

Abstract
Lead (Pb) poisoning is a major public health concern in environmental justice communities of the US and in many developing countries. There is no identified safety threshold for lead in blood, as low-level Pb exposures can lead to severe toxicity in highly susceptible individuals and late onset of diseases from early-life exposure. However, identifying “susceptibility genes” or “early exposure biomarkers” remains challenging in human populations. There is a considerable variation in susceptibility to harmful effects from Pb exposure in the general population, likely due to the complex interplay of genetic and/or epigenetic factors. This systematic review summarizes current state of knowledge on the role of genetic and epigenetic factors in determining individual susceptibility in response to environmental Pb exposure in humans and rodents. Although a number of common genetic and epigenetic factors have been identified, the reviewed studies, which link these factors to various adverse health outcomes following Pb exposure, have provided somewhat inconsistent evidence of main health effects. Acknowledging the compelling need for new approaches could guide us to better characterize individual responses, predict potential adverse outcomes, and identify accurate and usable biomarkers for Pb exposure to improve mitigation therapies to reduce future adverse health outcomes of Pb exposure.
Keywords: lead, in vivo, in vitro, epidemiology, pathways, genetics, epigenetics
The heavy metal lead (Pb) is a ubiquitous persistent environmental and occupational toxicant affecting human populations. Despite years of intensive research, educational efforts, and remedial measures, Pb poisoning remains a major environmental health concern. Human exposure to Pb occurs through inhalation of air contaminated with Pb dust, ingestion of contaminated food and/or water, or direct contact through the skin (Olympio et al. 2009). Although Pb has been removed from paints and gasoline, it can still be found in a number of consumer products used daily such as batteries, toys, food, and water (Olympio et al. 2009). The incidence of Pb poisoning is associated with numerous factors, including socioeconomic status, rurality, race, age, and the date one’s residence was built (Levin et al. 2008). Urban children in low socioeconomic status (SES) areas are at the highest risk, presumably due to the presence of Pb in older building materials and reduced access to healthy sources of nutrition (Mason et al. 2014; Tong et al. 2000). Additional sources of Pb in these areas are aging public infrastructure and mistreatment of water purification as occurred in Flint, Michigan (Masten et al. 2016).
Pb introduced into the bloodstream is excreted in urine and bile with a median biologic half-life of about 30 days (Barbosa et al. 2005), which mostly reflects acute exposure. The remaining Pb is distributed throughout soft tissues of the body, and eventually accumulates in bones with a half-life of approximately 25 to 30 years (Rădulescu and Lundgren 2019), which reflects one’s cumulative exposure over time (Pawlas et al. 2012). Although Pb is not essential for biological functions, it has the capacity to disrupt biological systems by altering molecular interactions, enzyme activities, and cell signaling and function (García-Lestón et al. 2012; Sanders et al. 2009). As a cumulative toxicant, acute and chronic exposure to Pb has irreversible toxicity in several human organs and systems, such as nervous, hematopoietic, and reproductive systems, as well as in kidneys and bones (Lopes et al. 2016; Shellenberger 1984). Although early life Pb exposure may have long-term negative impacts in adult health, children are more susceptible to Pb toxicity than adults as they undergo rapid growth and development, and windows of great plasticity and vulnerability accompany these processes. Moreover, children have heavier exposures because of their behavior, diet, and metabolic and physiologic characteristics (Moya et al. 2004). In fact, they can absorb a greater percentage of Pb from the gastrointestinal tract and inadequate nutrition, iron deficiency, and calcium deficiency can further influence Pb absorption (Hon et al. 2017). The levels of Pb considered safe for children have been repeatedly lowered by regulatory agencies over the past three decades as even low levels of Pb in blood result in adverse outcomes, which currently cannot be reversed (Vorvolakos et al. 2016). According to the latest guideline from the Centers for Disease Control and Prevention (CDC), a reference level of 5 μg/dL should be used to identify children with Pb toxicity (Hon et al. 2017). This reference value is based on the 97.5 percentile of the blood Pb level in U.S. population children aged 1–5 years. Additionally, the European Food Safety Authority (EFSA) concluded that there is no safe exposure to Pb based on international studies (Authority 2012). There is a considerable variation in the severity of harmful effects of Pb exposure in both the general population and occupational workers (Kim et al. 2014). Differences in health outcome may be affected by nutritional and physiological factors, degree of exposure, and/or host factors such as age, sex, genetic differences, and microbiome composition that can modify adsorption, distribution, metabolism and excretion (ADME), or the sensitivity to the toxic effects of Pb (Gundacker et al. 2010).
Experimental and epidemiological studies have shown gene-environment interactions and epigenetic changes to have a plausible role in variation to adverse health effects of Pb (Mitra et al. 2017). Such gene-environment interactions are still poorly understood as epidemiological studies in humans are confounded by many other factors such as co-morbidities, health and physiological status, and exposure to a wide variety of chemical mixtures over the lifetime of an individual (Harrill and McAllister 2017). Additionally, only surrogate measures of Pb exposure, that may not accurately represent body burden, are used in human studies (Barbosa et al. 2005). Challenges and limitations also exist in preclinical animal studies, the primary limitation being the use of inbred rodent strains that lack genetic diversity and thus the differential susceptibility that exists in humans. In fact, many recent studies have shown that genetic background can profoundly affect adverse outcomes of exposures to environmental contaminants (Harrill and McAllister 2017). On the other hand, analyses of animal models exposed to Pb have the potential to disclose unpredicted outcomes and, most importantly, mechanisms of toxicity relevant to human and environmental health risk assessment. Here we review available published information on Pb toxicity, genetic and epigenetic studies that underly variable Pb-induced adverse health outcomes in animal models and human cohorts.
Health effects from Pb exposure
Pb as cumulative toxicant can cause many deleterious systemic effects and yet their underlying mechanisms have not been completely unfolded. Information on Pb toxicity has been reviewed below from epidemiological studies in humans. The most extensively studied adverse health outcomes are neurological, renal, cardiovascular, hematological, reproductive, and developmental effects (Flora et al. 2012).
Research provides substantial evidence that Pb exposure alters the central nervous system with the brain being the most sensitive organ. Specifically, Pb can disrupt key molecules during neuronal migration and differentiation; interfere with synapse formation; premature differentiation of glial cells; interfere with neurotransmitter release (Mason et al. 2014). Pb effects on glutamatergic transmission can affect both development and neuronal plasticity (Sanders et al. 2009). Early-life exposure to Pb has short- and long-term consequences on cognitive function (e.g., lower IQ, lowered learning and memory, poor fine motor skills, antisocial and delinquent behaviors, and attention-deficit hyperactivity disorder (Caito and Aschner 2017). Behavioral problems, including attention-deficit hyperactivity disorder have been associated with disruption of dopaminergic functioning (Sanders et al. 2009). Moreover, recent evidence links developmental Pb poisoning with the etiology of disorders that appear much later in life, such as Alzheimer’s disease, Parkinson’s disease, and schizophrenia (Ordemann and Austin 2016). The underlying mechanism for neurotoxicity is not fully understood and is being actively explored. Oxidative stress, altered cell signaling, and neurotransmission play a major role in Pb neurotoxicity.
Adverse renal effects of Pb have been reported in numerous epidemiological studies. Despite the nephropathy caused by Pb exposure, the mechanisms by which Pb enters target cells in the kidneys are not well understood. Early-life exposure to low-levels of Pb has been shown to cause glomerular hypertrophy, which may result in renal insufficiency later in life. Furthermore, acute Pb exposure affects renal tubules determining defects in solute and amino acid transport culminating in Fanconi syndrome, whereas chronic Pb exposure results in progressive nephritis (Mitra et al. 2017). Pb-induced kidney injury has been reported to be associated with increased oxidative stress, leading to alterations of the mitochondrial permeability transition pore (MPTP) which can initiate apoptosis in kidney cells. Additionally, Pb can impair calcium distribution in renal cells leading to activation of signaling pathways that trigger apoptosis (Orr and Bridges 2017). Overall, the Pb role in kidney function is very inconsistent suggesting that individuals might have different responses to Pb poisoning. Effects on blood pressure are the most studied cardiovascular outcome. Epidemiological studies suggest a link between childhood Pb exposure and development of hypertension (Mitra et al. 2017). However, Pb exposure, as a causal factor of hypertension is still controversial (Mitra et al. 2017). High concentrations of Pb are toxic to both heart and vascular smooth muscle (Prozialeck et al. 2008) increasing the risk for mortality (Lanphear et al. 2018). Furthermore, nephrotoxicity Pb-induced can indirectly affect the cardiovascular system by modulating renin and angiotensin production.
Pb toxicity to the hematopoietic system has been confirmed in numerous studies. Exposure to Pb can result in hemolytic or Frank anemia as a consequence of the decrease in hemoglobin synthesis and in erythrocytes lifespan. In fact, Pb severely affects the heme biosynthesis by down-regulating three vital enzymes: delta-aminolevulinic acid dehydratase (ALAD), aminolevulinic acid synthetase (ALAS) and ferrochelatase. Anemia is usually observed in chronic Pb poisoning in adults and in case of iron deficiency, especially kids and women (Mitra et al. 2017). An increase in oxidative stress can damage or destabilize red blood cell (RBC) membranes.
Exposure to Pb has been reported to alter immune functions. Epidemiological and experimental studies suggest that Pb can influence levels of immunoglobulins, alter the number of lymphocytes, peripheral blood mononuclear cells (PBMCs) and macrophages, and depression neutrophil functions (Fenga et al. 2017; Mishra et al. 2003). In particular, Pb can affect both cellular and humoral immune response by altering Th cell function and increasing susceptibility to the development of autoimmunity and hypersensitivity (Mishra 2009). Pb can also deregulate cytokines production and tryptophan degradation which could produce depression of immune responses and contribute to the development of several immunological pathologies in individuals occupationally exposed (García-Lestón et al. 2012).
Reproductive studies in humans suggest Pb as a contributing factor to infertility in both men and women (Kumar 2018). Health effects on male reproductive system have been largely investigated in comparison to the female system. Specifically, it has been reported damage to sperm and potential alterations in serum levels of reproductive hormones (Naha et al. 2005; Taha et al. 2013). On the other hand, epidemiological studies conducted in women show an increased risk for miscarriages (Vigeh et al. 2010), prematurity, and earlier age at onset of menopause (Eum et al. 2014; Irgens et al. 1998). However, results on female reproductive effects from Pb are inconsistent. Similarly, Pb exposure has been associated with developmental outcomes such as decreased birth size and child growth (Berkowitz et al. 2006; Zhu et al. 2010) and yet results are also conflicting.
In addition, adverse health outcomes from Pb exposure to other organ systems have been reported as well. Pb can be considered an endocrine disruptor compound as it can interfere with the endocrine system. Early-life Pb exposure has been associated with low levels of vitamin D (Chang et al. 2014), delayed puberty and early menopause (Eum et al. 2014). Associations have also been reported between Pb exposure and decreased lung function, increased bronchial hyperreactivity, and increased risk of respiratory diseases (Taylor et al. 2019).
Although the skeleton is the major reservoir of lead in the body, its effect on it has not been widely studied. Early-life Pb exposure impacts skeletal development through unknown mechanisms which may affect bone growth and remodeling. From in vitro data and clinical observations, it appears that Pb may influence cartilage differentiation at two stages: chondrogenesis enhancement and arrest of transition to bone (Puzas et al. 2004). Given the effect, lead could be an unrecognized factor in osteoporosis as it accelerates bone maturation but determining a reduction in bone mass (Puzas et al. 2004). However, human studies are limited.
In severe cases of Pb poisoning, children or adults may experience severe abdominal pain, nausea, vomiting and constipation, as the gastrointestinal tract is responsible for Pb intake and uptake (Fenga et al. 2017).
Genetic susceptibility to Pb poisoning
The results of the strength-of-evidence of association between genetic factors and Pb susceptibility, including health outcomes are summarized in Figure 1. Generally, factors influencing susceptibility to Pb toxicity include variants and polymorphisms in genes involved in Pb ADME (Onalaja and Claudio 2000).
