measurements and extrapulmonary factors (eg, oral fibrosis, thoracic fibrosis, myopathy, fatigue, and cachexia) can influence the results. Proposed definitions of progressive interstitial lung disease based on PFT and HRCT trends can help identify patients with a progressive fibrosing phenotype who could benefit from more aggressive or alternate therapies.55 A clinical practice guideline defined progressive pulmonary fibrosis as fulfilling at least two of three criteria (worsening symptoms, radiological progression, and physiological progression) within the past year with no alternative explanation in a patient with an interstitial lung disease other than idiopathic pulmonary fibrosis.56 Involvement of experienced thoracic radiologists is central to identifying more subtle progression of interstitial lung disease on HRCT. Invasive procedures, including both broncho-alveolar lavage and lung biopsy, are typically only done in cases of diagnostic uncertainty.
Renal involvement
The prevalence of renal crisis in systemic sclerosis ranges from 1% to 14% depending on the geographical region, with the USA, the UK, and Australia reporting the highest rates of renal crisis.57–59 Characterised by rapid deterioration of renal function and malignant hypertension, renal crisis can progress to terminal disease requiring renal replacement within a few weeks from first clinical signs if untreated.60 1-year mortality rates have decreased considerably since the introduction of angiotensin-converting enzyme inhibitors.61
Measurement of systolic blood pressure is paramount to the timely detection of this complication. Risk factors for renal crisis include diffuse cutaneous systemic sclerosis, early disease course, African American race, pericardial effusion, tendon friction rubs at clinical examination, recent usage of prednisolone higher than 15 mg/day, and RNA polymerase 3 autoantibodies.62 Microangiopathic haemolytic anaemia together with thrombocytopenia is common in this disease and initial investigations should include blood smear analysis. Typical changes on kidney biopsy have been described, but this procedure should be preserved for patients with diagnostic uncertainty.62 A proportion of patients with scleroderma renal crisis are normotensive, which might reflect poor cardiovascular reserve, and these patients have a poor prognosis.63
Cardiac involvement
Although the advent of cardiac MRI has furthered our understanding of cardiac involvement in systemic sclerosis, no standardised definition of systemic sclerosis cardiac involvement exists.64 Systemic sclerosis is associated with severe cardiac abnormalities, including myocarditis, congestive heart failure, arrythmia, asymptomatic focal fibrosis, and impaired ventricular relaxation. Systemic sclerosis cardiac involvement accounts for a substantial proportion of systemic sclerosis-related deaths.4 Regardless of symptoms, patients with systemic sclerosis should be evaluated with electrocardiogram and echocardiogram. MRI should be considered when the findings of the aforementioned studies suggest cardiac involvement.65 Holter echocardiogram and implantable-loop recorder should be considered in patients at risk for malignant arrythmia.66
Pulmonary arterial hypertension
Pulmonary arterial hypertension is a serious complication of systemic sclerosis that has a 3-year reported mortality between 21% and 48%.67–69 Advances in both the treatment and timely diagnosis of this complication have improved its prognosis. Although all patients with systemic sclerosis are at an increased risk for this complication, it is more prevalent in patients with anti-centromere antibodies, extensive telangiectasias, and longer disease duration. Emerging evidence also suggests a higher prevalence of pulmonary arterial hypertension in patients with Th/To autoantibodies.70 Compared with patients with idiopathic pulmonary arterial hypertension, systemic sclerosis associated with pulmonary arterial hypertension has a poorer prognosis, even in patients with similar haemodynamic profiles.71
In addition to pulmonary arterial hypertension (group 1), patients with systemic sclerosis can exhibit elevated pulmonary artery pressure because of other causes (eg, left-heart dysfunction [group 2], interstitial lung disease [group 3], and, less commonly, pulmonary venoocclusive disease [group 1]).72 Right-heart catheterisation is the gold standard to diagnose this pulmonary arterial hypertension, although echocardiogram can be used in conjunction with PFTs to help screen for this disease. A decline in the DLCO disproportionate to a decline in FVC or an isolated decline in the DLCO can also suggest pulmonary hypertension.73 Given the invasive nature of right-heart catheterisation, algorithms exist to help select appropriate patients for this procedure, including the DETECT algorithm and the Australian Scleroderma Interest Group algorithm.74–77
Gastrointestinal tract involvement
At least 90% of patients with systemic sclerosis have gastrointestinal tract involvement.78 Systemic sclerosis gastrointestinal manifestations are heterogeneous and can occur at any time in the disease course. Oesophageal dysfunction is common and can manifest with reflux disease with or without oesophagitis. Oesophagitis can be asymptomatic and correct diagnosis requires endoscopic examination.79 Dysphagia commonly occurs secondary to oesophageal dysmotility.
Gastric antral vascular ectasias is a potentially life-threating complication, which can result in rapid or slow blood loss over time. Upper endoscopy is therefore paramount in patients with systemic sclerosis who have anaemia.80 In addition, patients can have delayed gastric emptying, resulting in abdominal distension and bloating, particularly after meals.
Involvement of the lower gastrointestinal tract occurs in up to 50% of patients with systemic sclerosis and is associated with increased morbidity and mortality.81 Manifestations include small intestinal bacterial over-growth, malabsorption, constipation, diarrhoea, recurrent pseudo-obstruction, and faecal incontinence.82–84 Intestinal pseudo-obstruction is a serious clinical manifestation of systemic sclerosis, affecting between 4% and 10% of patients.85–89 Associated with delayed colonic transit, pseudo-obstruction presents with the inability to move intestinal luminal contents forward, but in the absence of a mechanical true obstructive process.90–92 This condition is painful, often recurrent and, at times, life-threatening.93
Anorectal dysfunction affects 22–77% of patients with systemic sclerosis, and faecal incontinence is the most common symptom.94,95 Rectal prolapse can also occur manifesting with a bulging sensation in the anus and chronic stool leakage.
In severe cases of lower gastrointestinal involvement, patients might require total parenteral nutrition. In addition to general malnutrition, micronutrient deficiency can also occur and require specific testing.96,97
Musculoskeletal involvement
Musculoskeletal involvement in systemic sclerosis is common, but might be overlooked when internal organs are severely affected. Although arthralgia is common, some patients with systemic sclerosis will develop erosive arthritis. Clinical assessment of joint effusion and range of motion might be challenging because of coexistent skin fibrosis or subcutaneous oedema. Tendon friction rubs (ie, a crepitus originating from the moving tendon inside the tendon sheath) occur predominantly in patients with diffuse cutaneous disease early in their disease course and is a poor prognostic sign.98
Patients with systemic sclerosis can have myopathy of various origins. Early in the course of diffuse cutaneous systemic sclerosis, patients can exhibit mild elevations in creatine kinase because of a systemic sclerosis-specific myopathy.99 These abnormalities typically resolve once a patient starts immunomodulatory therapy for their diffuse cutaneous sclerosis. However, a subgroup of patients will have a true overlap immune-mediated myositis condition, manifesting with marked elevations in muscle enzymes.
Clinical challenges
Because systemic sclerosis affects several organ systems, a single treatment algorithm cannot be applied to all patients as an individual patient will have unique needs over the course of their disease. In addition, secondary complications can arise because of the underlying disease, therapeutic interventions, or both. For example, malnutrition can impair immune function, enhancing the risk of opportunistic infection, especially in patients receiving immunomodulatory therapies that further diminish immune response. Oesophageal dysfunction might hinder the ability of a patient to swallow prescribed medications. Depression is also common among patients with systemic sclerosis, and this can reduce compliance with treatment plans, and adversely affect quality of life.100
There are also manifestations of systemic sclerosis that are poorly understood with limited treatment options. For instance, many male patients with systemic sclerosis have severe erectile dysfunction, for which current therapies have limited success.101 Moreover, fatigue remains a common and often refractory symptom of systemic sclerosis.102
Pregnancy in systemic sclerosis is associated with an increased risk of complications for both the mother and fetus.103 Although historically patients with systemic sclerosis were dissuaded from pregnancy, an increasing number of fertility options are available (eg, surrogacy, donor eggs, and in-vitro fertilisation). Therefore, family planning discussions are important to initiate early in the disease course.
Finally, studies have demonstrated that patients with systemic sclerosis are at heightened risk for the development of certain malignancies, particularly patients who possess the anti-RNA polymerase 3 antibody.104 Although no universally accepted malignancy-screening protocols exist in systemic sclerosis, age-appropriate malignancy screening is vital for all patients with systemic sclerosis, and malignancy screening (regardless of age) has been suggested for patients who are positive for anti-RNA polymerase 3 at the time of diagnosis.105
Therapeutic strategies: immunomodulation
The distinguishing pathological hallmark of systemic sclerosis is aberrant and excessive deposition of extracellular matrix, resulting in fibrosis in several organ systems.106 Various factors contribute to excessive matrix accumulation in fibrosis, including increased myofibroblast activation, abnormal fibrotic signalling, and increased production and crosslinking of extracellular matrix molecules.107–109 Specific cytokines, chemokines, and growth factors can induce myofibroblast differentiation and dysregulation. For example, transforming growth factor-β (TGF-β) is a potent profibrotic mediator.110 Historically, therapeutic agents for systemic sclerosis aimed to curtail inflammation, targeting both the innate and adaptive immune system. Although the term immunosuppressant is often used to describe these agents, this might represent an oversimplification of drug mechanism, given that certain immunosuppressant agents (eg, mycophenolate and tocilizumab) can exert anti-fibrotic effects, whereas anti-fibrotic agents, such as nintedanib, can possess anti-inflammatory properties. Therefore, the present Seminar aims to avoid these superficial labels and instead focus on the therapeutic agent, its purported biological target or targets, and associated systemic sclerosis-related clinical outcomes (table 2). We focus our Seminar on therapies with approved indications or therapies widely used in clinical practice on the basis of robust clinical trial data (appendix p 1).
