

Summary
Neddylation has been implicated in various cellular pathways and in the pathophysiology of numerous diseases. We identified four individuals with bi-allelic variants in NAE1, which encodes the neddylation E1 enzyme. Pathogenicity was supported by decreased NAE1 abundance and overlapping clinical and cellular phenotypes. To delineate how cellular consequences of NAE1 deficiency would lead to the clinical phenotype, we focused primarily on the rarest phenotypic features, based on the assumption that these would best reflect the pathophysiology at stake. Two of the rarest features, neuronal loss and lymphopenia worsening during infections, suggest that NAE1 is required during cellular stress caused by infections to protect against cell death. In support, we found that stressing the proteasome system with MG132—requiring upregulation of neddylation to restore proteasomal function and proteasomal stress—led to increased cell death in fibroblasts of individuals with NAE1 genetic variants. Additionally, we found decreased lymphocyte counts after CD3/CD28 stimulation and decreased NF-κB translocation in individuals with NAE1 variants. The rarest phenotypic feature—delayed closure of the ischiopubic rami—correlated with significant downregulation of RUN2X and SOX9 expression in transcriptomic data of fibroblasts. Both genes are involved in the pathophysiology of ischiopubic hypoplasia. Thus, we show that NAE1 plays a major role in (skeletal) development and cellular homeostasis during stress. Our approach suggests that a focus on rare phenotypic features is able to provide significant pathophysiological insights in diseases caused by mutations in genes with pleiotropic effects.
Keywords: ubiquitination, neddylation, proteasome, post-translational protein modification, phenotypic specificity, neurodegeneration, NAE1, ocurrence ratio, lymphopenia
Graphical abstract
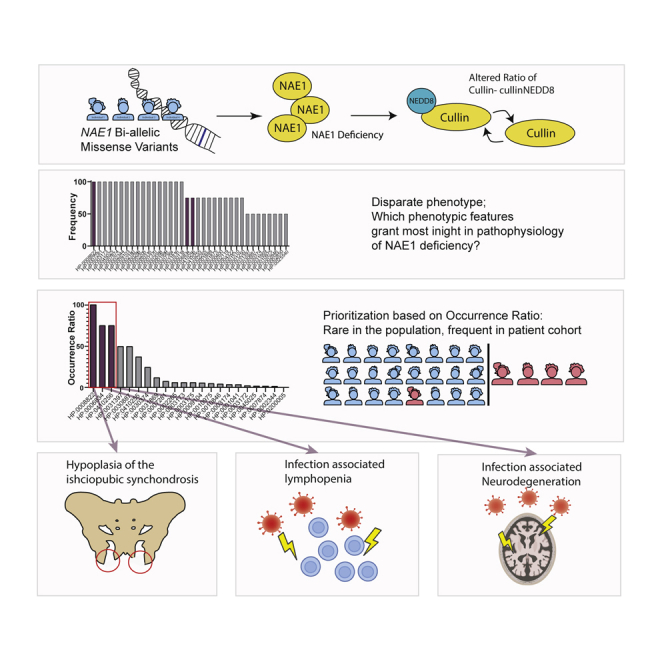
The authors describe four individuals with bi-allelic variants in NAE1, which encodes the NEDD8-activating enzyme E1 regulatory subunit and is essential for neddylation. By focusing on the biology underlying the rarest observed clinical features, they unveil the importance of neddylation for skeletal development and for maintaining cellular integrity during stress.
Introduction
Covalent attachment of ubiquitin, NEDD8, and SUMO proteins to target proteins (called ubiquitination, neddylation, and sumoylation, respectively) is an important mechanism for eukaryotic protein regulation.1 Neddylation involves the attachment of NEDD8 to a protein target through a sequential, three-step process, that is facilitated by so-called E1, E2, and E3 enzymes.2,3,4 The E1 complex consists of APPBP1—encoded by NAE1 (MIM: 603385)—and UBA3.5,6,7 While UBA3 acts as the activating enzyme, APPBP1 accelerates the kinetics of this first step.6 The E2 enzyme aids a consecutive transthiolation reaction of NEDD8. E3 enzymes catalyze the transfer of NEDD8 from the E2 enzyme onto the neddylation target. The best-known targets of neddylation are cullins, that form cullin-RING ligase complexes upon neddylation (CRLs),8,9 which can tag proteins for degradation via the 26S proteasome. Additionally, non-cullin neddylation targets have been described.2,10
Neddylation is thought to be involved in the pathophysiology of many diseases, including Alzheimer disease,11,12 Parkinson disease,13 certain auto-immune diseases,14 malignancies,15,16,17 and hepatic fibrosis.18 This is not surprising, given that neddylation is a key regulator of a wide array of essential intracellular pathways, including NF-κB,14,19 HIF-1α,20 β-catenin,21 and XPC.22,23 Despite neddylation’s broad involvement in disease, the exact pathophysiological role of neddylation in these diseases remains elusive, partly due to the large variety of cellular pathways affected by neddylation.
Through GeneMatcher,24 we identified four individuals harboring NAE1 genetic variants, that led to decreased NAE1 abundance and altered neddylation dynamics. Next, we aimed to study the cellular impact, but the large variety of cellular pathways that could be affected by faulty neddylation called for prioritization. Based on the rationale of information theory, which states that the information content of rare events is highest,25 we studied the underlying cellular mechanisms of phenotypic features that were present in all individuals but rarely present in individuals with other genetic diseases.26
The three rarest phenotypic features consisted of ischiopubic synchondrosis hypoplasia, infection-triggered lymphopenia, and infection-triggered neurodegeneration. In vitro biochemical analysis of these features helped to delineate the most important functions of NAE1 for human health: facilitating (skeletal) development and maintaining homeostasis during cellular stress, through a subset of regulatory pathways.
Material and methods
Ethics
Individuals 1 and 2 were recruited in the Wilhelmina Children’s Hospital Utrecht. Informed consent was obtained to use residual material collected for diagnostic purposes, to include in the Wilhelmina Children’s Hospital metabolic biobank (TCBio 19-489/B, https://tcbio.umcutrecht.nl). By using the same biobank, we included residual material of pediatric healthy fibroblast lines. Healthy adult donor peripheral blood mononuclear cells (PBMCs) were obtained through the Minidonor Service, an ethics review board-approved blood donation facility at the UMC Utrecht (protocol number 18-774). To compare the clinical phenotype of individuals with NAE1 variants to a pediatric reference population, clinical data from the Utrecht Patient-Oriented Database (UPOD) was used. UPOD is an infrastructure of relational databases comprising data on affected individuals that visited the University Medical Center Utrecht (UMC Utrecht) since 2004. UPOD data acquisition and management are in accordance with Dutch regulations concerning privacy and ethics. The structure and content of UPOD have been described in more detail elsewhere.27 Pelvic X-rays were analyzed using the picture archiving and communication system (PACS IDS7 22.1.5). The storage and handling of medical data was recorded in a data management plan which can be consulted via https://dmponline.dcc.ac.uk/plans/66973. Individual 3 was recruited via the Dravet Center and Comprehensive Epilepsy Center in New York, and individual 4 was recruited via the University of Arkansas for Medical Sciences. They were recruited through the PhenomeCentral repository. Funding for PhenomeCentral was provided by Genome Canada and Canadian Institute of Health Research (CIHR).28 From all four individuals, informed consent was obtained for publication of facial images and medical information. All procedures performed in studies involving human participants are in accordance with national and local institutional review boards (IRBs) of the participating centers.
