

Abstract
Cerebrospinal fluid (CSF) proteins and their structures have been implicated repeatedly in aging and neurodegenerative diseases. Limited proteolysis-mass spectrometry (LiP-MS) is a method that enables proteome-wide screening for changes in both protein abundance and structure. To screen for novel aging-associated changes in the CSF proteome, we performed LiP-MS on CSF from young and old mice with a modified analysis pipeline. We found 38 protein groups change in abundance with aging, most dominantly immunoglobulins of the IgM subclass. We discovered six high-confidence candidates that appeared to change in structure with aging, of which Kng1, Itih2, Lp-PLA2, and 14-3-3 proteins have binding partners or proteoforms known previously to change in the brain with Alzheimer’s disease. Intriguingly, using orthogonal validation by Western blot we found the LiP-MS hit Cd5l forms a covalent complex with IgM in mouse and human CSF whose abundance increases with aging. SOMAmer probe signals for all six LiP-MS hits in human CSF, especially 14-3-3 proteins, significantly associate with several clinical features relevant to cognitive function and neurodegeneration. Together, our findings show that LiP-MS can uncover age-related structural changes in CSF with relevance to neurodegeneration.
Aging is a major contributing factor to neurodegenerative diseases such as Alzheimer’s disease (AD).1 A better understanding of brain aging would aid in our understanding of the pathogenesis of these diseases. Many of the most successful biomarkers and therapeutic targets for aging-associated neurological diseases such as AD are specific proteoforms2,3 and complexes of proteins, such as the Aβ42 oligomer4 (Supplementary Figure 1), phospho-tau5 and others.6,7,8,9
Although the structures of proteins are critically relevant to aging, disease, and biology in general,10 a high-throughput screen for changes in protein structures has never been performed in a mammalian system. In vitro, limited proteolysis-mass spectrometry (LiP-MS) has provided a way to observe changes in both protein abundances and structures on a proteome-wide scale (Figure 1a).11,12,13,14,15 Observing that cerebrospinal fluid (CSF) contains the AD-relevant proteoforms and complexes referenced above,6,7,8,9 and that mechanistic studies in mice are able to reveal the biology of aging-associated proteins in biological fluids,16,17,18,19 we set out to use LiP-MS to screen for novel aging-associated proteoforms and complexes in CSF.
Figure 1. Limited Proteolysis-Mass Spectrometry (LiP-MS) Experimental and Analytical Workflow.
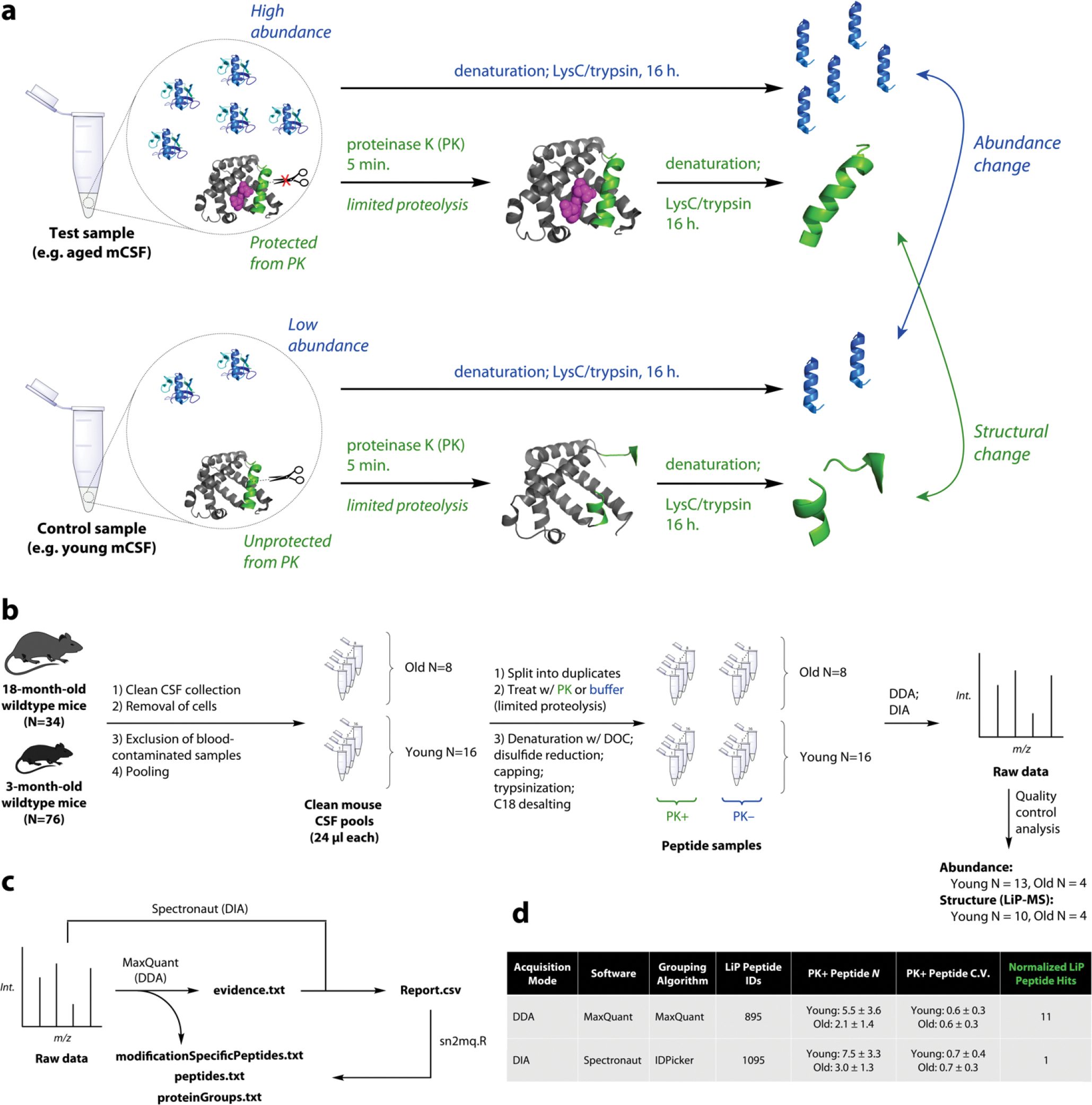
a. Schematic of limited proteolysis-mass spectrometry (LiP-MS). In LiP-MS, proteins in their native state are treated briefly with a nonspecific protease such as proteinase K (PK) which induces non-tryptic cleavages. The resulting peptides’ intensities relative to protein abundances reflect changes in the chemical structures or states of proteins.
b. Clean CSF was collected from 76 young and 34 old B6 wildtype mice. Cells were removed and samples were put through multiple quality control steps to remove contaminated samples. 24-μl pools were generated, each from a distinct combination of individuals, and LiP-MS was performed in both data-dependent acquisition (DDA) and data-independent acquisition (DIA) mode. Quality control analysis of raw and searched data resulted in final sample sizes 13, 10, and 4 for aging analysis.
c. Raw DDA data were searched with MaxQuant in order to generate a quantitative DDA dataset (.txt files) and to generate a DIA library. Raw DIA data were analyzed with this library in Spectronaut and the output (Report.csv) was converted using an R script to match the format of the MaxQuant output.
d. Although DIA performed better by fundamental metrics than DDA, DIA yielded fewer raw hits when the data were analyzed in the traditional manner, leading us to consider whether raw hits were genuine. Note: hardware malfunctions resulted in some unusable data files, resulting in lower N for processed data.
Results
LiP-MS Analysis of Young and Old Mouse CSF Using DIA
We first collected rigorously clean CSF from 76 3-month-old mice and 34 18-month-old mice (Figure 1b). We pooled CSF to generate 16 24-μl samples of young mouse CSF and 8 24-μl samples of old mouse CSF. Each sample was split in half and one duplicate was treated with proteinase K (“PK+”) while the other was treated with water (“PK−“), and then both duplicates were subjected to standard sample preparation including digestion with trypsin and LysC.20 LC-MS/MS was performed on all peptides in both data-dependent acquisition (DDA) and data-independent acquisition (DIA) modes for DIA library construction and DIA analysis respectively.21 Quality control analysis resulted in the inclusion of Nyoung = 13 and Nold = 4 for abundance analysis and Nyoung = 10 and Nold = 4 for LiP-MS analysis (Supplementary Figure 2–3).
Mouse CSF Protein Abundance in Aging
We analyzed the non-PK-treated DIA data for protein abundance changes with aging and found 38 out of 379 protein groups (PGs) significantly changed (Figure 2a, Supplementary Table 1–2). We found a striking increase in immunoglobulins, especially IgM (Ighm). To contextualize these findings, we performed gene ontology (GO) analysis and weighted gene correlation network analysis (WGCNA) on 338 PGs in young adult mouse CSF (Figure 2b–c).22,23 Six modules of correlated PGs emerged, highlighting B- and T-cell-derived factors and multiple families of enzymes. The abundance hits assemble into modules of interrelated proteins according to STRING-DB (Figure 2d). Changes in extracellular matrix (ECM) proteins collagen A1, collagen A2, and Efemp1 may reflect a transformation of the brain ECM with aging.24 Relationships between IgG (Ighg2c), B2m, inflammatory proteases such as lysozyme 2 (Lyz2) and cathepsin D (Ctsd) and protease inhibitor cystatin 3 (Cst3) suggest interrelated changes in the immunological state of the brain with aging.
