

Abstract
A modular synthetic enables two or four shielding arms to be appended strategically over the fluorochromes of near-infrared cyanine heptamethine dyes to create hydrophilic analogs of clinically approved Indocyanine Green. A key synthetic step is facile substitution of a heptamethine 4’-Cl atom by a phenol bearing two triethylene glycol chains. The lead compound is a heptamethine dye with four shielding arms, and a series of comparative spectroscopy studies showed that the shielding arms, (a) increased dye photostability and chemical stability, and (b) inhibited dye self-aggregation and association with albumin protein. In mice, the dye cleared from the blood primarily through the renal pathway, rather than the biliary pathway for ICG. This change in biodistribution reflects the much smaller hydrodynamic diameter of the shielded hydrophilic ICG analog compared to the 67 kDa size of the ICG/albumin complex. An attractive feature of the versatile synthetic chemistry is the capability to systematically alter the dye’s hydrodynamic diameter. The sterically shielded hydrophilic ICG dye platform is well-suited for immediate incorporation into Dynamic Contrast Enhanced (DCE) spectroscopy or imaging protocols using the same cameras and detectors that have been optimized for ICG.
Graphical Abstract
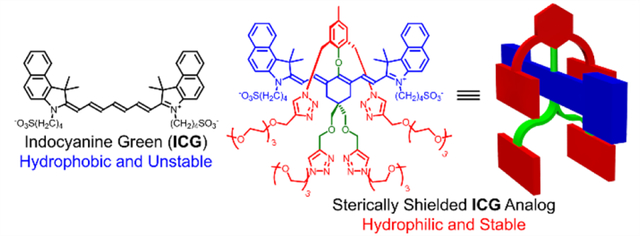
INTRODUCTION
Indocyanine green (ICG, Scheme 1) is a near-infrared (NIR) fluorescent heptamethine cyanine dye that has been used clinically for more than sixty years in an increasing number of biomedical diagnostic and imaging applications.1 In blood, ICG absorbs at ~800 nm and fluoresces at ~820 nm which makes it an attractive optical contrast agent due to minimal background absorption and autofluorescence by endogenous biomolecules. After injection into the bloodstream, ICG binds rapidly and strongly to blood proteins, (albumin, globulins, α-lipoproteins), and thus remains confined to the vascular system until blood passage through the liver, where the ICG is efficiently translocated, without chemical modification, into the bile and subsequently into the intestines. Roughly speaking, the wide range of clinical methods using ICG can be classified into two groups, near-infrared spectroscopy (NIRS), an absorption-based technique,2,3 or near-infrared fluorescence (NIRF).4 The expanding availability of detectors and cameras is promoting new developments in Dynamic Contrast Enhanced (DCE) techniques, which track time-dependent changes in the ICG signal to generate valuable pharmacokinetic information for a specific patient. For example, recent studies have used ICG for DCE spectroscopy or imaging to assess colon perfusion,5,6 bone perfusion,7,8 cerebral perfusion,9–13 and vascular permeability.14–18 Since ICG binds strongly to albumin, its effective diameter as a dynamic contrast agent is the size of the 67 kDa protein. There is emerging evidence that important additional pharmacokinetic insight can be gained by changing the size of the NIR contrast agent to interrogate different dynamic regimes.9,10,18–23
Scheme 1.