Table 2:
Evidence-based treatment options for systemic sclerosis
| Mechanism of action | Benefit in systemic sclerosis | Safety issues | |
|---|---|---|---|
| Diffuse cutaneous systemic sclerosis: mycophenolate is commonly used as first-line therapy for diffuse cutaneous systemic sclerosis. Rituximab, cyclophosphamide, and haematopoietic stem-cell transplantation are most often used for treatment-refractory diffuse cutaneous systemic sclerosis | |||
| Mycophenolate | Inhibits de-novo production of guanosine nucleotides, impairing both T-cell and B-cell proliferation | Clinically meaningful decrease in modified Rodnan skin score | Generally well tolerated and myelosuppression occurs rarely |
| Cyclophosphamide | Modulates regulatory T cells, leading to the decreased secretion of interferon γ and IL-12 | Clinically meaningful decrease in mRSS and decrease in mRSS compared with placebo | Myelosuppression occurs commonly and haematuria and malignancy are also concerns |
| Rituximab | Anti-CD20 monoclonal antibody that depletes peripheral B cells | Decrease in mRSS compared with placebo | Opportunistic infection and increased risk of severe COVID-19 infection and inadequate response to COVID-19 vaccination |
| Haematopoietic stem-cell transplantation | Stem-cell extraction and chemotherapy, followed by transplantation of multipotent haematopoietic stem cells to reconstitute immune system | Greater decrease in mRSS compared with cyclophosphamide | Opportunistic infection, infertility, secondary malignancies, and increased mortality in patients with certain comorbidities |
| Interstitial lung disease: mycophenolate is commonly used as first-line therapy for interstitial lung disease. Nintedanib, rituximab, cyclophosphamide, and HSCT are most often used as second-line therapies for interstitial lung disease. Tocilizumab can be considered as first-line or second-line therapy for patients with early interstitial lung disease with signs of active systemic inflammation | |||
| Mycophenolate | Inhibits de-novo production of guanosine nucleotides, impairing both T-cell and B-cell proliferation | Clinically meaningful improvement in forced vital capacity, radiographic fibrosis, and self-reported dyspnoea | Generally well tolerated and myelosuppression occurs rarely |
| Cyclophosphamide | Selectively modulates regulatory T cells, leading to the decreased secretion of interferon γ and IL-12 | Clinically meaningful improvement in FVC, radiographic fibrosis, and self-reported dyspnoea | Myelosuppression occurs commonly, and haematuria and malignancy are also concerns |
| Nintedanib | Inhibits several tyrosine kinases | Slowed the decline in FVC compared with placebo | Diarrhoea occurs commonly and baseline systemic sclerosis gastrointestinal involvement should be evaluated before introducing this agent |
| Rituximab | Anti-CD20 monoclonal antibody that depletes peripheral B cells | Stabilised FVC compared with a decline in FVC in placebo | Opportunistic infection, increased risk of severe COVID-19 infection, and inadequate response to COVID-19 vaccination |
| Tocilizumab | Humanised monoclonal antibody that blocks the IL-6 receptor | Stabilised FVC compared with a decline in FVC in placebo | Opportunistic infection and might cause dyslipidaemia |
| Haematopoietic stem-cell transplantation | Stem-cell extraction and chemotherapy, followed by transplantation of multipotent haematopoietic stem cells to reconstitute immune system | Less of a decline in FVC and fewer cases of respiratory failure compared with cyclophosphamide | Opportunistic infection, infertility, and secondary malignancies |
| Pulmonary arterial hypertension: monotherapy or combination therapy with sildenafil and tadalafil and bosentan and ambrisentan are commonly used as first-line therapy for systemic sclerosis associated with pulmonary arterial hypertension. Selexipag, riociguat, and prostacyclin therapy are most often used as second-line therapies | |||
| Sildenafil and tadalafil | Promote vasodilation via nitric oxide-cyclic guanosine monophosphate enhancement | Improved exercise capacity, haemodynamics, and functional class | Hypotension, particularly when used in combination with other vasodilator therapies |
| Bosentan and ambrisentan | Inhibit endothelin-A and endothelin-B receptor signalling | Improved exercise capacity, haemodynamics, and functional class | Lower extremity oedema and liver-function test abnormalities |
| Selexipag | Selective oral prostacyclin receptor agonist that results in vasodilation of the pulmonary vascular bed | Reduction in the number of hospitalisations and disease progression | Headache, diarrhoea, and nausea |
| Riociguat | Stimulates soluble guanylate cyclase, regardless of nitric oxide concentrations, to produce more cyclic guanosine monophosphate, resulting in vasodilation | Improved exercise capacity, haemodynamics, and functional class | Gastro-oesophageal reflux disease, diarrhoea, nausea, dizziness, and headache |
| Prostacyclin therapy (eg, reprostinil, epoprostenol, iloprost, and beraprost) | Prostacyclin agonist that promotes relaxation of smooth muscle, inhibition of platelet aggregation, and vasodilation of pulmonary arteries | Improved exercise capacity, haemodynamics, and functional class | Headache, diarrhoea, and nausea |
| Raynaud phenomenon and digital ulcers: calcium channel blockers are commonly used as first-line therapy for Raynaud phenomenon. Sildenafil and tadalafil or bosentan are often used as second-line therapies for Raynaud phenomenon and first-line therapies for digital ulcers. Fluoxetin and iloprost are often considered for patients with treatment-refractory Raynaud phenomenon and digital ulcers | |||
| Calcium channel blockers | Inhibit calcium entry into cells resulting in vascular smooth-muscle relaxation and reduction in systemic vascular resistance | Reduction in frequency and severity of Raynaud phenomenon | Lower-extremity oedema |
| Fluoxetine | Promotes vasodilation and disruption of normal platelet aggregation | Reduction in frequency and severity of Raynaud phenomenon | Sexual dysfunction, dry mouth, and anxiety |
| Sildenafil and tadalafil | Promote vasodilation via nitric oxide-cyclic guanosine monophosphate enhancement | Varying evidence to support their use in the treatment and prevention of Raynaud phenomenon and digital ulcers | Hypotension, particularly when used in combination with other vasodilator therapies |
| Iloprost | Promotes vasodilation via prostacyclin pathway | Prevention of new and healing of existing digital ulcers | Headache and hypotension |
| Bosentan | Inhibits endothelin-A and endothelin-B receptor signalling | Reduction in development of new digital ulcers | Lower-extremity oedema and liver-function test abnormalities |
Randomised controlled trials in systemic sclerosis have largely focused on the treatment for systemic sclerosis skin disease, systemic sclerosis associated with interstitial lung disease, systemic sclerosis associated with pulmonary arterial hypertension, systemic sclerosis-related Raynaud phenomenon, and digital ulcers. FVC=forced vital capacity. IL=interleukin. mRSS=modified Rodnan skin score.
Cyclophosphamide
Cyclophosphamide is a type of nitrogen mustard drug that when administered in lower dosages modulates regulatory T cells, leading to the decreased secretion of interferon γ and IL-12.111 The seminal randomised controlled trial (RCT) on cyclophosphamide for systemic sclerosis was the Scleroderma Lung Study (SLS) I,112 which demonstrated that 12 months of cyclophosphamide was associated with significant treatment benefits with respect to FVC%-predicted, radiographic fibrosis, health-related quality of life and cutaneous fibrosis.112 However, adverse events (eg, leukopenia, neutropenia, and haematuria) occurred more commonly in patients randomly assigned to cyclophosphamide than placebo. This was a high-quality study done at 13 scleroderma centres for excellence across the USA. Although cyclophosphamide is still administered (more commonly intravenously) today for the treatment of diffuse cutaneous systemic sclerosis and systemic sclerosis associated with interstitial lung disease, particularly in regions with limited access to mycophenolate mofetil (MMF), it is typically not a first-line agent because of its unfavourable safety profile, including the heightened risk of malignancy.113
Mycophenolate
MMF is a prodrug of mycophenolic acid, an inhibitor of inosine monophosphate dehydrogenase. Through inhibiting de-novo production of guanosine nucleotides, MMF impairs both T-cell and B-cell proliferation.114 In the SLS 2 (MMF for 24 months vs oral cyclophosphamide for 12 months, followed by 12 months of placebo), treatment with MMF was associated with significant improvements in FVC%-predicted, radio graphic fibrosis, self-reported dyspnoea, and the extent of cutaneous sclerosis in patients with systemic sclerosis associated with interstitial lung disease.115–118 No between-treatment differences were observed between MMF and cyclophosphamide with regard to efficacy endpoints; however, MMF was better tolerated than cyclophosphamide.115 The most commonly reported adverse events among patients randomly assigned to MMF (reported in eight [6%] of 142 patients) was anaemia. Otherwise, adverse events were infrequent among MMF users. SLS 2115 was a high-quality study done at 14 scleroderma centres for excellence across the USA. On the basis of the findings of SLS 2, MMF has emerged as a first-line therapy for systemic sclerosis associated with interstitial lung disease and diffuse cutaneous sclerosis; as a result, subsequent systemic sclerosis trials have permitted background use of MMF. These studies have provided additional evidence supporting the efficacy of MMF for the treatment of systemic sclerosis-related skin and lung disease, and the safety of this agent when used in combination with other systemic sclerosis therapies, such as nintedanib.119–121
Tocilizumab
Tocilizumab is a humanised monoclonal antibody that blocks the IL-6 receptor. IL-6 concentrations are elevated in some patients with systemic sclerosis and correlate with the extent of skin involvement.122 The findings of a phase 2 trial of tocilizumab for the treatment of relatively early diffuse cutaneous systemic sclerosis suggested a possible treatment benefit on mRSS; however, neither the phase 2 nor phase 3 trial found a significant difference in mRSS at 48 weeks (primary endpoint) between patients randomly assigned to tocilizumab versus placebo.28,123 The latter study28 showed a substantial difference in the change from baseline in FVC%-predicted at 48 weeks, favouring tocilizumab.28 Tocilizumab appeared safe and well tolerated in both studies, with similar proportions of patients having infection in the intervention and placebo groups. Furthermore, a post-hoc analysis of the participants in this trial who had interstitial lung disease (136 [65%] of 210 total participants) reported that treatment with tocilizumab was associated with stabilisation of FVC%-predicted.124
The aforementioned tocilizumab trials were of high quality and suggest that tocilizumab might effectively treat systemic sclerosis associated with interstitial lung disease in patients with relatively early (<5 years disease duration) diffuse cutaneous systemic sclerosis with elevated acute-phase reactants and signs of progressively worsening skin disease. The generalisability of the study findings to other populations of patients with systemic sclerosis (eg, patients with limited cutaneous disease, patients that do not have elevated acute-phase reactants, and patients with stable diffuse cutaneous disease) is unknown. It is unknown how treatment with tocilizumab compares with treatment with existing therapies for systemic sclerosis-associated interstitial lung disease, such as mycophenolate and nintedanib.