Whole-exome sequencing
Exomes were enriched using Agilent SureSelect XT Human All Exon kit V5 and sequenced on a HiSeq sequencing system (Illumina). Reads were aligned to hg19 using a Burrows-Wheeler Aligner. Variants were called using Genome Analysis Toolkit Variant Caller and annotated, filtered, and prioritized using the Bench NGS Lab platform (Agilent-Cartagenia) and/or an in-house designed “variant interface” and manual curation. The minimal coverage of the full target was >15 × 95%. All common polymorphisms with a minor allele frequency (MAF) higher than 0.25 were filtered out using several public databases including 1,000 genomes database,29 Ensembl GRCh37 genome browser,30 exome aggregation consortium database (ExAC),31 genome aggregation database (gnomAD),32 and database of single nucleotide polymorphisms (dbSNP). Variant calling was performed using the complete human reference genome (hg19, NCBI release GRCh37).33
RNA sequencing
RNA sequencing was performed as previously described.34 Briefly, total RNA was isolated from fibroblast cultures using Trizol LS reagent (Invitrogen). mRNA was isolated using Poly(A) Beads (NEXTflex). Sequencing libraries were prepared using the Rapid Directional RNA-Seq Kit (NEXTflex) and sequenced on a NextSeq500 (Illumina) to produce 75 base long reads (Utrecht DNA Sequencing Facility). Sequencing reads were mapped against the reference genome (hg19 assembly, NCBI37) using BWA35 package (mem –t 7 –c 100 –M –R). RPKM values were calculated using the rnaseq_countgeneread function from Cisgenome v.233 and log2 transformed for further analysis. To perform statistical enrichment in a ranked gene list, the differential gene expression score was calculated: (overall mean RPKM) × abs(log2 FC) × –log10(adjusted p value). Subsequent analysis was performed using the Gene Set Enrichment Analysis (GSEA) tool from Broad Institute with the following settings: GSEA pre-ranked, 1000 permutations, GO v.7.1 (updated on: 30-Mar-2021) and KEGG cellular processes.36
Statistical overrepresentation in Gene Ontology (GO) terms among differentially expressed downregulated genes (Log2Fold <−2 and adjusted p value < 0.05) were analyzed using R (v.4.0.3) and R Studio (v.1.3.1093) using the ClusterProfiler package.37 A list of all expressed genes within control subjects and the affected individuals with a base mean >2 was used as background.
Plasmid cloning of NAE1 shRNA and NAE1 rescue construct
Restriction enzymes (Age1, EoRi-Hf, Nhe1, Mlu1) were purchased from New England Biosciences. NAE1 shRNA was cloned into Tet-pLKO-puro (Addgene). Targeting sequences were obtained using the TRC sequencing database38 and can be found in Table S4. To generate shRNA-expressing plasmids, the stuffer DNA was removed from pLKO-Tet-On by an AgeI/EcoRI digest and replaced with double-stranded oligonucleotides encoding the desired shRNA and AgeI/EcoRI sites. This product was transformed to Stellar competent bacteria (Takara Bio). The bacteria were midi prepped according to the manufacturer’s protocol (Invitrogen) to obtain plasmid DNA. The sequence was verified by sequencing the entire insert. Lentiviruses were generated by co-transfecting HEK293 cells with 6 μg of the NAE1 shRNA-encoding plasmid and lentiviral packaging plasmids pMD2.G (Addgene) and psPAX (Addgene). After three days, lentiviruses were obtained by spinning down the filtered HEK293 medium at 50,000 × g for 120 min. The pellet contained the lentiviruses. Fibroblasts were transduced by incubation of 8 μg/mL polybrene with lentivirus containing medium for 24 h. The transduced fibroblasts with build-in construct were selected with Puromycin (Santa Cruz Biotechnology). shRNA expression was induced by addition of 1 μg/mL doxycycline (Sigma) for 7 days.39,40
The WT NAE1 cDNA of isoform a (GenBank: NM_003905.4) was cloned from human cDNA using flanking primers that contained complimentary 5′ and 3′ coding sequences and either Mlu1 or Nhe1 restriction sites, respectively. The pLenti CMV Puro vector (Addgene) was digested with Nhe1 and Mlu1 and then ligated with the insert. Plasmid DNA and lentiviruses were generated similar to the generation of NAE1 shRNA-containing plasmids.
Fibroblast viability assays
Fibroblasts were obtained from individual 1, individual 2, both parents of individual 1, and healthy control subjects. Cells were cultured in fibroblast culture medium (HAM F12 with 10% fetal bovine serum, penicillin [100 UI/mL] and streptomycin [100 μg/mL]), in a humidified incubator at 37°C and 5% CO2. Medium was changed every 3–4 days. Cells were split at 80% confluency.
For the viability assays, cells were seeded (2,000 cells/well) in a 96-well plate. The following day, normal culture medium was removed, and medium with different stressors—MG132 (Cayman Chemicals) or MLN4924 (Focus Biomolecules)—was added at the indicated concentrations. After 24 h, culture medium was removed and replaced with 1:2,000 HOECHST 33342 (Sigma) and 1:20 propidium iodine (ThermoFisher). Fluorescent images were obtained with the EVOS XL Imaging System (ThermoFisher). The percentage of dead cells was calculated by dividing the number of PI-positive cells by the number of HOECHST-positive cells.
Western blotting
Western blots of cullin 1, cullin 3, and NAE1 were performed in the Max Planck Institute. Cell lysates were prepared by adding RIPA lysis buffer (50 mM TrisHCl [pH 8.0], 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitors (Sigma), Phosphostop (Roche), 20 μM N-ethylmaleimide (NEM) (Sigma-Aldrich), and 1,10-orthophenathroline (OPT) (Sigma-Aldrich) to the cells, scraping into a 1.5 mL tube, sonicating, and centrifuging. Protein concentrations were determined by Bradford protein assay (Biorad). Equal amounts of protein (6 μg) were loaded into each lane of an 8%–15% SDS-PAGE gel, subjected to electrophoresis, and transferred onto a PVDF membrane (Millipore). Binding of secondary horseradish peroxidase-conjugated antibodies was visualized by chemiluminescent substrate (Millipore). Western blotting membranes were probed with NAE1 (dilution 1:1,000, Novus Biologicals), cullin 1 (dilution 1:1,000, Thermofisher Scientific), cullin 3 (dilution 1:1,000, Cell Signaling Technology), β-actin (dilution 1:1,000, Cell Signaling Technology). The intensity values were normalized against total protein using stain-free detection gels (Bio Rad). Western blots of NAE1 in fibroblasts transduced with NAE1 shRNA or cDNA were performed in the UMC Utrecht, as described previously.41 β-actin (dilution 1:1,000, Cell Signaling Technology) or HSP90 (dilution 1:1,000, Cell Signaling Technology) were used as a housekeeper for normalization.
Flow cytometry
PBMC’s were isolated from the whole blood fraction using Ficoll (GE Healthcare). CD3+ cells were separated from the PBMC fraction using Pan T cell isolation kit (Miltenyi). Cells were stimulated with soluble anti-CD3+ (Life Technologies, clone OKT3) and anti-CD28+ (Thermofisher) at 1 μg/mL for 1 day. Cells were stained with CD3 (dilution 1:50, Biolegend), CD4 (dilution 1:200, eBioscience), CD8 (dilution 1:25, BD), CD45RA (dilution 1:500, Biolegend), CD45RO (dilution 1:12.5, Beckman Coulter) for 20 min at 4°C, followed by incubation with Annexin-V and 7-AAD (dilution 1:20, BD Biosciences) for 15 min at room temperature.
For the analysis of double-negative (CD3+, CD4−, CD8−) T cell counts, number of naive T cells, and the MLN4924 assay, PBMCs were cultured in RPMI with 10% heat-inactivated FCS, 1% penicillin/streptomycin, and 1% L-glutamine for 1 day with or without MLN4924, on a plate coated with 0.1 μg/mL anti-CD3. Afterward, cells were stained with CD3, CD4, CD8, CD45RA, CD45RO, Fixable Viability Dye eFluor 506, fixed with formaldehyde, and permeabilized with methanol. Cells were immediately analyzed using BD LSRFortessa. Flow cytometry analysis was performed using FlowJo (v.10.6.2, Becton, Dickinson and Company; 2019). Gating strategy is shown in Figure S2.
Imagestream NF-κB translocation
CD3+ T cells were stimulated with anti-CD3+/CD28+ at 2 μg/mL for the indicated time points (0, 15, and 35 min). After stimulation, cells were fixed immediately using 4% paraformaldehyde. Fixed cells were permeabilized with 0.1% Triton X-100 for 10 min and stained with p65 (dilution 1:16,000, Cell Signaling Technology) and DRAQ5 (dilution 1:8,000, Biolegend) antibodies for 20 min at 4°C, followed by incubation with FITC-anti Rabbit (Jackson) for 20 min at 4°C. Data were collected with Amnis Imagestream MkII Imaging Flow Cytometrer (Luminex) and data analysis was performed with IDEAS v.6.0 software. Nuclear area was determined using the morphology mask. Cells with a similarity score >0 were considered translocated. Gating strategy can be found in Figure S3.
