

Abstract
FGFR2 rearrangements resulting in dysregulated signaling are drivers of cholangiocarcinoma (CCA) tumorigenesis, and occur almost exclusively in intrahepatic CCA. Pemigatinib, a selective, potent, oral inhibitor of FGFR1–3, has demonstrated efficacy and safety in a Phase II study of patients with previously treated locally advanced/metastatic CCA harboring FGFR2 fusions/rearrangements. We describe the study design of FIGHT-302, an open-label, randomized, active-controlled, multicenter, global, Phase III study comparing the efficacy and safety of first-line pemigatinib versus gemcitabine plus cisplatin in patients with advanced CCA with FGFR2 rearrangements (NCT03656536). The primary end point is progression-free survival; secondary end points are objective response rate, overall survival, duration of response, disease control rate, safety and quality of life.
Clinical Trial Registration: NCT03656536 (ClinicalTrials.gov)
Keywords: : cholangiocarcinoma, FGFR, INCB054828, pemigatinib
Cholangiocarcinomas (CCAs) are heterogeneous tumors originating from cholangiocytes in the epithelial layer of the biliary tract that account for 3% of all gastrointestinal malignancies [1]. CCA is classified anatomically as extrahepatic CCA (eCCA; which includes perihilar CCA [pCCA] and distal CCA) and intrahepatic CCA (iCCA) [2].
The incidences of CCA subtypes vary across different regions of the world [3]; in Western countries, the incidence of iCCA has generally increased, whereas the incidence of eCCA has remained relatively stable over the past decades [1]. iCCA is the second most common primary liver cancer after hepatocellular carcinoma, accounting for approximately 10–15% of primary liver cancers [4]. Mortality due to iCCA has increased, whereas mortality from eCCA has declined worldwide [5]. Whereas improvements in diagnostic capabilities and treatments have resulted in declining mortality rates in several other tumor types, iCCA remains a diagnostic challenge because it is generally asymptomatic in early stages or presents with nonspecific symptoms, such as night sweats, cachexia, abdominal pain and/or general malaise [2]. Thus, diagnosis of iCCA is incidental in approximately 20–45% of patients [1], and the majority of patients are already at an advanced disease stage at diagnosis (American Joint Committee on Cancer [AJCC] stage III = 20%; stage IV = 35%) [6]. The prognosis for patients diagnosed with locally advanced or metastatic iCCA is especially poor, with a median overall survival (OS) of only 14 and 4.5 months for patients with AJCC stage III and IV iCCA, respectively, compared with 23 and 25 months for patients with stage I or II iCCA, respectively [7].
Currently, surgery with complete resection is the only curative treatment option for newly diagnosed patients with CCA; however, less than a third of these patients qualify for surgery [8] and approximately 60% subsequently relapse within 5 years of surgery [9]. The prognosis of patients following resection remains poor, with reported 5-year OS rates of only 30% for both iCCA and pCCA [10].
Systemic chemotherapy
The standard of care (SoC) for first-line treatment of patients with unresectable, recurrent or metastatic biliary tract cancer (BTC) is cisplatin and gemcitabine combination therapy. This was based on results from the pivotal Advanced Biliary Cancer (ABC)-02 Phase III study, which demonstrated a significant, but modest, survival advantage versus gemcitabine alone (median OS, 11.7 vs 8.1 months; p < 0.001; median progression-free survival [PFS], 8.0 vs 5.0 months; p < 0.001) [11]. However, the survival advantage associated with cisplatin plus gemcitabine is modest and the prognosis of these patients remains poor. The potential for increasing treatment benefit by adding cetuximab to gemcitabine plus oxaliplatin for first-line treatment was assessed in a Phase II trial enrolling 150 patients with locally advanced or metastatic BTC [12]; no survival advantage was observed in patients receiving cetuximab combined with gemcitabine plus oxaliplatin. The Phase II ABC-03 study examined the potential benefit of adding cediranib (VEGFR1–3 inhibitor) or placebo to first-line gemcitabine plus cisplatin in 124 patients with advanced BTC [13]; the results showed only a modest prolongation of PFS by approximately 1 month in patients receiving cediranib combined with gemcitabine plus cisplatin versus the placebo cohort. Because of the lack of survival benefit observed in these combination studies, cisplatin plus gemcitabine remains the SoC first-line treatment for patients with advanced BTC. However, clinical research to identify first-line combinations with gemcitabine plus cisplatin that extend survival in these patients remains ongoing. For example, a Phase II/III trial is currently enrolling to assess modified FOLFIRINOX (oxaliplatin, leucovorin, 5-fluorouracil and irinotecan) versus gemcitabine plus cisplatin as first-line treatments in patients with locally advanced unresectable BTC (NCT02591030) [14]. The prospective, randomized, Phase III ABC-06 trial assessed modified FOLFOX (oxaliplatin, leucovorin and 5-fluorouracil) plus active symptom control (ASC) versus ASC alone in 162 patients with advanced BTC, who had relapsed after first-line cisplatin plus gemcitabine. The adjusted hazard ratio (HR) for OS in ASC plus modified FOLFOX versus ASC alone was 0.69 (95% CI, 0.50–0.97; p = 0.031); however, median OS with ASC plus modified FOLFOX was only 1 month longer than ASC alone (6.2 vs 5.3 months) [15]. Locoregional treatment approaches including radioembolization and continuous hepatic arterial infusion chemotherapy are showing promise in patients with iCCA [16,17]. Nevertheless, currently available systemic chemotherapy regimens are associated with only modest survival and disease control irrespective of administration route, and there is a clear and unmet need for improved treatment options for patients with advanced CCA in both the first- and second-line settings.
Genetic landscape & targeted therapies in CCA
Advances in genomic profiling techniques are continuing to identify genetic alterations that drive carcinogenesis across multiple tumor types, which may include activating point mutations, fusions or rearrangements, amplifications and/or deletions. Therapies targeted toward activating genetic alterations are providing new therapeutic approaches that are improving the prognosis for patients with various solid tumors and hematologic cancers.
Based on promising preclinical data, several clinical trials have investigated therapies (either as monotherapy or in combination with chemotherapy) for advanced BTC targeting a number of receptors and signaling elements including VEGF (e.g., sorafenib [18,19] and cediranib [13]), EGFR (e.g., cetuximab [12], erlotinib [20] and panitumumab [21]), MET proto-oncogene (e.g., cabozantinib) [22], panErbB family (e.g., afatinib) [23], MEK (e.g., selumetinib) [24], VEGFR2, EGFR/HEGFR1 and RET kinase (e.g., vandetanib) [25]. However, these study results have thus far been discouraging, both in first- and second-line treatments for advanced BTC. These negative results might be due to heterogeneity in patient populations; for example, these trials recruited patients with BTCs encompassing iCCA, eCCA or gallbladder cancer, and most of the trials recruited patients who were not molecularly selected.
Therapies targeting the immune system have vastly improved the prognosis of patients with various hematologic and solid tumors. Among these are the immune checkpoint inhibitors, which include pembrolizumab, an anti-PD1 antibody approved for the tissue agnostic treatment of solid tumors harboring microsatellite instability-high (MSI-H) or mismatch repair deficiency (dMMR) that have progressed following prior treatment and who have no satisfactory alternative treatment options. The Phase II KEYNOTE-158 study evaluating pembrolizumab in previously treated advanced noncolorectal MSI-H/dMMR cancer showed an objective response rate (ORR) of 40.9%, median PFS of 4.2 months and median OS of 24.3 months in the CCA cohort of 22 patients [26]. However, incidences of dMMR and/or MSI-H are reported to be low in CCA (MSI-H, ∼1–2% [27,28]; dMMR, ∼3% [29]), and the success of immunotherapy as monotherapy in CCA has thus far been limited in patients with anatomically and molecularly unselected advanced BTC [30]. Combinatorial immunotherapeutic approaches with chemotherapy, local ablative therapy, molecularly targeted therapy and other agents are being explored [31].