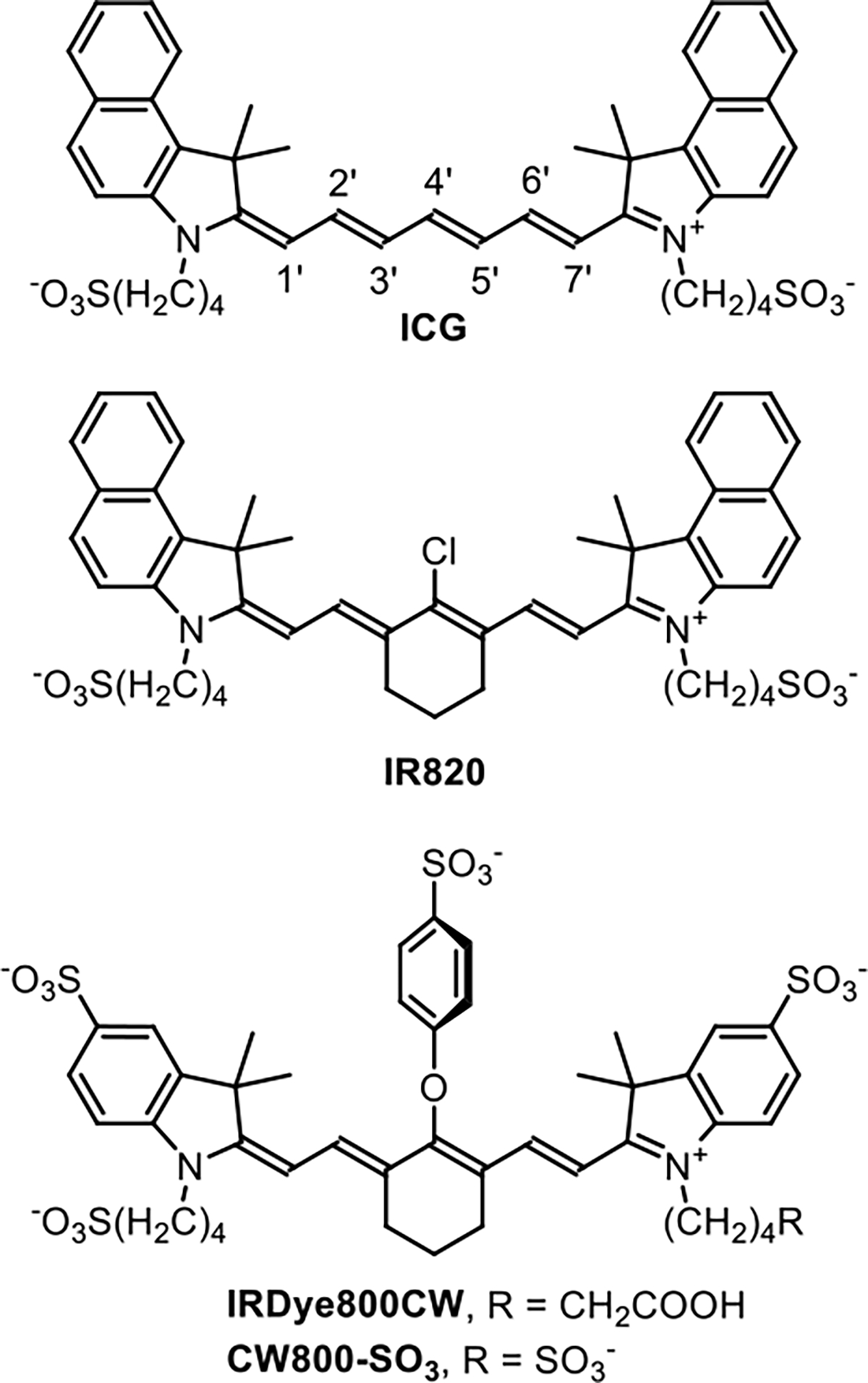
Chemical Structures of ICG, IR820, IRDye800CW, and CW800-SO3
Pursuit of this appealing multiscale DCE imaging paradigm requires a new set of small, hydrophilic NIR dyes that do not associate with blood proteins and can be detected using the same cameras and detectors that have been optimized for ICG. The inherent hydrophobicity of ICG makes it a synthetic challenge to create hydrophilic analogs that do not self-aggregate or associate with biological surfaces.24,25 At present, the most common rational design strategy is to create highly charged NIR heptamethine cyanine dyes, usually with multiple sulfonate groups.26,27 A common synthetic method starts with a commercial dye, such as IR820 (Scheme 1), whose reactive 4’-Cl atom can be readily substituted by O, N, or S nucleophiles.28 One hydrophilic NIR fluorescent dye that has been used for DCE experiments is IRDye800CW, a commercial dye with multiple sulfonates and a 4’-phenoxy group.10,18 IRDye800CW is reported to not produce acute toxicity,29 but a chemical drawback with this family of heptamethine cyanine dyes is susceptibility to covalent substitution of the 4’-phenoxy group by biological nucleophiles to give undesired dye degradation products that may diminish DCE imaging performance.30–32 A related concern is the polyanionic charge of dyes like IRDye800CW which can produce altered biodistribution patterns due to electrostatic interactions with blood components or biological surfaces.21,33–35
As part of an ongoing effort to improve the properties of NIR fluorescent dyes for biomedical imaging, we recently prepared a heptamethine cyanine dye with a C-C bonded 4’-aryl ring that projected two shielding arms over both faces of the dye.36–38 The success of this steric protection concept motivated us to expand the scope of shielded dye structures and the synthetic methodology to create them. Here we describe a new class of heptamethine dyes that have near-identical electrostatic and spectral profiles as ICG but they possess different numbers of shielding arms that sterically protect the chromophore. More specifically, we have conducted a systematic comparison of new dyes 1, 2 and 3, each with a 4’-phenoxy group projecting zero, two, or four shielding arms, respectively (Scheme 2). We find that the protecting arms block undesired dye self-aggregation or dye association with albumin protein and thus the new dyes have much smaller hydrodynamic diameters in blood. In addition, the protecting arms greatly enhance chemical and photochemical stability by inhibiting undesired intermolecular degradation reactions.