Figure 2. Mouse Cerebrospinal Fluid (CSF) Protein Abundance in Aging.
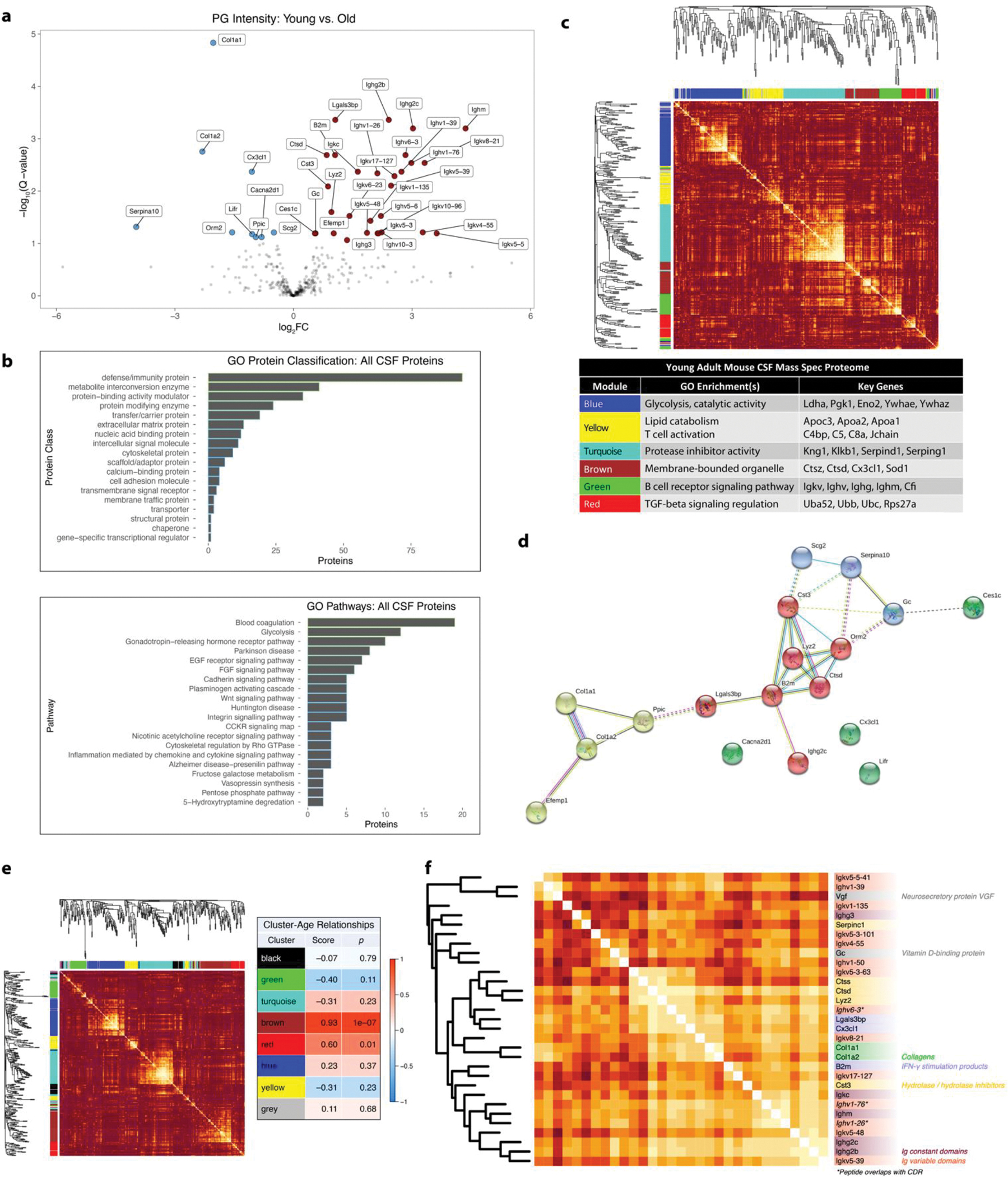
a. Volcano plot of 357 Spectronaut IDPicker PG intensities compared between young vs. old mouse CSF. Cutoff: FDR = 0.1.
b. GO annotation for all mouse CSF proteins sequenced in this study.
c. Weighted gene co-expression network analysis (WGCNA) of young adult mouse CSF (338 IDPicker PGs).
d. STRING-DB network analysis of hits. Cluster coloring by STRING algorithm. Dotted lines represent links between clusters.
e. WGCNA of 342 PG abundances from young and old mouse CSF, with the strength of relationship between each module and mouse age. Relationship score is Pearson coefficient of correlation between module eigengene and mouse age.
f. Annotated heatmap of a sub-module of the brown module in Extended Data Figure 1E. Asterisks denote germline chains with partially or wholly sequenced complementarity-determining regions (CDRs). All heatmap colors range from |Pearson correlation coefficient| = 0 (dark orange) to |Pearson correlation coefficient| = 1 (white).
To include pairwise correlational information in our aging analysis, we then performed WGCNA on our whole PG abundance dataset and calculated Pearson correlation coefficients between cluster eigengenes and age (Figure 2e).23 The “brown” module overrepresented aging-related proteins. Figure 2f shows a highly interrelated sub-module within “brown.” Relationships between cathepsins S (Ctss) and D (Ctsd), lysozyme C2 (Lyz2), cystatin C (Cst3), and Serpin C1 confirmed proteolytic network changes. IgG 3, IgM, IgG 2C, and IgG 2B, all genetically and clonally distinct, changed with aging with varying levels of intercorrelation. Sometimes, levels were highly correlated between a constant and variable chain, suggesting these may have arisen from the same clone and therefore the same antigen. Germline complementary determining regions (CDRs) in Ighv6-3, Ighv1-76, and Ighv1-26 were partially or wholly sequenced, indicating three possible age-associated IgM clones reactive to particular antigens (Figure 2f, Supplementary Figure 4).
Initial Structural Analysis: DDA vs. DIA
When we initially analyzed the DIA LiP-MS data (Spectronaut output) as described elsewhere,20 we found that only 1 normalized LiP peptide significantly changed with aging. Surprisingly, DDA data (MaxQuant output) analyzed in the same manner produced 11 hits, though DIA outperformed DDA in LiP peptide identifications and measurement counts (Figure 1d, Supplementary Figure 2) and critical evaluations showed that the DIA data were of high quality (see Supplementary Figures 5–10 and captions). We hypothesized that the LiP peptide normalization step may have created false positives in the DDA data.
LiP Peptide Normalization by PK− Protein Group Abundance
Peptide-level LiP data must be normalized by dividing PK− PG fold changes (FCs) (Supplementary Figure 11).20 We derived an expression for the normalized LiP peptide fold change, which we call the LiP Ratio Fold Change (FC), in terms of proteoform and peptide concentrations and PK cleavage rate constants (Extended Data Figure 1a). We calculated LiP Ratios across the whole LiP-peptidome and then conducted T tests on log-transformed LiP Ratios to screen for evidence of structural changes.
The grouping algorithms in MaxQuant and Spectronaut allow mismatches between protein mappings of peptides in the PG, so the numerator peptide may not originate from the same protein(s) as the denominator peptides (Extended Data Figure 1a–b).25,26,27 We tested the peptide-normalizing abilities of MaxQuant and IDPicker PGs by performing normalization on PK− peptides, whose changes are assumed to be due to protein abundance (Supplementary Figure 11). In our datasets, division by MaxQuant and IDPicker PG intensities failed to neutralize the effect of protein abundance change in individual peptides, in part because PGs contain large groups of peptides with diverse FCs (Supplementary Figure 12).
For LiP Ratio denominators in this study, we implemented a protein grouping algorithm where all of the peptides’ protein matches are exactly the same within the PG (Extended Data Figure 1b, Supplementary Code). We call this method the “metapeptide method.”27 The result is a larger number of PGs each containing fewer peptides with more similar behavior in aging (Supplementary Figure 13). In our dataset, the metapeptide method resulted in a higher number of significant LiP Ratios compared to IDPicker, perhaps due to improved numerator-denominator matching. Using peptide-level analysis of PK− samples, more hits were identified as potential false positives in MaxQuant PG-normalized data (Supplementary Figure 12) than metapeptide-normalized data (see below).
