

Summary
Variant interpretation remains a major challenge in medical genetics. We developed Meta-Domain HotSpot (MDHS) to identify mutational hotspots across homologous protein domains. We applied MDHS to a dataset of 45,221 de novo mutations (DNMs) from 31,058 individuals with neurodevelopmental disorders (NDDs) and identified three significantly enriched missense DNM hotspots in the ion transport protein domain family (PF00520). The 37 unique missense DNMs that drive enrichment affect 25 genes, 19 of which were previously associated with NDDs. 3D protein structure modeling supports the hypothesis of function-altering effects of these mutations. Hotspot genes have a unique expression pattern in tissue, and we used this pattern alongside in silico predictors and population constraint information to identify candidate NDD-associated genes. We also propose a lenient version of our method, which identifies 32 hotspot positions across 16 different protein domains. These positions are enriched for likely pathogenic variation in clinical databases and DNMs in other genetic disorders.
Keywords: mutational hotspot detection, homologous protein domains, disease-gene identification, de novo mutations, developmental disorders, variant interpretation, pathogenicity, function-altering, gain-of-function, gene expression
Graphical abstract
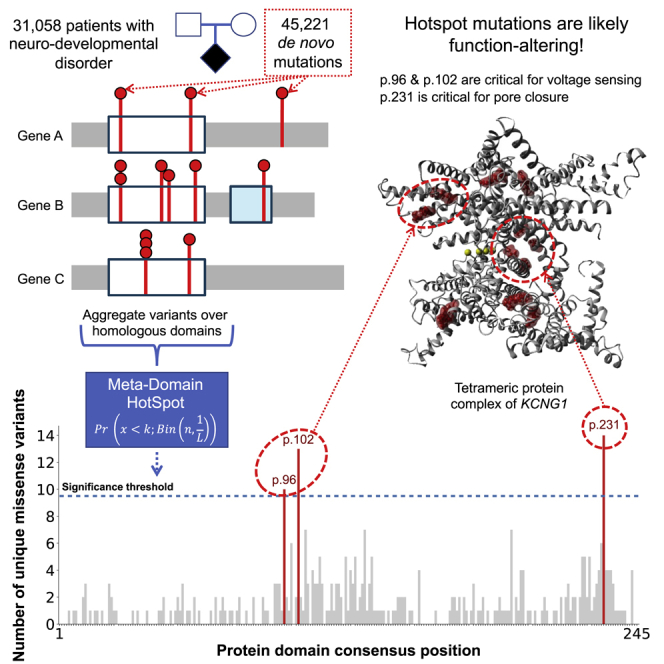
We developed MDHS which utilizes homologous protein domains to identify domain-based variant hotspots. Applying MDHS on de novo mutations from 31,058 patients with neurodevelopmental disorders (NDDs) identified three missense hotspots across 25 genes, of which 19 genes were previously associated with NDD. The identified missense mutations at the hotspots are suggested to alter function.
Introduction
The interpretation of sequence variation in the context of disease remains one of the biggest challenges in genetics. De novo mutations (DNMs) in protein-coding genes are an established cause of neurodevelopmental disorders (NDDs),5 and roughly ∼45% of NDDs are caused by a DNM in a protein-coding gene.6,7 By modeling the probability of DNMs occurring in specific genes, one can identify genes that are enriched for DNMs in patient cohorts, provided that large-enough cohorts are available. This statistical identification of NDD-associated genes requires ever-larger collections of affected individuals.1,7,8,9,10 A recent study of DNMs in 31,058 individuals with NDDs concluded that NDD-association of genes still is far from saturated and that over a thousand NDD-associated genes are still to be identified.1
Several studies of NDD-affected individuals have found that for specific genes, missense DNMs cluster in functional regions, and that this fact can be used to identify disease-associated genes.1,11,12 Conserved protein domains are of particular interest, because they harbor ∼71%13 of all curated disease-causing missense variants in the Human Gene Mutation Database (HGMD)14 and ClinVar.15 Indeed, missense DNMs in NDD genes are almost three times more likely to be located in protein domains.1 Clustered missense DNMs in these genes may act not through haploinsufficiency, but rather through dominant-negative or gain-of-function effects.11,12 The detection of mutation clusters, or hotspots, can be a crucial step toward associating genes with NDDs16 and for gaining insight into underlying disease mechanisms.12
Aggregation of variation across homologous domains can be a useful method to gain insight into patterns of variation13,17,18 and can increase statistical power to detect mutation hotspots. Methods such as mCluster19 and the DS-Score20 have been developed to detect re-occurrence of missense mutations at equivalent positions in protein domains. However, these methods cannot robustly be applied to population datasets. Therefore, we developed Meta-Domain HotSpot (MDHS), a method to detect mutation clustering at evolutionary equivalent positions across homologous protein domains. We applied this method to DNMs from a large cohort of NDD-affected individuals to identify protein consensus positions enriched for missense variation.
Material and methods
Dataset of de novo mutations
We obtained the set of 45,221 DNMs from the Kaplanis et al. study.1 These DNMs were identified in 31,058 DD-affected individuals combined across three centers. The genetic testing approach of these patients were described previously per center: DDD,7 GeneDX,21 and Radboudumc.8 All individuals that underwent genetic testing provided informed consent.1 Subsets of these patients have been analyzed and reported in previous publications.7,21,22,23
Developmental disorder diagnostic gene lists
We use the diagnostic lists of DD-associated genes from the Kaplanis et al. study. We consider all genes statistically associated with NDDs in this study to be NDD genes (novel, consensus, and discordant genes, total genes = 1,010).1
Additionally, we used the Deciphering Developmental Disorders Genotype2Phenotype (DDG2P, accessed 22-04-2021) list to assess the burden of function-altering mutational mechanisms already described in hotspot gene families. We considered activating, gain-of-function, dominant-negative, and increased gene dosage mutation consequences in this list to be function altering.
Annotation of transcript details, protein and meta-domain position annotation
The DNMs (Data S1) were annotated with corresponding GENCODE24 transcripts from release 19 GRCh37.p13 Basic set, protein information from UniProtKB/Swiss-Prot25 Release 2016_09, Pfam-A26 v30.0 protein domains information, and meta-domain13 positions using a local version of the MetaDome17 web server (code available at https://github.com/cmbi/metadome). Meta-domains are multiple sequence alignments of regions within human protein-coding genes that correspond to Pfam protein domain families. The DNMs that correspond to Pfam consensus positions are annotated with the corresponding Pfam domain ID and consensus position.
Filtering the annotated DNMs
The annotation process can result in multiple GENCODE gene transcripts per DNM. To ensure a single GENCODE transcript per gene we performed a filtering step by the following order of criteria:
-
1.
Filter to variants with transcript consequence: missense, synonymous, or stop-gained
-
2.
The transcript corresponds to a human canonical or isoform entry in Swiss-Prot
-
3.
This transcript contains all (or most) of the de novo mutations for the corresponding gene
-
4.
The transcript translates to the longest protein sequence length
-
5.
If multiple transcripts remain for a gene, one of these is selected
-
6.