Data analysis
Statistical analysis was performed using GraphPad Prism v.6 for Windos (GraphPad Software). Final counts are presented as the mean percentages ± SEM; one- or two-way ANOVA were performed for multiple comparisons. As post hoc, uncorrected Fisher’s least discriminant analysis was used. To assess normality, Shapiro-Wilk test was used. Unpaired two-tailed Student’s t tests or Mann-Whitney U were used for dual comparisons. p < 0.05 was considered significant: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Occurrence ratio
The occurrence ratio was calculated by dividing the percentage of individuals showing a rare trait by the number of associated genes, extracted from the Human Phenotype Ontology database (HPO) at 20-02-2021.42
Results
Clinical features of individuals with NAE1 variants
The index proband (individual 1) was born as the second child to healthy non-consanguineous parents. She presented with a ventricular septal defect, coarctation of the aorta, and failure to thrive. At one year of age, she was admitted to the ICU with a severe viral respiratory tract infection that led to subsequent loss of developmental milestones and profound therapy-resistant epilepsy. Brain MRI before ICU admission was normal, but after the ICU admission it showed brain atrophy, as evidenced by enlarged ventricles and diminished white matter volume. A subsequent viral respiratory infection a year later resulted in a similar loss of milestones. During these and other infections, she exhibited lymphopenia, which normalized upon recovery. Pelvic X-ray taken at age four showed absence of the os pubis ramus inferior and hip dysplasia. Extensive etiological workup did not lead to a diagnosis. Whole-exome sequencing revealed several genetic variants of uncertain significance (Table S1). Of these variants, the NAE1 variant was considered the most promising candidate, based on conservation, constraint metrics, and predictions: GenBank: NM_003905.4: c.[147G>C]; [254G>A], p.[Leu49Phe]; [Arg85Gln].
Through GeneMatcher,24 three additional individuals with similar phenotypic features, harboring homozygous variants in NAE1, were identified (Table 1). Additionally, individual 4 has a pathogenic genetic variant in SCN1A (MIM: 182389) and displays phenotypic features associated with Dravet syndrome43 (MIM: 607208) as well as NAE1 deficiency (Table S2).
Table 1.
Overview of symptoms of individuals with NAE1 variants
| NAE1 variant | Individual 1 c.[147G>C]; [254G>A]p.[Leu49Phe]; [Arg85Gln] | Individual 2c.[254G>A]; [254G>A]p.[Arg85Gln]; [Arg85Gln] | Individual 3 c.[1289 G>A]; [1289G>A] p.[Arg430Gln]; [Arg430Gln] | Individual 4 c.[882C>G]; [882C>G]p.[Cys294Trp]; [Cys294Trp] |
|---|---|---|---|---|
| Other variants identified with WES | full list in Table S1 | no | no | SCN1A:c.[1624C>T]; [=], p.[Arg542∗]; [=], |
| Current age (years) | 12 | 19 | 4 | 9 |
| Gender | F | M | M | F |
| Pregnancy term (weeks) | 40 | 36 | 37 | 40 |
| Birth weight (g) | 2,615 (−1.8 SDS) | 2,767 (0.05 SDS) | 2,140 (−1.6 SDS) | 2,300 (−2.48 SDS) |
| Head circumference (cm) | 51 (−0.47 SDS) (8 y) | 54 (−0.59 SDS) (13 y) | 47.3 (−1.5 SDS) (2 y 5 m) | 48.7 (−1.73 SDS) (6 y 6 m) |
| Height (cm) | 116 (−2.53 SDS) (8 y) | 150 (−1.5 SDS) (13 y) | 91 (−1 SDS) (2 y 5 m) | 97 (−5.9 SDS) (7 y) |
| Cleft palate | – | + | – | – |
| Asymmetrical palate | + | + | – | N/A |
| Heart defects | + ventricular septal defect and coarctation of the aorta | – | – | – |
| Developmental delay | severe | moderate | moderate | severe |
| Seizures | + | – | + | + |
| Hypotonia during infancy | + | – | + | + |
| Underdeveloped corpus callosum | + | + | + | + |
| Decreased myelination | + | + | + | + |
| Enlarged ventricles | + | – | + | + |
| Loss of milestones after infections | + | + | + | + |
| Neurodegeneration | + | + | + | + |
| Decreased bone density (DEXA scan) | + | + | + | + |
| Joint dislocation | + | + | – | – |
| Joint hyperextensibility | + | + | – | + |
| Joint stiffness | – | – | – | + |
| Resistant to sunburns | + | + | + | – |
| Recurrent infections | + respiratory, urinary tract, skin | + respiratory, skin | + respiratory | + respiratory, urinary tract |
| Hepatomegaly at infancy | + | – | + | – |
| Splenomegaly at infancy | + | + | + | – |
| Decreased amount of immunoglobulins | + IgG | + IgG, IgM | – | – |
| Leukopenia | + | + | – | + |
| Periodic lymphopenia | + | + | – | + |
| AST/ALT increase | + during infections | – | – | – |
| Delayed closure of ischiopubic rami | + | + | N/A | N/A |
| Increased size of ischiopubic rami | + | + | + | + |
All individuals showed similar facial features (Figure 1): broad forehead (individuals 1, 2, 4), downslant palpebral fissures (individuals 1, 2, 3), epicanthus/telecanthus (individuals 1, 3), round face (individuals 1, 3), prominent cheeks (individuals 1, 3, 4), broad nasal tip (individuals 1, 3, 4), pointed chin (individuals 1, 2, 4), and full lower lip (individuals 2, 4).
Figure 1.

Facial features of the cohort and structural analysis of NAE1 variants
(A) Representative photographs illustrating facial features of individuals with NAE1 variants.
(B) Graphical representation of UBA3 (space filling, yellow) in complex with NAE1 (light gray), the E2 of neddylation (EM2, blue), and two molecules of NEDD8 (light and dark green). The positions of Leu49, Arg85, Arg430, and Cys294 are marked in red. ATP is space filling in magenta. The color coding is used throughout the entire figure.
(C) The local environment of Leu49. NAE1 is represented in part as space filling and in part as backbone trace.
(D–F). Local environments of Arg85, Arg430, and Cys294. Dotted lines indicate hydrogen bonds. Figures were generated by use of programs Molscript and Raster3D.44,45
All individuals developed epilepsy and experienced exacerbation of epilepsy and/or developmental decline during infectious periods. All individuals showed moderate to severe developmental delay. All individuals showed underdeveloped corpus callosum and enlarged lateral ventricles in MRI. During infancy, three out of four individuals showed splenomegaly. All individuals had frequent infections, which were mostly respiratory tract infections, but urinary tract infections (2/4) and skin infections (2/4) were also observed. Three out of four individuals had documented lymphopenia during infections, that normalized during recovery. All individuals showed hypoplastic ischiopubic rami and decreased bone mineral density (assessed using dual energy X-ray absorptiometry [DEXA] scans). Fibroblasts and PBMCs were obtained from two individuals (individuals 1 and 2) for biochemical analyses. A summary of clinical features can be found in Table 1 and the supplemental note.
NAE1 abundance and the ratio of neddylated to non-neddylated cullin are altered in individuals with NAE1 variants
The amino acid positions where the NAE1 missense variants are located are highly conserved in other species (Figure S5). All variants cluster together in the three-dimensional structure of the protein, even though they are located at the N- and C-terminal portions of the protein (Figure 1B). Leu49 (individual 1) is part of the hydrophobic core of NAE1 (Figure 1C). A phenylalanine at this position is sterically not tolerated and is expected to result in (local) perturbation of the fold. This may reduce the ability of NAE1 to interact with UBA3 and impacts the positioning of Arg15. Arg15 is in close proximity to the UBA3-bond ATP and is required for efficient catalysis. Arg85 is partially surface exposed but the guanidine group is engaged in hydrogen bonds with Asp61 and Asp69. Substitution of Arg85 by glutamine (individual 1 and 2) disturbs this network of hydrogen bonds, which would destabilize the local fold, which may extend to the interaction surface with UBA3 in proximity to Asp69 (Figure 1D). Cys294 (individual 4) points into the hydrophobic core (Figure 1E). A substitution by a sterically very demanding tryptophan is incompatible with the local fold and would result in destabilization of the entire protein.
The side chain of Arg430 (individual 3) points inwards, and a substitution by a glutamine might thus destabilize the local fold (Figure 1F).
Western blot analysis of fibroblasts from individuals 1 and 2 with NAE1 genetic variants showed a reduction of NAE1 by almost 80% compared to healthy control subjects (Figures 2A, S4E, and S4F), whereas heterozygous carriers of NAE1 variants (parents of individual 1) exhibited a reduction of approximately 50% of NAE1 abundance (Figure 2A).
Figure 2.