P-Value Combination for Protein-Level Structural Change
A single structural change can affect multiple LiP sites (Extended Data Figure 1c). If a protein undergoing a change has many significant p-values, this change may not be detected without a significant q-value (Extended Data Figure 1d, Supplementary Figure 14). We employed the Fisher method28 to combine p-values of LiP peptides from the same PG before Benjamini-Hochberg correction, allowing us to test for structural change at the protein level (Extended Data Figure 1e). We validated results further by additional hit filtering, structural mapping, and follow-up experiments where possible as discussed below.
Limited Proteolysis Test-Control Screen (LiP-TeCS) Software
We built a flexible R script that implements all of these concepts for any LiP-MS dataset with test/control and PK−/PK+ groups and processed in MaxQuant or converted from Spectronaut using sn2mq.R (Extended Data Figure 1f, Supplementary Code). The script produces data tables and plots for quality control and biological interpretation. We wrote an open-source Windows desktop app in C# that runs the R script and allows the user to easily save, load, and visualize their computational parameters (Extended Data Figure 1g, Supplementary Code). We call this the Limited Proteolysis Test-Control Screen (LiP-TeCS) platform.
LiP-TeCS Reveals Protein Structures that Change with Aging
Using the LiP-TeCS pipeline on our aging data (Figure 1b–c), we found 16 statistically significant hits at the protein level (Supplementary Table 3–5). We performed two additional steps to refine the hit list: removal of hits whose PK− peptides differed from the PG log2FC by more than 0.5 and removal of hits that had significant PK− peptides with aging after dividing by PG intensity (Figure 3a). We identified 6 high-confidence candidates that appear to undergo structural changes during aging. Each had 1 significant LiP site. Labeled MS2 spectra for these peptides are shown in Supplementary Figure 15.29
Figure 3. Aging-Associated Changes in Structure Revealed by LiP-TeCS.
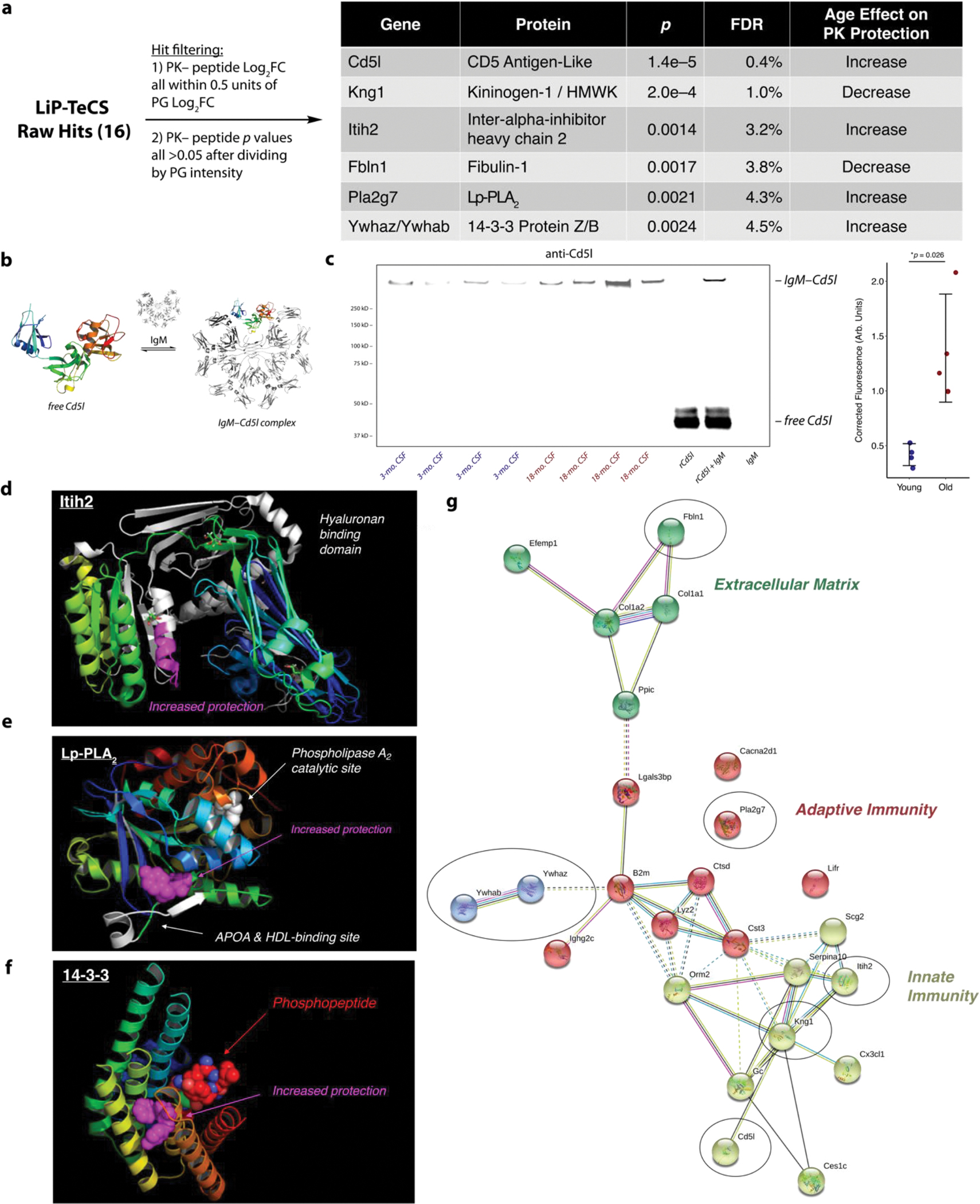
a. High-confidence LiP-TeCS hits.
b. Structures of free Cd5l and covalent Cd5l-IgM, known to coexist in plasma.
c. Nonreducing Western blot (anti-mCd5l, R&D Systems AF2834) of N=4 young vs. N=4 old biologically distinct CSF pools. rCd5l = recombinant mouse Cd5l (R&D Systems 2834-CL). IgM = mouse IgM (ThermoFisher MGM00). Error bars: mean +/− standard deviation. P value: two-sided T test.
d. Significant Itih2 LiP site is located in the hyaluronan binding domain.
e. Significant LiP site in Pla2g7/Lp-PLA2 is located between the known binding site for apolipoprotein A (APOA) / high-density lipoprotein (HDL) and the catalytic site where phosphatidylcholine (PC) is converted to lysophosphatidylcholine (LPC).
f. Significant LiP site on 14-3-3 protein Z/B is in the binding site of phosphoproteins involved in signaling.
g. STRING network with abundance and LiP-TeCS hits. LiP-TeCS hits in black ovals. Cluster coloring by STRING algorithm.
CD5 antigen-like protein (Cd5l) is known to circulate in the bloodstream predominantly as a covalent complex with IgM that can play a harmful role in autoimmunity (Figure 3b).30 The increased protection from PK in old mouse CSF may reflect a predominance of the IgM-Cd5l complexed state of Cd5l in old mouse CSF, which would agree with the strong upregulation of IgM with aging (Figure 2a). Though this complex has been detected several times in mouse and human plasma, it has not yet been shown to exist in CSF. We showed with nonreducing Western blots that the IgM-Cd5l complex exists in CSF and increases in abundance with aging (Figure 3c).
Other candidates have been implicated in AD. Levels of bradykinin, the inflammatory cleaved peptide of Kng1,31 are elevated in CSF of AD patients; decreased protection in old mouse CSF could reflect an increase in plasma kallikrein action, resulting in predominance of the cleaved states of Kng1.6 Hyaluronan, a ligand of inter-alpha-inhibitor heavy chain 2 (Itih2) (Figure 3d), is increased in CSF from patients with AD7 and vascular dementia; increased protection in old mouse CSF could reflect a predominance of the hyaluronan-bound form of Itih2.8 Lysophosphatidylcholine, the product of the lipoprotein-associated phospholipase A2 (Lp-PLA2/Pla2g7) enzyme (Figure 3e), is known to increase in abundance relative to PC with AD in CSF; increased protection in old mouse CSF could reflect a predominance of the LPC-bound state.9 Though not statistically significant, we found a trend toward increased Lp-PLA2 enzymatic activity with aging (Supplementary Figure 16), which could produce higher LPC and give this structural change. Finally, 14-3-3 proteins bind to several phosphoproteins in kinase signaling; increased PK protection with aging could arise from increased phosphoproteins (Figure 3f), which is known to occur in AD.32
Observing that these hits are involved in pathways that were implicated by our protein abundance data, we mined the STRING database for known connections between our protein abundance and our LiP-TeCS hits (Figure 3g). The presence of fibulin-1 (Fbln1) in the ECM corroborates the possible transformation of ECM with aging discussed above. Meanwhile, Cd5l, Kng1, and Itih2 confirm inflammatory protease activity changes.
LiP-MS Hits Correlate With Clinical Features in Humans
To investigate the potential relevance of these hit proteins to human physiology, we first performed a nonreducing Western blot against human CD5L on CSF samples from AD patients and age-matched people with no cognitive impairment (NCI). Strikingly, the CD5L-IgM form of CD5L exists in human CSF and is predominant (Figure 4a). Though not statistically significant, a preliminary analysis suggests a possible increase in CSF CD5L-IgM during human aging (Figure 4a).