Filter variants only to those that are in a Pfam protein domain
MDHS: Detection of variant hotspots in homologous protein domains
The Pfam domain ID and consensus position allows for aggregation of genetic variants through meta-domain positions. To identify meta-domain positions that are significantly enriched with variants, we created the MDHS (Meta-Domain HotSpot) p value as follows:
| (Equation 1) |
In the context of meta-domains, n corresponds to the total number of aggregated genetic variants for the Pfam domain ID, L is the total number of possible consensus positions for a Pfam domain ID, k is the total number of genetic variants aggregated at a single consensus position, and x = k − 1, which depicts the chance of finding less than observed genetic variants at the consensus position. The MDHS p value is adapted from the mCluster19 and DS-Score.20 In line with these methods, variants are assumed to follow a binomial distribution. We correct the MDHS p value via the Bonferroni method for the total number of Pfam protein domain IDs considered. If a Bonferroni corrected MDHS p value < 0.05, we consider it to be a significant mutational hotspot.
Our code to analyze the MDHS p value was optimized to compute only for domains which can have significant hotspots. It implements a filter for domain families which works as follows: (1) Count the number of domain consensus positions with one or more variant as n_hotspot_candidates. (2) Count the number of DNMs that span at least more than one unique protein position at each consensus position and sum them up to represent n_hotspots_with_variation_from_more_then_one_protein_position. (3) Apply the filter criteria such that each domain family abides:
See the value in column “hotspot_uniqueness” in Data S2, S3, and S4.
Stringent and lenient counting of variants in MDHS
We use two ways to determine variable k in the MDHS p value (Equation 1). Unless otherwise specified, we count unique variants by considering mutated chromosomal positions only once, thereby reducing the impact of recurrent mutations in a single gene (stringent). Alternatively, we refer to a lenient way of variant counting when we count every mutation equally (including recurrent mutations). For a schematic, see Figure 1.
Figure 1.

Workflow of how mutations are extracted from homologous domain regions within genes and aggregated to meta-domain positions
By clockwise orientation, starting in the upper left there are three protein representations of hypothetical genes A, B, and C with the mutations identified within a cohort are displayed as red lollipops, the domains as blue and white boxes. The white boxes represent domains that are homologous and are extracted and aligned, including their mutations, and displayed on the upper right part of this image as domains A, B, and C. The mutations within a codon are then aggregated over corresponding homologous domain positions based on sequence alignments to form a meta-domain mutation profile (bottom right). Here, the recurring mutations are counted only once for unique counts (for stringent hotspot identification). The unique counts are the input for variable k to compute a positional MDHS p value (Equation 1). Together with the total number of mutations n in the meta-domain mutation profile, the significance threshold (red dotted line left bottom) can be determined which indicates a meta-domain hotspot if the mutational count exceeds it.
Functional characterization
We used the Ensembl Variant Effect Predictor (VEP)27 to annotate all DNMs at hotspot sites (Data S5) with gnomAD allele frequency (AF),28 SIFT,29 Polyphen-2,30 MPC,31 and the CADD_Phred.32 MetaDome17 tolerance indication is a gene-based regional dN/dS based on gnomAD missense and synonymous mutation counts and was obtained manually. ACMG33 classification was obtained through variant curation by a laboratory specialist. Available phenotype information for individuals with missense mutations in hotspot positions can be found in Data S6.
Protein 3D structure modeling of the genes with identified hotspots
For each of the 25 genes with a DNM located at one of the hotspots, we submitted the corresponding protein sequence (based on the transcript of Filtering the annotated DNMs) to the YASARA & WHAT IF Twinset34,35 homology modeling script using the default settings. The regions corresponding to the PF00520 were extracted from all resulting homology structures and combined in a single YASARA scene and then structurally aligned using the MOTIF script. The structures are available in Data S7 and can be accessed through the freely available YASARA View software. The protein structure effects of mutations have been reported in Data S8.
Population constraint
Constraint information, including observed and expected counts, z-scores, and pLI, pNull, and pRec were calculated on gnomAD v.2.1.1.28
Regional missense constraint
Genes with regions of differential missense constraint were identified as described by Samocha et al.31 in the ExAC36 dataset. In brief, the fraction of observed missense variation along a transcript was tested for uniformity using a likelihood ratio test. If the distribution was not uniform, the transcript was considered to have evidence of regional missense constraint. 2,700 genes showed evidence of at least two regions of distinct missense constraint using this method.
Expression data
Pre-computed tissue expression values in transcripts per million (TPM) were taken from GTEx v.8 (GTEx_Analysis_2017-06-05_v8_RNASeQCv1.1.9_gene_median_tpm.gct.gz).37,38
Gene sets for constraint and expression analysis
The set of 56,200 genes for which median TPM values were available was divided into four sets: proposed novel hotspot genes, hotspot genes, NDD-associated genes, and control genes. Genes containing a mutation hotspot were divided into two categories: hotspot genes (n = 19) and proposed novel hotspot genes (n = 6). These categories were distinguished by their presence on the DD gene list (see Developmental disorder diagnostic gene lists); hotspot genes are on this list while proposed novel hotspot genes are not. The remaining genes were divided into NDD-associated genes (n = 992, excluding hotspot genes) and control genes not statistically associated with intellectual disability or developmental delay (n = 55,183). For some analyses, control genes present in DDG2P (n = 1,250, accessed 22-04-2021) or OMIM (n = 3,402, accessed 31-08-2021) but not statistically associated with NDDs were considered separate classes. Additionally, only some of these genes had population constraint information available from gnomAD v.2.1.1 (n = 19,658). Genes in all sets can be found in Data S9.
Of the 56,200 genes described above, 105 contained a PF00520 domain in MetaDome v.1.0.1. These 105 genes represented 19 hotspot genes, 6 proposed novel hotspot genes, 12 NDD-associated genes not containing a missense DNM at a hotspot position in our cohort, and 68 control genes (of which 6 are present in DDG2P and 26 in OMIM; Data S10).
Proportion of expressed genes across GTEx tissues
A fixed level of TPM > 1 was used to define expression in each tissue. NDD-associated and control genes were randomly sampled 1,000 times into sets of 19 genes, and the proportion of expressed genes (number of genes with TPM > 1/total number of genes) was calculated for each set. This generated a distribution of proportions across 54 GTEx tissues. The proportion of expressed hotspot genes per tissue was computed without sampling.
TPM differences between tissue groups
In order to assess expression differences between brain and other tissues, GTEx tissues were divided into two groups. We considered the amygdala, anterior cingulate cortex (BA24), caudate (basal ganglia), cerebellar hemisphere, cerebellum, cortex, frontal cortex (BA9), hippocampus, hypothalamus, nucleus accumbens (basal ganglia), putamen (basal ganglia), spinal cord (cervical c-1), and substantia nigra brain tissues (n = 12). All non-brain tissues were included in the “other tissues” set (n = 42). The TPM value for each tissue set was defined as the median TPM of all tissues in the set. Based on these differences, we modeled the brain TPM distribution in hotspot genes and control genes and the other tissue TPM distribution in hotspot genes and NDD-associated genes as normal distributions. For each set of distributions, a likelihood ratio of belonging to each distribution was calculated. Proposed novel hotspot genes were considered to have evidence for association with NDDs if they were more likely to belong to the hotspot gene distribution across both tests.
