

Abstract
The vast majority (≥90%) of literature reports agree on the regiochemical outcomes of Pd-catalyzed cross-coupling reactions for most classes of dihalogenated N-heteroarenes. Despite a well-established mechanistic rationale for typical selectivity, several examples reveal that changes to the catalyst can switch site selectivity, leading to the unconventional product. In this Perspective, we survey these unusual cases in which divergent selectivity is controlled by ligands or catalyst speciation. In some cases, the mechanistic origin of inverted selectivity has been established, but in others the mechanism remains unknown. This Perspective concludes with a discussion of remaining challenges and opportunities for the field of site-selective cross-coupling. These include developing a better understanding of oxidative addition mechanisms, understanding the role of catalyst speciation on selectivity, establishing an explanation for the influence of ring substituents on regiochemical outcome, inverting selectivity for some “stubborn” classes of substrates, and minimizing unwanted over-reaction of di- and polyhalogenated substrates.
Keywords: cross-coupling, site selectivity, oxidative addition, heterocycles, mechanism
Graphical Abstract
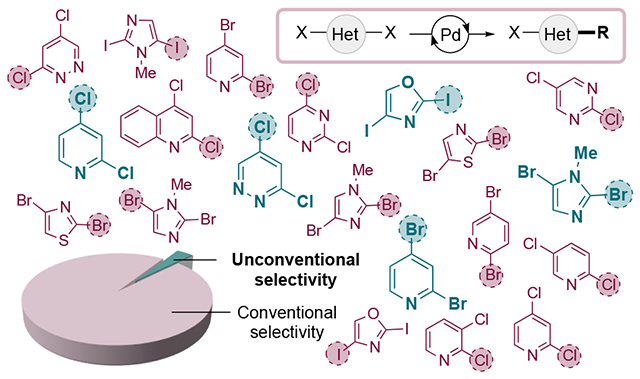
INTRODUCTION
Overview.
The challenge of regiocontrol is central to functionalizing heteroarenes, particularly in the context of ring substitution reactions including C—H functionalization, electrophilic aromatic substitution, nucleophilic aromatic substitution (SNAr), and—relevant to this Perspective—cross-coupling. For heteroaromatic substrates bearing two or more halides, Pd-catalyzed cross-coupling reactions tend to proceed selectively, with a bias for reaction at sites adjacent to a heteroatom (Table 1).1 This reactivity bias often enables straightforward synthesis of a single regioisomer. However, accessing target molecules with substitution patterns that are disfavored by cross-coupling is more challenging. A substrate-controlled approach to achieving unusual substitution patterns involves the use of substrates bearing mixed (nonidentical) halides. In this scenario, cross-coupling often takes place at the heavier halide even when there is a lighter halide at a privileged position α to a heteroatom. However, greater synthetic effort is required to prepare heteroarenes with mixed dihalides compared to identical dihalides, which translates into increased cost. Alternatively, unconventional site selectivity can be achieved through the use of catalyst-controlled approaches. Although the topic of catalyst-controlled site selectivity2 is at the forefront of other research areas, such as C—H functionalization,3 relatively little is known about the relationship between catalyst structure and cross-coupling site selectivity.4 This Perspective surveys the handful of known examples in which unconventional or divergent site selectivity is achieved by changing the catalyst structure (e.g., through changing the ligand). In some cases, the mechanistic basis for the switch in selectivity is known, although others remain unexplained. Developing a better understanding of these reactions could lead to improved versatility of cross-coupling chemistry.
Table 1.
Conventional Site Selectivity for Substrates Discussed in this Perspective.
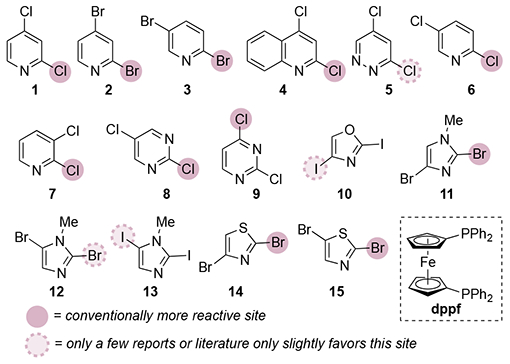
| |||
|---|---|---|---|
| Substrate | Total Examplesa | Percentage of Examples with Conventional Selectivity (%)b | Percentage of Examples with PPh3 or dppf (%)c |
| 1 | 186 | 81 | 43 |
| 2 | 143 | 92 | 81 |
| 3 | 822 | 97 | 77 |
| 4 | 77 | 94 | 52 |
| 5 | 68 | (56) | 50 |
| 6 | 164 | 93 | 49 |
| 7 | 223 | 99 | 52 |
| 8 | 162 | 88 | 36 |
| 9 | 1284 | 99 | 92 |
| 10 | 23 | (57) | 22 |
| 11 | 15 | 100 | 40 |
| 12 | 11 | (55) | 36 |
| 13 | 1 | (100) | 0 |
| 14 | 59 | 100 | 78 |
| 15 | 222 | 100 | 66 |
Total number of examples of Pd-catalyzed cross-coupling to form a C—C or C—N bond, according to a SciFinder search.5
Percentage of examples for which the major product is indicated to result from coupling at the highlighted position. Values in parentheses indicate that the literature is near-evenly divided in describing selectivity or the number of examples is too small to evaluate.
Percentage of examples for which PPh3 or dppf were included in the reaction conditions.
For the purposes of this Perspective, we define ‘conventional’ (or ‘typical’) selectivity as the regiochemical outcome that is reported in the large majority of literature examples for a given substrate class, to date. However, it has recently become clear that ligand choice, among other factors, can significantly affect selectivity (vide infra). As such, our perception of what is ‘typical’ is heavily influenced by a historical bias toward the use of certain ligands, especially PPh3 and the related bisphosphine dppf (Table 1). As methods for achieving divergent selectivity become better established, the line between conventional and unconventional regioselectivity may eventually blur.
Multiple excellent reviews already provide a comprehensive picture of conventional selectivity.1 The focus of this Perspective is the minority of cases in which the regiochemical outcome deviates from the norm, some of which are more recent than existing reviews. These deviations are rare. A SciFinder search suggests that, for many substrate classes, only 1-8% of cross-coupling reactions provide atypical selectivity (Table 1).5 Furthermore, in many of these reports, the putative exceptional selectivity is not discussed nor is it supported by structural characterization. In other cases, the exceptions catalogued by SciFinder may reflect minor rather than major products. As such, the number of actual exceptions to conventional selectivity is likely lower than shown in Table 1. In this Perspective, we largely limit our discussion to unsubstituted dihaloheteroarenes. Substituents can lead to directing effects (see Outlook section), 6,7,8 but here we focus on cases in which unconventional site selectivity is achieved through changes to the catalyst rather than changes to the substrate.
Origin of Conventional Site-Selectivity.
In the vast majority of reports, a C—X bond α to a heteroatom, if present, is favored to undergo cross-coupling. Handy correlated conventional selectivity with positional electrophilicity as inferred from the 1H NMR spectrum of the parent heteroarene.9 The most deshielded proton of the parent heteroarene (α to one or more heteroatoms) usually corresponds to the most reactive site of the analogous halogenated substrate. The correlation is not perfect, however. For example, the most reactive site of 2,4-dichloropyrimidine (9) is C4, but the most deshielded proton of pyrimidine is attached to C2.10 Through a series of computational studies, Houk and Merlic later showed that selectivity is often inversely correlated with C—X bond dissociation energy (BDE); that is, the most reactive C—X bond is usually the weakest one (distortion-controlled selectivity).11 For example, the C4—X bond of 9 is weaker than C2—X (Figure 1). However, exceptions to the bond strength trend were noted when (1) the weaker C—X bond is on a carbocycle (16); (2) the two C—X bonds have identical or very similar BDEs (17); and (3) on a substituted ring in which the two halides are α to two different heteroatoms (18). In these cases, selectivity was better explained by a stronger attractive interaction between Pd and the more reactive site of the substrate (interaction-controlled selectivity). This attractive interaction is influenced by frontier molecular orbital distribution. For example, Houk’s DFT calculations indicate a stronger interaction between Pd and C2 of 16, which can be explained by the much larger LUMO coefficient at C2 compared to C5.
Figure 1.
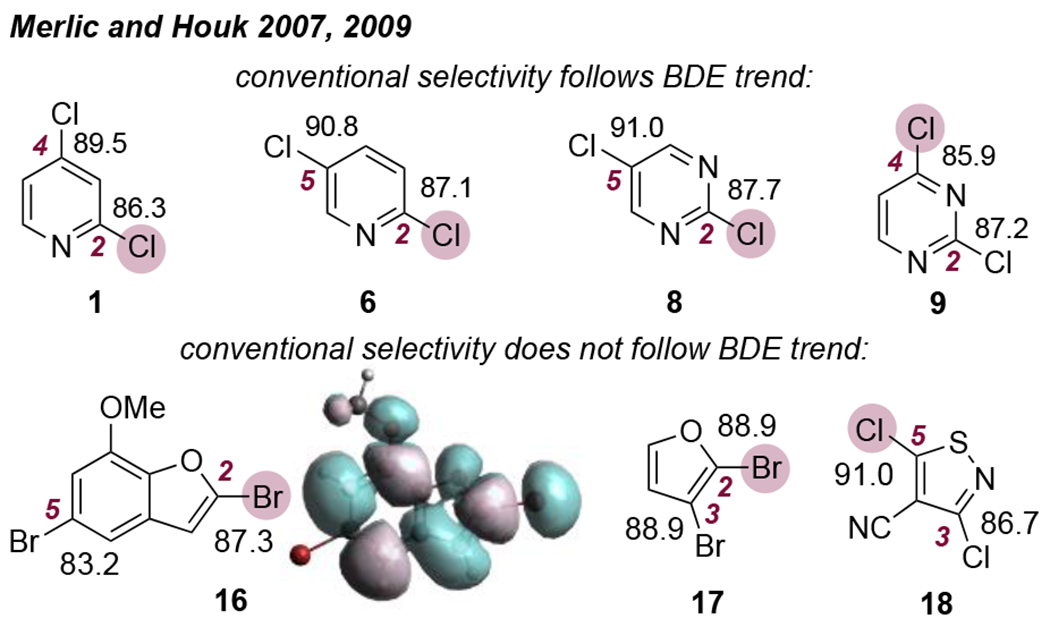
Bond dissociation energies (BDEs) for selected dihalogenated substrates. BDEs calculated with B3LYP/6-31G(d). The highlighted halide corresponds to the conventional site of reactivity.11
Recent work by Leitch et al. contributes to a more complete picture of conventional selectivity.12 Based on experimental results using Pd(PCy3)2, Leitch developed a model to predict substrate reactivity toward oxidative addition based on several molecular descriptors. This model revealed that C—X bond strength, on its own, is a poor predictor of the rate of oxidative addition. Instead, the electrostatic potential (ESP) at the ipso carbon was determined to be the most important descriptor in this model, followed by the ESP at the more negative ortho atom. The model was demonstrated to accurately predict conventional selectivity for a wide range of dihalogenated heterocycles.
Despite the importance of Houk’s and Leitch’s work, these models do not directly suggest an explanation for the reported exceptions to conventional selectivity, wherein changes to reaction conditions lead to the atypical product. However, for Houk’s distortion/interaction model, transition structures were optimized in the gas phase with a simple Pd-bisphosphine model Pd(PH3)2.11a For Leitch’s molecular descriptor model, parameterization was based on results using Pd(PCy3)2 in a single solvent (THF).12 It is known that the calculated mechanism for oxidative addition can change with palladium’s coordination number, with ligand identity, and with the use of solvation models.13,14,15 Thus it is reasonable to conclude that, in the exceptional situations where unconventional selectivity is observed, the transition states for C—X cleavage look significantly different from those that were the basis for Houk’s and Leitch’s models.
LIGAND-CONTROLLED SELECTIVITY
6-Membered Heterocycles.
Six-membered nitrogen heterocycles (azines) are ubiquitous in metabolites,16 therapeutics,17 agrochemicals,18 and functional materials.19 From metalation and SNAr to catalytic C—H functionalization by transition metals, regioselective elaborations of 6-membered nitrogen-containing heteroarenes have been the subject of extensive investigation. In discovery chemistry settings, dihalogenated azines are among the mainstay building blocks for the construction of di- or polysubstituted nitrogen heterocycles via iterative cross-coupling or other substitution methods.1
In the context of cross-coupling, the regioselectivity outcomes with dihalogenated 6-membered N-heteroarenes—especially dihalopyridine derivatives—are now the most studied and best understood. As discussed above, for dihalogenated pyridines, pyridazines, and quinolines, halides α to nitrogen are conventionally more reactive than distal halides. Because of the polarity of the C—N bond of pyridine, C2 has more positive charge than the more distal carbons, making it more reactive toward Pd(0). Furthermore, α C—X bonds are weaker than other C—X bonds due to nitrogen’s lone pair, which is in the same plane as C—X.
However, a few recent reports describe deviations from the conventional reactivity patterns when using sterically hindered ligands. In 2013, Dai, Chen, and coworkers demonstrated the first clear example of ligand-controlled inversion of site-selectivity.20 Although dppf, a popular bidentate phosphine ligand, exclusively promotes coupling of 3,5-dichloropyridazine at the expected C3-site, the bulky monophosphine QPhos promotes reaction at the unconventional C5-site (C5:C3 = 20:1, Scheme 1, top). Under the optimized conditions, a broad scope of aryl, alkenyl, and heteroaryl boron nucleophiles were selectively coupled at C5 of 3,5-dichloropyridazines. Additionally, QPhos was shown to mediate unconventional C4-arylation of 2,4-dichloropyridine, albeit with modest selectivity and yield (Scheme 1, bottom). Although a rationale for the unusual behavior of QPhos was not discussed at the time, the structural differences between QPhos and dppf provide a hint that palladium’s ligation state could be a deciding factor in the regiochemical outcome. Hartwig had previously established that, in the presence of QPhos, Pd is monoligated during oxidative addition of chloro- and bromoarenes,21,22 whereas Pd would almost certainly be bisligated when supported by the diphosphine dppf. Interestingly, RuPhos (2-dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl) was noted to promote diarylation of 3,5-dichloropyridazine despite a 1:1 ratio of the coupling partners.
Scheme 1.
