

Abstract
The abnormal expression of the c-Met tyrosine kinase has been linked to the proliferation of several human cancer cell lines, including non-small-cell lung cancer (NSCLC). In this context, the identification of new c-Met inhibitors based on heterocyclic small molecules could pave the way for the development of a new cancer therapeutic pathway. Using multiple linear regression (MLR)-quantitative structure–activity relationship (QSAR) and artificial neural network (ANN)-QSAR modeling techniques, we look at the quantitative relationship between the biological inhibitory activity of 40 small molecules derived from cyclohexane-1,3-dione and their topological, physicochemical, and electronic properties against NSCLC cells. In this regard, screening methods based on QSAR modeling with density-functional theory (DFT) computations, in silico pharmacokinetic/pharmacodynamic (ADME-Tox) modeling, and molecular docking with molecular electrostatic potential (MEP) and molecular mechanics-generalized Born surface area (MM-GBSA) computations were used. Using physicochemical (stretch–bend, hydrogen bond acceptor, Connolly molecular area, polar surface area, total connectivity) and electronic (total energy, highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels) molecular descriptors, compound 6d is identified as the optimal scaffold for drug design based on in silico screening tests. The computer-aided modeling developed in this study allowed us to design, optimize, and screen a new class of 36 small molecules based on cyclohexane-1,3-dione as potential c-Met inhibitors against NSCLC cell growth. The in silico rational drug design approach used in this study led to the identification of nine lead compounds for NSCLC therapy via c-Met protein targeting. Finally, the findings are validated using a 100 ns series of molecular dynamics simulations in an aqueous environment on c-Met free and complexed with samples of the proposed lead compounds and Foretinib drug.
1. Introduction
A significant amount of research has been devoted to medicinal chemistry, specifically the design and discovery of drugs based on small molecules derived from heterocyclic compounds. This is due to their various biological activities, particularly their anticancer activity. Several findings suggest that the heterocyclic compound containing the radicals pyran, pyrazole, thiophene, and thiazole has antiparasitic,1 antimicrobial,2 anxiolytic,3 arrhythmic,4 antifungal,5 and anticancer6 activities. According to an in vitro study carried out by Mohareb et al.,5 heterocyclic compounds bearing pyran, pyrazole, thiophene, and thiazole radicals are prepared based on the core structure of cyclohexane-1,3-dione. According to the study carried out by Mohareb et al., 40 cyclohexane-1,3-dione-based compounds were synthesized, and then their inhibitory activity was evaluated against six cancer cell lines, including the non-small-cell lung cancer (NSCLC) cell lines (H460 and A549), human colorectal cancer cell line (HT29), gastric carcinoma cell line (MKN-45), malignant glioma cell line (U87MG), and hepatocellular carcinoma cell line (SMMC-7721). Through in vitro screening, 19 molecules were selected as the most cytotoxic candidates against the six targeted cancer cell lines. On this basis, the inhibitory activity of the 19 selected molecules against receptor tyrosine kinases (c-Kit, Flt-3, VEGFR-2, EGFR, PDGFR) was scored. Then, six molecules were removed because of their half-maximal inhibitory concentration (IC50 > 1 nM), and the remaining 13 molecules were evaluated against the Pim-1 protein kinase. Also, the in vitro screening of the synthesized cyclohexane-1,3-dione derivatives revealed a strong correlation between the structure and activity of these molecules. Variations in the biological activity of the compounds as a result of the substitutions introduced on the structure of cyclohexane-1,3-dione translate this relationship. Overall, in vitro trials reported that the small molecules derived from cyclohexane-1,3-dione have the potential to become anticancer agents. This is evidenced by a more thorough formulation of the candidacy of these agents in targeted cancer therapy. However, it is difficult to judge if the hypothesis is likely to be valid or not based on the in vitro bioactivity screening data alone. Most especially in the absence of other more important data such as evaluations of bioavailability (drug-like) properties, pharmacokinetics (absorption, distribution, metabolism, and elimination), and pharmacodynamics (toxicity), stability and compatibility patterns of molecules with target protein receptors and other critical evaluations are required as part of successful drug discovery in the early phase of development. However, the high cost, significant effort, and time required to perform these evaluations in living models often lead to a halt in the research at the stage of synthesis and in vitro biological evaluations. Therefore, it is necessary to search for novel and reliable methods to enrich the traditional strategies of molecular screening, introducing the opportunity of molecular modeling methods as an alternative to develop, screen, and improve candidate molecules as promising agents for drug design as a model.
In this regard, advances in X-ray crystallography and computational power have rapidly improved the progress of the application of computer-aided drug design (CADD) methods in the pharmaceutical industry. The application of CADD approaches has led to the discovery of several marketed drugs (“saquinavir”, “nelfinavir”, “norfloxacin”, “zanamivir”, “amprenavir”, “zolmitriptan”, “dorzolamide”),6 leading to a reduction in the cost of drug discovery and development by more than 50% compared to the old strategies based on screening a large number of compounds through experimental testing.7 This means that experimental strategies are time-consuming and require large-scale investments. Therefore, the application of CADD approaches to screening chemical compounds in the near-early phase of the screening process will significantly reduce the number of compounds that will be tested in vitro and in vivo. In our current study, the biological activity of cyclohexane-1,3-diene derivatives against the six reported cancer cell lines and the three-dimensional structure of the receptor tyrosine kinase provided two sufficient insights for performing the CADD approach based on the ligand-based and structure-based drug design. This is to rationalize the screening of cyclohexane-1,3-dione compound candidates for use as anticancer agents. To this end, a multistage in silico screening based on advanced molecular modeling techniques aided in drug design and discovery was performed. This screening included the structure–activity relationship study of 40 cyclohexane-1,3-dione molecules based on the quantitative structure–activity relationship (QSAR) and density-functional theory (DFT) approaches, investigation of the drug-like and pharmacokinetic properties of the candidate drug molecules, simulation of protein–ligand interactions via molecular docking and molecular mechanics-generalized Born surface area (MM-GBSA) modeling, evaluation of various structural parameters related to the stability and compatibility of protein–drug systems via molecular dynamics simulation.
2. Materials and Methods
According to statistics provided by the World Health Organization in 2020, 10.44 million cancer deaths were recorded, including 2.26 million cases of breast cancer, 2.21 million cases of lung cancer, 1.93 million cases of colorectal cancer, 1.41 million cases of prostate cancer, 1.20 million cases of non-melanoma skin cancer, and 1.09 million cases of gastric cancer.8 Among these deaths, 50% were due to breast and lung cancer.9 Due to the high mortality rate caused by these two types of cancer, which are the most prevalent and deadly to human health, we focus in our current research on lung cancer as a model for therapeutic targeting by small molecules derived from cyclohexane-1,3-dione. Among lung cancer victims, approximately 85% of lung cancer cases are diagnosed as non-small-cell lung cancer.10 The cell lines H460 and A549 represent the most popular in vitro models for non-small-cell lung cancer (NSCLC) assays. In the present study, we developed QSAR models based on the values of IC50 (μM) of various molecules against the H460 cell line. The reason for using this cell line is that the surface of H460 cells contained over 50% of proteins compared to A549 cells.11 This may be a factor contributing to the much faster growth of H460 cells than A549 cells. On the other hand, receptor tyrosine kinases can be selected as a potential therapeutic choice for non-small-cell lung cancer cell growth inhibition through the use of cyclohexane-1,3-dione derivatives. This is due to the inhibitory effect of cyclohexane-1,3-dione against receptor tyrosine kinases (c-Kit, Flt-3, VEGFR-2, EGFR, PDGFR, and Pim-1).5,12 Overexpression of Pim-1 kinase has been reported to be strongly associated with c-MET expression in the diagnosis of non-small-cell lung cancer, although its molecular mechanisms underlying this context remain unclear.13,14 Thus, further investigation of the molecular interaction’s patterns between the c-Met receptor kinase and small molecules may provide insight into mechanisms of cancer therapeutic response. Following this background and using a structure-based drug design approach, in the present work, we evaluate suitable affinity profile conformations of cyclohexane-1,3-dione derivatives for their prospective activity as inhibitors of c-Met kinase enzymatic activity.
The tyrosine kinase c-Met is considered a suitable pathway for targeting EGFR-TKD for many cancer therapies, including NSCLC.15,16 We implemented a comprehensive approach to search in the PubMed database to identify current research on c-Met-targeted drugs for lung cancer.17,18 Search results reflected the prevalence of keywords “non-small-cell lung cancer”,19 “tivantinib”,20 “crizotinib”,21 “carbozantinib”,22 “foretinib”,23 and “onartuzumab”.24 Mohareb et al.5 examined the inhibitory effect of cyclohexane-1,3-dione derivatives in vitro against non-small-cell lung cancer cell lines (H460 and A549) using the standard drug Foretinib. In this study, we use the three-dimensional (3D) structure of the c-Met protein complexed with Foretinib (PDB code: 3LQ8)25 as a therapeutic target for viral screening across molecular docking, MM-GBSA, and molecular dynamics simulations. Foretinib and other c-Met tyrosine kinase inhibitors have shown promising results in clinical trials for cancer treatment, with patients having a better chance of survival.26 While the most serious side effects of these drugs were the lack of a significant effect on local tumor response and the occurrence of numerous side effects in chemotherapy patients.27 As a result, more research is needed to discover new compounds with structural properties suitable for safe drug use that are less toxic and more effective against the growth of cancer cell lines caused by the enzymatic activity of the c-Met protein than currently available drugs. For this reason, based on QSAR modeling, in our current work, we characterize the structures of small molecules based on cyclohexane-1,3-dione and identify the most important structural properties of these molecules that influence their biological activity against NSCLC.28,29 For QSAR modeling, we used a combination of topological, physicochemical, and electronic DFT molecular descriptors that are commonly used for geometrical structural characterization.30,31 The DFT computations were used because of their precision in providing precise indications on the electronic properties of the studied molecules, allowing for the generation of confident QSAR models.32−34 Furthermore, the drug-like and pharmacokinetic absorption, distribution, metabolism, excretion, toxicity (ADMET) properties of the candidate drug molecules will be examined.35−37 On the other hand, we performed molecular docking simulations to evaluate the binding potential of the examined small molecules toward the c-Met protein active pocket. This is due to the importance of this procedure for predicting potential interactions between ligands and active amino acid residue sites inside the target protein receptor pocket.38,39 The interactions between the investigated heterocyclic compounds and c-Met can result in a strong noncovalent binding between the two ends, which can provide a strong inhibition of c-Met protein enzymatic activity. As a result, c-Met activity induction can be inhibited; thus, the proliferation of cancer cells can be inhibited.40 In parallel, we perform a detailed validation regarding the stability of candidate drug molecules in the c-Met active pocket. To this end, we evaluated MM-GBSA free binding energy (BE), molecular dynamics, and thermodynamic profiles of protein–ligand systems.
2.1. Investigated Compound Library
We obtained the structures of 40 compounds derived from cyclohexane-1,3-dione and their inhibitory activity values (IC50) against a non-small-cell lung cancer cell line (H460) from the experimental study conducted by Mohareb et al.5 The biological inhibitory activity observed in vitro (pIC50 = −Log10[IC50]) of cyclohexane-1,3-dione derivatives against the non-small-cell lung cancer cell line H460 is shown in Table S1 in the Supporting Information. The structures of the studied compounds were carefully sketched by GaussView 5.0 software41 and optimized using the following software: ChemSketch,42 Chem3D,43 and Gaussian 09W,41 respectively, to calculate the topological, physicochemical, and quantum chemical molecular descriptors.
2.2. Quantitative Structure–Activity Relationship Modeling
For the purpose of describing the quantitative relationship between cyclohexane-1,3-dione derivative structures and their biological inhibitory activity against the non-small-cell lung cancer cell line H460, we characterize the structures of all 40 investigated molecules by two-dimensional (2D) and 3D molecular descriptors (Table 1). Table 1 shows the molecular descriptors computed in this study for modeling the structure–activity relationship of cyclohexane-1,3-dione-derived small molecules. The values of the molecular descriptors computed for each individual molecule are listed in Table S2. The database of calculated molecular descriptors was screened by the principal component analysis (PCA) technique to identify molecular descriptors that may correlate with the pIC50 biological activity of cyclohexane-1,3-dione derivatives.44 These analyses permit to evaluate the level of correlation between the molecular descriptors, thanks to the calculation of the correlation coefficient (R) between each of the two molecular descriptors.45 Through the PCA analyses, a correlation matrix (Pearson) is obtained, whereby the data have been automatically normalized or standardized before the calculations to avoid exaggerating the effect of variables with high variances on the output (Table S3).
Table 1. Molecular Descriptors Used for QSAR Modeling.
| descriptors | class | optimization methods |
|---|---|---|
| molecular weight (MW, Da), molar volume (MV, cm3), index of refraction (n), density (d, g/cm3), polarizability (αe, cm3) | topological | 3D optimization available in ChemSketch46 |
| stretch, bend, stretch–bend (S–B), torsion (Torr), non-1,4 VDW (NVDW), 1,4 VDW (VDW), dipole/dipole (dipole) (kcal/mol), hydrogen bond acceptor (HBA), hydrogen bond donor (HBD), rotatable bonds (RoB), partition coefficient (Log P), Connolly accessible area (CAA, Å2), Connolly molecular area (CMA, Å2), mol refractivity (MR), water solubility (Log S), Balaban index (BI), molecular topological index (MTI), polar surface area (PSA, Å2), total connectivity (TC), Wiener index (WI) | physicochemical | MM2 method force field method available in Chem3D47 |
| energy total (ET, eV), dipole moment (DM, eV), energy highest occupied molecular orbital (HOMO) (EHOMO, eV), energy lowest unoccupied molecular orbital (LUMO) (ELUMO, eV), energy gap (EGap, eV) electronegativity (x, eV), chemical potential (μ, eV), chemical hardness (η, eV), index of electrophilicity (w, eV) | DFT-based quantum chemical descriptors | B3LYP hybrid three-parameter Lee–Yang–Parr–Becke method using 6-31G(d,p) basis available in Gaussian 09W software48 |
After improving the developed database and limiting the number of less related structural descriptors, the data is divided into two sets (training and test). The training set is used to develop predictive QSAR models, while the test set is used to test the performance of the models developed by statistical modeling. The division of the database in this work was done by a univariate clustering technique (UV).49,50 After applying the UV algorithm and adapting it to classify the database into eight classes, we randomly select one molecule from each class to form the elements of the test set (20% of the database elements) while selecting the remaining 80% as inputs for the training set. The multiple linear regression (MLR)-QSAR model is generated and validated in terms of internal and external statistical significance and predictive power using the multiple linear regression (MLR) technique. This procedure is carried out in accordance with the principles of the OECD QSAR model and the Golbraikh and Tropsha criteria (Table 2).51,52 Also, the stability and applicability of the MLR-QSAR model are evaluated by a Y-randomization test and applicability domain (AD) techniques.53,54 Fifty iterations of the Y-randomization test were performed, and the AD of the proposed model was presented by Williams plots.55 A further external test was carried out to estimate the adequacy of the molecular descriptors selected by the MLR-QSAR model to describe the biological activity (pIC50) of the 40 cyclohexane-1,3-dione compounds examined. We performed this test using the feed-forward neural network (FFNN) algorithm to develop a nonlinear artificial neural network (ANN)-QSAR model using the same strategy employed in our previous work.32 In this study, ANN is used to improve the accuracy of predicting the biological activity of the compounds studied based on the molecular descriptors selected using the MLR method. This is done to show that the MLR-QSAR model can predict the activity of exogenic molecules containing the scaffold cyclohexane-1,3-dione. In this case, a three-layer feed-forward neural network algorithm (input layer, hidden layer, and output layer) was used. To train and test the ANN-QSAR model, the Levenberg–Marquardt training function was used in the input layer, which contains the eight molecular descriptors, the sigmoid transfer function in the hidden layer, and the linear transfer function in the output layer.32
Table 2. Golbraikh and Tropsha Acceptable Model Criteriaa.
| parameters | calculation model | threshold |
|---|---|---|
| R2 | 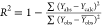 |
>0.6 |
| Radj2 | 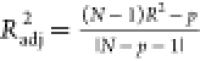 |
>0.6 |
| MSE | 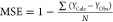 |
low value |
| Ftest | 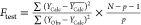 |
high value (>0.3) |
| Rtest2 | 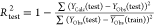 |
>0.6 |
| Qcv2 | 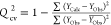 |
>0.5 |
| RRand2 | RRand2 average over 50 random iterations | <R2 |
| Qcv LOO(Rand)2 | Qcv loocv2 average over 50 random iterations | <Qcv2 |
| cRp2 | 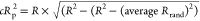 |
>0.5 |
Yobs and Ycalc: observed and predicted pIC50 values.  and
and  average of observed and predicted pIC50 values. N: number of individual (compound)
datasets and p: number of selected descriptors. Rand
refers to the values obtained by the Y-randomization
test.
average of observed and predicted pIC50 values. N: number of individual (compound)
datasets and p: number of selected descriptors. Rand
refers to the values obtained by the Y-randomization
test.
In this work, XLSTAT 2019 software is used to perform PCA analyses, partition the database, and develop a linear MLR-QSAR model.56 MATLAB R2015a software is used to scope the MLR-QSAR model applicability domain as well as to develop an ANN-QSAR model.57
2.3. In Silico Pharmacokinetic–Pharmacodynamic Modeling
The in silico screening of a drug candidate’s bioavailability, pharmacokinetics, and pharmacodynamic toxicity profiles (ADME-Tox) is a critical step before synthesis and in vivo/clinical trials.58,59 The implementation of experimental in vitro tests as part of a real screening strategy often leads to a large margin of error between the in vitro evaluations and the desired in vivo results.60,61 This makes the success rate of screened molecules as medicinal agents relatively low. The main problems that may be encountered include: (1) the structural properties of the molecule are not compatible with the bioavailability properties of the drug, (2) failure to achieve appropriate pharmacokinetics, (3) occurrence of undesirable side effects, (4) lethal dose concentration, (5) risk of toxicity, and (6) inability to achieve the therapeutic goal and biological response. Thus, to avoid these potential difficulties that could disrupt the process of drug discovery, it is necessary to perform pharmacophore-based virtual screening of candidate drug molecules before proceeding to in vivo trials. At this point in our research, we have focused on determining whether the candidate molecules’ drug-like properties are compatible with oral bioavailability using Lipinski, Veber, and Egan’s bioavailability rules.62 In addition, the pharmacokinetic and pharmacodynamic properties (ADME-Tox) of the candidate drug molecules are assessed. This study used the SwissADME and pkCSM web servers to implement pharmacophore-based virtual screening.63,64
2.4. Molecular Electrostatic Potential (MEP) and Molecular Docking Simulations
Using the molecular electrostatic potential (MEP) visualizations,65,66 we identified the reactive sites favorable for electrophilic and nucleophilic actions at the structure surface of the template compound (6d). 3D contour map analysis of the MEP simulation results was performed using GaussView 5.0 software. Furthermore, we performed molecular docking simulations to investigate prospective binding patterns between the examined molecules and the active amino acid residues inside the c-Met protein pocket and also to characterize the structural properties of cyclohexane-1,3-dione derivatives favorable to achieve noncovalent interactions inside the c-Met active pocket. In this work, molecular docking and MEP modeling are used to develop a deep insight into the conformations of the ligand with optimal binding affinity and structural compatibility toward the c-Met protein candidate for targeting cancer.
The structure of the c-Met protein complexed with the standard drug Foretinib served as the foundation for the structure-based drug design strategy used in this study. The 3D shape of the c-Met–Foretinib complex was obtained using the PDB database (code: 3LQ8) (Figure 1).67
Figure 1.