Filtering and annotation of additional de novo mutation cohorts
We analyzed the enrichment of missense and synonymous DNMs in lenient hotspot positions across a total of three additional published DNM cohorts. We used an autism-spectrum disorder (ASD) cohort published by Satterstrom et al.2 (35,584 total individuals, 11,986 with ASD), a congenital heart defect (CHD) cohort published by Jin et al.3 (2,645 trios), and a cohort of healthy individuals sequenced by Jonsson et al.4 (1,548 trios). To increase our power to find significant differences at lenient hotspot positions, we pooled the Jonsson et al.4 healthy individuals with unaffected siblings from the Satterstrom et al.3 ASD cohort (1,740 siblings) for a total of 3,288 unaffected individuals.
Annotation of meta-domain protein consensus positions for these datasets was done as previously described for Kaplanis et al.1 using MetaDome. The number of PTV, missense, and synonymous SNVs in Pfam protein domains in each dataset can be found in Table S1.
Variant annotation
Variants at protein consensus positions were checked for clinical interpretation across four curated variant databases: ClinVar, HGMDPro, Swiss-Prot, and VKGL, all accessed 21-08-2021. Mapping of protein consensus positions to GRCh37 genomic positions for each gene was done using MetaDome.
For analysis on stringent hotspot positions, ClinVar data were unfiltered on evidence level or review status. We classified missense variation as LP (pathogenic or likely pathogenic, ACMG Class V or IV) or VUS (variant of uncertain significance, ACMG Class III) based on the most severe class across all four databases (Data S6).
For the enrichment analysis on lenient hotspot positions, only ClinVar and VKGL data were used because these two databases include likely benign variants. ClinVar data were filtered on review status (required to be one of “practice guideline,” “reviewed by expert panel,” “criteria provided, multiple submitters, no conflicts,” “criteria provided, single submitter”). Variants were classified as LP (ACMG class V or IV), VUS (ACMG class III), or LB (benign or likely benign, ACMG class II or I). Variants with conflicting interpretations of pathogenicity within or between databases (LP and LB annotations) were removed, as were variants with only VUS annotations.
Results
General description of the data and the processing steps
To identify hotspots of de novo mutations in homologous protein domains, we computed MDHS (Equation 1) based on unique DNMs in NDD-affected individuals (Figure 1). These unique DNMs are aggregated over homologous protein domains to form a domain-based variation profile or a “meta-domain.”13 Next, MDHS assigns a p value to each position, in each meta-domain, based on how closely that position’s aggregated DNMs abide a binomial distribution in perspective of the entire meta-domain (material and methods). This was done for each variant type separately (missense, synonymous, nonsense). We first mapped 45,221 DNMs resulting from 31,058 individuals with developmental disorders1 onto gene transcripts using MetaDome17 (material and methods). Then, we aggregated these to 12,389 meta-domain13 positions (Data S1). The final 15,322 DNMs represent 73.7% missense (n = 11,288), 21.1% synonymous (n = 3,229), and 5.3% stop-gained mutations (n = 805) (Table S2).
Stringent DNM hotspots identified using MDHS
We initially used a stringent approach where recurrent DNMs were counted only once to prevent hotspots driven by DNMs in a single gene. Using all 11,288 missense DNMs in 2,032 protein domain families, our method identified three significant hotspots (Data S2 and S5) comprising 37 unique missense DNMs (57 total) in 25 different genes (Data S2, Table S3). Strikingly, all three hotspots are in domains belonging to the ion transport protein domain family (PF00520) (Figure 2). As a sanity check and to validate our approach, we also performed the stringent method separately for the 3,229 synonymous and 805 stop-gained DNMs in our cohort and identified no significant hotspots (Data S3 and S4).
Figure 2.

The count distribution of missense DNMs aggregated over the ion transport protein domain family (PF00520)
The total consensus length of this domain is 245 and the sum of the count distribution is 350. The significance threshold is displayed as a dotted black line, computed via the MDHS (Equation 1). The bars that exceeded the significance threshold are colored in red and represent the mutational hotspots p.96, p.102, and p.231.
The three significant missense hotspots we identify are located on domain consensus positions p.96 (10 unique DNMs, p = 3.6 × 10−2), p.102 (13 unique DNMs, p = 7.1 × 10−5), and p.231 (14 unique DNMs, p = 7.5 × 10−6) of the ion transport domain family. The ion transport protein domain family is one of four protein domain families that we previously found to be significantly enriched with missense DNMs in NDD-associated genes.1 Specifically, this Pfam domain family consists of sodium, potassium, and calcium ion channels and has six transmembrane helices in which the last two helices determine ion selectivity. Of the 25 genes identified with a missense DNM at a hotspot, 19 are known NDD-associated genes, or hotspot genes, representing a 3.17-fold enrichment of known NDD-associated genes (p = 1.11 × 10−13 chi-square test, Table S4). The remaining 6 genes, proposed novel hotspot genes, have not yet been associated with NDDs (Table 1).
Table 1.
Overview of the genes with missense variants at hotspot positions together with evidence of candidate association to DDs
| Gene | Variant | Functional NDD association | gnomAD AF/SIFT/Polyphen-2/MPC/CADD/MetaDome score | DNMs in other NDD-associated genes | Clinical interpretationa |
|---|---|---|---|---|---|
| TRPM5 (MIM: 604600) |
NC_000011.9:g.2432929C>T; ENST00000452833:c.2549G>A; p.Arg850Gln; PF00520:p.102 |
unknown | 1.20E−02//deleterious (0)//probably damaging (1)//-//28.7//intolerant (0.49) |
2: SLC9A1 ADNP |
uncertain (class 3) |
| TPCN2 (MIM: 612163) | ENST00000294309:c.1633C>A; p.Arg545Ser; PF00520:p.96 | part of the mTOR complex39 | -//deleterious (0.02)//probably damaging (0.965)//0.80//23.5//slightly intolerant (0.67) | 0 | – |
| TPCN1 (MIM: 609666) | ENST00000550785:c.794G>A; p.Arg265Gln; PF00520:p.96 | part of the mTOR complex39 | 7.97E−06//tolerated (0.1)//possibly damaging (0.903)//2.35//26.1//tolerant (1.03) | 0 | likely benign (class 2) |
| KCNH5 (MIM: 605716) | ENST00000322893:c.980G>A; p.Arg327His; PF00520:p.102 | identical VUS (p.327R>H) in unrelated individual with epileptic encephalopathy40 | -//deleterious (0)//probably damaging (0.999)//1.93//32//intolerant (0.19) | 0 | – |
| KCNG1 (MIM: 603788) | ENST00000371571:c.1046G>A; p.Arg349His; PF00520:p.102 | involved in neuronal differentiation41 | -//deleterious (0)//probably damaging (1)//2.74//32//highly intolerant (0.13) | 0 | – |
| CACNA1B (MIM: 601012) |
NC_000009.12:g.137984223G>A; ENST00000371372:c.1742G>A; p.Arg581His; PF00520:p.102 |
nonsense DNMs in CACNA1B lead to an NDD with seizures and nonepileptic hyperkinetic movements (MIM: 618497)42 | 4.58E−03//deleterious (0)//probably damaging (0.999)//1.32//26.1//highly intolerant (0.13) | 0 | – |
Missense variants in these genes have previously not been associated with NDDs. First column indicates the gene and OMIM identifier. Second column indicates the identified DNM at the hotspot position. Third column indicates a previously described functional association of the gene or variant to NDD. Fourth column indicates different prediction scores of variant pathogenicity, the fifth indicates if the same individual has any DNMs located in known NDD-associated genes, and sixth column is ACMG classification. The last three rows (KCNH5, KCNG1, and CACNA1B) are novel candidate NDD genes based on additional evidence as described in this paper. Evidence for ACMG classification is provided in Table S12. Genomic positions and additional phenotype information for all variants where available can be found in Data S6 and S17.