In recent years, molecular-profiling techniques, including whole exome and next-generation sequencing (NGS) of tumor tissue specimens, have identified a wide range of genetic alterations in BTC [32–34]. Notably, these studies have found that different anatomic and clinical subtypes of CCA exhibit different genomic profiles, suggesting different underlying mechanisms driving oncogenesis. There may also be differences in outcomes based on anatomical subtype. A post hoc analysis of the ABC-01, -02 and -03 studies showed a longer OS in patients with iCCA or liver-only disease versus those with other BTCs after treatment with gemcitabine plus cisplatin combinations [35].
Most frequent gene alterations in tumor specimens from patients with CCA include those in TP53, KRAS, CDKN2A/B, ERBB2, BRAF, BAP1, PI3KCA, ARID1A, IDH1/2, FGFR1–3, PBRM1, SMAD4 and MCL1 (Table 1) [32–34]. Among these alterations, those in ARID1A, CDKN2A/B, TP53, IDH1, BAP1 and FGFR2 are more commonly found in iCCA compared with eCCA; notably, alterations in IDH1, BAP1 and FGFR2 occur almost exclusively in iCCA [32–34]. BAP1 was the most frequent concurrent mutation with FGFR2 (21.6%), followed by CDKN2A/B (18.9%) and TP53 (17.6%) [36]. In addition, there was a tendency toward mutual exclusivity between FGFR2 fusions and KRAS, IDH1 or BRAF mutations [34,36].
Table 1. . Genes that are commonly altered in cholangiocarcinoma.
| Gene | Prevalence in iCCA | Prevalence in eCCA | Prevalence in iCCA relative to eCCA |
|---|---|---|---|
| ARID1A | 20%† 18%‡ |
5%† 12%‡ |
iCCA > eCCA |
| BAP1 | 9.1%† 15%‡,§ 19%¶ |
10%† 6%‡,§ 0¶ |
iCCA > eCCA |
| IDH1/2 | 23.6%† 20%‡,# 29%¶ |
0† 0‡,# 5%¶ |
iCCA > eCCA |
| FGFR1–3 | 11%‡,†† 13%¶ |
0‡,†† 0¶ |
iCCA > eCCA |
| CDKN2A/B | 27%‡,‡‡ | 17%‡,‡‡ | iCCA > eCCA |
| MET | 2%‡,§§ | 0‡,§§ | iCCA > eCCA |
| EGFR | 1%‡,§§ | 0‡,§§ | iCCA > eCCA |
| TP53 | 29.1%† 27%‡,# 18%¶ |
45%† 40%‡,# 49%¶ |
eCCA > iCCA |
| KRAS | 23.6%† 22%‡,# 7%¶ |
40%† 42%‡,# 38%¶ |
eCCA > iCCA |
| ERBB2 | 1.8%† 3%‡,§§ |
20%† 11%‡,§§ |
eCCA > iCCA |
| FBXW7 | 5.5%† | 15%† | eCCA > iCCA |
| SMAD4 | 3.6%† 5%¶ |
25%† 30%¶ |
eCCA > iCCA |
| STK11 | <1%¶ | 11%¶ | eCCA > iCCA |
| PI3KCA | 5%‡,# | 7%‡,# | iCCA ≈ eCCA |
| BRAF | 5%‡,# | 3%‡,# | iCCA ≈ eCCA |
Next-generation sequencing-based testing on 55 patients with iCCA and 20 patients with eCCA [32].
Hybrid capture-based comprehensive genomic profiling on 412 patients with iCCA and 57 patients with eCCA [33].
Substitutions/truncations.
Next-generation sequencing-based testing on 158 patients with iCCA and 37 patients with eCCA [34].
Substitutions.
Fusions and amplifications.
Loss.
Amplification.
eCCA: Extrahepatic cholangiocarcinoma; iCCA: Intrahepatic cholangiocarcinoma.
Genomic data suggest that approximately 40% of patients with CCA have potentially actionable alterations [34,37], highlighting the importance of routine comprehensive genomic profiling of patients with CCA to inform the treatment strategy. Many targeted therapies are currently under clinical investigation for treating cancers harboring the genetic alterations listed above, including trials enrolling patients with CCA harboring specific genetic alterations (Table 2). Preliminary clinical data evaluating IDH and FGFR2 inhibitors have shown encouraging antitumor activity in molecularly selected patients with CCA harboring the respective genetic alterations [38].
Table 2. . Active Phase II or III clinical trials of agents targeting FGFR alterations in patients with cholangiocarcinoma†.
| Agent | Phase | LOT | Monotherapy | Combination therapy | Comparator | ClinicalTrials.gov identifier‡ |
|---|---|---|---|---|---|---|
| Pemigatinib | 2 3 |
2L+ 1L |
✓ ✓ |
None None |
None Gemcitabine + cisplatin |
NCT02924376 NCT03656536 |
| Derazantinib (ARQ-087) | 3 Expanded access |
2L+ 2L+ |
✓ ✓ |
None None |
None None |
NCT03230318 NCT04087876 |
| E7090 | 2 | 1L | ✓ | None | None | NCT04238715 |
| Erdafitinib | 2 | 2L+ | ✓ | None | None | NCT02699606 |
| Futibatinib (TAS-120) | 3 | 1L | ✓ | None | Gemcitabine + cisplatin | NCT04093362 |
| Infigratinib (BGJ398) | 2 3 |
2L+ 1L |
✓ ✓ |
None None |
None Gemcitabine + cisplatin |
NCT02150967 NCT03773302 |
Table is focused on genes that are more frequently altered in iCCA than eCCA, and includes trials that either focus exclusively on CCA or include a specified cohort of CCA patients. ClinicalTrials.gov searched on 20 March 2020.
Bold text for clinical trial identifier numbers represent CCA-focused clinical trials.
1L: First line; 2L+: Second line and later line; CCA: Cholangiocarcinoma; eCCA: Extrahepatic cholangiocarcinoma; iCCA: Intrahepatic cholangiocarcinoma; LOT: Line of therapy.
The FGF/FGFR pathway is involved in a number of cellular processes that underlie malignancies including cell proliferation, differentiation, migration, angiogenesis and survival [39]. Genomic alterations in FGF/FGFR have been implicated as driver events in multiple cancer types (e.g., FGFR1 amplification in breast, lung, head and neck and esophageal cancers; FGFR2 amplification in breast and gastric cancers – FGFR2 fusions or rearrangements in CCA occurring almost exclusively in iCCA, and in 13–20% of patients [34,36,40]; FGFR3 fusions and mutations in bladder cancer; and FGFR4 amplification in colorectal cancer) [39].
Although the natural history of patients with FGFR2 alterations remains to be determined, a retrospective study of 377 patients with CCA reported that patients with FGFR aberrations were more likely to present with earlier stage disease, had significantly better OS regardless of treatment compared with those without FGFR aberrations, and had significantly better outcomes with FGFR-targeted therapy compared with standard treatment [36].