Rituximab
B-cell infiltration exists across several organ systems in systemic sclerosis, including the skin, lungs, and gastrointestinal tract. Although B cells have various important roles in immune function and homoeostasis in systemic sclerosis, B cells can promote profibrotic T-helper-cell 2 responses through the induction of dendritic cell maturation.125 Moreover, the discovery of several functional autoantibodies (eg, autoantibodies against angiotensin 2 receptor type-1 and endothelin-1 receptor type A) in systemic sclerosis has illuminated novel insights into systemic sclerosis pathogenesis.126
Rituximab is an anti-CD20 monoclonal antibody that depletes peripheral B cells, but spares plasma cells and haematopoietic stem cells that do not express the CD20 surface antigen.127 A 2021, relatively small (n=56) RCT demonstrated that treatment with rituximab at a dosage of 375 mg/m2 once per week for 4 weeks led to a significant improvement in mRSS compared with placebo (−6·3 vs +2·14) at 6 months in patients with both limited cutaneous systemic sclerosis and diffuse cutaneous systemic sclerosis.128 Most of the patients in this study (89%) had underlying interstitial lung disease, and treatment with rituximab also had a favourable effect on the change in FVC%-predicted at 6 months.129
Another recent RCT compared rituximab (1 g administered intravenously weekly for 2 weeks) to cyclopho sphamide (administered every month at a dosage of 600 mg per m2 body surface area for 20 weeks) in patients with interstitial lung disease caused by systemic sclerosis, myositis, or mixed connective-tissue disease.130 This study showed that treatment with rituximab and cyclophosphamide led to similar improve ments in FVC and quality of life; however, rituximab was better tolerated than cyclophosphamide.131 Although the quality of the aforementioned rituximab trials was limited because of their small sample sizes, these findings provide compelling evidence that rituximab is a viable treatment option for systemic sclerosis associated with interstitial lung disease and systemic sclerosis-associated skin disease. Rituximab should be used judiciously in patients at high risk for COVID-19 complications, because this agent is associated with impaired humoral responses to COVID-19 vaccination.132
Nintedanib
Nintedanib inhibits several tyrosine-kinase receptors, including platelet-derived growth factor receptors, fibroblast growth factor receptors, and vascular endothelial growth factor receptors.133 In a large RCT for systemic sclerosis associated with interstitial lung disease (n=576), nintedanib slowed the rate of decline of FVC over 52 weeks compared with placebo (−52·4 mL per year vs −93·3 mL per year).134 Approximately half of all patients were receiving mycophenolate at a stable dosage for at least 6 months before enrolment. Patients receiving mycophenolate at baseline who were randomly assigned to nintedanib experienced the slowest decline in lung function. Treatment with nintedanib was not associated with improvements in skin score nor self-reported dyspnoea. Changes in the radiographic extent of interstitial lung disease were not reported in this trial. Consistent with the known safety profile of nintedanib, the majority of patients (76%) randomly assigned to nintedanib had diarrhoea. This study was a high-quality trial that enrolled a large population of patients with systemic sclerosis and interstitial lung disease from across the world. However, because treatment with nintedanib was not associated with improvements in patient-reported outcomes or cutaneous fibrosis, nintedanib is not often used as a first-line therapy for systemic sclerosis associated with interstitial lung disease. Caution is needed when prescribing this medication to patients with systemic sclerosis with underlying diarrhoea caused by the disease.
Haematopoietic stem-cell transplantation
Among all of the immunomodulatory strategies available for treating systemic sclerosis, haematopoietic stem-cell transplantation (HSCT) may lead to the most profound improvements in both cutaneous sclerosis and interstitial lung disease. The Autologous Stem Cell Transplantation International Scleroderma trial135 demonstrated a significant decrease in mRSS at 2 years in patients with early diffuse cutaneous systemic sclerosis randomly assigned to autologous HSCT compared with 1 year of intravenous cyclophosphamide (−20 vs −9 decrease in mRSS score). Another HSCT RCT (the Scleroderma: Cyclophosphamide or Transplantation trial),136 used a novel composite endpoint, which included both objective (eg, death, event-free, survival, and FVC endpoints) and subjective measures (eg, patient-reported quality of life and mRSS), to compare outcomes between autologous HSCT and 1 year of intravenous cyclophosphamide. Not only did this study demonstrate superiority of HSCT compared with cyclophosphamide on the basis of the primary endpoint, but a strikingly low number of patients randomly assigned to HSCT initiated disease-modifying anti-rheumatic therapies by 54 months compared with those randomly assigned to cyclophosphamide (9% vs 44%).
A highly costly procedure associated with life-threatening adverse events, HSCT is generally considered for patients with early diffuse cutaneous systemic sclerosis as a second-line approach when other therapies fail or as a first-line approach in carefully selected patients when the anticipated benefit of HSCT outweighs the potential risks.137,138
Therapeutic strategies: vascular modulation
Microvascular damage and endothelial cell dysfunction are crucial pathogenic mediators of vascular modulation.139 Early in systemic sclerosis, perturbation of the endothelium promotes the overproduction of vasoactive factors, such as the vasoconstrictor endothelin 1, along with platelet activation and increased expression of adhesion molecules.140,141 The result is recurrent ischaemiareperfusion injury, abnormal regulation of reactive oxygen species, and microvascular damage.
Therapeutic strategies for addressing the vascular dimensions of systemic sclerosis (eg, Raynaud phenomenon, digital ulcers, pulmonary arterial hypertension, erectile dysfunction, and scleroderma renal crisis) have largely centred on vasodilation. However, there is a growing recognition that vascular proliferation, rather than vasoconstriction, is a major driver of the pathogenesis of the serious vascular manifestations of systemic sclerosis (eg, pulmonary arterial hypertension).142 For instance, dysfunction of bone morphogenetic protein receptor 2 (a member of the TGF-β superfamily) signalling is associated with the development of pulmonary arterial hypertension.143 Novel therapies that act as a ligand trap for members of the TGF-β superfamily (eg, sotatercept) have been shown to reduce pulmonary vascular resistance in patients with pulmonary arterial hypertension, probably via vascular remodelling.144,145
Among the vascular therapeutic options for systemic sclerosis, the dihydropyridines, amlodipine and nifedi-pine, are commonly used as first-line agents to manage Raynaud phenomenon.146,147 Endothelin receptor antagonists (eg, ambrisentan, bosentan, and macitentan) are approved for the treatment of pulmonary arterial hypertension and, among these, bosentan might also prevent digital ulcers.148–150 Among the commercially available phosphodiesterase isoenzyme 5 inhibitors, sildenafil and tadalafil are used for systemic sclerosis associated with pulmonary arterial hypertension, with varying amounts of evidence supporting their use for the treatment of Raynaud phenomenon and digital ulcers.151,152 Studies have demonstrated that combination therapy with a phosphodiesterase isoenzyme 5 inhibitor and endothelin receptor antagonist is associated with improved survival compared with monotherapy for pulmonary arterial hypertension.153,154 As such, early combination therapy for systemic sclerosis associated with pulmonary arterial hypertension is often favoured over sequential add-on therapy.
Analogues of prostaglandin 2 (eg, intravenous iloprost and epoprostenol or oral treprostinil), are viable treatment considerations for patients with severe refractory Raynaud phenomenon with digital ischaemia.155 Treatment with intravenous epoprostenol, subcutaneous treprostinil, and selexipag, a prostaglandin 2 receptor agonist, is associated with improved outcomes in systemic sclerosis associated with pulmonary arterial hypertension.156–161 Treatment with the soluble guanylate cyclase stimulator, riociguat, is associated with improvements in haemodynamics, functional class, and 6 min walk distance in patients with connective-tissue disease with pulmonary arterial hypertension (66 [59%] of 111 patients with connective-tissue disease and pulmonary arterial hypertension had systemic sclerosis in this trial), and those with Raynaud phenomenon with diffuse cutaneous systemic sclerosis.162,163
Treatment for systemic sclerosis associated with pulmonary hyper tension often involves a multi-disciplinary approach that could include pulmonologists and cardiologists to help identify the underlying cause or causes of pulmonary hypertension and to establish a personalised treatment strategy. For example, patients with systemic sclerosis associated with pulmonary hypertension caused by left-heart disease might derive minimal benefit from pulmonary arterial hypertension-targeted therapies. However, patients with systemic sclerosis associated with pulmonary hypertension and interstitial lung disease might have increased risk of pulmonary veno-occlusive disease-like lesions (ie, obstructive intimal fibrosis of small veins and venules) and, in such patients, starting vasodilators might precipitate pulmonary oedema.164 However, certain vasodilators can be safe to use in such patients as a bridge to lung transplantation.165
Early intervention with angiotensin-converting enzyme inhibitors (ACEIs) has substantially reduced the risk of mortality associated with scleroderma renal crisis, improving survival from 10% to 85% at 1 year.63,166 Paradoxically, studies investigating the prophylactic use of ACEIs for prevention of scleroderma renal crisis have reported that previous exposure to ACEIs is associated with worse outcomes after the onset of scleroderma renal crisis and might even increase the risk for the development of scleroderma renal crisis.167,168 However, this is a controversial topic because these data are based on observation trials subject to confounding.