Model of the c-Met protein complexed with inhibitor Foretinib (PDB code: 3LQ8).
Several missing fragments (such as hydrogens, side chains, a portion of the backbone chain, similar side chain alignments, non-protein entities, a lack of ligand identification, etc.) may require adjustment of the protein structure extracted from a PDB database, so the input file must be meticulously prepared before molecular docking modeling can initiate. Using the Discovery Studio 2016 software package, the structure of the c-Met protein was optimized and refined via the CHARMM force field, which was sufficient to prepare the protein for molecular docking simulations.68 Meanwhile, ligand candidates were prepared as inputs for molecular docking with c-Met after optimizing their geometries via DFT(B3LYP)/6-31G(d,p), SYBYL force field optimization geometry.69 Thus, we obtained optimal ligand conformations perfect for molecular docking. Recognition of the active pocket of the c-Met protein where the standard drug Foretinib interacts was fulfilled using the AUTOGRID algorithm available in AutoDockTools-1.5.6,70 the coordinates defined (x = 0.20 Å, y = 4.04 Å, z = 28.96 Å with a distance of 0.375 Å and a size of 30 × 20 × 20 Å3) as a grid box for ligand binding. The molecular docking results related to 2D and 3D interactions were visualized using Discovery Studio 2016. Molecular docking simulations were performed using AutoDock Vina software, docking was set to record the binding energies of the first 10 conformations with the active site of c-Met, and all other parameters were adapted to the default settings of AutoDock Vina.71 Molecular docking results and protein–ligand interaction profiles obtained from the molecular docking simulations were generated by Discovery Studio 2016.
2.5. Molecular Mechanics-Generalized Born Surface Area (MM-GBSA)
In this work, the molecular mechanics-generalized Born surface area (MM-GBSA) approach was used to rescale the docking patterns obtained by molecular docking simulation.72 We perform this procedure to assess the free binding energies of ligands toward the active pocket of c-Met. This was done to estimate the binding ability of the proposed drug ligands and the target receptor (c-Met) and also to predict the most favorable interactions to achieve the lowest free binding energy (ΔGbind). The prime MM-GBSA simulation was carried out by minimizing the energies of the complexes using the OPLS3e force field and the VSGB solvate model at pH 7 ± 2 through the MM-GBSA Prime package in Schrodinger 2020-3.73 During this setup, the energy of the protein–ligand complexes, as well as that of the ligands, is minimized. As a result, the free binding energy (ΔGbind) of the examined systems can be calculated. The binding energy (ΔGbind) of the complexes can be calculated according to eq 1(74,75)
| 1 |
2.6. Molecular Dynamics Simulations
We perform molecular dynamics simulations to evaluate the molecular dynamics and stability of the c-Met protein in an aqueous environment. This procedure is due to its great interest in the analysis of the chemical and physical basis of the molecular structure and its functional property variations in terms of time.76−78 In this study, the stability of proposed drug compounds in the c-Met pocket was examined by evaluating a set of dynamics parameters such as root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF) timelines, protein–ligand contact, ligand properties, and thermodynamic properties. The time course of MD simulation was fixed in 100 ns using the Desmond package of Schrödinger 2020-3 software.79 The examined systems were solvated using the orthorhombic single point charge (SPC) explicit water model.74,75 To improve and regenerate the protein–ligand systems, the optimized potentials for liquid simulations extended (OPLSe) force field was used.74,80 By adding sodium and chloride ions to the total complexes explored, the charge was neutralized. In addition, the energies of the systems were minimized by 2000 steps before running the MD simulation along a 100 ns path in the NPT ensemble.81 In parallel, the particle mesh Ewald (PME) function was adapted to take into account and preserve long-range electrostatic interactions in the 0.8 Å grid.75 The Nose–Hoover thermal algorithm and the Martyna–Tobias–Klein method were used to generate slow heating of the systems under 300 K and 1.013 bar pressure.82,83 The Simulation Interaction Diagram tool was used in the Desmond package to extract and analyze the detailed interactions between the target protein 3LQ8 and proposed ligands, and the simulation quality analysis package implemented in Desmond was used to generate and analyze all of the thermodynamic properties of the systems such as total and potential energies, temperature, pressure, and volume as a function of time.84 The thermodynamic properties of the systems investigated were evaluated by the Simulation Interaction Diagram tool in the Desmond package, as the latter is a powerful tool for computing energies and force fields compatible with various force field models used in biochemical and quantum research, including CHARMM, AMBER, and OPLS.79
3. Results and Discussion
Based on the correlation matrix obtained from the PCA analyses (Table S3), the 11 molecular descriptors [index of refraction (n), stretch–bend (S–B), hydrogen bond acceptor (HBA), Connolly molecular area (CMA), polar surface area (PSA), total connectivity (TC), energy total (ET), dipole moment (DM), energy HOMO (EHOMO), energy LUMO (ELUMO), and electrophilicity index (w)] are selected as the best outputs that show the lowest correlation with each other (R < 0.5). Therefore, we use these descriptors as entry data for QSAR modeling of cyclohexane-1,3-dione derivatives.
Using the multiple linear regression (MLR) technique applied to the optimized database, we try to find the molecular descriptors that affect the biological inhibitory activity (pIC50) of the studied molecules. For this, we aim to develop the optimal MLR-QSAR model in which the relationship (pIC50 = f(molecular descriptors)) can be defined. To achieve this goal, we split the database into two sets (training and test) using the univariate analysis method. Table 3 shows the split results obtained. Based on the structural diversity and pIC50 range of the 40 molecules investigated, we extracted the eight molecules (6b, 6d, 10a, 10c, 12b, 19b, 20a, and 22b) as components of the test set of QSAR models. However, the remaining 32 molecules were selected as components of the training set of QSAR models.
Table 3. Univariate Clustering.
| results
by class | ||||||||
|---|---|---|---|---|---|---|---|---|
| class | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| sum of wts | 2 | 5 | 9 | 1 | 11 | 5 | 4 | 3 |
| observation | ||||||||
| 6b | 6d | 10a | 10c | 12b | 19b | 20a | 22b | |
| 14 | 6a | 10b | 5d | 5a | 21b | 21a | ||
| 3b | 12a | 10d | 5c | 22a | 21c | |||
| 6c | 16b | 16a | 17b | 22c | ||||
| 3a | 16c | 17d | 5b | |||||
| 17a | 17f | |||||||
| 17c | 19a | |||||||
| 17e | 8a | |||||||
| 20b | 19c | |||||||
| 8b | ||||||||
| 20c | ||||||||
3.1. MLR-QSAR Modeling
Equation 2 signifies the MLR-QSAR model obtained from the training set of 32 molecules and 11 molecular descriptors used as inputs. The power of prediction of the resulting model is examined using the external test set of eight molecules.
| 2 |
Through eq 2, we can notice that the biological inhibitory activity (pIC50) correlated with eight molecular descriptors (physicochemical and electronic) that are stretch–bend (S–B), hydrogen bond acceptor (HBA), Connolly molecular area (CMA), polar surface area (PSA), total connectivity (TC), energy total (ET), energy HOMO (EHOMO), and energy LUMO (ELUMO). This means that the physicochemical and electronic structural features of heterocyclic compounds derived from cyclohexane-1,3-dione are closely related to their biological inhibitory activity against NSCLC cell lines. This strong correlation can be confirmed by high values of the correlation coefficient (R = 0.92, R2 = 0.85, Radj2 = 0.81, Q2 = 0.65, Rtest = 0.82, and F = 13.357) and low values of the MSE (0.04) and P-value (<0.0001). The high value of the correlation coefficient (R = 0.92) indicates the strong concordance between the observed pIC50 values and those predicted by the MLR-QSAR model. This is confirmed by the regular linear distribution of observed versus predicted pIC50 values, as shown in Figure 2A.
Figure 2.

(A) Distribution of observed and predicted pIC50 values. (B) William plot of standard residuals versus leverage.
3.1.1. Performance of MLR-QSAR Modeling
The high level of coefficient of determination of leave-one-out cross-validation (loocv) (Q2 = 0.65 > 0.5) obtained means that the developed MLR-QSAR model is more stable and is not affected by the loocv process. Also, the high value of the coefficient of determination (Rtest2 = 0.82) obtained by the external test indicates that the selected MLR-QSAR model has high predictive power. From Table S4, the Y-randomization indicates that the values of the correlation coefficients (RRand and QRand2) obtained by the Y-randomization test are lower than those obtained by the original MLR-QSAR model. Also, the significant gap between the coefficients of correlation of the original and randomized model (cRp = 0.70 > 0.5) indicates the good stability of the proposed MLR-QSAR model. This means that the predictive power of the MLR-QSAR model developed in this study is not random but rather that there is a logical correlation between the structure of each molecule and its interpreted biological activity. To complement the internal and external test indicators that confirmed the performance of the MLR-QSAR model, we performed another test related to the applicability domain (AD) of this model. This last test is performed to identify molecules that are probably outside the applicability domain of the MLR-QSAR model. In this work, the purpose of this test is to avoid the adoption of a molecular structure that is outside the applicability domain of the QSAR model during the design of novel compounds. Figure 2B presents the Williams plot of the applicability domain of the MLR-QSAR model developed in this work. Figure 2B shows that all molecules of the training and test sets did not violate the leverage threshold (h* = 2.5 × (k + 1)/n = 0.7), where k = 8 and n = 32. Similarly, the normalized residual values for all molecules in the training and test sets did not deviate outside the range ±x (x = 2.5). This means that the predictions of the MLR-QSAR model are accurate and reliable and that there is a high agreement between the pIC50 biological activities of all of the molecules examined in vitro with those predicted in silico. The successful placement of all molecules of the test and training sets in the AD indicates the validity of the MLR-QSAR model in the prediction of the activities of new molecules that can be designed on the basis of the screened cyclohexane-1,3-dione scaffolds.
We developed a nonlinear QSAR model based on the ANN technique32 to validate the suitability and ability of the eight molecular descriptors (S–B, HBA, CMA, PSA, TC, ET, EHOMO, and ELUMO) to describe precisely the biological inhibitory activity (pIC50), as well as to adopt them as a valid structural basis for improving pIC50 of the investigated molecules. Moreover, a feed-forward neural network model composed of three layers of neurons (input layer, hidden layer, and output layer) allows us to evaluate the correlation between pIC50 (output layer) and the molecular descriptors selected by the linear MLR model (input layer).32Figure 3A shows the architecture of the feed-forward neural network (FFNN) used in this study to develop the ANN-QSAR model, and Table 4 presents the statistical significance indexes for this model.
Figure 3.

(A) FFNN architecture (8-3-1). (B) Observed pIC50 values versus those predicted by the ANN-QSAR model.
Table 4. Statistical Parameters of the ANN-QSAR Model.
| ANN-QSAR
model | ||||||
|---|---|---|---|---|---|---|
| parameters | R2 | Radj2 | MSE | Rtest2 | Qcv2 | ρ85 |
| values | 0.98 | 0.94 | 0.01 | 0.97 | 0.76 | 1.03 |
| threshold | >0.6 | >0.6 | a low value | >0.6 | >0.5 | 1 < ρ < 3 |
The statistical parameters of the ANN-QSAR model presented in Table 4 indicate the significant correlation between the eight selected molecular descriptors and pIC50 biological inhibitory activity of the examined cyclohexane-1,3-dione derivatives. The high coefficient of correlation (98%) can be explained by the homogeneous distribution of pIC50 values observed and predicted by the nonlinear ANN-QSAR model (Figure 3B). This confirms that the transformation of the input data (molecular descriptors) into a new functional layer-level hidden layer space in the nonlinear ANN model provides more accurate output than the linear model that depends on only two layers (input layer and output layer). This can be confirmed by the observed gaps in the statistical significance indices of both MLR-QSAR (R2 = 0.85, Q2 = 0.65, Rtest2 = 0.82, MSE = 0.04) and ANN-QSAR (R2 = 0.98, Q2 = 0.76, Rtest = 0.97, MSE = 0.01) models. However, all internal and external validation parameters evaluated for both models indicate their high and adequate predictive ability. Besides, the MLR-QSAR model offers a clearer insight regarding the description of the relationship between molecular descriptors and biological activity, and this is represented in its output, which is a linear equation that can be analyzed and described based on the coefficients of each molecular descriptor. While the nonlinear ANN-QSAR model provides good predictions in the output layer, the description of the molecular descriptor parameter effects remains unclear for follow-up and analysis. Therefore, the application of the MLR-QSAR model to predict the biological activity of molecules based on the selected molecular descriptors is more rational in describing the structure–activity correlation.
Through the overall results of the QSAR modeling analyses developed in this work, it can be concluded that the biological inhibitory activity pIC50 against NSCLC of the investigated heterocyclic molecules is strongly correlated with the eight molecular descriptors (S–B, HBA, CMA, PSA, TC, ET, EHOMO, and ELUMO). Therefore, we can use these descriptors as structural keys of cyclohexane-1,3-dione derivatives useful to design new compounds with higher biological activity against NSCLC. Table S5 presents the computed physicochemical and quantum descriptors as well as the observed and predicted pIC50 values for the investigated molecules.
3.1.2. Molecular Descriptor Interpretations
Through the normalization coefficient diagram shown in Figure 4, we can describe the effect of the eight selected molecular descriptors on the pIC50 biological inhibitory activity.
Figure 4.