Recently, pemigatinib (INCB054828), a selective FGFR1–3 inhibitor, was approved by the US FDA for the treatment of adults with previously treated, unresectable locally advanced or metastatic CCA with an FGFR2 fusion or other rearrangement as detected by an FDA-approved test; this is the first targeted therapy to receive regulatory approval for the treatment of CCA. There are several other FGFR-selective inhibitors in clinical development as targeted therapies for solid tumor and hematologic malignancies harboring FGFR alterations including Debio 1347 (FGFR1–3 inhibitor) [41], futibatinib (TAS-120; irreversible FGFR1–4 inhibitor) [42], derazantinib (ARQ 087; pan-FGFR inhibitor) [43] and infigratinib (BGJ398; FGFR1–3 inhibitor) [44,45].
In addition to pemigatinib, derazantinib, infigratinib and futibatinib have also shown promising antitumor efficacy in patients with CCA with FGFR2 alterations (Table 3). A Phase I/II study of derazantinib in 29 patients with FGFR2-fusion positive, advanced, inoperable iCCA regardless of line of therapy showed an ORR of 21%, disease control rate (DCR) of 83%, and median PFS of 5.7 months [43]. A post hoc analysis of the same study showed an ORR of 20% in the first- or second-line group of 15 patients (DCR, 80%), and 21% in the post second-line group of 14 patients (DCR, 86%) [43]. In another post hoc analysis based on data from patients with iCCA expressing FGFR mutations/amplifications (n = 6) or no FGFR aberration (n = 9) as compared with previously reported data of patients with iCCA (n = 29) expressing FGFR2 fusions, derazantinib showed similar antitumor efficacy across the groups of patients with different FGFR2 aberrations [46]. A larger Phase II registration trial of derazantinib in previously treated patients with FGFR2 gene fusion-, mutation- or amplification-positive inoperable or advanced iCCA is ongoing (FIDES-01; NCT03230318). A Phase II study of infigratinib in 71 patients with advanced CCA-harboring FGFR2 fusions, showed an ORR of 31.0% (confirmed ORR, 26.9%), DCR of 83.6% and a manageable safety profile [44]. An interim analysis of data from a Phase II study of futibatinib reported preliminary efficacy (ORR, 34.3%; DCR, 76.1%) and tolerability in 67 patients with locally advanced or metastatic unresectable iCCA with FGFR2 fusions or other rearrangements [47]. Based on the promising Phase II results, Phase III studies for both infigratinib (PROOF trial; NCT03773302) and futibatinib (FOENIX-CCA3; NCT04093362) are currently recruiting or planned in the first-line setting versus gemcitabine plus cisplatin in patients with advanced CCA and FGFR2 rearrangements or fusions.
Table 3. . Efficacy data from Phase II studies of derazantinib, infigratinib and pemigatinib in cholangiocarcinoma with FGFR2 alterations.
| Parameter | Pemigatinib† (n = 107; FGFR2 fusions/rearrangements) |
Derazantinib‡ (n = 29; FGFR2 fusions) |
Infigratinib§ (n = 71; FGFR2 fusions) |
|---|---|---|---|
| Median follow-up, months (range) | 15.4 (7.0–24.7) | 6.7 (2.9–19.4) | 8.4 (NR) |
| Objective response rate, % (95% CI) | 35.5 (26.5–45.4) | 20.7 (NR) | 31 (20.5–43.1)¶ |
| Disease control rate, % (95% CI) | 82 (74–89) | 82.8 (NR) | 83.6 (72.5–91.5) |
| Median duration of response, months (95% CI) | 7.5 (5.7–14.5) | 4.6 (2.3–8.9) | 5.4 (3.7–7.4) |
| Median progression-free survival, months (95% CI) | 6.93 (6.2–9.6) | 5.7 (4.0–9.2) | 6.8 (5.3–7.6) |
| Median overall survival, months (95% CI) | 21.1 (14.8–not estimable)# | Not reached; median follow-up, 20 months |
12.5 (9.9–16.6) |
A Phase II study of pemigatinib in patients with previously treated locally advanced or metastatic cholangiocarcinoma (NCT02924376) [50].
A Phase I/II study of derazantinib in patients with advanced or inoperable FGFR2-gene positive intrahepatic cholangiocarcinoma (NCT01752920) [43].
A Phase II study of infigratinib in previously treated patients with advanced cholangiocarcinoma harboring FGFR2 fusions (NCT02150967) [44].
Unconfirmed objective response rate. Confirmed objective response rate was 26.9% (95% CI, 16.8–39.1).
Not mature at data cutoff.
NR: Not reported.
Here, we review data from a Phase II study of pemigatinib in patients with locally advanced or metastatic CCA that had progressed following prior treatment, and describe the design of FIGHT-302 (ClinicalTrials.gov: NCT03656536), a global, Phase III, randomized, active-controlled trial currently recruiting patients to evaluate pemigatinib compared with gemcitabine plus cisplatin chemotherapy as first-line therapy for unresectable and/or metastatic CCA with FGFR2 rearrangements.
Background & rationale
Pemigatinib is a selective, potent, orally active inhibitor of FGFR1, 2 and 3 [48]. Data from a Phase I/II dose-finding study (FIGHT-101; NCT02393248) demonstrated preliminary efficacy and manageable toxicities after treatment with pemigatinib in a subpopulation of patients with advanced CCA and FGFR alterations [49]. These promising results prompted the Phase II FIGHT-202 study, which assessed the efficacy and safety of pemigatinib in patients with locally advanced or metastatic CCA, who had progressed after at least one prior systemic therapy (NCT02924376) [50]. A total of 146 patients with documented FGF/FGFR status were enrolled into cohorts A (FGFR2 fusions or rearrangements), B (other FGF/FGFR alterations) or C (no FGF/FGFR alterations). In the 107 patients in cohort A with FGFR2 fusions or rearrangements, pemigatinib yielded an ORR of 35.5% (95% CI, 26.5–45.4), with three complete responses; median duration of response (DOR) was 7.5 (95% CI, 5.7–14.5) months, DCR was 82.0% (95% CI, 74–89), median PFS and median OS were 6.9 (95% CI, 6.2–9.6) and 21.1 (14.8–not estimable) months, respectively; OS was not mature at the time of primary data cutoff. No responses occurred among patients in cohorts B or C. Overall, the most common all-cause/grade adverse events (AEs) (>45%) were hyperphosphatemia (60%), alopecia (49%) and diarrhea (47%). All hyperphosphatemia events were of low severity (grade 1/2) and were managed with a low-phosphate diet, phosphate-lowering therapy or if needed, dose modifications. Hypophosphatemia was the most common all-cause grade ≥3 AE (12%), and none of these events were clinically significant or deemed serious. Other clinically notable AEs such as nail toxicities and serous retinal detachment were mostly grade 1 or 2 and with no clinical sequelae. On the basis of these positive results, pemigatinib received approval by the FDA for the treatment of adults with previously treated, unresectable locally advanced or metastatic CCA with an FGFR2 fusion or other rearrangement as detected by an FDA-approved test.
Based on the antitumor activity and manageable toxicities associated with pemigatinib observed in previously treated patients with CCA in FIGHT-101 [49] and FIGHT-202 [50], it is hypothesized in FIGHT-302 that pemigatinib would also provide benefit as a first-line treatment for patients with unresectable or metastatic CCA selected on the basis of FGFR2 fusions or rearrangements.
FIGHT-302
FIGHT-302 is a global, Phase III, open-label, randomized, active-controlled, multicenter study to evaluate the efficacy and safety of pemigatinib versus gemcitabine plus cisplatin chemotherapy in the first-line treatment of patients with unresectable and/or metastatic CCA with FGFR2 rearrangements (NCT03656536).
