

Abstract
Background:
The persistence of HIV-1-infected cells during antiretroviral therapy is well documented but may be modulated by early initiation of antiretroviral therapy in infants.
Methods:
Here, we longitudinally analyzed the proviral landscape in nine infants with vertical HIV-1 infection from Mozambique over a median period of 24 months, using single-genome, near full-length, next-generation proviral sequencing.
Results:
We observed a rapid decline in the frequency of intact proviruses, leading to a disproportional under-representation of intact HIV-1 sequences within the total number of HIV-1 DNA sequences after 12–24 months of therapy. In addition, proviral integration site profiling in one infant demonstrated clonal expansion of infected cells harboring intact proviruses and indicated that viral rebound was associated with an integration site profile dominated by intact proviruses integrated into genic and accessible chromatin locations.
Conclusion:
Together, these results permit rare insight into the evolution of the HIV-1 reservoir in infants infected with HIV-1 and suggest that the rapid decline of intact proviruses, relative to defective proviruses, may be attributed to a higher vulnerability of genome-intact proviruses to antiviral immunity. Technologies to analyze combinations of intact proviral sequences and corresponding integration sites permit a high-resolution analysis of HIV-1 reservoir cells after early antiretroviral treatment initiation in infants.
Keywords: Reservoir, Evolution, Infants, HIV-1, Integration sites
Introduction
With more than 1.7 million infected children worldwide, pediatric HIV-1 infection remains a global health challenge; in 2020 alone, current estimates suggest that more than 150,000 children were newly infected with HIV-1 [1]. Most of these children are infected through breastfeeding, but vertical mother-to-child transmission during pregnancy remains an important source of infection, despite the widespread introduction of effective prevention programs. A substantial proportion (46%) of infected infants and children do not have access to antiretroviral treatment (ART) [2], and in those who do, long-term toxicity may be a concern, as well as insufficient adherence to antiretroviral drugs. If successful in durably suppressing HIV-1 replication, ART in children is highly effective in restoring immune function and normalizing many of the immune perturbations that occur in infected infants [3]. However, even when initiated extremely early or within hours after birth, ART does not eliminate all HIV-1 infected cells, and a persisting reservoir of virally infected cells, frequently termed “HIV-1 reservoir cells” can persist life-long [4–9]. These cells harbor chromosomally integrated HIV-1 DNA that has traditionally been regarded as transcriptionally silent, but recent findings suggest that a considerable number of HIV-1 proviruses, specifically when integrated into accessible euchromatin, can actively transcribe HIV-1 DNA. Due to the high error rate of the viral polymerase (the reverse transcriptase), chromosomally integrated HIV-1 DNA frequently displays important sequence defects that preclude viral replication; such defective HIV-1 DNA typically accounts for more than 90% of all HIV-1 proviruses detectable in a given patient treated with ART but cannot give rise to replication-competent viral particles, as observed in previous adult cohorts [9, 10].
Over the recent years, significant progress has been made in profiling HIV-1 reservoir cells at a high-resolution, frequently enabling single-molecule analysis of individual HIV-1 DNA species in single infected cells [11]. Additional technical advances now permit us to characterize proviral sequences in conjunction with their chromosomal location site, which is likely to influence the transcriptional behavior of a given HIV-1 provirus [12]. Yet, very little is currently known about the dynamics of HIV-1 reservoir cells in pediatric patients with HIV-1, partially because peripheral blood mononuclear cell (PBMC) samples from infants infected with HIV-1 are difficult to obtain.
In this study, we assessed the evolution of HIV-1 DNA in nine infants from Mozambique who were perinatally infected with clade C HIV-1, started ART within the first months of life, and were followed up for 2 years while on ART. We used single-genome near full-length proviral sequencing to investigate the longitudinal dynamics of intact and defective proviruses and determine the integration sites of intact proviruses. Applying such high-resolution technologies, we evaluated the evolutionary trajectory of HIV-1 reservoir cells in infected infants from Maputo, Mozambique.
Methodology
Study population
Study patients were enrolled in the Towards AIDS Remission Approaches cohort (TARA) in Maputo Province, Mozambique. Infants were perinatally infected with HIV-1 subtype C. ART consisting of zidovudine (AZT) or abacavir (ABC), lamivudine (3TC), and ritonavir-boosted lopinavir (LPVr) (AZT/ABC+3TC+LPVr) in weight-adjusted doses was initiated at the time of HIV diagnosis within a median age of 33 days (interquartile range [IQR] 31.5–52). The viral load and clusters of differentiation 4 (CD4) T cell counts were measured using commercial assays during the follow-up time. PBMCs were isolated from whole blood drawn at defined time points between 2017 and 2021, cryopreserved, and sent to Miami, USA for research studies. Thereafter, the cells were shipped to Boston for reservoir cell analysis.
DNA extraction and HIV-1 copy number estimation
DNA was extracted from cryopreserved PBMCs using the DNeasy Blood & Tissue kit (Qiagen, Germantown, Maryland, USA), following the manufacturer’s instructions. Droplet digital polymerase chain reaction (PCR; Bio-Rad, Hercules, California, USA) was used to estimate the total HIV copy number by amplifying HIV-1 using primers and probes as previously described [13] (127bp 5’LTRGag amplicon; HXB2 coordinates 684–810). The number of cells was estimated through simultaneous amplification of the RPP30 host gene.
RNA extraction
In one of the infants, RNA was extracted from plasma samples using the QIAmp Viral RNA mini kit (Germantown, MD, USA), following the manufacturer’s instructions. Complementary DNA (cDNA) was further synthesized from RNA using a SuperScript IV commercial kit according to the manufacturer’s instructions (Thermofisher, Waltham, MA, USA). The HIV copy number was estimated as described previously. The cDNA was then subjected to a nested PCR to amplify the near full-length HIV genome.