Scheme 2.
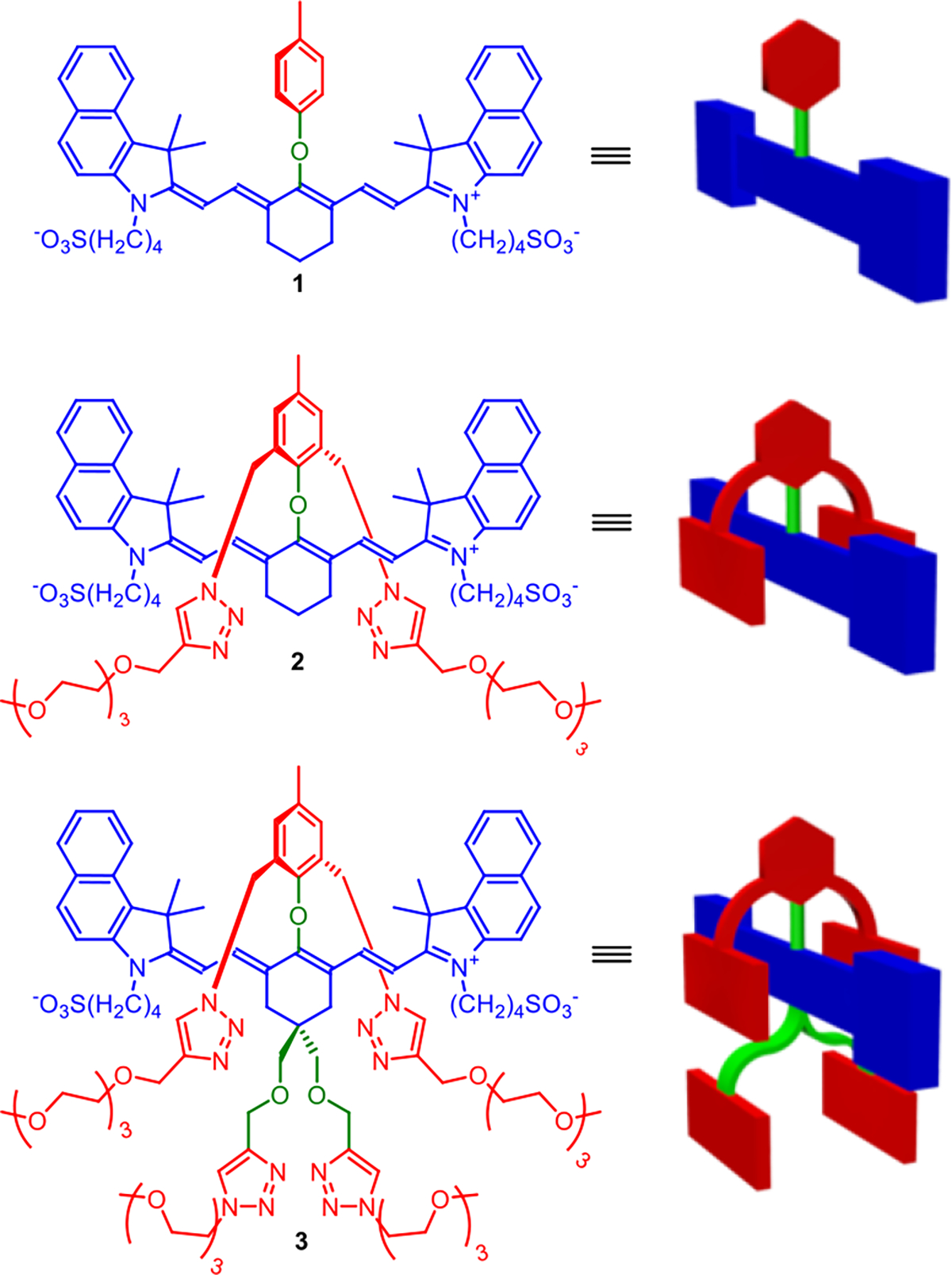
Chemical Structures and Cartoon Representations of ICG Analogs in This Study
RESULTS AND DISCUSSION
Synthesis.