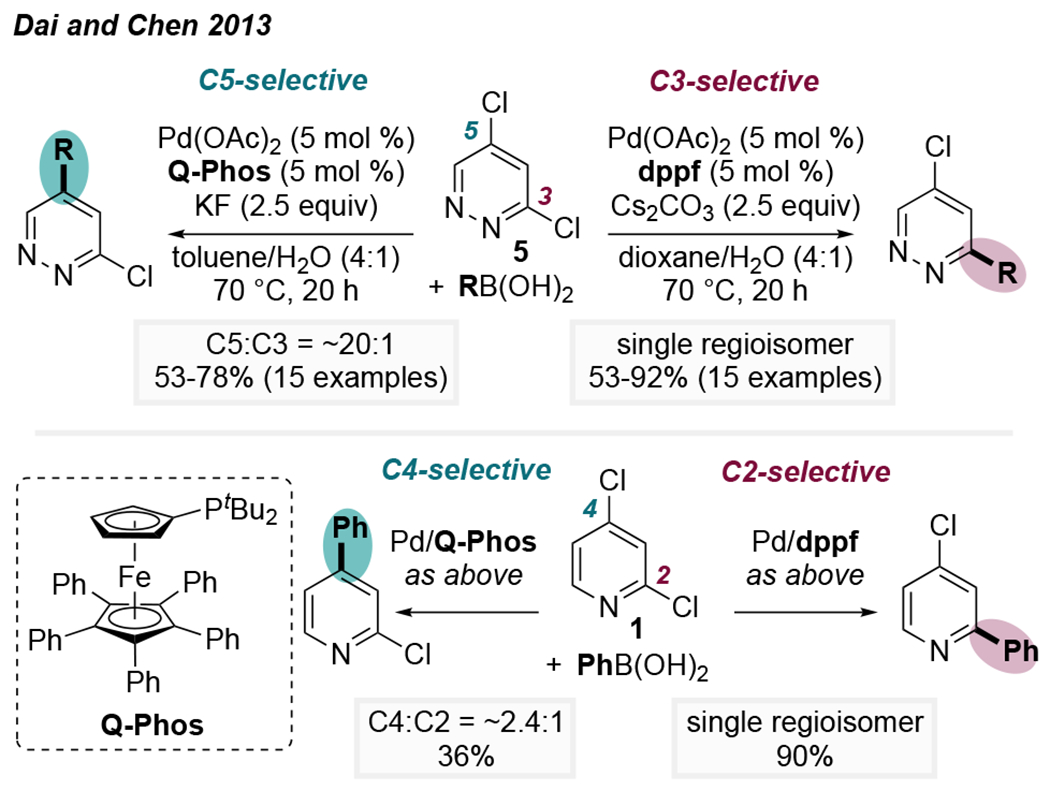
First Examples of Ligand-Controlled Inversion of Conventional Selectivity for 3,5-Dichloropyridazine and 2,4-Dichloropyridine20
Recently, hindered N-heterocyclic carbene (NHC) ligands have emerged as ligands that promote Pd-catalyzed cross-coupling at a distal site of dihaloazines. In 2019, Willans, Hardie, et al. reported that cyclotriveratrylene-tethered trinuclear Pd(II)-NHC complexes mediate Suzuki coupling at the 4-position of 2,4-dibromopyridine (2, C4:C2 = 6.5-10.7 : 1, Scheme 2), albeit with significant overarylation (2c).23 Additionally, these ligands promote a greater proportion of C5-arylation at 2,5-dibromopyridine (3, C5:C2 = 1 : 1.3-1.6) compared to PPh3 (C5:C2 = 1 : 14). The origin of selectivity was not determined in this report, although the authors suggested that it is unlikely to result from cooperative multinuclear catalysis.
Scheme 2.
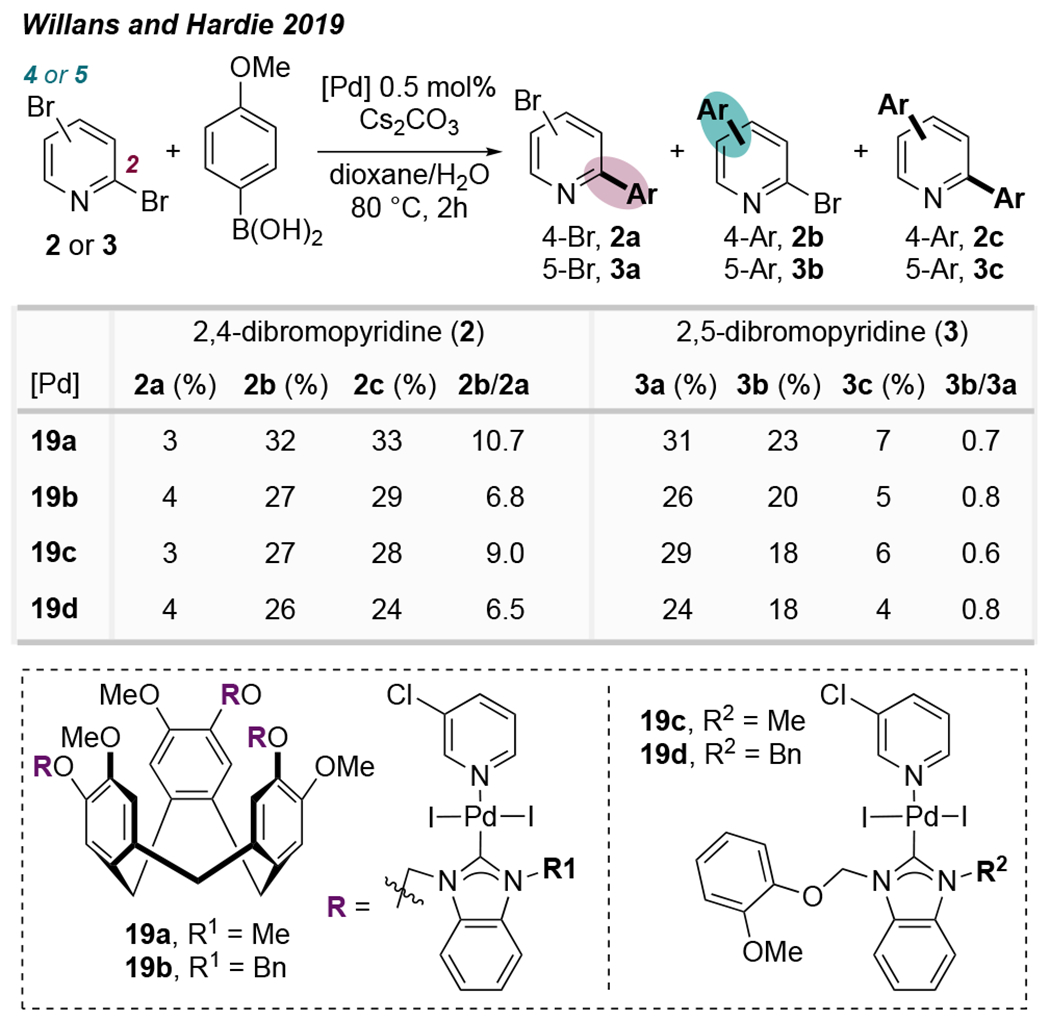
Ligand-Controlled Inversion of Conventional Selectivity for 2,4-Dibromopyridine and Erosion of Conventional Selectivity for 2,5-Dibromopyridine23a
In 2020, Yang et al. reported a C4-selective Suzuki-Miyaura coupling of 2,4-dichloropyridine (1) under Pd-catalyzed conditions in the presence of the hindered NHC ligand IPr.24 Throughout most of the authors’ optimization efforts (Scheme 3), Pd-PEPPSI-IPr was found to afford the C4-arylated product 1b with site-selectivity ranging from 2.5:1 to 10.4:1, depending on reaction solvent and base. Notably, the hindered ligands PtBu3 and PAd2(n-Bu) effected modest selectivity for the C4-site in a dioxane/H2O mixture. In this solvent system, RuPhos and IPr gave the largest proportion of unwanted diarylated product, although switching to PEG400 decreased diarylation with IPr. Remarkably, a drastic improvement in C4-selectivity (to ~99:1) was observed upon changing the reaction conditions to include KI and NaOAc (not shown in Scheme 3). However, it was recently found through ligand-free control reactions that selectivity under these latter conditions does not require IPr; as such, these results are discussed in the Speciation-Controlled Selectivity section below.
Scheme 3.
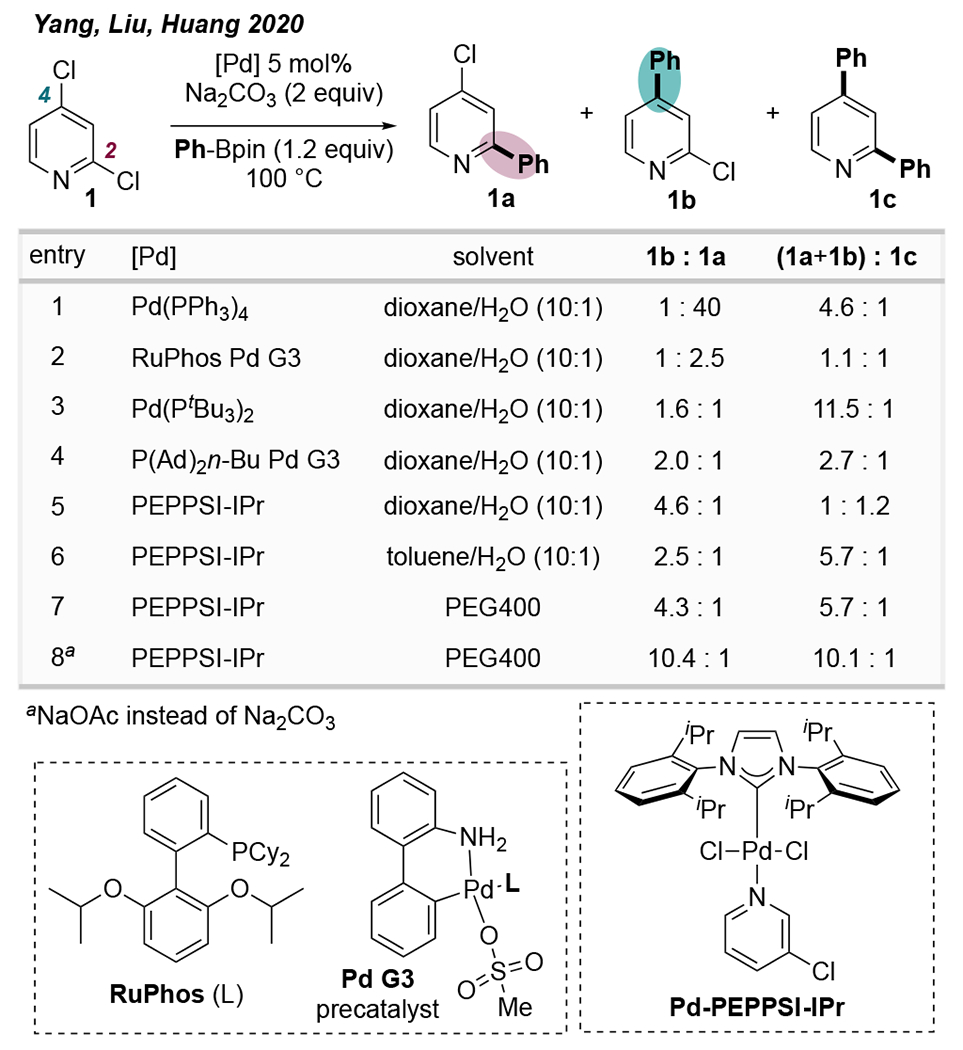
Salient Optimization Data from Yang et al. Showing that IPr Promotes C4-Selective Cross-Coupling of 2,4-Dichloropyridine24
Recently, we further explored the role of ligand sterics on the site-selectivity of Suzuki couplings of 2,4-dichloropyridine derivatives.15,25 In agreement with earlier reports, a systematic analysis of ligand trends revealed a clear correlation between increased steric congestion around the metal center and increased selectivity for the distal C—Cl site (Scheme 4A). The highest selectivity for reaction at the distal position was achieved with bulky NHC ligands. However, there is a trade-off that accompanies NHC ligand size: the proportion of diarylated product increases concurrently with the C4:C2 ratio of monoarylated products. This unwanted reactivity is especially notable with the ligand IPent. As such, IPr offers the best compromise between high C4:C2 selectivity and low diarylation:monoarylation, thus maximizing the yield of the C4-monoarylated product. Under our optimized room-temperature conditions, no reaction is observed in the absence of IPr. A detailed scope investigation showed that Pd catalysis with IPr is general to Suzuki, Kumada, and Negishi cross-coupling at the distal site of diverse 2,4-dichloropyridines and -quinolines as well as 3,5-dichloropyridazines (Scheme 4B). This methodology allows installation of diverse sp2 and sp3-carbon substituents at a distal site while retaining a chloride α to nitrogen.
Scheme 4.