Fig. 1.
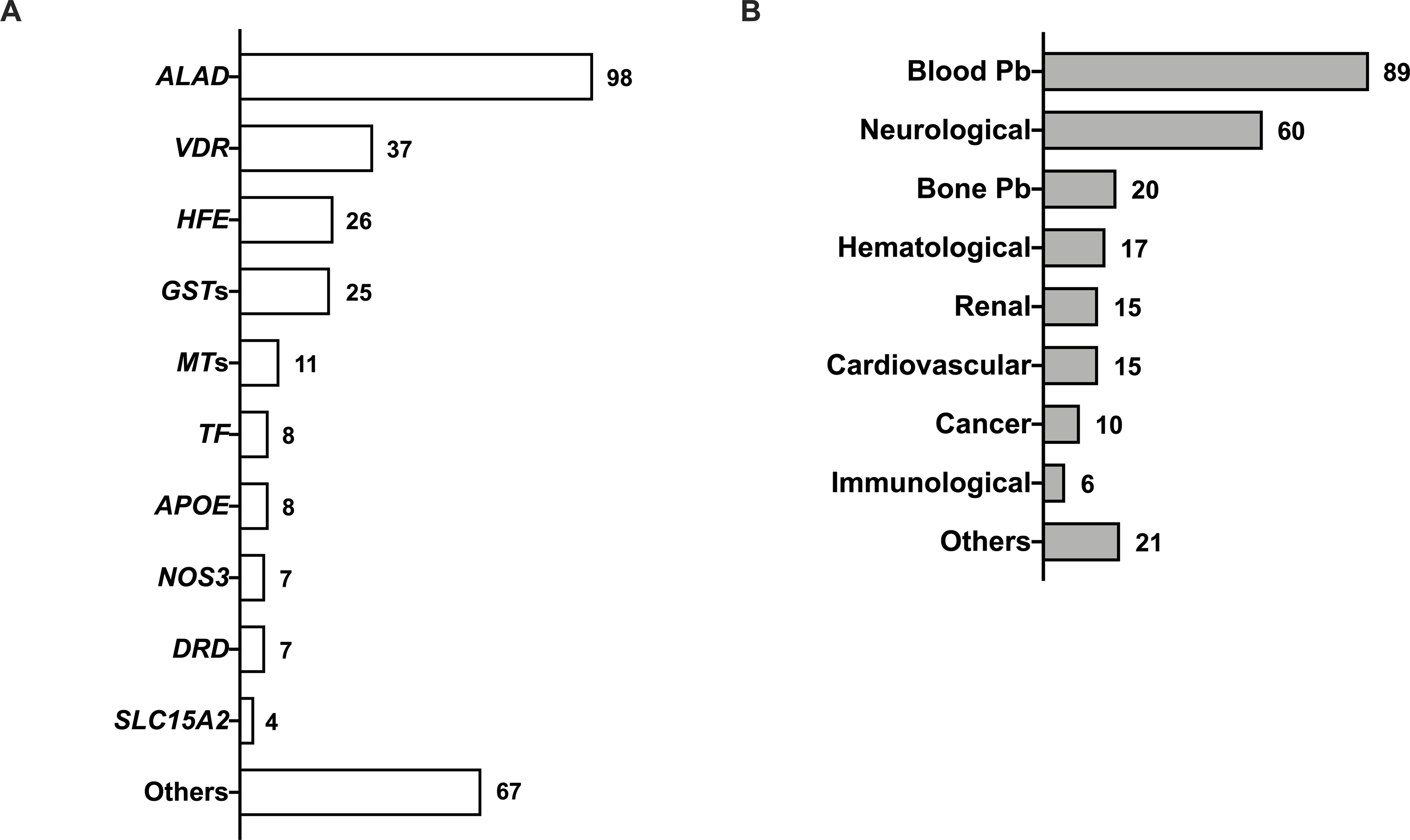
Gene polymorphisms in Pb susceptibility (A) and phenotype associations (B). Numbers indicate the number of studies for a given genetic factor and adverse health outcomes associated with them when reported.
Epidemiological studies
Three genes have been widely reported to be involved in Pb metabolism: δ-aminolevulinic acid dehydratase (ALAD), the vitamin D receptor (VDR), and the hemochromatosis (HFE). Polymorphisms in ALAD, whose genotype frequencies vary by geography and ethnic origin, have been implicated in susceptibility to Pb toxicity (Onalaja and Claudio 2000). ALAD is the second enzyme in the heme synthesis pathway and the primary binding protein in erythrocytes (Zheng et al. 2011). Human ALAD, encoded by a single gene on chromosome (Chr) 9q34, has eight variants that have been described. The most studied polymorphism is a G → C transversion at position 177 in the coding region of ALAD (rs1800435), substituting a neutral asparagine (Asn) for a positively charged lysine (Lys) at amino acid 59, which results in three distinct phenotypic variants (ALAD1-1, ALAD1-2, and ALAD2-2) (Battistuzzi et al. 1981; Wetmur et al. 1991). The ALAD1 (59Asn) gene product is more electronegative than that of ALAD2 (59Lys) suggesting that ALAD1-2/2–2 may bind Pb more tightly than ALAD1-1. Consistent with this prediction, ALAD2 carriers seem to retain more Pb in blood than ALAD1 homozygotes, while there is no difference at low exposure (Scinicariello et al. 2007). Occasionally, ALAD2 has also been reported to be associated with higher concentration of Pb in urine. Furthermore, ALAD polymorphisms can influence urinary excretion of δ-aminolevulinic acid (U-ALA) in children with ALA being one of the contributors to Pb-induced neurotoxicity (Tasmin et al. 2015). Furthermore, exposure to Pb negatively impacted achieved growth in children carrying ALAD1-2 and ALAD2-2 genotypes (Kerr et al. 2019). Other ALAD polymorphisms have been investigated as well, however their role in Pb accumulation is not clear due to study limitations (Mani et al. 2019). Since Pb exerts toxic effects on numerous organ systems, ALAD genotype might affect inter-individual variation in susceptibility to these toxic effects through differential sequestering of Pb by ALAD, making it less bioavailable for pathogenetic process. Although it has been shown that the ALAD genotype alters the toxicokinetics of Pb, the association between adverse health outcomes of Pb and ALAD genotype is not well established.
Through an entirely different pathway, another much less studied genetic variant also impacts blood Pb burden and brain ALA. The solute carrier family 15 member 2 gene (SLC15A2), also known as PEPT2, protects the brain from excess peptide-bound amino acids and potentially limits Pb-induced neurotoxicity (Hu et al. 2007). Two PEPT2 single nucleotide polymorphisms, PEPT2-1 and PEPT2-2, predominate. At lowest levels of Pb exposure, males but not females homozygous for PEPT2-2 have significantly increased blood Pb level (Sobin et al. 2011).
Variants of the vitamin D receptor gene (VDR), involved in intestinal Ca2+ absorption and storage in the bone, have been suggested to play a role in modifying Pb absorption since it can mimic calcium at the calcium binding site of transporters. VDR plays a major role in maintaining calcium and phosphate homeostasis, regulating bone metabolism, and anti-proliferative, pro-apoptotic, and immunosuppressive activities. VDR, located on human Chr. 12, is regulated by the active form of vitamin D3 (1,25-dihydroxyvitamin D3). Many polymorphisms have been identified in VDR, mostly defined by restriction fragment length polymorphisms (RFLPs). Previous studies showed that the Bsm I RFLP polymorphism in intron 8 is associated with uptake of Pb (Schwartz et al. 2000; Weaver et al. 2005). There are indications that carriers of the Bsm I VDR allele are predisposed to significantly higher Pb concentrations in blood, bones, and urine compared with non-carriers of the Bsm I allele. Moreover, the Fok 1 VDR RFLP may modify the relationship of Pb exposure and blood Pb levels during the first two years of life, when children are most susceptible to Pb ingestion and absorption. The Fok 1 VDR allele is associated with posture disturbance as well as hearing impairment in children exposed to very low Pb levels (Pawlas et al. 2014; Pawlas et al. 2015). Little information is available about the association between blood Pb levels and other polymorphisms defined by Apa I and Taq I RFLPs of VDR. Overall, modification of both ALAD and VDR genes have shown to have an impact on cognition in children exposed to Pb (Pawlas et al. 2012). However, the role of VDR polymorphisms in Pb uptake from bone tissues remains unclear.
There is strong evidence that excessive Pb uptake affects iron metabolism and may be an important cause of iron deficiency. Thus, iron metabolism genes are potential players in Pb absorption and storage. The hemochromatosis gene (HFE) is responsible for accumulation of iron in humans, variants in which can lead to severe liver failure, heart disease, or diabetes (Fleming et al. 2005). Two common missense mutations (H63D and C282Y) result in HFE deficiency, leading to upregulation of transferrin receptors and divalent metal transporters (DMTs) followed by increased iron uptake in the gut and also perhaps Pb (Hollerer et al. 2017). Pregnant mothers with H63D variants have higher Pb levels in placental tissue, umbilical cord, and blood compared to the homozygote typical (HH) (Kayaalti et al. 2015a). A strong association between Pb exposure and amyotrophic lateral sclerosis (ALS) has been reported among HFE C282Y variant carriers (Eum et al. 2015). However, the mechanistic basis for the relationship of increased Pb absorption and HFE deficiency has not been proven. Furthermore, there is no strong evidence for the influence of HFE variants on Pb body burden. In addition, several association studies revealed the influence of transferrin (TF) mutations, a monomeric glycoprotein that facilitates the transport of iron, in modifying the impact of cumulative Pb exposure on factors such childhood neurocognition, placental transfer, and homocysteine levels (Karwowski et al. 2014; Roy et al. 2013). Apart from HFE and TF, divalent metal transporter 1 (DMT1) has been implicated as a modulator of Pb toxicity in one study. A single nucleotide polymorphism (IVS4þ44C/A) in DMT1 was associated with higher blood Pb levels in 486 unrelated and healthy individuals in a Turkish population (Kayaaltı et al. 2015b). However, the study only suggested a potential link between the genetic variant and increased disease risk. Since these polymorphisms could potentially determine several phenotypic outcomes from Pb exposure due to its crosstalk with iron, additional studies are warranted.
Several polymorphisms in the glutathione-S-transferase genes GST (GSTM1, GSTT1, and GSTP1), key players in metal-induced oxidative stress defenses, can contribute to phenotypic variation in enzymatic production and activity, and thus also may play a role in susceptibility to Pb toxicity. GSTP1 Ile105Val and GSTM1 null genotypes have been associated with modified Pb-induced cognitive function (Eum et al. 2015). Moreover, GST polymorphisms like GSTT1 have been linked to increased blood pressure in Pb-exposed Korean male workers and inflammatory responses associated with Pb toxicity (Bozina et al. 2009; Lee et al. 2012).
Interaction with proteins is an important mechanism of Pb toxicity, especially interactions with high-affinity metal-binding proteins that have a high content of sulfhydryl groups. Metallothioneins (MTs) are cysteine-rich low molecular weight proteins with numerous functions including metal detoxification. Four major isoforms have been identified in mammals (MT1-MT4) (Raudenska et al. 2014). An inverse association between the MT2a variant (rs10636) and blood Pb levels has been reported (Fernandes et al. 2016; Gundacker et al. 2009; Whitfield et al. 2007). However, these studies report high variation in outcomes making the results ambiguous.
Several studies have associated cognitive impairment and/or Alzheimer’s disease (AD) in adults with low-level Pb exposure (Basha et al. 2005; Weisskopf et al. 2004). However, few studies have evaluated the role of genetic susceptibility on the cognitive effects of environmental Pb exposures. Apolipoprotein E (APOE) epsilon-4 allele (e4) is a known genetic risk marker for cognitive function impairment and AD (Liu et al. 2013). In fact, individuals with APOE e4 show age-related reduction in hippocampal volume (Lind et al. 2006), decreased antioxidant capacity (Miyata and Smith 1996), and altered use of nutrients (Reiman et al. 2004). Therefore, APOE e4 carriers may be more susceptible to Pb-induced neuroinflammation and impaired adult hippocampal neurogenesis contributing to these cognitive deficits (Engstrom et al. 2017; Prada et al. 2016).