The three dyes (1, 2 and 3) were prepared in modular fashion using synthetic pathways in Scheme 3. The first key building block, phenol 6, was prepared in two straightforward steps from starting material 4. A second important building block, the Vilsmeier reagent 10, was prepared in three steps from the known intermediate 7 which in turn was prepared in four literature steps.39 Reaction of 10 with two equivalents of benzoindole 12 afforded the 4’-Cl bisalkyne heptamethine 13 in 73% yield. The 4’-Cl atom in 13 was smoothly substituted by phenol 6 under mild conditions to give bisalkyne heptamethine 14 in 93% yield which was subsequently converted into dye 3 with four shielding arms. The 4’-Cl atom on commercial IR820 was substituted with phenol 6 or p-cresol to give dye 2 with two shielding arms or unshielded control dye 1. Interestingly, the 4’-Cl of IR820 could not be displaced by the less sterically hindered p-cresol under the same mild conditions used for shielded 6. Instead, forcing conditions (higher temperature, a large excess of p-cresol and strong base) were needed to react IR820 with p-cresol and generate dye 1. There is evidence that the 4’-Cl substitution reaction in room temperature, organic solvent proceeds by an SRN1 mechanism with radical intermediates;40 thus, we tentatively attribute the enhanced reactivity of phenol 6 with IR820 to through space stabilization of radical structures by the two triazole rings on 6. The expediency of the high yielding, single-step, substitution reaction using phenol 6 compares favorably with the three step process developed by Schnermann and coworkers that installed an 4’-alkoxy substituent to create a stabilized heptamethine cyanine dye.30
Scheme 3.
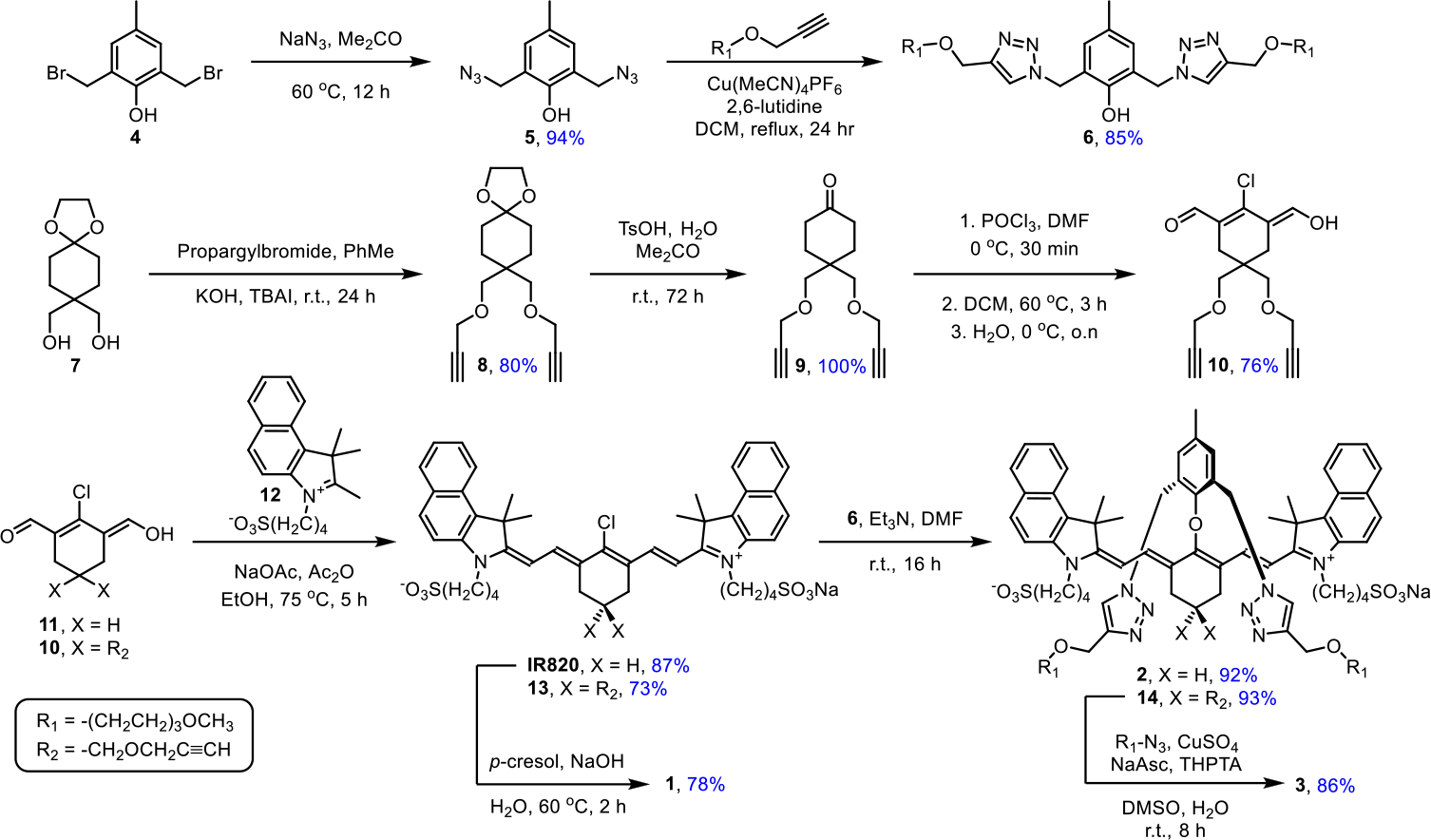
Synthesis of ICG Analogs
Photophysical Properties.