Full-length individual proviral sequencing
The near full-length proviral sequences were generated from DNA/cDNA samples as previously described [13]. Briefly, to isolate single proviral sequences, the DNA samples were diluted according to Poisson distribution statistics based on the droplet digital PCR estimates of HIV DNA copy numbers. A dilution of <30% of PCR-positive wells on a 96-well plate is associated with an approximately 90% chance of isolating single proviral sequences. The diluted samples were subjected to a nested PCR using primers spanning near full-length HIV-1 (HXB2 coordinates 638–9632, 8994bp). HIV-1 sequences from each infant were subjected to Illumina MiSeq sequencing, with a median sequencing depth of 2500 reads per base. A previously designed in-house analysis pipeline was used to analyze the sequences and determine whether they are intact or defective proviral sequences [13]. Multiple sequence alignments and phylogenetic distance between sequences were examined using the maximum-likelihood trees in MEGA (https://megasoftware.net/) [14, 15]. The aligned sequences were further visualized using highlighter plots included in the Los Alamos HIV Sequence Database (www.hiv.lanl.gov) [16].
Matched integration site and proviral sequencing
Integration sites of intact proviruses were determined using a previously described methodology known as matched integration site and proviral sequencing (MIP-Seq) [12]. Briefly, HIV-1 DNA diluted to single-genome levels was subjected to a whole-genome amplification, using multiple displacement amplification with phi29 polymerase. Individual whole-genome amplification products were split and separately subjected to near full-length proviral sequencing and chromosomal integration site analysis using the ISLA protocol [17]. Chromosomal coordinates were aligned to the human genome (Hg38) using the genome browser (http://genome.ucsc.edu) [18].
Analysis of HIV-1 tropism and drug resistance mutations
Further analysis of the proviruses for co-receptor usage was conducted using the Geno2pheno tool (https://coreceptor.geno2pheno.org) [19]. In addition, sequence mutations associated with antiviral drug resistance were determined using the Stanford HIV Drug resistance database tool available online (https://hivdb.stanford.edu) [20].
Statistics and data analysis
Data analysis was performed using GraphPad PRISM7 software. Data are presented as pie charts and data plots. The differences were tested for statistical significance using the Mann-Whitney U test, Wilcoxon matched signed rank test, or chi-square tests, as appropriate. A P-value of < 0.05 was considered significant.
Sequence data availability
Raw sequences were deposited in GenBank (accession numbers OK649265–OK649295).
Results
Longitudinal dynamics of HIV-1 proviruses in infected infants
Nine infants with pre- or perinatally acquired HIV-1 clade C infection were enrolled in the TARA cohort in Maputo, Mozambique. The TARA cohort had enrolled 43 infants, and the nine infants in this substudy were selected based on the availability of longitudinal PMBCs at two or more time points. The nine infants were followed up for a median of 24 (IQR 24–27.5) months after birth. The time of diagnosis was approximately 1 month after birth, and ART was initiated at a median age of 33 (IQR 31.5–52) days after birth. All infants were receiving combination therapy with weight-adjusted zidovudine or abacavir, lamivudine, and ritonavir-boosted lopinavir (AZT+3TC+LPV/r or ABC+3TC+LPV/r). The median baseline plasma viral load at the time of diagnosis was 238,347 (IQR 20,200–2,400,109) HIV-1 RNA copies/ml (Table 1). In our study, viral suppression was defined as a viral load measure of 100 HIV-1 RNA copies/ml or less at least five consecutive time points, whereas viral rebound was defined as an increase in viral load to more than 1000 HIV-1 RNA copies/ml at any time point. Six (66.6%) infants were virally suppressed during the time of follow-up, whereas three (33.3%) infants experienced transient viral rebound at a single visit during the time of follow-up (Supplementary Figure 1), likely due to medication non-adherence.
Table 1.
Clinical characteristics of study participants.
| Participant ID | Age at ART (days) | Duration of follow-up (months) | ART regimen | Baseline viral load (HIV copies/ml) | Baseline CD4 (mm3) |
|---|---|---|---|---|---|
| Infant 1 | 31 | 24 | AZT+3TC+NVP | 10,000,000 | 1,155 |
| Infant 2 | 33 | 24 | AZT+3TC+LVPr | 718,996 | 2,324 |
| Infant 3 | 73 | 24 | AZT+3TC+LVPr | 36,965 | n/a |
| Infant 4 | 30 | 25 | AZT+3TC+LVPr | 3,434 | 4,033 |
| Infant 5 | 61 | 24 | AZT+3TC+LVPr | 1,978,332 | 2,081 |
| Infant 6 | 35 | 25 | AZT+3TC+LVPr | 176,970 | 5,713 |
| Infant 7 | 43 | 36 | AZT+3TC+LVPr | 2,821,886 | 1,620 |
| Infant 8 | 32 | 19 | ABC+3TC+LVPr | 695 | n/a |
| Infant 9 | 32 | 30 | ABC+3TC+LVPr | 238,347 | 2,384 |
| Median (IQR) | 33 (31.5 – 52) | 24 (24–27.5) | 238,347 (20,200–2,400,109) | 2,324 (1,620–4,033) |
Abbreviations: ART, antiretroviral treatment; AZT, zidovudine; ABC, abacavir; CD, clusters of differentiation; LVPr, ritonavir-boosted lopinavir; 3TC, lamivudine.
To assess the genetic composition of the reservoir, near full-length individual proviral sequencing was performed, using DNA extracted from PBMCs as a template. Longitudinal quantification of HIV-1 DNA was conducted at three time points throughout the duration of the follow-up. The time point 1 (TP1) analysis was performed at the age of 2–3 months, time point 2 (TP2) was analyzed at the age of 5–6 months, and time point 3 (TP3) was analyzed at the age of 19–36 months. Collectively, we analyzed a total of 230 proviruses from a total of 68 million PBMCs; of these, 87% (199 proviruses) were defective and 13% (31 proviruses) were genome-intact. It is important to note that intact proviruses were only detected in eight of the nine infants, likely because the frequency of intact HIV-1 proviruses was below the threshold of detection in the available numbers of PBMC in the ninth infant. The numbers of proviruses identified from each infant are summarized in Supplementary Table 1.