The endothelial nitric oxide synthase gene (NOS3) is also of interest with respect to Pb. It has a common polymorphism NOS3 Glu298Asp in exon 7 that involves a G → T conversion at nucleotide position 298, which is associated with Pb susceptibility to cardiovascular disease (Vaziri and Ding 2001; Vaziri et al. 2001; Weaver et al. 2003).
A population-based study suggested that genetic polymorphisms within the dopamine receptor D2 gene (DRD2) TAQ IA increases susceptibility to intelligence deficits in Indian children due to Pb exposure (Roy et al. 2011). Conversely, another population study found no evidence of an interaction between the DRD2 TAQ IA polymorphism and Pb exposure on cognition in Mexican children (Kordas et al. 2011). The discrepancy could be due to the differences in the degree of Pb exposure and cognitive domains analyzed, as well as the genetic constitution of the exposed populations.
N-Methyl-D-aspartate receptors (NMDARs) mediate neuronal maturation and excitatory neurotransmission in mammalian brain, which affects learning and memory (Endele et al. 2010; Gavazzo et al. 2008; Omelchenko et al. 1997; Toscano and Guilarte 2005). Polymorphisms in its subunit genes GRIN2A and GRIN2B have been reported to contribute to Pb-induced toxicity. A population-based study showed that GRIN2A decreased more than 30% after Pb exposure and its SNP rs2650429 is associated with Pb-induced neurotoxicity (Wu et al. 2017). Additionally, genetic variants of GRIN2A (rs727605 and rs1070503) and GRIN2B (rs7301328 and rs1806201) have been associated with multiple adverse cognitive phenotypes in children (Rooney et al. 2018).
Two SNPs (rs10503970 and rs9642758) were identified on Chr 8 in a genomic region of Unc-5 netrin receptor D gene (UNC5D) in humans that have a potential impact on neurodevelopmental outcomes in response to prenatal Pb-exposure (Wang et al. 2017). UNC5D controls neuronal cell survival, cell-cell adhesion, cell guidance (e.g., axon attraction or repulsion), and migration. It also acts as a dependence receptor required for apoptosis induction when not associated with netrin (Takemoto et al. 2011).
Experimental studies: in vivo
So far, only few reports have examined the role of genetic factors in Pb toxicity in rodent populations and have been published. Studies in animal models have shown an association between Pb-induced neurotoxicity and Grin2a, which encodes for an important subunit of the N-methyl-D-aspartate receptor (NMDAR) as mentioned previously. Pb exposure can alter GRIN2A concentrations in the brain overall and specifically in the hippocampus of rats and, consequently, can cause synaptic morphological and functional changes in hippocampal CA1 pyramidal neurons (Tüzmen et al. 2015), resulting in behavioral changes (Neal et al. 2011; Wang et al. 2016).
Additionally, adverse effects from Pb are mitigated by MTs whose synthesis has been shown to be Pb-induced in rat liver and kidney (Jurczuk et al. 2006; Stacchiotti et al. 2009). MT null mice and cells are more sensitive to Pb toxicity than wildtype equivalents (Qu et al. 2002).
Epigenetic susceptibility to Pb poisoning
The results of the strength-of-evidence of association between epigenetic factors and Pb susceptibility, including health outcomes are summarized in Figure 2.
Fig. 2.
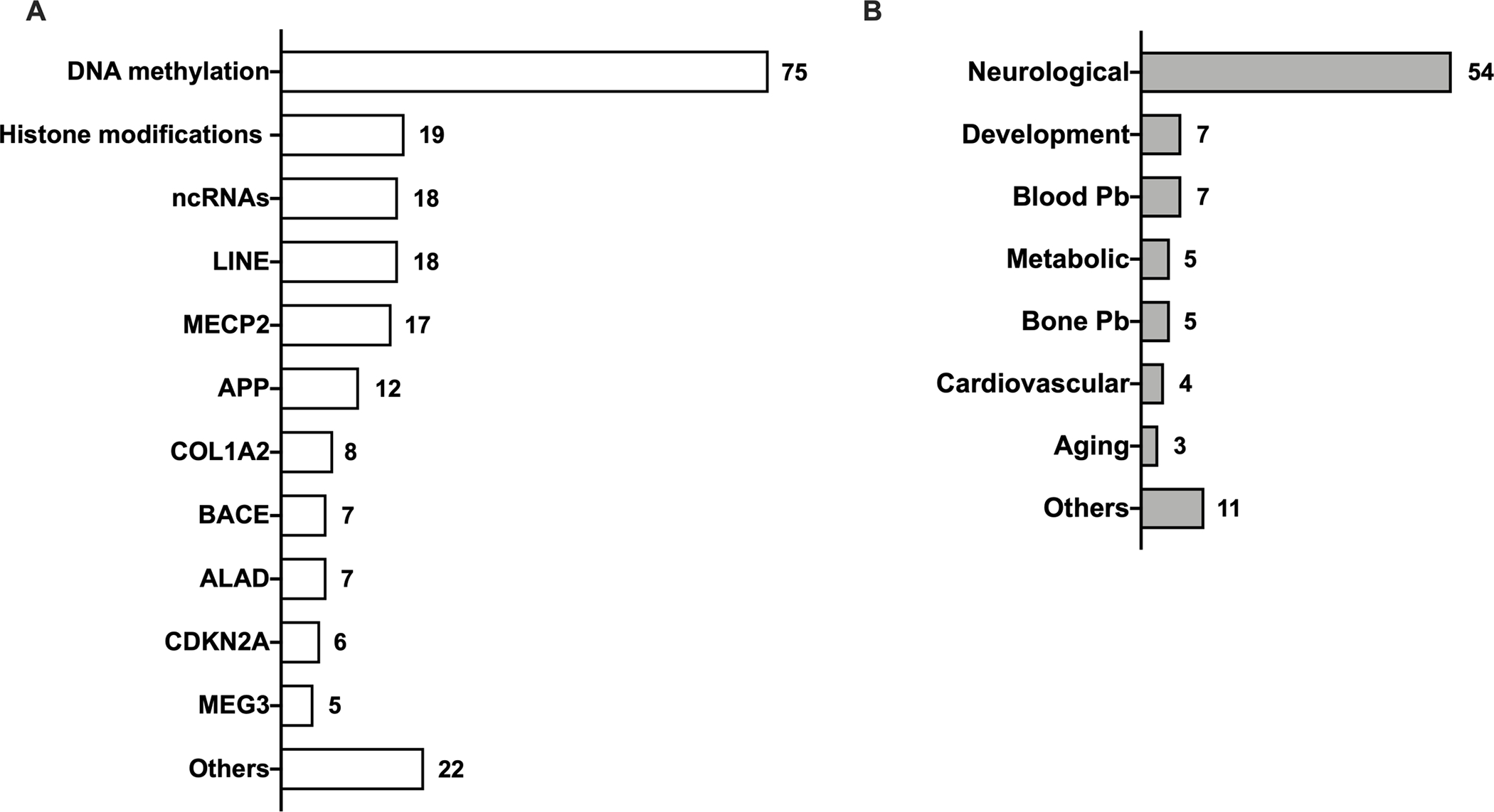
Epigenetic factors in Pb susceptibility (A) and phenotype associations (B). Numbers indicate the number of studies for a given epigenetic factor and adverse health outcomes associated with them when reported.
Epidemiological studies
Studies of Pb exposure in occupational cohorts have identified DNA promoter methylation changes, where individuals have hypomethylation in long interspersed nuclear elements-1 (LINE-1) (Li et al. 2013) and hypermethylation of ALAD (Li et al. 2011) and CDKN2A (Kovatsi et al. 2010), that are associated with increased risk of toxicity from Pb exposure. LINE-1 is a repetitive DNA retrotransposon containing numerous CpG islands and its methylation helps maintain genomic stability and integrity (Li and Zhang 2014). LINE-1 methylation represents a biomarker of past Pb exposure in elderly men as it inversely correlates with bone Pb accumulation (Wright et al. 2010). Besides inhibiting ALAD enzyme activity, Pb can increase the level of ALAD methylation and thus decrease ALAD transcription (Li et al. 2011). Together these changes have been suggested as risk factors of ASD (Keil and Lein 2016). Interestingly, the dual role of the ALAD gene in Pb toxicity may suggest a link between epigenetic regulation and genetic polymorphisms contributing to Pb susceptibility. The tumor suppressor gene CDKN2A, overexpressed during neurodegeneration, is hypermethylated in individuals with high blood Pb levels, whereas individuals with lower blood Pb levels show only partial methylation (Kovatsi et al. 2010). Additionally, Pb-exposed workers have higher CpG island methylation associated with increased DNA damage making them susceptible to tumorigenesis (Yu et al. 2018). Alteration of DNA methylation within the promoter region of the collagen type 1 alpha-2 (COL1A2) gene, an important component of the connective tissues linked to preterm birth in humans, has been associated with blood Pb level in women undergoing in vitro fertilization (Hanna et al. 2012).
Lastly, Pb-induced changes in methylation have also been recently reported as transgenerational, with data indicating that a mother’s blood Pb levels and methylation status can directly impact that of her children and potentially grandchildren (Sen et al. 2015b). Moreover, exposure to Pb during pregnancy can alter methylation patterns of DNA repair and antioxidant genes in newborns, which could be a risk factor for developing diseases later in life (Montes-Castro et al. 2019). For instance, Pb has been associated with decreased methylation of the glycoprotein VI platelet (GP6) gene (Engström et al. 2015), a key factor in platelet aggregation and thrombus formation, and with hypermethylation at the MEG3 (Nye et al. 2016) regulatory region indicating a link between Pb and onset of cardiometabolic diseases later in life. Another study reported sex-specific changes in DNA methylation from blood of children exposed to Pb in early-life (Sen et al. 2015a). In addition, children that were exposed to Pb in utero exhibited irregular methylation patterns in imprinted genes, which may determine increased risk of childhood obesity and cardiometabolic disease in adulthood (Goodrich et al. 2015; Nye et al. 2016).
Pb toxicity has also been reported to be associated with miRNAs/lncRNAs dysregulation. In particular, exportin-5 (XPO5) polymorphism rs2257082 is strongly associated with the susceptibility to Pb poisoning. XPO5 is a key element of the miRNA biogenesis pathway as it is required for the transport of small RNAs and double-stranded RNA-binding proteins from the nucleus to cytoplasm in a Ran-GTP dependent process. Workers harboring the C allele tend to have higher blood Pb levels (Zhang et al. 2016). However, specific molecular mechanisms for how this SNP modifies susceptibility to Pb is not known.
The reported studies show that environmentally relevant Pb exposure in humans, some resulting in even lower blood Pb levels than what recommended from CDC guidelines, can elicit changes in the epigenome that could translate into onset of diseases. However, adverse health effects from such changes can only be suggested since data from most of the human cohort studies lack phenotypic data addressing the overall health status.
Experimental studies: in vitro and in vivo
Studies performed in human cell lines and experimental animals provide evidence that prenatal Pb exposure is inversely associated with genomic DNA methylation in cord blood, suggesting that Pb can alter the fetal epigenome in humans, leading to long-term reprogramming of the DNA methylation profile and increased disease risk (Pilsner et al. 2009). Specifically, Pb exposure in human neural progenitor cells alters DNA methylation status of genes involved in neuronal growth (Senut et al. 2014). Additionally, Pb exposure in human neuroblastoma cultures disrupts IGF1 stimulated methionine synthase activity, an enzyme that regulates DNA methylation (Waly et al. 2004). In these studies, cell line exposure to Pb can be considered relevant to humans since Pb concentrations of 120–1200 nM correspond to blood Pb levels of 1–10 μg/dL. In addition, several in vivo studies described below used a wide range of Pb concentrations through different routes of exposures to better recapitulate human exposure and its link to disease onset.