The spectral properties of the three shielded dyes (1, 2 and 3) and two control heptamethine cyanine dyes (IR820 and CW800-SO3) are listed in Table 1 (additional data in Table S1 and Figures S7 and S17). Shown in Figure 1a are absorption spectra of dyes 1, 2 and 3 in PBS at the same concentration with additional spectra at multiple concentrations in PBS and DMSO in Figure S2–S6. Inspection of the spectra reveals the following trends for the dyes in PBS: the absorption spectrum for unshielded dye 1 exhibits a major non-fluorescent H-aggregate band at 680 nm and a small monomer band at 805 nm; most of dye 2 with two shielding arms is in a monomeric state, but there is a minor H-aggregate absorption peak at 738 nm; dye 3 with four appended shielding arms exists completely in the monomeric state. A computational molecular model of 3 (Figure 2) shows that the plane of the 4’-phenoxy ring is essentially orthogonal to the plane of the heptamethine chain and strong experimental support for this conformation is provided by the 1H NMR spectra for the different dyes (Figure S1).
Table 1.
Photophysical properties at room temperature
| Dye | Solventa | (nm) | (nm) | ε (M−1cm−1) | ΦF (%)b | Stable to thiol |
|---|---|---|---|---|---|---|
|
| ||||||
| ICG | PBS | 780 | 805 | 64,180 | 2.9 | Yes |
| FBS | 800 | 819 | 159,000 | 9.7 | ||
| DMSO | 794 | 825 | 159,000 | 19.0 | ||
| CW800-SO 3 | PBS | 774 | 791 | 190,870 | 10.5 | No |
| 1 | PBS | 805 | 813 | 19,790 | 1.0 | No |
| DMSO | 818 | 840 | 160,310 | 15.4 | ||
| 2 | PBS | 809 | 827 | 172,980 | 2.3 | Yes |
| DMSO | 817 | 841 | 214,800 | 11.9 | ||
| 3 | PBS | 803 | 823 | 180,540 | 2.3 | Yes |
| FBS | 804 | 826 | 179,000 | - | ||
| DMSO | 809 | 837 | 191,320 | 10.8 | ||
PBS = pH 7.4 1X phosphate buffered saline; FBS = fetal bovine serum; DMSO = dimethyl sulfoxide.
Absolute fluorescence quantum yield measured by photon counting.
Figure 1.