Overall, we observed a reduction in the frequency of total HIV-1 copies per million PBMCs (Figure 1a) during the observation time in all infants, except for infant 5. This longitudinal decrease was particularly notable for intact proviruses, which declined from a median of 0.62 copies/million PBMC to detection levels below the threshold of our assay in all study participants (Figure 1b). Defective proviruses also declined during the follow-up period, from a median of 4.86 to 1.65 defective HIV-1 copies per million PBMCs (Figure 1c). Corresponding to these findings, we noted that the relative contribution of intact proviruses to the total number of all detectable proviruses showed a significant decline over time from 25% at TP1 to 11% at TP2 and finally, 7% at TP3 (Figure 1d). In infant 5, who had experienced viral rebound, an expansion of the frequency of intact proviruses was noted; in contrast, there was no expansion of the reservoir size in the two other infants (infant 2 and 7) who also experienced viral rebound.
Figure 1.
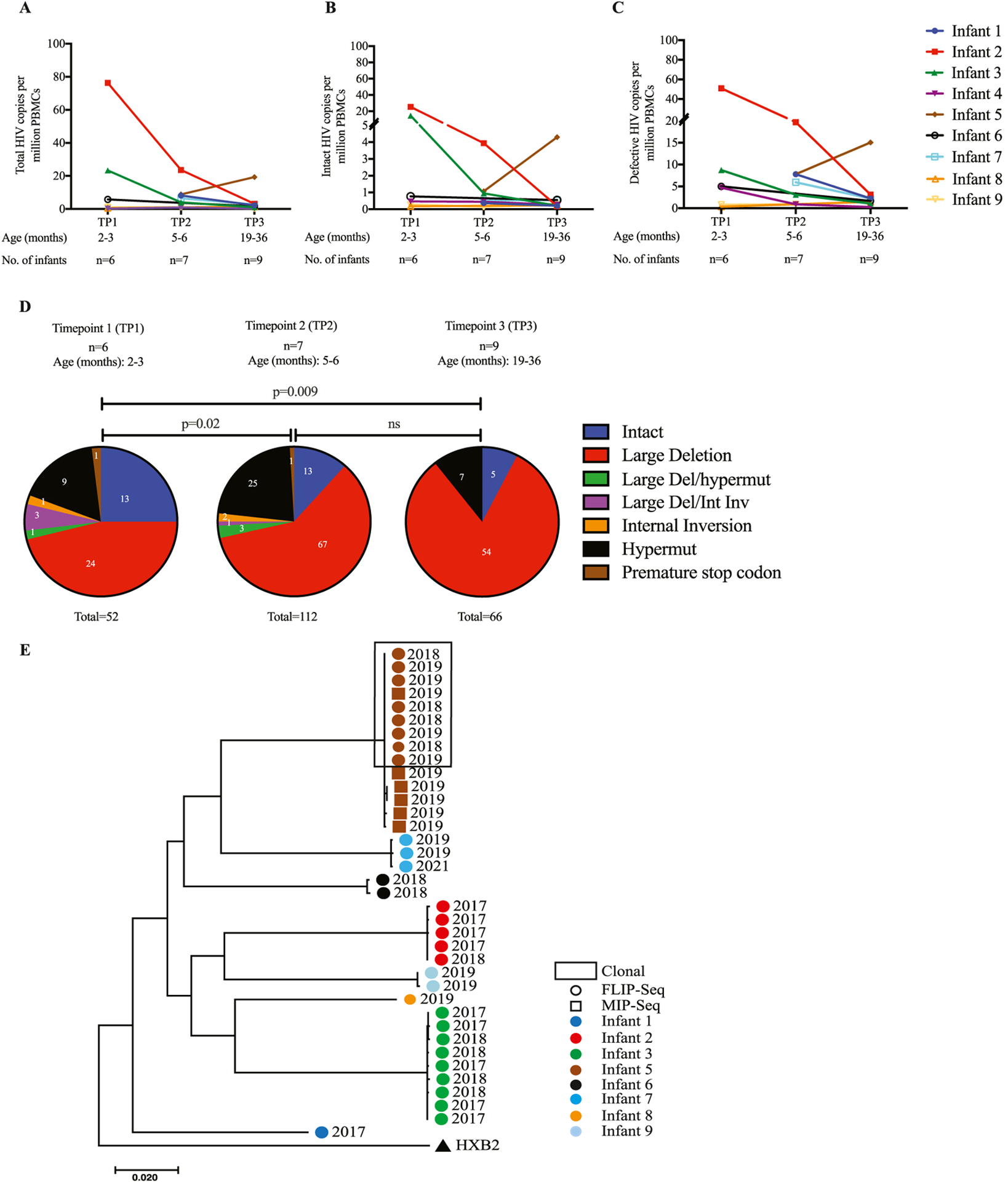
Longitudinal quantification of the proviral reservoir size in nine infants over the period of 24 months. (a) Longitudinal trends of total HIV copies per million PBMCs. Infant trends are distinguished by color. (b) Longitudinal absolute copies of genome-intact HIV per million PBMCs. (c) Longitudinal absolute copies of genome-defective HIV per million PBMCs. (d) Cumulative proportions of different proviruses detected from the nine infants at three different TPs grouped by age: TP1 = 2–3 months old, TP2 = 5–6 months old and TP3 = 19–36 months old. Statistical significance was tested using the chi-square test. Proportions of intact (Blue), large deletion (Red), large deletion with hypermut (Green), large deletion with internal inversion (Orange), hypermut (Black) and premature stop codon (Brown). (e) Maximum-likelihood phylogenetic tree of HIV-1 DNA intact genomes from eight infants. Proviral sequences generated from infants’ samples are differentiated by color coding: infant 1 (Blue), infant 2 (Red), infant 3 (Green), infant 5 (Brown), infant 6 (Black), infant 7 (Turquise), infant 8 (Tangerine/Orange), infant 9 (Sky blue).