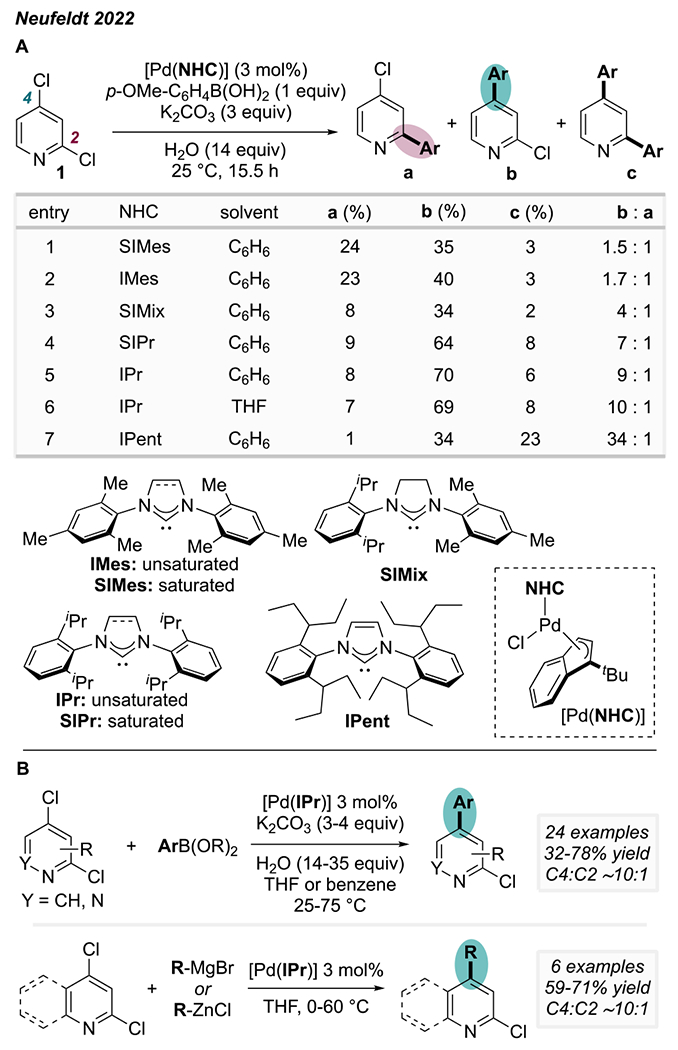
(A) C4-Selectivity with 1 Correlates with Ligand Sterics; (B) IPr Enables C4-Selective Suzuki, Kumada, and Negishi Coupling of Dichloroazines.25
Taken together, the reports described so far in this section demonstrate an unmistakable relationship between ligand sterics and unconventional site-selectivity for 2,4-dihalopyridines and pyridazines. We recently proposed that this correlation can be explained by the tendency of larger ligands to promote low-coordinate (i.e., 12 e−) PdL during the selectivity-determining oxidative addition step.15 Less hindered ligands, especially those that are better π-acceptors, favor 14 e− PdL2 during the oxidative addition step. DFT calculations with a model system comprising PhCl and Pd(PMe3)n (n = 1 or 2) suggest that 12 e− and 14 e− palladium may be biased toward different mechanisms for oxidative addition (Figure 2, top). Monoligated Pd(PMe3) prefers to react through a classic, 3-centered concerted mechanism in which Pd interacts with both chloride and the ipso carbon of the substrate. In contrast, bisligated Pd(PMe3)2 prefers a displacement-type mechanism in which Pd interacts with both the ipso and the ortho carbons, but does not interact directly with chloride. A displacement-type mechanism for oxidative addition has been proposed in several previous instances, and is also referred to as an SNAr-like mechanism14,26 or a dissociative process.13 Our analysis of frontier molecular orbitals suggests that the distinct mechanisms arise from differences in orbital symmetries when comparing the HOMOs of mono- vs. bis-ligated Pd. PdL has a σ-type HOMO, whereas the HOMO of PdL2 has π-type symmetry once it is bent into the geometry required for forming the pre-oxidative addition π complex with the substrate (Figure 2, bottom). The HOMO σ-symmetry of PdL enables palladium to donate into a single ring atom of PhCl (a single lobe of the substrate’s LUMO), consistent with the 3-centered concerted mechanism. On the other hand, the HOMO π-symmetry of PdL2 enables palladium to donate into two ring atoms, consistent with the displacement mechanism.
Figure 2.
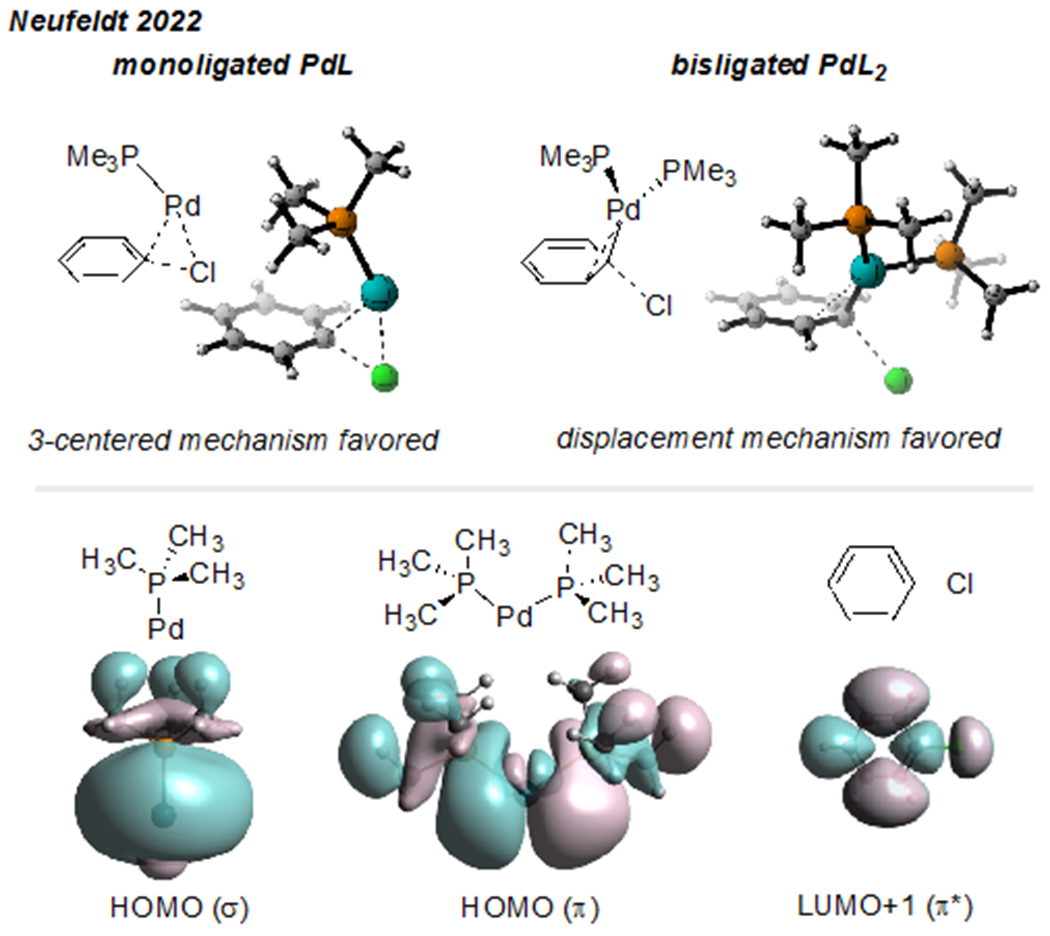
PdL and PdL2 can favor different mechanisms for oxidative addition due to differences in orbital symmetry.15
For the substrate 2,4-dichloropyridine (1), the percent contribution to the LUMO is substantially greater at the C4 site (26% vs 8%, Figure 3A), which predicts more favorable orbital overlap between Pd and substrate at this site if Pd can only interact with a single ring atom, as is the case for monoligated PdL (e.g., L = IPr). Indeed, Pd/IPr-catalyzed cross-couplings of other dihaloheteroarenes also reveal a correlation between selectivity for a distal site and a relatively larger LUMO coefficient at that site (Figure 3B). In contrast to PdL, because PdL2 can interact with two ring atoms, the LUMO coefficient at individual carbon atoms is less important. Instead, conventional selectivity for oxidative addition of the C2—Cl bond at PdL2 is determined by other factors such as C—Cl bond strength and the polarity of the C2—N bond.
Figure 3.
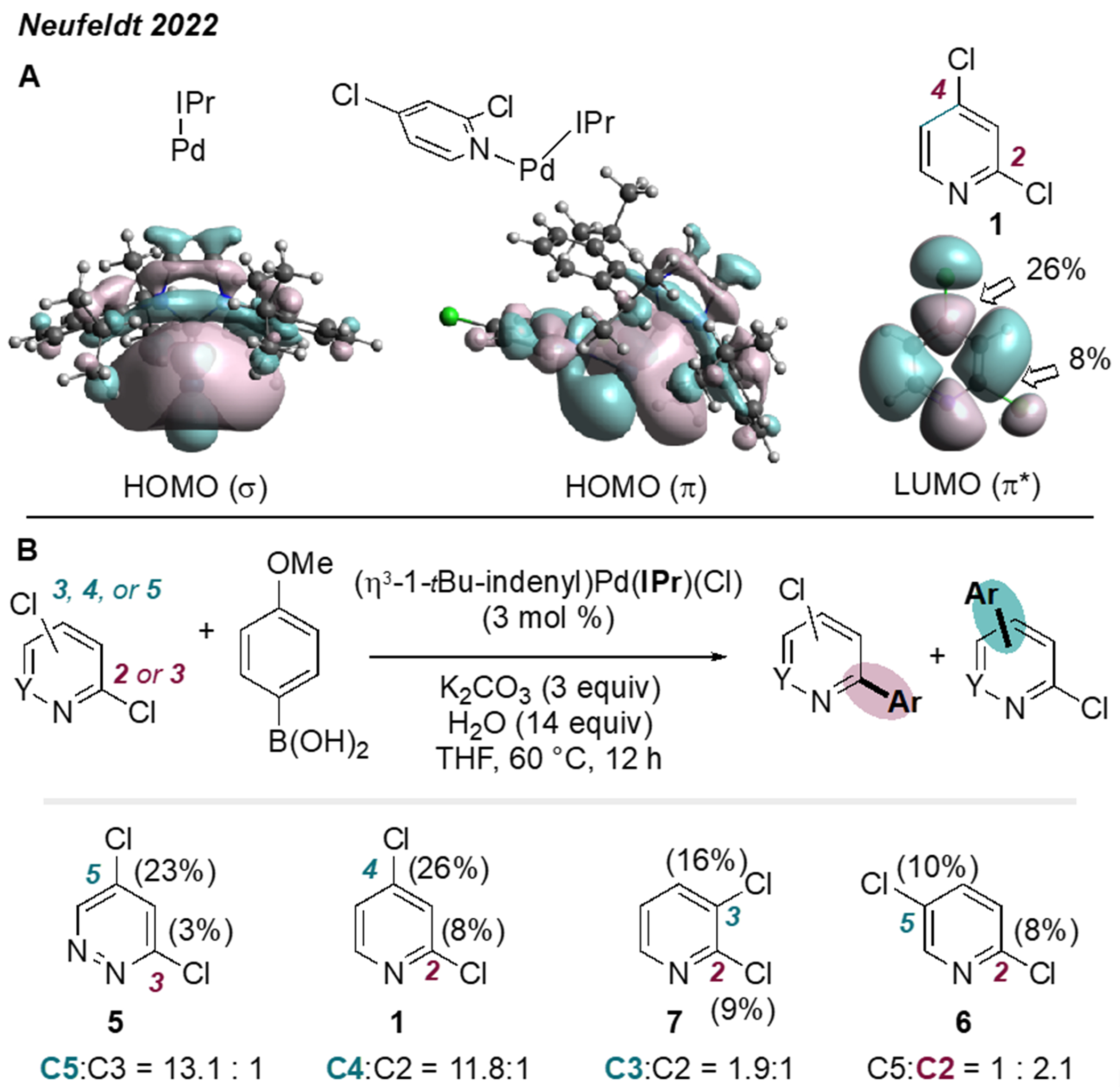
(A) Frontier molecular orbitals of 12 e− Pd(IPr), bent 14 e− Pd(IPr)(1), and 2,4-dichloropyridine (1). (B) The distal-selectivity of Pd/IPr-catalyzed Suzuki-coupling correlates with the difference in LUMO coefficients between the two sites (% contribution to LUMO is indicated in parentheses).15
Consistent with this reasoning, calculated transition structures for the reaction of 12 e− Pd(IPr) with 2,4-dichloropyridine suggest that the lowest energy pathway for reaction at either C2 or C4 involves a 3-centered concerted transition state (Figure 4, top). Reaction at C4 is predicted to be favored by 1.7 kcal/mol, which shows good agreement with experiment. In contrast, oxidative addition involving bisligated Pd(IPr)(1) is predicted to proceed through a displacement-type mechanism at either site, with reaction at C2 favored by this mechanism. However, the structures involving Pd(IPr)(1) are markedly higher in energy than those involving Pd(IPr), and thus unlikely to be experimentally relevant. Similar observations are made with Pd(PtBu3) and Pd(PtBu3)(1), although Pd(PtBu3) is less C4-selective than Pd(IPr), likely due to the weaker σ-donicity of phosphines compared to NHC ligands (Figure 4, middle). On the other hand, parallel calculations using the less hindered ligand IMes predict that a displacement mechanism for reaction at C2 via Pd(IMes)(1) is energetically competitive with the concerted mechanism for reaction at C4 via monoligated Pd(IMes) (Figure 4, bottom). This prediction is consistent with the poor selectivity observed experimentally with IMes, and with the observation that selectivity skews toward the C2-coupled product when the Pd/IMes-catalyzed reactions employ higher concentrations of 1.15
Figure 4.