NAE1 abundance and neddylated to non-neddylated cullin ratio are reduced in individuals with NAE1 variants
(A) Western blots showing NAE1, neddylated, and non-neddylated cullin 1/cullin 3 abundance in dermal fibroblasts of healthy control subjects (2 healthy controls [C1 and C2]; 2 biological replicates per healthy control [C1.1, C1.2, C2.1, C2.2]; 6 technical replicates each), heterozygous carriers (father and mother of individual 1; 5 technical replicates each), and the fibroblasts of individual 1 with bi-allelic NAE1 genetic variants (6 technical replicates). Bar graphs represent the average band intensity of all technical replicates per donor ± SEM. All band intensities were normalized to total protein content. Quantification was performed using ImageJ. One way ANOVA, Post-hoc: Dunnet’s multiple comparisons test (∗∗p < 0.01, ∗∗∗p < 0.001).
(B) Graphic showing the neddylation and deneddylation cycles of cullins.
(C) Gene set enrichment analysis of the pre-ranked (based on differential expression score) transcriptomic dataset of individual 1- and 2-derived fibroblasts compared to 3 healthy control fibroblast lines. For this analysis, all genes were taken into account. The top 10 most significantly enriched (based on FDR (Q-value); blue, downregulated; red, upregulated) pathways are shown. Pathways in bold are associated with protein degradation and proteasome function. Pathways were aligned to the GO Ontology database (https://doi.org/10.5281/zenodo.4495804, released 2021-02-01).
(D) Cell death in fibroblasts derived from individuals 1 and 2, parents of individual 1, and healthy control subjects, after treatment with MG132 for 24 h at the indicated concentrations (in μM). Cell death was assessed by dividing the amount of PI-positive cells by the total amount of cells. Error bars represent SEM of 3 technical replicates of 2 donors for each group. two-way ANOVA. Post-hoc: Fisher’s least significance difference test (LSD) (∗∗∗∗p < 0.0001).
(E) Cell death in fibroblasts derived from individuals 1 and 2 (NAE1−/−) with and without a vector expressing WT NAE1 after treatment with MG132 for 24 h at the indicated concentrations (in μM). Cell death was assessed by dividing the amount of PI-positive cells by the total amount of cells. Errors bars represent SEM of 3 technical replicates of 2 donors for each group. Two-way ANOVA. Post-hoc: Fisher’s LSD (∗∗∗∗p < 0.0001).
(F) Cell death in fibroblasts derived from parents of individual 1 (NAE1+/−) with and without addition of NAE1 shRNA after treatment with MG132 for 24 h at the indicated concentrations (in μM). Cell death was assessed by dividing the amount of PI-positive cells by the total amount of cells. Error bars represent SEM of 3 technical replicates of 2 donors for each group. Two-way ANOVA. Post-hoc: Fisher’s LSD (∗∗p < 0.01).
Next, we quantified both neddylated (cullinNEDD8) and non-neddylated cullin 1 and cullin 3 ( Figure 2B). Even though the amount of neddylated cullins was normal, the ratio of neddylated to non-neddylated cullin 1 was significantly decreased in individual 1 and showed a trend toward a decreased ratio in individual 2 (Figures 2A, S4E, and S4F). The ratio of cullin 3 similarly showed a trend toward a decreased ratio in both individuals (Figures 2A, S4E, and S4F). CUL1 and CUL3 mRNA levels were unchanged, indicating that the altered ratio does not result from transcriptional upregulation (Figures S4C and S4D). These results indicate that NAE1 deficiency appears to alter the neddylated to non-neddylated cullin ratio.
To determine the effects of the altered neddylated to non-neddylated cullin ratio on proteasome function, we studied the transcriptomic profile of dermal fibroblast lines of individuals with NAE1 variants and healthy pediatric dermal fibroblast lines (Figure S1, Table S5). Four out of ten most significantly upregulated enriched pathways (highest false discovery rate) found with gene set enrichment analysis included pathways involved in protein degradation. This is probably the result of proteasome dysfunction, causing compensatory upregulation of alternative cellular protein degradation pathways (Figure 2C). Functionally, this was evidenced by increased sensitivity to proteasome inhibitor MG132 (Figures 2D and S7). Addition of a wild-type NAE1 cDNA expressing plasmid to cells from individuals with NAE1 variants decreased MG132 sensitivity (Figures 2E and S4B), while addition of NAE1-shRNA to cells from heterozygous carriers mimicked the phenotype of individuals with NAE1 variants (Figures 2F and S4A). These results suggest that proteasome function might be affected as a result of decreased NAE1 abundance.
Characterization of rare phenotypic features in individuals with NAE1 variants
After determining the pathogenicity of NAE1 variants, we studied the cellular and clinical consequences of NAE1 deficiency. To specify our search, we delineated the rarest phenotypic features by calculating the occurrence ratio of all phenotypic traits. The occurrence ratio takes both the prevalence within the cohort and the rareness of a feature within the population into account.26 This approach identified the three rarest phenotypic features, shared by at least 75% of the cohort: hypoplastic ischiopubic rami, infection-associated lymphopenia, and neurodevelopmental setback during infections (Figure 3, Table S3).
Figure 3.

Rarest clinical features in individuals with NAE1 variants
Dot plot showing features with occurrence ratio >0.5 in individuals with NAE1 variants. Color coding refers to the percentage of individuals in our cohort experiencing a specific symptom. Dot size refers to the number of associated genes in HPO, transformed with −10log to give features with the lowest amount of associated genes the largest dot size. The occurrence ratio was calculated by dividing the percentage of individuals experiencing a specific symptom by the number of associated genes.
Individuals with NAE1 variants show hypoplasia of the os pubis ramus inferior
In individual 1, we observed increased size of the ischiopubic synchondrosis (Figure 4A), which prompted us to study the pelvic X-rays of the rest of the cohort. For comparison, we determined the size of the ischiopubic synchondrosis at different ages using pelvic X-rays of a pediatric reference population (Figures 4A and 4B). All individuals with NAE1 variants showed increased size of the ischiopubic synchondrosis compared to the reference population. In transcriptomic data, Gene Ontologies (Biological Function) aligned to significantly downregulated genes (>2-fold, adjusted p value < 0.05) in fibroblasts of individuals with NAE1 variants compared to healthy control fibroblasts and were enriched for cartilage development, ossification, and chondrocyte differentiation (Figure 4C). Two of the main drivers for these pathways included SOX9 (MIM: 608160) (log2fold change −2, adjusted p value = 0.03) and RUNX2 (MIM: 600211) (log2fold change −2.3, adjusted p value = 0.002) (Figure 4D). Both SOX9 and RUNX2 are associated with congenital skeletal disorders (campomelic and cleidocranial dysplasia [MIM: 114290 and MIM:119600]) that result in hypoplastic ischiopubic rami.46,47,48 Thus, NAE1 might influence expression of these genes, resulting in altered endochondral bone development.
Figure 4.

Individuals with NAE1 variants show hypoplasia of the ischiopubic synchondrosis
(A) Size of the ischiopubic synchondrosis was measured in n = 200 pediatric individuals that had pelvic X-rays taken (black dots). The blue line represents the mean size of the ischiopubic synchondrosis in this population with a 95% confidence interval depicted in gray. Red/pink colored dots are the values of the individuals with NAE1 variants.
(B) Pelvic X-rays of individuals with NAE1 variants showing increased size of the ischiopubic synchondrosis.
(C) Graph showing the 23 most overrepresented pathways in fibroblasts of individuals with NAE1 variants using significantly downregulated genes (adjusted p value < 0.05, log2foldchange >2 and <2, Table S5) as input. Five out of nine pathways (bold) that are significantly enriched are involved in endochondral bone development and might explain delayed closure of ischiopubic synchondrosis in individuals with NAE1 variants. The X axis shows the GeneRatio, defined as the number of genes associated with the pathway divided by the total number of selected genes.
(D) Volcano plot showing differentially expressed genes in individual 1 and 2 fibroblasts versus three healthy age-matched control subjects. RUNX2 and SOX9 (in purple) are involved in bone development and are linked to disease presenting with os pubis ramus inferior hypoplasia and are both downregulated in individuals with NAE1 variants.
Individuals with NAE1 variants experience lymphopenia and decreased NF-κB translocation during infections
The second rarest phenotypic feature was the lymphopenia, which worsened during infections (Figure 5A). This clinical observation could be mimicked in vitro, where we found that individuals with NAE1 variants showed a trend toward decreased CD3+ T cell counts without stimulation, which became significant during stimulation with CD3/CD28 for 24 h (Figure 5B). We found the lymphopenia was not the result of a proliferation defect, as the proliferation rates of T cells in healthy control subjects and affected individuals were similar (Figure S6).
Figure 5.