FLIP-Seq, full-length individual proviral sequencing; MIP-Seq, Matched integration site and proviral sequencing; PBMC, peripheral blood mononuclear cell; TP, timepoint.
Antiretroviral drug resistance mutations in intact proviruses
A phylogenetic analysis of intact HIV-1 proviruses using maximum-likelihood trees demonstrated relatively little intraindividual sequence diversity among intact proviruses, likely because of the rapid treatment initiation after birth; however, strong phylogenetic differences were observed between sequences derived from the individual study patients, likely reflecting infection of patients with distinct founder viruses (Figure 1e). Further analysis of the proviruses using the Geno2pheno tool indicated that all intact proviruses sequenced from these infants were CCR5-tropic. Using the Stanford HIV Drug resistance database tool, we detected multiple non-nucleoside reverse transcriptase inhibitor (NNRTI) resistance mutations in the patients’ sequences in seven of the nine infants—namely, V106M, K103N, V106A, G190A, E138A, and Y181C (Table 2). These mutations were detected in genome-intact proviral sequences at all time points. The K103M and Y181C variations were found to be the most dominant NNRTI-associated mutations detected in the four infants. These mutations are unlikely to have been selected within these infants because none of these infants had been treated with NNRTIs; instead, they are more likely to reflect transmission of resistance mutations within founder viruses from their mothers. No protease inhibitor-associated mutations were detected in genome-intact proviral sequences at all time points in all infants. However, in the genome-intact proviral sequences of infant 7 (one of the three infants who experienced viral rebound), we detected NRTI-associated mutations, including A62V, K65R, M184V, and K219E. All these mutations were detected at the two time points analyzed (TP2 at age 6 months and TP3 at age 36 months). These resistance mutations may explain or contribute to the viral rebound experienced by this child who was treated with AZT+3TC+LPVr.
Table 2.
Antiretroviral drug resistance mutations in intact proviruses.
| Patient ID | NNRTI mutations | NRTI mutations |
|---|---|---|
| Infant 1 | E138A | none |
| Infant 2 | V106M, Y181C | none |
| Infant 3 | K103N | none |
| Infant 4 | Sequences N/A | Sequences N/A |
| Infant 5 | K103N, Y181C | none |
| Infant 6 | Y181C, V106A | none |
| Infant 7 | V106M, Y181C, G190A | A62V, K65R, M184, K219E |
| Infant 8 | none | none |
| Infant 9 | K103N, Y181C | none |
Abbreviations: N/A, not available; NNRTI, non-nucleoside reverse transcriptase inhibitor; NRTI, nucleoside reverse transcriptase inhibitor.
One of the nine infants (infant 5) experienced viral rebound at age 10 months (Figure 2a), which is best explained by insufficient adherence to antiretroviral medication; although NNRTI-associated resistance mutations to ART (K103N and Y181C) were detected in this infant’s genome-intact proviral sequences, no resistance was noted to protease inhibitors and NRTI, which were part of the patient’s treatment regimen. As indicated in Figures 1a, b, and c, this infant displayed an increase in the frequency of total, intact, and defective HIV-1 copies per million PBMCs after 19 months of follow-up, relative to baseline (at 6 months); this coincided with rebound of plasma viremia to 229,626 HIV-1 RNA copies/ml. The majority (84%) of proviruses generated from this infant at the different time points were defective, and the relative contribution of intact and defective proviruses to the total reservoir in this infant did not change over time (Figure 2b).