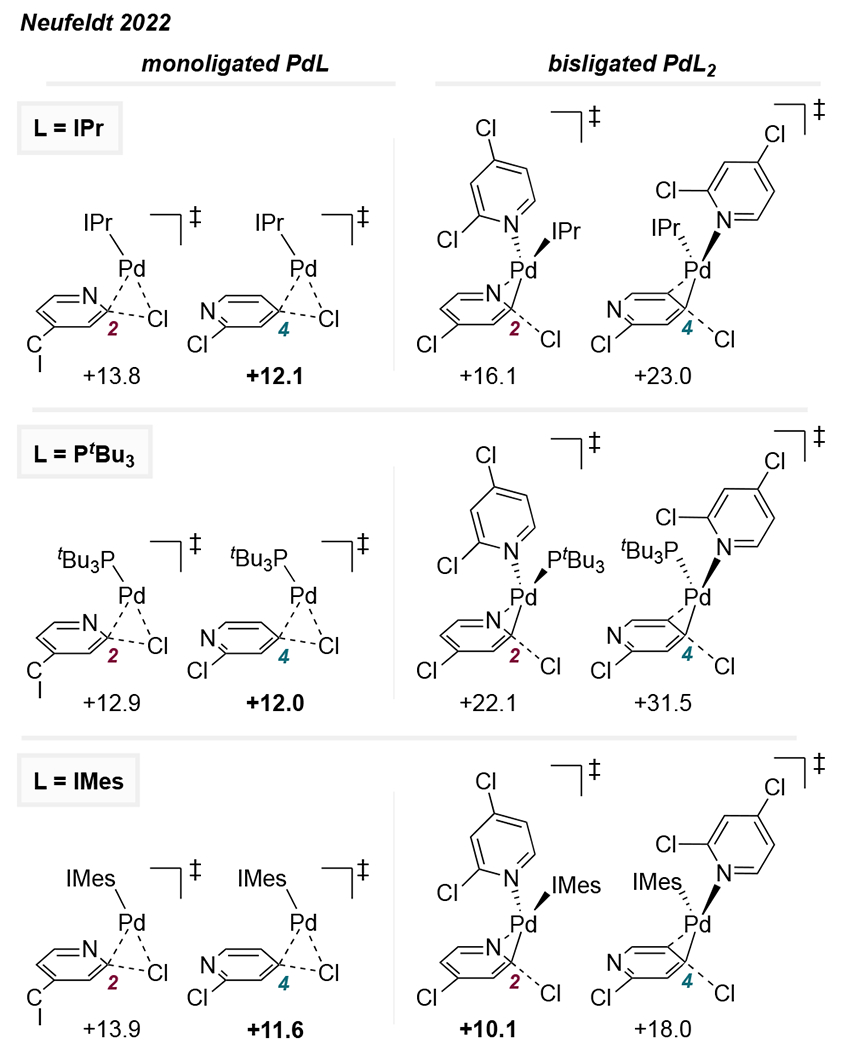
Mono- and bisligated transition structures for oxidative addition of 2,4-dichloropyridine at Pd(IPr), Pd(PtBu3), and Pd(IMes). Free energies in kcal/mol relative to the lowest-energy preceding π-complex.15
In summary, it appears that the ligand-controlled examples of unconventional site-selectivity in cross-couplings of dihalogenated azines can largely be explained by the ability of bulky ligands to promote 12 e− Pd. Although more traditional 14 e− Pd prefers to react α to nitrogen via a displacement mechanism involving a Pd•••N interaction, 12 e− Pd cannot benefit from this interaction because its HOMO symmetry precludes such a mechanism. Instead, 12 e− Pd reacts through a concerted mechanism at the site with a significantly larger LUMO coefficient, which can be a site distal to nitrogen. A similar mechanistic phenomenon may be at play in related systems wherein ligands control the chemoselectivity of cross-coupling of bromochloroazines. Sigman and Tan reported that several bulky ligands favor amination at the C5—Br of 5-bromo-2-chloropyridine (Scheme 5A).27 However, most bidentate phosphines, in particular 20, promote reaction at C2—Cl. In the Suzuki coupling of 21, Ashcroft and Fussell found that several bulky monophosphines including QPhos favor reaction at Cl, while 22 (as well as PPh3 and dppf) gave more reaction at Br (Scheme 5B).28 These observations are consistent with the hypothesis that bisligated Pd reacts α to nitrogen via a displacement mechanism, whereas monoligated Pd—which doesn’t benefit from a Pd•••N interaction during a 3-centered transition state—instead reacts with the weaker C—Br bond.
Scheme 5.
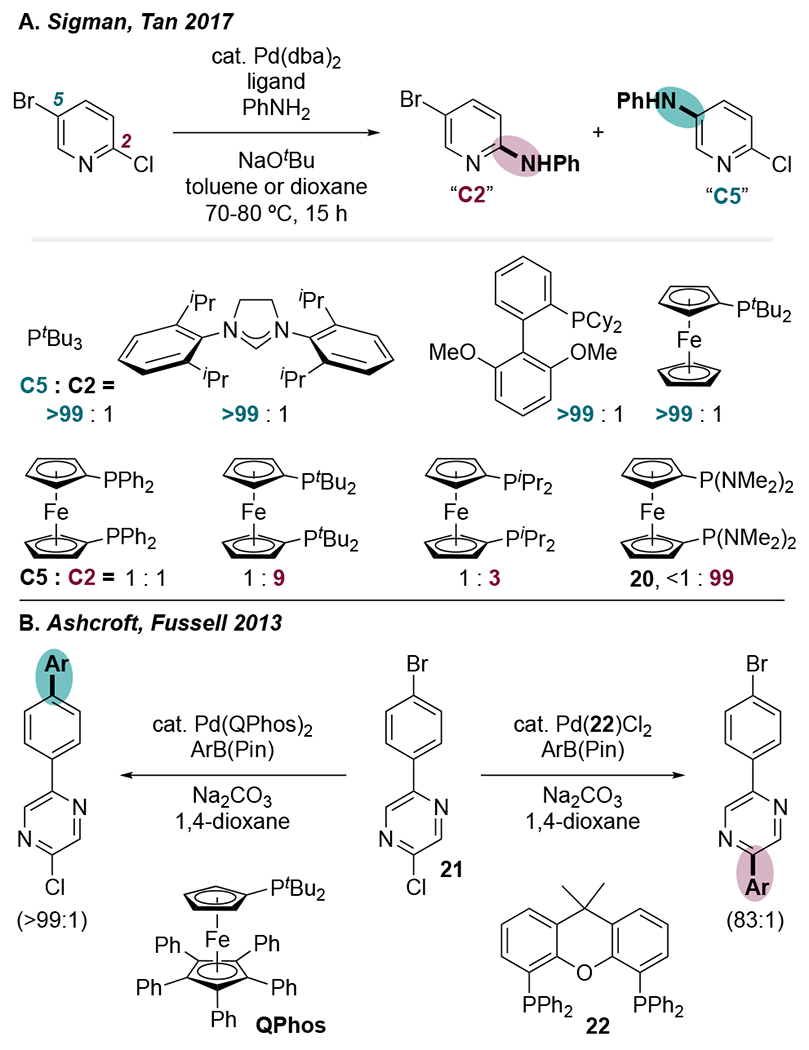
Ligand-Switchable Chemoselectivity Between Cl and Br on 6-Membered Heteroarenes Reported by (A) Sigman, Tan, et al.27 and (B) Ashcroft, Fussell, and Wilford.28
5-Membered Heterocycles.
Compared to 6-membered heteroarenes, relatively few cross-couplings of 5-membered heteroarenes have been reported (for examples, see Table 1). This might be explained by the more electron-rich character of 5-membered arenes, making them less reactive electrophiles. Because of the small number of publications on this topic and the diversity of 5-membered heteroarenes, it is difficult to define ‘conventional’ selectivity for these substrates. Handy’s NMR-based model predicts that the C2-site of oxazoles, thiazoles, and imidazoles should be the most reactive based on a more downfield chemical shift for the corresponding hydrogen in the 1H NMR spectra of the parent compounds (Figure 5, top). Houk’s computations show that a C2—X bond of these substrates is weaker than a C4—X or C5—X bond, suggesting that the C2 site may be more reactive on the basis of distortion energies.11b The C2 carbon of oxazole, thiazole, and N-methylimidazole also has a more positive electrostatic potential than the other ring carbons,29 which might predict greater reactivity at this site based on the selectivity principles described by Leitch et al.12 Nevertheless, a literature survey suggests that selectivity for these substrate classes is not necessarily predictable by established models. In particular, systematic studies by Strotman, Chobanian, and coworkers on 2,4- and 2,5-dihaloimidazoles, -oxazoles, and -thiazoles revealed that selectivity with some of these substrates depends on the phosphine ligand and/or the reaction solvent, as discussed below (Figure 5, bottom).30
Figure 5.
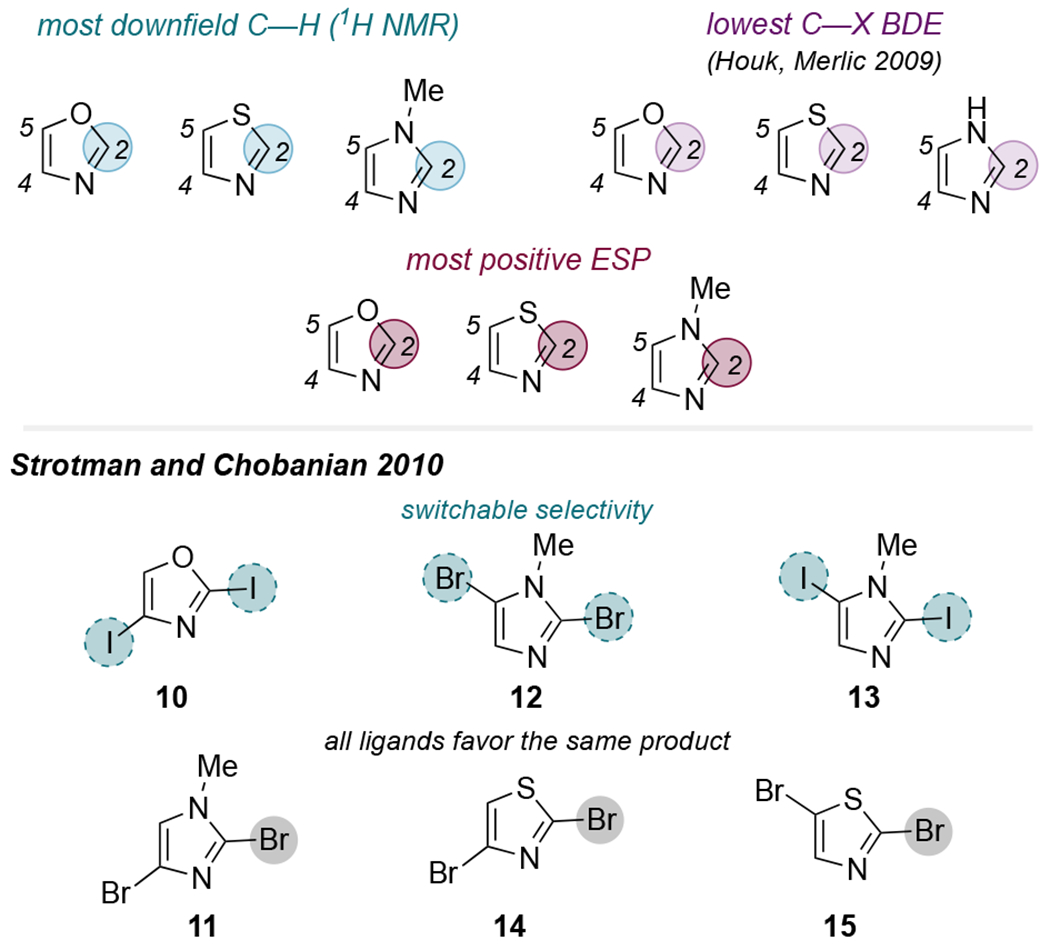
(Top) Highlighted positions represent the expected site of highest reactivity for 5-membered heteroarenes based on Handy’s NMR model,9 Houk and Merlic’s BDE calculations,11b and electrostatic potentials at carbon,29 which are related to Leitch’s model.12 (Bottom) Summary of the actual selectivity behavior for the substrates studied by Strotman and Chobanian.30
The C2-site of 10 is expected to be more reactive based on Handy’s model, C—I bond dissociation energies, and electrostatic potentials at C2 vs. C4. However, a preliminary ligand screen carried out by Strotman et al. resulted in C4-selective Suzuki coupling when using ligands such as PPh3, dppf, XPhos (23), and Xantphos (22) in THF (Scheme 6A). Among these, 22 promotes the highest C4-selectivity (13:1) as well as high selectivity for the monoarylated product over the diarylated product (~7:1). In contrast, PtBu3 enables inverted selectivity in THF, modestly favoring reaction at C2 by about 2:1. Because hindered ligands are known to promote low-coordinate PdL, one could speculate that this selectivity switch relates to palladium’s ligation state during oxidative addition, similar to other examples discussed above in the context of 6-membered heteroarenes. However, a subsequent exhaustive ligand screen of ~200 achiral phosphines led Strotman et al. to identify 1,3,5-triaza-7-phosphaadamantane (26) as a ligand which promotes high selectivity for the C2-site in acetonitrile (~13:1). With its very small cone angle of 102°, even smaller than that of PMe3 (116°),31 26 has little in common with the bulky ligand PtBu3. Furthermore, the bidentate phosphine 25 also slightly favors reaction at C2 in THF. These results seem to suggest against a hypothesis that low-coordinate PdL is responsible for C2-selectivity with this substrate. Importantly, the use of MeCN as solvent is critical to obtaining high C2-selectivity in the reaction of 10 using 26. In THF, the reaction catalyzed by Pd/26 is unselective (1:1). The selectivity with other ligands in MeCN was not reported.
Scheme 6.
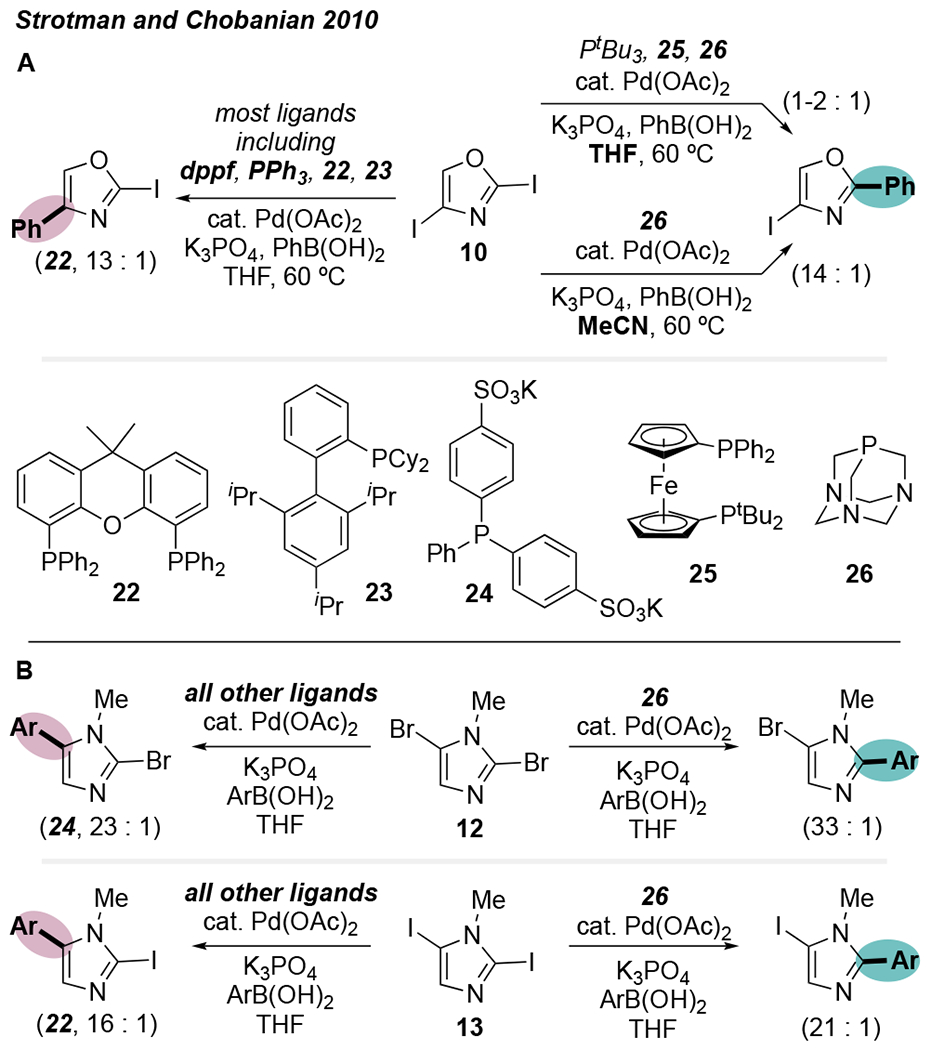
Ligand-Switchable Site Selectivity for 5-Membered Heteroarene Substrates: (A) 2,4-Diiodooxazole and (b) 2,5-Dihaloimidazoles.30
Based on the anomalous selectivity effected by 26 in the reaction of 10, Strotman compared 26 to other ligands for Suzuki couplings of several additional classes of substrates. Once again, 26 was unique in its ability to promote cross-coupling at the C2-site of 12 and 13 (Scheme 6B). Other ligands led to a mixture of products that were biased toward reaction at C5. The higher reactivity of C5—X with most ligands is contrary to the predicted reactivity with Handy or Houk’s model: the 1H NMR chemical shifts of imidazole and N-methylimidazole, 32 and the relative BDEs of C2—Cl and C5—Cl for chloroimidazoles,11b predict that C2—X should be the most reactive (Figure 5, top). Interestingly, other reports in the literature—employing either Pd(dppf)Cl2 or Pd(PPh3)4—suggest that C2 is the preferred reaction site for 2,5-dibromoimidazoles. 33 However, none of these reports include detailed regiochemical characterization, whereas Strotman et al. substantiated the regiochemical assignments of their observed products with NOE experiments.