Eligibility criteria
Key inclusion and exclusion criteria are described in Table 4. In brief, men and women aged 18 years or older (a legally minor participant from Japan needs written parental consent) are eligible if they have histologically or cytologically confirmed CCA that is previously untreated and considered unresectable and/or metastatic (stage IV per the AJCC Cancer Staging Manual [51]), radiographically measurable or evaluable disease by computed tomography or MRI per Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1), with a documented FGFR2 rearrangement and Eastern Cooperative Oncology Group performance status ≤1.
Table 4. . Inclusion and exclusion criteria for FIGHT-302.
| Inclusion criteria |
|---|
| • Signed informed consent form • Men and women aged ≥18 years (a legally minor participant from Japan needs written parental consent) • Histologically or cytologically confirmed CCA considered unresectable and/or metastatic (stage IV per the AJCC Cancer Staging Manual); no prior treatment for advanced disease • Documented FGFR2 rearrangement • ECOG performance status 0–1 • Radiographically measurable or evaluable disease by CT or MRI per RECIST v1.1 |
| Exclusion criteria |
| • Previous systemic therapy (excluding adjuvant/neoadjuvant treatment completed ≥6 months before enrollment) • Child–Pugh score for cirrhosis mortality, B and C • Known evidence of clinically significant corneal/retinal disorder confirmed by ophthalmologic examination • History of calcium and phosphate homeostasis disorder or systemic mineral imbalance with ectopic soft tissue calcification • Presence of known untreated CNS metastases or history of uncontrolled seizures • Radiation therapy administered within 4 weeks of enrollment/randomization/first dose of study treatment • Toxicities related to prior therapies must be CTCAE v5.0 ≤ grade 1 at the time of screening • Concurrent anticancer therapy, other than the therapies being tested in this study • Must not be a candidate for potentially curative surgery • Known additional malignancy that is progressing or requires active treatment (except basal cell or squamous cell carcinoma of the skin, or in situ cervical cancer that has undergone potentially curative therapy) • Laboratory values at screening outside the protocol-defined range • Gastrointestinal conditions/disorders that may raise gastric and/or small intestinal pH that could interfere with absorption, metabolism or excretion of pemigatinib • Clinically significant or uncontrolled cardiac disease • History or presence of an abnormal ECG, which, in the investigator’s opinion, is clinically meaningful • Chronic or current active infectious disease requiring systemic antibiotic, antifungal or antiviral treatment within 2 weeks prior to enrollment • Use of any potent CYP3A4 inhibitors or inducers or moderate CYP3A4 inducers within 14 days or five half-lives (whichever is longer) before the first dose of study treatment Note: Moderate CYP3A4 inhibitors are not prohibited • Known hypersensitivity or severe reaction to pemigatinib, gemcitabine, cisplatin or their excipients • Inadequate recovery from toxicity and/or complications from a major surgery before starting therapy |
AJCC: American Joint Committee on Cancer; CCA: Cholangiocarcinoma; CT: Computed tomography; CTCAE: Common terminology criteria for adverse events; ECOG: Eastern Cooperative Oncology Group; RECIST: Response evaluation criteria in solid tumors.
Before screening for other eligibility criteria, patients will be prescreened for FGFR2 rearrangement by central assessment using FoundationOne NGS (FoundationOne®, Foundation Medicine Inc., MA, USA). Patients with a Clinical Laboratory Improvement Amendments–certified local assessment will also be included. Patients can also be considered for enrollment if they had an existing FoundationOne report. FGFR2 status documented locally will be confirmed retrospectively by central assessment with FoundationOne; however, this will not impact on the timing of randomization or initiation of treatment.
In contrast to classical methods like fluorescence in situ hybridization, in which fusion status is determined by the uncoupling of collocated probes, NGS allows for precise identification of the underlying genomic breakpoint. Based on the level of detail provided by DNA-based NGS assays, the FGFR2 fusion partners and their resulting reading frame can be determined. With this granularity, the FoundationOne assay delineates two types of FGFR2 alteration classifications: first, FGFR2 rearrangements, which are typically classified as fusions if the genomic breakpoint is within the intron 17 or exon 18 hotspot and the fusion gene partner is known or is predicted to be in-frame with FGFR2 (i.e., both genes are translated); second, other FGFR2 rearrangements including those with genomic breakpoint within the FGFR2 intron 17 or exon 18 hotspot and with either a novel partner gene predicted to be out-of-strand or out-of-frame with FGFR2, or no identifiable partner gene (designated as intron 17 rearrangement or partner N/A).
Study design
432 patients (target enrollment) will be randomized in a 1:1 ratio to receive pemigatinib at a starting dose of 13.5 mg once daily administered continuously on a 3-week cycle (Group A) or gemcitabine (1000 mg/m2) plus cisplatin (25 mg/m2) administered intravenously on days 1 and 8 of a 3-week cycle for up to eight cycles (Group B; Figure 1). Patients will be stratified by geographic region (Western [North America and European Union] vs Asia Pacific vs rest of the world) and tumor burden (locally advanced vs distant metastases).
Figure 1. . FIGHT-302 study schema.
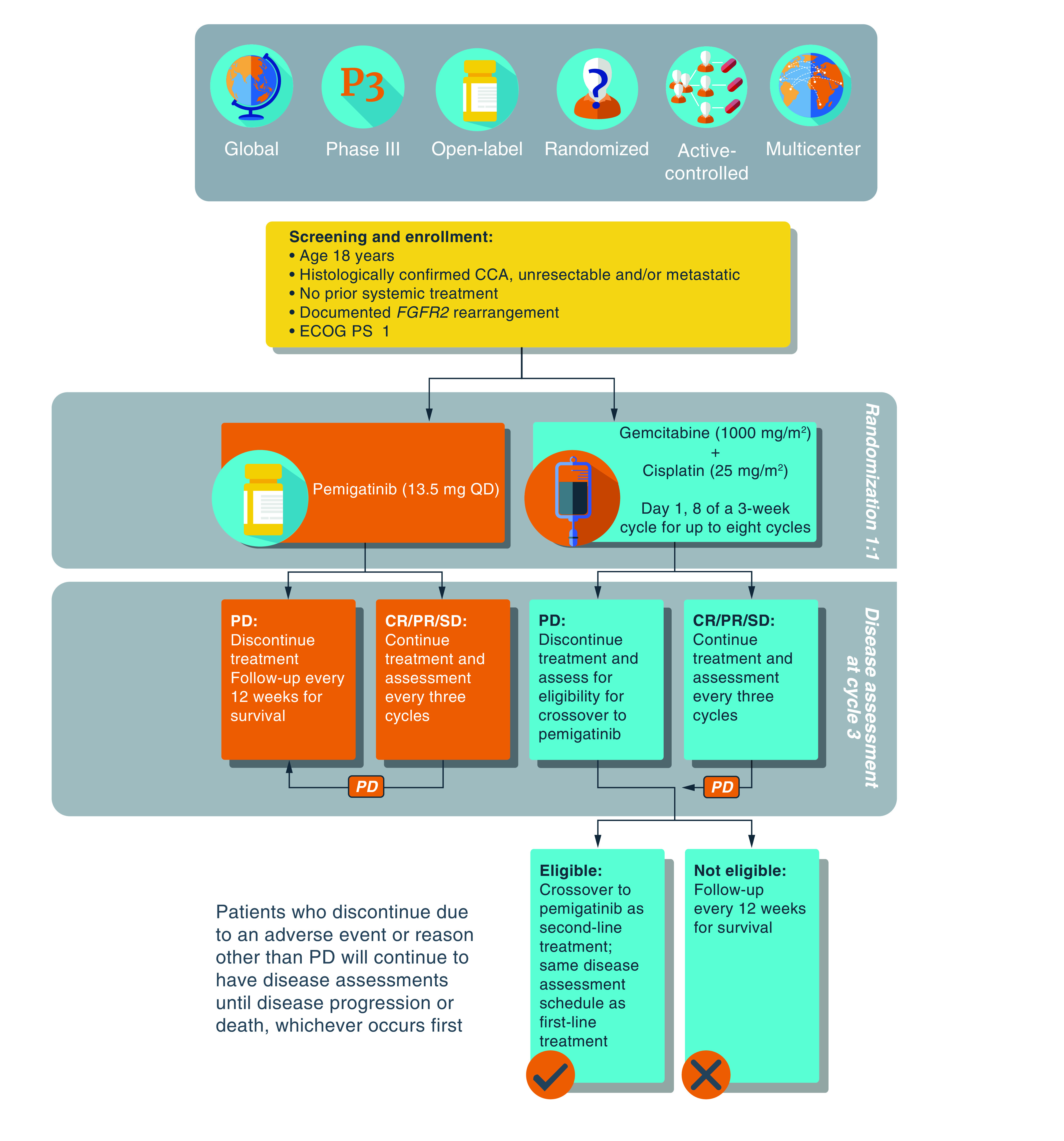
CCA: Cholangiocarcinoma; CR: Complete response; ECOG PS: Eastern Cooperative Oncology Group performance status; PD: Progressive disease; PR: Partial response; QD: Once daily; SD: Stable disease.