Individuals with NAE1 variants experience lymphopenia during inflammation
(A) Lymphocyte counts (combined B cell and T cell counts) during outpatient clinic visits and during infections for which individuals visited the emergency room (ER visits). All counts were normalized to the lower reference value of the corresponding age and depicted as a percentage. Individual data points of individuals (dots) are shown, line and error bars reflect the mean ± SD, two-way ANOVA. Post Hoc: uncorrected Fisher’s LSD (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
(B) Flow cytometry analysis of absolute CD3+ counts after treatment for 1 day with 1 μg/ml anti-CD3/CD28 of individuals with NAE1 variants (−/−) (2 donors, 1 technical replicate) versus healthy controls (3 donors, 1 technical replicate). Bars represent mean value ± SD two-way ANOVA, Fisher’s LSD (∗∗p < 0.01).
(C) Percentage of NF-κB (p65) nuclear translocation in individuals with NAE1 variants (2 donors, 1 technical replicate) and healthy controls (3 donors, 1 technical replicate) CD3+ cells after stimulation with 2 μg/ml anti-CD3/CD28 for different timepoints (minutes) measured with Imagestream. Lines represent mean ± SD two-way ANOVA repeated measurement, without correction. Post-Hoc: Fisher’s LSD (∗∗p < 0.01). The images on the right show an example of cytoplasmic (untranslocated) and nuclear (translocated) location of p65 in the cell. The graph shows how translocation was quantified, by using the similarity score (pixel similarity p65/nucleus). All cells with similarity scores higher than zero were considered as “translocated” cells.
(D) Graph showing the intensity of CD45RA (Y axis) and CD45RO (X axis) of all cells within the CD4+ population in healthy controls (3 donors, 1 technical replicate) and individuals with NAE1 variants (2 donors, 1 technical replicate) without anti-CD3/CD28 stimulation.
(E) Bar graph showing the percentages of CD45RA positive cells within the CD4 population in the healthy control (3 donors, 1 technical replicate) and individuals with NAE1 variants (2 donors, 1 technical replicate), bar graph represents the mean value ±SD Students’ T-test (∗p < 0.05).
(F) Bar graphs showing percentage of CD3 positive but CD4 and CD8 negative T cells in individuals with NAE1 variants (2 donors, 1 technical replicate) and healthy controls (4 donors, 1 technical replicate each) without anti-CD3/CD28 stimulation, bars represent mean ± SD; Student’s t test (∗∗p < 0.01).
Neddylation is an important regulator of NF-κB,19,49 a signaling molecule known to promote lymphocyte survival.50 Therefore, we studied NF-κB activation by measuring p65 nuclear translocation in CD3+ T cells upon stimulation with anti-CD3/CD28. Indeed, p65 nuclear translocation in response to CD3/CD28 stimulation was reduced in individuals with NAE1 variants (Figure 5C).
Another rare feature involved the significantly decreased percentages of naive (CD45RA-positive) CD4+ cells in steady-state conditions (Figures 5D and 5E). Additionally, when performing the gating of CD4 and CD8 subpopulations within CD3, we noticed increased percentages (40%) of double-negative (CD4−CD8−) CD3+ cells (Figure 5F). Double-negative T cells are immature thymic T cells which are normally found in low (<10%) percentages in peripheral blood, and their physiological role remains poorly understood.51 Gene set enrichment analysis using the KEGG database showed several pathways that could potentially link neddylation dysfunction to the observed immunologic phenotype, including downregulated WNT/β-catenin and MAP-kinase signaling (Figure S1).
Together, these findings indicate that lymphocytes of individuals with NAE1 variants have different immunological characteristics at baseline, indicated by their decreased naive T cell percentages and increased double-negative cell counts. Additionally, their defective neddylation leads to perturbed NF-κB signaling during infections, which might limit NF-κB-mediated lymphocyte survival, causing lymphopenia.
Individuals with NAE1 variants experience neurodegeneration and increased seizure frequency during infections
All individuals with NAE1 variants showed brain loss and setbacks in development, mostly related to major hospital admissions due to severe viral respiratory tract infections (Figures 6A and 6B). Similarly, seizure frequency worsened after these severe infections. Neddylation is upregulated during several types of cellular stress, including DNA damage,52,53 hypoxia,54 and proteotoxic stress.55 Therefore, we hypothesized that neddylation is sufficient in homeostatic conditions in individuals with NAE1 variants, but insufficient in conditions that require upregulated neddylation such as physical stress caused by infections, and attempted to test this hypothesis in fibroblasts. It proved difficult to mimic infectious conditions in fibroblasts, and therefore we tried to downregulate the neddylation system instead, to a level where it would already be impaired in homeostatic conditions. We did so by using the neddylation inhibitor MLN4924. We found that NAE1 deficiency leads to increased sensitivity to MLN4924-mediated cell death in fibroblasts. In PBMCs, we found that while MLN4924 caused decreased cell counts in all donors, this resulted in significantly lower cell count only in individuals with NAE1 genetic variants (Figures 6C and 6D). The increased MLN4924 sensitivity suggests that individuals with NAE1 variants cannot adequately upregulate their neddylation.
Figure 6.

Individuals with NAE1 variants experience neurodegeneration and seizure frequency during infections
(A) Timeline showing the development and seizure frequency (red line) in individual 1 and 2. Development and seizure frequency were quantified by parents of individuals 1 and 2 (arbitrary units). If the red line drops below the 0-point (the black line), this indicates that development decelerated. Circles indicate infections, massive red circles indicate infections requiring admissions. The bigger the circle, the longer the time of admission.
(B) MRI images of individual 1, individual 3, and individual 4 are shown. For individual 1, MRI was taken at 10 months of age and at 20 months of age (after ICU admission). Note the brain loss occurring between this time. The MRI of individual 3 was taken at approximately 29 months of age, 6–7 months after his seizures began which were treated with ACTH. For individual 4, brain MRI was obtained at the age of 2.5 years, showing severe brain loss, resulting from multiple severe infections.
(C) Bar graph showing the lymphocyte count after 1 day of anti-CD3 (0.1 μg/mL) stimulation with and without the addition of MLN4924 (200 nM) in healthy controls (3 donors, 1 technical replicate each) and lymphocytes of individuals with NAE1 variants (2 donors, 1 technical replicate each). Bars represent mean value ± SD, two-way ANOVA, post hoc: Fisher’s LSD (∗p < 0.05).
(D) Bar graph showing the percentage of death cells (PI positive) over the total amount of cells (counted using Hoechst) in fibroblasts, with or without MLN4924 treatment (500 nM) in healthy controls (3 donors, 1 technical replicate each, gray bars) and individuals with NAE1 variants (2 donors, 1 technical replicate each, black bars). Bars represent mean value ± SD, two-way ANOVA, post hoc: Fisher’s LSD. (not significant, p > 0.05).
Discussion
In this work we describe four unrelated individuals with intellectual disability harboring bi-allelic variants in NAE1. Reduced NAE1 abundance and decreased ratios of neddylated to non-neddylated cullins in fibroblasts supported pathogenicity. Subsequent phenotypic specificity analysis revealed ischiopubic synchondrosis hypoplasia, infection-triggered lymphopenia, and infection-triggered neurodegeneration as the rarest phenotypic features. We found several pathways involved in endochondral bone remodeling that showed altered expression and downregulated RUNX2 and SOX9 expression. Moreover, we found enhanced cell death in fibroblasts after stressing the system with MG132 and MLN4924. In contrast to healthy controls subject, CD3/CD28 signaling led to decreased lymphocyte counts in individuals with NAE1 variants, which was accompanied by decreased NF-κB translocation. These results provide in-depth support for the pathogenicity of the genetic variants and suggest that NAE1 deficiency impairs homeostasis during cellular stress and leads to disturbances in (skeletal) development.
The pathogenicity of the NAE1 variants was supported by the significant reduction of NAE1 abundance, resulting in a lower cullin 1NEDD8/cullin 1 ratio. The unaltered CUL1 mRNA levels argue against transcriptional regulation, and thus provide indirect support for increased stability of non-neddylated cullins, which are exposed to fewer neddylation cycles.56 Since cullin function is controlled by cycles of cullin neddylation and deneddylation, maintaining a proper cullin 1NEDD8/cullin 1 ratio is crucial to ensure cullin-mediated functions.57 As fibroblasts of individuals with NAE1 genetic variants were more sensitive to MG132 treatment, we speculate that the altered cullin 1NEDD8/cullin 1 ratio results in proteasome dysfunction. However, it should be noted that non-cullin neddylation targets, which we did not study in this research, might also be involved in the pathophysiology.