Despite the observed ligand-dependent divergent selectivity using 2,5-dihaloimidazoles and 2,4-diiodooxazole, Strotman et al. found that other dihalogenated 5-membered heteroarenes are not sensitive to ligand effects. 2,4- and 2,5-Dibromothiazoles (14 and 15) and 2,4-dibromoimidazole (11) were found to undergo C2-selective Suzuki coupling with all of the ~200 ligands screened by Strotman. The mechanistic origin of the ligand-controlled divergent selectivity with some 5-membered heteroarenes—and not with others—has not yet been reported.
SPECIATION-CONTROLLED SELECTIVITY
Thus far, this Perspective has highlighted examples in which atypical site selectivity is achieved due to unique steric or electronic properties of an ancillary phosphine or NHC ligand. However, very recent reports suggest that changes in catalyst speciation (i.e., from mono- to multinuclear) can have an equal or even more powerful influence on site selectivity compared to changes to the ligand environment at mononuclear Pd. Catalyst speciation can be complicated, and in many circumstances it is reasonable to expect a mixture of homogeneous, heterogeneous, and soluble nanoparticle species.34 In particular, higher reaction temperatures can promote the conversion from mono- to multinuclearity, and some additives (e.g., tetralkylammonium halides or -hydroxides) can stabilize nanoparticles by preventing their aggregation into higher-order species.35
The conversion of mononuclear catalytic species into higher-order ones can sometimes translate into fundamental changes in the mechanism of cross-coupling. For example, Li and coworkers reported that the trinuclear Pd3X cluster 27 may catalyze Suzuki cross-coupling through a mechanism in which transmetallation occurs first (Scheme 7).36 The resulting species Pd3Ar (28) then reacts further with the aryl halide electrophile via a σ-bond metathesis mechanism, furnishing the cross-coupled product and regenerating Pd3X. This mechanism stands in contrast to the classic Suzuki reaction in that the two coupling partners enter the catalytic cycle in the reverse order, and there are no traditional redox steps (oxidative addition or reductive elimination). The likelihood of divergent mechanisms for multi- vs. mononuclear Pd is consistent with recent reports of divergent site selectivity for cross-couplings of dihaloheteroarenes catalyzed by multi- vs. mononuclear Pd, as discussed below.
Scheme 7.
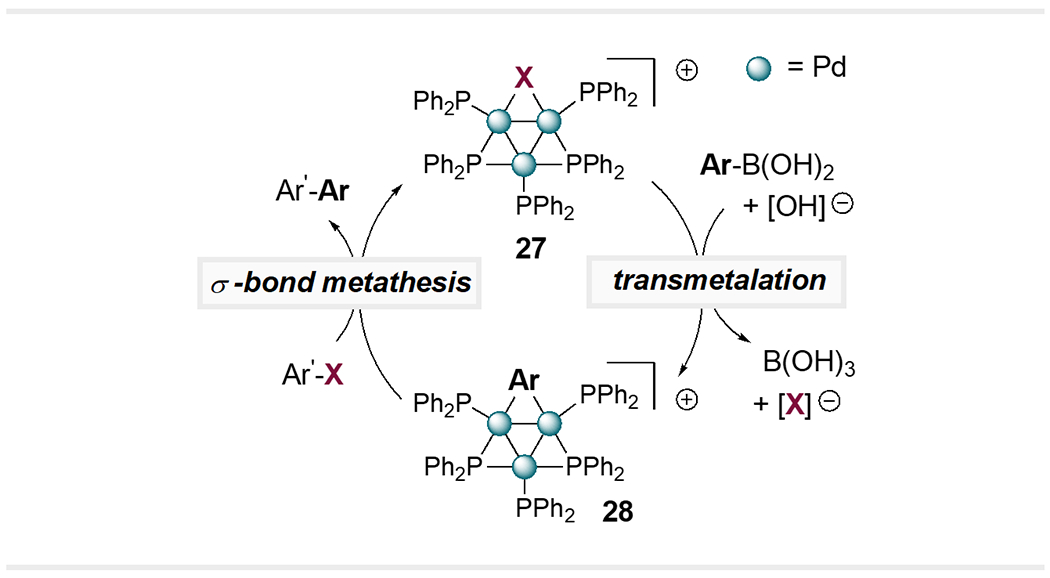
Proposed Catalytic Cycle for Pd3X-Catalyzed Suzuki Cross-Coupling.36
2,4-Dibromopyridine.
Fairlamb et al. demonstrated that the regiochemical outcome of Suzuki and Kumada couplings of 2,4-dibromopyridine can be dependent on the ratio of PPh3:Pd. Conventional C2-selectivity is observed with PPh3:Pd(OAc)2 ratios ≥3:1 (Scheme 8A).37 In contrast, atypical C4-selectivity is seen with PPh3:Pd(OAc)2 ratios ≤2.5:1 or when using the preformed trimer [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]Cl, providing product 2b with up to 13:1 selectivity. The inclusion of tetraalkylammonium bromide or hydroxide salts was necessary to achieve high yield and selectivity for the C4-product, hinting toward the involvement of Pd nanoparticles (PdNPs) under conditions with low PPh3:Pd ratios. Indeed, Fairlamb’s group had previously shown that the combination of PPh3 and Pd(OAc)2 in a 2:1 ratio can lead to the in situ generation of Pd3X clusters (Scheme 8B),38 and related clusters were shown to be intermediates en route to nanoparticle formation.39 As such, the evidence suggests that multinuclear Pd (likely PdNPs) is responsible for the atypical C4-selectivity at low PPh3:Pd ratios. The mechanistic rationale for the unusual site preference of PdNPs is currently unknown, but Fairlamb suggests that one possible explanation could involve a switch in mechanism similar to that reported by Li (vide supra).
Scheme 8.
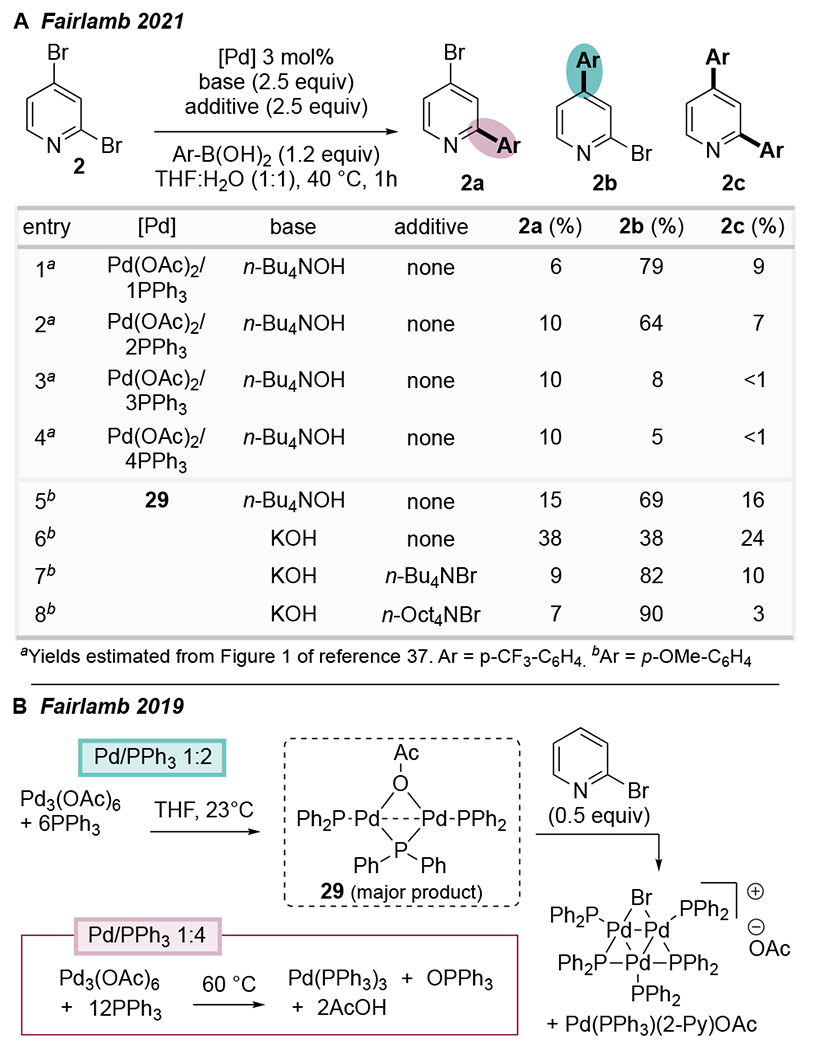
(A) Changes in Speciation can Modulate Site-Selectivity; (B) Modified Pd:PPh3 Ratios Change Catalyst Speciation.37,38
Dichloropyridines, quinolines, and pyrimidines.
In 2022, we reported that ligand-free “Jeffery” conditions facilitate exquisitely selective C4-coupling in the Pd-catalyzed Suzuki reactions of 2,4-dichloropyridine (>99:1) and 2,4-dichloroquinoline (53:1, Scheme 9). Remarkably, these conditions also favor reaction at the C5 position of 2,5-dichloropyridine (6) and 2,5-dichloropyrimidine (8). Preferential reaction at C5 of these substrates, with spectroscopically substantiated regiochemical assignments, had not been previously reported. Prior evidence supports the formation of nanoparticles under such Jeffery conditions involving a simple palladium salt (PdCl2) in the presence of a tetralkylammonium halide.35a,40 As such, it is likely that the unusual site-selectivity in this system is due to PdNPs, similar to the observations made by Fairlamb using 2,4-dibromopyridine. A direct comparison between the Jeffery conditions and Pd/IPr conditions for the Suzuki coupling of 2,4-dichloropyridine 1 indicates that speciation-control has the potential to be much more powerful than ligand-control. Whereas the Pd/IPr catalytic system typically provides ~10:1 selectivity, the ligand-free conditions lead to an order-of-magnitude improvement in selectivity (~99:1).
Scheme 9.
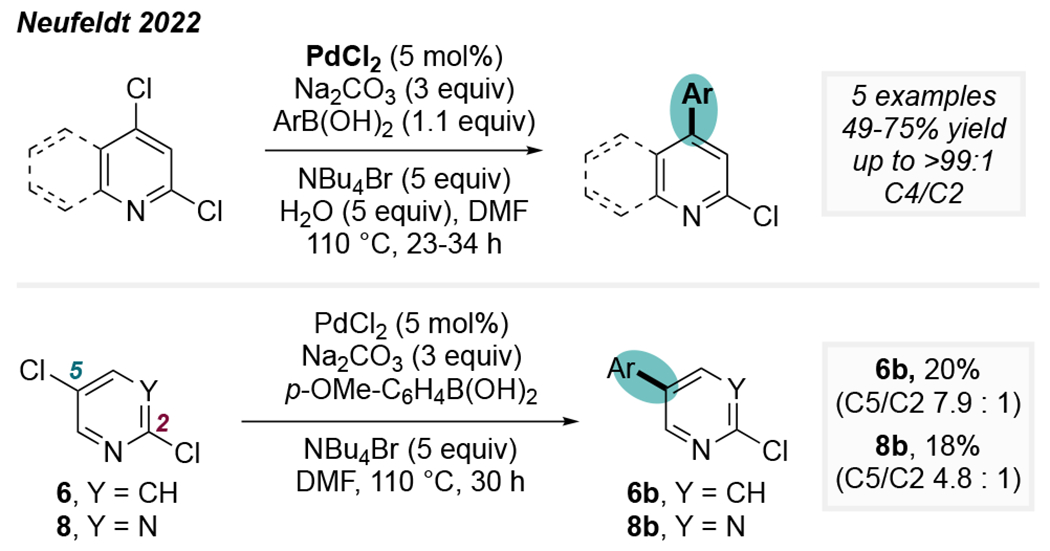
Ligand-free ‘Jeffery’ conditions give unconventional selectivity.25
The remarkable improvement in selectivity under ligand-free conditions compared to Pd/IPr conditions led us to re-examine a previous report by Yang et al. As discussed above, Yang had shown that a PEPPSI-IPr catalytic system gives ~10:1 selectivity for the C4-position during Suzuki cross-coupling of 2,4-dichloropyridine under fairly routine reaction conditions.24 However, they described a surprising jump in C4-selectivity upon modifying their high-temperature reaction conditions to include KI, along with two bases (NaOAc and Na2CO3) added at different times and a higher catalyst loading (Scheme 10A). These conditions are effective for achieving ~99:1 C4-selectivity for Suzuki coupling of a wide range of substituted dichloropyridines. Although the catalyst used by Yang was Pd-PEPPSI-IPr, the exquisite selectivity under their unusual reaction conditions resembles the selectivity that we observe under ligand-free conditions. As such, we investigated this system more closely with a ligand-free control experiment using PdCl2 instead of Pd-PEPPSI-IPr (Scheme 10B). Indeed, the high selectivity was maintained in the absence of IPr, indicating that selectivity under Yang’s optimized conditions is not ligand-controlled. Rather, higher-order Pd species may be responsible for the observed unconventional selectivity.