Figure 4. Studies on LiP-TeCS Hits in Human CSF.
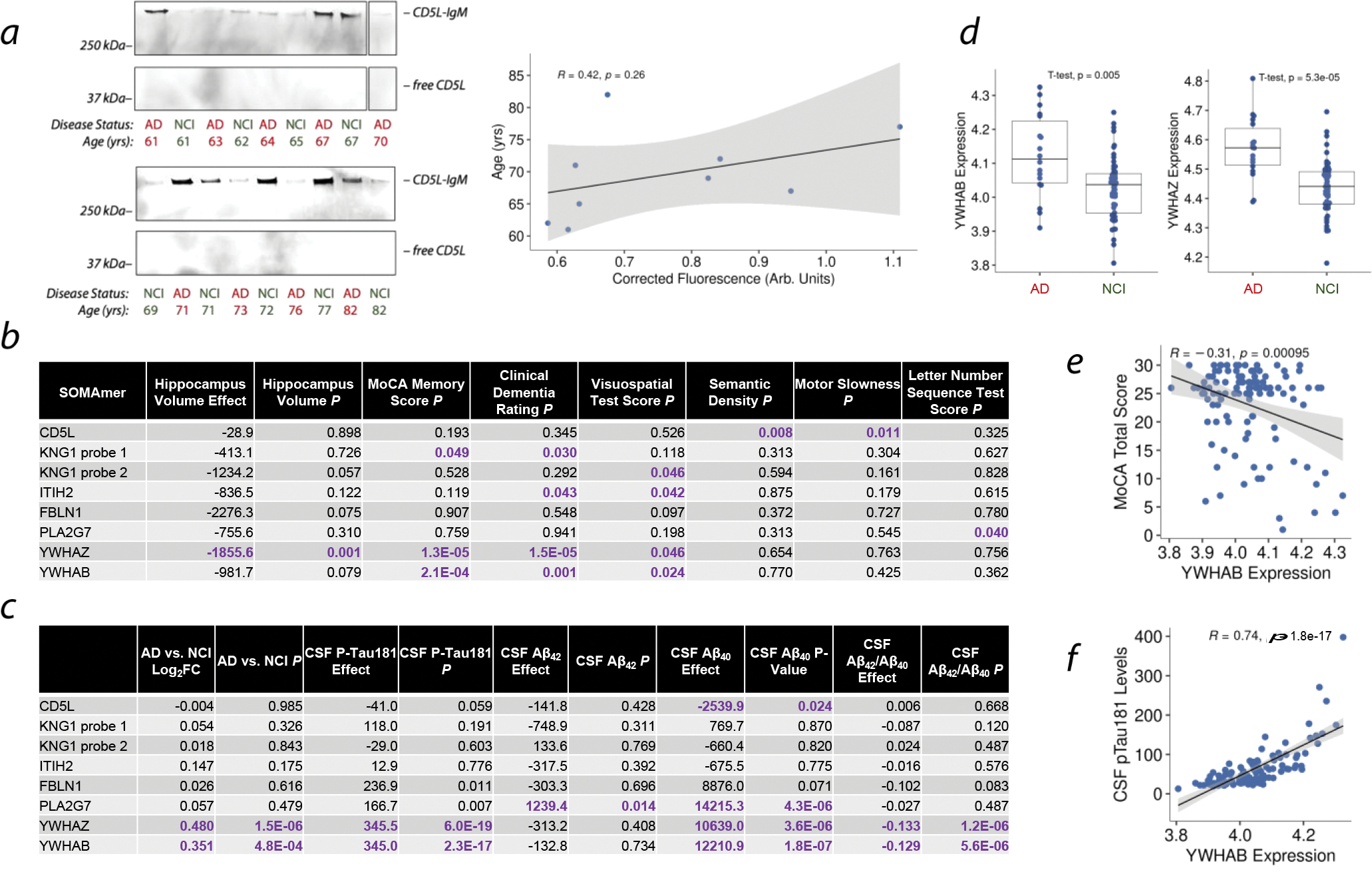
a. Nonreducing Western blot against human CD5L in older adults with no cognitive impairment (NCI) and age-matched patients with AD. Scatter plot shows Western blot intensity versus age in the NCI cohort. Error band: 95% CI for line slope. P value: linear regression.
b. Associations between human CSF protein abundances measured by SOMAmer array and clinical measures of neurological health.
c. Associations between human CSF protein abundances measured by SOMAmer array and disease state or AD biomarker.
d. Comparisons of YWHAZ and YWHAB abundances between AD patients and NCI controls by SOMAmer array.
e. Relationship between Montreal Cognitive Assessment (MoCA) Memory Score (MOCAREGI) and YWHAZ abundance. Error band: 95% CI for line slope. P value: linear regression.
f. Relationship between CSF phospho-tau 181 and YWHAB abundance. Error band: 95% CI for line slope. P value: linear regression.
Next, we analyzed protein abundance data from 114 individuals’ CSF using the SOMAscan assay platform (SomaLogic Inc.) and analyzed the relationships between the LiP-MS hit proteins and various clinical and pathological measurements (Figure 4b–f). All six proteins were found to associate with features relevant to brain aging, neurological function, and/or AD, such as hippocampal volume, memory, clinical dementia, neurological function, and the AD pathological biomarkers Aβ40, Aβ42, and phospho-tau 181.33,34 Interestingly, different probes for Kng1, which recognize different epitopes of Kng1, associated with different features (Figure 4b). Most strikingly, 14-3-3 proteins YWHAZ and YWHAB were strongly associated with all of these types of features and are more abundant in CSF of AD patients compared to NCI controls (Figure 4d–f).
Discussion
This is the first example of a protein abundance screen in mouse CSF with aging, and it is the first example of a LiP-MS screen for structural changes in mammalian samples. We identified 374 MaxQuant PGs by DDA and used the spectra to build a DIA library that was used to identify 379 IDPicker PGs; these numbers compare favorably to precedent in spite of a fastidious collection and quality control procedure that eliminates extraneous proteins from blood and cells.35,36,37
The striking changes in immunoglobulin abundances give rise to questions about the corresponding antigens; the behavior of some germline variable chains, in some cases with fully sequenced CDRs, is further evidence of age-associated increases in particular IgM clones. We have previously found that B cells expressing Igj proliferate dramatically with aging;38 others have found shifts in meningeal B cell identities with aging39 and described aging-associated IgM+ B-cells.40 Since IgM is known to play a role in autoimmunity and multiple sclerosis,41,42 further work may provide neurologically relevant autoantigens for these antibodies.
The metapeptide method we developed here is designed to improve LiP Ratio integrity by matching all protein mappings within each PG.43,44 The additional filtering steps that remove hits based on PK− peptide behavior (Figure 3a) may be excessively conservative because of common, benign outlier peptides;45 methods to identify true positives among such rejected proteins will be the focus of future work.
A single high-abundant protein such as albumin can produce numerous peptides, raising the likelihood of outlier peptides appearing significant by chance. Such a protein will also contribute to a high number of peptides, making Benjamini-Hochberg correction harsher. The Fisher method for p-value combination addresses both of these issues simultaneously (Supplementary Figure 15 & 17). The development of LiP-TeCS enabled the side-by-side analyses necessary to illuminate these problems and test the solutions, and now LiP-TeCS can be used by anyone aiming to use LiP-MS to study proteoforms and complexes on a proteome-wide scale.
The coincidences of proteins in our LiP hit list with previous studies of the CSF in AD reveal at least four potential novel links between mouse aging and human AD. Further analyses of clinical data strengthened this link, revealing 14-3-3 proteins as particularly interesting. We also discovered a new age-associated protein complex: Cd5l-IgM. Although the low amounts of protein present in CSF prevented us from detecting monomeric Cd5l by Western blot, LiP-MS suggests that the Cd5l-IgM complex may increase to a greater degree than monomeric Cd5l. The Cd5l-IgM complex is a specific proteoform of IgM with different biological activities from IgM alone, e.g., enhanced autoantigen presentation in obesity.30,46 This known relevance to autoimmunity begs further questions about the potential roles of autoantigens in brain aging. A Western blot using human samples revealed the existence of human CSF CD5L-IgM and the predominance of this form over monomeric CD5L. Although other biochemical assays lacked the sensitivity to confirm structural changes on other hit proteins in mouse CSF, future work will expand further into human biology, where larger amounts of CSF can be obtained. Together, these results showcase the power of LiP-MS screens to reveal novel proteoforms and complexes that are relevant to biology and disease.