Treatment will continue in 3-week cycles until disease progression per RECIST v1.1, as assessed by an independent central reviewer (ICR), or if unacceptable toxicity is reported. Patients receiving gemcitabine plus cisplatin with centrally confirmed disease progression may be considered for crossover to pemigatinib.
Hyperphosphatemia (defined as serum phosphate >5.5 mg/dl) is an expected on-target pharmacologic effect associated with FGFR inhibition and is associated with FGFR inhibitors, including pemigatinib (data not shown). In prior studies, some patients who received pemigatinib did not experience hyperphosphatemia and were found to have lower serum concentrations of pemigatinib, as also observed previously in patients receiving infigratinib [52] or erdafitinib [53]. Therefore, uptitration of pemigatinib from 13.5 to 18 mg at cycle 2 will be implemented for patients who do not experience hyperphosphatemia at any time during cycle 1 and do not have ongoing grade ≥2 treatment-related AEs. A similar approach was used by a Phase II trial of erdafitinib in patients with advanced urothelial carcinoma (NCT02365597). Hyperphosphatemia will be managed by dietary modifications, phosphate binders and diuretics or dose reduction or interruption if needed.
The primary endpoint of the study is PFS, defined as the time from date of randomization until date of disease progression (per RECIST v1.1 and assessed by ICR) or death, whichever occurs first. Secondary endpoints are ORR, OS, DOR and DCR per RECIST v1.1 by ICR, safety and tolerability and health-related quality of life assessed using the European Organization for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30), the EuroQol 5D (EQ-5D) questionnaire and the EORTC QLQ-BIL21 questionnaire. Exploratory endpoints include association of specific FGFR2 rearrangements with PFS, OS and ORR, and additional biomarker assessments.
Other study procedures
Radiographic tumor assessments (computed tomography/MRI) are performed at baseline, every 9 weeks (every three cycles), starting from cycle 3, and until progression is noted by the central reviewer. After the end of treatment if progression has been noted, patient enters the safety period and then survival follow-up. AEs are graded and recorded per the National Cancer Institute Common Terminology Criteria for Adverse Events version 5.0. Because ocular toxicities can occur with FGFR inhibitors, comprehensive ophthalmic examinations will be conducted at screening, every three cycles, or as clinically indicated, during study. Quality-of-life assessments are conducted on day 1 of each cycle.
Statistical methods
For the primary end point, assuming a 5% lost to follow-up rate, a total of 432 patients are needed to observe 338 PFS events in two combined treatment groups to ensure 90% power to test the null hypothesis (PFS, HR = 1) versus the alternative hypothesis (PFS, HR = 0.7), assuming analysis by a one-sided log-rank test at the overall 0.025 level of significance.
Efficacy will be assessed in the intent-to-treat (ITT) population (all randomized patients in the study) and analyzed by randomized treatment group. Safety will be assessed in the safety population (all randomized patients who received at least one dose of study drug [pemigatinib, gemcitabine or cisplatin]) and will be analyzed by treatment received.
The primary analysis of PFS (based on the ITT population) and sensitivity analysis of PFS (based on the ITT population who are compliant with the study protocol [per-protocol population]) will compare PFS difference between treatment groups using log-rank test stratified by geographic region and tumor burden. ORR between treatment groups will be compared using Cochran–Mantel–Haenszel test, stratified by geographic region and tumor burden. For PFS, OS and DOR, the HR and its 95% CI will be estimated based on the Cox regression model with geographic region and tumor burden as covariates using Ephron’s method accounting for ties. The rank-preserving structural failure time model will be applied as a sensitivity analysis for OS to minimize estimation bias in the presence of crossover.
Ethical considerations
The protocol has been approved by each participating institutional ethics review board. The study will be performed in accordance with the ethical principles of the Declaration of Helsinki and conducted in adherence to the study protocol, applicable Good Clinical Practices, and applicable laws and country-specific regulations in which the study is being conducted. Informed consent will be obtained from all patients before any study-related procedures are conducted.
Conclusion
Most patients with CCA are diagnosed with advanced disease when potentially curative surgery is not an option [1]. OS in patients receiving first-line SoC cisplatin plus gemcitabine is poor (median OS, 11.7 months) [11], and trials with other chemotherapies alone or in combination with other agents have not demonstrated improved outcomes. Therefore, there is an unmet need for better first-line treatment options for patients with advanced CCA. Recent advances in molecular profiling techniques have provided greater insight into the mutational landscape of CCAs, identifying potentially targetable genetic alterations [32–34]. The high rate of actionable alterations found in CCA underscores the need for biology-driven strategies and the value of genetic testing to inform treatment options. The discovery of FGFR2 gene fusions and rearrangements in CCA led to studies investigating the efficacy and safety of FGFR inhibitors in patients with advanced CCA; these studies have demonstrated promising clinical benefit of FGFR inhibitors in the second-line setting in these patients [41–44,50]. Based on the promising efficacy and manageable safety of pemigatinib observed in the second-line setting [50], FIGHT-302 will assess pemigatinib versus gemcitabine plus cisplatin chemotherapy as first-line therapy in patients with unresectable or metastatic CCA with FGFR2 rearrangements, with the aim of potentially providing these patients with a new first-line treatment option.
Executive summary.
Background
Cholangiocarcinomas (CCAs) are heterogeneous tumors classified anatomically as extrahepatic (which includes perihilar and distal), and intrahepatic. Most patients are diagnosed with advanced disease.
The standard-of-care first-line treatment for advanced CCA is cisplatin plus gemcitabine.
In patients with advanced biliary tract cancer, including CCA, cisplatin plus gemcitabine demonstrated an objective response rate (ORR) of 26%, median overall survival (OS) of 11.7 months and median progression-free survival (PFS) of 8.0 months.
There is no standard of care for patients with advanced biliary cancer who have relapsed or are refractory to first-line chemotherapy, and current second-line chemotherapy regimens remain limited in their treatment efficacy.
With emerging technologies including next-generation sequencing, several potentially targetable gene alterations have been identified in CCA, including alterations in the FGFR gene.