Variants were clinically interpreted where proband phenotype information was available (see Data S6).
Effects of missense mutations at stringent hotspots on protein structure
Mutations that cluster in genes have previously been associated with likely function-altering effects.12 We find that 6 out of 16 hotspot genes present in the Developmental Disorder Genotype2Phenotype (DDG2P) gene list are known to have an activating or gain-of-function mutation consequence (p = 0.0008, Fisher’s exact test), underscoring that missense mutations at hotspot positions are likely function altering (Table S5). To further investigate this, we created 3D protein structure homology models for each of the 25 genes (Data S7, material and methods). Ion transport protein domain 3D structures are 3-fold more identical to each other in conformation than their protein sequences would suggest (CATH-Gene3D ID: 1.20.120.350).43 This structural overlap encouraged us to investigate whether molecular effects of missense variants at these hotspots are likely to have similar impact on domain function across the 25 genes (Data S8).
In the 25 homology models, we find that hotspot p.96 (Figure 3A) and p.102 (Figure 3B) are part of the voltage-sensing helix that is important for the channel (in-)activation.44 These results are in line with functional studies that have been performed for missense mutations at two of these hotspots.45,46 Hotspot p.231 (Figure 3C) is part of the channel gate at the end of a transmembrane helix (Data S8). In addition, we find that missense mutations follow a specific pattern for each of these hotspots. Of the 13/16 missense DNMs located at hotspot p.96 and 20/20 at p.102 change the positively charged wild-type residue to lose the positive charge. Losing positive charges at these locations has previously been described to trigger a function altering disease-mechanism (Figures 3A and 3B).45,46 At hotspot p.231, 20/21 of the missense DNMs changes the wild-type residue from a small into a larger residue. This change in residue size likely impacts the pore closure. This hypothesis is shared by Kortüm et al. who suggest this likely causes a steric hindrance and result into a function-altering mechanism of disease (Figure 3C).47 Lastly, all hotspots are located at the surface of the protein structure, a feat that was previously observed to be characteristic for clustered missense DNMs in NDDs that likely act through non-haploinsufficiency.12 Overall, this shows that missense mutations at the identified hotspots are likely deleterious to domain function.
Figure 3.

Changes in structure caused by missense DNMs in NDD-associated genes for each hotspot
(A) Homology model of the KCNQ3 (MIM: 602232) complex with missense DNM ENST00000388996:c.680G>A (p.Arg227Gln) marked as a green to red change. The KCNQ3 complex is a tetramer constructed from four copies of the KCNQ3 monomer. All monomers are marked in different color shades. This DNM is located at identified hotspot p.96. The wild-type arginine residue is part of the voltage-sensing helix and changed into a glutamine. This change causes it to lose the positive charged that was previously found to cause a function-altering mechanism of disease.45
(B) Homology model of CACNA1A (MIM: 601011) with missense DNM ENST00000360228:c.4988G>A (p.Arg1663Gln) marked as a green to red change. This DNM is located at identified hotspot p.102. The wild-type arginine residue is part of the voltage-sensing helix and changed into a glutamine. This change causes it to lose the positive charge that was previously found to cause a function-altering mechanism of disease.46
(C) Homology model of the KCNH1 (MIM: 603305) complex with missense DNM ENST00000271751:c.1486G>A (p.Gly496Arg) marked as a green to red change. The KCNH1 complex is a tetramer constructed from four copies of the KCNH1 monomer. All monomers are marked in different color shades. This DNM is located at identified hotspot p.231. The wild-type glycine residue is near the pore-closing region and changed into a much larger arginine. This may impact pore closure and was previously reported to result into a function-altering mechanism of disease.47
Stringent hotspot genes are constrained against missense and loss of function variation
Dominant NDD genes are characterized by population constraint against damaging genetic variation.23,28 We compared observed counts of loss of function, missense, and synonymous variants in control, NDD-associated, hotspot, and proposed novel hotspot genes in gnomAD v.2 to expected counts based on a null mutational model.31 Both hotspot and novel hotspot genes are constrained against loss-of-function and missense variation (Figure 4A). Novel hotspot genes have lower constraint against loss-of-function variation than hotspot genes (Data S11). We also considered whether hotspots were located in regions of particular constraint against missense variation within genes. In total, 2,700 genes have statistical evidence of regional differences in missense constraint.31 Of these, 16 are hotspot genes, representing a significant enrichment compared to control genes (Fisher exact test p < 2.2 × 10−16) and NDD-associated genes (Fisher exact test p = 0.02, Figure S1). Three are proposed novel hotspot genes (KCNH5 [MIM: 605716], CACNA1B [MIM: 601012], TPCN1 [MIM: 609666]). Using regional missense constraint information, we show that PF00520 domains in hotspot genes are significantly more constrained against missense variation than PF00520 domains in control genes (p = 1.4 × 10−7, Wilcoxon rank-sum test; Figure 4B), but similarly constrained compared to NDD-associated genes without a hotspot that also contain a PF00520 domain (p = 0.65, Wilcoxon rank-sum test).
Figure 4.

Hotspot genes are constrained against loss-of-function and missense variation
(A) Constraint in hotspot and proposed novel hotspot genes. The observed variant counts for loss of function (red), missense (orange), and synonymous (pink) variants from the gnomAD v.2 release were compared to the expected counts based on a null mutational model.31 Points represent the mean observed/expected ratios for all genes in each set and bars denote the mean upper and lower bound fractions for these ratios. The dashed line at observed/expected = 1 indicates perfect adherence to the null mutational model (observed counts = expected counts); values that fall below this line are constrained.
(B) Mutation hotspots occur in missense constrained regions within genes. Regional missense constraint was compared across PF00520 domain-containing control genes (blue), PF00520 domain-containing NDD-associated genes (green), and hotspot genes (yellow). Boxes represent the lower and upper quartiles of the distribution, and whiskers represent the distance from 1.5× the interquartile range to the lower/upper quartiles.
Brain-specific expression of stringent hotspot genes
We analyzed the expression of the 19 known hotspot genes in approximately 948 donors across 54 tissues from the GTEx v8 release.37 We observed that NDD genes and control genes have distinct gene expression patterns, with a higher proportion of NDD genes constitutively expressed across all tissues (p < 2.2 × 10−16, Fisher exact test; Table S6). Hotspot genes share a characteristic expression pattern compared to these two groups (Figure 5A), with a significantly higher proportion of hotspot genes expressed in the brain compared to control genes and significantly lower proportion expressed in all other tissues compared to NDD genes (in 40/42 non-brain tissues, Data S12). Given this tissue-specific expression pattern, we grouped GTEx tissues into two tissue groups (brain and other tissues, material and methods). The hotspot gene set is significantly enriched for genes with higher expression in brain compared to control genes (89.4% versus 19.8% expressed higher in brain, p = 2.985 × 10−5, Fisher exact test) and NDD genes (89.4% versus 31.3%, p = 0.002, Fisher exact test) (Figure S2). Only two hotspot genes do not have higher expression in brain: SCN10A (MIM: 604427), which is constitutively unexpressed across tissues in GTEx samples, and CACNA1C (MIM: 114205) (median TPM in brain = 2.94, median TPM in other tissues = 4.35). We further show that this expression pattern is not characteristic of all genes containing an ion transport domain, but only the subset of these genes statistically associated with NDDs (Data S13, Figures S3 and S4).