Normalized coefficients of each selected molecular descriptor.
According to the coefficient normalization diagram shown in Figure 4, the stretch–bend (S–B) descriptor has a positive coefficient, which means that the investigated compounds are more bioactive the higher the stretch–bend energy. Additionally, we can see that the hydrogen bond acceptor (HBA) coefficient is positive, indicating that the more hydrogen bonds a molecule can accept, the more active it will be biologically against NSCLC. The creation of noncovalent interactions through hydrogen bonds with the target protein’s receptor pocket’s active amino acid sites can account for this. Additionally, the CMA descriptor coefficient’s negative sign shows that the pIC50 of the molecule decreases as the molecular surface area increases. This can be explained by the rise in energy required to get past the solvent molecules’ coherent interactions and create a cavity for the solute molecules. This may be because water molecules interact more cohesively than lipid molecules do, which explains why larger molecules exhibit lipid-like properties. Therefore, we can draw the conclusion that small-size groups should be added to the modeled structures or large-size groups should be replaced by smaller-size groups to improve the biological activity of the molecular structures studied in this work. The PSA descriptor coefficient has a negative sign, which means that as the polar surface area of the biomolecule structure increases, its biological activity decreases. This might be because molecules with a high PSA index find it challenging to pass through cell membranes. As a result, in the current work, we check that the PSA index is low for the molecules that can be modeled to enhance the biological activity of the studied biomolecular structures and facilitate the absorption of auxiliary biomolecular structures. Moreover, the negative sign of the TC coefficient indicates that the more TC is increased, the more the biological activity of the molecule is decreased. The TC index has been interpreted as a steric parameter associated with the difficulty degree of molecule passage in cell membranes.86 Molecules with a low TC index can easily penetrate the cell membrane and gain access to living organisms. Therefore, compounds with small TC values will lead to high biological activity.
In terms of how electronic quantum descriptors affect pIC50, the negative total energy coefficient (ET) shows that a molecule has higher biological activity if its total energy is more stable (more negative). The symmetries of the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO), according to Frontier molecular orbital (FMO) theory, are also crucial for foretelling molecular reactivity.87 The biological inhibitory activity of the molecule increases with the number of stable electrons (more negative energy) in the HOMO level of the molecule, as shown by the negative sign of the descriptor coefficient (EHOMO) in Figure 4.
Conversely, the higher the number of stable electrons in the LUMO level (more negative energy), the lower the biological activity of the molecule will be, according to the positive value of the descriptor coefficient (ELUMO). Therefore, compared to molecules with high LUMO energy, those with low LUMO energy have a better ability to accept electrons. On this basis, electron-donating groups should be replaced by electron-accepting groups to reduce the LUMO energy of the studied molecules and increase their biological activity. By examining the structural features of heterocyclic compounds based on cyclohexane-1,3-dione that influence their biological inhibitory activities toward non-small-cell lung cancer cell lines, it is significant that the bioactivity of these molecules is highly correlated with their physicochemical and electronic molecular descriptors (S–B, HBA, CMA, PSA, TC, ET, EHOMO, and ELUMO). To generate novel heterocyclic small molecules and predict their inhibitory activity against NSCLC before moving forward with synthesis and in vitro tests, we can adopt the predictions of the MLR-QSAR model adjusted by machine.
3.1.3. Screening Appropriate Scaffold for Ligand-Based Drug Design
We can screen the bioavailability and pharmacodynamic parameters of the entire database of 40 cyclohexane-1,3-dione derivatives at this stage of screening to identify candidate scaffolds for drug design.58,88 To simplify the procedure, we excluded all molecules that demonstrated weak biological inhibitory activities against the cancer cell lines tested in vitro. As a result, we investigated the drug-like properties and toxicity risks of 19 molecules that demonstrated the most powerful inhibitory activity against the cancer cell lines evaluated in vitro.5 This in silico pharmacokinetic model included a screening of the 19 compounds (3b, 5c, 5d, 6b, 6c, 6d, 8a, 8b, 10c, 10d, 12a, 12b, 16c, 17d, 17e, 17f, 19c, 20c, and 22c) using the standard drug Foretinib as a control for comparison and validation.
Table 5 presents the in silico drug-like property profile obtained in this work for the selected 19 molecules. In this in silico drug-like property screening, we evaluate the bioavailability parameters of 19 synthesized molecules based on the rules of Lipinski, Veber, and Egan.89−91 This is due to the importance of these rules in the discovery of appropriate drugs for oral use. In addition, we evaluated the TPSA and n-ROTB indices due to their importance in predicting the flexibility level of the molecules.92 We also evaluated the synthetic accessibility index of the molecules in vitro (SA)93 and predicted the toxicity risks of the synthesized molecules by AMES and hepatotoxicity estimations.94,95
Table 5. Drug-Like Property Screeninga.
| drug-likeness |
toxicity |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| entry | MW (Da) | n-ROTB | n-HBA | n-HBD | TPSA | Lipinski | Veber | Egan | Log P | SA | AMES | hepatotoxicity |
| rule | <500 | <10 | <10 | ≤5 | <140 Å2 | yes/no | ≤5 | 0 < SA < 10 | yes/no | |||
| 3b | 206.26 | 1 | 2 | 0 | 62.38 | yes | yes | yes | 1.25 | 2.74 | no | no |
| 5c | 286.37 | 1 | 2 | 1 | 123.36 | yes | yes | yes | 1.25 | 3.27 | yes | yes |
| 5d | 333.43 | 4 | 3 | 1 | 125.87 | yes | yes | yes | 1.99 | 3.63 | yes | yes |
| 6b | 257.24 | 1 | 5 | 1 | 83.46 | yes | yes | yes | –0.17 | 3.78 | no | no |
| 6c | 258.34 | 1 | 2 | 1 | 87.28 | yes | yes | yes | 1.59 | 3.97 | yes | yes |
| 6d | 259.32 | 1 | 3 | 1 | 81.49 | yes | yes | yes | 1.59 | 3.61 | no | no |
| 8a | 339.43 | 2 | 2 | 0 | 95.47 | yes | yes | yes | 2.02 | 3.57 | yes | no |
| 8b | 355.50 | 2 | 1 | 0 | 110.57 | yes | yes | yes | 2.90 | 3.60 | yes | no |
| 10c | 232.30 | 1 | 2 | 2 | 95.30 | yes | yes | yes | 1.10 | 3.01 | yes | no |
| 10d | 384.50 | 4 | 2 | 1 | 70.45 | yes | yes | yes | 4.10 | 3.71 | yes | yes |
| 12a | 218.21 | 1 | 5 | 1 | 71.76 | yes | yes | yes | 0.27 | 3.49 | yes | yes |
| 12b | 234.27 | 1 | 4 | 1 | 86.86 | yes | yes | yes | 1.10 | 3.51 | yes | no |
| 16c | 245.27 | 3 | 3 | 1 | 55.40 | yes | yes | yes | 0.83 | 2.26 | no | no |
| 17d | 356.44 | 5 | 3 | 2 | 109.66 | yes | yes | yes | 2.10 | 3.72 | no | no |
| 17e | 325.38 | 3 | 3 | 2 | 116.38 | yes | yes | yes | 0.82 | 3.30 | yes | no |
| 17f | 372.44 | 6 | 4 | 2 | 118.89 | yes | yes | yes | 1.55 | 3.71 | no | yes |
| 19c | 320.79 | 3 | 2 | 2 | 100.43 | yes | yes | yes | 2.15 | 2.96 | yes | yes |
| 20c | 387.84 | 6 | 4 | 2 | 127.30 | yes | yes | yes | 1.48 | 3.37 | yes | yes |
| 22c | 437.96 | 2 | 2 | 1 | 110.48 | yes | yes | yes | 4.36 | 3.73 | yes | no |
| Foretinib | 632.65 | 14 | 10 | 2 | 111.25 | yes | no | yes | 2.62 | 4.05 | no | yes |
MW: molecular weight, n-ROTB: number of rotatable bonds, n-HBA: number of hydrogen bond acceptors, n-HBD: number of hydrogen bonds donors, TPSA: topological polar surface area, drug-likeness: applicable (yes) or not applicable (no), Log P: logarithm of partition coefficient of compound between n-octanol, SA: synthetic accessibility, AMES: AMES toxicity test, and hepatotoxicity: hepatotoxicity index.
From the screening results presented in Table 5, we can note that all 19 molecules fulfill Lipinski, Veber, and Egan rules regarding the bioavailability of candidate drug molecules. It is also indicated by the TPSA < 140 Å and n-ROTB < 10 scores that the molecules have a high ability to easily penetrate the brain barrier and flexibly interact with the therapeutic target. Also, we can notice that the SA values of the examined molecules were between 2.26 and 3.73, while the synthetic accessibility value of Foretinib was 4.05. This means that small molecules derived from cyclohexane-1,3-dione can be synthesized easily compared to the synthesis of Foretinib.
On the other hand, according to the predictions of molecule toxicity, it appears that the majority of the synthesized molecules are toxic according to AMES and hepatotoxicity tests, except for the molecules (3b, 6b, 6d, 16c, and 17d) that are nontoxic. Also, we can notice that the standard drug Foretinib is predicted to be hepatotoxic. Therefore, we can conclude that the in vitro screening of the 19 molecules based on the IC50 range of compounds is not sufficient to describe the drug-like properties of the compounds. As a result, when the 19 molecules are examined in silico, we observe only five molecular structures among the 19 possess drug-like properties. Therefore, for the purpose of predicting suitable biomolecular structures to design new biomolecules in this work, all toxic and hepatotoxic molecules are filtered out. Table 6 shows the SMILES structure of the compounds selected in silico with their pIC50 values observed in vitro and predicted by the QSAR models. We have kept the Foretinib scaffold for future comparison, although it has potential hepatotoxicity.
Table 6. Biological Inhibitory Activity of Scaffold Candidates.
| obs(pIC50) | pred(pIC50) MLR | pred(pIC50) ANN | ||
|---|---|---|---|---|
| com. | SMILES | in vitro | in silico | |
| 3b | O=C1C(C(CCC1)=O)=CC2=CC=CS2 | 6.41 | 6.035 | 6.598 |
| 6b | O=C1C([C@H](C2=CC=CO2)C(C#N)=C(O)O3)=C3CCC1 | 6.09 | 5.950 | 6.200 |
| 6d | OC(O1)=C(C#N)[C@@H](C2=CC=CS2)C3=C1CCCC3 | 6.38 | 6.888 | 6.354 |
| 16c | O=C(C1=CNC2=CC=C(OC)C=C2)CCCC1=O | 6.17 | 5.983 | 6.33 |
| 17d | O=C(OCC)C1=C(N)SC2=C1/C(C(CC2)=O)=C/NC3=CC=C(C)C=C3 | 5.78 | 5.886 | 5.64 |
| Foretinib | COC1=CC2=C(C=CN=C2C=C1OCCCN3CCOCC3)OC4=C(C=C(C=C4)NC(=O)C5(CC5)C(=O)NC6=CC=C(C=C6)F)F | 6.67 | ||
To select the best scaffold candidate structure for modeling new heterocyclic compounds against NSCLC, we count on the criterion of observed and predicted pIC50 values. Table 6 shows that the molecular structures 3b (6.41, 6.035, 6.598) and 6d (6.38, 6.88, 6.354) have higher pIC50 biological inhibitory activity than the other three molecules 6b (6.09, 5.95, 6.20), 16c (6.17, 5.983, 6.33), and 17d (5.78, 5.88, 5.64). As the observed pIC50 values are very close for molecules 3b (6.41) and 6d (6.38), it is difficult to favor one over the other. Therefore, we base this on a comparison between the pIC50 values predicted by the MLR-QSAR/ANN-QSAR models developed in this study. Table 6 shows that molecule 6d (pIC50 = 6.88) has a higher predicted biological activity than 3b (pIC50 = 6.035). This means that the molecular descriptors proposed in the MLR-QSAR model are appropriate to improve the pIC50 biological activity of molecular 6d structure. Therefore, we adopt molecule 6d (Figure 5) as a scaffold for designing novel heterocyclic molecules as inhibitors of NSCLC by targeting the c-Me protein.
Figure 5.
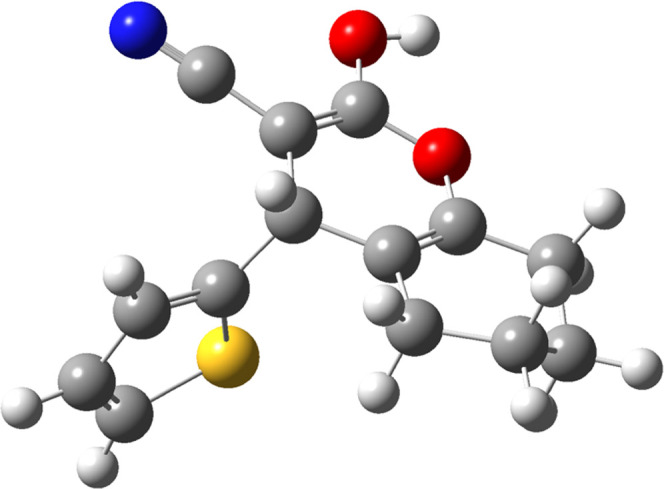
Molecular structure of scaffold 6d (2-hydroxy-4-(thiophen-2-yl)-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile).
3.2. Structural Characterization of the Scaffold Design
We perform molecular electrostatic potential (MEP) computations and molecular docking simulations based on the 6d scaffold structure in this section of this study with the main goal of highlighting the reactive sites in the 6d scaffold structure that are likely to interact with the active amino acid residues in the c-Met target protein pocket.
3.2.1. Molecular Electrostatic Potential Contour Maps
The 3D molecular electrostatic potential (MEP) contour maps can be used to identify reactive sites for potential electrophilic and nucleophilic attacks inside the c-Met active pocket. The MEP of the template compound (6d) is presented in Figure 6. The red color indicates negative electrostatic potential, which means that these sites are suitable for electrophilic attacks. The blue color indicates positive electrostatic potential, which means that these sites are suitable for nucleophilic attacks, while there are no potential reactive sites in the green contours.96
Figure 6.
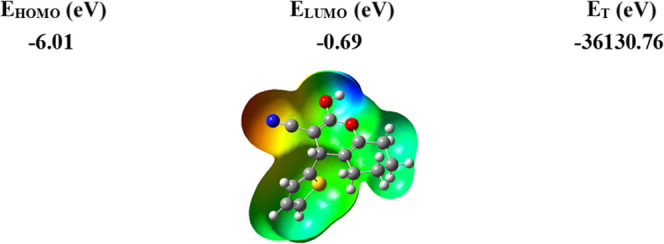
Molecular electrostatic potential map of the geometrically optimized compound 6d.
Through the 3D visualization (Figure 6), we can notice that the most negative potential in red is centered around the radical (CN), which means that this region is favorable for electrophilic attacks; this region is coded with (R1) on the structure of template molecule 6d. On the other hand, we can notice that the most positive potential in blue is arranged on the atoms of hydroxide H and oxygen O, indicating that these regions are favorable to nucleophilic attacks. These regions are coded with (R2) and (R3), respectively, on the structure of the template molecule 6d. Therefore, to enhance the molecular reactivity of the compound 6d, the (R1) and (R2, R3) sites must be substituted with groups whose properties favor electrophilic and nucleophilic attacks, respectively.
3.2.2. Molecular Docking Simulation
The structure of the target protein will be validated prior to performing molecular docking simulations between the scaffold (6d) and the c-Met protein (PDB code: 3LQ8). This is done to improve the accuracy and reliability of protein–ligand interaction predictions and to identify the active binding site in the target protein’s structure. This was done by redocking the ligand Foretinib into the active pocket of 3LQ8, evaluating the root mean square deviation (RMSD) of docking ligand superposition, binding energies (BE), and ligand–protein interactions.97Figure 7A,B shows the structure of the c-Met protein complexed with the native inhibitor Foretinib. Figure 7C–F shows the obtained redocking results based on the optimal conformation of Foretinib redocked inside the c-Met pocket (BE = −6.8 kcal/mol, RMSD = 0.2292 Å).
Figure 7.

