

To the Editor:
First-degree relatives of patients with sporadic and familial pulmonary fibrosis have been demonstrated to have high rates of interstitial lung abnormalities (ILA) and interstitial lung disease (ILD) (1). However, less is known about the rates of progression in these relatives (2).
Methods
Relatives enrolled as described previously in the CGS-PF (Clinical Genetics and Screening for Pulmonary Fibrosis) study (1) had baseline pulmonary function tests and chest computed tomography (CT) scans that were repeated 2 years after enrollment. Relatives underwent prone volumetric chest CT scans at full inspiration, and CTs were assessed for the presence of ILA defined by Fleischner Society recommendations (3) and subtyped as previously described (4, 5). All relatives with ILA on either baseline or 2-year CT had both sets of images simultaneously compared in order to determine imaging progression as previously defined (6). For comparison, relatives were divided into two groups: 1) those with ILA at either baseline or 2-year follow-up; and 2) those without ILA (no ILA or indeterminate) at both time points. Progression was assessed using thresholds of lung function decline alone (5% and 10%) or in combination with radiologic changes, including an adaptation of criteria used by the INBUILD trial of either an FVC loss of greater than 10% or 5–10% with progression on CT (7). Continuous variables were compared with Wilcoxon rank-sum and categorical variables with Fisher exact tests. Multivariable models were adjusted for age, sex, and history of ever smoking. Two-sided P values less than 0.05 were considered statistically significant. All analyses were performed using Statistical Analysis Software version 9.4 (SAS Institute).
Results
Of the 107 relatives in the original CGS-PF study, 73 had 2-year follow-up CTs, of which 20 had ILA at baseline and 53 did not. There were no statistically significant differences in baseline characteristics between relatives who did and did not participate in the 2-year follow-up. At 2 years, 21 had ILA, including 19 who had ILA at baseline and 2 additional cases with incident ILA, 51 relatives were without ILA at both baseline and 2-year follow-up, and 1 participant with ILA at baseline that was not present at the 2-year follow-up. Table 1 presents baseline characteristics for those with ILA at either time point and those without ILA at both time points. Compared with those without ILA, relatives with ILA were more likely to be male and, at baseline, had higher absolute monocyte counts and lower FEV1/FVC ratio and percent predicted measures of FVC, TLC, and DlCO.
Table 1.
Characteristics of Relatives with Interstitial Lung Abnormalities at Either Baseline or 2 Years versus without Interstitial Lung Abnormalities at Baseline and 2 Years
| Variable | Without ILA at Baseline and 2 Years (n = 51 [70%]) | ILA at Baseline or at 2 Years (n = 22 [30%]) | Unadjusted P Value |
|---|---|---|---|
| Baseline | |||
| Demographic characteristics | |||
| Age (yr), median (IQR) | 58.0 (53.0 to 63.0) | 59.0 (56.0 to 65.0) | 0.4 |