Figure 5.

Hotspot genes have a distinct gene expression pattern
(A) Tissue expression of hotspot genes compared to control and other NDD genes. Expression of the 19 established NDD genes containing missense DNM(s) at a stringent mutation hotspots (hotspot genes, yellow) were evaluated across 54 GTEx tissues (x axis). Hotspot gene expression was compared to NDD genes (green) and control genes (blue). The y axis depicts the proportion of expressed genes (material and methods). Squares and bars depict the median and SD, respectively, of NDD and control gene distributions.
(B) TPM distribution in brain and other tissues varies across gene sets. Control (blue) and NDD-associated (green) genes are represented by 2D density distributions. Hotspot genes (yellow) are shown as points, as are NDD-associated genes containing a PF00520 domain (gray). Proposed novel hotspot genes are marked by their gene name in red text.
We also compared the TPM distribution in brain and other tissues for control genes, NDD-associated genes, hotspot genes, and the six proposed novel hotspot genes (material and methods; Figure 5B). Both hotspot and NDD-associated genes had significantly higher TPM in brain tissues than control genes (p = 0.0039 and p < 2.2 × 10−16, Wilcoxon rank-sum test), and both hotspot genes and control genes had significantly lower TPM in other tissues than NDD-associated genes (p < 2.2 × 10−16 and p = 2.08 × 10−8, Wilcoxon rank-sum test; Figure S5). Modeling suggests that CACNA1B and KCNG1 (MIM: 603788) belong to the hotspot gene distribution by odds ratio (Data S14). Additionally, we find 6 NDD-associated PF00520 domain-containing genes (HCN1 [MIM: 602780], TRPV3 [MIM: 607066], KCNQ5 [MIM: 607357], KCNC1 [MIM: 176258], KCNC3 [MIM: 176264], TRPM3 [MIM: 608961]) that also belong to the hotspot gene distribution by odds ratio. We hypothesize that missense mutations at hotspot positions in these six genes may also cause NDDs.
Missense mutations at lenient DNM hotspots are enriched in clinical databases
We also implemented a lenient version of MDHS that counts all missense variants at protein consensus positions, even if they recur between individuals (see material and methods, Figure S6). Counting recurrent missense variants gives us more power to detect hotspot positions, but these hotspot positions may be driven by missense variation in a single domain. Applying this lenient method to our cohort identified 32 significant missense hotspots across 16 Pfam protein domain families (Data S2 and S15) and no significant hotspots for synonymous or nonsense mutations (Data S3 and S4). 12 protein domain families had hotspots spanning multiple gene-codons based on 245 DNMs from 67 genes. 48 of these 67 genes (72%) are statistically associated with NDDs, representing a 2.53-fold enrichment (p = 1.26−31 chi-square test; Table S7) and showing the merit of this approach. We find a significant enrichment of genes statistically associated with NDD (Fisher’s exact p < 2.2 × 10−16) and DDG2P (Fisher’s exact p < 2.2 × 10−16) genes at lenient hotspot positions (Figure S7).
We also find that missense variants at these positions are significantly more likely to be pathogenic or likely pathogenic in clinical databases (VKGL, Figure 6A; ClinVar, Figure 6B). We compared the proportion of reported likely pathogenic (LP) missense variants at hotspot positions to those at other protein consensus positions across the 16 Pfam domain families with a lenient hotspot (Figures 6A and 6B). We find a significant enrichment of LP variants at hotspot positions when we consider all positions (Fisher’s exact p < 2.2 × 10−16, ClinVar; p < 2.2 × 10−16, VKGL), only positions without a DNM in our cohort (Fisher’s exact p < 2.2 × 10−16, ClinVar; p < 2.2 × 10−16, VKGL), and only codons without a DNM in our cohort (Fisher’s exact p < 2.2 × 10−16, ClinVar; p = 3.08 × 10−13, VKGL; Table S8).
Figure 6.

Lenient hotspots are enriched for likely pathogenic missense variation in clinical databases
Counts of likely pathogenic (LP, red), uncertain (VUS, gray), and likely benign (LB, blue) missense variants in VKGL (A) and ClinVar (B) in domains containing a lenient hotspot position. The proportion of LP missense variants (LP/(LP + LB), see material and methods) was compared between mutation hotspots (purple) and all other protein consensus positions within the domain (orange). This comparison was done for all possible missense variants in these domains (row 2) and with positions containing DNMs in our cohort excluded (row 3).
Lenient hotspots are enriched for missense variation in autism-spectrum disorders
We investigated whether we could find further evidence for the identified lenient hotspots in a combined cohort of publicly available de novo mutation datasets for autism-spectrum disorders (ASD; 11,986 ASD probands, 35,584 individuals) and congenital heart defects (CHD; 2,654 trios) alongside DNMs from unaffected individuals (1,740 ASD siblings, 1,548 population control subjects; see material and methods).
We observe a significant enrichment of missense DNMs at hotspot positions in NDD and ASD cohorts compared to unaffected individuals (Fisher’s exact p = 3.5 × 10−13, NDD; Fisher’s exact p = 0.007, ASD; Fisher’s exact p = 0.07, CHD; Figure 7A, Table S9). We observe no significant enrichment in synonymous DNMs at hotspot positions in any cohort (Table S10). However, we predict that some lenient hotspot positions are driven by mutational processes or ascertainment bias in particular genes and not necessarily by the cumulative effect of pathogenic mutations across several genes. To correct for this, we also tested for an enrichment of missense variants unique to ASD and CHD probands at lenient hotspots (Figure 7B, Table S11). We find a significant enrichment of these unique missense variants in ASD probands (Fisher’s exact p = 0.047) but not in CHD probands (Fisher’s exact p = 1). The majority (10/13, 77%) of the missense variants driving this enrichment in ASD probands are in genes statistically associated with NDDs.1
Figure 7.

Affected individuals are enriched for missense variants in lenient hotspot positions compared to healthy population control subjects
(A) NDD and ASD are enriched for missense DNMs in lenient hotspot positions compared to unaffected individuals (green) but are not enriched for synonymous DNMs (gray). Only DNMs within protein consensus positions were used for this comparison (see material and methods).
(B) ASD probands are enriched for missense DNMs in lenient hotspot positions (green) not present in our NDD cohort.
Discussion
By exploiting homology within the human genome, we were able to identify mutational clustering of DNMs at evolutionarily conserved positions across genes that share protein domains. We identify three stringent (p.96, p.102, p.231) and 32 lenient mutational hotspots across 16 Pfam domain families using our MDHS method. Missense DNMs at stringent hotspots are located in 25 genes within our cohort. Structural and functional work by us and others suggest that missense mutations at these positions may be function altering.48,49 Functional work for hotspots in each of the 25 genes would be necessary to confirm this.