Methods
Animals
Aged C57BL/6JN male wild-type mice (18 months old) were obtained from the National Institute on Aging, while young C57BL/6J males (3 months old) which have maximum genetic similarity to C57BL/6JN mice were purchased from The Jackson Laboratory, and young C57BL/6NCrl males (3 months old) which have maximum environmental similarity to C57BL/6JN mice were purchased from Charles River. Mice were housed under a 12-h light-dark cycle in pathogen-free conditions at 68–73 °F under 40–60% humidity in accordance with the Guide for Care and Use of Laboratory Animals of the National Institutes of Health. Old and young mice were simultaneously housed in an on-site facility for at least 2 weeks prior to surgical operation for environmental acclimation. All animal procedures were approved by the VA Palo Alto Committee on Animal Research and the institutional administrative panel of laboratory animal care at Stanford University.
CSF Collection and Sample Quality Control (for more details see Supporting Information)
Preliminary experiments yielded the conclusion that abundances and structural changes could be detected with N=3 in each group in aging mouse CSF using LiP-MS. Sample sizes were maximized within reasonable operational limits for this study.
Glass capillaries for puncturing the cisterna magna membrane were prepared from single-barrel borosilicate glass tubes without filament, inner diameter 1.30 mm, outer diameter 1.70 mm, length 4.00 in (King Precision Glass). Each tube was loaded onto a P-97 Flaming/Brown Micropipette Puller (Sutter Instrument Co.) and pulled into two tapered capillaries using an in-house optimized method. Each capillary was then gently trimmed with a cotton swab.
All CSF collections were performed in an alternating young-old pattern to ensure identical treatment and to eliminate time-of-day effects in the analysis (which were also addressed by the pooling algorithm, see below). All surgical tools were sterilized prior to the day of procedure, and between surgeries they were all scrubbed with 70% ethanol.
The surgical procedure was performed on each animal as follows. The animal was anesthetized for about 2 minutes in an isoflurane chamber and then loaded onto the mouthpiece of a Small Animal Stereotactic Instrument (David Kopf Instruments). After exposure of muscle tissue, Blunt Retractors (Fine Science Tools) attached to the Stereotactic Instrument with Elastomer (Fine Science Tools) were used to separate the upper layer of muscle. Blunt forceps were then used to detach the lower layer of muscle from the skull, exposing the cisterna magna membrane, using sterile cotton swabs to remove blood as necessary. The cisterna magna membrane was wiped assiduously with alternating wet and dry sterile cotton swabs at least 2x each in a center-out pattern to remove any remaining blood or tissue. Once the membrane was shiny and colorless, a capillary was used to puncture the cisterna magna membrane with the Stereotactic Instrument and was slightly retracted to avoid making contact with the brain while intracranial volume decreased. The capillary was allowed to fill with cerebrospinal fluid for up to 5 minutes. If any color, brain tissue, or turbidity was observed in the sample, it was immediately discarded. The fluid was transferred to an Axygen™ MaxyClear microcentrifuge tube (Fisher) on ice. The fluid was then centrifuged at 420g for 15 minutes at 4 °C. If any pellet was visible by eye, the sample was immediately discarded. The supernatant was decanted and the invisible pellet was resuspended in 6 μl pure water to lyse any cells. The resulting pellet lysate was analyzed on a Nanodrop (Themo) for absorbance at 415 nm to screen for blood contamination.47 Any CSF samples corresponding to pellet lysates absorbing past a stringent absorbance threshold (0.02 absorbance units) were discarded.
In this way, CSF was collected from 76 3-month old male C57BL/6JN mice and 34 3-month old male C57BL/6JN mice. CSF samples were pooled from biologically distinct sets of mice of the same age to generate 24-μl pools.
Limited Proteolysis (more details in Supporting Information)
Biologically distinct pools of 24 μl mouse CSF were made from 3-month-old and 18-month-old wild-type mice (see above) such that CSF from collections occurring at the earliest times of day were combined with samples occurring at the latest times of day. CSF from mice from different sources (Jackson vs. Charles River vs. NIA, see above) were never combined.
The protein content of each sample was measured using a BCA assay kit (ThermoFisher Scientific) with a scaled-down procedure using 2 μl of sample analyzed on a NanoDrop spectrophotometer (ThermoFisher Scientific). The samples were split into duplicates and one replicate in each pair was treated with limited proteolysis (LiP) by proteinase K (PK) for 5 minutes at 1:50 E:S ratio, while the other replicate was treated with water. A standard sample preparation followed, featuring denaturation, reduction, alkylation, digestion with LysC and Trypsin, and final detergent and salt removal.20 Desalted, lyophilized peptides were stored at −78°C until being resuspended in 0.1% formic acid in HPLC-grade water (Fisher) and being stored at 4 °C for no longer than 2 weeks before they were analyzed by LC-MS/MS.
LC-MS/MS
LC-MS/MS analysis of all samples was performed on a Q Exactive HF-X (ThermoFisher Scientific) with an ACQUITY UPLC M-Class System performing chromatographic separation (Waters) using XCalibur (ThermoFisher Scientific) version 4.1.31.9. Solvent A was 0.2% formic acid in HPLC-grade water (Fisher) and Solvent B was 0.2% formic acid in HPLC-grade acetonitrile (Fisher). In all cases, an 80-minute gradient was used: 2–65% Solvent B with a 10-minute 98% Solvent B wash at the end of the method. The flow rate was 300 nl/min on a uPAC 50-cm column (Pharmafluidics).
LC-MS/MS analysis of all samples was performed in DDA mode as follows. Full MS scans were performed at a resolution of 60,000 units, with an automatic gain control (AGC) target of 3e6 and maximum injection time (IT) of 20 ms and scan range 300 to 1650 m/z. Spectrum data type was Centroid. MS2 scan parameters were as follows: Resolution 15,000; AGC target 1e5; maximum IT 54 ms; Loop count 15; TopN 15; Isolation window 1.4 m/z; fixed first mass 100.0 m/z; normalized collision energy (NCE) 28 units. Spectrum data type was Centroid; minimum AGC target 2.90e3; charge exclusion unassigned, 1, 6–8, >8; peptide match preferred; exclude isotopes on; and fragmented m/z values were dynamically excluded from further selection for a period of 45 s.
LC-MS/MS analysis of all samples was performed in DIA mode with the following parameters. The inclusion list included m/z values of 365.5, 394.5, 420, 445, 470, 494, 518, 541.5, 565.5, 591.5, 619, 647.5, 677.5, 710, 746.5, 787.5, 835.5, 895.5, 979, and 1263.5. CS values were 2 for all, and polarity was positive. Full MS scans were performed at a resolution of 60,000 units, with an AGC target of 3e6 and maximum IT of 20 ms and scan range 300 to 1650 m/z. The DIA parameters were as follows: default charge state 2; resolution 30,000; AGC target 1e6; loop count 1; MSX count 1; MSX isochronous ITs were enabled; Isolation window corresponded to the inclusion list; fixed first mass 100.0 m/z; NCE 28 units.
Raw Data Processing
The following numbers of runs were successful in each experiment, resulting in the corresponding N in downstream analysis: in DDA, all 8 samples in the old PK− and old PK+ group were successful, and all 16 samples in the young PK− and old PK+ group were successful. Due to chromatographic issues, in DIA, 13 young PK− samples were successfully run; 11 young PK+ samples; 5 old PK− samples; and 5 old PK+ samples. For LiP analysis, pairwise comparisons were used to identify LiP peptides, so samples without a corresponding PK−/PK+ counterpart were excluded. For side-by-side comparisons of DDA vs. DIA, a DDA run was only analyzed if it had a corresponding successful DIA run, to match statistical power.
Raw DDA data were processed using the MaxQuant software48 version 1.6.3.4 using the following parameters: Oxidation (M), Acetyl (Protein N-term), and Deamidation (N) were included as variable modifications; Carbamidomethyl (C) was the only fixed modification; maximum number of modifications per peptide 5; Orbitrap main search peptide tolerance 4.5 ppm; individual peptide mass tolerance enabled; isotope match tolerance 2 ppm; centroid match tolerance 8 ppm; centroid half width 35 ppm; maximum charge 7; intensity determination by total sum; semispecific trypsin/P digestion; label-free quantification disabled; Uniprot’s mouse and mouse additional reference proteomes used as Fasta files (UP000000589_10090, accessed July 22, 2018); include contaminants enabled; minimum peptide length 7; maximum peptide mass 4600 Da; peptides for quantification unique + razor; FTMS MS/MS match tolerance 20 ppm, de novo tolerance 10 ppm, deisotoping tolerance 7 ppm, top peaks per Da interval 12, top x mass window 100 Da; decoy mode revert; PSM FDR, protein FDR and site decoy fraction all set to 0.01; minimum peptides 1; minimum razor + unique peptides 1; minimum unique peptides 0.
The MaxQuant output was imported into Spectronaut (Biognosys) version 13.15.200430.43655 and used to prepare a DIA library using the following parameters: all search tolerances were dynamic; all MS1/MS2 correction factors were 1; MaxQuant FDR threshold was 0.01; protein inference was performed with semi-specific trypsin/P digestion; toggling N-terminal M was enabled; spectral library filters were set to default, i.e.: ions with fewer than 3 amino acids were filtered out; m/z maximum was 1800 and minimum was 300; relative intensity minimum was 5; the maximum best fragments per peptide was 6 and the minimum was 3; all other filters were disabled; iRT reference strategy was empirical iRT database; deep learning assisted iRT regression was enabled as a backup; minimum Rsquare was set to 0.8; use RT as iRT was disabled; use stripped sequence to identify reference peptides was disabled; fragment ion selection strategy was intensity based; in-silico generation of missing channels was disabled.