Therapeutic strategies: symptomatic management
Several clinical manifestations of systemic sclerosis do not have disease-modifying treatment options, and management strategies largely focus on improving symptoms, but with very scarce evidence (table 3). Future studies are needed to investigate whether immunomodulatory and vasoactive therapies are disease modifying for these manifestations, particularly for manifestations associated with high morbidity and mortality (eg, myocardial disease and gastrointestinal disease).
Table 3:
Symptom-directed adjuvant treatment options for systemic sclerosis
| Intervention | |
|---|---|
| Dyspnoea | Pulmonary rehabilitation and supplemental oxygen |
| Cough | Antitussives |
| Dry mouth | Cholinergic agonists, sugar-free lozenges, and frequent dental hygiene visits |
| Dry eyes | Artificial tears, cyclosporine eye drops, and lymphocyte function-associated antigen-1 antagonists |
| Microstomia | Mouth stretching and oral augmentation exercises and botulinum toxin or hyaluronidase injections |
| Pruritic rash | Limit time in baths and showers and avoid hot water, minimise use of harsh soaps, particularly in dry areas of the body, use simple oils (eg, olive oil and jojoba oil) to moisturise the skin, and avoid moisturisers with fragrance |
| Telangiectasias | Camouflage techniques, including specialised make-up, pulse dye laser, and intense pulse light |
| Heartburn and reflux | Proton pump inhibitors, histamine type-2 receptor antagonists, and dietary adjustments |
| Distension and bloating | Gastrointestinal promotility agents (eg, prucalopride), antibiotics if small intestinal bacterial overgrowth present, and small, frequent meals |
| Diarrhoea | Dietary adjustments, antibiotics if small intestinal bacterial overgrowth present, fluid resuscitation if dehydration is present, adjustment of medications known to cause diarrhoea, and anti-diarrhoeal agents (eg, loperamide) |
| Constipation | Dietary adjustments, gastrointestinal promotility agents (eg, prucalopride), and laxatives |
| Faecal incontinence | Physiotherapy, biofeedback, and sacral nerve stimulation |
| Joint contractures | Physiotherapy, occupational therapy, and infrared heat |
| Calcinosis | Surgical excision, bisphosphonates, and diltiazem |
Rigorous trials are scarce for these important clinical dimensions of systemic sclerosis. The proposed therapies are recommended on the basis of evidence from observational studies and the expert clinical opinion of the authors, and therefore should be used judiciously.
COVID-19: emerging research
The ongoing COVID-19 pandemic has presented unique challenges for patients with systemic sclerosis and their providers.169,170 Patients with systemic sclerosis represent a vulnerable patient population with respect to COVID-19 infection given their propensity for interstitial lung disease, immunocompromised status, and need for regular in-person visits to monitor and manage their systemic sclerosis.171 Although research on COVID-19-related outcomes in patients with systemic sclerosis is largely limited to single-centre, observational experiences, studies have demonstrated that the presence of interstitial lung disease and certain medications (eg, rituximab) are associated with more severe COVID-19 infection, including a higher risk of mortality.172,173 Reassuringly, patients with systemic sclerosis appear to tolerate COVID-19 vaccinations, and the implementation of telehealth has improved continuity of care and expanded the reach of speciality centres for systemic sclerosis in many areas.174,175 Future research is needed to better understand the long-term sequelae of previous COVID-19 infection, and the impact of the pandemic on the mental health of all patients with systemic sclerosis.176
Summary
Although systemic sclerosis is a heterogeneous disease, this condition should be suspected in any patient with Raynaud phenomenon that starts or worsens near the time of antinuclear antibody detection, particularly in patients who also possess systemic sclerosis-specific autoantibodies, abnormal nailfold capillaries, puffy fingers, or all symptoms. The most common organ systems affected in systemic sclerosis include the skin, gastrointestinal tract, and lungs. In addition to a careful history and physical examination, all patients with newly diagnosed systemic sclerosis should undergo comprehensive serological assessment for systemic sclerosis-specific autoantibodies, HRCT of the chest to screen for interstitial lung disease, and echocardiogram to provide a baseline estimation of pulmonary artery pressure. In patients with interstitial lung disease, pulmonary function tests are also important to obtain at diagnosis to establish a baseline for monitoring interstitial lung disease pro gression over time. If feasible, referral to a scleroderma centre might not only improve patient care through early diagnosis and risk stratification, but it might also afford patients the opportunity to participate in clinical and translational research studies.
Future directions
Despite the substantial advances made in (very) early disease detection and drug discovery for systemic sclerosis over the past 5 years, several unmet needs still exist. Clinical evaluation has largely driven risk stratification early in the systemic sclerosis disease course; however, integration of biological data (ie, genomics, proteomics, and intestinal and skin micro-biome) might improve our ability to refine disease phenotypes and personalise treatment options for patients. With approved therapies targeting different facets of the immune system, an opportunity has emerged to study the safety and efficacy of combination therapy, a cornerstone to the management of other complex rheumatic diseases. Studies are specifically needed to better understand the risks and benefits of upfront combination therapies versus sequential therapy for individual systemic sclerosis manifestations.
There is also a growing need to understand why and how systemic sclerosis disproportionately affects specific patient populations (eg, female patients and African American patients), as these insights could illuminate new pathogenic targets and lead to the application of precision medicine for managing this condition. Along these lines, it is paramount to ensure that trials strive to enrol balanced proportions of male and female sexes, different genders, and racial subgroups to ensure that we fully understand the safety and efficacy of investigational products in all patients with systemic sclerosis.
The development of valid trial endpoints that are both meaningful to the patient and capture the impact of the multiple clinical dimensions of systemic sclerosis is also greatly needed. This development is particularly important as we consider recent trials that did not detect significant differences in specific primary endpoints, such as the mRSS.28,123,177 It is conceivable that certain patients with systemic sclerosis may actually benefit from therapies that failed to demonstrate significant treatment effects in clinical trials.
Finally, new therapeutic strategies are essential to prevent the development of organ involvement in patients with very early systemic sclerosis. The validation of the very early criteria provides an opportunity to identify patients with very early disease to study preventive therapies. Lastly, as in all rare diseases, evidence-based, clinical practice guidelines will help to improve the standardisation of multidisciplinary care across centres.
Supplementary Material
Search strategy and selection criteria.
Following the Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines for scoping reviews, we searched PubMed for studies published between Jan 15, 1990, and May 15, 2022, using the search terms “systemic sclerosis” or “scleroderma” in combination with the terms “treatment” or “management.” We largely selected publications from the past 3 years; however, high-quality older publications were included. No PubMed filters or limits were applied to maintain a broad search strategy. We also did a manual search of references cited in original research studies and reviewed articles on systemic sclerosis to identify additional relevant articles. Although primary research publications were prioritised, review articles are also cited to provide opportunities for further reading on specific topics. An in-depth review of randomised controlled trials was provided if the inclusion criteria for the study included systemic sclerosis diagnosis, and if the outcome of the trial led to either approval of a systemic sclerosis indication or widespread use of the medication in clinical practice.
Acknowledgments
ERV receives funding from the National Institute of Health and National Heart Lung and Blood Institute (K23 HL150237). KA receives funding from Ulla och Roland Gustafssons Donationsfond and the Swedish Medical Society. VS is Senior Clinical Investigator of the Research Foundation of Flanders (Belgium; 1.8.029.20N). VS is supported by an unrestricted educational chair on systemic sclerosis of Janssen-Cilag. These funding sources had no role in the literature review and the synthesis and interpretation of the data, in the writing of the report, or in the decision to submit the paper for publication. VS is Chair of the European Alliance of Associations for Rheumatology study group on microcirculation in rheumatic diseases. None of the authors have been paid to write this article by a pharmaceutical company or other agency. We acknowledge Nathalie Berghen and Tessa Dufour for their assistance organising the references for this mansucript.
Declaration of interests
ERV has received grant and research support to her institution from Boehringer Ingelheim, Forbius, Kadmon, and Horizon, and consulting and speaking fees from Boehringer Ingelheim (payments made to herself). KA has received grant and research support to his institution from Ulla och Roland Gustafssons Donationsfond and the Swedish Medical Society. VS has received grant and research support to her institution from the Research Foundation Flanders, Belgian Fund for Scientific Research in Rheumatic Diseases, Janssen-Cilag, and Boehringer Ingelheim, consulting fees from Boehringer Ingelheim (payments made to herself and her institution) and Janssen-Cilag (payments made to institution), speaker fees from UCB (payments made to her institution), Boehringer Ingelheim (payments made to self and her institution), and Janssen-Cilag (payments made to her institution), and support for attending meetings or travel expenses from Boehringer Ingelheim (payments made to her institution).
Contributor Information
Elizabeth R Volkmann, Department of Medicine, Division of Rheumatology, University of California, Los Angeles, CA, USA; David Geffen School of Medicine, Los Angeles, CA, USA.
Kristofer Andréasson, Department of Rheumatology, Lund University, Lund, Sweden.
Vanessa Smith, Department of Internal Medicine and Department of Rheumatology, Ghent University (Hospital), Ghent, Belgium; Unit for Molecular Immunology and Inflammation, VIB Inflammation Research Centre, Ghent, Belgium.