(A) Crystalline 3D model of the c-Met protein (PDB: 3LQ8). (B) 2D visualization of native Foretinib structure interactions with c-Met active amino acid residues. (C–E) 3D visualizations of the superimposed pose between the original (black) and the redocked (yellow) Foretinib ligand in the c-Met pocket. (F) 2D visualization of the most important interactions between redocked Foretinib and amino acid residues of the c-Met receptor.
From Figure 7, we can notice that the original Foretinib ligand (Figure 7B) interacted with these amino acid residues: Phe1134, Ala1221, Ala1108, Met1160, Met1211, Gly1163, Tyr1159, Ile1084, Phe1223, Lys1110, and Asp1222. On the other hand, we can notice that the redocked Foretinib ligand (Figure 7F) interacts with all of the identical 11 residues with which the original ligand interacted. In addition, the redocked Foretinib ligand interacts with another four new residues, i.e., Met1131, His1202, Leu1157, and Pro1158. Also, the superimposed modes (RMSD = 0.2292 < 2 Å) shown in Figure 7C–E indicate the accurate predictions obtained by the molecular redocking simulation. Prediction results obtained through validation of the molecular docking protocol indicate the adaptation of the c-Met protein model for docking novel molecular structures to it. Therefore, in the rest of the work, molecular docking simulations can be reliably performed using the 3D model of c-Met (3LQ8) as well as the AutoDock Vina algorithms.
Figure 8 shows the docking pattern of ligand (6d) inside the active pocket of the c-Met receptor, as well as the most important interactions predicted in the most stable 6d–3LQ8 complex conformation (BE = −7.4 kcal/mol).
Figure 8.

(A) Ligands 6d and Foretinib in the c-Met receptor pocket. (B) 3D and 2D views of interactions between ligand 6d and the predicted active sites.
From Figure 8A, we can clearly notice that the 6d biomolecular structure is successfully docked into the pocket of the c-Met protein. Also, from Figure 8B, we can notice that the carbonitrile (R1) group interacted with active site Lys1110 (5.28 Å) by one conventional hydrogen bond. The hydroxide group (R2) and the oxygen atom (R3) interact with the active site Asp1222 with two hydrogen bonds (2.46 and 2.04 Å, respectively). Also, the thiophene ring interacted with the Glu1127 site (4.09 Å) through the π–anion electrostatic bond. Also, the thiophene ring interacted with both Glu1127 (4.09 Å) and Asp1222 (5.27 Å) sites through the two electrostatic π–anion bonds. Moreover, we can notice that the ring structure (tetrahydro-4H-chromene) interacted with the active sites Phe1200 (4.90 Å), Phe1134 (4.85 Å), His1202 (5.41 Å), and Met1131 (4.28 Å) through hydrophobic alkyl and π–alkyl interactions. Moreover, we can notice that ligand 6d interacted with five referential active sites in the inhibition of c-Met enzymatic activity by Foretinib, namely, Phe1134, His1202, Asp1222, Met1131, and Lys1110. Also, the binding energy of scaffold 6d (−7.4 kcal/mol) is higher than that of Foretinib (−6.8 kcal/mol) to the c-Met receptor, which means that the structure of ligand 6d can be more suitable for c-Met activity inhibition compared to Foretinib.
As a result, the 6d scaffold structure may provide novel cyclohexane-1,3-dione derivatives as candidates for inhibiting NSCLC growth by targeting c-Met enzymatic activity. To develop a better understanding of the functional groups of the 6d scaffold that are most likely to bind to the active site of the c-Met protein, we visualized the surface of the c-Met–6d complex generated by molecular docking in 3D (Figure 9).
Figure 9.

Graphical description of (A) aromatic, (B) H-bond, (C) charge, (D) hydrophobicity, (E) ionizability, and (F) solvent accessibility surface area of c-Met around 6d.
Figure 9A–F shows the surfaces of the receptors in terms of the aromatic edge/surface, H-bond, charge, hydrophobicity, solvent access surface (SAS), and ionization susceptibility. Figure 9A representing the aromatic edge/face surface (blue (edge)/orange (face)) indicates that blue predominates over orange in the 6d ligand structure. This means that the position of the aromatic cycles in the 6d molecule is favorable for the edge alignment.
Also, the hydrogen bond donor/acceptor surface (Figure 9B, purple (donor)/green (acceptor)) is shown. We can notice the sites (R1, R2, R3) located in the purple-colored zone, which means that these sites are favorable for hydrogen bond donor groups.
The charge surface is shown (Figure 9C, colors blue (positive charge)/red (negative charge)). We can notice that the structure of ligand 6d surrounded by the colors close to blue means that the structure of ligand 6d is favorable to the positive charge.
The hydrophobic surface (Figure 9D, blue (hydrophilic)/brown (hydrophobic)) has been shown. We can notice that the R1, R2, and R3 sites on the 6d ligand structure are surrounded by brown, which means that these sites are favorable for hydrophobic groups.
The ionization surface is shown (Figure 9E, colors blue (basic)/red (acid)). We can notice that the structure of ligand 6d is favorable to basic conditions. The solvent accessibility surface (SAS) is shown (Figure 9F, blue (high SAS)/green (low SAS)). We can notice that the blue color predominates over the green color in the structure of ligand 6d. This means that the solvent accessibility surface area (SASA) is high, which means that the amino acid residues on the biosurface of the c-Met protein possess a high SAS tendency.
The overall results obtained from the structural characterization of the 6d molecule via QSAR, MEP, drug-like, and molecular docking indicate that the structure of the molecule 6d is suitable as a reference for the design of novel NSCLC inhibitors targeting the enzymatic activity of c-Met. Figure 10 summarizes the most important 2D/3D structural properties of cyclohexane-1,3-dione derivatives to enhance their biological inhibitory activity against NSCLCs and their binding to the c-Met tyrosine kinase receptor.
Figure 10.

Rationalization of key structural properties needed to design new drug molecules derived from cyclohexane-1,3-dione.
3.3. Design, Optimization, and Screening of Novel Cyclohexane-1,3-dione Derivatives
Based on the summary of favorable structural properties for improved biological activity against non-small-cell lung cancer shown in Figure 10, a series of 36 heterocyclic compounds were designed based on the chemical reactions of cyclohexane-1,3-dione with radicals that had favorable properties to enhance the biological activity against NSCLC (Figure S1). We have designed these molecules by suggesting different reaction mechanisms, including the Michael reaction appropriate for the synthesis of such compounds, following the same synthesis protocol used in some of our previous work.98,99Figure 11 shows the synthetic pathways adopted in the design of the proposed new molecular structures.
Figure 11.