The hotspots we statistically identify in our cohort may have broader clinical relevance. We hypothesize that the 19 hotspot genes are examples of a broader class of ion transport domain-containing genes and that missense mutations at hotspot positions in these genes are generally damaging. Our finding that clinical databases contain many pathogenic missense mutations at hotspot positions in other monogenic disease genes (Data S16) supports this hypothesis. Other studies have shown that missense mutations in ion transport domain-containing genes may have position-specific functional effects.50,51 Several of these mutations occur in genes not statistically associated with NDDs, indicating that missense mutations at hotspot positions could be pathogenic across a variety of disorders. In line with this, we observe that some PF00520 domain-containing NDD-associated genes have lower expression in brain but have a similar level of tissue-specific expression in a non-brain tissue. SCN4A (MIM: 603967), for example, is not expressed in brain and is predominantly expressed in skeletal muscle. Although the expression pattern of SCN4A is different from the hotspot genes presented in our analysis, hotspot positions in SCN4A are similarly constrained against missense variation (Figure 5B). We hypothesize that phenotypes associated with pathogenic mutations at hotspot positions may vary depending on where the mutated gene is expressed. For example, of the four PF00520 domain-containing genes predominantly expressed in skeletal muscle (SCN4A, CACNA1S [MIM: 114208], RYR1 [MIM: 180901], and KCNA7 [MIM: 176268]), three of these (SCN4A, RYR1, and CACNA1S) have pathogenic missense variation at hotspot positions in clinical databases (Data S16). Individuals with these mutations present with disorders of the skeletal muscle, including myotonia (MIM: 608390), paramyotonia (MIM: 168300), and hyperkalemic paralysis (MIM: 170500). Our work suggests that missense mutations at hotspot positions in KCNA7 may also result in skeletal muscle disorders based on the tissue expression of KCNA7 and the conservation of hotspot positions in the PF00520 domain of this gene.
Our method also identified six genes with a mutation at the hotspot location that have not previously been associated with NDDs. Three genes—KCNH5, CACNA1B, and KCNG1—have evidence supporting NDD association (Table 1). In KCNH5, the same DNM was described as a variant of unknown significance (VUS) in an individual with an epileptic encephalopathy,40 and very recently a study of a cohort of NDD-affected individuals with KCNH5 DNMs was published, including nine individuals with recurrent p.Arg327His mutations.52 CACNA1B was recently established as an NDD-associated gene on the basis of LoF DNM enrichment.42 In line with this, CACNA1B is the only proposed novel hotspot gene that is predicted to be intolerant to heterozygous loss of function by population constraint (pLI = 1; Data S11). However, our work suggests that the missense variants we identify at hotspot positions in CACNA1B may be function altering. KCNG1 has been implicated in neuronal development,41 and the expression profile matches well with that of the other NDD genes that have hotspot mutations. There is also some circumstantial evidence for two of the other three genes. Both TPCN1 and TPCN2 (MIM: 612163) have no prior NDD association, but both genes are part of the mTOR complex, which has previously been associated with NDDs.39 Phenotypic data for the individual with the missense DNM in TPCN1 shows that this person has macrocephaly and severe ASD (Data S6), which is in line with the fact that mTOR genes have been associated with intracranial volume and intellectual disability.53 However, the only TPCN1 missense mutation presently described in literature at a hotspot position (p.102) is associated with early-onset cardiomyopathy.54
In this analysis, we initially identified stringent mutation hotspots statistically using unique missense mutation counts. While MDHS analysis on even larger cohorts may identify additional hotspots, we believe our method could be used on smaller datasets by also considering recurrent mutations. Applying this lenient method to our cohort identified 32 significant missense hotspots (Data S2). Even though the inclusion of recurrent mutations allows hotspots to be driven by proliferative advantages of single mutations in the germline or soma, CpG hypermutability, or biases in clinical ascertainment, it also increases power to detect robust hotspot positions (Figure S6). In support of this, we find an enrichment of likely pathogenic missense variants at lenient hotspot positions in clinical databases even if we look only at codons without a mutation in our cohort, showing the merit of this approach. However, additional filtering may be required to remove hotspots driven solely by recurrent missense mutations. Additionally, there is in principle no reason to restrict this method to de novo mutations; it could easily be applied to rare inherited variants in large patient cohorts.
Kaplanis et al.1 estimate that approximately 350,000 parent-offspring trios would be required to detect the majority of remaining haploinsufficient genes associated with NDDs. Genes enriched for function-altering mutations are predicted to be even more difficult to detect, even in very large cohorts. Methods based on homology, like MDHS, are an approach to the identification of disease genes and mechanisms in existing datasets without increasing cohort size. Additionally, the systematic identification of function-altering mutations in large population datasets will have fundamental impact on our understanding of disease biology and may lead to improvements in patient care. In NDD-associated haploinsufficient genes, we observe that function-altering mutations can have substantially different phenotypes and severities than mutations resulting in loss of function. In the future, affected individuals could be stratified for targeted therapies or counseled about their prognosis based on the mutational mechanism of their disease-causing variant. More broadly, function-altering mutations will provide insight into the molecular function of protein domains. The way haploinsufficiency causes disease is not domain specific, whereas the function-altering mutations we identify are a specific property of the domain in which they occur. New approaches are required to understand the role of function-altering mutations in the human germline, and we provide compelling evidence that the aggregation of mutations over homologous protein domains could be one of these approaches.
Acknowledgments
We thank Dr. Torti and Dr. Retterer from GeneDX for connecting us with Prof. Mefford. We thank Prof. Mefford for information regarding the likely NDD-association of KCHN5. We thank Elke de Boer for useful discussions. This work was in part financially supported by grants from the Dutch Research Council (NWO) (916-14-043 to C.G. and 918-15-667 to J.A.V.) and from the Radboud Institute for Molecular Life Sciences, Radboud University Medical Center (R0002793 to G.V.).
Declaration of interests
The authors have no competing interests.
Published: December 22, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2022.12.001.
Web resources
CATH-Gene3D, http://www.cathdb.info/
DECIPHER, https://www.deciphergenomics.org/
MetaDomainHotspot analyses repository, https://github.com/cmbi/MetaDomainHotSpot
MetaDome GitHub repository, https://github.com/cmbi/metadome
MetaDome web server, https://stuart.radboudumc.nl/metadome/
Swiss-Prot, https://www.uniprot.org/
Variant Effect Predictor, https://www.ensembl.org/Tools/VEP
VKGL, https://www.vkgl.nl/nl/diagnostiek/vkgl-datashare-database
YASARA, http://www.yasara.org/
Supplemental information
Data and code availability
The published article includes all data generated during this study in Data S1, S2, S3, S4, S5, S6, S7, S8, S9, S10, S11, S12, S13, S14, S15, S16, and S17.
The code and docker configurations generated during this study are available at GitHub (https://github.com/cmbi/MetaDomainHotSpot). Data to reproduce figures and all parts of the analyses in this study are included in this repository.
A local version of MetaDome (https://stuart.radboudumc.nl/metadome/) was used to annotate genomic data with meta-domain information and MetaDome tolerance scores. The original MetaDome source code is available on Github (https://github.com/cmbi/metadome) and all data underlying the MetaDome web server is available on Zenodo (https://zenodo.org/record/6625251).
External de novo mutation (DNM) dataset used in this this study are publicly available in the original publications: developmental disorder (NDD) DNMs, Kaplanis et al.1; autism spectrum disorder (ASD) DNMs, Satterstrom et al.2; congenital heart defect (CHD) DNMs, Jin et al.3; DNMs from unaffected individuals, Jonsson et al.,4 Satterstrom et al.2
Expression data used for this work is publicly available through GTEx (https://gtexportal.org/home/), and we have made use GTEx release v.8 for this study.