DNA methylation is maintained by DNA methyltranferases (DNMTs) including DNMT1 and DNMT3a and 3b, and involves the recruitment of methyl-CpG binding protein 2 (MECP2) and other proteins (Bestor 2000). DNMT1 is responsible for maintaining methylation patterns following DNA replication whereas DNMT3a and 3b are required for de novo methylation and are essential for the establishment of DNA methylation patterns during development (Okano et al. 1999). MECP2 is an important epigenetic regulator as it acts as a transcriptional repressor by recruiting histone deacetylases and other factors to silence target genes (Im et al. 2010). Another study reported a significant decrease in DNMT1 levels across the lifespan of mice developmentally exposed to Pb and altered expression of MECP2, which could contribute to cognitive deficits observed in early-life exposure to Pb (Stansfield et al. 2012).
DNA promoter methylome analysis from a cortical neuron-specific population of cells of male adult mice exposed to Pb in utero showed methylation changes, albeit small, in regions connected to neurodevelopment and cognitive functions (Dou et al. 2019). Another study reported hypomethylation of cadherin 7, type 2 (Chd7), a gene essential for neural crest migration and patterning, and its potential link to autism spectrum disorders (ASD) (Hill et al. 2015).
Early-life Pb exposure in rodents was associated with altered expression of genes coding for amyloid-beta precursor protein (App) and beta-site APP-cleaving enzyme 1 (Bace1) later in life, dysregulating biological pathways important to late-onset AD pathogenesis (Kim et al. 2014). Amyloid β (Aβ) plays a crucial role in the pathogenesis of neurodegeneration and its increase can generate ROS and modulate epigenetic patterns of cytosines interfering with repair and oxidation potential of adjacent oxidized guanines (Zawia et al. 2009). This observation is consistent with the role of APOE e4 in AD and Pb susceptibility due to its role in the process of Aβ deposition contributing to the formation of aggregates or clearance alteration (Dodart et al. 2002). It is possible that the individuals carrying the APOE e4 variant and exposed to Pb in early life could be even more susceptible to the AD onset.
Epigenetic outcomes from Pb exposure can also happen at the histone level (Schneider et al. 2016). Developmental exposure to Pb can induce aberrant histone modifications in the hippocampi of rats resulting in attention-deficit/hyperactivity disorder (ADHD) (Luo et al. 2014). In addition, mice exposed to Pb during post-natal day (PND) 1–20 showed both decreased levels of specific epigenetic marks such as H3K9Ac and H3K4me2, which are associated with gene activation, and increased levels of H3K27me3 indicative of gene repression (Eid et al. 2016).
The analysis of rats’ hippocampi chronically exposed to Pb revealed changes in the expression of miRNAs (Mir204, Mir211, Mir448, Mir449a, Mir34b, and Mir34c) associated with neuronal injury and neurodegeneration (An et al. 2014). The SNP rs 7958904 in the lncRNA HOTAIR, involved in oxidative stress, has been linked to Pb susceptibility in workers with high blood Pb levels (Chen et al. 2018). Another study reported an increased expression of the lncRNA lncRpa and the circRNA circRar1, which directly regulate Mir671 expression to promote neuronal apoptosis in the hippocampus and cerebral cortex of a mouse model of Pb-induced neurotoxicity (Nan et al. 2017). Moreover, early postnatal exposure to Pb alters the expression of miRNAs that target epigenetic mediators and neurotoxic AD-related proteins (Masoud et al. 2016). Undoubtedly, Pb can affect ncRNA expression, but further studies are needed to unveil molecular mechanisms and consequences of such change following exposure.
Discussion
The severity of a toxic effect depends on dose, duration, and windows of exposure (i.e. pre- and post-natal development) (Sexton and Hattis 2007). However, humans show great variability in response to environmental exposures even under similar exposure settings (Dornbos and LaPres 2018). The high variability in clinical consequences of Pb exposure may be explained, in part, by the genetic background and the epigenetic changes that can alter susceptibility to Pb. The mechanisms by which Pb, interacting with genetics, trigger epigenetic changes remain to be clarified, and the causal relationship between such alterations and potential aberrant phenotypes needs to be further investigated. Therefore, it is crucial to understand how genetics and epigenetics influence the wide range of possible outcomes from Pb exposure within populations. This review presents the status of current knowledge on associations between adverse outcomes from Pb exposure and specific genetic and epigenetic factors involved in individual susceptibility to Pb toxicity (Figure 3). At least three polymorphic genes are well-established susceptibility markers in humans: ALAD, VDR, and HFE. Moreover, many other genes reviewed here also modulate Pb absorption and toxicokinetics, but their role remains to be explored as their impact on health effects of Pb are still unclear (Mani et al. 2019). Unlike genotoxic factors, which lead to permanent changes of genes, epigenetic changes are reversible and responsive to different environmental factors including lifestyle, diet, and other exposures (Crews et al. 2014; Foley et al. 2009). Epigenetic modifications occurring early in life have the potential to alter later life events. In fact, disturbances in critical developmental windows can lead to increased risk of adult chronic diseases, including neurodevelopmental disorders that can manifest in childhood, adulthood, or even transgenerationally (Perera and Herbstman 2011). Epigenetic modifications are mediated through a series of interconnected pathways that include DNA methylation, histone modifications, and ncRNAs (Handy et al. 2011). All three epigenetic mechanisms are critical players in the development of a healthy brain. Although it is no coincidence that the previously mentioned Pb-induced epigenetic changes have been primarily associated with neurotoxicity (Senut et al. 2012), it is likely they have roles in other adverse health outcomes in other organ systems as well.
Fig. 3.
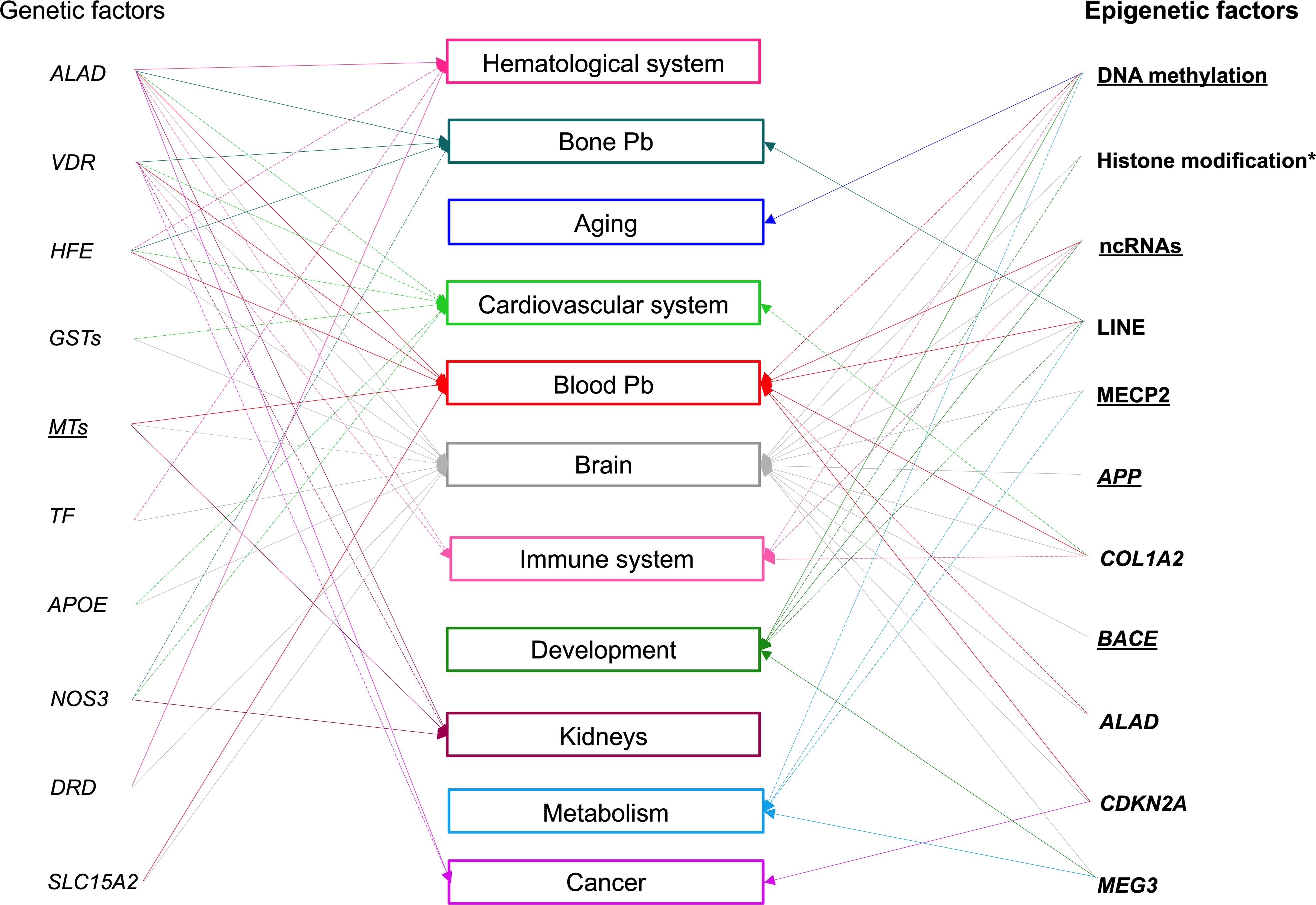
An illustrative summary of well-known or suggested associations between genetic or epigenetic factors and health adverse outcomes from Pb exposure in humans. Italic font style indicates genetic factors, bold font style illustrates epigenetic factors and rectangles identify targets/adverse health outcomes. Underlined factors have been reported in both humans and rodents, asterisks delineate factors reported solely in rodents. The solid lines connecting either genetic or epigenetic factors represent a well-established relation with adverse health outcomes from Pb exposure and dotted lines a potential relation.
Most of the literature reviewed here relies on epidemiological studies which can address the exposure-response relationship and susceptibility to Pb, but they have been challenging due to the variety of clinical symptoms, uncontrolled environments, the use of safety measurements in occupational environments, difficulty in relating phenotypic effect to dose, uncontrolled genetic backgrounds, and modifying effects of other chemicals. Furthermore, most human exposure studies are underpowered, lacking sufficient information on timing and duration of exposure, and require additional validation in multiple independent sample sets or functional analyses to further elucidate the gene-phenotype relationship. The heterogenous results of these studies is likely due to the difficulty of characterizing gene-environment interactions.
On the other hand, we retrieved very few studies utilizing in vitro and/or in vivo models particularly regarding the genetic susceptibility to Pb-induced adverse health outcomes.
Although animal models cannot completely mimic real-world exposures, results from animal studies can still provide valuable information to the body of evidence linking genetic and epigenetic factors with susceptibility to Pb toxicity. Animal studies can also shed light on biological mechanisms critical for determining adverse responses from Pb exposure. Understanding these processes as they relate to Pb susceptibility may help identify prevention and/or treatment strategies.
In conclusion, the examination of the available literature on associations between genetic and epigenetic factors and human health outcomes from Pb exposure indicate that both factors can influence an individual’s response to Pb and likely the variation in the types of responses. Moreover, the discrepancy of some reports reveals gaps in knowledge within the field which would benefit from utilizing new approaches to validate, perhaps by utilizing more in vitro and in vivo models, and help find additional genetic factors or modifiers contributing to Pb susceptibility. Such knowledge could allow the identification of biomarkers for early diagnosis, disease susceptibility, and design of novel therapeutic or mitigation strategies to prevent long-lasting, Pb-induced effects.
Methods
This study used the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines to report the results (Figure 4).
Fig. 4.