Synthetic pathways adopted to design new molecular structures.
The new molecular designs are prepared and optimized using the same approach as used in this work. The molecular descriptors (S–B, HBA, CMA, PSA, TC, ET, EHOMO, and ELUMO) are calculated after optimized structures of the proposed compounds (H01–H36) by DFT/B3LYP/6-31G(d,p) method. The inhibitory activity values predicted for the 36 new molecular structures are shown in Table S6 in the Supporting Information.
The pIC50 values corresponding to the engineered molecular structures were predicted by the MLR-QSAR model, which validated in this study its success in describing the structure–activity relationship for cyclohexane-1,3-dione derivatives. From Table S6, we can notice that the predicted pIC50 values for the new molecules ranged from 5.28 to 7.92. Among the 36 new molecules, we notice that the biological inhibitory activity against NSCLC was improved for 11 new molecules H01(pIC50 = 7.06), H06(pIC50 = 7.22), H12(pIC50 = 7.19), H18(pIC50 = 7.01), H20(pIC50 = 7.49), H21(pIC50 = 7.02), H24(pIC50 = 7.32), H30(pIC50 = 7.62), H31(pIC50 = 7.05), H32(pIC50 = 7.31), and H36(pIC50 = 7.92). All 11 molecules showed a higher predicted pIC50 activity compared to the activity observed for both reference structure 6d (pIC50 = 6.38, pred(pIC50 = 6.88)) and Foretinib (pIC50 = 6.67). On this basis, 11 new compounds were identified as strong inhibitors against NSCLC cell proliferation compared to scaffold 6d and Foretinib.
3.3.1. In Silico ADME/Tox Screening
To filter molecules for medicinal use, they must have favorable absorption, distribution, metabolism, excretion, and toxicity (ADME-Tox) properties.100 For this purpose, the drug-like and ADME-Tox models of the 11 proposed compounds were predicted using the SwissADME and pkCSM online servers. In view of the importance of drug-like property assessment as an initial step in the drug development pathway, this procedure measures the level of correlation between the physical and chemical properties of the compound with its bioavailability properties in the human body.101 Therefore, drug-like assessment perspectives allow us to predict the level of influence of drug candidate molecules on oral bioavailability. Table 7 shows the drug-like predictions based on the Lipinski, Veber, and Egan criteria (such as absorption, topological polar surface area, number of rotatable bonds, molecular weight, Log P), the bioavailability score, and the synthetic accessibility (SA) index.
Table 7. Predicted Drug-Like Property Profile of the Candidate Compounds.
| entry | TPSA(Å2) | n-ROTB | MW (Da) | WLog P | n-HBA | n-HBD | Lipinski’s violations | Veber violations | Egan violation | bioavailability score | SA | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rule | ABS | <140 | <10 | <500 | ≤5 | <10 | <5 | <2 | <2 | <2 | F > 0.1 | pains | 0 < SA < 10 |
| H01 | high | 89.25 | 1 | 256.26 | 2.09 | 4 | 1 | 0 | 0 | 0 | 0.56 | 0 | 3.92 |
| H06 | high | 76.11 | 1 | 300.74 | 3.15 | 3 | 1 | 0 | 0 | 0 | 0.56 | 0 | 3.77 |
| H12 | high | 78.62 | 4 | 347.79 | 3.19 | 4 | 1 | 0 | 0 | 0 | 0.56 | 0 | 4.09 |
| H18 | high | 70.32 | 1 | 301.72 | 3.75 | 4 | 1 | 0 | 0 | 0 | 0.85 | 0 | 3.62 |
| H20 | high | 101.07 | 4 | 320.36 | 3.20 | 5 | 1 | 0 | 0 | 0 | 0.56 | 0 | 4.01 |
| H21 | high | 72.83 | 4 | 314.33 | 3.14 | 5 | 1 | 0 | 0 | 0 | 0.85 | 0 | 3.96 |
| H24 | high | 72.83 | 4 | 348.78 | 3.79 | 5 | 1 | 0 | 0 | 0 | 0.85 | 0 | 3.92 |
| H30 | high | 73.98 | 1 | 298.72 | 3.50 | 4 | 1 | 0 | 0 | 0 | 0.55 | 0 | 2.59 |
| H31 | high | 89.63 | 4 | 301.29 | 2.74 | 6 | 1 | 0 | 0 | 0 | 0.55 | 0 | 3.08 |
| H32 | high | 104.73 | 4 | 317.36 | 3.21 | 5 | 1 | 0 | 0 | 0 | 0.55 | 0 | 3.04 |
| H36 | high | 76.49 | 4 | 345.78 | 3.80 | 5 | 1 | 0 | 0 | 0 | 0.55 | 0 | 2.95 |
The drug-like property assessments presented in Table 7 indicate that all 11 proposed molecules meet the Lipinski, Viber, and Egan criteria suitable for use as a drug. The absence of the pain (0 alert) index also indicates that the positive biological results of the 11 proposed structures are not false.63
Also, the score (F > 10%) of the proposed molecules indicates that the bioavailability properties of the proposed structures match the five Lipinski rules.102 The synthetic accessibility index score of the proposed structures indicates that the SA values range from 2.95 to 4.09 (far from 10), which means that the synthetic potential of the proposed molecules in vitro is very easy.103
The overall drug-like properties evaluated indicate a strong correlation between bioavailability and the proposed molecular structures. Thus, the 11 new molecular structures are likely to be suitable for medicinal use. To test this hypothesis, we evaluate the pharmacokinetic and pharmacodynamic properties of these molecules in silico through the prediction of ADME-Tox modeling. The ADME-Tox profile of the 11 compounds that passed the oral bioavailability screening is shown in Table 8. The ADME-Tox properties of these candidate drugs were predicted using the scaffold 6d structure and the standard drug Foretinib as references. Oral drugs are absorbed in the human small intestine, so assessment of the absorption index of drugs is important in predicting the fullness of an oral drug. Molecules with an absorption index of less than 30% are poorly absorbed in the human intestine.64 Based on the predictions of the absorption index obtained in Table 8, we can notice that all examined molecular structures have absorption rates above 90%. This means that the absorption potential of these molecules in the human intestine is high, thus increasing the potential for therapeutic access by these molecules. Also, the skin permeability index (log Kp) was evaluated for its critical significance in predicting skin sensitivity to chemical compounds. The lower Kp (in log cm/s) indicates the lower skin permeability of substances; therefore, the risk of contact with these substances decreases and the security of their use as drugs increases.81 Predictions of the Kp profile of the investigated molecules indicate that all Kp values are less than −2.5; therefore, all proposed molecules and 6d and Foretinib have low skin permeability and no potential dermatological effect.
Table 8. In Silico Predicted ADMET Properties.
| ADME-Tox properties | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
|
metabolism |
|||||||||||
| |
|
CYP |
|||||||||||
| absorption |
distribution |
substrate |
inhibitor |
||||||||||
| models | intestinal absorption (human) | skin permeability | VDss (human) | fraction unbound (human) | BBB permeability | CNS permeability | 2D6 | 3A4 | 1A2 | 2C19 | 2C9 | 2D6 | 3A4 |
| unity | numeric (%absorbed) | numeric (log Kp) | numeric (Log L/kg) | numeric (Fu) | numeric (Log BB) | numeric (Log PS) | categorical (yes/no) | ||||||
| Predicted Values | |||||||||||||
| Fore | 92.836 | –2.735 | –0.026 | 0.326 | –2.02 | –3.728 | no | yes | no | no | no | no | yes |
| 6d | 90.719 | –2.714 | 0.26 | 0.159 | 0.047 | –1.749 | no | yes | yes | yes | no | no | no |
| H01 | 95.431 | –3.304 | –0.145 | 0.307 | –0.209 | –3.933 | no | yes | no | no | no | no | no |
| H06 | 93.642 | –3.086 | 0.102 | 0.09 | –0.094 | –2.913 | no | yes | no | yes | no | no | no |
| H12 | 94.507 | –3.913 | –0.094 | 0.078 | –0.258 | –3.175 | no | yes | no | yes | no | no | no |
| H18 | 92.339 | –3.581 | 0.049 | 0.04 | –0.155 | –2.819 | no | yes | yes | yes | no | no | no |
| H20 | 93.067 | –3.676 | –0.102 | 0.123 | –0.385 | –3.197 | no | yes | no | yes | no | no | no |
| H21 | 94.658 | –3.51 | –0.056 | 0.078 | –0.167 | –3.173 | no | yes | no | no | no | no | no |
| H24 | 92.998 | –3.479 | –0.082 | 0.042 | –0.319 | –3.059 | no | yes | no | no | no | no | no |
| H30 | 93.241 | –2.981 | 0.089 | 0.041 | –0.233 | –2.893 | no | yes | yes | yes | yes | no | no |
| H31 | 95.908 | –2.766 | –0.365 | 0.205 | –0.577 | –3.082 | no | no | yes | no | no | no | no |
| H32 | 93.356 | –2.782 | –0.324 | 0.027 | –0.451 | –3.367 | no | yes | no | yes | no | no | no |
| H36 | 93.408 | –2.764 | –0.289 | 0 | –0.383 | –3.224 | no | yes | no | yes | yes | no | no |
| ADME-Tox
properties | ||||||
|---|---|---|---|---|---|---|
| excretion | toxicity |
|||||
| models | total clearance | AMES toxicity | oral rat acute toxicity (LD50) | oral rat chronic toxicity (LOAEL) | hepatotoxicity | skin sensitization |
| unity | numeric (log mL/(min kg)) | categorical (yes/no) | numeric (mol/kg) | numeric (log mg/kg_bw/day) | categorical (yes/no) | categorical (yes/no) |
| Predicted Values | ||||||
| Fore | 1.145 | no | 2.665 | 2.832 | yes | no |
| 6d | 0.015 | no | 2.728 | 1.988 | no | no |
| H01 | 0.585 | no | 2.866 | 0.888 | no | no |
| H06 | –0.091 | no | 2.758 | 1.278 | no | no |
| H12 | –0.024 | no | 2.905 | 1.121 | no | no |
| H18 | 0.05 | no | 2.946 | 1.308 | no | no |
| H20 | 0.194 | no | 3.032 | 1.113 | no | no |
| H21 | 0.504 | no | 2.870 | 1.172 | no | no |
| H24 | 0.121 | no | 3.102 | 1.106 | no | no |
| H30 | 0.025 | no | 2.664 | 0.823 | no | no |
| H31 | 0.822 | no | 3.077 | 1.393 | yes | no |
| H32 | 0.194 | yes | 2.683 | 0.816 | yes | no |
| H36 | 0.122 | no | 2.645 | 0.800 | no | no |
In terms of the distribution index, the volume of distribution (VDss), the unbound fraction (FU), the blood–brain barrier (BBB), and central nervous system (CNS) permeability were predicted. The larger the volume of distribution, the more widespread drug distribution in body tissues. The higher the FU levels of the molecules, the more likely they are to bind to plasma rather than the target protein, resulting in low pharmacological activity of the molecules. The negative VDss indices obtained indicate that most of the proposed molecules, including Foretinib, show a limited distribution at the bloodstream pathway in the body. Except for the molecules 6d, H18, and H30, their VDss values are positive, which means that these molecules are likely to spread to the tissues of the body.104
The unbound fractions (FU) of the molecules examined in human plasma ranged from 0 to 0.3, which greatly increases the possibility of binding the proposed molecular structures to the c-Met protein and thus achieving the desired therapeutic target. The capacity of the investigated molecules on the permeability of the blood–brain barrier (BBB) and the central nervous system (CNS) was evaluated. Due to the importance of these two parameters in the pharmaceutical industry, they are used to determine whether a drug affects the brain. A drug has the ability to cross the BBB if it has log BB > 0.3, whereas drugs that have log BB < −1 are poorly able to cross the BBB.64 Therefore, candidate drugs with log PS > −2 are considered able to penetrate the CNS, while those with log PS < −3 are considered poorly able to penetrate the CNS. Fortunately, the results shown in Table 8 indicate that all of the proposed new compounds and Foretinib, with the exception of the reference compound 6d, have an acceptable range and are negative in the blood. Although compound 6d showed a positive log BBB value, it did not reach a threshold of 0.3, so the possibility of this molecule crossing the blood–brain barrier remains relative.
In terms of metabolism, the cytochrome CYP450 enzymes are responsible for removing toxins from the body.105 This is done through the oxidation of intrusive foreign substances, including drugs. These enzymes facilitate the process of clearance, and cytochromes 1A2, 2C9, 2C19, 2D6, and 3A4 have been reported to be the most important enzymes responsible for the metabolism of over 90% of drugs after the first stage of treatment.32 Co-enzyme 3A4 is considered the most important cytochrome enzyme responsible for drug metabolism in the human body. If the drug is an inhibitor of 3A4, the ability of the enzyme to decompose the drug is reduced, resulting in increased side effects of the drug. Conversely, when the drug is a 3A4 substrate, the ability of the enzyme to metabolize the drug is increased, and the drug is not rapidly rejected, thus increasing the potential for therapeutic access and reducing unwanted side effects. The predicted inhibitory actions against CYP3A4 indicated that all of the investigated molecules did not have an inhibitory interaction against this enzyme as well as for CYP2D6. Furthermore, most of the candidate molecules were substrates of CYP3A4, with the exception of compound H31.
Regarding the excretion index, this index is very important in predicting the behavior of the liver in terms of detoxification.106 A lower excretion index (total clearance) means that the drug has a longer half-life of stability in the body, thus the higher the probability of achieving the therapeutic target by the drug.107Table 8 displays that the total clearance index of all proposed molecules was less than 1 (0.015–0.58), whereas the total clearance index of Foretinib was greater than 1 (1.145). This means that the stability of the 11 proposed molecules and the 6d molecule in the body may be better than Foretinib.
The assessment of a molecule’s toxicity potential is critical in pharmaceutical chemistry. The toxicity of the investigated molecules was assessed in this study by predicting AMES and acute oral toxicity for rats (LD50), chronic oral toxicity for rats (LOAEL), hepatotoxicity, and skin sensitivity. The predictions of the AMES test presented that the toxicity of H31 is positive, which means that H31 is mutagenic and has potential toxic symptoms. Similarly, the prediction of hepatotoxicity indicates that H31, H32, and Foretinib have hepatotoxic effects, while the rest of the examined compounds showed no potential toxicity according to the AMES test and hepatotoxicity. The LD50 and LOAEL indicators also indicate that the potential toxicity of the proposed compound may occur at very high doses, which is not likely to occur due to the micromolar IC50 dose values of the investigated compounds. The skin sensitivity test indicates the negativity of the investigated candidates, which confirms the safety of contact with the proposed drug candidates.
The ADME-Tox property evaluations obtained indicate the suitability of molecular structures (H01, H06, H12, H18, H20, H21, H24, H30, and H36) as potential reference structures in drug design for NSCLC therapy. The structure of the molecules (H31 and H32) was excluded due to the potential toxicity of these molecules. Based on the ADME-Tox predictions, we can conclude that the structures of the nine selected lead compounds are suitable for drug discovery and development targeted at NSCLC therapy. This also suggests that the structural properties of the scaffold 6d were appropriate for the design and optimization of the nine proposed lead compounds.
3.3.2. c-Met Targeting by Lead Compounds
We perform molecular docking simulations between the c-Met protein (PDB code: 3LQ8) and the structures of nine molecules screened as potential drugs for NSCLC. This is needed to evaluate the most likely interactions between the active pocket of the c-Met protein and candidate ligands as NSCLC cell growth inhibitors. The noncovalent interactions predicted by the molecular docking simulations and the recorded binding energy values are used to identify the optimal candidate ligand conformations required to interact with the target c-Met protein. In addition, we perform MM-GBSA simulations under the VSGB 2.0 solvation model to recalculate and estimate the free binding energy (ΔGbind) yielded by noncovalent cross-binding between c-Met and the proposed nine ligands (H01, H06, H12, H18, H20, H21, H24, H30, and H36). This allows us to reinforce the hypothesis regarding the most stable Protein–ligand systems and to cross-check the molecular docking predictions. Figure 12 presents 2D visualizations of key potential interactions between the nine candidate drug ligands and the active residue sites in the c-Met protein pocket (PDB code: 3LQ8). Table 9 presents summary results of the molecular docking and MM-GBSA simulations obtained in this section.
Figure 12.

Key potential interactions between 3LQ8 and designed compounds (A) H01, (B) H06, (C) H12, (D) H18, (E) H20, (F) H21, (G) H24, (H) H30, and (I) H36. (J) Superimposed poses of designed compounds with Foretinib inside the pocket of 3LQ8.
Table 9. Syntheses of Molecular Docking and MM-GBSA Free Binding Energy Simulations.
| complexes | binding affinity (kcal/mol) | ΔGbind (MM-GBSA) (kcal/mol) | ΔGbind vdW (MM-GBSA) (kcal/mol) | complex energy (kcal/mol) | ΔGbindH-bond(kcal/mol) | hydrogen-binding interaction | electrostatic interaction | van der Waals interaction | hydrophobic interaction |
|---|---|---|---|---|---|---|---|---|---|
| 3LQ8–H01 | –7.7 | –18.10 | –23.55 | –10 961.10 | –1.64 | Glu1127 (2.24 Å) | Asp1222 (3.56 Å) | His1202, Asp1204, Gln1123, Leu1225, Gly1224, Phe1200, Ile1130 | Arg1203 (5.19 Å) |
| 3LQ8–H06 | –7.9 | –20.05 | –34.28 | –10 975.14 | –1.64 | Asp1222 (2.73 Å), His1202 (2.25 Å) | Asp1222 (4.10 Å), Glu1127 (4.34 Å) | Val1201, Phe1200, Arg1203, Asp1204, Glu1224, Gln1123 | Met1131 (4.52 Å), Ile1130 (5.41 Å) |
| 3LQ8–H12 | –8.1 | –20.32 | –33.79 | –10 980.51 | –1.84 | His1202 (1.95 Å), Asp (1222), Arg1203 (3.12 Å) | Glu1127 (4.37 Å), Asp1222 (4.07 Å) | Glu1224, Gln1123, Val1201, Asp1204 | Phe1200 (4.87 Å), Ile1130 (3.79 Å), Met1131 (4.52 Å) |
| 3LQ8–H18 | –6.4 | –15.26 | –26.07 | –10 988.99 | –0.04 | Asp1222 (3.75 Å) | Gln1123, Gly1224, Glu1127, His1202, Arg1203, Asp1204, Tyr1235 | Ile1130 (3.71 Å), Leu1225 (4.96 Å) | |
| 3LQ8–H20 | –7.8 | –19.72 | –33.69 | –11 014.74 | –0.11 | Asp1164 (2.89 Å) | Tyr1159, His1094, Lys1161, His1162, Asn1167, Gly1163, Met1211, Val1092, Ala1108, Met1160 | Ile1084 (3.75 Å), Ile1084 (4.70 Å), Phe1223 (5.22 Å) | |
| 3LQ8–H21 | –6.9 | –27.95 | –33.32 | –11 037.18 | –0.58 | Asp1164 (2.95 Å) | Gly1085, Asp1228, Asn1167, Phe1223, Val1092, Gly1163, Met1160, His1162, Lys1161 | Met1211 (3.82 Å), Ala1108 (5.0 Å), Ile1084 (3.53 Å), Tyr1159 (5.90 Å) | |
| 3LQ8–H24 | –6.4 | –21.71 | –36.37 | –11 019.17 | –0.10 | Ala1108, Met1160, Tyr1159, Val1083, His1162, Asn1167, Asp1164, Gly1163 | His1094 (4.72 Å), Ile1084 (3.68 Å), Ile1084 (4.59 Å), Met1211 (5.46 Å), Val1092 (5.31 Å), Phe1223 (5.14 Å) | ||
| 3LQ8–H30 | –8.3 | –22.65 | –47.49 | –10 976.08 | –1.06 | Lys1110 (3.15 Å) | Gln1123, Glu1127, Asp1222, Gly1224, Leu1157, Leu1140, Val1139, Val1220, Leu1195, Phe1200 | Ile1130 (5.40 Å), Ile1130 (4.82 Å), His1202 (5.47 Å), Phe1134 (5.41 Å), Ala1221 (4.27 Å), Ala1221 (4.74 Å), Met1131 (5.09 Å), Met1131 (4.03 Å) | |
| 3LQ8–H36 | –8.9 | –43.65 | –53.80 | –11 019.61 | –0.67 | Lys1110 (3.37 Å) | Glu1127 (4.74 Å) | Val1139, Asp1222, Leu1140, Gly1128, Leu1157, Phe1223, Gly1224, Gln1123, Phe1200, Phe1134, Val1220 | His1202 (5.43 Å), Ile1130 (4.77 Å), Ile1130 (5.38 Å), Ala1221 (4.84 Å), Ala1221 (4.25 Å), Met1131 (5.09 Å), Met1131 (4.06 Å), Val1155 (3.60 Å), Lys1110 (4.68 Å) |
The docking pattern of the nine lead compounds in the c-Met active pocket shown in Figure 12A–I shows that the main protein–ligand interactions generated were hydrogen bonds, electrostatic interactions, van der Waals (vdW) interactions, and hydrophobic interactions. The presence of these weak bonds increases the likeliness of compatibility between the c-Met receptor and proposed computational drug ligands. This is due to the importance of weak binding (noncovalent interactions) in drug molecule clearance after achieving the therapeutic target. With strong binding such as covalent interactions, it is difficult to easily get rid of the drug molecule bound to the receptor, which can lead to an adverse drug effect represented by toxic risks. Also, the docking poses of the designed drug molecules aligned with the Foretinib inhibitor in the c-Met active binding target site indicate that all compounds were successfully binding in the active pocket of this receptor (Figure 12J). This supports the hypothesis that the conformational structures of cyclohexane-1,3-dione scaffold small molecules are highly compatible with the c-Met protein tyrosine kinase. This can be explained by the significantly lower binding free energies recorded by molecular docking (−8.9 to −6.4 kcal/mol) and prime MM-GBSA simulations (ΔGbind, ΔGbind vdW, and ΔGbind H-bond ranging from −43.65 to −15.26, −53.8 to −23.55, and −1.64 to −0.04 kcal/mol, respectively) (Table 9). Furthermore, from Figure S2, we can notice that all noncovalent interactions generated between the nine examined ligands and the c-Met active site were preserved after the prime MM-GBSA simulations. The stability of these predicted interactions can strongly contribute to modeling the enzymatic activity of the c-Met protein and achieving the intended therapeutic goal.
3.3.3. Molecular Dynamics Simulations
To further explore the stability status of the designed lead compounds inside the c-Met active pocket, we perform molecular dynamics simulations on the four complexes 3LQ8–H06 (−7.9 kcal/mol), 3LQ8–H12 (−8.1 kcal/mol), 3LQ8–H30 (−8.3 kcal/mol), and 3LQ8–H36 (−8.9 kcal/mol), which exhibited higher negative binding affinity than the scaffold 6d (−7.4 kcal/mol) and Foretinib drug (−6.8 kcal/mol). Figure 13 depicts the free binding energy calculated by prime MM-GBSA for samples 3LQ8–H06 (ΔGbind = −20.05 kcal/mol), 3LQ8–H12 (ΔGbind = −20.32 kcal/mol), 3LQ8–H30 (ΔGbind = −22.65 kcal/mol), and 3LQ8–H36 (ΔGbind = −43.65 kcal/mol) used as inputs for MD simulations.
Figure 13.