Both the neurological and immunological phenotype of individuals with NAE1 variants reflect impairments in development and stress response. During routine outpatient clinic visits, we found low to normal lymphocyte counts, but during infections, lymphopenia became more obvious. Lymphocyte survival in resting and activated conditions is partially regulated by NF-κB activation, which in turn is regulated by neddylation.50 In lymphocytes of individuals with NAE1 variants, NF-κB activation was severely decreased, which might explain the decreased lymphocyte counts both in resting and activated conditions. Anti-CD3/CD28 signaling requires upregulation of neddylation, which in turn influences activity of many other pathways in T cells.58 One example is WNT/β-catenin signaling, important for thymic function.21,59,60 Defective WNT signaling can explain the decreased numbers of naive T cells, as WNT signaling can keep T cells in a naive state, corresponding with low expression of AXIN2 in memory T cells.61 Moreover, defective WNT signaling and subsequent thymic dysfunction can explain the increased percentages of double-negative (CD4−CD8−) T cells, which might represent immature T cells leaving the thymus prior to complete maturation. Thus, similar to the neurodegeneration, the lymphocyte phenotype might be the result of developmental and stress-mediated defects.
While the stress-mediated neurodegeneration was the most severe phenotypic feature in our cohort, additional neurodevelopmental abnormalities were seen, not related to neurodegeneration. This is not surprising, given the fact that recent evidence indicates that NAE1 is essential for development, maintenance, and outgrowth of neuronal spines.10,59,62,63 Regarding the infection-mediated neurodegeneration, several mechanisms might be involved. First, during cellular stress caused by infections, unfolded proteins accumulate, requiring neddylation-mediated Parkin and PINK1 activation13,64 to break down these unfolded proteins and prevent neuronal apoptosis and brain damage. Dysfunction of these pathways has been implicated in the pathophysiology of Alzheimer and Parkinson disease.11,12,64 Other pathways are also involved in neuronal integrity, like the neddylation-controlled WNT/β-catenin pathway.59 Dysfunction of this pathway can result in loss of cell-cell adhesion and neuronal apoptosis.65 Not only the integrity but also the excitability of neurons during stress is under the control of neddylation.63 Mice with defective neddylation display decreased Nav1.1 stability during fever, leading to increased neuronal excitability and epilepsy.66 Finally, in mice, double-negative T cells can significantly enhance neuroinflammation during brain injury.67 Therefore, the high numbers of double-negative T cells found in individuals with NAE1 variants might have enhanced neuroinflammation. Additional experiments using different stressors—preferably in neurons—will be an important step to reliably establish the link between neddylation, stress, and cell death, and how this links to stress-mediated neurodegeneration.
Currently, there is no gold standard how to elucidate pathophysiologic cellular mechanisms leading to a specific phenotype caused by variants of uncertain significance (VUSs). As a result, researchers tend to focus on the most severe clinical phenotypes. Instead, we propose that focusing on information-dense phenotypes could provide more insight in the underlying pathophysiology. In our case, focusing on the most severe phenotypic features would have led us to focus on the neurodegeneration and epilepsy, which probably would have given insight in the mechanism of disease in the brain, but would have overlooked the influence of faulty neddylation on the immune system and (bone) development. Our overarching view led to a clear pattern in lymphocyte values, that dropped significantly during infections, which resembled the neurodegeneration, that similarly worsened during infections. This framework allowed us to delineate the most important pathophysiological mechanism: during stress, neddylation needs to be upregulated, protecting cells from stress. We suspect individuals with NAE1 variants are unable to adequately upregulate their neddylation for this purpose, resulting in stress-mediated cell death.
Naturally, the complexity and resources needed to discover the pathophysiology of VUSs limits the number of genetic diagnoses that can be made. When considering variants in pleiotropic genes, the wealth of pathways and phenotypic features that can be studied threatens to complicate this process even more. Therefore, smart prioritization is of the essence. This work exemplifies the power of a focus on the most informative features. If validated on a larger scale, this approach could be beneficial for complex VUSs in genes encoding pleiotropically acting proteins.
In conclusion, this study shows that NAE1 deficiency leads to developmental delay, epilepsy, ischiopubic synchondrosis hypoplasia, and infection-triggered lymphopenia and neurodegeneration. It thereby unveils the importance of proper NAE1 function in facilitating development and suggests a crucial role for neddylation in maintaining cellular homeostasis during cellular stress.
Acknowledgments
This work was supported by the Max Planck Society (D.R.), the VolkswagenStiftung (D.R.), the Agencia Nacional de Promoción Científica y Tecnológica, Argentina (D.R.) and the Fondo para la convergencia estructural del mercosur-FOCEM (COF 03/11) (D.R.). We would like to thank the parents of individual 1 for drafting Figure 6A.
Author contributions
Concept: P.M.v.H. and I.J.J.M. Designing experiments: I.J.J.M., I.F.S., M.M.v.d.W., C.B., M.L., D.R., F.v.W., S.A.F., E.E.S.N., and P.M.v.H. Conducting experiments: I.J.J.M., I.F.S., M.M.v.d.W., C.B., M.L., N.G.A. Acquiring (medical) data of study participants and cells: I.J.J.M., Y.A.Z., L.V.S., J.B., J.P., M.L.H., J.C.G., S.H., A.H., F.V., M.P.G.M., M.L., P.M.v.H. Analyzing data: I.J.J.M., I.F.S., M.P.G.M., M.M.v.d.W. Genetic testing and interpretation of variants: M.P.G.M., G.v.H. Molecular predictions: M.P.G.M., H.R. Writing the manuscript: I.J.J.M. and P.M.v.H. Supervision of experiments and writing process: P.M.v.H., S.A.F., F.v.W., E.E.S.N., D.R.
S.A.F., D.R., and F.v.W. spent equal time and effort on supervision of the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: January 5, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2022.12.003.
Supplemental information
Data code and availability
The authors declare that all data supporting the findings of this study are available within the paper and its supplemental information. As individuals in this study did not consent to publicly available access to their FastQ files, this will not be posted to a public repository such as the Gene Expression Omnibus, but their RNA sequencing data are available upon request. A list of significantly up- and downregulated genes identified with RNA sequencing in fibroblasts can be found in Table S5. The variants described in this study were entered in ClinVar (VCV001526415, VCV001526416, VCV001206325, VCV001676942).