Raw DIA data were analyzed in Spectronaut using the resulting DIA library with the following parameters: MS1/MS2 intensity extraction was by maximum intensity; MS1/MS2 mass tolerance strategy was dynamic with a correction factor of 1; XIC RT extraction window was dynamic with a correction factor of 1; source specific iRT calibration was enabled; calibration mode was automatic; MZ extraction strategy was by maximum intensity; precision iRT was enabled; deamidated peptides were excluded; RT regression type was local (non-linear); the “used Biognosys’ iRT kit” value was set to false; calibration carry-over was disabled; decoy method was inverse; single hit definition was by peptide precursor ID; single hit proteins were not excluded; PTMs were localized with a probability cutoff of 0.75; the p-value estimator was a kernel density estimator; no proteotypicity filter was used; major grouping was by protein group ID; minor grouping was by modified sequence; major group quantity was the sum of peptide quantities; major group top N was disabled; minor group quantity was the sum of precursor quantities; minor group top N was disabled; quantity MS-level was MS2; quantity type was area; data filtering was Q-value; cross-run normalization was enabled except to analyze non-normalized data for quality control; normalization strategy was global normalization on medians; row selection was automatic; in silico library optimization was disabled; multi-channel workflow definition was from library annotation; fallback option was labeled; profiling strategy was none; unify peptide peaks strategy was none; protein inference workflow was automatic.
In Spectronaut, the output was formatted as a run pivot report with the following columns: PG protein accessions, PG genes, PG protein descriptions, PG UniProt IDs, PG FASTA header, PG cellular component, PG biological process, PG molecular function, peptide stripped sequence, peptide “is proteotypic” value, peptide position, precursor ID, modified sequence, PG quantity, PG MS1 quantity, PG MS2 quantity, peptide quantity, peptide MS1 quantity, peptide MS2 quantity, target reference ratio, target quantity, reference quantity, and total quantity; with the “no decoy” filter enabled and “quantification data filtering” enabled. This output was exported in TSV format. The exported data were reformatted using an R script (mq2sn.R, Supplementary Code) to match the format of MaxQuant outputs. The reformatted data were analyzed using the LiP-TeCS platform in R and/or the LiP-TeCS desktop application.
Data Analysis
The LiP-TeCS analytical workflow was written in R and the desktop application was written in C# using Visual Studio. The LiP-TeCS platform was utilized to analyze the DDA and DIA datasets as described above and as implemented in the Supplementary Code. Normality of log-transformed protein and peptide intensities was tested qualitatively using Q-Q plots. For LiP peptide identification, two-sided T tests were performed and p-values were adjusted using the Benjamini-Hochberg method, retaining hits with a Q-value cutoff of 0.05. For LiP Ratio changes, two-sided T tests were performed, the p-values were combined by PG using the Fisher method, p-values were adjusted using the Benjamini-Hochberg method, and hits with a Q-value below 0.1 were retained. Structures were manually mapped onto structures in the RCSB PDB using Microsoft Excel, Uniprot (Uniprot.org), and MacPyMOL. Figures were made in RStudio, Adobe Illustrator, MacPyMOL, Chimera (UCSF), ChemDraw (PerkinElmer), Microsoft Powerpoint, and STRING (version 11.0) (string-db.org).
Nonreducing Western Blot (for more details see Supporting Information)
Western blots were performed as described in detail in the Supplementary Information. Pools of CSF were prepared from separate groups of C57BL/6 mice. Blotting was performed at 4 °C for 1.5 hours on a nitrocellulose membrane before blocking with Intercept Blocking Buffer in TBS (LiCor) and then staining with goat anti-mCd5l or anti-hCD5L (R&D Systems) at 0.1 μg/ml (dilution of 2,000x from 200 μg/ml reconstituted antibody) overnight at 4 °C. Blots were washed with tris-buffered saline (TBS) with 0.1% Tween-20 (TBST) five times before staining with a fluorescent donkey anti-goat-IgG (LiCor IRDye® 680RD) at 20,000x dilution (from 1.0 mg/ml reconstituted antibody) for 1 hour at room temperature. Blots were then washed three times with TBST and twice with TBS before imaging on an Odyssey blot scanner (LiCor). Results were processed and quantified in ImageJ. Background fluorescence on the blot at a consistent height was used as a control for fluorescence correction.
Enzyme Activity Assay
Lp-PLA2 activity was assayed using a PAF Acetylhydrolase Assay Kit (Cayman Chemical, Item No. 760901) as described in the user manual (©10/13/2017).
Human Study Participants
Samples were acquired through the NIA-funded Stanford Alzheimer’s Disease Research Center (ADRC). Collection of CSF was approved by the Institutional Review Board, and written consent was obtained from all participants. CSF was collected from a total of 114 participants in this study and data from 110 participants were used as described below. Diagnostic group was determined during a clinical consensus meeting after each participant completed a formal neurological and motor examination, comprehensive neuropsychological battery, and clinical history by a licensed neurologist. All participants were adjudicated as Alzhheimer’s disease (AD), mild cognitive impairment (MCI), Parkinson’s disease (PD), Lewy body dementia (LBD), or no cognitive impairment (NCI) based on published criteria. All participants were free from acute infectious diseases and in good physical condition. Group characteristics, including demographic, cognitive testing data, and biomarker data, are presented in Supplementary Table 6.
Human Tissue Collection
CSF was collected by lumbar puncture, then centrifuged at 300G to pellet immune cells. CSF samples were checked for blood contamination by resuspending the pelleted cells in 100 μl of CSF and mixing 10 μl (10%) CSF with 10 μl trypan blue to assess red blood cell content and viability. Cells were visualized on a TC20 automated cell counter (BioRad) and cell viability and presence/absence of red blood cells was recorded. CSF samples contaminated with blood were not used in the study. The resuspended cells were then mixed with 900 μl Recovery Cell Culture Freezing Medium (ThermoFisher Scientific). All samples were frozen overnight at −80°C in a Mr. Frosty freezing container (ThermoFisher Scientific) and transferred the following day to liquid nitrogen for storage.
SOMAScan Assay of Human CSF
We used the SOMAscan assay platform (SomaLogic Inc.) to measure the relative levels of 4985 human proteins in CSF. The SOMAScan platform technology and its performance characteristics have been previously described.49,50 The assay uses slow off-rate modified DNA aptamers (SOMAmers) capable of binding to specific protein targets with high sensitivity and specificity.
We collected 114 CSF samples from a multi-ethnic cohort of older American adults (age range 36–87, median 70) between 2015 and 2020. Samples were stored at −80 °C, and 150 μl aliquots of CSF were sent on dry ice to SomaLogic. CSF samples were analyzed via SOMAScan assay in five batches. To account for variation within and across batches, control, calibrator and buffer samples are added in each 96 well plate. Data normalization was conducted by the manufacturer following three stages, first Hybridization Control Normalization, hybridization control probes are used to remove individual sample variance. Second, Intraplate Median Signal Normalization, median normalization removed inter-sample differences within the plate. Last, Plate Scaling and Calibration, this final step removed variance across assay runs. Samples with normalizing factors outside of the typical assay range were removed before analysis. Additional quality control was performed to remove samples with a connectivity score greater than 3 standard deviations from the mean. Finally, to ensure that we analyzed only robustly detected proteins in the CSF, we computed the buffer-blank distribution of fluorescence values for each probe, and removed probes where greater than 50% of the sample fluorescence values fell within the buffer-blank range at a false-discovery rate of 5%. After filtering, 4178 proteins were robustly detected and analyzed in 110 samples.
Analysis of Core CSF AD Biomarkers
Core CSF AD biomarkers total tau, phospho-tau 181, Aβ40 and Aβ42 were measured using Lumipulse G assays on the fully automated Lumipulse G1200 Instrument (Fujirebio Diagnostics Inc, Malvern, PA). Briefly, CSF samples were thawed on wet ice and analyzed as previously described51 by investigators blinded to the diagnostic information of the samples.
Statistical Methods
All statistical analysis of human subject clinical features and SomaLogic proteomics was performed in R. Proteomics data was log transformed before analysis. Associations were drawn via linear modeling after accounting for the biological and technical covariates age, sex, CSF storage time, and the first 5 principal components calculated after regressing out all known sources of biological and technical variation, as previously published.52
Data Availability
Mass spectrometry data have been accepted by the ProteomeXchange Consortium via the PRIDE53 partner repository with the dataset identifier PXD031174. All other data are available from the corresponding author upon request.