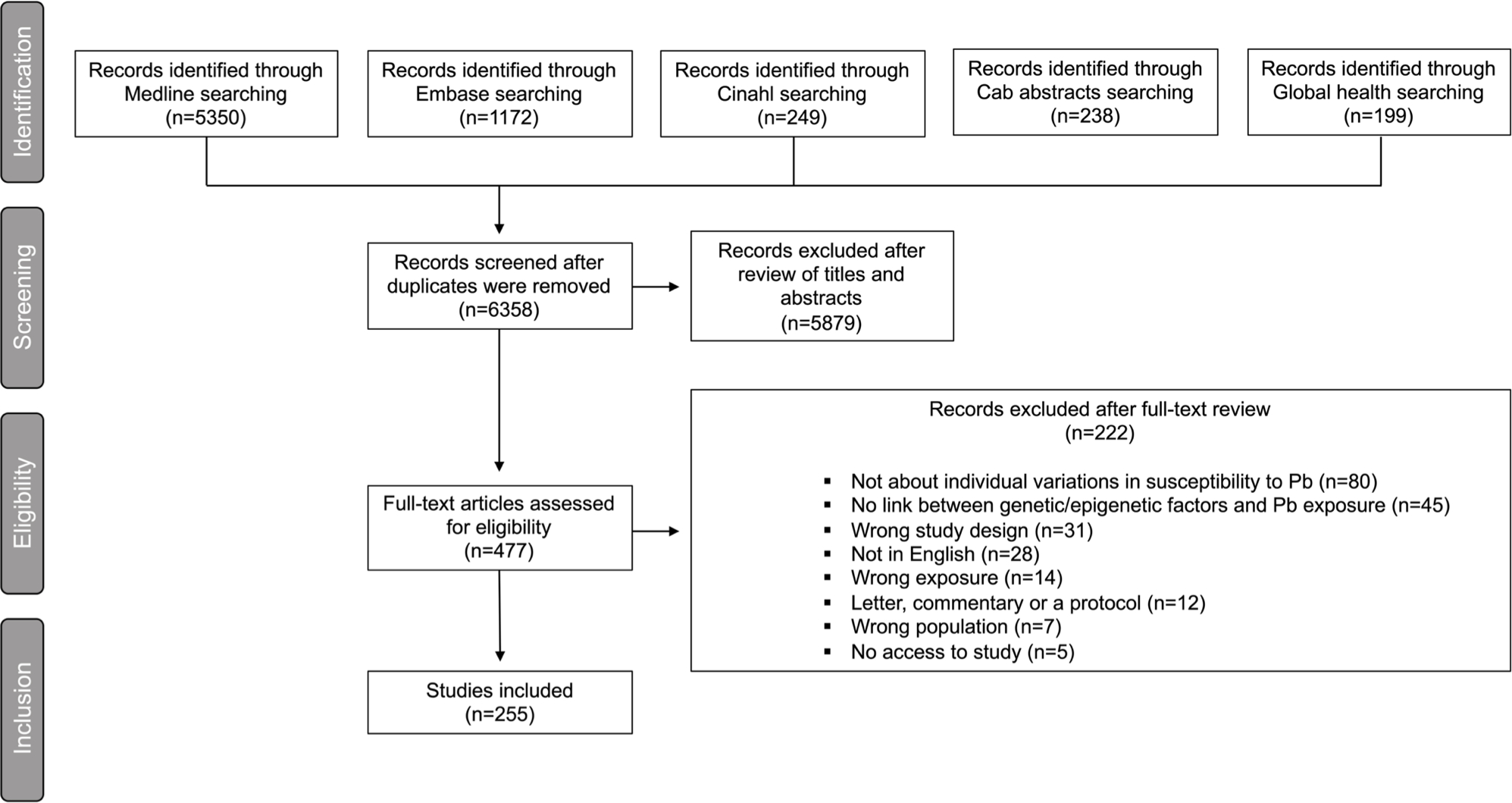
Literature search and screening results for studies reporting on the associations between genetic/epigenetic factors and susceptibility to Pb displayed as a PRISMA flow diagram (http://www.prisma-statement.org).
Search Strategy
Five databases were searched: Medline (Ovid), Global Health (Ovid), CINAHL (Ebsco), Cab Abstracts (Ovid), and Embase (Ovid) with the concepts of Pb, genetic susceptibility, and human or rats or mice. The search was developed by a medical librarian (M.J.F.) with expertise in systematic reviews. The search strings used to identify studies of interest are reported in Table 1. Results retrieved from the searches were uploaded to Covidence, which identifies duplicates and manages the screening process. Lastly, database searches were supplemented with studies identified through citation searching of included or highly relevant studies.
Table 1.
Search strategy for systematic review
| Databases searched | Search strings |
|---|---|
|
| |
| Medline | 1. exp Lead Poisoning/ 2. exp Lead/ or (lead or pb).ti,ab. 3. exp environmental exposure/ or exp food contamination/ or exp Occupational Exposure/ or exp Environmental Pollutants/ 4. (poison* or exposure* or contaminat* or toxic* or intoxic* or pollut*).ti,ab. 5. toxicity.fs. 6. 1 or (2 and (3 or 4 or 5)) 7. exp Genetic Predisposition to Disease/ or exp Genetic Variation/ or exp Disease Susceptibility/ or exp Gene-Environment Interaction/ or exp Epigenomics/ or exp genomics/ 8. (gene* adj2 (predispos* or suscept*)).ti,ab. 9. (epigen* or genomic*).ti,ab. 10. disease suscept*.ti,ab. 11. (genetic adj1 variation*).ti,ab. 12. or/7–8 13. exp animals/ not humans.sh. 14. (rats or rat or mouse or mice).ti,ab. or exp Rats/ or exp Mice/ 15. (6 and 12 and 14) or ((6 and 12) not 13) |
| Embase | 1. exp lead poisoning/ 2. exp lead/ or (lead or pb).ti,ab,kw. 3. exp environmental exposure/ or exp food contamination/ or exp Occupational Exposure/ or exp Environmental Pollutants/ 4. (poison* or exposure* or contaminat* or toxic* or intoxic* or pollut*).ti,ab. 5. 1 or (2 and (3 or 4)) 6. exp genetic predisposition/ or exp disease predisposition/ or exp genotype environment interaction/ or exp epigenetics/ or exp genomics/ or exp genetic variability/ or exp genetic variation/ 7. (gene* adj2 (predispos* or suscept*)).ti,ab. 8. (epigen* or genomic*).ti,ab,kw. 9. disease suscept*.ti,ab. 10. (genetic adj1 variation*).ti,ab. 11. or/6–7 12. exp animals/ not humans.sh. 13. (rats or rat or mouse or mice).ti,ab. or exp mouse/ or exp rat/ 14. (5 and 11 and 13) or ((5 and 11) not 12) 15. limit 14 to yr=“2000 -Current” |
| Cinhal | (MH “Lead Poisoning”) OR (((MH “Lead”) or AB (lead or pb) or TI (lead or pb)) AND ((MH “Environmental Pollution+”) OR (MH “Occupational Exposure”) OR (MH “Food Contamination+”) OR (MH “Toxins+”) OR (AB (poison* or exposure* or contaminat* or toxic* or intoxic* or pollut*)) OR ( TI (poison* or exposure* or contaminat* or toxic* or intoxic* or pollut*)))) AND ((MH “Epigenomics”) OR (MH “Genomics+”) ) OR (AB ((disease suscept* or epigen* or genomic*) or (gene* n2 (predispos* or suscept*)) or (genetic n1 variation*)) ) OR ( TI ( (disease suscept* or epigen* or genomic*) or (gene* n2 (predispos* or suscept*)) or (genetic n1 variation*))) AND (MH “Human”) OR (AB (rats or rat or mouse or mice or human*)) OR (TI (rats or rat or mouse or mice or human*)) |
| CAB Abstracts | 1. (lead or pb).ti,ab. 2. (poison* or exposure* or contaminat* or toxic* or intoxic* or pollut*).ti,ab. 3. (disease suscept* or epigen* or genomic* or (gene* adj2 (predispos* or suscept*)) or (genetic adj1 variation*)).ti,ab. 4. (rats or rat or mouse or mice or human*).ti,ab. 5. and/1–4 6. exp genomics/ 7. exp man/ 8. exp rats/ 9. exp mice/ 10. predisposition.sh. 11. exp lead poisoning/ 12. 11 or (1 and 2) 13. 3 or 10 14. 4 or 7 or 8 or 9 15. and/12–14 |
| Global Health | ((lead or pb) and (poison* or exposure* or contaminat* or toxic* or intoxic* or pollut*)).ti,ab. AND ((disease suscept* or epigen* or genomic*) or (gene* adj2 (predispos* or suscept*)) or (genetic adj1 variation*)).ti,ab. (rats or rat or mouse or mice or human*).ti,ab. |
Study Eligibility Criteria
To be included studies needed to examine the association between general environmental, occupational, and experimental exposures to Pb, genetic and/or epigenetic factors, and health outcomes. Table 2 was developed to identify relevant to health effects of exposure to Pb detailing the Populations of interest, Exposures, Comparators, and Outcomes (PECO). Studies that reported health outcomes from Pb exposure without any link to genetic or epigenetic variation were excluded. Conference abstracts, letters, and editorials were also excluded. Articles were limited to those published between 2000 and September 2020. Only English-language primary studies were eligible.
Table 2.
Population, Exposure, Comparator and Outcome (PECO) statement
| PECO element | Evidence |
|---|---|
|
| |
| Population | Human populations or rodent models. |
| Exposure | Pb exposure without any restrictions of the definition, measurement methods, length, or timing. Co-exposure studies that examined individual susceptibilities were only included if Pb effects could be extrapolated. |
| Comparators | Comparison group with lower exposure or no exposure. |
| Outcome | Dysfunction for any health outcome. |
Study Selection and Extraction
Study selection was conducted in two steps, first by title and abstract, then by full text. A standardized form was developed in Covidence to extract data from included studies. The form covered the following information: study type, risks of bias, location, population, route of exposure, window of exposure, exposure definition (i.e., general environmental, occupational, experimental, relevance to humans), research type (i.e., genetic or epigenetic variation), genetic variants, epigenetic variation, and health outcomes from genetic and epigenetic variants. The health outcomes were classified based upon organ dysfunction or Pb accumulation. The selection and extraction were completed by one reviewer in Covidence (D.C.) and data was exported from Covidence to Microsoft Excel and uploaded into the Texas Data Repository (https://doi.org/10.18738/T8/DTGDZB).
Supplementary Material
Funding
The work was funded by the National Human Genome Research Institute grant RM1 HG008529 and National Institute of Environmental Health Science grant P30 ES029067.
Footnotes
Conflicts of interest/Competing interests The authors have no conflicts or competing inetreests to declare.
Code availability Not applicable
Ethics approval Not applicable
Consent to participate Not applicable
Consent to publish All authors approved the final manuscript version.
Available of data and material
All data generated or analyzed during this study are included in this published article.