References
- 1.Bergamasco A, Hartmann N, Wallace L, Verpillat P. Epidemiology of systemic sclerosis and systemic sclerosis-associated interstitial lung disease. Clin Epidemiol 2019; 11: 257–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morrisroe K, Stevens W, Sahhar J, et al. The clinical and economic burden of systemic sclerosis related interstitial lung disease. Rheumatology (Oxford) 2020; 59: 1878–88. [DOI] [PubMed] [Google Scholar]
- 3.Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis 2007; 66: 940–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elhai M, Meune C, Boubaya M, et al. Mapping and predicting mortality from systemic sclerosis. Ann Rheum Dis 2017; 76: 1897–905. [DOI] [PubMed] [Google Scholar]
- 5.Smith V, Scire CA, Talarico R, et al. Systemic sclerosis: state of the art on clinical practice guidelines. RMD Open 2018; 4: e000782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lopez-Bastida J, Linertova R, Oliva-Moreno J, et al. Social/economic costs and health-related quality of life in patients with scleroderma in Europe. Eur J Health Econ 2016; 17 (suppl 1): 109–17. [DOI] [PubMed] [Google Scholar]
- 7.Decuman S, Smith V, Verhaeghe S, Deschepper E, Vermeiren F, De Keyser F. Work participation and work transition in patients with systemic sclerosis: a cross-sectional study. Rheumatology 2012; 51: 297–304. [DOI] [PubMed] [Google Scholar]
- 8.Talarico R, Aguilera S, Alexander T, et al. The added value of a European Reference Network on rare and complex connective tissue and musculoskeletal diseases: insights after the first 5 years of the ERN ReCONNET. Clin Exp Rheumatol 2022; 40 (suppl 134): 3–11. [DOI] [PubMed] [Google Scholar]
- 9.Assassi S, Leyva AL, Mayes MD, et al. Predictors of fatigue severity in early systemic sclerosis: a prospective longitudinal study of the GENISOS cohort. PloS One 2011; 6: e26061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.LeRoy EC, Medsger TA. Criteria for the classification of early systemic sclerosis. J Rheumatol 2001; 28: 1573–76. [PubMed] [Google Scholar]
- 11.van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis 2013; 72: 1747–55. [DOI] [PubMed] [Google Scholar]
- 12.Koenig M, Joyal F, Fritzler MJ, et al. Autoantibodies and microvascular damage are independent predictive factors for the progression of Raynaud’s phenomenon to systemic sclerosis: a twenty-year prospective study of 586 patients, with validation of proposed criteria for early systemic sclerosis. Arthritis Rheum 2008; 58: 3902–12. [DOI] [PubMed] [Google Scholar]
- 13.Smith V, Vanhaecke A, Herrick AL, et al. Fast track algorithm: how to differentiate a “scleroderma pattern” from a “nonscleroderma pattern”. Autoimmun Rev 2019; 18: 102394. [DOI] [PubMed] [Google Scholar]
- 14.Bellando-Randone S, Del Galdo F, Lepri G, et al. Progression of patients with Raynaud’s phenomenon to systemic sclerosis: a five-year analysis of the European Scleroderma Trial and Research group multicentre, longitudinal registry study for Very Early Diagnosis of Systemic Sclerosis (VEDOSS). Lancet Rheumatol 2021; 3: e834–43. [DOI] [PubMed] [Google Scholar]
- 15.Smith V, Herrick AL, Ingegnoli F, et al. Standardisation of nailfold capillaroscopy for the assessment of patients with Raynaud’s phenomenon and systemic sclerosis. Autoimmun Rev 2020; 19: 102458. [DOI] [PubMed] [Google Scholar]
- 16.Avouac J, Fransen J, Walker UA, et al. Preliminary criteria for the very early diagnosis of systemic sclerosis: results of a Delphi Consensus Study from EULAR Scleroderma Trials and Research Group. Ann Rheum Dis 2011; 70: 476–81. [DOI] [PubMed] [Google Scholar]
- 17.LeRoy EC, Black C, Fleischmajer R, et al. Scleroderma (systemic-sclerosis)–classification, subsets and pathogenesis. J Rheumatol 1988; 15: 202–05. [PubMed] [Google Scholar]
- 18.Nihtyanova SI, Schreiber BE, Ong VH, et al. Prediction of pulmonary complications and long-term survival in systemic sclerosis. Arthritis Rheumatol 2014; 66: 1625–35. [DOI] [PubMed] [Google Scholar]
- 19.Hachulla E, Agard C, Allanore Y, et al. French recommendations for the management of systemic sclerosis. Orphanet J Rare Dis 2021; 16: 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Homer KL, Warren J, Karayev D, et al. Performance of anti-topoisomerase I antibody testing by multiple-bead, enzyme-linked immunosorbent assay and immunodiffusion in a university setting. J Clin Rheumatol 2020; 26: 115–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamaguchi Y, Takehara K. Anti-nuclear autoantibodies in systemic sclerosis: news and perspectives. J Scleroderma Relat Disord 2018; 3: 201–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nihtyanova SI, Sari A, Harvey JC, et al. Using autoantibodies and cutaneous subset to develop outcome-based disease classification in systemic sclerosis. Arthritis Rheumatol 2020; 72: 465–76. [DOI] [PubMed] [Google Scholar]
- 23.Mitev A, Christ L, Feldmann D, et al. Inflammatory stays inflammatory: a subgroup of systemic sclerosis characterized by high morbidity and inflammatory resistance to cyclophosphamide. Arthritis Res Ther 2019; 21: 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jha M, Wang M, Steele R, et al. NT-proBNP, hs-cTnT, and CRP predict the risk of cardiopulmonary outcomes in systemic sclerosis: findings from the Canadian Scleroderma Research Group. J Scleroderma Relat Disord 2022; 7: 62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, Mayes MD, Pedroza C, et al. Does C-reactive protein predict the long-term progression of interstitial lung disease and survival in patients with early systemic sclerosis? Arthritis Care Res (Hoboken) 2013; 65: 1375–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pokeerbux MR, Giovannelli J, Dauchet L, et al. Survival and prognosis factors in systemic sclerosis: data of a French multicenter cohort, systematic review, and meta-analysis of the literature. Arthritis Res Ther 2019; 21: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Lauretis A, Sestini P, Pantelidis P, et al. Serum interleukin 6 is predictive of early functional decline and mortality in interstitial lung disease associated with systemic sclerosis. J Rheumatol 2013; 40: 435–46. [DOI] [PubMed] [Google Scholar]
- 28.Khanna D, Lin CJF, Furst DE, et al. Tocilizumab in systemic sclerosis: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med 2020; 8: 963–74. [DOI] [PubMed] [Google Scholar]
- 29.Volkmann ER, Tashkin DP, Kuwana M, et al. Progression of interstitial lung disease in systemic sclerosis: the importance of pneumoproteins krebs von den lungen 6 and CCL18. Arthritis Rheumatol 2019; 71: 2059–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elhai M, Hoffmann-Vold AM, Avouac J, et al. Performance of candidate serum biomarkers for systemic sclerosis-associated interstitial lung disease. Arthritis Rheumatol 2019; 71: 972–82. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Qin D, Qin L, Yang X, Luo Q, Wang H. Diagnostic value of cardiac natriuretic peptide on pulmonary hypertension in systemic sclerosis: a systematic review and meta-analysis. Joint Bone Spine 2022; 89: 105287. [DOI] [PubMed] [Google Scholar]
- 32.Assassi S, Volkmann ER, Zheng WJ, et al. Peripheral blood gene expression profiling shows predictive significance for response to mycophenolate in systemic sclerosis-related interstitial lung disease. Ann Rheum Dis 2022; 81: 854–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clements PJ, Lachenbruch PA, Seibold JR, et al. Skin thickness score in systemic sclerosis: an assessment of interobserver variability in 3 independent studies. J Rheumatol 1993; 20: 1892–96. [PubMed] [Google Scholar]
- 34.Moore TL, Lunt M, McManus B, Anderson ME, Herrick AL. Seventeen-point dermal ultrasound scoring system–a reliable measure of skin thickness in patients with systemic sclerosis. Rheumatology (Oxford, England) 2003; 42: 1559–63. [DOI] [PubMed] [Google Scholar]
- 35.Vanhaecke A, Cutolo M, Heeman L, et al. High frequency ultrasonography: reliable tool to measure skin fibrosis in SSC? A systematic literature review and additional pilot study. Rheumatology (Oxford, England) 2021; 61: 42–52. [DOI] [PubMed] [Google Scholar]
- 36.de Oliveira MFC, Leopoldo VC, Pereira KRC, et al. Durometry as an alternative tool to the modified Rodnan’s skin score in the assessment of diffuse systemic sclerosis patients: a cross-sectional study. Adv Rheumatol 2020; 60: 48. [DOI] [PubMed] [Google Scholar]
- 37.Vanhaecke A, Verschuere S, Vilela V, Heeman L, Cutolo M, Smith V. Durometry in SSc: the hard facts. A systematic literature review and additional pilot study. Rheumatology 2021; 60: 2099–108. [DOI] [PubMed] [Google Scholar]
- 38.Liu CH, Assassi S, Theodore S, et al. Translational optical coherence elastography for assessment of systemic sclerosis. J Biophotonics 2019; 12: e201900236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spierings J, Ong V, Denton CP. PASTUL questionnaire: a tool for self-assessment of scleroderma skin during the COVID-19 pandemic. Ann Rheum Dis 2021; 80: 819–820. [DOI] [PubMed] [Google Scholar]
- 40.Valenzuela A, Song P, Chung L. Calcinosis in scleroderma. Curr Opin Rheumatol 2018; 30: 554–61. [DOI] [PubMed] [Google Scholar]
- 41.De Almeida Chaves S, Porel T, Mounie M, et al. Sine scleroderma, limited cutaneous, and diffused cutaneous systemic sclerosis survival and predictors of mortality. Arthritis Res Ther 2021; 23: 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoffmann-Vold AM, Fretheim H, Halse AK, et al. Tracking impact of interstitial lung disease in systemic sclerosis in a complete nationwide cohort. Am J Respir Crit Care Med 2019; 200: 1258–66. [DOI] [PubMed] [Google Scholar]
- 43.Steele R, Hudson M, Lo E, Baron M. Clinical decision rule to predict the presence of interstitial lung disease in systemic sclerosis. Arthritis Care Res (Hoboken) 2012; 64: 519–24. [DOI] [PubMed] [Google Scholar]