References
- 1.Kaplanis J., Samocha K.E., Wiel L., Zhang Z., Arvai K.J., Eberhardt R.Y., Gallone G., Lelieveld S.H., Martin H.C., McRae J.F., et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. 2020;586:757–762. doi: 10.1038/s41586-020-2832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Satterstrom F.K., Kosmicki J.A., Wang J., Breen M.S., De Rubeis S., An J.Y., Peng M., Collins R., Grove J., Klei L., et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell. 2020;180:568–584. doi: 10.1016/j.cell.2019.12.036. e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jin S.C., Homsy J., Zaidi V., Lu Q., Morton S., DePalma S.R., Zeng X., Qi H., Chang W., Sierant M.C., et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49:1593–1601. doi: 10.1038/ng.3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jónsson H., Sulem P., Kehr B., Kristmundsdottir S., Zink F., Hjartarson E., Hardarson M.T., Hjorleifsson K.E., Eggertsson H.P., Gudjonsson S.A., et al. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–522. doi: 10.1038/nature24018. [DOI] [PubMed] [Google Scholar]
- 5.Veltman J. a, Brunner H.G. De novo mutations in human genetic disease. Nat. Rev. Genet. 2012;13:565–575. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- 6.Martin H.C., Jones W.D., McIntyre R., Sanchez-Andrade G., Sanderson M., Stephenson J.D., Jones C.P., Handsaker J., Gallone G., Bruntraeger M., et al. Quantifying the contribution of recessive coding variation to developmental disorders. Science. 2018;362:1161–1164. doi: 10.1126/science.aar6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deciphering Developmental Disorders Study Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433–438. doi: 10.1038/nature21062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Ligt J., Willemsen M.H., van Bon B.W.M., Kleefstra T., Yntema H.G., Kroes T., Vulto-van Silfhout A.T., Koolen D.A., de Vries P., Gilissen C., et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 9.Deciphering Developmental Disorders Study Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519:223–228. doi: 10.1038/nature14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner T.N., Yi Q., Krumm N., Huddleston J., Hoekzema K., F Stessman H.A., Doebley A.L., Bernier R.A., Nickerson D.A., Eichler E.E. a compendium of human de novo variants. Nucleic Acids Res. 2017;45:D804–D811. doi: 10.1093/nar/gkw865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geisheker M.R., Heymann G., Wang T., Coe B.P., Turner T.N., Stessman H.A.F., Hoekzema K., Kvarnung M., Shaw M., Friend K., et al. Hotspots of missense mutation identify neurodevelopmental disorder genes and functional domains. Nat. Neurosci. 2017;20:1043–1051. doi: 10.1038/nn.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lelieveld S.H., Wiel L., Venselaar H., Pfundt R., Vriend G., Veltman J.A., Brunner H.G., Vissers L.E.L.M., Gilissen C. Spatial clustering of de novo missense mutations identifies candidate neurodevelopmental disorder-associated genes. Am. J. Hum. Genet. 2017;101:478–484. doi: 10.1016/j.ajhg.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wiel L., Venselaar H., Veltman J.A., Vriend G., Gilissen C. Aggregation of population-based genetic variation over protein domain homologues and its potential use in genetic diagnostics. Hum. Mutat. 2017;38:1454–1463. doi: 10.1002/humu.23313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stenson P.D., Mort M., Ball E.V., Evans K., Hayden M., Heywood S., Hussain M., Phillips A.D., Cooper D.N. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017;136:665–677. doi: 10.1007/s00439-017-1779-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landrum M.J., Lee J.M., Benson M., Brown G., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Hoover J., et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–D868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schuurs-Hoeijmakers J.H.M., Oh E.C., Vissers L.E.L.M., Swinkels M.E.M., Gilissen C., Willemsen M.A., Holvoet M., Steehouwer M., Veltman J.A., de Vries B.B.A., et al. Recurrent de novo mutations in PACS1 cause defective cranial-neural-crest migration and define a recognizable intellectual-disability syndrome. Am. J. Hum. Genet. 2012;91:1122–1127. doi: 10.1016/j.ajhg.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wiel L., Baakman C., Gilissen D., Veltman J.A., Vriend G., Gilissen C. MetaDome: pathogenicity analysis of genetic variants through aggregation of homologous human protein domains. Hum. Mutat. 2019;40:1030–1038. doi: 10.1002/humu.23798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peterson T.A., Park D., Kann M.G. A protein domain-centric approach for the comparative analysis of human and yeast phenotypically relevant mutations. BMC Genom. 2013;14:S5. doi: 10.1186/1471-2164-14-S3-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yue P., Forrest W.F., Kaminker J.S., Lohr S., Zhang Z., Cavet G. Inferring the functional effects of mutation through clusters of mutations in homologous proteins. Hum. Mutat. 2010;31:264–271. doi: 10.1002/humu.21194. [DOI] [PubMed] [Google Scholar]
- 20.Peterson T.A., Nehrt N.L., Park D., Kann M.G. Incorporating molecular and functional context into the analysis and prioritization of human variants associated with cancer. J. Am. Med. Inform. Assoc. 2012;19:275–283. doi: 10.1136/amiajnl-2011-000655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Retterer K., Juusola J., Cho M.T., Vitazka P., Millan F., Gibellini F., Vertino-Bell A., Smaoui N., Neidich J., Monaghan K.G., et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 2016;18:696–704. doi: 10.1038/gim.2015.148. [DOI] [PubMed] [Google Scholar]
- 22.Wright C.F., Fitzgerald T.W., Jones W.D., Clayton S., McRae J.F., van Kogelenberg M., King D.A., Ambridge K., Barrett D.M., Bayzetinova T., et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2015;385:1305–1314. doi: 10.1016/S0140-6736(14)61705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lelieveld S.H., Reijnders M.R.F., Pfundt R., Yntema H.G., Kamsteeg E.J., de Vries P., de Vries B.B.A., Willemsen M.H., Kleefstra T., Löhner K., et al. Meta-analysis of 2, 104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 2016;19:1194–1196. doi: 10.1038/nn.4352. [DOI] [PubMed] [Google Scholar]
- 24.Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S., et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boutet E., Lieberherr D., Tognolli M., Schneider M., Bansal P., Bridge A.J., Poux S., Bougueleret L., Xenarios I. UniProtKB/Swiss-Prot, the manually annotated section of the UniProt KnowledgeBase: how to use the entry view. Methods Mol. Biol. 2016;1374:23–54. doi: 10.1007/978-1-4939-3167-5_2. [DOI] [PubMed] [Google Scholar]
- 26.Finn R.D., Coggill P., Eberhardt R.Y., Eddy S.R., Mistry J., Mitchell A.L., Potter S.C., Punta M., Qureshi M., Sangrador-Vegas A., et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–D285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P., Cunningham F. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122–214. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141, 456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 30.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samocha K.E., Kosmicki J.A., Karczewski K.J., O’Donnell-Luria A.H., Pierce-Hoffman E., MacArthur D.G., Neale B.M., Daly M.J. Regional missense constraint improves variant deleteriousness prediction. bioRxiv. 2017 doi: 10.1101/148353. Preprint at. [DOI] [Google Scholar]