References
- An J, Cai T, Che H, Yu T, Cao Z, Liu X, et al. 2014. The changes of mirna expression in rat hippocampus following chronic lead exposure. Toxicol Lett 229:158–166. doi: 10.1016/j.toxlet.2014.06.002 [DOI] [PubMed] [Google Scholar]
- Authority EFS. 2012. Lead dietary exposure in the european population. EFSA Journal 10:2831. doi: 10.2903/j.efsa.2012.2831 [DOI] [Google Scholar]
- Barbosa F Jr., Tanus-Santos JE, Gerlach RF, Parsons PJ. 2005. A critical review of biomarkers used for monitoring human exposure to lead: Advantages, limitations, and future needs. Environ Health Perspect 113:1669–1674. doi: 10.1289/ehp.7917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge YW, et al. 2005. The fetal basis of amyloidogenesis: Exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci 25:823–829. doi: 10.1523/jneurosci.4335-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battistuzzi G, Petrucci R, Silvagni L, Urbani FR, Caiola S. 1981. Delta-aminolevulinate dehydrase: A new genetic polymorphism in man. Ann Hum Genet 45:223–229. doi: 10.1111/j.1469-1809.1981.tb00333.x [DOI] [PubMed] [Google Scholar]
- Berkowitz Z, Price-Green P, Bove FJ, Kaye WE. 2006. Lead exposure and birth outcomes in five communities in shoshone county, idaho. In: Int j hyg environ health, Vol. 209. Germany, 123–132 doi: 10.1016/j.ijheh.2005.11.001 [DOI] [PubMed] [Google Scholar]
- Bestor TH. 2000. The DNA methyltransferases of mammals. Hum Mol Genet 9:2395–2402. doi: 10.1093/hmg/9.16.2395 [DOI] [PubMed] [Google Scholar]
- Bozina N, Bradamante V, Lovrić M. 2009. Genetic polymorphism of metabolic enzymes p450 (cyp) as a susceptibility factor for drug response, toxicity, and cancer risk. Arh Hig Rada Toksikol 60:217–242. doi: 10.2478/10004-1254-60-2009-1885 [DOI] [PubMed] [Google Scholar]
- Caito S, Aschner M. 2017. Developmental neurotoxicity of lead. Adv Neurobiol 18:3–12. doi: 10.1007/978-3-319-60189-2_1 [DOI] [PubMed] [Google Scholar]
- Chang L, Liu X, Shi H, Dai W, Wang H, Jiang Y. 2014. Association of 25-hydroxyvitamin d with hb and lead in children: A chinese population-based study. In: Public health nutr, Vol. 17. England, 827–832 doi: 10.1017/s1368980013001675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Zhao QN, Qian XR, Zhu BL, Ding EM, Wang BS, et al. 2018. Association between the hotair polymorphism and susceptibility to lead poisoning in a chinese population. Biomed Environ Sci 31:473–478. doi: 10.3967/bes2018.063 [DOI] [PubMed] [Google Scholar]
- Crews D, Gillette R, Miller-Crews I, Gore AC, Skinner MK. 2014. Nature, nurture and epigenetics. Mol Cell Endocrinol 398:42–52. doi: 10.1016/j.mce.2014.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Johnstone EM, Little SP, Paul SM. 2002. Apolipoprotein e alters the processing of the beta-amyloid precursor protein in app(v717f) transgenic mice. Brain Res 955:191–199. doi: 10.1016/s0006-8993(02)03437-6 [DOI] [PubMed] [Google Scholar]
- Dornbos P, LaPres JJ. 2018. Incorporating population-level genetic variability within laboratory models in toxicology: From the individual to the population. Toxicology 395:1–8. doi: 10.1016/j.tox.2017.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou JF, Farooqui Z, Faulk CD, Barks AK, Jones T, Dolinoy DC, et al. 2019. Perinatal lead (pb) exposure and cortical neuron-specific DNA methylation in male mice. Genes (Basel) 10. doi: 10.3390/genes10040274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid A, Bihaqi SW, Renehan WE, Zawia NH. 2016. Developmental lead exposure and lifespan alterations in epigenetic regulators and their correspondence to biomarkers of alzheimer’s disease. Alzheimers Dement (Amst) 2:123–131. doi: 10.1016/j.dadm.2016.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, et al. 2010. Mutations in grin2a and grin2b encoding regulatory subunits of nmda receptors cause variable neurodevelopmental phenotypes. Nat Genet 42:1021–1026. doi: 10.1038/ng.677 [DOI] [PubMed] [Google Scholar]
- Engstrom AK, Snyder JM, Maeda N, Xia Z. 2017. Gene-environment interaction between lead and apolipoprotein e4 causes cognitive behavior deficits in mice. Mol Neurodegener 12:14. doi: 10.1186/s13024-017-0155-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engström K, Rydbeck F, Kippler M, Wojdacz TK, Arifeen S, Vahter M, et al. 2015. Prenatal lead exposure is associated with decreased cord blood DNA methylation of the glycoprotein vi gene involved in platelet activation and thrombus formation. Environ Epigenet 1:dvv007. doi: 10.1093/eep/dvv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eum KD, Weisskopf MG, Nie LH, Hu H, Korrick SA. 2014. Cumulative lead exposure and age at menopause in the nurses’ health study cohort. Environ Health Perspect 122:229–234. doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eum KD, Seals RM, Taylor KM, Grespin M, Umbach DM, Hu H, et al. 2015. Modification of the association between lead exposure and amyotrophic lateral sclerosis by iron and oxidative stress related gene polymorphisms. Amyotroph Lateral Scler Frontotemporal Degener 16:72–79. doi: 10.3109/21678421.2014.964259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenga C, Gangemi S, Di Salvatore V, Falzone L, Libra M. 2017. Immunological effects of occupational exposure to lead (review). Mol Med Rep 15:3355–3360. doi: 10.3892/mmr.2017.6381 [DOI] [PubMed] [Google Scholar]
- Fernandes KC, Martins AC Jr., Oliveira A, Antunes LM, Cólus IM, Barbosa F Jr., et al. 2016. Polymorphism of metallothionein 2a modifies lead body burden in workers chronically exposed to the metal. Public Health Genomics 19:47–52. doi: 10.1159/000441713 [DOI] [PubMed] [Google Scholar]
- Fleming RE, Britton RS, Waheed A, Sly WS, Bacon BR. 2005. Pathophysiology of hereditary hemochromatosis. Semin Liver Dis 25:411–419. doi: 10.1055/s-2005-923313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flora G, Gupta D, Tiwari A. 2012. Toxicity of lead: A review with recent updates. Interdisciplinary toxicology 5:47–58. doi: 10.2478/v10102-012-0009-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley DL, Craig JM, Morley R, Olsson CA, Dwyer T, Smith K, et al. 2009. Prospects for epigenetic epidemiology. Am J Epidemiol 169:389–400. doi: 10.1093/aje/kwn380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Lestón J, Roma-Torres J, Mayan O, Schroecksnadel S, Fuchs D, Moreira AO, et al. 2012. Assessment of immunotoxicity parameters in individuals occupationally exposed to lead. J Toxicol Environ Health A 75:807–818. doi: 10.1080/15287394.2012.690327 [DOI] [PubMed] [Google Scholar]
- Gavazzo P, Zanardi I, Baranowska-Bosiacka I, Marchetti C. 2008. Molecular determinants of pb2+ interaction with nmda receptor channels. Neurochem Int 52:329–337. doi: 10.1016/j.neuint.2007.07.003 [DOI] [PubMed] [Google Scholar]
- Goodrich JM, Sánchez BN, Dolinoy DC, Zhang Z, Hernández-Ávila M, Hu H, et al. 2015. Quality control and statistical modeling for environmental epigenetics: A study on in utero lead exposure and DNA methylation at birth. Epigenetics 10:19–30. doi: 10.4161/15592294.2014.989077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundacker C, Wittmann KJ, Kukuckova M, Komarnicki G, Hikkel I, Gencik M. 2009. Genetic background of lead and mercury metabolism in a group of medical students in austria. Environ Res 109:786–796. doi: 10.1016/j.envres.2009.05.003 [DOI] [PubMed] [Google Scholar]
- Gundacker C, Gencik M, Hengstschläger M. 2010. The relevance of the individual genetic background for the toxicokinetics of two significant neurodevelopmental toxicants: Mercury and lead. Mutat Res 705:130–140. doi: 10.1016/j.mrrev.2010.06.003 [DOI] [PubMed] [Google Scholar]
- Handy DE, Castro R, Loscalzo J. 2011. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 123:2145–2156. doi: 10.1161/circulationaha.110.956839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna CW, Bloom MS, Robinson WP, Kim D, Parsons PJ, vom Saal FS, et al. 2012. DNA methylation changes in whole blood is associated with exposure to the environmental contaminants, mercury, lead, cadmium and bisphenol a, in women undergoing ovarian stimulation for ivf. Hum Reprod 27:1401–1410. doi: 10.1093/humrep/des038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrill AH, McAllister KA. 2017. New rodent population models may inform human health risk assessment and identification of genetic susceptibility to environmental exposures. Environ Health Perspect 125:086002. doi: 10.1289/ehp1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DS, Cabrera R, Wallis Schultz D, Zhu H, Lu W, Finnell RH, et al. 2015. Autism-like behavior and epigenetic changes associated with autism as consequences of in utero exposure to environmental pollutants in a mouse model. Behav Neurol 2015:426263. doi: 10.1155/2015/426263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollerer I, Bachmann A, Muckenthaler MU. 2017. Pathophysiological consequences and benefits of hfe mutations: 20 years of research. Haematologica 102:809–817. doi: 10.3324/haematol.2016.160432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon KL, Fung CK, Leung AK. 2017. Childhood lead poisoning: An overview. Hong Kong Med J 23:616–621. doi: 10.12809/hkmj176214 [DOI] [PubMed] [Google Scholar]
- Hu Y, Shen H, Keep RF, Smith DE. 2007. Peptide transporter 2 (pept2) expression in brain protects against 5-aminolevulinic acid neurotoxicity. J Neurochem 103:2058–2065. doi: 10.1111/j.1471-4159.2007.04905.x [DOI] [PubMed] [Google Scholar]
- Im HI, Hollander JA, Bali P, Kenny PJ. 2010. Mecp2 controls bdnf expression and cocaine intake through homeostatic interactions with microrna-212. Nat Neurosci 13:1120–1127. doi: 10.1038/nn.2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irgens A, Krüger K, Skorve AH, Irgens LM. 1998. Reproductive outcome in offspring of parents occupationally exposed to lead in norway. In: Am j ind med, Vol. 34. United States, 431–437 doi: [DOI] [PubMed] [Google Scholar]
- Jurczuk M, Moniuszko-Jakoniuk J, Brzóska MM. 2006. Involvement of some low-molecular thiols in the peroxidative mechanisms of lead and ethanol action on rat liver and kidney. Toxicology 219:11–21. doi: 10.1016/j.tox.2005.10.022 [DOI] [PubMed] [Google Scholar]
- Karwowski MP, Just AC, Bellinger DC, Jim R, Hatley EL, Ettinger AS, et al. 2014. Maternal iron metabolism gene variants modify umbilical cord blood lead levels by gene-environment interaction: A birth cohort study. Environ Health 13:77. doi: 10.1186/1476-069x-13-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayaalti Z, Kaya-Akyüzlü D, Söylemez E, Söylemezoğlu T. 2015a. Maternal hemochromatosis gene h63d single-nucleotide polymorphism and lead levels of placental tissue, maternal and umbilical cord blood. Environ Res 140:456–461. doi: 10.1016/j.envres.2015.05.004 [DOI] [PubMed] [Google Scholar]
- Kayaaltı Z, Akyüzlü DK, Söylemezoğlu T. 2015b. Evaluation of the effect of divalent metal transporter 1 gene polymorphism on blood iron, lead and cadmium levels. Environ Res 137:8–13. doi: 10.1016/j.envres.2014.11.008 [DOI] [PubMed] [Google Scholar]
- Keil KP, Lein PJ. 2016. DNA methylation: A mechanism linking environmental chemical exposures to risk of autism spectrum disorders? Environ Epigenet 2. doi: 10.1093/eep/dvv012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr BT, Ochs-Balcom HM, López P, García-Vargas GG, Rosado JL, Cebrián ME, et al. 2019. Effects of alad genotype on the relationship between lead exposure and anthropometry in a cohort of mexican children. Environ Res 170:65–72. doi: 10.1016/j.envres.2018.12.003 [DOI] [PubMed] [Google Scholar]