References
- 1.Kerscher O., Felberbaum R., Hochstrasser M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 2006;22:159–180. doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- 2.Enchev R.I., Schulman B.A., Peter M. Protein neddylation: Beyond cullin-RING ligases. Nat. Rev. Mol. Cell Biol. 2015;16:30–44. doi: 10.1038/nrm3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whitby F.G., Xia G., Pickart C.M., Hill C.P. Crystal structure of the human ubiquitin-like protein NEDD8 and interactions with ubiquitin pathway enzymes. J. Biol. Chem. 1998;273:34983–34991. doi: 10.1074/jbc.273.52.34983. [DOI] [PubMed] [Google Scholar]
- 4.Schulman B.A., Harper J.W. Ubiquitin-like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009;10:319–331. doi: 10.1038/nrm2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walden H., Podgorski M.S., Huang D.T., Miller D.W., Howard R.J., Minor D.L., Holton J.M., Schulman B.A. The structure of the APPBP1-UBA3-NEDD8-ATP complex reveals the basis for selective ubiquitin-like protein activation by an E1. Mol. Cell. 2003;12:1427–1437. doi: 10.1016/s1097-2765(03)00452-0. [DOI] [PubMed] [Google Scholar]
- 6.Malik-Chaudhry H.K., Gaieb Z., Saavedra A., Reyes M., Kung R., Le F., Morikis D., Liao J. Dissecting distinct roles of NEDDylation E1 ligase heterodimer APPBP1 and UBA3 reveals potential evolution process for activation of ubiquitin-related pathways. Sci. Rep. 2018;8:10108. doi: 10.1038/s41598-018-28214-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bohnsack R.N., Haas A.L. Conservation in the mechanism of Nedd8 activation by the human AppBp1-Uba3 heterodimer. J. Biol. Chem. 2003;278:26823–26830. doi: 10.1074/jbc.M303177200. [DOI] [PubMed] [Google Scholar]
- 8.Wada H., Kito K., Caskey L.S., Yeh E.T., Kamitani T. Cleavage of the C-terminus of NEDD8 by UCH-L3. Biochem. Biophys. Res. Commun. 1998;251:688–692. doi: 10.1006/bbrc.1998.9532. [DOI] [PubMed] [Google Scholar]
- 9.Mendoza H.M., Shen L.N., Botting C., Lewis A., Chen J., Ink B., Hay R.T. NEDP1, a highly conserved cysteine protease that deNEDDylates cullins. J. Biol. Chem. 2003;278:25637–25643. doi: 10.1074/jbc.M212948200. [DOI] [PubMed] [Google Scholar]
- 10.Vogl A.M., Phu L., Becerra R., Giusti S.A., Verschueren E., Hinkle T.B., Bordenave M.D., Adrian M., Heidersbach A., Yankilevich P., et al. Global site-specific neddylation profiling reveals that NEDDylated cofilin regulates actin dynamics. Nat. Struct. Mol. Biol. 2020;27:210–220. doi: 10.1038/s41594-019-0370-3. [DOI] [PubMed] [Google Scholar]
- 11.Dil Kuazi A., Kito K., Abe Y., Shin R.W., Kamitani T., Ueda N. NEDD8 protein is involved in ubiquitinated inclusion bodies. J. Pathol. 2003;199:259–266. doi: 10.1002/path.1283. [DOI] [PubMed] [Google Scholar]
- 12.Chen Y., Neve R.L., Liu H. Neddylation dysfunction in Alzheimer’s disease. J. Cell Mol. Med. 2012;16:2583–2591. doi: 10.1111/j.1582-4934.2012.01604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choo Y.S., Vogler G., Wang D., Kalvakuri S., Iliuk A., Tao W.A., Bodmer R., Zhang Z. Regulation of parkin and PINK1 by neddylation. Hum. Mol. Genet. 2012;21:2514–2523. doi: 10.1093/hmg/dds070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu K., Chen K., Zhang Q., Zhang L., Yan Y., Guo C., Qi J., Yang K., Wang F., Huang P., et al. TRAF6 neddylation drives inflammatory arthritis by increasing NF-κB activation. Lab. Invest. 2019;99:528–538. doi: 10.1038/s41374-018-0175-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schoppmann S.F., Vinatzer U., Popitsch N., Mittlböck M., Liebmann-Reindl S., Jomrich G., Streubel B., Birner P. Novel clinically relevant genes in gastrointestinal stromal tumors identified by exome sequencing. Clin. Cancer Res. 2013;19:5329–5339. doi: 10.1158/1078-0432.CCR-12-3863. [DOI] [PubMed] [Google Scholar]
- 16.Soucy T.A., Dick L.R., Smith P.G., Milhollen M.A., Brownell J.E. The NEDD8 conjugation pathway and its relevance in cancer biology and therapy. Genes Cancer. 2010;1:708–716. doi: 10.1177/1947601910382898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delgado T.C., Barbier-Torres L., Zubiete-Franco I., Lopitz-Otsoa F., Varela-Rey M., Fernández-Ramos D., Martínez-Chantar M.L. Neddylation, a novel paradigm in liver cancer. Transl. Gastroenterol. Hepatol. 2018;3:37. doi: 10.21037/tgh.2018.06.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zubiete-Franco I., Fernández-Tussy P., Barbier-Torres L., Simon J., Fernández-Ramos D., Lopitz-Otsoa F., Gutiérrez-de Juan V., de Davalillo S.L., Duce A.M., Iruzubieta P., et al. Deregulated neddylation in liver fibrosis. Hepatology. 2017;65:694–709. doi: 10.1002/hep.28933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Read M.A., Brownell J.E., Gladysheva T.B., Hottelet M., Parent L.A., Coggins M.B., Pierce J.W., Podust V.N., Luo R.S., Chau V., Palombella V.J. Nedd8 modification of cul-1 activates SCF(beta(TrCP))-dependent ubiquitination of IkappaBalpha. Mol. Cell Biol. 2000;20:2326–2333. doi: 10.1128/mcb.20.7.2326-2333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liakopoulos D., Büsgen T., Brychzy A., Jentsch S., Pause A. Conjugation of the ubiquitin-like protein NEDD8 to cullin-2 is linked to von Hippel-Lindau tumor suppressor function. Proc. Natl. Acad. Sci. USA. 1999;96:5510–5515. doi: 10.1073/pnas.96.10.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tripathi R., Kota S.K., Srinivas U.K. Cullin4B/E3-ubiquitin ligase negatively regulates β-catenin. J. Biosci. 2007;32:1133–1138. doi: 10.1007/s12038-007-0114-0. [DOI] [PubMed] [Google Scholar]
- 22.Sugasawa K., Okuda Y., Saijo M., Nishi R., Matsuda N., Chu G., Mori T., Iwai S., Tanaka K., Tanaka K., Hanaoka F. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell. 2005;121:387–400. doi: 10.1016/j.cell.2005.02.035. [DOI] [PubMed] [Google Scholar]
- 23.Liu L., Lee S., Zhang J., Peters S.B., Hannah J., Zhang Y., Yin Y., Koff A., Ma L., Zhou P. CUL4A abrogation augments DNA damage response and protection against skin carcinogenesis. Mol. Cell. 2009;34:451–460. doi: 10.1016/j.molcel.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sobreira N., Schiettecatte F., Valle D., Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 2015;36:928–930. doi: 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shannon C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948;27:623–656. [Google Scholar]
- 26.Haijes H.A., Jaeken J., van Hasselt P.M. Hypothesis: determining phenotypic specificity facilitates understanding of pathophysiology in rare genetic disorders. J. Inherit. Metab. Dis. 2020;43:701–711. doi: 10.1002/jimd.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ten Berg M.J., Huisman A., Van Den Bemt P.M.L.A., Schobben A.F.A.M., Egberts A.C.G., Van Solinge W.W. Linking laboratory and medication data: new opportunities for pharmacoepidemiological research. Clin. Chem. Lab. Med. 2007;45:13–19. doi: 10.1515/CCLM.2007.009. [DOI] [PubMed] [Google Scholar]
- 28.Buske O.J., Girdea M., Dumitriu S., Gallinger B., Hartley T., Trang H., Misyura A., Friedman T., Beaulieu C., Bone W.P., et al. PhenomeCentral: a portal for phenotypic and genotypic matchmaking of patients with rare genetic diseases. Hum. Mutat. 2015;36:931–940. doi: 10.1002/humu.22851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The 1000 Genomes Project Consortium. Auton A., Abecasis G.R., Altshuler D.M., Durbin R.M., Abecasis G.R., Bentley D.R., Chakravarti A., Clark A.G., Donnelly P., et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zerbino D.R., Achuthan P., Akanni W., Amode M.R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Girón C.G., et al. Ensembl 2018. Nucleic Acids Res. 2018;46:D754–D761. doi: 10.1093/nar/gkx1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lek M., Karczewski K., Minikel E., Samocha K., Banks E., Fennell T., et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang H., Wang F., Dyer N.P., Wong W.H. CisGenome browser: A flexible tool for genomic data visualization. Bioinformatics. 2010;26:1781–1782. doi: 10.1093/bioinformatics/btq286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mijnheer G., Lutter L., Mokry M., van der Wal M., Scholman R., Fleskens V., Pandit A., Tao W., Wekking M., Vervoort S., et al. Conserved human effector Treg cell transcriptomic and epigenetic signature in arthritic joint inflammation. Nat. Commun. 2021;12:2710. doi: 10.1038/s41467-021-22975-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H., Durbin R. Fast and accurate short read alignment with Burrows – Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu G., Wang L.G., Han Y., He Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. OMICS A J. Integr. Biol. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang X., Boehm J.S., Yang X., Salehi-Ashtiani K., Hao T., Shen Y., Lubonja R., Thomas S.R., Alkan O., Bhimdi T., et al. A public genome-scale lentiviral expression library of human ORFs. Nat Methods. 2011;8 doi: 10.1038/nmeth.1638. 659–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wiederschain D., Wee S., Chen L., Loo A., Yang G., Huang A., Chen Y., Caponigro G., Yao Y.M., Lengauer C., et al. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle. 2009;8:498–504. doi: 10.4161/cc.8.3.7701. [DOI] [PubMed] [Google Scholar]