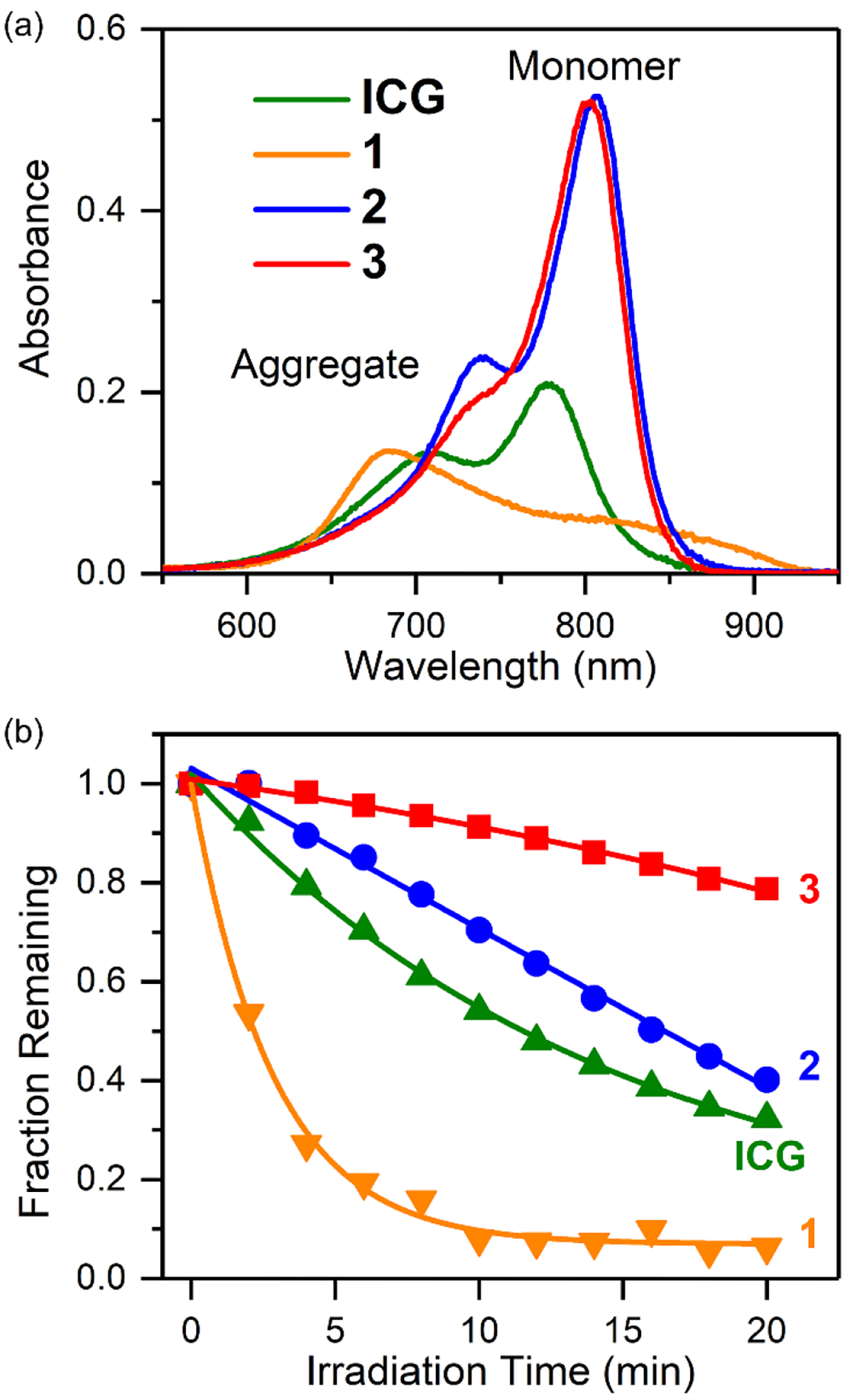
(a) Absorption spectra (3 μM in PBS, pH 7.4). (b) Photobleaching of ICG and its analogs (2 μM in pH 7.4 PBS irradiated by a 0.5 mW/cm2 Xenon lamp with a 620 nm long-pass filter).
Figure 2.