- Kim J, Lee Y, Yang M. 2014. Environmental exposure to lead (pb) and variations in its susceptibility. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 32:159–185. doi: 10.1080/10590501.2014.907461 [DOI] [PubMed] [Google Scholar]
- Kordas K, Ettinger AS, Bellinger DC, Schnaas L, Téllez Rojo MM, Hernández-Avila M, et al. 2011. A dopamine receptor (drd2) but not dopamine transporter (dat1) gene polymorphism is associated with neurocognitive development of mexican preschool children with lead exposure. J Pediatr 159:638–643. doi: 10.1016/j.jpeds.2011.03.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovatsi L, Georgiou E, Ioannou A, Haitoglou C, Tzimagiorgis G, Tsoukali H, et al. 2010. P16 promoter methylation in pb2+ -exposed individuals. Clin Toxicol (Phila) 48:124–128. doi: 10.3109/15563650903567091 [DOI] [PubMed] [Google Scholar]
- Kumar S 2018. Occupational and environmental exposure to lead and reproductive health impairment: An overview. Indian J Occup Environ Med 22:128–137. doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanphear BP, Rauch S, Auinger P, Allen RW, Hornung RW. 2018. Low-level lead exposure and mortality in us adults: A population-based cohort study. Lancet Public Health 3:e177–e184. doi: 10.1016/s2468-2667(18)30025-2 [DOI] [PubMed] [Google Scholar]
- Lee B-K, Lee SJ, Joo JS, Cho K-S, Kim NS, Kim H-J. 2012. Association of glutathione s-transferase genes (gstm1 and gstt1) polymorphisms with hypertension in lead-exposed workers. Molecular & Cellular Toxicology 8:203–208. doi: 10.1007/s13273-012-0025-5 [DOI] [Google Scholar]
- Levin R, Brown MJ, Kashtock ME, Jacobs DE, Whelan EA, Rodman J, et al. 2008. Lead exposures in u.S. Children, 2008: Implications for prevention. Environ Health Perspect 116:1285–1293. doi: 10.1289/ehp.11241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Xu M, Wang S, Yang X, Zhou S, Zhang J, et al. 2011. Lead exposure suppressed alad transcription by increasing methylation level of the promoter cpg islands. Toxicol Lett 203:48–53. doi: 10.1016/j.toxlet.2011.03.002 [DOI] [PubMed] [Google Scholar]
- Li C, Yang X, Xu M, Zhang J, Sun N. 2013. Epigenetic marker (line-1 promoter) methylation level was associated with occupational lead exposure. Clin Toxicol (Phila) 51:225–229. doi: 10.3109/15563650.2013.782410 [DOI] [PubMed] [Google Scholar]
- Li E, Zhang Y. 2014. DNA methylation in mammals. Cold Spring Harb Perspect Biol 6:a019133. doi: 10.1101/cshperspect.a019133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind J, Persson J, Ingvar M, Larsson A, Cruts M, Van Broeckhoven C, et al. 2006. Reduced functional brain activity response in cognitively intact apolipoprotein e epsilon4 carriers. Brain 129:1240–1248. doi: 10.1093/brain/awl054 [DOI] [PubMed] [Google Scholar]
- Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G. 2013. Apolipoprotein e and alzheimer disease: Risk, mechanisms and therapy. Nat Rev Neurol 9:106–118. doi: 10.1038/nrneurol.2012.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes AC, Peixe TS, Mesas AE, Paoliello MM. 2016. Lead exposure and oxidative stress: A systematic review. Rev Environ Contam Toxicol 236:193–238. doi: 10.1007/978-3-319-20013-2_3 [DOI] [PubMed] [Google Scholar]
- Luo M, Xu Y, Cai R, Tang Y, Ge MM, Liu ZH, et al. 2014. Epigenetic histone modification regulates developmental lead exposure induced hyperactivity in rats. Toxicol Lett 225:78–85. doi: 10.1016/j.toxlet.2013.11.025 [DOI] [PubMed] [Google Scholar]
- Mani MS, Kabekkodu SP, Joshi MB, Dsouza HS. 2019. Ecogenetics of lead toxicity and its influence on risk assessment. Hum Exp Toxicol 38:1031–1059. doi: 10.1177/0960327119851253 [DOI] [PubMed] [Google Scholar]
- Mason LH, Harp JP, Han DY. 2014. Pb neurotoxicity: Neuropsychological effects of lead toxicity. Biomed Res Int 2014:840547. doi: 10.1155/2014/840547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoud AM, Bihaqi SW, Machan JT, Zawia NH, Renehan WE. 2016. Early-life exposure to lead (pb) alters the expression of microrna that target proteins associated with alzheimer’s disease. J Alzheimers Dis 51:1257–1264. doi: 10.3233/jad-151018 [DOI] [PubMed] [Google Scholar]
- Masten SJ, Davies SH, McElmurry SP. 2016. Flint water crisis: What happened and why? J Am Water Works Assoc 108:22–34. doi: 10.5942/jawwa.2016.108.0195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra KP, Singh VK, Rani R, Yadav VS, Chandran V, Srivastava SP, et al. 2003. Effect of lead exposure on the immune response of some occupationally exposed individuals. In: Toxicology, Vol. 188. Ireland, 251–259 doi: 10.1016/s0300-483x(03)00091-x [DOI] [PubMed] [Google Scholar]
- Mishra KP. 2009. Lead exposure and its impact on immune system: A review. In: Toxicol in vitro, Vol. 23. England, 969–972 doi: 10.1016/j.tiv.2009.06.014 [DOI] [PubMed] [Google Scholar]
- Mitra P, Sharma S, Purohit P, Sharma P. 2017. Clinical and molecular aspects of lead toxicity: An update. Crit Rev Clin Lab Sci 54:506–528. doi: 10.1080/10408363.2017.1408562 [DOI] [PubMed] [Google Scholar]
- Miyata M, Smith JD. 1996. Apolipoprotein e allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat Genet 14:55–61. doi: 10.1038/ng0996-55 [DOI] [PubMed] [Google Scholar]
- Montes-Castro N, Alvarado-Cruz I, Torres-Sánchez L, García-Aguiar I, Barrera-Hernández A, Escamilla-Núñez C, et al. 2019. Prenatal exposure to metals modified DNA methylation and the expression of antioxidant- and DNA defense-related genes in newborns in an urban area. J Trace Elem Med Biol 55:110–120. doi: 10.1016/j.jtemb.2019.06.014 [DOI] [PubMed] [Google Scholar]
- Moya J, Bearer CF, Etzel RA. 2004. Children’s behavior and physiology and how it affects exposure to environmental contaminants. Pediatrics 113:996–1006. doi: [PubMed] [Google Scholar]
- Naha N, Bhar RB, Mukherjee A, Chowdhury AR. 2005. Structural alteration of spermatozoa in the persons employed in lead acid battery factory. Indian J Physiol Pharmacol 49:153–162. doi: [PubMed] [Google Scholar]
- Nan A, Chen L, Zhang N, Liu Z, Yang T, Wang Z, et al. 2017. A novel regulatory network among lncrpa, circrar1, mir-671 and apoptotic genes promotes lead-induced neuronal cell apoptosis. Arch Toxicol 91:1671–1684. doi: 10.1007/s00204-016-1837-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal AP, Worley PF, Guilarte TR. 2011. Lead exposure during synaptogenesis alters nmda receptor targeting via nmda receptor inhibition. Neurotoxicology 32:281–289. doi: 10.1016/j.neuro.2010.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nye MD, King KE, Darrah TH, Maguire R, Jima DD, Huang Z, et al. 2016. Maternal blood lead concentrations, DNA methylation of meg3 dmr regulating the dlk1/meg3 imprinted domain and early growth in a multiethnic cohort. Environ Epigenet 2. doi: 10.1093/eep/dvv009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E. 1999. DNA methyltransferases dnmt3a and dnmt3b are essential for de novo methylation and mammalian development. Cell 99:247–257. doi: 10.1016/s0092-8674(00)81656-6 [DOI] [PubMed] [Google Scholar]
- Olympio KP, Goncalves C, Gunther WM, Bechara EJ. 2009. Neurotoxicity and aggressiveness triggered by low-level lead in children: A review. Rev Panam Salud Publica 26:266–275. doi: 10.1590/s1020-49892009000900011 [DOI] [PubMed] [Google Scholar]
- Omelchenko IA, Nelson CS, Allen CN. 1997. Lead inhibition of n-methyl-d-aspartate receptors containing nr2a, nr2c and nr2d subunits. J Pharmacol Exp Ther 282:1458–1464. doi: [PubMed] [Google Scholar]
- Onalaja AO, Claudio L. 2000. Genetic susceptibility to lead poisoning. Environ Health Perspect 108 Suppl 1:23–28. doi: 10.1289/ehp.00108s123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordemann JM, Austin RN. 2016. Lead neurotoxicity: Exploring the potential impact of lead substitution in zinc-finger proteins on mental health. Metallomics 8:579–588. doi: 10.1039/c5mt00300h [DOI] [PubMed] [Google Scholar]
- Orr SE, Bridges CC. 2017. Chronic kidney disease and exposure to nephrotoxic metals. Int J Mol Sci 18. doi: 10.3390/ijms18051039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlas N, Broberg K, Olewińska E, Prokopowicz A, Skerfving S, Pawlas K. 2012. Modification by the genes alad and vdr of lead-induced cognitive effects in children. Neurotoxicology 33:37–43. doi: 10.1016/j.neuro.2011.10.012 [DOI] [PubMed] [Google Scholar]
- Pawlas N, Broberg K, Skerfving S, Pawlas K. 2014. Disturbance of posture in children with very low lead exposure, and modification by vdr foki genotype. Ann Agric Environ Med 21:739–744. doi: 10.5604/12321966.1129926 [DOI] [PubMed] [Google Scholar]
- Pawlas N, Broberg K, Olewińska E, Kozłowska A, Skerfving S, Pawlas K. 2015. Genetic modification of alad and vdr on lead-induced impairment of hearing in children. Environ Toxicol Pharmacol 39:1091–1098. doi: 10.1016/j.etap.2015.03.008 [DOI] [PubMed] [Google Scholar]
- Perera F, Herbstman J. 2011. Prenatal environmental exposures, epigenetics, and disease. Reprod Toxicol 31:363–373. doi: 10.1016/j.reprotox.2010.12.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilsner JR, Hu H, Ettinger A, Sánchez BN, Wright RO, Cantonwine D, et al. 2009. Influence of prenatal lead exposure on genomic methylation of cord blood DNA. Environ Health Perspect 117:1466–1471. doi: 10.1289/ehp.0800497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prada D, Colicino E, Power MC, Weisskopf MG, Zhong J, Hou L, et al. 2016. Apoe ε4 allele modifies the association of lead exposure with age-related cognitive decline in older individuals. Environ Res 151:101–105. doi: 10.1016/j.envres.2016.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prozialeck WC, Edwards JR, Nebert DW, Woods JM, Barchowsky A, Atchison WD. 2008. The vascular system as a target of metal toxicity. Toxicol Sci 102:207–218. doi: 10.1093/toxsci/kfm263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzas JE, Campbell J, O’Keefe RJ, Rosier RN. 2004. Lead toxicity in the skeleton and its role in osteoporosis. In: Nutrition and bone health. Totowa, NJ:Humana Press, 363–376 doi: 10.1007/978-1-59259-740-6_22 [DOI] [Google Scholar]