Prime MM-GBSA free binding energy parameters for the best complexes.
In this regard, protein–ligand systems were simulated in an aqueous environment, and the structural and dynamical stability of c-Met protein free and complexed with lead compounds and standard drug Foretinib was evaluated over an independent time cycle of 100 ns. In this part of the investigation, the timelines of RMSD and RMSF parameter variations, protein–ligand contact profiles, ligand properties (RMSF, RMSD, rGyr, MolSA, intraHB, SASA, and PSA), and thermodynamic properties (total energy, potential energy, temperature, pressure, and volume) were analyzed.
3.3.3.1. RMSD and RMSF Analyses
We used the two measures, root mean square deviation (RMSD) and root mean square fluctuation (RMSF), to investigate potential structural fluctuations in the examined systems of 3LQ8 (uncomplexed and complexed with ligands). Using these two indices, the average movement of all atoms in the α-carbon backbone structure for each residue in the systems examined is calculated relative to the first reference frame of the protein backbone (uncomplexed 3LQ8) across a time trajectory fixed at 100 ns (Figure 14).
Figure 14.

(A) RMSD of free protein 3LQ8, complexed with ligands H06, H12, H30, H36, and Foretinib. (B) RMSF of backbone atoms in free 3LQ8 protein, complexed with ligands H06, H12, H30, H36, and Foretinib. (C) RMSF of ligands H06, H12, H30, H36, and Foretinib complexed with the 6JQR protein.
From Figure 14A, the data indicate clearly that all systems (free 3LQ8 and its complexes) showed minor fluctuations at the start of the simulation and then became stable after 20 ns of MD simulation. This stability indicates that the systems reach equilibrium after about the first 20 ns and then remain stable during the whole simulation time period. The average RMSD for free 3LQ8 and complexed with H06, H12, H30, H36, and Foretinib were obtained as 1.824, 2.011, 1.876, 2.420, 2.259, and 1.790 Å, respectively. The low values of RMSD fluctuation observed indicate that all of the systems investigated have reached the desired equilibrium. Therefore, it can be said that the complexes examined have a stable nature under physiological conditions.
RMSF analyses of the α-carbon atom residues of 3LQ8 uncomplexed and complexed with the selected ligands H06, H12, H30, H36, and Foretinib indicated average values of 0.904, 0.940, 0.879, 1.086, 1.112, and 0.921 Å, respectively (Figure 14B). The average RMSF level values of the residue atoms in the systems examined indicate a very high stability level of 3LQ8 uncomplexed and complexed with the various ligands. Some minor fluctuations in RMSF can be observed for some residues (ILE1053, ILE1118, ASP1054, GLU1061, ASN1100, and ASP1231), especially the extreme residues outside the active reference residue sites in the c-Met protein pocket. The RMSFs of each ligand atom were also determined to define the dynamic profile of the ligand structures in the c-Met protein pocket. The data presented in Figure 14C indicate the presence of certain fluctuations that signal some conformational dynamics in the evolution of ligands in their active binding sites. These slight fluctuations are probably due to the flexible nature of the ligand structures in the c-Met pocket.
3.3.3.2. Dynamics of Protein–Ligand Contact and Ligand Properties
The visualizations of the protein–ligand interactions provide a better view of the interaction profiles that provide a stable environment for the ligands in the pocket of the target c-Met protein. Figure 15A shows the various contact interactions between the ligands H06, H12, H30, H36, and Foretinib to active residue sites in the 3LQ8 protein during the 100 ns pathway of MD simulation.
Figure 15.


Diagram report of the MD simulation interactions over the 100 ns time: (A) protein–ligand contacts, (B) ligand–protein contacts, and (C) ligand properties.
From 2D visualizations of the interactions between the examined ligands and amino acid residues shown in Figure 15B, we can notice that the most important interactions that contributed to the stability of the systems examined were of type H-bonds, hydrophobic, ionic, and water bridges. Furthermore, the (protein–ligand contacts) and (ligand–protein contacts) plots of interaction modes of examined ligands with the 3LQ8 protein clearly show that the predicted key contacts with the molecular docking were almost conserved throughout the MD simulation time of 100 ns.
Also, six properties of the investigated ligands were analyzed, namely, the RMSD, radius of gyration (rGyr), molecular surface area (MolSA), intramolecular hydrogen bonding (intraHB), solvent accessible surface area (SASA), and polar surface area (PSA), respectively (Figure 15C). All of the dynamics measurements obtained indicate the high stability of the investigated systems over the 100 ns period of the MD simulation time.
3.3.3.3. Thermodynamic Property Analysis
Moreover, the summary MD quality simulation was analyzed by calculating the variation of total energy (E), potential energy (EP), temperature (T), pressure (P), and volume (V) along the 100 ns of the simulation period, and these analyses were done by subjecting the studied systems to simulation quality analysis.108 The plots of the thermodynamic properties calculated along 100 ns of MD simulation time for 3LQ8 free and complexed with ligands are presented in Figure 16. Through the thermodynamic plots obtained, we can notice that the energy parameters of the six investigated systems are very close and showed perfect stability throughout the MD simulation time. This can be confirmed by the average values of the parameters (E, EP, T, P, and V) presented in Table 10.
Figure 16.