- 40.Wee S., Wiederschain D., Maira S.M., Loo A., Miller C., deBeaumont R., Stegmeier F., Yao Y.M., Lengauer C. PTEN-deficient cancers depend on PIK3CB. Proc. Natl. Acad. Sci. USA. 2008;105:13057–13062. doi: 10.1073/pnas.0802655105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muffels I.J.J., Wiame E., Fuchs S.A., Massink M.P.G., Rehmann H., Musch J.L.I., Van Haaften G., Vertommen D., van Schaftingen E., van Hasselt P.M. NAA80 bi-allelic missense variants result in high-frequency hearing loss, muscle weakness and developmental delay. Brain Commun. 2021;3:1–32. doi: 10.1093/braincomms/fcab256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Köhler S., Gargano M., Matentzoglu N., Carmody L.C., Lewis-Smith D., Vasilevsky N.A., Danis D., Balagura G., Baynam G., Brower A.M., et al. The human phenotype ontology in 2021. Nucleic Acids Res. 2021;49:D1207–D1217. doi: 10.1093/nar/gkaa1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ishii A., Watkins J.C., Chen D., Hirose S., Hammer M.F. Clinical implications of SCN1A missense and truncation variants in a large Japanese cohort with Dravet syndrome. Epilepsia. 2017;58:282–290. doi: 10.1111/epi.13639. [DOI] [PubMed] [Google Scholar]
- 44.Kraulis P.J. MOLSCRIPT. A program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 1991;24(pt 5):946–950. [Google Scholar]
- 45.Merritt E.A., Murphy M.E. Raster3D version 2.0 A program for photorealistic molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 46.Gao X., Li K., Fan Y., Sun Y., Luo X., Wang L., Liu H., Gong Z., Wang J., Wang Y., et al. Identification of RUNX2 variants associated with cleidocranial dysplasia. Hereditas. 2019;156:31. doi: 10.1186/s41065-019-0107-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matsushita M., Kitoh H., Kaneko H., Mishima K., Kadono I., Ishiguro N., Nishimura G. A novel SOX9 H169Q mutation in a family with overlapping phenotype of mild campomelic dysplasia and small patella syndrome. Am. J. Med. Genet. 2013;161A:2528–2534. doi: 10.1002/ajmg.a.36134. [DOI] [PubMed] [Google Scholar]
- 48.Wagner T., Wirth J., Meyer J., Zabel B., Held M., Zimmer J., Pasantes J., Bricarelli F.D., Keutel J., Hustert E., et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell. 1994;79:1111–1120. doi: 10.1016/0092-8674(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 49.Ehrentraut S.F., Curtis V.F., Wang R.X., Saeedi B.J., Ehrentraut H., Onyiah J.C., Kelly C.J., Campbell E.L., Glover L.E., Kominsky D.J., Colgan S.P. Perturbation of neddylation-dependent NF-κB responses in the intestinal epithelium drives apoptosis and inhibits resolution of mucosal inflammation. Mol. Biol. Cell. 2016;27:3687–3694. doi: 10.1091/mbc.E16-05-0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gerondakis S., Siebenlist U. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb. Perspect. Biol. 2010;2:a000182. doi: 10.1101/cshperspect.a000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liapis K., Tsagarakis N.J., Panitsas F., Taparkou A., Liapis I., Roubakis C., Tsokanas D., Vasileiou P., Grigoriou E., Kakiopoulos G., et al. Causes of double-negative T-cell lymphocytosis in children and adults. J. Clin. Pathol. 2020;73:431–438. doi: 10.1136/jclinpath-2019-206255. [DOI] [PubMed] [Google Scholar]
- 52.Cui D., Xiong X., Sun Y., Zhao Y. FBXW7 confers radiation survival by targeting p53 for degradation. SSRN Electron. J. 2019 doi: 10.1016/j.celrep.2019.12.032. [DOI] [PubMed] [Google Scholar]
- 53.Nishitani H., Shiomi Y., Iida H., Michishita M., Takami T., Tsurimoto T. CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during s phase and after UV irradiation. J. Biol. Chem. 2008;283:29045–29052. doi: 10.1074/jbc.M806045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ryu J.H., Li S.H., Park H.S., Park J.W., Lee B., Chun Y.S. Hypoxia-inducible factor α subunit stabilization by NEDD8 conjugation is reactive oxygen species-dependent. J. Biol. Chem. 2011;286:6963–6970. doi: 10.1074/jbc.M110.188706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maghames C.M., Lobato-Gil S., Perrin A., Trauchessec H., Rodriguez M.S., Urbach S., Marin P., Xirodimas D.P. NEDDylation promotes nuclear protein aggregation and protects the ubiquitin proteasome system upon proteotoxic stress. Nat. Commun. 2018;9:4376. doi: 10.1038/s41467-018-06365-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu J.T., Lin H.C., Hu Y.C., Chien C.T. Neddylation and deneddylation regulate Cul1 and Cul3 protein accumulation. Nat. Cell Biol. 2005;7:1014–1020. doi: 10.1038/ncb1301. [DOI] [PubMed] [Google Scholar]
- 57.Liu Q., Zhou Y., Tang R., Wang X., Hu Q., Wang Y., He Q. Increasing the Unneddylated Cullin1 Portion Rescues the csn Phenotypes by Stabilizing Adaptor Modules To Drive SCF Assembly. Mol. Cell Biol. 2018;38:e00109-17. doi: 10.1128/MCB.00667-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cheng Q., Liu J., Pei Y., Zhang Y., Zhou D., Pan W., Zhang J. Neddylation contributes to CD4+ T cell-mediated protective immunity against blood-stage Plasmodium infection. PLoS Pathog. 2018;14:e1007440. doi: 10.1371/journal.ppat.1007440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang L., Jing H., Li H., Chen W., Luo B., Zhang H., Dong Z., Li L., Su H., Xiong W.C., Mei L. Neddylation is critical to cortical development by regulating Wnt/β-catenin signaling. Proc. Natl. Acad. Sci. USA. 2020;117:26448–26459. doi: 10.1073/pnas.2005395117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weerkamp F., Baert M.R.M., Naber B.A.E., Koster E.E.L., De Haas E.F.E., Atkuri K.R., van Dongen J.J.M., Herzenberg L.A., Staal F.J.T. Wnt signaling in the thymus is regulated by differential expression of intracellular signaling molecules. Proc. Natl. Acad. Sci. USA. 2006;103:3322–3326. doi: 10.1073/pnas.0511299103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tiemessen M.M., Baert M.R.M., Kok L., van Eggermond M.C.J.A., van den Elsen P.J., Arens R., Staal F.J.T. T cell factor 1 represses CD8 + effector T cell formation and function. J. Immunol. 2014;193:5480–5487. doi: 10.4049/jimmunol.1303417. [DOI] [PubMed] [Google Scholar]
- 62.Vogl A.M., Brockmann M.M., Giusti S.A., MacCarrone G., Vercelli C.A., Bauder C.A., Richter J.S., Roselli F., Hafner A.S., Dedic N., et al. Neddylation inhibition impairs spine development, destabilizes synapses and deteriorates cognition. Nat. Neurosci. 2015;18:239–251. doi: 10.1038/nn.3912. [DOI] [PubMed] [Google Scholar]
- 63.Brockmann M.M., Döngi M., Einsfelder U., Körber N., Refojo D., Stein V. Neddylation regulates excitatory synaptic transmission and plasticity. Sci. Rep. 2019;9:17935. doi: 10.1038/s41598-019-54182-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xiong H., Wang D., Chen L., Choo Y.S., Ma H., Tang C., Xia K., Jiang W., Ronai Z., Zhuang X., Zhang Z. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J. Clin. Invest. 2009;119:650–660. doi: 10.1172/JCI37617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen Y.Z. APP induces neuronal apoptosis through APP-BP1-mediated downregulation of β-catenin. Apoptosis. 2004;9(4):415–422. doi: 10.1023/B:APPT.0000031447.05354.9f. [DOI] [PubMed] [Google Scholar]
- 66.Chen W., Luo B., Gao N., Li H., Wang H., Li L., Cui W., Zhang L., Sun D., Liu F., et al. Neddylation stabilizes Nav1.1 to maintain interneuron excitability and prevent seizures in murine epilepsy models. J. Clin. Invest. 2021;131:e136956. doi: 10.1172/JCI136956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meng H., Zhao H., Cao X., Hao J., Zhang H., Liu Y., Zhu M.S., Fan L., Weng L., Qian L., et al. Double-negative T cells remarkably promote neuroinflammation after ischemic stroke. Proc. Natl. Acad. Sci. USA. 2019;116:5558–5563. doi: 10.1073/pnas.1814394116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the paper and its supplemental information. As individuals in this study did not consent to publicly available access to their FastQ files, this will not be posted to a public repository such as the Gene Expression Omnibus, but their RNA sequencing data are available upon request. A list of significantly up- and downregulated genes identified with RNA sequencing in fibroblasts can be found in Table S5. The variants described in this study were entered in ClinVar (VCV001526415, VCV001526416, VCV001206325, VCV001676942).