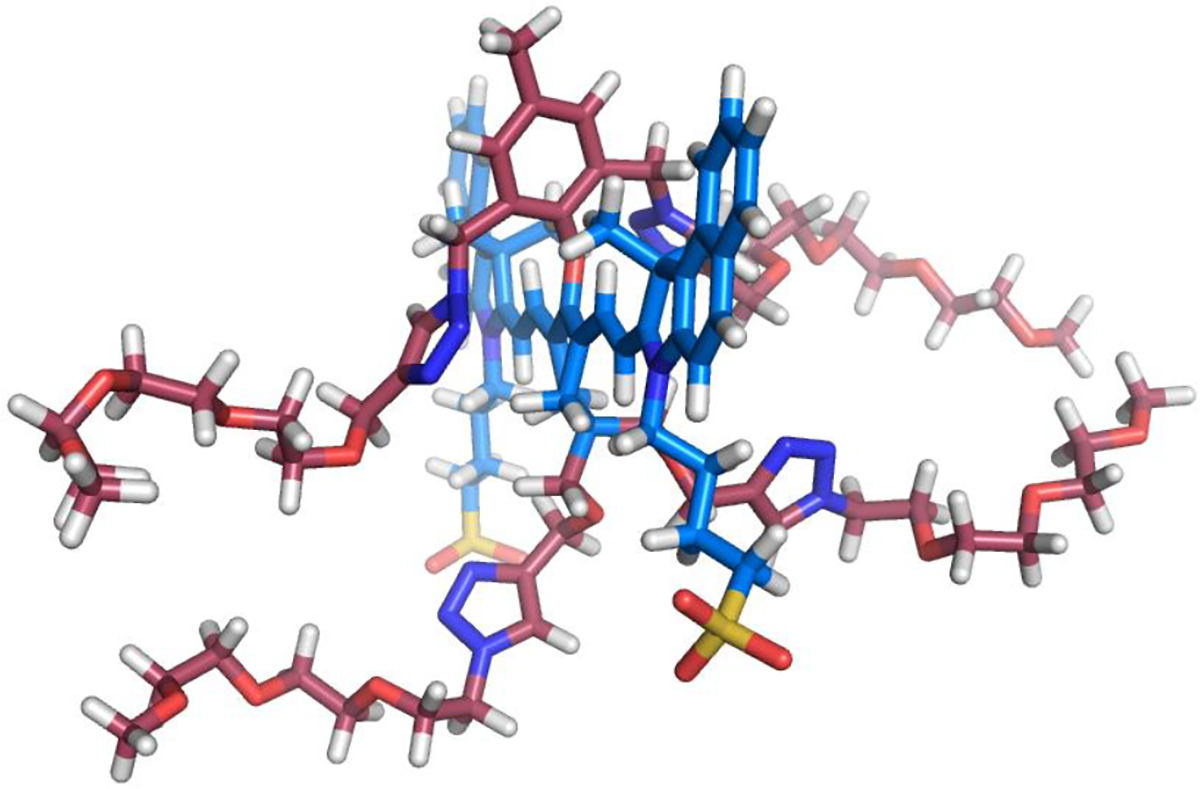
Energy-minimized molecular model of shielded dye 3, red bonds for shielding arms, blue bonds for heptamethine fluorochrome; H: white atoms; O: red atoms; N: deep blue atoms; S: yellow atoms.
A property that is related to dye-self aggregation in water is dye association with albumin protein in water. The expectation that the shielding arms of 3 would reduce affinity for albumin was confirmed by measuring the association with bovine serum albumin (BSA). Standard titration experiments measured quenching of the protein’s tryptophan fluorescence caused by incremental addition of dye.26 The titration data in Figure S18 produced Ka = 7.0 × 105 M−1 at 37 °C for ICG which matches the literature,26 and an 11-fold lower value of 6.0 × 104 M−1 for shielded dye 3 under the same condition. Thus, the four shielding arms in dye 3 are quite effective at blocking dye self-aggregation and association with albumin protein and by extension other blood proteins and related biological surfaces.