Figure 2.
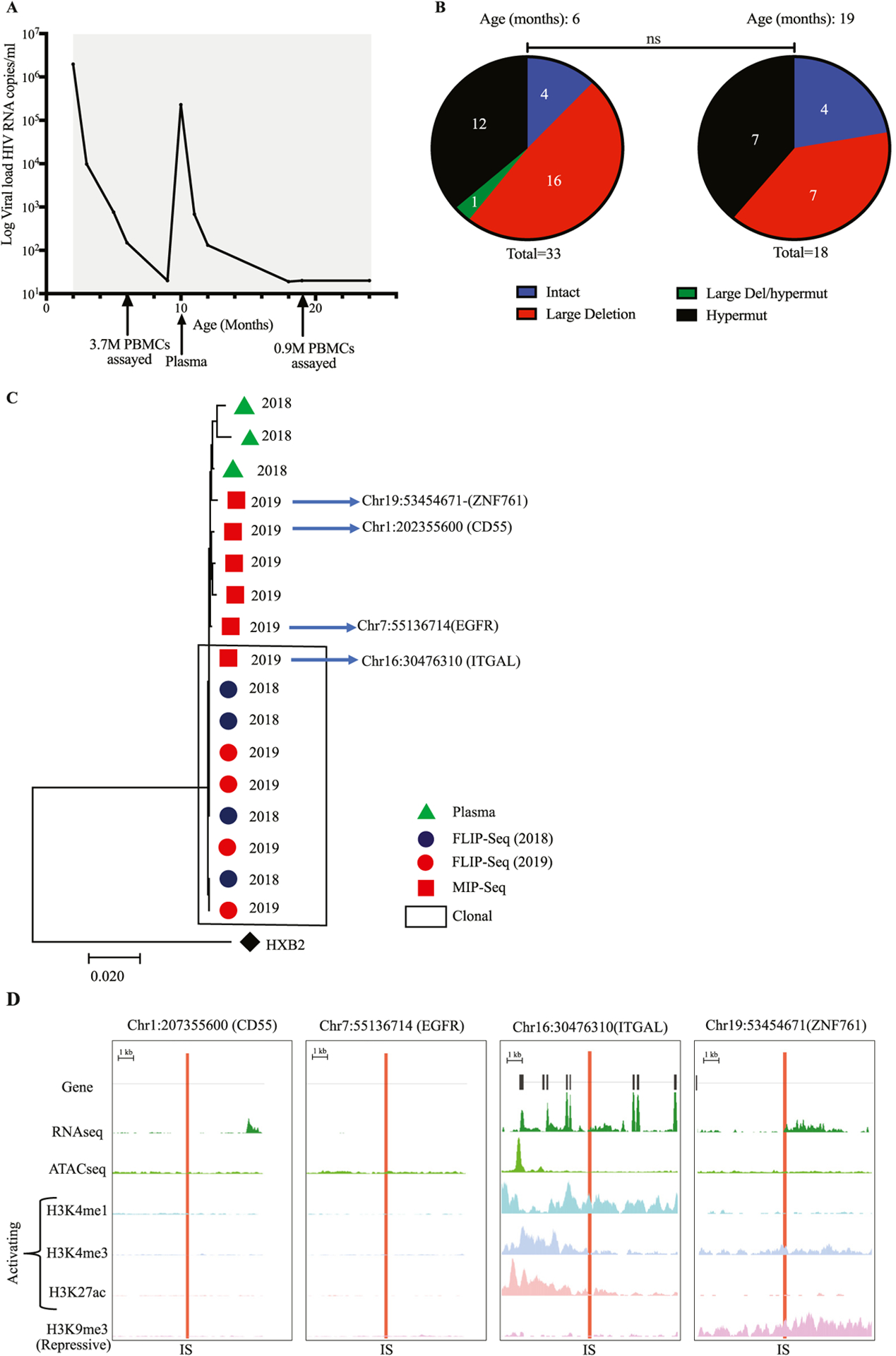
(a) Longitudinal viral load dynamics of infant 5 followed for a duration for 24 months. The gray bars indicate the duration of antiretroviral therapy. Arrows indicate the timepoint where specimens were collected: at age of 6 months old, 3.7 million PBMCs were assayed, at age of 10 months old, plasma specimen was analyzed and at age of 19 months old 900,000 PBMCs were assayed. (b) Proportions of different proviruses detected from infant 5 at two different TPs: The TP1 was at 6 months old and TP2 at 19 months old. Proportions of proviruses are differentiated by color: intact (Blue), large deletion (Red), large deletion with hypermut (Green), hypermut (Black). (c) Maximum-likelihood phylogenetic tree of HIV-1 DNA intact genomes from infant 5. Proviral sequences generated from the infant’s samples at different timepoints and using different assays are differentiated by color and shape. Red circles (FLIP-Seq) sequences from 2019, Blue circles (FLIP-Seq) sequences from 2018, Red squares (MIP-Seq) sequences from 2019 and Green Triangles represent sequences from plasma. (d) Chromosomal distance between integration sites of genome-intact proviral sequences and the most proximal transcriptional start sites, to the most proximal ATAC-seq peaks in total CD4+ T cells, numbers of DNA-sequencing reads in association with activating and repressive histone protein modifications in proximity to integration sites.
CD, clusters of differentiation; FLIP-Seq, full-length individual proviral sequencing; IS, integration site; MIP-Seq, Matched integration site and proviral sequencing; PBMC, peripheral blood mononuclear cell; TP, timepoint.
Clonal expansion of intact proviruses
Notably, at month 6 of follow-up in infant 5, we noted a cluster of four sequence-identical genome-intact proviruses; these sequences were again detected 13 months later at 19 months of age (Figure 2b). These data suggest the persistence of a large clone of infected CD4 T cells with a high rate of proliferative turnover. Integration site mapping indicated that intact proviruses from this clone were located in the ITGAL gene on chromosome 16 (Figure 2c), which can play a role in cell proliferation and, possibly, malignant transformation of cells [21,22]. Notably, other genome-intact proviruses that were independent of the large clone of intact proviruses with integration sites coordinates within the ITGAL gene were integrated into the following locations: Chr1:202355600 (CD55), Chr7:55136714 (EGFR), and Chr19:53454671 (ZNF761) (Figure 2c).
Phylogenetic analysis of rebound viremia
Having observed that infant 5 experienced viral rebound, we sought to determine which genome-intact proviruses were more likely driving plasma viremia during a time of possible non-adherence. For this purpose, we generated three near full-length HIV-1 RNA sequences from the plasma of this infant at the time of viral rebound (Figure 2c). We observed a high level of phylogenetic concordance between the HIV-1 plasma RNA sequence and the genome-intact proviral sequences isolated at the time of rebound; due to the low level of phylogenetic diversity among all intact proviruses, we were unable to directly identify which exact proviral sequence was responsible for the viral rebound. Overall, these results suggest that an integration site profile dominated by intact proviruses integrated into genic chromatin locations may be associated with rapid viral rebound; this represents an apparent contrast to individuals with natural, drug-free control of HIV-1, in whom genome-intact HIV-1 is predominantly integrated into heterochromatin locations [23].