FGFR2 rearrangements may result in aberrant FGFR signaling, leading to malignancy by promoting cell proliferation, migration, survival, invasion and angiogenesis.
Pemigatinib
Pemigatinib (INCB054828) is a selective, potent, oral FGFR1–3 inhibitor.
Data from the Phase II FIGHT-202 trial (NCT02924376) demonstrated that pemigatinib is associated with antitumor activity and manageable toxicities in patients with previously treated locally advanced or metastatic CCA with FGFR2 fusions or rearrangements (ORR, 35.5%; DOR, 7.5 months; median PFS, 6.9 months).
Pemigatinib has received approval by the US FDA for the treatment of adults with previously treated, unresectable locally advanced or metastatic CCA with an FGFR2 fusion or other rearrangement as detected by an FDA-approved test.
Based on the efficacy demonstrated in previously treated CCA patients harboring FGFR2 fusions/rearrangements, it is hypothesized that pemigatinib would also provide benefit as a first-line treatment for patients with locally advanced or metastatic CCA selected on the basis of FGFR2 rearrangements.
FIGHT-302
FIGHT-302 is an ongoing open-label, randomized, active-controlled, multicenter, global, Phase III study comparing the efficacy and safety of first-line pemigatinib versus gemcitabine plus cisplatin in patients with unresectable/metastatic CCA with FGFR2 rearrangements (NCT03656536).
Eligible patients are adults with histologically confirmed, unresectable and/or metastatic CCA with documented FGFR2 rearrangements who have received no prior systemic treatments. Patients must have radiographically measurable/evaluable disease (magnetic resonance imaging/computed tomography) per RECIST v1.1 criteria, and Eastern Cooperation Oncology Group performance status of 0–1.
Patients will be randomized (1:1; stratified by geographic region and tumor burden) to receive pemigatinib at a starting dose of 13.5 mg once daily administered continuously on a 3-week cycle or gemcitabine (1000 mg/m2) plus cisplatin (25 mg/m2) administered intravenously on days 1 and 8 of a 3-week cycle for up to eight cycles.
Patients receiving gemcitabine plus cisplatin with centrally confirmed disease progression may be considered for crossover to pemigatinib.
The primary end point is PFS; secondary end points are ORR, OS, DOR, disease control rate, safety and quality of life.
A total of 432 patients are anticipated to be enrolled.
Supplementary Material
Footnotes
Financial & competing interests disclosure
TS Bekaii-Saab received research funding (to their institution) from Abgenomics, Amgen, Array Biopharma, Bayer, Bristol-Myers Squibb, Boston Biomedical, Celgene, Clovis, Genentech, Incyte, Ipsen, Lilly, Merck and Seattle Genetics; consulting fees (to their institution) from Array Biopharma, Bayer, Genentech, Incyte, Ipsen and Merck; and is an Independent Data Monitoring Committee (IDMC)/Data Safety Monitoring Board (DSMB) member at 1Globe, AstraZeneca, Exelixis, Lilly and PanCan. JW Valle received honoraria from Ipsen; is a consultant for and/or is on the advisory board of Agios, AstraZeneca, Debiopharm, Delcath Systems, Genoscience Pharma, Imaging Equipment Limited, Incyte, Ipsen, Keocyt, Merck, Mundipharma EDO, Novartis, NuCana, Pfizer, PCI Biotech, Pieris Pharmaceuticals, QED Therapeutics, Servier and Wren Laboratories; served on a speaker’s bureau for Imaging Equipment Limited, Ipsen, Novartis and NuCana; received travel grant from Celgene, NuCana and Pfizer. E Van Cutsem is a consultant for and/or is on the advisory board of Astellas, AstraZeneca, Bayer, Bristol-Myers Squibb, Celgene, Incyte, Lilly, Merck Sharp & Dohme, Merck KGaA, Novartis, Roche and Servier; received research grant/funding from Amgen, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Ipsen, Lilly, Merck Sharp & Dohme, Merck KGaA, Novartis, Roche and Servier. L Rimassa is a consultant for and/or is on the advisory board of Amgen, ArQule, Basilea, Baxter, Bayer, Celgene, Eisai, Exelixis, Hengrui, Incyte, Ipsen, Italfarmaco, Lilly, MSD, Roche, Sanofi and Sirtex Medical; received honoraria (lecture fees) from AbbVie, Amgen, AstraZeneca, Gilead, Ipsen, Lilly, Roche and Sanofi; received travel expenses from ArQule and Ipsen; institutional research grant/funding from Agios, ARMO BioSciences, AstraZeneca, BeiGene, Eisai, Exelixis, Incyte, Ipsen, Lilly and MSD. J Furuse received honoraria from and is a consultant for and/or is on the advisory board of AbbVie, Astellas Pharma, AstraZeneca, Bayer Yakuhin, Chugai Pharma, Daiichi Sankyo, EA Pharma, Eisai, Eli Lilly Japan, Fujifilm, Fujifilm Toyama Chemical, J-Pharma, Kyowa Hakko Kirin Otsuka, Merck Serono, Mochida Pharmaceutical, MSD, Nihon Servier, Nippon Kayaku, Nobelpharma, Novartis, Ono Pharmaceutical, Pfizer, Sanofi, Sawai Pharmaceutical, Shionogi, Shire, Sumitomo Dainippon, Taiho Pharmaceutical, Takara Bio, Takeda, Teijin Pharma and Yakult Honsha; received research funding from Astellas Pharma, AstraZeneca, Bayer Yakuhin, Chugai Pharma, Daiichi Sankyo, Eli Lilly Japan, Eisai, Kyowa Hakko Kirin, Mochida Pharmaceutical, MSD, NanoCarrier, Ono Pharmaceutical, Pfizer, Sanofi, Sumitomo Dainippon, Taiho Pharmaceutical, Takeda and Yakult Honsha. T Ioka is a consultant for and/or is on the advisory board of Incyte, Otsuka and Taiho Pharmaceutical; received research grant/funding from Astellas, AstraZeneca, Eisai, Incyte, Nihon Zouki, Otsuka, Servier, Shire, Taiho Pharmaceutical and Takara Bio. D Melisi received research funding from Celgene, Evotec, Incyte and Shire; is a consultant for Baxter, Eli Lilly, Evotec, Incyte and Shire. T Macarulla is on the advisory board of Amgen, AstraZeneca, Celgene, Eisai, Incyte, Sanofi and Servier; received research funding from AstraZeneca, BeiGene and Celgene. J Bridgewater is a consultant for Bayer, Bristol-Myers Squibb, Incyte, Merck Serono and Roche; received travel support from Bristol-Myers Squibb and Merck Sharpe Dohme. H Wasan is on the advisory board of Array BioPharma, Celgene, ERYTECH Pharma, Incyte, Pierre Fabre, Roche/Genentech/ FM AG, Servier, Shire and Sirtex Medical; is a consultant for NICE and OncoSil Medical; is an expert for Bayer (uncompensated); and received honoraria, serves on the speaker’s bureau for, and received travel expenses from BTG/Biocompatibles, Bristol-Myers Squibb and Merck KGaA; received research funding from Merck Serono, MSD, Pfizer and Sirtex. MJ Borad received research grant/funding from Adaptimmune, Agios, ARIAD, Basilea, Bioline, Boston Biomed, Celgene, Dicerna, EMD Merck Serono, Halozyme Therapeutics, Incyte, Isis Pharmaceuticals, MedImmune, Mirna Therapeutics, Novartis, Pieris, PUMA, QED Therapeutics, Redhill Biopharma, Senhwa Biosciences, SillaJen, Sun Biopharma, Taiho Pharmaceutical and Toray; received honoraria from ADC Therapeutics, Exelixis, G1 Therapeutics, Immunovative Therapies, Inspyr Therapeutics, Lynx Group and Western Oncolytics; is a shareholder/stockholder at AVEO, Intercept and OncBioMune Pharmaceuticals; received travel/accommodation support from AstraZeneca. GK Abou-Alfa received research funding from ActaBiologica, Agios, Array, AstraZeneca, Bayer, BeiGene, Bristol-Myers Squibb, Casi, Celgene, Exelixis, Genentech, Halozyme, Incyte, Mab Vax Therapeutics, Polaris Puma, QED Therapeutics and Roche; and is a consultant for Agios, AstraZeneca, Autem, Bayer, BeiGene, Berry Genomics, Bioline, Celgene, CytomX, Debiopharm, Eisai, Eli Lilly, Flatiron, Genentech, Genoscience, Gilead, Incyte, Ipsen, LAM, Loxo Oncology, Merck, MINA Pharmaceuticals, QED Therapeutics, Redhill Biopharma, Roche, Silenseed, SillaJen, Sobi, Targovax, Therabionics, twoXAR and Yiviva. P Jiang, CF Lihou, H Zhen, E Asatiani and L Féliz are employees and stockholders of Incyte Corporation. A Vogel is a consultant for and/or is on the advisory board of AstraZeneca, Bayer, Delcath, Incyte, Lilly, Medac, Roche and Shire; serves on the speaker’s bureau for and/or received fees for expert testimony from AstraZeneca, Bayer, Delcath, Incyte, Lilly, Medac, Roche and Shire; and received travel/accommodation expenses from AstraZeneca, Bayer, Delcath, Incyte, Lilly, Medac, Roche and Shire. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Medical writing assistance was provided by Sneha DSilva, MD, CMPP (Envision Pharma Group, PA, USA), and funded by Incyte Corporation.