Code Availability
All code relevant to this study are included as supplementary files. In addition, the code can be accessed on Zenodo at https://doi.org/10.5281/zenodo.5884992.54
Extended Data
Extended Data Figure 1. Theoretical and Computational Development of the LiP-MS Test-Control Screen (LiP-TeCS) Platform.
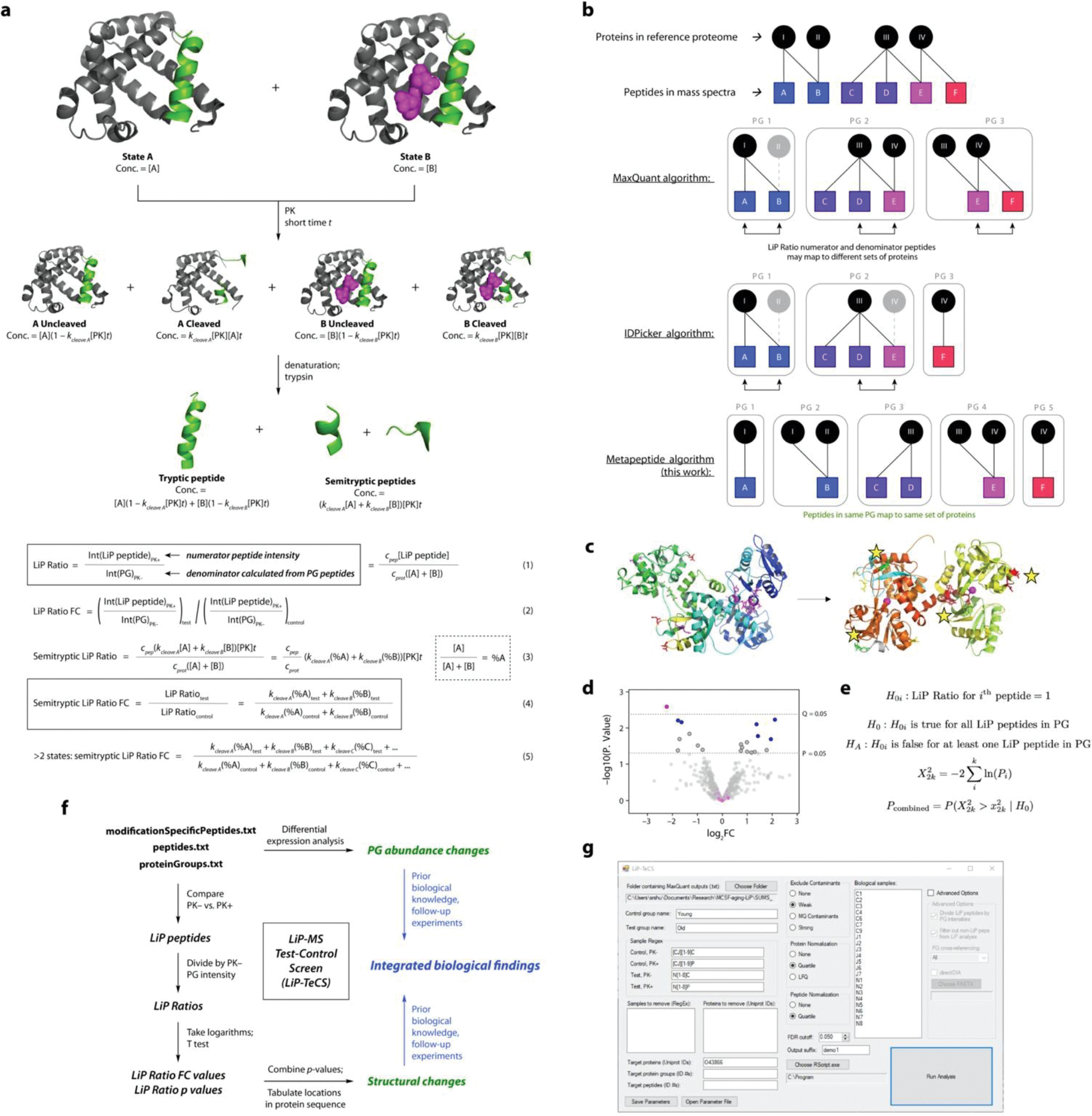
a. Derivation of an expression for the LiP Ratio fold change (FC) in terms of concentrations and rate constants. Assumptions and details of the model are discussed in the Supplementary Information. For tryptic peptides, coefficients kcleave X are replaced by coefficients f(kcleave X) = (1 − kcleave X[PK]t) in Equations 4 and 5.
b. Illustrations of protein grouping algorithms. In the indicated softwares, gray objects are omitted from the outputs to reduce redundancy and protein ID overestimation. The metapeptide method ensures that the LiP Ratio numerator peptide and all peptides in the denominator PG map to the same set of proteins and that all information is (matches are) reported.
c. In transferrin (Tf), a single structural change (Fe binding) results in significant LiP Ratio FCs at multiple sites on the same protein, shown here with yellow stars. Structural change occurs at the protein level.
d. Example peptide-level LiP-MS analysis. p-value from two-sided T test, q-value from Benjamini-Hochberg correction. A PG containing multiple significant peptides by p value but not q value (blue) would not be considered in the same dataset as a PG with one significant q value and numerous non-significant peptides (pink).
e. Formulation of the Fisher method in this experiment. Assumptions are discussed in the Supplementary Information.
f. Flow chart showing the implementation of these concepts, as well as the analysis described in Ref. 20, in a single workflow called LiP Test-Control Screen (LiP-TeCS) (black and green objects). Blue objects represent downstream interpretation.
g. Screenshot of the LiP-TeCS desktop app.
Supplementary Material
Acknowledgements
The authors thank Dr. Niclas Olsson, Dr. Joshua Elias, Dr. Dan Itzhak, and Sophia Shi for help with LCMS setup and maintenance; Dr. Paola Picotti and Dr. Marie Therese-Mackmull for training in LiP-MS methodology, advice on data analysis, and input on the manuscript; Dr. Ludovic Gillet and Dr. Paul Boersema for help with DIA instrument methods; Ummara Khan for literature searches; Dr. Lisa Eshun-Wilson for help with protein modeling; Dr. Art Owen, Dan Kluger, and Dr. Andreas Beyer for advice and feedback on statistics and meta-analysis; Dr. Edward Wilson and Dr. Katrin Andreasson for CSF biomarker data; Dr. Victor Henderson and the Stanford Alzheimer’s Disease Research Center team for clinical data; and members of the Wyss-Coray Lab, Elias Lab, and Picotti Lab for feedback and support. All raw data for mass spectrometry experiments discussed in the main text were acquired at Stanford University Mass Spectrometry by R.D.L., Kratika Singhal, Fang Liu, and Rowan Matney. Other data necessary for the completion of the project and presented in the Supplementary Information were acquired by S.R.S. in the Picotti Lab or Elias Lab on instruments owned by the Picotti Lab or Wyss-Coray Lab. This work was funded by the NOMIS Foundation (T.W.-C.), the Paul F. Glenn Center for the Biology of Aging (T.W.-C.), the NIA-funded Stanford Alzheimer’s Disease Research Center (P50 AG047366), the BioX Stanford Interdisciplinary Graduate Fellowship (S.R.S.), the Stanford Center for Molecular Analysis and Design Graduate Fellowship (S.R.S.), and the Stanford Graduate Fellowship (SGF) (J.R.).
Footnotes
Competing Interests
The authors declare no competing interests.