Scheme 10.
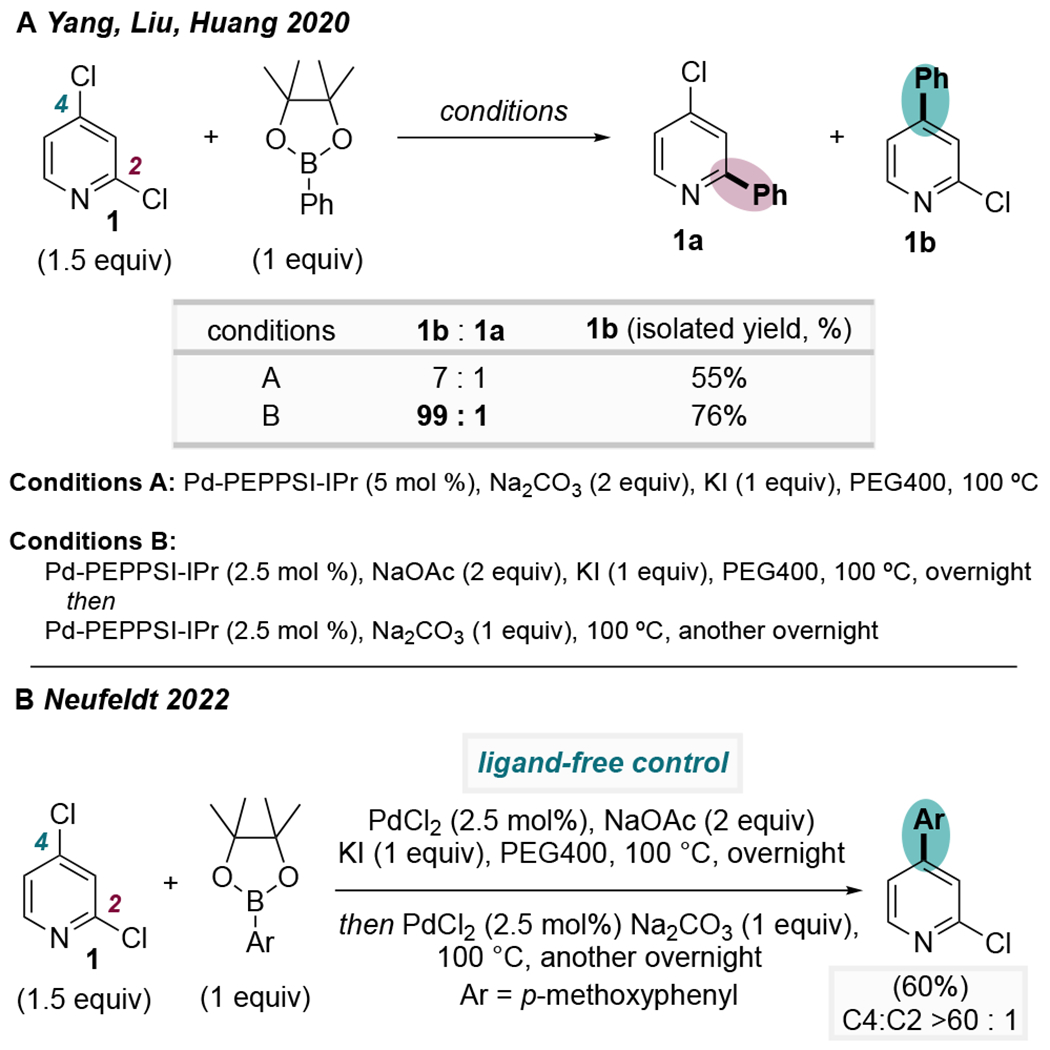
(A) Modified Reaction Conditions Give Unusually High Selectivity with Pd/IPr.24 (B) Control Reaction in the Absence of IPr Shows that Selectivity is Not Ligand-Controlled Under These Conditions.25
OUTLOOK
As described above, unconventional or divergent site-selectivity has been established for a few classes of N-heteroarenes. Nevertheless, catalyst-controlled site-selective cross-couplings remain largely unexplored. Machine-learning algorithms for predicting selectivity are likely to be biased toward conventional selectivity patterns, promoted especially by dppf and PPh3, due to the prevalence of these ligands in literature.41 As such, the development of new methods may rely on improving our mechanistic understanding of selectivity. In particular, we consider the mechanistic picture to be lacking with respect to (1) the significance of two possible mechanisms for oxidative addition (concerted vs. displacement) and the relationship between the preferred mechanism and ligand/substrate identity, (2) the role of catalyst speciation, specifically in cases where multinuclear catalysts promote unconventional site selectivity, and (3) the influence of ring substituents on the regiochemical outcome of cross-coupling reactions. Filling in these mechanistic gaps may lay the groundwork for overcoming additional challenges that face the field of site-selective cross-coupling, including inverting selectivity for some “stubborn” classes of substrates and minimizing unwanted over-reaction of di- and polyhalogenated substrates (e.g., avoiding diarylation). Here we summarize our perspective on these current challenges and opportunities.
Mechanism of Oxidative Addition.
Although oxidative addition of aryl halides at Pd(0) is traditionally thought of as proceeding through a 3-centered concerted mechanism, the existence of a second mechanistic possibility has been well-established. In particular, oxidative addition can also proceed through a pathway that has been termed “SNAr-like”, “dissociative”, or “displacement” (Scheme 11A).13,14,15,26,42 In this pathway, C—X cleavage does not involve interaction between Pd and X in the transition state; instead, Pd interacts with the ipso carbon and an ortho atom of the ring while X dissociates as an anion. Several computational studies have indicated that use of a solvation model is necessary to locate displacement-type transition structures, and that this pathway is more favorable in polar solvents or an externally applied electric field.13,14,43 Calculations by Maseras et al. show that the preferred mechanism for the reaction between PhBr and PdLn depends on the ligand identity and the value of n (1 or 2).14 Palladium’s coordination number during oxidative addition can be influenced by ligand sterics44 or even by substrate concentration.45 In our own work, we found that the preferred mechanism for oxidative addition is intimately related to the divergent site selectivity observed in Pd-catalyzed couplings of 2,4-dichloropyridines using bulky ligands versus smaller or bidentate ligands.15 Developing a better understanding of the circumstances under which these mechanisms (or other mechanisms) are relevant for other classes of dihaloheteroarenes may shed light on strategies for controlling site selectivity in less-explored substrate classes.
Scheme 11.
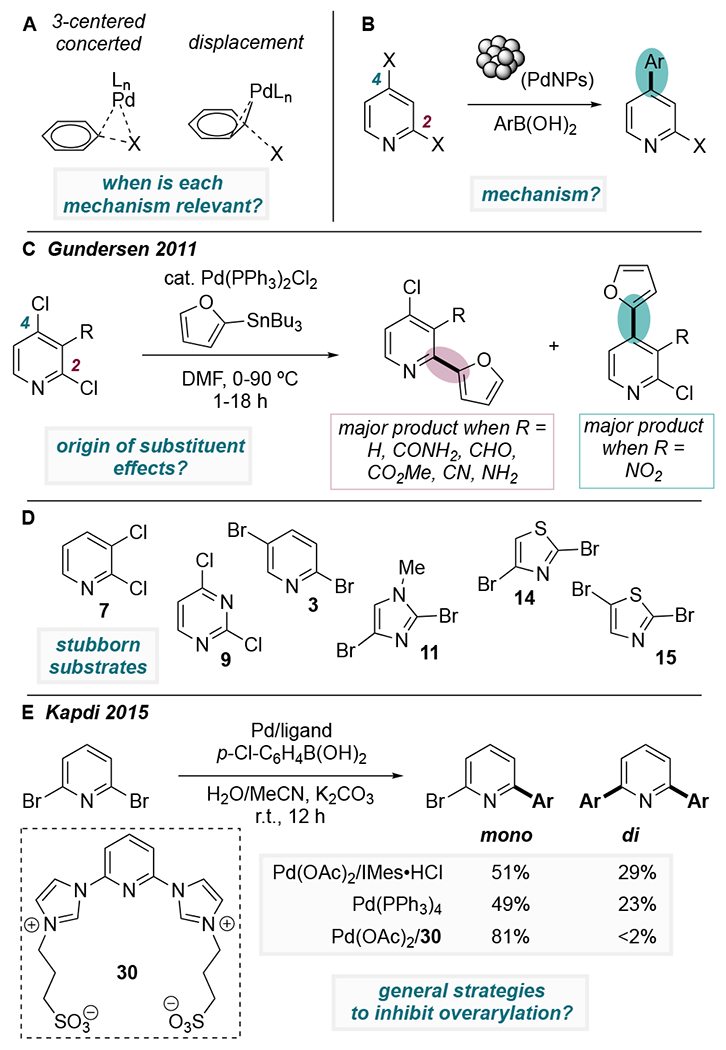
Challenges and Opportunities: (A) Understanding Significance of Different Mechanisms for Oxidative Addition, (B) Mechanistic Origin of Selectivity with Multinuclear Pd, (C) Substituent Effects on Selectivity, (D) Inverting Selectivity with “Stubborn” Substrates, (E) Minimizing Unwanted Difunctionalization.
Catalyst Speciation.
As discussed above, the relevance of catalyst speciation (mono- vs. multinuclear) on site selectivity was discovered by Fairlamb in the context of couplings of 2,4-dibromopyridine.37 Our group later provided evidence that multinuclear Pd also gives unique selectivity in reactions of 2,4-dichloropyridine, as well as 2,5-dichloropyridine and -pyrimidine.25 Remarkably, it appears that multinuclear palladium has the potential to give much higher selectivities than systems in which selectivity is ligand-controlled. Despite the robust evidence for multinuclear speciation, especially in Fairlamb’s system, there is currently no explanation for why multinuclear Pd prefers to react at the distal site (Scheme 11B). Developing a deeper understanding of the mechanism of C—X cleavage at multinuclear Pd is expected to catalyze the development of new selective coupling methods which complement those involving mononuclear catalysis. Much is already known about the size and shape of PdNPs in Suzuki cross-coupling reactions.46 At this time, however, the effect of these properties on site selectivity is unknown. Exploiting the current knowledge base about PdNP size and shape might enable the active sites for selective catalysis to be determined.
Ring Substituents.
For dihalogenated heteroarenes, the effect of substituent sterics and electronics on the site-selectivity of cross-coupling has largely been a matter of empirical study. In a few reports, there is evidence to support the coordination of a substituent (most often carboxylates) to Pd, directing oxidative addition to take place at an adjacent site.6 However, other substituent effects cannot be explained by this phenomenon. In 2011, Khoje and Gundersen summarized the effects of six distinct substituents located at the C3-position of 2,4-dichloropyridine in Pd-catalyzed Stille couplings (Scheme 11C).7 Consistent with its established conventional selectivity, Pd/PPh3 effects C2-arylation in nearly every example. However, when the C3-substituent is a nitro group, arylation occurs almost exclusively at C4. There is currently no published rationale for this phenomenon, yet anomalous examples such as this one provide an opportunity to identify new mechanistic features which could complement our current understanding of selectivity.
“Stubborn” Substrate Classes.
For the majority of di- and polyhalogenated N-heteroarenes, strategies for inverting conventional site selectivity remain undeveloped or underdeveloped (examples in Scheme 11D). For many heteroaryl substrate classes, little selectivity information has been established due to poor reactivity. These substrates include substituted pyrazoles and fused 5,6-heterocycles such as benzimidazoles and indoles.1g,47 For other substrates, existing literature seems to indicate that selectivity always follows a particular pattern. For instance, Strotman’s extensive ligand screening for the Suzuki couplings of 2,4- and 2,5-dibromothiazoles (14 and 15) and a 2,4-dibromoimidazole (11) resulted in C2-selective cross-coupling in all cases, leaving no option to access the opposite regioisomer through cross-coupling.30 As an another example, 2,4-dichloropyrimidines present a particular challenge. These substrates generally react preferentially at C4 (both in cross-couplings and in SNAr reactions), but a method to cross-couple selectively at C2 would be quite desirable due to the prevalence of the resulting motifs in bioactive compounds. However, it is difficult to envision a robust foothold for biasing reactivity toward C2, since multiple factors seem to favor reaction at C4. The C4—Cl bond is weaker, and the C4 carbon has a much larger LUMO coefficient. Thus, a method to successfully achieve C2-selective coupling may require a fundamentally different strategy than those that have been previously reported for other substrates.
Diarylation.
Undesired “overarylation” is common in cross-coupling reactions of dihaloheteroarenes when using bulky NHC or biarylphosphine ligands, despite a 1:1 ratio of substrate to nucleophile.20,23a,24,25 This trend is consistent with literature reports involving halogenated aromatic carbocycles. For example, Larosa et al. found that IPent enables efficient “exhaustive arylation” of various dihalogenated arenes despite a deficit of nucleophile.48 Further, Hein et al. have reported that both IPr and RuPhos are effective di- or polyamination catalysts for regioselective elaborations of tetrabromospirobifluorenes.49 When a monofunctionalized product is desired, competitive overarylation detracts from the yield, in part because diarylated products can be challenging to separate from monofunctionalized product. Furthermore, the accumulation of diarylated product in a reaction mixture can complicate an evaluation of site-selectivity if either monoarylated isomer undergoes a second arylation more rapidly than the other. Formation of diarylated product can thus artificially inflate or deflate the observed ratio of monoarylated products. In the context of Pd-catalyzed cross-couplings of dihalo(hetero)arenes, the mechanism of diarylation may mirror that of catalytic chain growth polymerizations in which oxidative addition of a nascent monoarylated product takes place more rapidly than dissociation of the catalyst from product at the end of a catalytic cycle. This mechanism is referred to as a chain-walking mechanism.50 Kapdi et al. reported that several sulfonated alkylimidazolium ligands markedly disfavor diarylation of 2,6-dibromopyridine compared to IMes, PPh3, and other ligands under Pd-catalyzed SMC conditions (Scheme 11E), hinting that ligand bifunctionality may play a role in assisting the dissociation step.23 Further mechanistic study into the relationship between over-functionalization and ligand environment could guide catalyst design to minimize unwanted overfunctionalization.