Epigenetic features in chromosomal proximity of intact proviruses
In a subsequent exploratory analysis, we evaluated the epigenetic features in proximity to intact proviruses from study participant 5, using reference genome-wide ATAC-Seq and RNA-Seq data from primary CD4 T cells of patients treated with ART [24]. We observed that the large clone of genome-intact proviruses, integrated into the ITGAL gene, was located in immediate proximity to accessible chromatin and host transcriptional start sites (Figure 2d). Using ChIP-Seq data from primary memory CD4+ T cells available from the ROADMAP Epigenomics Project [25], we noted that the integration site of this clone was surrounded by the activating histone features H3K4me1, H3K4me3, and H3K27ac. The integration of intact proviruses in immediate proximity to activating histone modifications and accessible chromatin has previously been associated with high-level proviral transcriptional activity [24]; for that reason, it is reasonable to hypothesize that this proviral clone may most likely be responsible for the viral rebound in infant 5. Our data support the hypothesis that the presence of intact proviral clones integrated into an accessible and highly permissive chromatin location is associated with a rapid viral rebound during insufficient antiretroviral adherence.
Discussion
In this report, we analyzed the proviral reservoir evolution in infants from Mozambique who were vertically infected with HIV-1 subtype C, began ART within a median of 33 days of life, and were subsequently followed up for a period of 24 months. We observed that (i) six infants were virally suppressed throughout the entire time of follow-up; (ii) using high-resolution single-genome viral sequencing assays, we analyzed the individual proviral species, of which the majority were defective; (iii) we observed a decrease in the number of proviral sequences during longitudinal ART and this decline was more pronounced for intact proviruses, leading to a progressively increasing contribution of defective proviruses to the overall proviral landscape; (iv) in one of the infants who experienced viral rebound, we observed a viral reservoir profile dominated by clonal expansion of genome-intact proviruses integrated into the ITGAL gene on chromosome 16, in immediate proximity to accessible chromatin and activating histone modifications.
The detection of genome-intact proviral sequences is frequently difficult due to their low frequency, especially in individuals who initiated ART very early [26], such as the infants described here. Longitudinal analysis of the proviral reservoir conducted over the period of 24 months showed a decline in the size of the reservoir over time, which was more pronounced for the intact proviruses than the defective proviruses and resulted in a disproportionate over-representation of defective proviruses after more advanced analysis time points. These findings are consistent with earlier studies evaluating the longitudinal changes in the proviral reservoir in adults [27–29] and in neonates from Botswana [3,30]. We propose that the more pronounced decline of intact proviruses might be because cells harboring intact HIV-1 may be more vulnerable to host immune responses, arguably because such cells can produce complete viral particles that may be sensed by host immune recognition mechanisms. Future studies evaluating the susceptibility of HIV-1 reservoir cells to host immunity will be helpful to clarify this further.
In one of the nine infants studied (infant 5), we observed clonal expansion of genome-intact proviruses at month 6 and month 19, indicating that clonal proliferation of HIV-1-infected cells can occur early in life. We observed that genome-intact proviruses integrated into the following locations: Chr1(CD55), Chr7(EGFR), Chr19(ZNF761), and Chr16(ITGAL). The clonally expanded genome-intact proviruses were found to be located in the ITGAL gene on chromosome 16; this gene has previously been associated with a role for contributing to the regulation of cell proliferation and may have a pathogenetic function in a number of malignant diseases [21,22]. It is possible that proviral integration into such a proliferation-/cancer-associated gene might be driving proliferative turnover of the infected cells through insertional mutagenesis [17,31]. Generally speaking, insertional mutagenesis is a phenomenon where a foreign base pair is added to a host DNA sequence. When HIV-1 DNA integrates into the host DNA, it may alter the expression or function of the gene harboring the chromosomal integration site; this can in certain cases influence the phenotype or proliferative behavior of the infected cell.
The same infant (infant 5) who displayed clonally-expanded intact proviruses also experienced viral rebound at age 10 months. Although we were unable to identify the specific proviral species responsible for rebound, due to the low phylogenetic diversity among all identified intact proviruses, we propose that viral rebound may have most likely derived from the genome-intact proviral clone that integrated into the ITGAL gene on Chr16, which was located in immediate proximity to activating chromatin features; viral rebound originating from clonally expanded proviruses has indeed been postulated previously [32,33]. Although one of the identified intact proviruses was located in a ZNF gene on chromosome 19, a genetic region associated with heterochromatin features [34], we noted that the integration site landscape of intact proviruses in infant 5 was dominated by genic locations in euchromatin positions, supporting the hypothesis that such an integration site profile is readily able to fuel viral rebound in case of treatment interruptions. In contrast, an integration site profile characterized by the predominance of intact proviruses in heterochromatin locations, previously described in elite controllers, may contribute to the ability to control HIV-1 in the absence of suppressive therapy [23].
A weakness of our study was the relatively limited availability of PBMCs, related to the fact that PBMC sample quantities that can be safely collected from neonates and infants are typically restricted to around 1–5 million PBMC. Nonetheless, this study provides useful information on the impact of early ART initiation on the evolution and persistence of proviral reservoirs in HIV-1 subtype C infection in the first 2 years of life. An in-depth analysis focusing on single-genome sequencing of the proviral DNA, combined with integration site determination, may allow for a better understanding of the dynamics of proviral reservoir cell evolution in future studies.
Conclusion
In this study, we demonstrated that early ART initiation in infants perinatally infected with HIV-1 clade C resulted in a rapid decline of intact proviral sequences, likely due to immune mechanisms that can successfully eliminate a considerable proportion of cells infected with genome-intact HIV-1. In addition, the integration of genome-intact proviruses in locations that are in close proximity to activating chromatin marks may contribute to fueling viral rebound. Technologies to analyze both intact proviral sequences and integration sites permit a high-resolution evaluation of HIV-1 reservoir cells after early ART initiation in infants.