| Gender (female), n (%) | 35 (69) | 9 (41) | 0.04 |
| BMI, median (IQR) | 27.8 (24.9 to 32.8) | 27.2 (22.2 to 31.2) | 0.3 |
| Ever-smoker, n (%) | 20 (39) | 12 (54) | 0.3 |
| Pack-years smoking, median (IQR)* | 0 (0 to 2.3) | 0.4 (0 to 23) | 0.06 |
| Comorbidities, n (%) | |||
| Obstructive lung disease† | 2 (4) | 1 (5) | 1.0 |
| Cancer | 6 (12) | 1 (5) | 0.7 |
| Heart failure | 0 (0) | 0 (0) | — |
| Kidney disease | 3 (5) | 0 (0) | 0.5 |
| Liver disease | 0 (0) | 0 (0) | — |
| Relative with familial pulmonary fibrosis, n (%) | 25 (49) | 7 (32) | 0.4 |
| Baseline ILA status, n (%) | 0 (0) | 20 (91) | — |
| Lymphocyte telomere length <10th percentile for age, n (%) | 14 (27) | 10 (45) | 0.2 |
| MUC5B promoter variant, n (%) | 21 (41) | 14 (64) | 0.1 |
| Monocyte count (K/μl), median (IQR)‡ | 0.50 (0.42 to 0.60) | 0.62 (0.48 to 0.69) | 0.01 |
| Pulmonary function | |||
| FEV1/FVC, median (IQR) | 0.78 (0.75 to 0.80) | 0.81 (0.78 to 0.83) | 0.02 |
| FEV1% predicted, median (IQR) | 111 (97 to 117) | 103 (91 to 110) | 0.07 |
| FVC% predicted, median (IQR) | 112 (98 to 123) | 100 (89 to 108) | 0.009 |
| TLC% predicted, median (IQR) | 104 (97 to 116) | 96 (87 to 102) | 0.004 |
| DlCO% predicted, median (IQR) | 87 (79 to 99) | 78 (63 to 88) | 0.006 |
| 2-year follow-up | |||
| CT change, n (%) | |||
| Progression | — | 15 (68) | — |
| Stable | — | 7 (32) | — |
| FEV1% predicted, median (IQR)§ | 107 (98 to 122) | 99 (92 to 107) | 0.049 |
| FEV1 change (ml), median (IQR)§ | −75 (−135 to 40) | −130 (−260 to −80) | 0.02 |
| FEV1 change (%), median (IQR)§ | −2.4 (−4.9 to 1.2) | −4.7 (−7.6 to −2.7) | 0.03 |
| FVC% predicted, median (IQR)ǁ | 107 (100 to 120) | 92 (89 to 104) | 0.01 |
| FVC change (ml), median (IQR)ǁ | −90 (−240 to 0) | −175 (−330 to −30) | 0.2 |
| FVC change (%), median (IQR)ǁ | −3.1 (−6.3 to 0.0) | −4.4 (−9.0 to −0.7) | 0.2 |
| FVC change (⩾10% loss), n (%)ǁ | 6 (12) | 5 (22) | 0.1 |
| FVC change (⩾5% loss), n (%)ǁ | 17 (35) | 10 (45) | 0.4 |
| DlCO% predicted, median (IQR)ǁ | 83 (77 to 99) | 76 (62 to 84) | 0.007 |
| DlCO change (ml/min/mm Hg), median (IQR)ǁ | −1.1 (−2.1 to 0.0) | −0.6 (−1.3 to 0.2) | 0.3 |
| DlCO change (%), median (IQR)ǁ | −6.3 (−9.2 to 0.1) | −3.1 (−10.9 to −1.2) | 0.4 |
| DlCO change (⩾10% loss), n (%)ǁ | 12 (24) | 6 (27) | 0.8 |
| DlCO change (⩾5% loss), n (%)ǁ | 27 (55) | 8 (36) | 0.2 |
Definition of abbreviations: BMI = body mass index; CT = computed tomography; ILA = interstitial lung abnormality; IQR = interquartile range.
For unadjusted analyses, a comparison of categorical variables was made using Fisher exact tests and continuous variables with Wilcoxon rank-sum.
Missing pack-year smoking history data for nine relatives (two with ILA and seven without ILA).
Obstructive lung disease, defined as FEV1/FVC < 70%.
Missing monocyte count data for six relatives (three with ILA and three without ILA).
Missing 2-year FEV1 for three relatives (one with ILA and two without ILA); change values are compared with baseline.
Missing 2-year FVC and DlCO data for two relatives (two without ILA); change values are compared with baseline.