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Blechacz B. Cholangiocarcinoma: current knowledge and new developments. Gut Liver 11(1), 13–26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huguet JM, Lobo M, Labrador JM et al. Diagnostic-therapeutic management of bile duct cancer. World J. Clin. Cases 7(14), 1732–1752 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Florio AA, Ferlay J, Znaor A et al. Global trends in intrahepatic and extrahepatic cholangiocarcinoma incidence from 1993 to 2012. Cancer 126(11), 2666–2678 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mukkamalla SKR, Naseri HM, Kim BM, Katz SC, Armenio VA. Trends in incidence and factors affecting survival of patients with cholangiocarcinoma in the United States. J. Natl Compr. Canc. Netw. 16(4), 370–376 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Bertuccio P, Malvezzi M, Carioli G et al. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma. J. Hepatol. 71(1), 104–114 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Meng ZW, Pan W, Hong HJ, Chen JZ, Chen YL. Macroscopic types of intrahepatic cholangiocarcinoma and the eighth edition of AJCC/UICC TNM staging system. Oncotarget 8(60), 101165–101174 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waseem D, Tushar P. Intrahepatic, perihilar and distal cholangiocarcinoma: management and outcomes. Ann. Hepatol. 16(1), 133–139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banales JM, Cardinale V, Carpino G et al. Expert consensus document: cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 13(5), 261–280 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Kitano Y, Yamashita YI, Nakagawa S et al. Effectiveness of surgery for recurrent cholangiocarcinoma: a single center experience and brief literature review. Am. J. Surg. 219(1), 175–180 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Cillo U, Fondevila C, Donadon M et al. Surgery for cholangiocarcinoma. Liver Int. 39(Suppl. 1), 143–155 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valle J, Wasan H, Palmer DH et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 362(14), 1273–1281 (2010). [DOI] [PubMed] [Google Scholar]; •• Pivotal Phase III study that establishes cisplatin and gemcitabine combination therapy as standard of care for first-line treatment of patients with advanced biliary cancer.
- 12.Malka D, Cervera P, Foulon S et al. Gemcitabine and oxaliplatin with or without cetuximab in advanced biliary-tract cancer (BINGO): a randomised, open-label, non-comparative Phase II trial. Lancet Oncol. 15(8), 819–828 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valle JW, Wasan H, Lopes A et al. Cediranib or placebo in combination with cisplatin and gemcitabine chemotherapy for patients with advanced biliary tract cancer (ABC-03): a randomised Phase II trial. Lancet Oncol. 16(8), 967–978 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phelip JM, Edeline J, Blanc JF et al. Modified FOLFIRINOX versus CisGem first-line chemotherapy for locally advanced non resectable or metastatic biliary tract cancer (AMEBICA)-PRODIGE 38: study protocol for a randomized controlled multicenter Phase II/III study. Dig. Liver Dis. 51(2), 318–320 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Lamarca A, Palmer DH, Wasan HS et al. ABC-06 | A randomised Phase III, multi-centre, open-label study of active symptom control (ASC) alone or ASC with oxaliplatin/5-FU chemotherapy (ASC+mFOLFOX) for patients (pts) with locally advanced/metastatic biliary tract cancers (ABC) previously-treated with cisplatin/gemcitabine (CisGem) chemotherapy. J. Clin. Oncol. 37(Suppl. 15), 4003 (2019). [Google Scholar]
- 16.Edeline J, Touchefeu Y, Guiu B et al. Radioembolization plus chemotherapy for first-line treatment of locally advanced intrahepatic cholangiocarcinoma: a Phase II clinical trial. JAMA Oncol. 6(1), 51–59 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cercek A, Boerner T, Tan BR et al. Assessment of hepatic arterial infusion of floxuridine in combination with systemic gemcitabine and oxaliplatin in patients with unresectable intrahepatic cholangiocarcinoma: a Phase II clinical trial. JAMA Oncol. 6(1), 60–67 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El-Khoueiry AB, Rankin CJ, Ben-Josef E et al. SWOG 0514: a Phase II study of sorafenib in patients with unresectable or metastatic gallbladder carcinoma and cholangiocarcinoma. Invest. New Drugs 30(4), 1646–1651 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moehler M, Maderer A, Schimanski C et al. Gemcitabine plus sorafenib versus gemcitabine alone in advanced biliary tract cancer: a double-blind placebo-controlled multicentre Phase II AIO study with biomarker and serum programme. Eur. J. Cancer 50(18), 3125–3135 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Lee J, Park SH, Chang HM et al. Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: a multicentre, open-label, randomised, Phase III study. Lancet Oncol. 13(2), 181–188 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Vogel A, Kasper S, Bitzer M et al. PICCA study: panitumumab in combination with cisplatin/gemcitabine chemotherapy in KRAS wild-type patients with biliary cancer – a randomised biomarker-driven clinical Phase II AIO study. Eur. J. Cancer 92, 11–19 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Goyal L, Zheng H, Yurgelun MB et al. A Phase II and biomarker study of cabozantinib in patients with advanced cholangiocarcinoma. Cancer 123(11), 1979–1988 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moehler M, Maderer A, Ehrlich A et al. Safety and efficacy of afatinib as add-on to standard therapy of gemcitabine/cisplatin in chemotherapy-naive patients with advanced biliary tract cancer: an open-label, Phase I trial with an extensive biomarker program. BMC Cancer 19(1), 55 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bridgewater J, Lopes A, Beare S et al. A Phase Ib study of selumetinib in combination with cisplatin and gemcitabine in advanced or metastatic biliary tract cancer: the ABC-04 study. BMC Cancer 16, 153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santoro A, Gebbia V, Pressiani T et al. A randomized, multicenter, Phase II study of vandetanib monotherapy versus vandetanib in combination with gemcitabine versus gemcitabine plus placebo in subjects with advanced biliary tract cancer: the VanGogh study. Ann. Oncol. 26(3), 542–547 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Marabelle A, Le DT, Ascierto PA et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the Phase II KEYNOTE-158 study. J. Clin. Oncol. 38(1), 1–10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonneville R, Krook MA, Kautto EA et al. Landscape of microsatellite instability across 39 cancer types. JCO Precis. Oncol. 2017, (2017) (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Javle MM, Murugesan K, Shroff RT et al. Profiling of 3,634 cholangiocarcinomas (CCA) to identify genomic alterations (GA), tumor mutational burden (TMB), and genomic loss of heterozygosity (gLOH). J. Clin. Oncol. 37(Suppl. 15), 4087 (2019). [Google Scholar]
- 29.Le DT, Durham JN, Smith KN et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357(6349), 409–413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ueno M, Chung HC, Nagrial A et al. Pembrolizumab for advanced biliary adenocarcinoma: results from the multicohort, Phase II KEYNOTE-158 study [abstract 625PD]. Ann. Oncol. 29(Suppl. 8), viii210 (2018). [Google Scholar]
- 31.Loeuillard E, Conboy CB, Gores GJ, Rizvi S. Immunobiology of cholangiocarcinoma. JHEP Reports 1(4), 297–311 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Churi CR, Shroff R, Wang Y et al. Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PLoS ONE 9(12), e115383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Javle M, Bekaii-Saab T, Jain A et al. Biliary cancer: utility of next-generation sequencing for clinical management. Cancer 122(24), 3838–3847 (2016). [DOI] [PubMed] [Google Scholar]; •• Large, multicenter series of intrahepatic cholangiocarcinoma, extrahepatic cholangiocarcinoma and gallbladder cancer cases that noted a large number of actionable mutations.