Supplementary Material
Funding
C.K.K. was supported by the Campbell Foundation, Fogarty International Center of the National Institutes of Health (D43 TW009610, D43 TW010543), and the Harvard University Center for AIDS Research, a National Institute of Health funded program (P30 AI060354). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. M.L. is supported by National Institutes of Health grants AI135940, AI114235, AI117841, AI120008, AI152979, AI130005, DK120387, K24 AI155233 (to M.L. and C.G.), American Foundation for AIDS Research (amfAR) (110181-69-RGCV), and the Campbell Foundation. M.L. is a member of the Delaney AIDS Research Enterprise (DARE), the Enterprise for Research & Advocacy to Stop & Eradicate HIV (ERASE), the Pediatric Adolescent Virus Elimination (PAVE), and the BEAT-HIV Martin Delaney Collaboratories (UM1 AI164560, AI164562, AI164566, AI164570). M.L., P.P., S.P., and N.C. are members of the Early Treated Perinatally HIV Infected Individuals: Improving Children’s Actual Life (EPIICAL) Consortium, funded by ViiV Healthcare. S.P. was supported by the National Institutes of Health grant AI127347. S.P. is a member of the PAVE HIV Martin Delaney Collaboratory (AI164566), the International Maternal Pediatric Adolescent AIDS Clinical Trials Network (IMPAACT), and the Multi AIDS cohort Study/Women’s Interagency HIV Study - Combined Cohort Study (MACS/WIHS - CCS) (U01HL146203).
Footnotes
Declaration of competing interest
The authors have no competing interests to declare.
Ethical approval
The legal caregivers of study patients consented to study participation; this study was approved by the National Ethics Committee in Mozambique and the permission for secondary use of collected PBMC samples was obtained from the Institutional Review Board of the University of Miami, Leonard M. Miller School of Medicine, the Massachusetts General Hospital, and the Brigham and Women’s Hospital.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi: 10.1016/j.ijid.2022.11.042.
CRediT authorship contribution statement
Catherine K. Koofhethile: Conceptualization, Methodology, Investigation, Writing – original draft, Writing x review & editing. Stefano Rinaldi: Investigation, Writing – review & editing. Yelizaveta Rassadkina: Investigation, Writing – review & editing. Vinh B. Dinh: Investigation, Writing – review & editing. Ce Gao: Investigation, Writing – original draft, Writing – review & editing. Suresh Pallikkuth: Writing – review & editing. Pilar Garcia-Broncano: Investigation, Writing – review & editing. Lesley R. de Armas: Writing – review & editing. Rajendra Pahwa: Writing – review & editing. Nicola Cotugno: Writing – review & editing. Paula Vaz: Writing – review & editing, Resources. Maria Grazia Lain: Conceptualization, Methodology, Writing – original draft, Writing – review & editing, Resources. Paolo Palma: Writing – review & editing, Resources. Xu G. Yu: Writing – review & editing, Supervision. Roger Shapiro: Writing – review & editing. Savita Pahwa: Writing – review & editing. Mathias Lichterfeld: Writing – review & editing, Resources, Supervision.
References
- [1].UNAIDS Global HIV statistics. Geneva: UNAIDs; 2020. [Google Scholar]
- [2].UNAIDS UNAIDS data 2021. Geneva: UNAIDs; 2021. [Google Scholar]
- [3].Hartana CA, Garcia-Broncano P, Rassadkina Y, et al. Immune correlates of HIV-1 reservoir cell decline in early-treated infants. Cell Rep 2022;40:111126. doi: 10.1016/j.celrep.2022.111126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Luzuriaga K, Tabak B, Garber M, YaH Chen, Ziemniak C, McManus MM, Murray D, et al. HIV Type 1 (HIV-1) proviral reservoirs decay continuously under sustained virologic control in HIV-1-infected children who received early treatment. J Infect Dis 2014;210:1529–38. doi: 10.1093/infdis/jiu297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Van Zyl GU, Bedison MA, van Rensburg AJ, Laughton B, Cotton MF, Mellors JW. Early antiretroviral therapy in South African children reduces HIV-1-infected cells and cell-associated HIV-1 RNA in blood mononuclear cells. J Infect Dis 2015;212:39–43. doi: 10.1093/infdis/jiu827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Katusiime MG, Halvas EK, Wright I, Joseph K, Bale MJ, Kirby-McCullough B, Engelbrecht S, et al. Intact HIV proviruses persist in children seven to nine years after initiation of antiretroviral therapy in the first year of life. J Virol 2020;94:e01519. doi: 10.1128/JVI.01519-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kuhn L, Paximadis M, Da Costa Dias B, Loubser S, Strehlau R, Patel F, Shiau S, Coovadia A, Abrams EJ, Tiemessen CT. Age at antiretroviral therapy initiation and cell-associated HIV-1 DNA levels in HIV-1-infected children. PLoS One 2018;13:e0195514. doi: 10.1371/journal.pone.0195514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Josefsson L, Von Stockenstrom S, Faria NR, Sinclair E, Bacchetti P, Maudi Killian, Epling L, et al. The HIV-1 reservoir in eight patients on long-term suppressive antiretroviral therapy is stable with few genetic changes over time. Proc Natl Acad Sci U S A 2013;110:E4987–96. doi: 10.1073/pnas.1308313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DIS, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013;155:540–51. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hiener B, Horsburgh BA, Eden JS, Barton K, Schlub TE, Lee E, von Stockenstrom S, Odevall L, Milush JM, Liegler T, Sinclair E, Hoh R, Boritz EA, Douek D, Fromentin R, Chomont N, Deeks SG, Hecht FM, Palmer S. Identification of genetically intact HIV-1 proviruses in specific CD4+ T cells from effectively treated participants. Cell Rep 2017;21:813–22. doi: 10.1016/j.celrep.2017.09.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lee GQ, Reddy K, Einkauf KB, Gounder K, Chevalier JM, Dong KL, Walker BD, Yu XG, Ndung’u T, Lichterfeld M. HIV-1 DNA sequence diversity and evolution during acute Subtype C infection. Nat Commun 2019;10:2737. doi: 10.1038/s41467-019-10659-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Einkauf KB, Lee GQ, Gao Ce, Sharaf R, Sun X, Hua S, MY Chen S, et al. Intact HIV-1 proviruses accumulate at distinct chromosomal positions during prolonged antiretroviral therapy. J Clin Invest 2019;129:988–98. doi: 10.1172/JCI124291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lee GQ, Orlova-Fink N, Einkauf K, Chowdhury FZ, Sun X, Harrington S, Kuo HH, et al. Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+ T cells. J Clin Invest 2017;127:2689–96. doi: 10.1172/JCI93289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 2018;35:1547–9. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Stecher G, Tamura K, Kumar S. Molecular evolutionary genetics analysis (MEGA) for MacOS. Mol Biol Evol 2020;37:1237–9. doi: 10.1093/molbev/msz312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].HIV Sequence Compendium 2018, Abfalterer Werner, Giori Elena E., Fischer Will, Hraber Peter, Macke Jennifer, Szinger James J., Kshitij Wagh, Yoon Hyejin., Los Alamos National Laboratory HIV Sequence Database (2018).