References
- 1.Reitz C; Brayne C; Mayeux R “Epidemiology of Alzheimer disease.” Nature Rev. Neurol. 2011, 7, 137–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith LM; Kelleher NL “Proteoform: a single term describing protein complexity.” Nature Methods 2013, 10, 186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kelleher NL; et al. “How many human proteoforms are there?” Nature Chem. Biol. 2018, 14, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selkoe DJ; Hardy J “The amyloid hypothesis of Alzheimer’s disease at 25 years.” EMBO Mol. Med. 2016, 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sebastián-Serrano Á; de Diego-García L; Díaz-Hernández M “The Neurotoxic Role of Extracellular Tau Protein.” Int. J. Mol. Sci. 2018, 19, 998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergamaschini L; Parnetti L; Pareyson D; Canziani S; Cugno M; Agostoni A “Activation of the Contact System in Cerebrospinal Fluid of Patients with Alzheimer Disease.” Alzheimer Disease and Associated Disorders 1998, 12, 102–108. [DOI] [PubMed] [Google Scholar]
- 7.Nielsen H; Palmqvist S; Minthon L; Londos E; Wennström M “Gender-dependent levels of hyaluronic acid in cerebrospinal fluid of patients with neurodegenerative dementia.” Curr. Alzheimer Res. 2012, 9, 257–266. [DOI] [PubMed] [Google Scholar]
- 8.Nägga K; Hansson O; van Westen D; Minthon L; Wennström M “Increased Levels of Hyaluronic Acid in Cerebrospinal Fluid in Patients with Vascular Dementia.” J. Alzheimer’s Disease 2014, 42, 1435–1441. [DOI] [PubMed] [Google Scholar]
- 9.Fonteh AN; Chiang J; Cipolla M; Hale J; Diallo F; Chirino A; Arakaki X; Harrington MG “Alterations in cerebrospinal fluid glycerophospholipids and phospholipase A2 activity in Alzheimer’s disease.” J. Lipid Res. 2013, 54, 2884–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.López-Otín C; Blasco MA; Partridge L; Serrano M; Kroemer G “The Hallmarks of Aging.” Cell 2013, 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng Y; De Franceschi G; Kahraman A; Soste M; Melnik A; Boersema PJ; de Laureto PP; Nikolaev Y; Oliveira AP; Picotti P “Global analysis of protein structural changes in complex proteomes.” Nature Biotech. 2014, 10, 1036–1044. [DOI] [PubMed] [Google Scholar]
- 12.Piazza I; Picotti P; et al. “A Map of Protein-Metabolite Interactions Reveals Principles of Chemical Communication.” Cell 2018, 172, 358–372. [DOI] [PubMed] [Google Scholar]
- 13.Cappelletti V; et al. “Dynamic 3D proteomes reveal protein functional alterations at high resolution in situ.” Cell 2021, 184, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geiger R; Picotti P; Mann M; Lanzavecchia A; et al. “L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity.” Cell 2016, 167, 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zampieri M; Szappanos B; Buchieri MV; Picotti P; Sauer U; et al. “High-throughput metabolomic analysis predicts mode of action of uncharacterized antimicrobial compounds.” Science Transl. Med. 2018, 10, eaal3973, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wyss-Coray T “Ageing, neurodegeneration and brain rejuvenation.” Nature 2016, 539, 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castellano JM; et al. “Human umbilical cord plasma proteins revitalize hippocampal function in aged mice.” Nature 2017, 544, 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yousef H; et al. “Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1.” Nature Med. 2019, 25, 988–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pluvinage JV; et al. “CD22 blockade restores homeostatic microglial phagocytosis in ageing brains.” Nature 2019, 568, 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schopper S; Kahraman A; Leuenberger P; Picotti P; et al. “Measuring protein structural changes on a proteome-wide scale using limited proteolysis-coupled mass spectrometry.” Nature Protocols 2017, 12, 2391–2410. [DOI] [PubMed] [Google Scholar]
- 21.Ludwig C; Gillet L; Rosenberger G; Amon S; Collins BC; Aebersold R “Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial.” Mol. Systems Biol. 2018, 14, e8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith JS; Smith RD; et al. “Characterization of individual mouse cerebrospinal fluid proteomes.” Proteomics 2014, 14, 1102–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang B; Horvath S “A General Framework for Weighted Gene Co-Expression Network Analysis.” Statistical Applications in Genetics and Mol. Biol. 2005, 4, 17. [DOI] [PubMed] [Google Scholar]
- 24.Morawski M; Vargova L; et al. “ECM in brain aging and dementia.” Prog. Brain Res. 2014, 214, 207–227. [DOI] [PubMed] [Google Scholar]
- 25.Cox J; Mann M “MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification.” Nature Biotech. 2008, 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- 26.Tyanova S; Temu T; Cox J “The MaxQuant computational platform for mass spectrometry–based shotgun proteomics.” Nature Protocols 2016, 11, 2301–2319. [DOI] [PubMed] [Google Scholar]
- 27.Zhang B; Chambers MC; Tabb DL “Proteomic Parsimony through Bipartite Graph Analysis Improves Accuracy and Transparency.” J. Proteome Res. 2007, 6, 3549–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borenstein M; Hedges LV; Higgins JPT; Rothstein HR Introduction to Meta-Analysis. John Wiley & Sons, Ltd: 2009. ISBN: 978–0-470–05724-7. [Google Scholar]
- 29.Brademan DR; Riley NM; Kwiecien NW; Coon JJ “Interactive Peptide Spectral Annotator: A Versatile Web-based Tool for Proteomic Applications.” Mol. Cell. Proteomics 2019, 18, S193–S201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arai S; Miyazaki T; et al. “Obesity-Associated Autoantibody Production Requires AIM to Retain the Immunoglobulin M Immune Complex on Follicular Dendritic Cells.” Cell Reports 2013, 3, 1187–1198. [DOI] [PubMed] [Google Scholar]
- 31.Schmaier AH “The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities.” J. Thrombosis and Haemostasis 2016, 14, 28–39. [DOI] [PubMed] [Google Scholar]
- 32.Sathe G; Pandey A; et al. “Multiplexed Phosphoproteomic Study of Brain in Patients with Alzheimer’s Disease and Age-Matched Cognitively Healthy Controls.” OMICS 2020, 24, 216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thijssen EH; Boxer AL; et al. “Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration.” Nature Medicine 2020, 26, 387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Janelidze S; Hansson O; et al. “Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia.” Nature Medicine 2020, 26, 379–386. [DOI] [PubMed] [Google Scholar]
- 35.Cunningham R; Jany P; Messing A; Li L “Protein Changes in Immunodepleted Cerebrospinal Fluid from a Transgenic Mouse Model of Alexander Disease Detected Using Mass Spectrometry.” J. Proteome Res. 2013, 12, 719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith JS; et al. “Characterization of individual mouse cerebrospinal fluid proteomes.” Proteomics 2014, 14, 1102–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dislich B; et al. “Label-free Quantitative Proteomics of Mouse Cerebrospinal Fluid Detects β-Site APP Cleaving Enzyme (BACE1) Protease Substrates In Vivo.” Mol. Cell. Proteomics 2015, 14, 2550–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schaum N; Lehallier B; Hahn O; Wyss-Coray T; et al. “Ageing hallmarks exhibit organ-specific temporal signatures.” Nature 2020, 583, 596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mrdjen D; Becher B; et al. “High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease.” Immunity 2018, 48, 380–395. [DOI] [PubMed] [Google Scholar]
- 40.Ratliff M; Riley RL “In senescence, age-associated B cells secrete TNFa and inhibit survival of B-cell precursors.” Aging Cell 2013, 12, 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pfuhl C; Ruprecht K; Oechtering J; et al. “Intrathecal IgM production is a strong risk factor for early conversion to multiple sclerosis.” Neurology 2019, 93, e1440–e1451. [DOI] [PubMed] [Google Scholar]
- 42.Negi N; Das BK “Decoding intrathecal immunoglobulins and B cells in the CNS: their synthesis, function, and regulation.” Intl. Rev. Immunol. 2020, 39, 67–79. [DOI] [PubMed] [Google Scholar]
- 43.Nesvizhskii AI; Aebersold R “Interpretation of Shotgun Proteomic Data: The Protein Inference Problem.” Mol. Cell. Proteomics 2005, 4, 1419–1440. [DOI] [PubMed] [Google Scholar]
- 44.Jin S; Daly DS; Springer DL; Miller JH “The Effects of Shared Peptides on Protein Quantitation in Label-Free Proteomics by LC/MS/MS.” J. Proteome Res. 2008, 7, 164–169. [DOI] [PubMed] [Google Scholar]
- 45.Forshed J; et al. “Enhanced Information Output From Shotgun Proteomics Data by Protein Quantification and Peptide Quality Control (PQPQ).” Mol. Cell. Proteomics 2011, 10, M111.010264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miyazaki T; Yamazaki T; Sugisawa R; Gershwin ME; Arai S “AIM associated with the IgM pentamer: attackers on stand-by at aircraft carrier.” Cell. Mol. Immunol. 2018, 15, 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aasebø E; Berven FS; et al. “Effects of Blood Contamination and the Rostro-Caudal Gradient on the Human Cerebrospinal Fluid Proteome.” PLoS ONE 2014, 9, e90429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cox J; Mann M “MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification.” Nature Biotechnology 2008, 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- 49.Gold L; et al. “Aptamer-based multiplexed proteomic technology for biomarker discovery.” PLoS ONE 2010, 5, e15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim CH; et al. “Stability and reproducibility of proteomic profiles measured with an aptamer-based platform.” Sci. Rep. 2018, 8, 8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilson EN; Swarovski MS; Linortner P; Shahid M; Zuckerman AJ; Wang Q; et al. “Soluble TREM2 is elevated in Parkinson’s disease subgroups with elevated CSF tau.” Brain 2020,143, 932–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee S; Sun W; Wright FA; Zou F “An improved and explicit surrogate variable analysis procedure by coefficient adjustment.” Biometrika 2017, 104, 303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perez-Riverol Y; et al. “The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences.” Nucleic Acids Res. 2022, 50, D543–D552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shuken Steven R.; Wyss-Coray T “Aging-Associated Changes in CSF Protein Abundances and Structures Revealed by a Modified LiP-MS Screen.” Zenodo. DOI: 10.5281/zenodo.5884992. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass spectrometry data have been accepted by the ProteomeXchange Consortium via the PRIDE53 partner repository with the dataset identifier PXD031174. All other data are available from the corresponding author upon request.