- 32.Kircher M., Witten D.M., Jain P., O'Roak B.J., Cooper G.M., Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krieger E., Koraimann G., Vriend G. Increasing the precision of comparative models with YASARA NOVA—a self-parameterizing force field. Proteins. 2002;47:393–402. doi: 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
- 35.Vriend G. WHAT IF: A molecular modeling and drug design program. J. Mol. Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 36.Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O'Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., et al. Analysis of protein-coding genetic variation in 60, 706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aguet F., et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.GTEx Consortium. Battle A., Brown C.D., Engelhardt B.E., Montgomery S.B. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cang C., Zhou Y., Navarro B., Seo Y.J., Aranda K., Shi L., Battaglia-Hsu S., Nissim I., Clapham D.E., Ren D. mTOR regulates lysosomal ATP-sensitive two-pore Na(+) channels to adapt to metabolic state. Cell. 2013;152:778–790. doi: 10.1016/j.cell.2013.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Veeramah K.R., Johnstone L., Karafet T.M., Wolf D., Sprissler R., Salogiannis J., Barth-Maron A., Greenberg M.E., Stuhlmann T., Weinert S., et al. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia. 2013;54:1270–1281. doi: 10.1111/epi.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chiocchetti A.G., Haslinger D., Stein J.L., de la Torre-Ubieta L., Cocchi E., Rothämel T., Lindlar S., Waltes R., Fulda S., Geschwind D.H., Freitag C.M. Transcriptomic signatures of neuronal differentiation and their association with risk genes for autism spectrum and related neuropsychiatric disorders. Transl. Psychiatry. 2016;6 doi: 10.1038/tp.2016.119. e864–e864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gorman K.M., Meyer E., Grozeva D., Spinelli E., McTague A., Sanchis-Juan A., Carss K.J., Bryant E., Reich A., Schneider A.L., et al. Bi-allelic Loss-of-Function CACNA1B Mutations in Progressive Epilepsy-Dyskinesia. Am. J. Hum. Genet. 2019;104:948–956. doi: 10.1016/j.ajhg.2019.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sillitoe I., Lewis T., Orengo C. Using CATH-Gene3D to analyze the sequence, structure, and function of proteins. Curr. Protoc. Bioinformatics. 2015;50:1.28.1–1.28.21. doi: 10.1002/0471250953.bi0128s50. [DOI] [PubMed] [Google Scholar]
- 44.Bezanilla F. How membrane proteins sense voltage. Nat. Rev. Mol. Cell Biol. 2008;9:323–332. doi: 10.1038/nrm2376. [DOI] [PubMed] [Google Scholar]
- 45.Sands T.T., Miceli F., Lesca G., Beck A.E., Sadleir L.G., Arrington D.K., Schönewolf-Greulich B., Moutton S., Lauritano A., Nappi P., et al. Autism and developmental disability caused by KCNQ3 gain-of-function variants. Ann. Neurol. 2019;86:181–192. doi: 10.1002/ana.25522. [DOI] [PubMed] [Google Scholar]
- 46.Luo X., Rosenfeld J.A., Yamamoto S., Harel T., Zuo Z., Hall M., Wierenga K.J., Pastore M.T., Bartholomew D., Delgado M.R., et al. Clinically severe CACNA1A alleles affect synaptic function and neurodegeneration differentially. PLoS Genet. 2017;13:e1006905. doi: 10.1371/journal.pgen.1006905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kortüm F., Caputo V., Bauer C.K., Stella L., Ciolfi A., Alawi M., Bocchinfuso G., Flex E., Paolacci S., Dentici M.L., et al. Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat. Genet. 2015;47:661–667. doi: 10.1038/ng.3282. [DOI] [PubMed] [Google Scholar]
- 48.Daniil G., Fernandes-Rosa F.L., Chemin J., Blesneac I., Beltrand J., Polak M., Jeunemaitre X., Boulkroun S., Amar L., Strom T.M., et al. CACNA1H mutations are associated with different forms of primary aldosteronism. EBioMedicine. 2016;13:225–236. doi: 10.1016/j.ebiom.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang X.Y., Wen J., Yang W., Wang C., Gao L., Zheng L.H., Wang T., Ran K., Li Y., Li X., et al. Gain-of-function mutations in SCN11A cause familial episodic pain. Am. J. Hum. Genet. 2013;93:957–966. doi: 10.1016/j.ajhg.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heyne H.O., Singh T., Stamberger H., Abou Jamra R., Caglayan H., Craiu D., De Jonghe P., Guerrini R., Helbig K.L., Koeleman B.P.C., et al. De novo variants in neurodevelopmental disorders with epilepsy. Nat. Genet. 2018;50:1048–1053. doi: 10.1038/s41588-018-0143-7. [DOI] [PubMed] [Google Scholar]
- 51.Heyne H.O., Baez-Nieto D., Iqbal S., Palmer D.S., Brunklaus A., May P., Epi25 Collaborative. Johannesen K.M., Lauxmann S., Lemke J.R., et al. Predicting functional effects of missense variants in voltage-gated sodium and calcium channels. Sci. Transl. Med. 2020;12:eaay6848. doi: 10.1126/scitranslmed.aay6848. [DOI] [PubMed] [Google Scholar]
- 52.Happ H.C., Sadleir L.G., Zemel M., de Valles-Ibáñez G., Hildebrand M.S., McConkie-Rosell A., et al. Neurodevelopmental and Epilepsy Phenotypes in Individuals With Missense Variants in the Voltage Sensing and Pore Domain of KCNH5. Neurology. Published online October. 2022;28:2022. doi: 10.1212/WNL.0000000000201492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reijnders M.R.F., Kousi M., van Woerden G.M., Klein M., Bralten J., Mancini G.M.S., van Essen T., Proietti-Onori M., Smeets E.E.J., van Gastel M., et al. Variation in a range of mTOR-related genes associates with intracranial volume and intellectual disability. Nat. Commun. 2017;8:1052. doi: 10.1038/s41467-017-00933-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reuter M.S., Chaturvedi R.R., Liston E., Manshaei R., Aul R.B., Bowdin S., Cohn I., Curtis M., Dhir P., Hayeems R.Z., et al. The cardiac genome clinic: implementing genome sequencing in pediatric heart disease. Genet. Med. 2020;22:1015–1024. doi: 10.1038/s41436-020-0757-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all data generated during this study in Data S1, S2, S3, S4, S5, S6, S7, S8, S9, S10, S11, S12, S13, S14, S15, S16, and S17.
The code and docker configurations generated during this study are available at GitHub (https://github.com/cmbi/MetaDomainHotSpot). Data to reproduce figures and all parts of the analyses in this study are included in this repository.
A local version of MetaDome (https://stuart.radboudumc.nl/metadome/) was used to annotate genomic data with meta-domain information and MetaDome tolerance scores. The original MetaDome source code is available on Github (https://github.com/cmbi/metadome) and all data underlying the MetaDome web server is available on Zenodo (https://zenodo.org/record/6625251).
External de novo mutation (DNM) dataset used in this this study are publicly available in the original publications: developmental disorder (NDD) DNMs, Kaplanis et al.1; autism spectrum disorder (ASD) DNMs, Satterstrom et al.2; congenital heart defect (CHD) DNMs, Jin et al.3; DNMs from unaffected individuals, Jonsson et al.,4 Satterstrom et al.2
Expression data used for this work is publicly available through GTEx (https://gtexportal.org/home/), and we have made use GTEx release v.8 for this study.