Thermodynamic property analysis: (A) 3LQ8 uncomplexed, (B) 3LQ8–H06, (C) 3LQ8–H12, (D) 3LQ8–H30, (E) 3LQ8–H36, and (F) 3LQ8–Foretinib.
Table 10. Average Thermodynamic Property Scores for Systems Examined.
| thermodynamic properties |
|||||
|---|---|---|---|---|---|
| protein–ligand complex | total energy (kcal/mol) | potential energy (kcal/mol) | temperature (K) | pressure (bar) | volume (Å3) |
| 3LQ8 | –90 302.277 | –110 373.609 | 298.685 | 1.207 | 327 802.838 |
| 3LQ8–H06 | –90 109.333 | –110 165.406 | 298.674 | 0.707 | 327 539.865 |
| 3LQ8–H12 | –90 087.429 | –110 144.368 | 298.683 | 0.653 | 327 645.373 |
| 3LQ8–H30 | –90 340.770 | –110 444.722 | 298.685 | 1.157 | 328 390.322 |
| 3LQ8–H36 | –90 340.788 | –110 448.703 | 298.691 | 1.204 | 328 435.906 |
| 3LQ8–Foretinib | –89 995.713 | –110 065.429 | 298.683 | 0.797 | 327 581.987 |
The full MD analysis results demonstrated that the structures of the computational drug compounds (H06, H12, H30, and H36) were highly stable inside the c-Met protein pocket. This further validates the reason for screening the nine novel designed compounds as prime candidates for c-Met enzyme activity inhibition and drug use against NSCLC.
4. Conclusions
Cyclohexane-1,3-dione derivatives’ cytotoxicity against a variety of cancer cell lines, including non-small-cell lung cancer (NSCLC), makes them highly attractive for targeted cancer therapy. In this paper, we conduct a large-scale computational study on 40 heterocyclic small molecules with cydohexane-1,3-dione scaffolds that have inhibitory biological activity in vitro against the non-small-cell lung cancer cell line H460. The goal is to discover the most important structural features and properties of these molecules that are related to their biological activity against NSCLC, as well as to gain insights into how to optimize these molecules to become successful lead agents for drug design. To achieve the desired goals, we take a hybrid approach that combines ligand-based and structure-based drug design approaches.
In this context, based on statistical modeling techniques: principal component analysis (PCA), univariate analysis (UV), multiple linear regression (MLR), artificial neural network (ANN), Y-randomization, applicability domain (AD), and the structure–activity relationship between the studied compounds is modeled and interpreted by the QSAR modeling approach. Predictions of the MLR-QSAR model trained, validated internally and externally, and supported by ANN-QSAR modeling indicated that the biological inhibitory activity of cyclohexane-1,3-dione derivatives is strongly correlated with physicochemical and electronic molecular descriptors (S–B, HBA, CMA, PSA, TC, ET, EHOMO, and ELUMO). The compound 6d is identified as the optimal scaffold for the further design and optimization of new lead compounds based on QSAR modeling predictions and in silico pharmacokinetic and pharmacodynamic (ADME-Tox) evaluations of synthesized compounds.
Abnormal tyrosine kinase enzyme activity is most likely associated with cancer cell growth and proliferation; specifically, c-Met enzymatic activity was a significant factor associated with the cell growth of NSCLC that was focused on as part of this study. Thus, in the context of fostering the search for novel small-molecule tyrosine kinase inhibitors. In this study, we use the protein tyrosine kinase c-Met as a focal point for the structure-based drug design. In this context, the 6d scaffold is docked to the c-Met active pocket (PDB code: 3LQ8) and generates the most predicted protein–ligand interactions and the most important functional groups involved. Simultaneously, from the DFT calculations, the MEP contour maps generated on the 6d scaffold are analyzed. As a result, we obtain a well-defined and in-depth mapping of the pharmacokinetic sites favorable for the modifications to achieve strong noncovalent interactions with the receptor tyrosine kinase c-Met.
Based on the properties of the scaffold 6d of cyclohexane-1,3-dione derivatives and the pharmacophore models developed in this study, 36 novel small molecules are designed, optimized, and screened as new candidates for cancer targeting. Among the newly designed lead compounds, nine new compounds are selected as the best drug candidates against NSCLC, compared to the synthesized compounds and the standard drug Foretinib. This screening is performed on the basis of QSAR, ADME-Tox, molecular docking, and prime MM-GBSA simulations.
To further validate the obtained results, we investigated the stability dynamics of the designed drug molecules inside the c-Met protein active pocket by performing molecular dynamics simulations. For this, we consider the four systems 3LQ8–H06 (ΔGbind = −20.05 kcal/mol), 3LQ8–H12 (ΔGbind = −20.32 kcal/mol), 3LQ8–H30 (ΔGbind = −22.65 kcal/mol), and 3LQ8–H36 (ΔGbind = −43.65 kcal/mol), which showed that more negative free binding energy (ΔGbind) were used as validation samples. The MD simulation along the 100 ns trajectory showed high stability for all four c-Met complexes examined, alongside c-Met free and crystallized with Foretinib. Finally, the use of the hybrid drug design approach carried out in this study led to the successful development of a new class of heterocyclic small molecules based on the cyclohexane-1,3-dione scaffold for drug design against NSCLC and other types of cancer. Retrosynthesis of the newly designed compounds and evaluation of their inhibitory activity toward the enzymatic activity of c-Met and other receptor tyrosine kinases could be useful for further experimental and theoretical studies.
Acknowledgments
The author A.B.S. would like to acknowledge St. Mary’s College of Pharmacy and St. Mary’s Group of Institutions, Guntur, Andhra Pradesh, India. R.R.B. would like to thank the Deanship of Graduate Studies and Research, Ajman University, UAE, for their support in providing assistance in the article processing charges of this manuscript.
Data Availability Statement
All data used in this work are private.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c07585.
Structures of cyclohexane-1,3-dione derivatives and their biological inhibitory activities (pIC50) against NSCLC observed in vitro and predicted in silico; computed data of the molecular descriptors used in QSAR modeling, and correlation matrix data obtained via PCA for the molecular descriptors; statistical data supporting the Y-randomization validation test, as well as the newly designed structures of cyclohexane-1,3-dione derivatives (PDF)
Accession Codes
The codes used in this work are not available.
Author Contributions
O.D.: study design, writing—review and editing, investigation, formal analysis, conceptualization, lab work, and methodology. S.E.: supervision, software, validation, lab work, review and editing. M.B. and S.N.M.: data curation, review and editing. S.B., R.R.B., and A.B.S.: visualization and resources. S.C.: review and editing, software, methodology, validation.
The authors declare that this research received no financial support for research. However, funding for APC charges is received from the Deanship of Graduate Studies and Research, Ajman University, UAE.
The authors declare no competing financial interest.
Supplementary Material
References
- Pagliero R. J.; Lusvarghi S.; Pierini A. B.; Brun R.; Mazzieri M. R. Synthesis, Stereoelectronic Characterization and Antiparasitic Activity of New 1-Benzenesulfonyl-2-Methyl-1, 2, 3, 4-Tetrahydroquinolines. Bioorg. Med. Chem. 2010, 18, 142–150. 10.1016/j.bmc.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Barbuceanu S.-F.; Saramet G.; Almajan G. L.; Draghici C.; Barbuceanu F.; Bancescu G. New Heterocyclic Compounds from 1,2,4-Triazole and 1,3,4-Thiadiazole Class Bearing Diphenylsulfone Moieties. Synthesis, Characterization and Antimicrobial Activity Evaluation. Eur. J. Med. Chem. 2012, 49, 417–423. 10.1016/j.ejmech.2012.01.031. [DOI] [PubMed] [Google Scholar]
- Gümüş M.; Yakan M.; Koca İ. Recent Advances of Thiazole Hybrids in Biological Applications. Future Med. Chem. 2019, 11, 1979–1998. 10.4155/fmc-2018-0196. [DOI] [PubMed] [Google Scholar]
- Amr A. E.-G. E.; Sherif M. H.; Assy M. G.; Al-Omar M. A.; Ragab I. Antiarrhythmic, Serotonin Antagonist and Antianxiety Activities of Novel Substituted Thiophene Derivatives Synthesized from 2-Amino-4, 5, 6, 7-Tetrahydro-N-Phenylbenzo [b] Thiophene-3-Carboxamide. Eur. J. Med. Chem. 2010, 45, 5935–5942. 10.1016/j.ejmech.2010.09.059. [DOI] [PubMed] [Google Scholar]
- Mohareb R. M.; Milad Y. R.; Masoud A. A. New Approaches for the Synthesis of Heterocyclic Compounds Derived from Cyclohexan-1, 3-Dione with Anti-Proliferative Activities. Acta Chim. Slov. 2021, 68, 72–87. 10.17344/acsi.2020.6182. [DOI] [PubMed] [Google Scholar]
- Anwar T.; Kumar P.; Khan A. U.. Modern Tools and Techniques in Computer-Aided Drug Design. Molecular Docking for Computer-Aided Drug Design; Elsevier, 2021; pp 1–30. [Google Scholar]
- Surabhi S.; Singh B. K. Computer Aided Drug Design: An Overview. J. Drug Delivery Ther. 2018, 8, 504–509. 10.22270/jddt.v8i5.1894. [DOI] [Google Scholar]
- Ferlay J.; Colombet M.; Soerjomataram I.; Parkin D. M.; Piñeros M.; Znaor A.; Bray F. Cancer Statistics for the Year 2020: An Overview. Int. J. Cancer 2021, 149, 778. 10.1002/ijc.33588. [DOI] [PubMed] [Google Scholar]
- Dyba T.; Randi G.; Bray F.; Martos C.; Giusti F.; Nicholson N.; Gavin A.; Flego M.; Neamtiu L.; Dimitrova N.; Negrão Carvalho R.; Ferlay J.; Bettio M. The European Cancer Burden in 2020: Incidence and Mortality Estimates for 40 Countries and 25 Major Cancers. Eur. J. Cancer 2021, 157, 308–347. 10.1016/j.ejca.2021.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina J. R.; Yang P.; Cassivi S. D.; Schild S. E.; Adjei A. A. Non-Small Cell Lung Cancer: Epidemiology, Risk Factors, Treatment, and Survivorship. Mayo Clin. Proc. 2008, 83, 584–594. 10.1016/S0025-6196(11)60735-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend M. H.; Anderson M. D.; Weagel E. G.; Velazquez E. J.; Weber K. S.; Robison R. A.; O’Neill K. L. Non-Small-Cell Lung Cancer Cell Lines A549 and NCI-H460 Express Hypoxanthine Guanine Phosphoribosyltransferase on the Plasma Membrane. OncoTargets Ther. 2017, 10, 1921–1932. 10.2147/OTT.S128416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megally Abdo N. Y.; Milad Mohareb R.; Halim P. A. Uses of Cyclohexane-1,3-Dione for the Synthesis of 1,2,4-Triazine Derivatives as Anti-Proliferative Agents and Tyrosine Kinases Inhibitors. Bioorg. Chem. 2020, 97, 103667 10.1016/j.bioorg.2020.103667. [DOI] [PubMed] [Google Scholar]
- Cao L.; Wang F.; Li S.; Wang X.; Huang D.; Jiang R. PIM1 Kinase Promotes Cell Proliferation, Metastasis and Tumor Growth of Lung Adenocarcinoma by Potentiating the c-MET Signaling Pathway. Cancer Lett. 2019, 444, 116–126. 10.1016/j.canlet.2018.12.015. [DOI] [PubMed] [Google Scholar]
- Cen B.; Xiong Y.; Song J. H.; Mahajan S.; DuPont R.; McEachern K.; DeAngelo D. J.; Cortes J. E.; Minden M. D.; Ebens A.; Mims A.; LaRue A. C.; Kraft A. S. The Pim-1 Protein Kinase Is an Important Regulator of MET Receptor Tyrosine Kinase Levels and Signaling. Mol. Cell. Biol. 2014, 34, 2517–2532. 10.1128/MCB.00147-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.-D.; Wu T.-T.; Yu P.; Wang Z.-Z.; Xiao W.; Jiang Y.; Zhao L.-G. Molecular Dynamics Analysis of Binding Sites of Epidermal Growth Factor Receptor Kinase Inhibitors. ACS Omega 2020, 5, 16307–16314. 10.1021/acsomega.0c02183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Liu X.; Gan W.; Wu J.; Zhou H.; Yang Z.; Zhang Y.; Liao M.; Yuan P.; Xu S.; Zheng P.; Zhu W. Discovery of Triazolo-Pyridazine/-Pyrimidine Derivatives Bearing Aromatic (Heterocycle)-Coupled Azole Units as Class II c-Met Inhibitors. ACS Omega 2020, 5, 16482–16490. 10.1021/acsomega.0c00838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquini G.; Giaccone G. C-MET Inhibitors for Advanced Non-Small Cell Lung Cancer. Expert Opin. Invest. Drugs 2018, 27, 363–375. 10.1080/13543784.2018.1462336. [DOI] [PubMed] [Google Scholar]
- Tuğcu G.; Bayram F. E. Ö.; Sipahi H. In Silico Modeling and Toxicity Profiling of a Set of Quinoline Derivatives as C-MET Inhibitors in the Treatment of Human Tumors. Turk. J. Pharm. Sci. 2021, 18, 738. 10.4274/tjps.galenos.2021.54815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Yang S.; Wang K.; Sun S.-Y. MET Inhibitors for Targeted Therapy of EGFR TKI-Resistant Lung Cancer. J. Hematol. Oncol. 2019, 12, 63 10.1186/s13045-019-0759-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scagliotti G. V.; Shuster D.; Orlov S.; von Pawel J.; Shepherd F. A.; Ross J. S.; Wang Q.; Schwartz B.; Akerley W. Tivantinib in Combination with Erlotinib versus Erlotinib Alone for EGFR-Mutant NSCLC: An Exploratory Analysis of the Phase 3 MARQUEE Study. J. Thorac. Oncol. 2018, 13, 849–854. 10.1016/j.jtho.2017.12.009. [DOI] [PubMed] [Google Scholar]
- Shaw A. T.; Riely G. J.; Bang Y.-J.; Kim D.-W.; Camidge D. R.; Solomon B. J.; Varella-Garcia M.; Iafrate A. J.; Shapiro G. I.; Usari T.; Wang S. C.; Wilner K. D.; Clark J. W.; Ou S.-H. I. Crizotinib in ROS1-Rearranged Advanced Non-Small-Cell Lung Cancer (NSCLC): Updated Results, Including Overall Survival, from PROFILE 1001. Ann. Oncol. 2019, 30, 1121–1126. 10.1093/annonc/mdz131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo H.-N.; Liu P. Targeting MET in Cancer Therapy. Chronic Dis. Transl. Med. 2017, 3, 148–153. 10.1016/j.cdtm.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leighl N. B.; Tsao M.-S.; Liu G.; Tu D.; Ho C.; Shepherd F. A.; Murray N.; Goffin J. R.; Nicholas G.; Sakashita S.; Chen Z.; Kim L.; Powers J.; Seymour L.; Goss G.; Bradbury P. A. A Phase I Study of Foretinib plus Erlotinib in Patients with Previously Treated Advanced Non-Small Cell Lung Cancer: Canadian Cancer Trials Group IND.196. Oncotarget 2017, 8, 69651–69662. 10.18632/oncotarget.18753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B. J.; Kim D.; Kim J. H.; Kim H. S.; Jang H. J. The Efficacy and Safety of Onartuzumab in Patients with Solid Cancers: A Meta-Analysis of Randomized Trials. Indian J. Cancer 2021, 58, 232–240. 10.4103/ijc.IJC_797_18. [DOI] [PubMed] [Google Scholar]
- Qian F.; Engst S.; Yamaguchi K.; Yu P.; Won K.-A.; Mock L.; Lou T.; Tan J.; Li C.; Tam D.; et al. Inhibition of Tumor Cell Growth, Invasion, and Metastasis by EXEL-2880 (XL880, GSK1363089), a Novel Inhibitor of HGF and VEGF Receptor Tyrosine Kinases. Cancer Res. 2009, 69, 8009–8016. 10.1158/0008-5472.CAN-08-4889. [DOI] [PubMed] [Google Scholar]
- Ye S.; Li J.; Hao K.; Yan J.; Zhou H. The Efficacy and Risk Profile of C-Met Inhibitors in Non-Small Cell Lung Cancer: A Meta-Analysis. Sci. Rep. 2016, 6, 35770 10.1038/srep35770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garajová I.; Giovannetti E.; Biasco G.; Peters G. J. C-Met as a Target for Personalized Therapy. Transl. Oncogenomics 2015, 7, 13–31. 10.4137/TOG.S30534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratov E. N.; Bajorath J.; Sheridan R. P.; Tetko I. V.; Filimonov D.; Poroikov V.; Oprea T. I.; Baskin I. I.; Varnek A.; Roitberg A.; Isayev O.; Curtalolo S.; Fourches D.; Cohen Y.; Aspuru-Guzik A.; Winkler D. A.; Agrafiotis D.; Cherkasov A.; Tropsha A. QSAR without Borders. Chem. Soc. Rev. 2020, 49, 3525–3564. 10.1039/D0CS00098A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khatabi K.; Aanouz I.; El-Mernissi R.; Singh A.; Ajana M.; Lakhlifi T.; Kumar S.; Bouachrine M. Integrated 3D-QSAR, Molecular Docking, and Molecular Dynamics Simulation Studies on 1,2,3-Triazole Based Derivatives for Designing New Acetylcholinesterase Inhibitors. Turk. J. Chem. 2021, 45, 647–660. 10.3906/kim-2010-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khamouli S.; Belaidi S.; Bakhouch M.; Chtita S.; Hashmi M. A.; Qais F. A. QSAR Modeling, Molecular Docking, ADMET Prediction and Molecular Dynamics Simulations of Some 6-Arylquinazolin-4-Amine Derivatives as DYRK1A Inhibitors. J. Mol. Struct. 2022, 1258, 132659 10.1016/j.molstruc.2022.132659. [DOI] [Google Scholar]
- Abchir O.; Daoui O.; Belaidi S.; Ouassaf M.; Qais F. A.; ElKhattabi S.; Belaaouad S.; Chtita S. Design of Novel Benzimidazole Derivatives as Potential α-Amylase Inhibitors Using QSAR, Pharmacokinetics, Molecular Docking, and Molecular Dynamics Simulation Studies. J. Mol. Model. 2022, 28, 106 10.1007/s00894-022-05097-9. [DOI] [PubMed] [Google Scholar]
- Daoui O.; Elkhattabi S.; Chtita S.; Elkhalabi R.; Zgou H.; Benjelloun A. T. QSAR, Molecular Docking and ADMET Properties in Silico Studies of Novel 4,5,6,7-Tetrahydrobenzo[D]-Thiazol-2-Yl Derivatives Derived from Dimedone as Potent Anti-Tumor Agents through Inhibition of C-Met Receptor Tyrosine Kinase. Heliyon 2021, 7, e07463 10.1016/j.heliyon.2021.e07463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra R. K.; El-ajaily M. M.; Alassbaly F. S.; Sarangi A. K.; Das D.; Maihub A. A.; Ben-Gweirif S. F.; Mahal A.; Suleiman M.; Perekhoda L.; Azam M.; Al-Noor T. H. DFT, Anticancer, Antioxidant and Molecular Docking Investigations of Some Ternary Ni(II) Complexes with 2-[(E)-[4-(Dimethylamino)Phenyl]Methyleneamino]Phenol. Chem. Pap. 2021, 75, 1005–1019. 10.1007/s11696-020-01342-8. [DOI] [Google Scholar]
- Daoui O.; Elkhattabi S.; Chtita S. Rational Identification of Small Molecules Derived from 9,10-Dihydrophenanthrene as Potential Inhibitors of 3CLpro Enzyme for COVID-19 Therapy: A Computer-Aided Drug Design Approach. Struct. Chem. 2022, 33, 1667–1690. 10.1007/s11224-022-02004-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar B. H.; Manandhar S.; Mehta C. H.; Nayak U. Y.; Pai K. S. R. Structure-Based Docking, Pharmacokinetic Evaluation, and Molecular Dynamics-Guided Evaluation of Traditional Formulation against SARS-CoV-2 Spike Protein Receptor Bind Domain and ACE2 Receptor Complex. Chem. Pap. 2022, 76, 1063–1083. 10.1007/s11696-021-01917-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorat B.; Purohit V. P.; Yamgar R.; Bhagat D.; Wavhal S. D.; Mali S. N. Structural Insight into 2-Aryl-4-Quinoline Carboxylic Acid-Based Dihydroorotate Dehydrogenase (DHODH) and Its Potential Anti-SARS-CoV-2 Activity Through Pharmacophore Modeling, Multidimensional QSAR, ADME, and Docking Studies. Phys. Chem. Res. 2023, 11, 783–800. 10.22036/pcr.2022.365408.2218. [DOI] [Google Scholar]