- Qu W, Diwan BA, Liu J, Goyer RA, Dawson T, Horton JL, et al. 2002. The metallothionein-null phenotype is associated with heightened sensitivity to lead toxicity and an inability to form inclusion bodies. Am J Pathol 160:1047–1056. doi: 10.1016/s0002-9440(10)64925-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rădulescu A, Lundgren S. 2019. A pharmacokinetic model of lead absorption and calcium competitive dynamics. Scientific Reports 9:14225. doi: 10.1038/s41598-019-50654-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudenska M, Gumulec J, Podlaha O, Sztalmachova M, Babula P, Eckschlager T, et al. 2014. Metallothionein polymorphisms in pathological processes. Metallomics 6:55–68. doi: 10.1039/c3mt00132f [DOI] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, et al. 2004. Functional brain abnormalities in young adults at genetic risk for late-onset alzheimer’s dementia. Proc Natl Acad Sci U S A 101:284–289. doi: 10.1073/pnas.2635903100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney JPK, Woods NF, Martin MD, Woods JS. 2018. Genetic polymorphisms of grin2a and grin2b modify the neurobehavioral effects of low-level lead exposure in children. Environ Res 165:1–10. doi: 10.1016/j.envres.2018.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Hu H, Bellinger DC, Mukherjee B, Modali R, Nasaruddin K, et al. 2011. Hemoglobin, lead exposure, and intelligence quotient: Effect modification by the drd2 taq ia polymorphism. Environ Health Perspect 119:144–149. doi: 10.1289/ehp.0901878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Ettinger AS, Hu H, Bellinger D, Schwartz J, Modali R, et al. 2013. Effect modification by transferrin c2 polymorphism on lead exposure, hemoglobin levels, and iq. Neurotoxicology 38:17–22. doi: 10.1016/j.neuro.2013.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders T, Liu Y, Buchner V, Tchounwou PB. 2009. Neurotoxic effects and biomarkers of lead exposure: A review. Rev Environ Health 24:15–45. doi: 10.1515/reveh.2009.24.1.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JS, Anderson DW, Kidd SK, Sobolewski M, Cory-Slechta DA. 2016. Sex-dependent effects of lead and prenatal stress on post-translational histone modifications in frontal cortex and hippocampus in the early postnatal brain. Neurotoxicology 54:65–71. doi: 10.1016/j.neuro.2016.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz BS, Stewart WF, Kelsey KT, Simon D, Park S, Links JM, et al. 2000. Associations of tibial lead levels with bsmi polymorphisms in the vitamin d receptor in former organolead manufacturing workers. Environ Health Perspect 108:199–203. doi: 10.1289/ehp.00108199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scinicariello F, Murray HE, Moffett DB, Abadin HG, Sexton MJ, Fowler BA. 2007. Lead and delta-aminolevulinic acid dehydratase polymorphism: Where does it lead? A meta-analysis. Environ Health Perspect 115:35–41. doi: 10.1289/ehp.9448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen A, Heredia N, Senut MC, Hess M, Land S, Qu W, et al. 2015a. Early life lead exposure causes gender-specific changes in the DNA methylation profile of DNA extracted from dried blood spots. Epigenomics 7:379–393. doi: 10.2217/epi.15.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen A, Heredia N, Senut MC, Land S, Hollocher K, Lu X, et al. 2015b. Multigenerational epigenetic inheritance in humans: DNA methylation changes associated with maternal exposure to lead can be transmitted to the grandchildren. Sci Rep 5:14466. doi: 10.1038/srep14466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senut MC, Cingolani P, Sen A, Kruger A, Shaik A, Hirsch H, et al. 2012. Epigenetics of early-life lead exposure and effects on brain development. Epigenomics 4:665–674. doi: 10.2217/epi.12.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senut MC, Sen A, Cingolani P, Shaik A, Land SJ, Ruden DM. 2014. Lead exposure disrupts global DNA methylation in human embryonic stem cells and alters their neuronal differentiation. Toxicol Sci 139:142–161. doi: 10.1093/toxsci/kfu028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton K, Hattis D. 2007. Assessing cumulative health risks from exposure to environmental mixtures - three fundamental questions. Environ Health Perspect 115:825–832. doi: 10.1289/ehp.9333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shellenberger MK. 1984. Effects of early lead exposure on neurotransmitter systems in the brain. A review with commentary. Neurotoxicology 5:177–212. doi: [PubMed] [Google Scholar]
- Sobin C, Parisi N, Schaub T, Gutierrez M, Ortega AX. 2011. Δ-aminolevulinic acid dehydratase single nucleotide polymorphism 2 and peptide transporter 2*2 haplotype may differentially mediate lead exposure in male children. Arch Environ Contam Toxicol 61:521–529. doi: 10.1007/s00244-011-9645-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacchiotti A, Morandini F, Bettoni F, Schena I, Lavazza A, Grigolato PG, et al. 2009. Stress proteins and oxidative damage in a renal derived cell line exposed to inorganic mercury and lead. Toxicology 264:215–224. doi: 10.1016/j.tox.2009.08.014 [DOI] [PubMed] [Google Scholar]
- Stansfield KH, Pilsner JR, Lu Q, Wright RO, Guilarte TR. 2012. Dysregulation of bdnf-trkb signaling in developing hippocampal neurons by pb(2+): Implications for an environmental basis of neurodevelopmental disorders. Toxicol Sci 127:277–295. doi: 10.1093/toxsci/kfs090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taha EA, Sayed SK, Ghandour NM, Mahran AM, Saleh MA, Amin MM, et al. 2013. Correlation between seminal lead and cadmium and seminal parameters in idiopathic oligoasthenozoospermic males. Cent European J Urol 66:84–92. doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto M, Hattori Y, Zhao H, Sato H, Tamada A, Sasaki S, et al. 2011. Laminar and areal expression of unc5d and its role in cortical cell survival. Cereb Cortex 21:1925–1934. doi: 10.1093/cercor/bhq265 [DOI] [PubMed] [Google Scholar]
- Tasmin S, Furusawa H, Ahmad SA, Faruquee MH, Watanabe C. 2015. Delta-aminolevulinic acid dehydratase (alad) polymorphism in lead exposed bangladeshi children and its effect on urinary aminolevulinic acid (ala). Environ Res 136:318–323. doi: 10.1016/j.envres.2014.08.045 [DOI] [PubMed] [Google Scholar]
- Taylor MP, Isley CF, Glover J. 2019. Prevalence of childhood lead poisoning and respiratory disease associated with lead smelter emissions. In: Environ int, Vol. 127. Netherlands:© 2019 The Authors. Published by Elsevier Ltd., 340–352 doi: 10.1016/j.envint.2019.01.062 [DOI] [PubMed] [Google Scholar]
- Tong S, McMichael AJ, Baghurst PA. 2000. Interactions between environmental lead exposure and sociodemographic factors on cognitive development. Archives of Environmental Health: An International Journal 55:330–335. doi: 10.1080/00039890009604025 [DOI] [PubMed] [Google Scholar]
- Toscano CD, Guilarte TR. 2005. Lead neurotoxicity: From exposure to molecular effects. Brain Res Brain Res Rev 49:529–554. doi: 10.1016/j.brainresrev.2005.02.004 [DOI] [PubMed] [Google Scholar]
- Tüzmen MN, Yücel NC, Kalburcu T, Demiryas N. 2015. Effects of curcumin and tannic acid on the aluminum- and lead-induced oxidative neurotoxicity and alterations in nmda receptors. Toxicol Mech Methods 25:120–127. doi: 10.3109/15376516.2014.997947 [DOI] [PubMed] [Google Scholar]
- Vaziri ND, Ding Y. 2001. Effect of lead on nitric oxide synthase expression in coronary endothelial cells: Role of superoxide. Hypertension 37:223–226. doi: 10.1161/01.hyp.37.2.223 [DOI] [PubMed] [Google Scholar]
- Vaziri ND, Ding Y, Ni Z. 2001. Compensatory up-regulation of nitric-oxide synthase isoforms in lead-induced hypertension; reversal by a superoxide dismutase-mimetic drug. J Pharmacol Exp Ther 298:679–685. doi: [PubMed] [Google Scholar]
- Vigeh M, Yokoyama K, Kitamura F, Afshinrokh M, Beygi A, Niroomanesh S. 2010. Early pregnancy blood lead and spontaneous abortion. In: Women health, Vol. 50. United States, 756–766 doi: 10.1080/03630242.2010.532760 [DOI] [PubMed] [Google Scholar]
- Vorvolakos T, Arseniou S, Samakouri M. 2016. There is no safe threshold for lead exposure: Α literature review. Psychiatriki 27:204–214. doi: 10.22365/jpsych.2016.273.204 [DOI] [PubMed] [Google Scholar]
- Waly M, Olteanu H, Banerjee R, Choi SW, Mason JB, Parker BS, et al. 2004. Activation of methionine synthase by insulin-like growth factor-1 and dopamine: A target for neurodevelopmental toxins and thimerosal. Mol Psychiatry 9:358–370. doi: 10.1038/sj.mp.4001476 [DOI] [PubMed] [Google Scholar]
- Wang T, Guan R-L, Liu M-C, Shen X-F, Chen JY, Zhao M-G, et al. 2016. Lead exposure impairs hippocampus related learning and memory by altering synaptic plasticity and morphology during juvenile period. Molecular Neurobiology 53:3740–3752. doi: 10.1007/s12035-015-9312-1 [DOI] [PubMed] [Google Scholar]
- Wang Z, Claus Henn B, Wang C, Wei Y, Su L, Sun R, et al. 2017. Genome-wide gene by lead exposure interaction analysis identifies unc5d as a candidate gene for neurodevelopment. Environ Health 16:81. doi: 10.1186/s12940-017-0288-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver VM, Schwartz BS, Ahn KD, Stewart WF, Kelsey KT, Todd AC, et al. 2003. Associations of renal function with polymorphisms in the delta-aminolevulinic acid dehydratase, vitamin d receptor, and nitric oxide synthase genes in korean lead workers. Environ Health Perspect 111:1613–1619. doi: 10.1289/ehp.6116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver VM, Schwartz BS, Jaar BG, Ahn KD, Todd AC, Lee SS, et al. 2005. Associations of uric acid with polymorphisms in the delta-aminolevulinic acid dehydratase, vitamin d receptor, and nitric oxide synthase genes in korean lead workers. Environ Health Perspect 113:1509–1515. doi: 10.1289/ehp.7927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisskopf MG, Wright RO, Schwartz J, Spiro A 3rd, Sparrow D, Aro A, et al. 2004. Cumulative lead exposure and prospective change in cognition among elderly men: The va normative aging study. Am J Epidemiol 160:1184–1193. doi: 10.1093/aje/kwh333 [DOI] [PubMed] [Google Scholar]
- Wetmur JG, Lehnert G, Desnick RJ. 1991. The delta-aminolevulinate dehydratase polymorphism: Higher blood lead levels in lead workers and environmentally exposed children with the 1–2 and 2–2 isozymes. Environ Res 56:109–119. doi: 10.1016/s0013-9351(05)80001-5 [DOI] [PubMed] [Google Scholar]
- Whitfield JB, Dy V, McQuilty R, Zhu G, Montgomery GW, Ferreira MA, et al. 2007. Evidence of genetic effects on blood lead concentration. Environ Health Perspect 115:1224–1230. doi: 10.1289/ehp.8847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright RO, Schwartz J, Wright RJ, Bollati V, Tarantini L, Park SK, et al. 2010. Biomarkers of lead exposure and DNA methylation within retrotransposons. Environ Health Perspect 118:790–795. doi: 10.1289/ehp.0901429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Wang Y, Wang M, Sun N, Li C. 2017. Grin2a polymorphisms and expression levels are associated with lead-induced neurotoxicity. Toxicol Ind Health 33:332–339. doi: 10.1177/0748233716647636 [DOI] [PubMed] [Google Scholar]
- Yu LB, Tu YT, Huang JW, Zhang YN, Zheng GQ, Xu XW, et al. 2018. Hypermethylation of cpg islands is associated with increasing chromosomal damage in chinese lead-exposed workers. Environ Mol Mutagen 59:549–556. doi: 10.1002/em.22194 [DOI] [PubMed] [Google Scholar]
- Zawia NH, Lahiri DK, Cardozo-Pelaez F. 2009. Epigenetics, oxidative stress, and alzheimer disease. Free Radic Biol Med 46:1241–1249. doi: 10.1016/j.freeradbiomed.2009.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Xu M, Zhao Q, Sun K, Gong W, Zhang Q, et al. 2016. Association between polymorphism of exportin-5 and susceptibility to lead poisoning in a chinese population. Int J Environ Res Public Health 14. doi: 10.3390/ijerph14010036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Tian L, Liang Y, Broberg K, Lei L, Guo W, et al. 2011. Δ-aminolevulinic acid dehydratase genotype predicts toxic effects of lead on workers’ peripheral nervous system. Neurotoxicology 32:374–382. doi: 10.1016/j.neuro.2011.03.006 [DOI] [PubMed] [Google Scholar]
- Zhu M, Fitzgerald EF, Gelberg KH, Lin S, Druschel CM. 2010. Maternal low-level lead exposure and fetal growth. Environ Health Perspect 118:1471–1475. doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article.