At the 2-year follow-up, the majority (13 [65%]) of those with ILA at baseline had radiologic progression, and 2 (4%) of those without ILA at baseline developed ILA (examples shown in Figure 1). Of the 15 total relatives with radiologic progression, 4 (27%) were from families with familial pulmonary fibrosis; the remaining 11 (73%) had a single first-degree relative with IPF. Of the 20 relatives with baseline ILA, 6 (30%) had definite fibrosis, of which 5 (83%) had 2-year radiologic progression, whereas 8 of the 14 (57%) without baseline fibrosis progressed. At 2 years, FEV1, FVC, and DlCO remained reduced in those with ILA compared to without ILA. Although there were no statistically significant differences in the loss of FVC and DlCO from baseline to 2 years between relatives with and without ILA, those with ILA had greater loss of FEV1 in both unadjusted analyses (Table 1) and after adjusting for covariates (−145 ml; 95% confidence interval, −249 ml to −40 ml; P = 0.007) when compared with those without ILA. Almost half of the relatives with ILA (10 of the 22 [45%] with ILA at either time point; 9 of the 20 [45%] with ILA at enrollment) demonstrated adapted INBUILD trial criteria for progression (Table 1).
Figure 1.
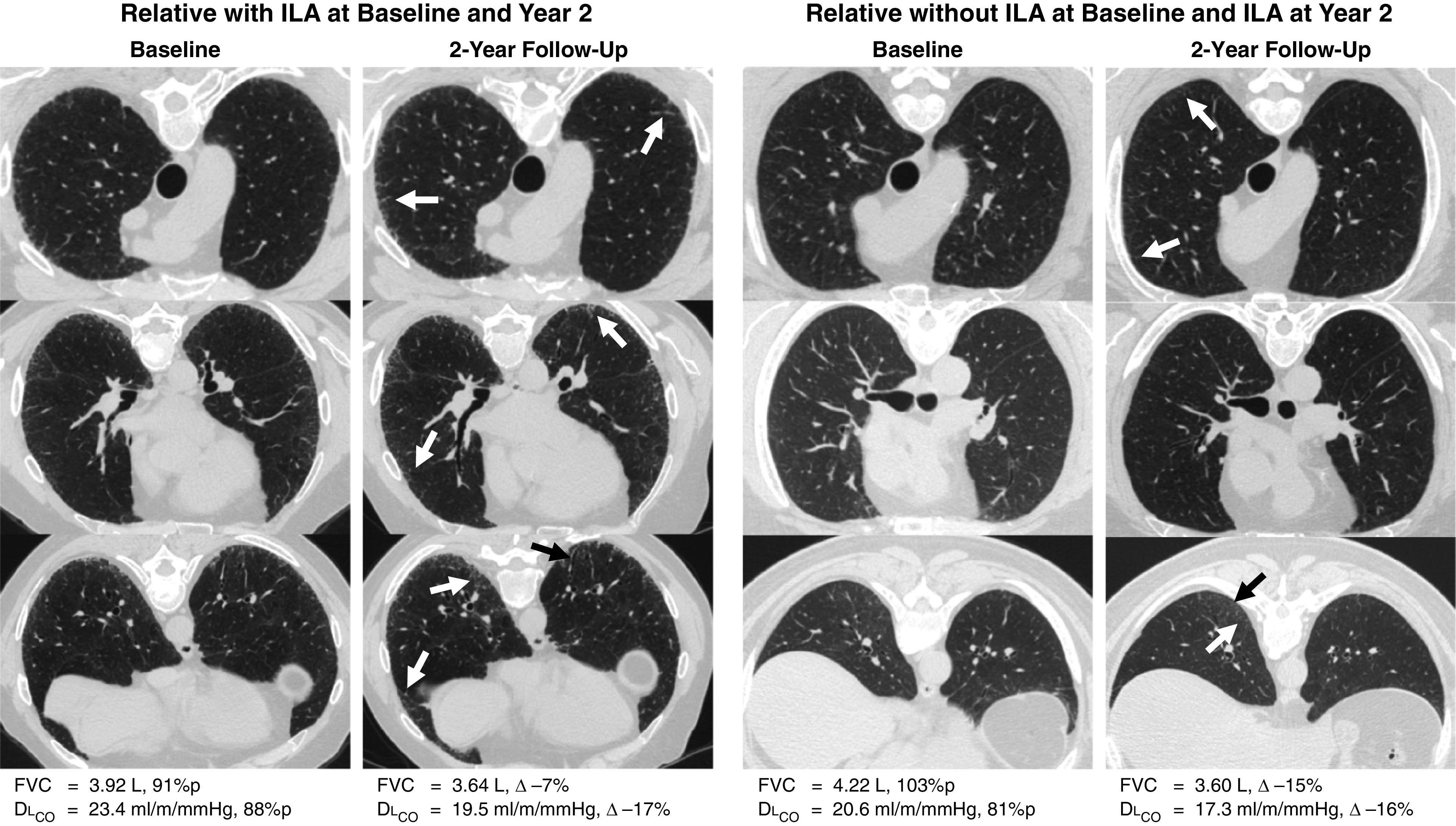
Computed tomography scans of the chest from two relatives with interstitial lung abnormalities (ILA) and progression at the 2-year follow-up. The first participant with ILA at baseline and 2-year follow-up had radiologic progression, including worsening reticulations throughout the lungs (white arrows) and increased basilar traction bronchiolectasis (black arrow). At the 2-year follow-up, FVC had decreased by 7% and DlCO by 17%. The second participant was indeterminate for ILA at baseline but had ILA at the 2-year follow-up and radiologic progression, including new and increased areas of upper lobe and basilar reticulation (white arrows) as well as new basilar traction bronchiolectasis (black arrow). At the 2-year follow-up, FVC had decreased by 15% and DlCO by 16%.
Discussion
In this 2-year follow-up study, disease progression, whether defined by imaging alone or in combination with loss of lung function, is common among first-degree relatives of patients with pulmonary fibrosis. Imaging progression was observed in 65% of relatives with ILA, and those with ILA were more likely to be male, had higher circulating monocyte counts, and had significantly greater loss of their FEV1 at 2 years compared with those without ILA.