ACKNOWLEDGMENT
This work was supported by the National Institute of General Medical Sciences (NIGMS) of the NIH under Award Number R35GM137971.
REFERENCES
- 1.(a) Schröter S; Stock C; Bach T Regioselective Cross-Coupling Reactions of Multiple Halogenated Nitrogen-, Oxygen-, and Sulfur-Containing Heterocycles. Tetrahedron 2005, 61, 2245–2267. [Google Scholar]; (b) Fairlamb IJS Regioselective (site-selective) functionalisation of unsaturated halogenated nitrogen, oxygen and sulfur heterocycles by Pd-catalysed cross-couplings and direct arylation processes. Chem. Soc. Rev 2007, 36, 1036–1045. [DOI] [PubMed] [Google Scholar]; (c) Bellina F; Rossi R; Regioselective Functionalization of the Imidazole Ring via Transition Metal-Catalyzed C-N and C-C Bond Forming Reactions. Adv. Synth. Catal 2010, 352, 1223–1276 [Google Scholar]; (d) Rossi R; Bellina F; Lessi M Selective Palladium-Catalyzed Suzuki–Miyaura Reactions of Polyhalogenated Heteroarenes. Adv. Synth. Catal 2012, 354, 1181–1255. [Google Scholar]; (e) Manabe K; Yamaguchi M Catalyst-Controlled Site-Selectivity Switching in Pd-Catalyzed Cross-Coupling of Dihaloarenes. Catalysts 2014, 4, 307–320. [Google Scholar]; (f) Kapdi AR; Prajapati D Regioselective palladium-catalysed cross-coupling reactions: a powerful synthetic tool. RSC Adv. 2014, 4, 41245–41259. [Google Scholar]; (g) Almond-Thynne J; Blakemore DC; Pryde DC; Spivey AC Site-selective Suzuki–Miyaura coupling of heteroaryl halides – understanding the trends for pharmaceutically important classes. Chem. Sci 2017, 8, 40–62. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Palani V; Perea MA; Sarpong R Site-Selective Cross-Coupling of Polyhalogenated Arenes and Heteroarenes with Identical Halogen Groups. Chem. Rev 2021, 122, 10126–10169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Mahatthananchai J; Dumas AM; Bode JW Catalytic Selective Synthesis. Angew. Chem. Int. Ed 2012, 51, 10954–10990. [DOI] [PubMed] [Google Scholar]; (b) Shang W; He B; Niu D Ligand-controlled, transition-metal catalyzed site-selective modification of glycosides. Carbohydrate. Res 2019, 474, 16–33. [DOI] [PubMed] [Google Scholar]; (c) Nájera C; Beletskaya IP; Yus M Metal-Catalyzed Regiodivergent Organic Reactions. Chem. Soc. Rev 2019, 48, 4515–4618. [DOI] [PubMed] [Google Scholar]; (d) Sakakibara Y; Murakami K Switchable Divergent Synthesis Using Photocatalysis. ACS Catal. 2022, 12, 1857–1878. [Google Scholar]; (e) Wang R; Yu Y Site-selective reactions mediated by molecular containers. Beilstein J. Org. Chem 2022, 18, 309–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selected reviews:; (a) Neufeldt SR; Sanford MS; Controlling Site Selectivity in Palladium-Catalyzed C–H Bond Functionalization. Acc. Chem. Res 2012, 45, 936–946. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gigant N; Chausset-Boissarie L; Gillaizeau I Direct Metal-Catalyzed Regioselective Functionalization of Enamides. Chem. Eur. J 2014, 20, 7548–756. [DOI] [PubMed] [Google Scholar]; (c) Yang J; Transition metal catalyzed meta-C–H functionalization of aromatic compounds. Org. Biomol. Chem, 2015, 13, 1930–1941. [DOI] [PubMed] [Google Scholar]; (d) Murakami K; Yamada S; Kaneda T; Itami K C–H Functionalization of Azines. Chem. Rev 2017, 117, 9302–9332. [DOI] [PubMed] [Google Scholar]; (e) Das R; Kapur M Transition-Metal-Catalyzed Site-Selective C–H Halogenation Reactions. Asian J. Org. Chem 2018, 7, 1524–1541. [Google Scholar]; (f) Kang D; Ahn K; Hong S Site-Selective C–H Bond Functionalization of Chromones and Coumarins. Asian J. Org. Chem 2018, 7, 1136–1150. [Google Scholar]; (g) Tiwari VK; Kapur M Catalyst-controlled positional-selectivity in C-H functionalizations. Org. Biomol. Chem 2019, 17, 1007–1026. [DOI] [PubMed] [Google Scholar]; (h) Jiao K-J; Xing Y-K; Yang Q-L; Qiu H; Mei T-S Site-Selective C–H Functionalization via Synergistic Use of Electrochemistry and Transition Metal Catalysis. Acc. Chem. Res 2020, 53, 300–310. [DOI] [PubMed] [Google Scholar]; (i) Chen H; Farizyan M; van Gemmeren M Regioselective Olefination of 3-Substituted Five-membered Heteroarenes. Eur. J. Org. Chem 2020, 6318–6327. [Google Scholar]; (j) Gramage-Doria R Steering Site-Selectivity in Transition Metal-Catalyzed C–H Bond Functionalization: the Challenge of Benzanilides. Chem. Eur. J 2020, 26, 9688–9709. [DOI] [PubMed] [Google Scholar]; (k) Kuninobu Y; Torigoe T Recent progress of transition metal-catalysed regioselective C-H transformations based on noncovalent interactions. Org. Biomol. Chem 2020, 18, 4126–4134. [DOI] [PubMed] [Google Scholar]; (l) Li B; Elsaid M; Ge H Transition-metal-catalyzed site-selective γ- and δ-C(sp3)–H functionalization reactions. Chem, 2022, 8, 1254–1360. [Google Scholar]; (m) Sinha SK; Guin S; Maiti S; Biswas JP; Porey S; Maiti D Toolbox for Distal C–H Bond Functionalizations in Organic Molecules. Chem. Rev 2022, 122, 5682–5841. [DOI] [PubMed] [Google Scholar]
- 4.Chemodivergent cross-coupling (i.e., when Pd chooses between two different functional groups such as chloride vs. triflate) has been somewhat better studied; for a review see:; Reeves EK; Entz ED; Neufeldt SR Chemodivergence Between Electrophiles in Cross-Coupling Reactions. Chem. Eur. J 2021, 27, 6161–6177. [DOI] [PubMed] [Google Scholar]
- 5.“Reaction” searches were performed in the CAS SciFindern database for each substrate in Table 1. A second reactant, “R1-R2, ” was included as the nucleophilic coupling partner where R1 = C or N atoms and R2 = H, B, C, Mg, Si, Cu, Zn, or Sn atoms. For each substrate, one search was performed for each regioisomeric product where one halogen was replaced by R1. All unchanged atoms in the reactant were “mapped” onto those in the product, and non-halogenated ring atoms were “locked” so that the search would not include substituted derivatives. Search results were refined by “catalyst” to only include conditions involving Pd. Separately, search results were refined by “catalyst” to include only Pd(PPh3)4, Pd(PPh3)2Cl2, PPh3, Pd(dppf)Cl2, Pd(dppf)Cl2•CH2Cl2, and dppf. In some cases, results were manually excluded when non-selective mixtures of products were reported. Date accessed: July 25, 2022.
- 6.For directing group effects via possible coordination of Pd or Ni to a carboxylate or amide, see:; (a) Yang W; Wang Y; Corte JR Efficient Synthesis of 2-Aryl-6-chloronicotinamides via PXPd2-Catalyzed Regioselective Suzuki Coupling. Org. Lett 2003, 5, 3131–3134. [DOI] [PubMed] [Google Scholar]; (b) Houpis IN; Liu R; Wu Y; Yuan Y; Wang Y; Nettekoven UJ Org. Chem 2010, 75, 6965–6968. [DOI] [PubMed] [Google Scholar]; (c) Nguyen T; Chiu W; Wang X; Sattler MO; Love JA Ligandless Nickel-Catalyzed Ortho-Selective Directed Trifluoromethylthiolation of Aryl Chlorides and Bromides Using AgSCF3. Org. Lett 2016, 18, 5492–5495. [DOI] [PubMed] [Google Scholar]
- 7.Khoje AD; Gundersen L-L Reactivity and regioselectivity in Stille couplings of 3-substituted 2,4-dichloropyridines. Tetrahedron Lett 2011, 52, 523–525. [Google Scholar]
- 8.For other examples of directing effects, see:; (a) Ahmed S; Sharif M; Shoaib K; Reimann S; Iqbal J; Patonay T; Spannenberg A; Langer P Synthesis of 2,6-diaryl-3-(trifluoromethyl)pyridines by regioselective Suzuki–Miyaura reactions of 2,6-dichloro-3-(trifluoromethyl)pyridine. Tetrahedron Lett. 2013, 54, 1669–1672. [Google Scholar]; (b) Blaise E; mmerle AE; Hammoud H; de Araújo-Júnior JX; Bihel F; Bourguignon J-J; Schmitt M Access to 4-Alkylaminopyridazine Derivatives via Nitrogen-Assisted Regioselective Pd-Catalyzed Reactions. J. Org. Chem 2014, 79, 10311–10322. [DOI] [PubMed] [Google Scholar]; (c) Zhao P; Young MD; Beaudry CM Regioselective Suzuki Couplings of Non-Symmetric Dibromobenzenes: Alkenes as Regiochemical Control Elements. Org. Biomol. Chem 2015, 13, 6162–6165. [DOI] [PubMed] [Google Scholar]
- 9.(a) Handy ST; Zhang Y A simple guide for predicting regioselectivity in the coupling of polyhaloheteroaromatics. Chem. Commun 2006, 3, 299–301; [DOI] [PubMed] [Google Scholar]; see also; (b) Zhang Y; Handy ST A Solvent-Induced Reversal of Regioselectivity in the Suzuki Coupling of Pyrrole Esters. Open Org. Chem. J 2008, 2, 58–64. [Google Scholar]; (c) Kashihara M; Gordon CP; Copéret C Reactivity of Substituted Benzenes toward Oxidative Addition Relates to NMR Chemical Shift of the Ipso-Carbon. Org. Lett 2020, 22, 8910–8915. [DOI] [PubMed] [Google Scholar]
- 10.Abraham RJ; Reid M 1H chemical shifts in NMR. Part 18. Ring currents and p-electron effects in hetero-aromatics. J. Chem. Soc., Perkin Trans 2 2002, 1081–1091. [Google Scholar]
- 11.(a) Legault CY; Garcia Y; Merlic CA; Houk KN Origin of Regioselectivity in Palladium-Catalyzed Cross-Coupling Reactions of Polyhalogenated Heterocycles. J. Am. Chem. Soc 2007, 129, 12664–12665. [DOI] [PubMed] [Google Scholar]; (b) Garcia Y; Schoenebeck F; Legault CY; Merlic CA; Houk KN Theoretical Bond Dissociation Energies of Halo-Heterocycles: Trends and Relationships to Regioselectivity in Palladium-Catalyzed Cross-Coupling Reactions. J. Am. Chem. Soc 2009, 131, 6632–6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu J; Donnecke S; Paci I; Leitch DC A reactivity model for oxidative addition to palladium enables quantitative predictions for catalytic cross-coupling reactions. Chem. Sci 2022, 13, 3477–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Senn HM; Ziegler T Oxidative Addition of Aryl Halides to Palladium(0) Complexes: A Density-Functional Study Including Solvation. Organometallics 2004, 23, 2980–2988. [Google Scholar]
- 14.Besora M; Maseras F The diverse mechanisms for the oxidative addition of C–Br bonds to Pd(PR3) and Pd(PR3)2 complexes. Dalton Trans. 2019, 48, 16242–16248. [DOI] [PubMed] [Google Scholar]
- 15.Norman JP; Larson NG; Neufeldt SR Different Oxidative Addition Mechanisms for 12- and 14-Electron Palladium(0) Explain Ligand-Controlled Divergent Site Selectivity. ACS Catal. 2022, 12, 8822–8828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walsh CT Nature loves nitrogen heterocycles. Tetrahedron Lett, 2013, 56, 3075–3081. [Google Scholar]
- 17.(a) Heterocyclic Chemistry in Drug Discovery, Li JJ, Ed. Wiley-VCH, Weinheim, 2013; [Google Scholar]; (b) Taylor AP; Robinson RP; Fobia YM; Blakemore DC; Jones LH; Fadeyi O Modern advances in heterocyclic chemistry in drug discovery. Org. Biomol. Chem 2016, 14, 6611–6637. [DOI] [PubMed] [Google Scholar]
- 18.Lamberth C Heterocyclic chemistry in crop protection. Pest Manage. Sci 2013, 69, 1106–1107. [DOI] [PubMed] [Google Scholar]
- 19.Chen D; Su S-J; Cao Y Nitrogen Heterocycle-Containing Materials for Highly Efficient Phosphorescent OLEDs with Low Operating Voltage J. Mater. Chem. C 2014, 2, 9565–9578. [Google Scholar]
- 20.Dai X; Chen Y; Garrell S; Liu H; Zhang L-K; Palani A; Hughes G; Nargund R Ligand-Dependent Site-Selective Suzuki Cross-Coupling of 3,5-Dichloropyridazines. J. Org. Chem 2013, 78, 7758–7763. [DOI] [PubMed] [Google Scholar]
- 21.Barrios-Landeros F; Hartwig FJ Distinct Mechanisms for the Oxidative Addition of Chloro-, Bromo-, and Iodoarenes to a Bisphosphine Palladium(0) Complex with Hindered Ligands. J. Am. Chem. Soc 2005, 127, 6944–6945. [DOI] [PubMed] [Google Scholar]
- 22.Additionally, Dai and Chen note that similar selectivities were observed regardless of whether QPhos was used in a 1:1 or 2:1 ratio with Pd, which is consistent with the inability of Pd to accommodate two QPhos ligands during oxidative addition, even when an excess of QPhos is available.