- [17].Wagner TA, McLaughlin S, Garg K, Cheung CYK, Larsen BB, Styrchak S, Huang HC, Edlefsen PT, Mullins JI, latency Frenkel LMHIV. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 2014;345:570–3. doi: 10.1126/science.1256304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lengauer T, Sander O, Sierra S, Thielen A, Kaiser R. Bioinformatics prediction of HIV coreceptor usage. Nat Biotechnol 2007;25:1407–10. doi: 10.1038/nbt1371. [DOI] [PubMed] [Google Scholar]
- [20].Liu TF, Shafer RW. Web resources for HIV Type 1 genotypic-resistance test interpretation. Clin Infect Dis 2006;42:1608–18. doi: 10.1086/503914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kang H, Tan M, Bishop JA, Jones S, Sausen M, Ha PK, Agrawal N. Whole-exome sequencing of salivary gland mucoepidermoid carcinoma. Clin Cancer Res 2017;23:283–8. doi: 10.1158/1078-0432.CCR-16-0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Boguslawska J, Kedzierska H, Poplawski P, Rybicka B, Tanski Z, Piekielko-Witkowska A. Expression of genes involved in cellular adhesion and extracellular matrix remodeling correlates with poor survival of patients with renal cancer. J Urol 2016;195:1892–902. doi: 10.1016/j.juro.2015.11.050. [DOI] [PubMed] [Google Scholar]
- [23].Jiang C, Lian X, Gao C, Sun X, Einkauf KB, Chevalier JM, MY Chen S, et al. Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nature 2020;585:261–7. doi: 10.1038/s41586-020-2651-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Einkauf KB, Osborn MR, Gao C, Sun W, Sun X, Lian X, Parsons EM, et al. Parallel analysis of transcription, integration, and sequence of single HIV-1 proviruses. Cell 2022;185:266–82 e15. doi: 10.1016/j.cell.2021.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Consortium Roadmap Epigenomics, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, et al. Integrative analysis of 111 reference human epigenomes. Nature 2015;518:317–30. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rainwater-Lovett K, Ziemniak C, Watson D, Luzuriaga K, Siberry G, Petru A, Chen YaH, et al. Paucity of intact non-induced provirus with early, long-term antiretroviral therapy of perinatal HIV infection. PLoS One 2017;12:e0170548. doi: 10.1371/journal.pone.0170548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Peluso MJ, Bacchetti P, Ritter KD, Beg S, Lai J, Martin JN, Hunt PW, et al. Differential decay of intact and defective proviral DNA in HIV-1–infected individuals on suppressive antiretroviral therapy. JCI Insight 2020;5:e132997. doi: 10.1172/jci.insight.132997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gandhi RT, Cyktor JC, Bosch RJ, Mar H, Laird GM, Martin A, Collier AC, et al. Selective decay of intact HIV-1 proviral DNA on antiretroviral therapy. J Infect Dis 2021;223:225–33. doi: 10.1093/infdis/jiaa532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Falcinelli SD, Kilpatrick KW, Read J, Murtagh R, Allard B, Ghofrani S, Kirchherr J, et al. Longitudinal dynamics of intact HIV proviral DNA and outgrowth virus frequencies in a cohort of individuals receiving antiretroviral therapy. J Infect Dis 2021;224:92–100. doi: 10.1093/infdis/jiaa718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Garcia-Broncano P, Maddali S, Einkauf KB, Jiang C, Gao C, et al. Early antiretroviral therapy in neonates with HIV-1 infection restricts viral reservoir size and induces a distinct innate immune profile. Sci Transl Med 2019;11:eaax7350. doi: 10.1126/scitranslmed.aax7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liu R, Yeh YJ, Varabyou A, Collora JA, Sherrill-Mix S, Talbot CC, Mehta S, et al. Single-cell transcriptional landscapes reveal HIV-1-driven aberrant host gene transcription as a potential therapeutic target. Sci Transl Med 2020;12. doi: 10.1126/scitranslmed.aaz0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Simonetti FR, Sobolewski MD, Fyne E, Shao W, Spindler J, Hattori J, Anderson EM, et al. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc Natl Acad Sci U S A 2016;113:1883–8. doi: 10.1073/pnas.1522675113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].De Scheerder MA, Vrancken B, Dellicour S, Schlub T, Eunok Lee, Shao W, Rutsaert S, et al. HIV rebound is predominantly fueled by genetically identical viral expansions from diverse reservoirs. Cell Host Microbe 2019;26:347–58 e7. doi: 10.1016/j.chom.2019.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lukic S, Nicolas JC, Levine AJ. The diversity of zinc-finger genes on human chromosome 19 provides an evolutionary mechanism for defense against inherited endogenous retroviruses. Cell Death Differ 2014;21:381–7. doi: 10.1038/cdd.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.