The rates of imaging progression among relatives with ILA at 2 years (65%) is high and greater than that reported in other cohorts over this time interval (e.g., the National Lung Cancer Screening Study) (8), possibly because of the unique risk of this population. Although the INBUILD study helped to define criteria for progression on the basis of patients with known pulmonary fibrosis (7), when these criteria are extrapolated to relatives with ILA at enrollment, 9 of the 20 (45%) had either a loss of FVC of ⩾10% or FVC loss of 5–10% plus imaging progression. Among all relatives, 21% had imaging progression, and 14% made a modified INBUILD criteria for progressive ILD overall.
Our findings are consistent with the conclusions of studies in the general population (not selected on the basis of family history of fibrosis) that ILA is associated with an accelerated loss of lung volume over time (6). In addition, lung function loss in relatives without ILA is somewhat higher than expected in the general population, which may be cohort-specific or because of other factors such as obstructive lung disease (given the lower FEV1/FVC ratio in those without ILA). Further longitudinal follow-up is necessary to understand if the rate of lung function loss in relatives is greater than that of other groups, if the rate of loss accelerates over time, and if lung function loss correlates with other adverse clinical outcomes.
There are several limitations to this work. Most notably, our small sample size may have limited statistical power to detect differences between those with and without ILA. This study is now actively recruiting additional relatives, which may help to address issues with power in future analyses. In addition, whereas the rates of progression at 2-year follow-up are quite high, longer follow-up will be needed to provide a more complete picture of the rates of progression and the risk of ILA and ILD in this population.
Conclusions
This study demonstrates that radiologic progression is common in relatives of patients with pulmonary fibrosis that have ILA during 2 years of follow-up and may be associated with accelerated loss of lung function. Future work will be needed to identify risk factors for progression, assess various criteria used to define progression, and evaluate clinical outcomes. The findings of this study add more weight to the argument for screening relatives of patients with pulmonary fibrosis and suggest that future studies to assess therapeutic interventions may be warranted.