- 44.Volkmann ER. Natural history of systemic sclerosis-related interstitial lung disease: how to identify a progressive fibrosing phenotype. J Scleroderma Relat Disord 2020; 5: 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 2010; 69: 1809–15. [DOI] [PubMed] [Google Scholar]
- 46.Jaafar S, Lescoat A, Huang S, et al. Clinical characteristics, visceral involvement, and mortality in at-risk or early diffuse systemic sclerosis: a longitudinal analysis of an observational prospective multicenter US cohort. Arthritis Res Ther 2021; 23: 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freitas R, Martins P, Dourado E, et al. Gender differences in clinical features and outcomes of a Portuguese systemic sclerosis cohort. Clin Rheumatol 2022; 41: 1139–44. [DOI] [PubMed] [Google Scholar]
- 48.Moore DF, Kramer E, Eltaraboulsi R, Steen VD. Increased morbidity and mortality of scleroderma in African Americans compared to non-African Americans. Arthritis Care Res (Hoboken) 2019; 71: 1154–63. [DOI] [PubMed] [Google Scholar]
- 49.Volkmann ER, Steen V, Li N, et al. Racial disparities in systemic sclerosis: short- and long-term outcomes among African American participants of SLS I and II. ACR Open Rheumatol 2021; 3: 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim DS, Yoo B, Lee JS, et al. The major histopathologic pattern of pulmonary fibrosis in scleroderma is nonspecific interstitial pneumonia. Sarcoidosis Vasc Diffuse Lung Dis 2002; 19: 121–7. [PubMed] [Google Scholar]
- 51.Fischer A, Swigris JJ, Groshong SD, et al. Clinically significant interstitial lung disease in limited scleroderma: histopathology, clinical features, and survival. Chest 2008; 134: 601–05. [DOI] [PubMed] [Google Scholar]
- 52.Fujita J, Yoshinouchi T, Ohtsuki Y, et al. Non-specific interstitial pneumonia as pulmonary involvement of systemic sclerosis. Ann Rheum Dis 2001; 60: 281–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoffmann-Vold AM, Maher TM, Philpot EE, et al. The identification and management of interstitial lung disease in systemic sclerosis: evidence-based European consensus statements. Lancet Rheumatol 2020; 2: e71–e83. [DOI] [PubMed] [Google Scholar]
- 54.Bernstein EJ, Jaafar S, Assassi S, et al. Performance characteristics of pulmonary function tests for the detection of interstitial lung disease in adults with early diffuse cutaneous systemic sclerosis. Arthritis Rheumatol 2020; 72: 1892–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Distler O, Assassi S, Cottin V, et al. Predictors of progression in systemic sclerosis patients with interstitial lung disease. Eur Respir J 2020; 55: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2022; 205: e18–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stern EP, Unwin R, Burns A, Ong VH, Denton CP. Exploring molecular pathology of chronic kidney disease in systemic sclerosis by analysis of urinary and serum proteins. Rheumatol Adv Pract 2021; 5: rkaa083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moinzadeh P, Kuhr K, Siegert E, et al. Scleroderma renal crisis: risk factors for an increasingly rare organ complication. J Rheumatol 2020; 47: 241–48. [DOI] [PubMed] [Google Scholar]
- 59.Cole A, Ong VH, Denton CP. Renal disease and systemic sclerosis: an update on scleroderma renal crisis. Clin Rev Allergy Immunol 2022; published online June 1. doi: 10.1007/s12016-022-08945-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Butler EA, Baron M, Fogo AB, et al. Generation of a core set of items to develop classification criteria for scleroderma renal crisis using consensus methodology. Arthritis Rheumatol 2019; 71: 964–71. [DOI] [PubMed] [Google Scholar]
- 61.Kim H, Lefebvre F, Hoa S, Hudson M. Mortality and morbidity in scleroderma renal crisis: a systematic literature review. J Scleroderma Relat Disord 2021; 6: 21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hudson M, Ghossein C, Steen V. Scleroderma renal crisis. Presse Med 2021; 50: 104063. [DOI] [PubMed] [Google Scholar]
- 63.Penn H, Howie AJ, Kingdon EJ, et al. Scleroderma renal crisis: patient characteristics and long-term outcomes. QJM 2007; 100: 485–94. [DOI] [PubMed] [Google Scholar]
- 64.Bissell LA, Anderson M, Burgess M, et al. Consensus best practice pathway of the UK Systemic Sclerosis Study group: management of cardiac disease in systemic sclerosis. Rheumatology 2017; 56: 912–21. [DOI] [PubMed] [Google Scholar]
- 65.Ross L, Paratz E, Baron M, La Gerche A, Nikpour M. Sudden cardiac death in systemic sclerosis: diagnostics to assess risk and inform management. Diagnostics (Basel) 2021; 11: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bruni C, Ross L. Cardiac involvement in systemic sclerosis: getting to the heart of the matter. Best Pract Res Clin Rheumatol 2021; 35: 101668. [DOI] [PubMed] [Google Scholar]
- 67.Lefevre G, Dauchet L, Hachulla E, et al. Survival and prognostic factors in systemic sclerosis-associated pulmonary hypertension: a systematic review and meta-analysis. Arthritis Rheum 2013; 65: 2412–23. [DOI] [PubMed] [Google Scholar]
- 68.Chauvelot L, Gamondes D, Berthiller J, et al. Hemodynamic response to treatment and outcomes in pulmonary hypertension associated with interstitial lung disease versus pulmonary arterial hypertension in systemic sclerosis: data from a study identifying prognostic factors in pulmonary hypertension associated with interstitial lung disease. Arthritis Rheumatol 2021; 73: 295–304. [DOI] [PubMed] [Google Scholar]
- 69.Chung L, Domsic RT, Lingala B, et al. Survival and predictors of mortality in systemic sclerosis-associated pulmonary arterial hypertension: outcomes from the pulmonary hypertension assessment and recognition of outcomes in scleroderma registry. Arthritis Care Res 2014; 66: 489–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Suresh S, Charlton D, Snell EK, et al. Development of pulmonary hypertension in over one-third of patients with Th/To antibody-positive scleroderma in long-term follow-up. Arthritis Rheumatol 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ramjug S, Hussain N, Hurdman J, et al. Idiopathic and systemic sclerosis-associated pulmonary arterial hypertension: a comparison of demographic, hemodynamic, and MRI characteristics and outcomes. Chest 2017; 152: 92–102. [DOI] [PubMed] [Google Scholar]
- 72.Haque A, Kiely DG, Kovacs G, Thompson AAR, Condliffe R. Pulmonary hypertension phenotypes in patients with systemic sclerosis. Eur Respir Rev 2021; 30: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Steen VD, Graham G, Conte C, Owens G, Medsger TA Jr. Isolated diffusing capacity reduction in systemic sclerosis. Arthritis Rheum 1992; 35: 765–70. [DOI] [PubMed] [Google Scholar]
- 74.Young A, Moles VM, Jaafar S, et al. Performance of the DETECT algorithm for pulmonary hypertension screening in a systemic sclerosis cohort. Arthritis Rheumatol 2021; 73: 1731–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hao Y, Thakkar V, Stevens W, et al. A comparison of the predictive accuracy of three screening models for pulmonary arterial hypertension in systemic sclerosis. Arthritis Res Ther 2015; 17: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bruni C, De Luca G, Lazzaroni MG, et al. Screening for pulmonary arterial hypertension in systemic sclerosis: a systematic literature review. Eur J Intern Med 2020; 78: 17–25. [DOI] [PubMed] [Google Scholar]
- 77.Ninagawa K, Kato M, Nakamura H, et al. Reduced diffusing capacity for carbon monoxide predicts borderline pulmonary arterial pressure in patients with systemic sclerosis. Rheumatol Int 2019; 39: 1883–87. [DOI] [PubMed] [Google Scholar]
- 78.McMahan ZH. Gastrointestinal involvement in systemic sclerosis: an update. Curr Opin Rheumatol 2019; 31: 561–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zampatti N, Garaiman A, Jordan S, et al. Performance of the UCLA Scleroderma Clinical Trials Consortium Gastrointestinal Tract 2.0 instrument as a clinical decision aid in the routine clinical care of patients with systemic sclerosis. Arthritis Res Ther 2021; 23: 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ghrenassia E, Avouac J, Khanna D, et al. Prevalence, correlates and outcomes of gastric antral vascular ectasia in systemic sclerosis: a EUSTAR case-control study. J Rheumatol 2014; 41: 99–105. [DOI] [PubMed] [Google Scholar]
- 81.Richard N, Hudson M, Wang M, et al. Severe gastrointestinal disease in very early systemic sclerosis is associated with early mortality. Rheumatology 2019; 58: 636–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McMahan ZH, Paik JJ, Wigley FM, Hummers LK. Determining the risk factors and clinical features associated with severe gastrointestinal dysmotility in systemic sclerosis. Arthritis Care Res 2018; 70: 1385–92. [DOI] [PubMed] [Google Scholar]
- 83.Nagaraja V, McMahan ZH, Getzug T, Khanna D. Management of gastrointestinal involvement in scleroderma. Curr Treatm Opt Rheumatol 2015; 1: 82–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jaovisidha K, Csuka ME, Almagro UA, Soergel KH. Severe gastrointestinal involvement in systemic sclerosis: report of five cases and review of the literature. Semin Arthritis Rheum 2005; 34: 689–702. [DOI] [PubMed] [Google Scholar]
- 85.Dein E, Kuo PL, Hong YS, Hummers LK, Mecoli CA, McMahan ZH. Evaluation of risk factors for pseudo-obstruction in systemic sclerosis. Semin Arthritis Rheum 2019; 49: 405–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gonzalez Z, McCallum R. Small bowel dysmotility, pseudoobstruction, and functional correlation with histopathology: lessons learned. Curr Gastroenterol Rep 2020; 22: 14. [DOI] [PubMed] [Google Scholar]