- 23.(a) Fowler JM; Britton E; Pask CM; Willans CE; Hardie MJ Cyclotriveratrylene-tethered trinuclear palladium(II)-NHC complexes; reversal of site selectivity in Suzuki-Miyaura reactions. Dalton Trans. 2019, 48, 14687–14695; [DOI] [PubMed] [Google Scholar]; (b) See also: [Google Scholar]; Prajapati D; Schulzke C; Kindermann MK; Kapdi AR Selective palladium-catalysed arylation of 2,6-dibromopyridine using N-heterocyclic carbene ligands. RSC Adv., 2015, 5, 53073–53085. [Google Scholar]
- 24.Yang M; Chen J; He C; Hu X; Ding Y; Kuang Y; Liu J; Huang Q Palladium-Catalyzed C-4 Selective Coupling of 2,4-Dichloropyridines and Synthesis of Pyridine-Based Dyes for Live-Cell Imaging. J. Org. Chem 2020, 85, 6498–6508. [DOI] [PubMed] [Google Scholar]
- 25.Norman JP; Larson NG; Entz ED; Neufeldt SR Unconventional Site-Selectivity in Palladium-Catalyzed Cross-Couplings of Dichloroheteroarenes under Ligand-Controlled and Ligand-Free Systems. J. Org. Chem 2022, 87, 7414–7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Portnoy M; Milstein D Mechanism of Aryl Chloride Oxidative Addition to Chelated Palladium(0) Complexes. Organometallics 1993, 12, 1665–1673; [Google Scholar]; (b) Maes BUW; Verbeek S; Verhelst T; Ekomié A; von Wolff N; Lefèvre G; Mitchell EA; Jutand A Oxidative Addition of Haloheteroarenes to Palladium(0): Concerted versus SNAr-Type Mechanism. Chem. Eur. J 2015, 21, 7858–7865. [DOI] [PubMed] [Google Scholar]
- 27.Keylor MH; Niemeyer ZL; Sigman MS; Tan KL Inverting Conventional Chemoselectivity in Pd-Catalyzed Amine Arylations with Multiply Halogenated Pyridines. J. Am. Chem. Soc 2017, 139, 10613–10616. [DOI] [PubMed] [Google Scholar]
- 28.Ashcroft CP; Fussell SJ; Wilford K Catalyst controlled regioselective Suzuki cross-coupling of 2-(4-bromophenyl)-5-chloropyrazine. Tetrahedron Lett. 2013, 54, 4529–4532. [Google Scholar]
- 29.Personal communication with Prof. David C. Leitch, July 29, 2022.
- 30.Strotman NA; Chobanian HR; He J; Guo Y; Dormer PG; Jones CM; Steves JE Catalyst-Controlled Regioselective Suzuki Couplings at Both Positions of Dihaloimidazoles, Dihalooxazoles, and Dihalothiazoles. J. Org. Chem 2010, 75, 1733–1739. [DOI] [PubMed] [Google Scholar]
- 31.DeLerno J; Trefonas L; Darensbourg M; Majeste RJ Molecular structure and spectral properties of phosphatriazaadamantanemolybdenum pentacarbonyl complex. Inorganic Chemistry 1976, 15, 816–819. [Google Scholar]
- 32.Kim J-W; Abdelaal SM; Bauer L; Heimer NE Synthesis of 1-(dimethylsulfamoyl)-2- and 5-imidazolecarboxaldehydes. Rearrangement of 1-(dimethylsulfamoyl)-5-imidazole-carboxaldehyde to the 4-carboxaldehyde. J. Heterocyclic Chem 1995, 32, 611–620. [Google Scholar]
- 33.(a) Attenni B; Ferrigno F; Jones P; Ingenito R; Kinzel O; Llauger B, Laura O, Jesus M; Pescatore G; Rowley M; Scarpelli R; Schultz C Preparation of heterocycle derivatives as histone deacetylase inhibitors. WO2006061638, 2006-June-15; [Google Scholar]; (b) Tamayo NA; Norman MH; Bartberger MD; Hong F-T; Bo Y; Liu L; Nishimura N; Yang KC; Tadesse S; Fotsch C; Chen J; Chmait S; Cupples R; Hale C; Jordan SR; Lloyd DJ; Sivits G; Van G; Jean DJ Small Molecule Disruptors of the Glucokinase-Glucokinase Regulatory Protein Interaction: 5. A Novel Aryl Sulfone Series, Optimization Through Conformational Analysis. J. Med. Chem 2015, 58, 4462–4482. [DOI] [PubMed] [Google Scholar]; (c) Bair KW; Herbertz T; Kauffman GS; Kayser-Bricker KJ; Luke GP; Martin MW; Millan DS; Schiller SER; Talbot AC Tetrahydroquinoline compositions as BET bromodomain inhibitors and their preparation. US20160256448. 2016-September-08. [Google Scholar]; (d) Wang B; Chao Q Triazolecarboxylic acid derivatives as glycolate oxidase inhibitors for the treatment of disease and their preparation. WO2019133770. 2019-July-04. [Google Scholar]; (e) Wang B; Crawford BE; Bhagwat S; Bai X Preparation of 1H-1,2,3-triazole-5-carboxylic acid derivatives as glycolate oxidase inhibitors for the treatment of hyperoxaluria and related diseases. WO2020257487. 2020-December-24. [Google Scholar]
- 34.(a) Widegren JA; Finke RG A review of the problem of distinguishing true homogeneous catalysis from soluble or other metal-particle heterogeneous catalysis under reducing conditions. J. Mol. Catal. A 2003, 198, 317–341. [Google Scholar]; (b) Phan NTS; Van Der Sluys M; Jones CW On the Nature of the Active Species in Palladium Catalyzed Mizoroki–Heck and Suzuki–Miyaura Couplings – Homogeneous or Heterogeneous Catalysis, A Critical Review Adv. Synth. Catal 2006, 348, 609–679. [Google Scholar]; (c) de Vries JG When Does Catalysis with Transition Metal Complexes Turn into Catalysis by Nanoparticles? Selective Nanocatalysts and Nanoscience, 2011; Wiley:VCH, pp 73–103. [Google Scholar]; (d) Fairlamb IJS; Lee AF Fundamental Pd0/PdII Redox Steps in Cross-coupling Reactions: Homogeneous, Hybrid Homogeneous–Heterogeneous to Heterogeneous Mechanistic Pathways for C–C Couplings in C–H and C–X Bond Functionalization: Transition Metal Mediation, ed. Ribas X; RSC Publishing, Cambridge, UK, 2013, ch. 3, p. 72–107. [Google Scholar]; (e) Cano R; Schmidt AF; McGlacken G Direct arylation and heterogeneous catalysis; ever the twain shall meet. Chem. Sci 2015, 6, 5338–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Fairlamb IJS; Scott NWJ Pd Nanoparticles in C–H Activation and Cross-coupling Catalysis. In Nanoparticles in Catalysis, ed. Kobayashi, S., Top. Organomet. Chem 2020, 474, 171–205. [Google Scholar]
- 35.(a) Chernyshev VM; Khazipov OV; Eremin DB; Denisova EA; Ananikov VP Formation and stabilization of nanosized Pd particles in catalytic systems: Ionic nitrogen compounds as catalytic promoters and stabilizers of nanoparticles. Coord. Chem. Rev 2021, 437, 213860. [Google Scholar]; (b) Ohtaka A. Recent Progress of Metal Nanoparticle Catalysts for C–C Bond Forming Reactions. Catalysts 2021, 11, 1266. [Google Scholar]
- 36.Fu F; Xiang J; Cheng H; Cheng L; Chong H; Wang S; Li P; Wei S; Zhu M; Li Y A Robust and Efficient Pd3 Cluster Catalyst for the Suzuki Reaction and Its Odd Mechanism. ACS Catal. 2017, 7, 1860–1867 [Google Scholar]
- 37.Scott NWJ; Ford MJ; Jeddi N; Eyles A; Simon L; Whitwood AC; Tanner T; Willans CE; Fairlamb IJS A Dichotomy in Cross-Coupling Site Selectivity in a Dihalogenated Heteroarene: Influence of Mononuclear Pd, Pd Clusters, and Pd Nanoparticles–the Case for Exploiting Pd Catalyst Speciation. J. Am. Chem. Soc 2021, 143, 9682–9693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott NWJ; Ford MJ; Schotes C; Parker RR; Whitwood AC; Fairlamb IJS The ubiquitous cross-coupling catalyst system ‘Pd(OAc)2’/2PPh3 forms a unique dinuclear PdI complex: an important entry point into catalytically competent cyclic Pd3 clusters. Chem. Sci 2019, 10, 7898–7906. [Google Scholar]
- 39.Appleby KM; Dzotsi E; Scott NWJ; Dexin G; Jeddi N; Whitwood AC; Pridmore NE; Hart S; Duckett SB; Fairlamb IJS Bridging the Gap from Mononuclear PdII Precatalysts to Pd Nanoparticles: Identification of Intermediate Linear [Pd3(XPh3)4]2+ Clusters as Catalytic Species for Suzuki–Miyaura Couplings (X = P, As). Organometallics 2021, 40, 3560–3570. [Google Scholar]
- 40.(a) Jeffery T Palladium-catalysed Vinylation of Organic Halides under Solid-Liquid Phase Transfer Conditions. J. Chem. Soc., Chem. Commun, 1984, 1287–1289; [Google Scholar]; (b) Jeffery T Highly stereospecific palladium-catalysed vinylation of vinylic halides under solid-liquid phase transfer conditions. Tetrahedron Lett. 1985, 26, 2667–2670; [Google Scholar]; (c) Jeffery T On the efficiency of tetraalkylammonium salts in Heck type reactions. Tetrahedron 1996, 52, 10113–10130. [Google Scholar]
- 41.Beker W; Roszak R; Wolos A; Angello NH; Rathore V; Burke MD; Grzybowksi BA Machine Learning May Sometimes Simply Capture Literature Popularity Trends: A Case Study of Heterocyclic Suzuki-Miyaura Coupling. J. Am. Chem. Soc 2022, 144, 4819–4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Entz ED; Russell JEA; Hooker LV; Neufeldt SR Small Phosphine Ligands Enable Selective Oxidative Addition of Ar—O over Ar—Cl Bonds at Nickel(0). J. Am. Chem. Soc 2020, 142, 15454–15463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Joy J; Stuyver T; Shaik S Oriented External Electric Fields and Ionic Additives Elicit Catalysis and Mechanistic Crossover in Oxidative Addition Reactions. J. Am. Chem. Soc 2020, 142, 3836–3850. [DOI] [PubMed] [Google Scholar]
- 44.(a) Schoenebeck F; Houk KN Ligand-Controlled Regioselectivity in Palladium-Catalyzed Cross Coupling Reactions. J. Am. Chem. Soc 2010, 132, 2496–2497. [DOI] [PubMed] [Google Scholar]; (b) Proutiere F; Lyngvi E; Aufiero M; Sanhueza IA; Schoenebeck F Combining the Reactivity Properties of PCy3 and PtBu3 into a Single Ligand, P(iPr)(tBu)2. Reaction via Mono- or Bisphosphine Palladium(0) Centers and Palladium(I) Dimer Formation. Organometallics 2014, 33, 6879–6884. [Google Scholar]
- 45.Mitchell EA; Jessop PG; Baird MC A Kinetics Study of the Oxidative Addition of Bromobenzene to Pd(PCy3)2 (Cy = cyclohexyl) in a Nonpolar Medium: The Influence on Rates of Added PCy3 and Bromide Ion. Organometallics 2009, 28, 6732–6738. [Google Scholar]
- 46.(a) Ellis PJ; Fairlamb I. J. s.; Hackett SFJ; Wilson K; Lee AF Evidence for the Surface-Catalyzed Suzuki–Miyaura Reaction overPalladium Nanoparticles: An Operando XAS Study. Angew. Chem. Int. Ed 2010, 49, 1820–1824. [DOI] [PubMed] [Google Scholar]; (b) Lee AF; Ellis PJ; Fairlamb IJS; Wilson K Surface catalysed Suzuki–Miyaura cross-coupling by Pd nanoparticles: an operando XAS study. Dalton Trans. 2010, 39, 10473–10482. [DOI] [PubMed] [Google Scholar]; (c) Collins G; Schmidt M; O’Dwyer C; Holmes JD; McGlacken GP The Origin of Shape Sensitivity in Palladium-Catalyzed Suzuki–Miyaura Cross Coupling Reactions. Angew. Chem. Int. Ed 2014, 53, 4142–4145. [DOI] [PubMed] [Google Scholar]
- 47.Düfert MA; Billingsley KL; Buchwald SL Suzuki-Miyaura Cross-Coupling of Unprotected, Nitrogen-Rich Heterocycles: Substrate Scope and Mechanistic Investigation. J. Am. Chem. Soc 2013, 135, 12877–12885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Groombridge BJ; Goldup SM; Larrosa I Selective and general exhaustive cross-coupling of dichloroarenes with a deficit of nucleophiles mediated by a Pd–NHC complex. Chem. Commun 2015, 51, 3832–3834. [DOI] [PubMed] [Google Scholar]
- 49.Deem MC; Derasp JS; Malig TC; Legard K; Berlinguette CP; Hein JE Ring walking as a regioselectivity control element in Pd-catalyzed C-N cross-coupling. Nat. Commun 2022, 13, 2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.(a) Kiriy A; Senkovskyy V; Sommer M Kumada Catalyst-Transfer Polycondensation: Mechanism, Opportunities, and Challenges. Macromol. Rapid Commun 2011, 32, 1503–1517. [DOI] [PubMed] [Google Scholar]; (b) Bryan ZJ; McNeil AJ Conjugated polymer synthesis via catalyst-transfer polycondensation (CTP): mechanism, scope, and applications. Macromolecules 2013, 46, 8395–8405. [Google Scholar]; (c) Bryan ZJ; McNeil AJ Evidence for a Preferential Intramolecular Oxidative Addition in Ni-Catalyzed Cross-Coupling Reactions and their Impact on Chain-Growth Polymerizations. Chem. Sci 2013, 4, 1620–1624. [Google Scholar]