- Nedeljkovic N.; Nikolic M.; Stankovic A.; Jeremic N.; Tomovic D.; Bukonjic A.; Radic G.; Mijajlovic M. Virtual Screening, Drug-Likeness Analysis, and Molecular Docking Study of Potential Severe Acute Respiratory Syndrome Coronavirus 2 Main Protease Inhibitors. Turk. J. Chem. 2022, 46, 116–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav J. K.; Yadav P.; Singh V. K.; Agarwal A. Molecular Docking and Density Functional Theory Studies of Potent 1,3-Disubstituted-9H-Pyrido[3,4-b]Indoles Antifilarial Compounds. Struct. Chem. 2021, 32, 1925–1947. 10.1007/s11224-021-01772-4. [DOI] [Google Scholar]
- Ullah H.; Liaqat A.; Khan Q. U.; Taha M.; Khan F.; Rahim F.; Uddin I.; Rehman Z. U. Synthesis, in Vitro Thymidine Phosphorylase Activity and Molecular Docking Study of Thiadiazole Bearing Isatin Analogs. Chem. Pap. 2022, 76, 213–224. 10.1007/s11696-021-01842-1. [DOI] [Google Scholar]
- Underiner T. L.; Herbertz T.; Miknyoczki S. J. Discovery of Small Molecule C-Met Inhibitors: Evolution and Profiles of Clinical Candidates. Anti-Cancer Agents Med. Chem. 2010, 10, 7–27. 10.2174/1871520611009010007. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.. et al. Gaussian 09, revision B.01.; Gaussian, Inc.: Wallingford, CT, 2010.
- Structure Drawing Software for Academic and Personal Use|ACD/ChemSketch, 2021. https://www.acdlabs.com/resources/freeware/chemsketch/index.php (accessed March 22, 2021).
- ChemOffice Download—ChemDraw and Chem3D, 2021. https://www.cambridgesoft.com/Ensemble_for_Chemistry/details/Default.aspx?fid=16 (accessed March 22, 2021).
- Yoo C.; Shahlaei M. The Applications of PCA in QSAR Studies: A Case Study on CCR5 Antagonists. Chem. Biol. Drug Des. 2018, 91, 137–152. 10.1111/cbdd.13064. [DOI] [PubMed] [Google Scholar]
- Daoui O.; Elkhattabi S.; Chtita S. Rational Design of Novel Pyridine-Based Drugs Candidates for Lymphoma Therapy. J. Mol. Struct. 2022, 1270, 133964 10.1016/j.molstruc.2022.133964. [DOI] [Google Scholar]
- ACDLABS 10; Advanced Chemistry Development, Inc.: Toronto, ON. Canada, 2015. Available: https://www.acdlabs.com/resources/free-chemistry-software-apps/chemsketch-freeware/ (accessed March 02, 2022).
- Allinger N. L. Conformational Analysis. 130. MM2. A Hydrocarbon Force Field Utilizing V1 and V2 Torsional Terms. J. Am. Chem. Soc. 1977, 99, 8127–8134. 10.1021/ja00467a001. [DOI] [Google Scholar]
- Becke A. D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Grootveld M.; Chowdhury C. R.; Khijmatgar S.; Chowdhury A.; Harding S.; Lynch E. Dental Anxiety in First- and Final-Year Indian Dental Students. BDJ Open 2019, 15 10.1038/s41405-019-0017-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chtita S.; Ghamali M.; Hmamouchi R.; Elidrissi B.; Bourass M.; Larif M.; Bouachrine M.; Lakhlifi T. Investigation of Antileishmanial Activities of Acridines Derivatives against Promastigotes and Amastigotes Form of Parasites Using Quantitative Structure Activity Relationship Analysis. Adv. Phys. Chem. 2016, 2016, 5137289 10.1155/2016/5137289. [DOI] [Google Scholar]
- Golbraikh A.; Tropsha A. Predictive QSAR Modeling Based on Diversity Sampling of Experimental Datasets for the Training and Test Set Selection. Mol. Diversity 2000, 5, 231–243. 10.1023/A:1021372108686. [DOI] [PubMed] [Google Scholar]
- Wehr M. M.; Sarang S. S.; Rooseboom M.; Boogaard P. J.; Karwath A.; Escher S. E. RespiraTox – Development of a QSAR Model to Predict Human Respiratory Irritants. Regul. Toxicol. Pharmacol. 2022, 128, 105089 10.1016/j.yrtph.2021.105089. [DOI] [PubMed] [Google Scholar]
- Abdulrahman H. L.; Uzairu A.; Uba S. Computer Modeling of Some Anti-Breast Cancer Compounds. Struct. Chem. 2021, 32, 679–687. 10.1007/s11224-020-01608-7. [DOI] [Google Scholar]
- Rücker C.; Rücker G.; Meringer M. Y-Randomization–a Useful Tool in QSAR Validation, or Folklore. J. Chem. Inf. Model. 2007, 47, 2345–2357. 10.1021/ci700157b. [DOI] [PubMed] [Google Scholar]
- Nour H.; Abchir O.; Belaidi S.; Qais F. A.; Chtita S.; Belaaouad S. 2D-QSAR and Molecular Docking Studies of Carbamate Derivatives to Discover Novel Potent Anti-Butyrylcholinesterase Agents for Alzheimer’s Disease Treatment. Bull. Korean Chem. Soc. 2021, 277. 10.1002/bkcs.12449. [DOI] [Google Scholar]
- XLSTAT, Your Data Analysis Solution, XLSTAT version 2019.1, 2021. https://www.xlstat.com/en/ (accessed March 22, 2021).
- Download Matlab—Best Software & Apps, 2021. https://en.softonic.com/downloads/matlab (accessed March 22, 2021).
- Dhaundiyal A.; Kumari P.; Kalra S.. Pharmacophore-Based Screening for Identification of Human Acyl-CoA Cholesterol Acyltransferase Inhibitors: An In-Silico Study. Lett. Drug Des. Discovery 202118 (), 816–829 10.2174/1570180818999201231200643. [DOI] [Google Scholar]
- Thangavel N.; Albratty M. Pharmacophore Model-Aided Virtual Screening Combined with Comparative Molecular Docking and Molecular Dynamics for Identification of Marine Natural Products as SARS-CoV-2 Papain-like Protease Inhibitors. Arabian J. Chem. 2022, 15, 104334 10.1016/j.arabjc.2022.104334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belhassan A.; Chtita S.; Zaki H.; Alaqarbeh M.; Alsakhen N.; Almohtaseb F.; Lakhlifi T.; Bouachrine M. In Silico Detection of Potential Inhibitors from Vitamins and Their Derivatives Compounds against SARS-CoV-2 Main Protease by Using Molecular Docking, Molecular Dynamic Simulation and ADMET Profiling. J. Mol. Struct. 2022, 1258, 132652 10.1016/j.molstruc.2022.132652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J.-B.; Feng Y.; Luo D.; Wang T.-H. 6-Amide-2-Aryl Benzoxazole/Benzimidazole Derivatives as VEFGR-2 Inhibitors in Two-and Three-Dimensional QSAR Studies: Topomer CoMFA and HQSAR. Chem. Pap. 2021, 75, 3551–3562. 10.1007/s11696-021-01588-w. [DOI] [Google Scholar]
- Azad I.; Nasibullah M.; Khan T.; Hassan F.; Akhter Y. Exploring the Novel Heterocyclic Derivatives as Lead Molecules for Design and Development of Potent Anticancer Agents. J. Mol. Graphics Modell. 2018, 81, 211–228. 10.1016/j.jmgm.2018.02.013. [DOI] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires D. E. V.; Blundell T. L.; Ascher D. B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouhi R. E.; Daoui O.; Fitri A.; Benjelloun A. T.; Khattabi S. E.; Benzakour M.; Mcharfi M.; Kurban M. A Strategy to Enhance VOC of π-Conjugated Molecules Based on Thieno[2,3-b] Indole for Applications in Bulk Heterojunction Organic Solar Cells Using DFT, TD-DFT, and 3D-QSPR Modeling Studies. New J. Chem. 2022, 812. 10.1039/D2NJ04281A. [DOI] [Google Scholar]
- Lakhera S.; Devlal K.; Ghosh A.; Chowdhury P.; Rana M. Modelling the DFT Structural and Reactivity Study of Feverfew and Evaluation of Its Potential Antiviral Activity against COVID-19 Using Molecular Docking and MD Simulations. Chem. Pap. 2022, 76, 2759. 10.1007/s11696-022-02067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bank R. P. D.RCSB PDB—3LQ8: Structure of the Kinase Domain of c-Met Bound to XL880 (GSK1363089), 2022. https://www.rcsb.org/structure/3LQ8 (accessed Jan 23, 2022).
- Dassault Systèmes BIOVIA Discovery Studio Modeling Environment, release 2017; Dassault Systèmes, 2016. https://discover.3ds.com/discovery-studio-visualizer-download (accessed May 07, 2021).
- Sybyl-X 2.0; Tripos International: St. Louis, Missouri, 2021. Software Informer. https://sybyl-x.software.informer.com/2.0/ (accessed July 17, 2021).
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang J.; Gao S.; Sun Z.; Yang G. Discovery of Small Molecule PLpro Inhibitor against COVID-19 Using Structure-Based Virtual Screening, Molecular Dynamics Simulation, and Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) Calculation. Struct. Chem. 2021, 32, 879–886. 10.1007/s11224-020-01665-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger . Prime, release 2020-3; Schrödinger, LLC, 2020. https://scholar.google.com/scholar_lookup?hl=en&publication_year=2020&author=Schr%C3%B6dinger+Release+2020-3&title=Prime (accessed Jan 09, 2022). [Google Scholar]
- Choudhary M. I.; Shaikh M.; tul-Wahab A.; ur-Rahman A. In Silico Identification of Potential Inhibitors of Key SARS-CoV-2 3CL Hydrolase (Mpro) via Molecular Docking, MMGBSA Predictive Binding Energy Calculations, and Molecular Dynamics Simulation. PLoS One 2020, 15, e0235030 10.1371/journal.pone.0235030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal K.; Varakumar P.; Aparna B.; Byran G.; Jupudi S. Identification of Some Novel Oxazine Substituted 9-Anilinoacridines as SARS-CoV-2 Inhibitors for COVID-19 by Molecular Docking, Free Energy Calculation and Molecular Dynamics Studies. J. Biomol. Struct. Dyn. 2021, 39, 5551–5562. 10.1080/07391102.2020.1798285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth S. A.; Dror R. O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. 10.1016/j.neuron.2018.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.-Z.; Dai C.; Shen Y.; Wang J.; Hu Y.; Wang Y.-Q.; Lin Z.-H. 3D-QSAR, Docking, and Molecular Dynamics Simulations Studies on Quinazoline Derivatives as PAK4 Inhibitors. Lett. Drug Des. Discovery 2021, 18, 1025–1038. 10.2174/1570180818666210602155310. [DOI] [Google Scholar]
- Munni Y. A.; Ali M. C.; Selsi N. J.; Sultana M.; Hossen M.; Bipasha T. H.; Rahman M.; Uddin M. N.; Hosen S. M. Z.; Dash R. Molecular Simulation Studies to Reveal the Binding Mechanisms of Shikonin Derivatives Inhibiting VEGFR-2 Kinase. Comput. Biol. Chem. 2021, 90, 107414 10.1016/j.compbiolchem.2020.107414. [DOI] [PubMed] [Google Scholar]
- Schrödinger . Desmond Molecular Dynamics System, release 2020-3; D. E. Shaw Research: New York, NY, 2020. Maestro-Desmond Interoperability Tools. Schrödinger. https://scholar.google.com/scholar_lookup?hl=en&publication_year=2020&author=Schr%C3%B6dinger+Release+2020-3&title=Desmond+Molecular+Dynamics+System%2C+D.+E.+Shaw+Research%2C+New+York%2C+NY%2C+2020.+Maestro-Desmond+Interoperability+Tools (accessed Jan 09, 2022).
- Gopinath P.; Kathiravan M. K. Docking Studies and Molecular Dynamics Simulation of Triazole Benzene Sulfonamide Derivatives with Human Carbonic Anhydrase IX Inhibition Activity. RSC Adv. 2021, 11, 38079–38093. 10.1039/D1RA07377J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Jumaili M. H. A.; Siddique F.; Abul Qais F.; Hashem H. E.; Chtita S.; Rani A.; Uzair M.; Almzaien K. A. Analysis and Prediction Pathways of Natural Products and Their Cytotoxicity against HeLa Cell Line Protein Using Docking, Molecular Dynamics and ADMET. J. Biomol. Struct. Dyn. 2021, 1–13. 10.1080/07391102.2021.2011785. [DOI] [PubMed] [Google Scholar]
- Martyna G. J.; Klein M. L.; Tuckerman M. Nosé–Hoover Chains: The Canonical Ensemble via Continuous Dynamics. J. Chem. Phys. 1992, 97, 2635–2643. 10.1063/1.463940. [DOI] [Google Scholar]
- Shinoda W.; Mikami M. Rigid-Body Dynamics in the Isothermal-Isobaric Ensemble: A Test on the Accuracy and Computational Efficiency. J. Comput. Chem. 2003, 24, 920–930. 10.1002/jcc.10249. [DOI] [PubMed] [Google Scholar]
- Genheden S.; Ryde U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discovery 2015, 10, 449–461. 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kůrková V. Kolmogorov’s Theorem and Multilayer Neural Networks. Neural Networks 1992, 5, 501–506. 10.1016/0893-6080(92)90012-8. [DOI] [Google Scholar]
- Jing G.; Zhou Z.; Zhuo J. Quantitative Structure–Activity Relationship (QSAR) Study of Toxicity of Quaternary Ammonium Compounds on Chlorella pyrenoidosa and Scenedesmus quadricauda. Chemosphere 2012, 86, 76–82. 10.1016/j.chemosphere.2011.09.021. [DOI] [PubMed] [Google Scholar]
- Afantitis A.; Melagraki G.; Sarimveis H.; Koutentis P. A.; Markopoulos J.; Igglessi-Markopoulou O. A Novel QSAR Model for Predicting Induction of Apoptosis by 4-Aryl-4H-Chromenes. Bioorg. Med. Chem. 2006, 14, 6686–6694. 10.1016/j.bmc.2006.05.061. [DOI] [PubMed] [Google Scholar]
- Maji D.; Samanta S.; Patil V. M.. In Silico ADMET and Docking Studies of Thiazolidinedione-Acetic-Acid Hybrids as Antidiabetics with Cardioprotection Lett. Drug Des. Discovery, 171475–1484. 10.2174/1570180817999200618103328. [DOI] [Google Scholar]
- Lipinski C. A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discovery Today: Technol. 2004, 1, 337–341. 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H.-Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- Egan W. J.; Merz K. M.; Baldwin J. J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. 10.1021/jm000292e. [DOI] [PubMed] [Google Scholar]
- Caron G.; Digiesi V.; Solaro S.; Ermondi G. Flexibility in Early Drug Discovery: Focus on the beyond-Rule-of-5 Chemical Space. Drug Discovery Today 2020, 25, 621–627. 10.1016/j.drudis.2020.01.012. [DOI] [PubMed] [Google Scholar]
- Fukunishi Y.; Kurosawa T.; Mikami Y.; Nakamura H. Prediction of Synthetic Accessibility Based on Commercially Available Compound Databases. J. Chem. Inf. Model. 2014, 54, 3259–3267. 10.1021/ci500568d. [DOI] [PubMed] [Google Scholar]
- Chtita S.; Belhassan A.; Aouidate A.; Belaidi S.; Bouachrine M.; Lakhlifi T. Discovery of Potent SARS-CoV-2 Inhibitors from Approved Antiviral Drugs via Docking and Virtual Screening. Comb. Chem. High Throughput Screening 2021, 24, 441–454. 10.2174/1386207323999200730205447. [DOI] [PubMed] [Google Scholar]
- Haloui R.; Daoui O.; Mkhayar K.; El Yaqoubi M.; Elkhattabi S.; Haoudi A.; Rodi Y. K.; Ouazzani F. C.; Chtita S. 3D-QSAR, Drug-Likeness, ADMET Prediction, and Molecular Docking Studies in Silico of Novel 5-Oxo-1-Thioxo-4,5-Dihydro-1H-Thiazolo[3,4-a]Quinazoline Derivatives as MALT1 Protease Inhibitors for the Treatment of B Cell Lymphoma. Chem. Pap. 2022, 10.1007/s11696-022-02627-w. [DOI] [Google Scholar]
- Hachi M.; Slimi A.; Fitri A.; Benjelloun A. T.; El khattabi S.; Benzakour M.; Benzakour M.; Mcharfi M.; Mcharfi M.; Khenfouch M.; Khenfouch M.; Zorkani I.; Zorkani I.; Bouachrine M. Theoretical Design and Characterization of D-A1-A Based Organic Dyes for Efficient DSSC by Altering Promising Acceptor (A1) Moiety. J. Photochem. Photobiol., A 2021, 407, 113048 10.1016/j.jphotochem.2020.113048. [DOI] [Google Scholar]
- Daoui O.; Elkhattabi S.; Chtita S. Design and Prediction of ADME/Tox Properties of Novel Magnolol Derivatives as Anticancer Agents for NSCLC Using 3D-QSAR, Molecular Docking, MOLCAD and MM-GBSA Studies. Lett. Drug Des. Discovery 2022, 19, 1–25. [Google Scholar]
- Bakhouch M.; Houari G. A.; Daoudi M.; Kerbal A.; Yazidi M. E. Michael Addition of Active Methylene Compounds to (Z)-2-Arylidenebenzo[b]Thiophen-3(2H)-Ones. Mediterr. J. Chem. 2015, 4, 9–17. 10.13171/mjc.4.1.2015.16.02.23/elyazidi. [DOI] [Google Scholar]
- Bakhouch M.; Es-Sounni B.; Nakkabi A.; El Yazidi M. Thioaurones: Recent Advances in Synthesis, Reactivity, and Biological Activity. Mini-Rev. Org. Chem. 2021, 18, 313–327. 10.2174/1570193X17999200719135019. [DOI] [Google Scholar]
- Daoui O.; Mazoir N.; Bakhouch M.; Salah M.; Benharref A.; Gonzalez-Coloma A.; Elkhattabi S.; Yazidi M. E.; Chtita S. 3D-QSAR, ADME-Tox, and Molecular Docking of Semisynthetic Triterpene Derivatives as Antibacterial and Insecticide Agents. Struct. Chem. 2022, 33, 1063–1084. 10.1007/s11224-022-01912-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daoui O.; Nour H.; Abchir O.; Elkhattabi S.; Bakhouch M.; Chtita S. A Computer-Aided Drug Design Approach to Explore Novel Type II Inhibitors of c-Met Receptor Tyrosine Kinase for Cancer Therapy: QSAR, Molecular Docking, ADMET and Molecular Dynamics Simulations. J. Biomol. Struct. Dyn. 2022, 1–18. 10.1080/07391102.2022.2124456. [DOI] [PubMed] [Google Scholar]
- Ezeh M. I.; Okonkwo O. E.; Okpoli I. N.; Orji C. E.; Modozie B. U.; Onyema A. C.; Ezebuo F. C. Chemoinformatic Design and Profiling of Derivatives of Dasabuvir, Efavirenz, and Tipranavir as Potential Inhibitors of Zika Virus RNA-Dependent RNA Polymerase and Methyltransferase. ACS Omega 2022, 7, 33330–33348. 10.1021/acsomega.2c03945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin Y. C. A Bioavailability Score. J. Med. Chem. 2005, 48, 3164–3170. 10.1021/jm0492002. [DOI] [PubMed] [Google Scholar]
- Chtita S.; Belhassan A.; Bakhouch M.; Taourati A. I.; Aouidate A.; Belaidi S.; Moutaabbid M.; Belaaouad S.; Bouachrine M.; Lakhlifi T. QSAR Study of Unsymmetrical Aromatic Disulfides as Potent Avian SARS-CoV Main Protease Inhibitors Using Quantum Chemical Descriptors and Statistical Methods. Chemom. Intell. Lab. Syst. 2021, 210, 104266 10.1016/j.chemolab.2021.104266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams I. S.; Gatchie L.; Bharate S. B.; Chaudhuri B. Biotransformation, Using Recombinant CYP450-Expressing Baker’s Yeast Cells, Identifies a Novel CYP2D6.10A122V Variant Which Is a Superior Metabolizer of Codeine to Morphine Than the Wild-Type Enzyme. ACS Omega 2018, 3, 8903–8912. 10.1021/acsomega.8b00809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szakács G.; Váradi A.; Özvegy-Laczka C.; Sarkadi B. The Role of ABC Transporters in Drug Absorption, Distribution, Metabolism, Excretion and Toxicity (ADME–Tox). Drug Discovery Today 2008, 13, 379–393. 10.1016/j.drudis.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Fatima S.; Gupta P.; Sharma S.; Sharma A.; Agarwal S. M. ADMET Profiling of Geographically Diverse Phytochemical Using Chemoinformatic Tools. Future Med. Chem. 2020, 12, 69–87. 10.4155/fmc-2019-0206. [DOI] [PubMed] [Google Scholar]
- Jakhar R.; Khichi A.; Kumar D.; Dangi M.; Chhillar A. K. Discovery of Novel Inhibitors of Bacterial DNA Gyrase Using a QSAR-Based Approach. ACS Omega 2022, 7, 32665–32678. 10.1021/acsomega.2c04310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used in this work are private.