Footnotes
Supported by the National Institutes of Health (NIH) grants K08 HL140087 (R.K.P.); T32 HL007633 (J.A.R.); R01 ES031252 (M.B.R.); R01 CA203636, U01CA209414, R01 HL111024, R01 HL135142, and R01 HL130974 (H.H.); R01 HL130974, R01 HL118455, P01 HL132825, and U01 TR001810 (B.A.R.); U01 HL133232 and R01 HL130974 (I.O.R.); and R01 HL111024, R01 HL130974, and R01 HL135142 (G.M.H.). This paper is subject to the NIH public access policy: https://publicaccess.nih.gov/policy.htm.
Author Contributions: Study design: J.A.R., B.A.R., I.O.R., and G.M.H. Acquisition, analysis, or interpretation of the data: J.A.R., M.A.P.F., A.H.M., M.F.P.G., N.E.C., S.G., M.B.R., H.J.G., R.K.P., H.H., B.A.R., I.O.R., and G.M.H. Critical revision of the manuscript for important intellectual content: J.A.R., M.A.P.F., A.H.M., M.F.P.G., N.E.C., S.G., M.B.R., H.J.G., R.K.P., H.H., B.A.R., I.O.R., and G.M.H. Statistical analysis: J.A.R., B.A.R., I.O.R., and G.M.H. Obtained funding: B.A.R., I.O.R., and G.M.H.
Originally Published in Press as DOI: 10.1164/rccm.202208-1470LE on September 13, 2022
Author disclosures are available with the text of this letter at www.atsjournals.org.
References
- 1. Hunninghake GM, Quesada-Arias LD, Carmichael NE, Martinez Manzano JM, Poli De Frías S, Baumgartner MA, et al. Interstitial lung disease in relatives of patients with pulmonary fibrosis. Am J Respir Crit Care Med . 2020;201:1240–1248. doi: 10.1164/rccm.201908-1571OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Salisbury ML, Hewlett JC, Ding G, Markin CR, Douglas K, Mason W, et al. Development and progression of radiologic abnormalities in individuals at risk for familial interstitial lung disease. Am J Respir Crit Care Med . 2020;201:1230–1239. doi: 10.1164/rccm.201909-1834OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hatabu H, Hunninghake GM, Richeldi L, Brown KK, Wells AU, Remy-Jardin M, et al. Interstitial lung abnormalities detected incidentally on CT: a position paper from the Fleischner Society. Lancet Respir Med . 2020;8:726–737. doi: 10.1016/S2213-2600(20)30168-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Washko GR, Hunninghake GM, Fernandez IE, Nishino M, Okajima Y, Yamashiro T, et al. COPDGene Investigators Lung volumes and emphysema in smokers with interstitial lung abnormalities. N Engl J Med . 2011;364:897–906. doi: 10.1056/NEJMoa1007285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Putman RK, Gudmundsson G, Axelsson GT, Hida T, Honda O, Araki T, et al. Imaging patterns are associated with interstitial lung abnormality progression and mortality. Am J Respir Crit Care Med . 2019;200:175–183. doi: 10.1164/rccm.201809-1652OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Araki T, Putman RK, Hatabu H, Gao W, Dupuis J, Latourelle JC, et al. Development and progression of interstitial lung abnormalities in the Framingham heart study. Am J Respir Crit Care Med . 2016;194:1514–1522. doi: 10.1164/rccm.201512-2523OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, et al. INBUILD Trial Investigators Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med . 2019;381:1718–1727. doi: 10.1056/NEJMoa1908681. [DOI] [PubMed] [Google Scholar]
- 8. Jin GY, Lynch D, Chawla A, Garg K, Tammemagi MC, Sahin H, et al. Interstitial lung abnormalities in a CT lung cancer screening population: prevalence and progression rate. Radiology . 2013;268:563–571. doi: 10.1148/radiol.13120816. [DOI] [PMC free article] [PubMed] [Google Scholar]