- 87.Downes TJ, Cheruvu MS, Karunaratne TB, De Giorgio R, Farmer AD. Pathophysiology, diagnosis, and management of chronic intestinal pseudo-obstruction. J Clin Gastroenterol 2018; 52: 477–89. [DOI] [PubMed] [Google Scholar]
- 88.Valenzuela A, Li S, Becker L, et al. Intestinal pseudo-obstruction in patients with systemic sclerosis: an analysis of the Nationwide Inpatient Sample. Rheumatology 2016; 55: 654–58. [DOI] [PubMed] [Google Scholar]
- 89.Muangchan C, Baron M, Pope J. The 15% rule in scleroderma: the frequency of severe organ complications in systemic sclerosis. A systematic review. J Rheumatol 2013; 40: 1545–56. [DOI] [PubMed] [Google Scholar]
- 90.McMahan ZH, Tucker AE, Perin J, et al. Relationship between gastrointestinal transit, medsger gastrointestinal severity, and university of California-Los Angeles scleroderma clinical trial consortium gastrointestinal tract 2.0 symptoms in patients with systemic sclerosis. Arthritis Care Res 2022; 74: 442–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zapatier JA, Ukleja A. Intestinal obstruction and pseudo-obstruction in patients with systemic sclerosis. Acta Gastroenterol Latinoam 2013; 43: 227–30. [PubMed] [Google Scholar]
- 92.Mecoli C, Purohit S, Sandorfi N, Derk CT. Mortality, recurrence, and hospital course of patients with systemic sclerosis-related acute intestinal pseudo-obstruction. J Rheumatol 2014; 41: 2049–54. [DOI] [PubMed] [Google Scholar]
- 93.Valenzuela A, Li S, Becker L, et al. Intestinal pseudo-obstruction in patients with systemic sclerosis: an analysis of the Nationwide Inpatient Sample. Rheumatology 2016; 55: 654–8. [DOI] [PubMed] [Google Scholar]
- 94.Trezza M, Krogh K, Egekvist H, Bjerring P, Laurberg S. Bowel problems in patients with systemic sclerosis. Scand J Gastroenterol 1999; 34: 409–13. [DOI] [PubMed] [Google Scholar]
- 95.Garros A, Marjoux S, Khouatra C, et al. Prevalence of fecal incontinence in a cohort of systemic sclerosis patients within a regional referral network. United European Gastroenterol J 2017; 5: 1046–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Paolino S, Pacini G, Schenone C, et al. Nutritional status and bone microarchitecture in a cohort of systemic sclerosis patients. Nutrients 2020; 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nguyen AM, McMahan Z, Volkmann ER. Micronutrient deficiencies in systemic sclerosis: a scoping review. Open Access Rheumatol: Res Rev 2022; Accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Barbacki A, Baron M, Wang M, et al. Damage trajectories in systemic sclerosis using group-based trajectory modeling. Arthritis Care Res 2022; published online Feb 28. DOI: 10.1002/acr.24873 [DOI] [PubMed] [Google Scholar]
- 99.Lefebvre F, Giannini M, Ellezam B, et al. Histopathological features of systemic sclerosis-associated myopathy: a scoping review. Autoimmun Rev 2021; 20: 102851. [DOI] [PubMed] [Google Scholar]
- 100.Thombs BD, Taillefer SS, Hudson M, Baron M. Depression in patients with systemic sclerosis: a systematic review of the evidence. Arthritis Rheum 2007; 57: 1089–97. [DOI] [PubMed] [Google Scholar]
- 101.Jaeger VK, Walker UA. Erectile dysfunction in systemic sclerosis. Curr Rheumatol Rep 2016; 18: 49. [DOI] [PubMed] [Google Scholar]
- 102.Sandusky SB, McGuire L, Smith MT, Wigley FM, Haythornthwaite JA. Fatigue: an overlooked determinant of physical function in scleroderma. Rheumatology 2009; 48: 165–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Clark KE, Etomi O, Ong VH. Systemic sclerosis in pregnancy. Obstet Med 2020; 13: 105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shah AA, Casciola-Rosen L. Cancer and scleroderma: a paraneoplastic disease with implications for malignancy screening. Curr Opin Rheumatol 2015; 27: 563–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lazzaroni MG, Cavazzana I, Colombo E, et al. Malignancies in patients with anti-RNA polymerase III antibodies and systemic sclerosis: analysis of the EULAR scleroderma trials and research cohort and possible recommendations for screening. J Rheumatol 2017; 44: 639–47. [DOI] [PubMed] [Google Scholar]
- 106.Cutolo M, Soldano S, Smith V. Pathophysiology of systemic sclerosis: current understanding and new insights. Expert Rev Clin Immunol 2019; 15: 753–64. [DOI] [PubMed] [Google Scholar]
- 107.Soldano S, Trombetta AC, Contini P, et al. Increase in circulating cells coexpressing M1 and M2 macrophage surface markers in patients with systemic sclerosis. Ann Rheum Dis 2018; 77: 1842–45. [DOI] [PubMed] [Google Scholar]
- 108.Truchetet ME, Brembilla NC, Chizzolini C. Current concepts on the pathogenesis of systemic sclerosis. Clinic Rev Allerg 2021; published online Sept 6. DOI: 10.1007/s12016-021-08889-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cutolo M, Gotelli E, Montagna P, et al. Nintedanib downregulates the transition of cultured systemic sclerosis fibrocytes into myofibroblasts and their pro-fibrotic activity. Arthritis Res Ther 2021; 23: 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ayers NB, Sun CM, Chen SY. Transforming growth factor-beta signaling in systemic sclerosis. J Biomed Res 2018; 32: 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ahlmann M, Hempel G. The effect of cyclophosphamide on the immune system: implications for clinical cancer therapy. Cancer Chemother Pharmacol 2016; 78: 661–71. [DOI] [PubMed] [Google Scholar]
- 112.Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med 2006; 354: 2655–66. [DOI] [PubMed] [Google Scholar]
- 113.Faurschou M, Mellemkjaer L, Voss A, Keller KK, Hansen IT, Baslund B. Prolonged risk of specific malignancies following cyclophosphamide therapy among patients with granulomatosis with polyangiitis. Rheumatology 2015; 54: 1345–50. [DOI] [PubMed] [Google Scholar]
- 114.Allison AC, Eugui EM. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology 2000; 47: 85–118. [DOI] [PubMed] [Google Scholar]
- 115.Tashkin DP, Roth MD, Clements PJ, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med 2016; 4: 708–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Goldin JG, Kim GHJ, Tseng CH, et al. Longitudinal changes in quantitative interstitial lung disease on computed tomography after immunosuppression in the scleroderma lung study II. Ann Am Thorac Soc 2018; 15: 1286–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Volkmann ER, Tashkin DP, LeClair H, et al. Treatment with mycophenolate and cyclophosphamide leads to clinically meaningful improvements in patient-reported outcomes in scleroderma lung disease: results of scleroderma lung study II. ACR Open Rheumatol 2020; 2: 362–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Namas R, Tashkin DP, Furst DE, et al. Efficacy of mycophenolate mofetil and oral cyclophosphamide on skin thickness: post hoc analyses from two randomized placebo-controlled trials. Arthritis Care Res 2018; 70: 439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Spiera R, Hummers L, Chung L, et al. Safety and efficacy of lenabasum in a phase II, randomized, placebo-controlled trial in adults with systemic sclerosis. Arthritis Rheumatol 2020; 72: 1350–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Spiera R, Khanna D, Kuwana M, et al. A randomised, double-blind, placebo-controlled phase 3 study of lenabasum in diffuse cutaneous systemic sclerosis: RESOLVE-1 design and rationale. Clin Exp Rheumatol 2021; 39 (suppl 131): 124–33. [DOI] [PubMed] [Google Scholar]
- 121.Distler O, Gahlemann M, Maher TM. Nintedanib for systemic sclerosis-associated interstitial lung disease. Reply. N Engl J Med 2019; 381: 1596–97. [DOI] [PubMed] [Google Scholar]
- 122.Khan K, Xu S, Nihtyanova S, et al. Clinical and pathological significance of interleukin 6 overexpression in systemic sclerosis. Ann Rheum Dis 2012; 71: 1235–42. [DOI] [PubMed] [Google Scholar]
- 123.Khanna D, Denton CP, Jahreis A, et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet 2016; 387: 2630–40. [DOI] [PubMed] [Google Scholar]
- 124.Roofeh D, Lin CJF, Goldin J, et al. Tocilizumab prevents progression of early systemic sclerosis-associated interstitial lung disease. Arthritis Rheumatol 2021; 73: 1301–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sanges S, Guerrier T, Launay D, et al. Role of B cells in the pathogenesis of systemic sclerosis. Rev Med Interne 2017; 38: 113–24. [DOI] [PubMed] [Google Scholar]
- 126.Kill A, Riemekasten G. Functional autoantibodies in systemic sclerosis pathogenesis. Curr Rheumatol Rep 2015; 17: 34. [DOI] [PubMed] [Google Scholar]
- 127.Weiner GJ. Rituximab: mechanism of action. Semin Hematol 2010; 47: 115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ebata S, Yoshizaki A, Oba K, et al. Safety and efficacy of rituximab in systemic sclerosis (DESIRES): a double-blind, investigator-initiated, randomised, placebo-controlled trial. Lancet Rheumatol 2021; 3: e489–97. [DOI] [PubMed] [Google Scholar]
- 129.Goswami RP, Ray A, Chatterjee M, Mukherjee A, Sircar G, Ghosh P. Rituximab in the treatment of systemic sclerosis-related interstitial lung disease: a systematic review and meta-analysis. Rheumatology 2021; 60: 557–67. [DOI] [PubMed] [Google Scholar]
- 130.Saunders P, Tsipouri V, Keir GJ, et al. Rituximab versus cyclophosphamide for the treatment of connective tissue disease-associated interstitial lung disease (RECITAL): study protocol for a randomised controlled trial. Trials 2017; 18: 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Maher TM, Tudor V, Saunders P, et al. Rituximab versus cyclophosphamide for the treatment of connective tissue disease associated interstitial lung disease (RECITAL): a multi-centre randomized controlled trial. San Fransisco: ATS, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Arnold J, Winthrop K, Emery P. COVID-19 vaccination and antirheumatic therapy. Rheumatology 2021; 60: 3496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wollin L, Wex E, Pautsch A, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J 2015; 45: 1434–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Distler O, Highland KB, Gahlemann M, et al. Nintedanib for systemic sclerosis-associated interstitial lung disease. N Engl J Med 2019; 380: 2518–28. [DOI] [PubMed] [Google Scholar]