- 34.Lowery MA, Ptashkin R, Jordan E et al. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: potential targets for intervention. Clin. Cancer Res. 24(17), 4154–4161 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Demonstrates the therapeutic and prognostic inferences of comprehensive genomic analysis of patients with advanced cholangiocarcinoma.
- 35.Lamarca A, Ross P, Wasan HS et al. Advanced intrahepatic cholangiocarcinoma: post hoc analysis of the ABC-01, -02 and -03 clinical trials. J. Natl Cancer Inst. 112(2), 200–210 (2020). [DOI] [PubMed] [Google Scholar]; •• A post hoc analysis of patient data collected from the prospective ABC-01, -02 and -03 clinical trials, providing reference survival data in order to inform the design of such studies.
- 36.Jain A, Borad MJ, Kelley RK et al. Cholangiocarcinoma with FGFR genetic aberrations: a unique clinical phenotype. JCO Precis. Oncol. 2, 1–12 (2018) (Epub ahead of print). [DOI] [PubMed] [Google Scholar]; •• Clinical evidence showing that patients with FGFR genetic aberrations had significantly better survival, regardless of treatment, compared with those without FGFR genetic aberrations, and had significantly better outcomes with FGFR-targeted therapy compared with standard treatment.
- 37.Silverman IM, Murugesan K, Lihou CF et al. Comprehensive genomic profiling in FIGHT-202 reveals the landscape of actionable alterations in advanced cholangiocarcinoma. J. Clin. Oncol. 37(Suppl. 15), 4080 (2019). [Google Scholar]
- 38.Tella SH, Kommalapati A, Borad MJ, Mahipal A. Second-line therapies in advanced biliary tract cancers. Lancet Oncol. 21(1), e29–e41 (2020). [DOI] [PubMed] [Google Scholar]; •• A comprehensive review of second-line therapies including all FGFR-targeted drugs.
- 39.Babina IS, Turner NC. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 17(5), 318–332 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Farshidfar F, Zheng S, Gingras MC et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 18(11), 2780–2794 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ng MCH, Goyal L, Bang YJ et al. Debio 1347 in patients with cholangiocarcinoma harboring an FGFR gene alteration: preliminary results [abstract AB065. P-36]. Hepatobiliary Surg. Nutr. 8(Suppl. 1), AB065 (2019). [Google Scholar]
- 42.Goyal L, Arkenau H-T, Tran B et al. Early clinical efficacy of TAS-120, a covalently bound FGFR inhibitor, in patients with cholangiocarcinoma [abstract O-020]. Ann. Oncol. 28(Suppl. 3), 145 (2017). [Google Scholar]
- 43.Droz Dit Busset M, El-Rayes BF, Harris WP et al. Derazantinib (DZB) provides antitumor efficacy regardless of line of therapy in patients (pts) with FGFR2-fusion positive advanced intrahepatic cholangiocarcinoma (iCCA). J. Clin. Oncol. 37(Suppl. 15), e15607 (2019). [Google Scholar]
- 44.Javle M, Kelley RK, Roychowdhury S et al. AB051. P-19. A Phase II study of infigratinib (BGJ398) in previously-treated advanced cholangiocarcinoma containing FGFR2 fusions. Hepatobiliary Surg. Nutr. 8(Suppl. 1), AB051 (2019). [Google Scholar]
- 45.Javle M, Lowery M, Shroff RT et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J. Clin. Oncol. 36(3), 276–282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Proof-of-concept study of an FGFR-targeted therapy for cholangiocarcinoma.
- 46.Droz Dit Busset M, Braun S, El-Rayes B et al. Efficacy of derazantinib (DZB) in patients (pts) with intrahepatic cholangiocarcinoma (iCCA) expressing FGFR2-fusion or FGFR2 mutations/amplifications [abstract 721P]. Ann. Oncol. 30(Suppl. 5), v253–v324 (2019). [Google Scholar]
- 47.Goyal L, Meric-Bernstam F, Hollebecque A et al. FOENIX-CCA2: a Phase II, open-label, multicenter study of futibatinib in patients (pts) with intrahepatic cholangiocarcinoma (iCCA) harboring FGFR2 gene fusions or other rearrangements. J. Clin. Oncol. 38(Suppl. 15), 108 (2020). [Google Scholar]
- 48.Liu PCC, Koblish H, Wu L et al. INCB054828 (pemigatinib), a potent and selective inhibitor of fibroblast growth factor receptors 1, 2, and 3, displays activity against genetically defined tumor models. PLoS ONE 15(4), e0231877 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Subbiah V, Barve M, Iannotti NO et al. FIGHT-101: a Phase I/II study of pemigatinib, a highly selective fibroblast growth factor receptor (FGFR) inhibitor, as monotherapy and as combination therapy in patients with advanced malignancies [abstract A078]. Mol. Cancer Ther. 18(Suppl. 12), A078 (2019). [Google Scholar]
- 50.Abou-Alfa GK, Sahai V, Hollebecque A et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, Phase II study. Lancet Oncol. 21(5), 671–684 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Amin MB, Edge S, Greene F et al. AJCC Cancer Staging Manual (8th Edition). Springer International Publishing, (2017). [Google Scholar]
- 52.Lyou Y, Grivas P, Rosenberg JE et al. Relationship between hyperphosphatemia with infigratinib (BGJ398) and efficacy in FGFR3-altered advanced/metastatic urothelial carcinoma (aUC). J. Clin. Oncol. 38(Suppl. 6), 576 (2020).31800340 [Google Scholar]
- 53.Tagawa ST, Siefker-Radtke AO, Dosne A et al. Hyperphosphatemia due to erdafitinib and anti-tumor activity among patients with advanced urothelial cancer. Ann. Oncol. 30(Suppl. 5), v356–v402 (2019). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.