- 135.van Laar JM, Farge D, Sont JK, et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA 2014; 311: 2490–8. [DOI] [PubMed] [Google Scholar]
- 136.Sullivan KM, Goldmuntz EA, Keyes-Elstein L, et al. Myeloablative autologous stem-cell transplantation for severe scleroderma. N Engl J Med 2018; 378: 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Spierings J, Chiu YH, Voortman M, van Laar JM. Autologous stem-cell transplantation in systemic sclerosis-associated interstitial lung disease: early action in selected patients rather than escalation therapy for all. Ther Adv Musculoskelet Dis 2021; 13: 1759720X211035196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Daikeler T, Tichelli A, Passweg J. Complications of autologous hematopoietic stem cell transplantation for patients with autoimmune diseases. Pediatr Res 2012; 71: 439–44. [DOI] [PubMed] [Google Scholar]
- 139.Cutolo M, Smith V. Detection of microvascular changes in systemic sclerosis and other rheumatic diseases. Nat Rev Rheumatol 2021; 17: 665–77. [DOI] [PubMed] [Google Scholar]
- 140.Mostmans Y, Cutolo M, Giddelo C, et al. The role of endothelial cells in the vasculopathy of systemic sclerosis: a systematic review. Autoimmun Rev 2017; 16: 774–86. [DOI] [PubMed] [Google Scholar]
- 141.Romano E, Rosa I, Fioretto BS, Matucci-Cerinic M, Manetti M. New insights into profibrotic myofibroblast formation in systemic sclerosis: when the vascular wall becomes the enemy. Life 2021; 11: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rubin LJ. It’s time to put the term “pulmonary vasodilators” to rest. J Am Coll Cardiol Basic Trans Science 2021; 6: 870–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Guignabert C, Humbert M. Targeting transforming growth factor-beta receptors in pulmonary hypertension. Eur Respir J 2021; 57: 1. [DOI] [PubMed] [Google Scholar]
- 144.Andre P, Joshi SR, Briscoe SD, Alexander MJ, Li G, Kumar R. Therapeutic approaches for treating pulmonary arterial hypertension by correcting imbalanced TGF-beta superfamily signaling. Front Med 2021; 8: 814222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Humbert M, McLaughlin V, Gibbs JSR, et al. Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med 2021; 384: 1204–15. [DOI] [PubMed] [Google Scholar]
- 146.Herrick AL, Wigley FM. Raynaud’s phenomenon. Best Pract Res Clin Rheumatol 2020; 34: 101474. [DOI] [PubMed] [Google Scholar]
- 147.Rirash F, Tingey PC, Harding SE, et al. Calcium channel blockers for primary and secondary Raynaud’s phenomenon. Cochrane Database Syst Rev 2017; 12: CD000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Correale M, Ferraretti A, Monaco I, Grazioli D, Di Biase M, Brunetti ND. Endothelin-receptor antagonists in the management of pulmonary arterial hypertension: where do we stand? Vasc Health Risk Manag 2018; 14: 253–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Korn JH, Mayes M, Matucci Cerinic M, et al. Digital ulcers in systemic sclerosis: prevention by treatment with bosentan, an oral endothelin receptor antagonist. Arthritis Rheum 2004; 50: 3985–93. [DOI] [PubMed] [Google Scholar]
- 150.Matucci-Cerinic M, Denton CP, Furst DE, et al. Bosentan treatment of digital ulcers related to systemic sclerosis: results from the RAPIDS-2 randomised, double-blind, placebo-controlled trial. Ann Rheum Dis 2011; 70: 32–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Barnes H, Brown Z, Burns A, Williams T. Phosphodiesterase 5 inhibitors for pulmonary hypertension. Cochrane Database Syst Rev 2019; 1: CD012621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Impens AJ, Phillips K, Schiopu E. PDE-5 inhibitors in scleroderma Raynaud phenomenon and digital ulcers: current status of clinical trials. Int J Rheumatol 2011; 2011: 392542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pestana-Fernandez M, Rubio-Rivas M, Tolosa-Vilella C, et al. Longterm efficacy and safety of monotherapy versus combination therapy in systemic sclerosis-associated pulmonary arterial hypertension: a retrospective RESCLE registry study. J Rheumatol 2020; 47: 89–98. [DOI] [PubMed] [Google Scholar]
- 154.Coghlan JG, Galie N, Barbera JA, et al. Initial combination therapy with ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH): subgroup analysis from the AMBITION trial. Ann Rheum Dis 2017; 76: 1219–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hinze AM, Wigley FM. Pharmacotherapy options in the management of Raynaud’s phenomenon. Curr Treatm Opt Rheumatol 2018; 4: 235–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 1996; 334: 296–301. [DOI] [PubMed] [Google Scholar]
- 157.Badesch DB, Tapson VF, McGoon MD, et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med 2000; 132: 425–34. [DOI] [PubMed] [Google Scholar]
- 158.Simonneau G, Barst RJ, Galie N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 2002; 165: 800–04. [DOI] [PubMed] [Google Scholar]
- 159.Oudiz RJ, Schilz RJ, Barst RJ, et al. Treprostinil, a prostacyclin analogue, in pulmonary arterial hypertension associated with connective tissue disease. Chest 2004; 126: 420–27. [DOI] [PubMed] [Google Scholar]
- 160.Gaine S, Chin K, Coghlan G, et al. Selexipag for the treatment of connective tissue disease-associated pulmonary arterial hypertension. Eur Respir J 2017; 50: 1602493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sitbon O, Channick R, Chin KM, et al. Treprostinil, a prostacyclin analogue, in pulmonary arterial hypertension associated with connective tissue disease. N Engl J Med 2015; 373: 2522–33.26699168 [Google Scholar]
- 162.Humbert M, Coghlan JG, Ghofrani HA, et al. Riociguat for the treatment of pulmonary arterial hypertension associated with connective tissue disease: results from PATENT-1 and PATENT-2. Ann Rheum Dis 2017; 76: 422–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Khanna D, Allanore Y, Denton CP, et al. Riociguat in patients with early diffuse cutaneous systemic sclerosis (RISE-SSc): randomised, double-blind, placebo-controlled multicentre trial. Ann Rheum Dis 2020; 79: 618–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gupta S, Gupta A, Rehman S, et al. Pulmonary veno-occlusive disease is highly prevalent in scleroderma patients undergoing lung transplantation. ERJ Open Res 2019; 5: 00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Montani D, Jaïs X, Price LC, et al. Cautious epoprostenol therapy is a safe bridge to lung transplantation in pulmonary veno-occlusive disease. Eur Respir J 2009; 34: 1348–56. [DOI] [PubMed] [Google Scholar]
- 166.Zanatta E, Polito P, Favaro M, et al. Therapy of scleroderma renal crisis: state of the art. Autoimmun Rev 2018; 17: 882–89. [DOI] [PubMed] [Google Scholar]
- 167.Hudson M, Baron M, Tatibouet S, Furst DE, Khanna D, International Scleroderma Renal Crisis Study I. Exposure to ACE inhibitors prior to the onset of scleroderma renal crisis-results from the International Scleroderma Renal Crisis Survey. Semin Arthritis Rheum 2014; 43: 666–72. [DOI] [PubMed] [Google Scholar]
- 168.Butikofer L, Varisco PA, Distler O, et al. ACE inhibitors in SSc patients display a risk factor for scleroderma renal crisis-a EUSTAR analysis. Arthritis Res Ther 2020; 22: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Denton CP, Campochiaro C, Bruni C, Distler O, Iagnocco A, Matucci Cerinic M. COVID-19 and systemic sclerosis: rising to the challenge of a pandemic. J Scleroderma Relat Disord 2021; 6: 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hoffmann-Vold AM, Distler O, Bruni C, et al. Systemic sclerosis in the time of COVID-19. Lancet Rheumatol 2022; 4: e566–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Orlandi M, Lepri G, Bruni C, et al. The systemic sclerosis patient in the COVID-19 era: the challenging crossroad between immunosuppression, differential diagnosis and long-term psychological distress. Clin Rheumatol 2020; 39: 2043–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.de Oliveira SM, Martins LVO, Lupino-Assad AP, et al. Severity and mortality of COVID-19 in patients with systemic sclerosis: a Brazilian multicenter study. Semin Arthritis Rheum 2022; 55: 151987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Loarce-Martos J, García-Fernández A, López-Gutiérrez F, et al. High rates of severe disease and death due to SARS-CoV-2 infection in rheumatic disease patients treated with rituximab: a descriptive study. Rheumatol Int 2020; 40: 2015–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Gordon JK, Showalter K, Wu Y, et al. Systemic sclerosis and COVID-19 vaccines: a SPIN Cohort study. Lancet Rheumatol 2022; 4: e243–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Spinella A, Magnani L, De Pinto M, et al. Management of systemic sclerosis patients in the COVID-19 era: the experience of an expert specialist reference center. Clin Med Insights Circ Respir Pulm Med 2021; 15: 11795484211001349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wu Y, Kwakkenbos L, Henry RS, et al. Factors associated with fears due to COVID-19: a Scleroderma Patient-centered Intervention Network (SPIN) COVID-19 cohort study. J Psychosom Res 2021; 140: 110314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Khanna D, Spino C, Johnson S, et al. Abatacept in early diffuse cutaneous systemic sclerosis: results of a phase II investigator-initiated, multicenter, double-blind, randomized, placebo-controlled trial. Arthritis Rheumatol 2020; 72: 125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.