Photochemical stability is highly desired for NIR spectroscopy and fluorescence imaging but electron-rich polymethine fluorochromes react readily with photogenerated singlet oxygen.41 The relative photostability of dyes 1, 2 and 3 was assessed with a set of comparative photobleaching experiments that monitored the decrease in the dye absorption maxima signal (Figures S8–S11). The observed photostability trend of 3 > 2 > 1 (Figure 1b, Table S2) correlates with the number of shielding arms which suggests that the shielding arms sterically hinder the bimolecular reaction of photogenerated singlet oxygen with the polymethine fluorochrome. A second major reactivity question with dyes 1, 2 and 3 was susceptibility of the 4’-phenoxy group to covalent substitution by biological nucleophiles such as glutathione (GSH). Substitution of the 4’-phenoxy group by GSH is easily tracked by absorption spectroscopy since the 4’-thiol substitution product exhibits a characteristic red-shifted absorbance band.28,40,42 Shown in Figure S12–S15 are the changes in absorbance spectra for four separate dye solutions in the presence of 1 mM GSH (pH 7.4 PBS, room temperature).43 The spectra for unshielded dye 1 and control dye CW800-SO3 revealed time-dependent appearance of red-shifted absorption bands caused by GSH thiol substitution of the 4’-phenoxy group. In the case of CW800-SO3 this conclusion was confirmed by isolating and characterizing the GSH substitution product (Scheme 4) by 1H NMR, mass spectrometry, absorption, and fluorescence spectroscopy (Figure S16). The absorption spectra for shielded dyes 2 and 3 showed no change in dye absorbance after 10 hours (Figure S12 and S13), strongly suggesting no reaction with the GSH. The results indicate that the shielding arms in dyes 2 and 3 sterically inhibit nucleophilic attack of the thiol group of GSH at the 4’-position of the dye. Combined, the broadly reduced reactivity of shielded dyes 2 and 3 with electrophiles and nucleophiles is a compelling chemical demonstration of the significant steric protection provided by the shielding arms.
Scheme 4.
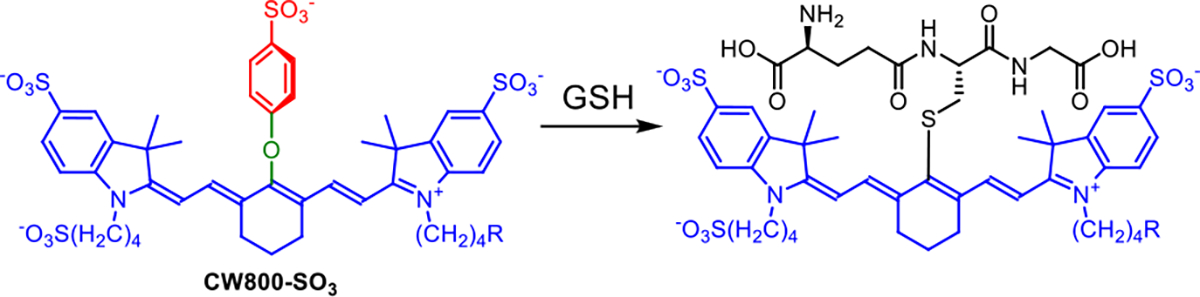
Substitution of the 4’-phenoxy group of CW800-SO3 by GSH
Mouse Biodistribution.
The very similar absorption/emission wavelengths of shielded dye 3 and ICG in fetal bovine serum (FBS) (Figure S7 and S17) prompted a preliminary set of biological studies that measured cytotoxicity and compared in vivo biodistribution profiles. Standard MTT assays determined the metabolic activity of four different cell-lines in the presence and absence of added dye and detected no evidence for cytotoxicity (Figure S19). A preliminary comparison of dye biodistribution in living mice used a protocol that was approved by the Institutional Animal Care and Use Committee. Healthy female BALB/c mice (N = 4) were injected (retro-orbital) with either dye 3 or ICG (100 μL, 10 nmol/mouse) and sacrificed 2 hours later. The major organs were harvested and NIR fluorescence images were acquired using a commercial in vivo imaging station(Figure 3a). Standard region-of-interest analysis of the ex vivo NIR fluorescence images provided quantitative bar graph plots of normalized mean pixel intensity for each organ and tissue sample (Figure 3b). As expected, there was extensive accumulation of ICG in the intestines consistent with its biliary clearance pathway. In contrast, shielded dye 3 (MW = 1.9 kDa) cleared primarily through the kidneys as indicated by the green color of the mouse urine, with some concurrent clearance through the biliary pathway. As indicated by the images in Figure S20, the amount of ICG in the intestines was more than three times dye 3. The large difference in the mouse biodistribution of dye 3 and ICG shows that the shielding arms in dye 3 greatly alter the pharmacokinetics and clearance pathways.25 The biological data supports the feasibility of this molecular probe platform for future next-generation NIR fluorescence DCE imaging and spectroscopy paradigms.21–23
Figure 3.
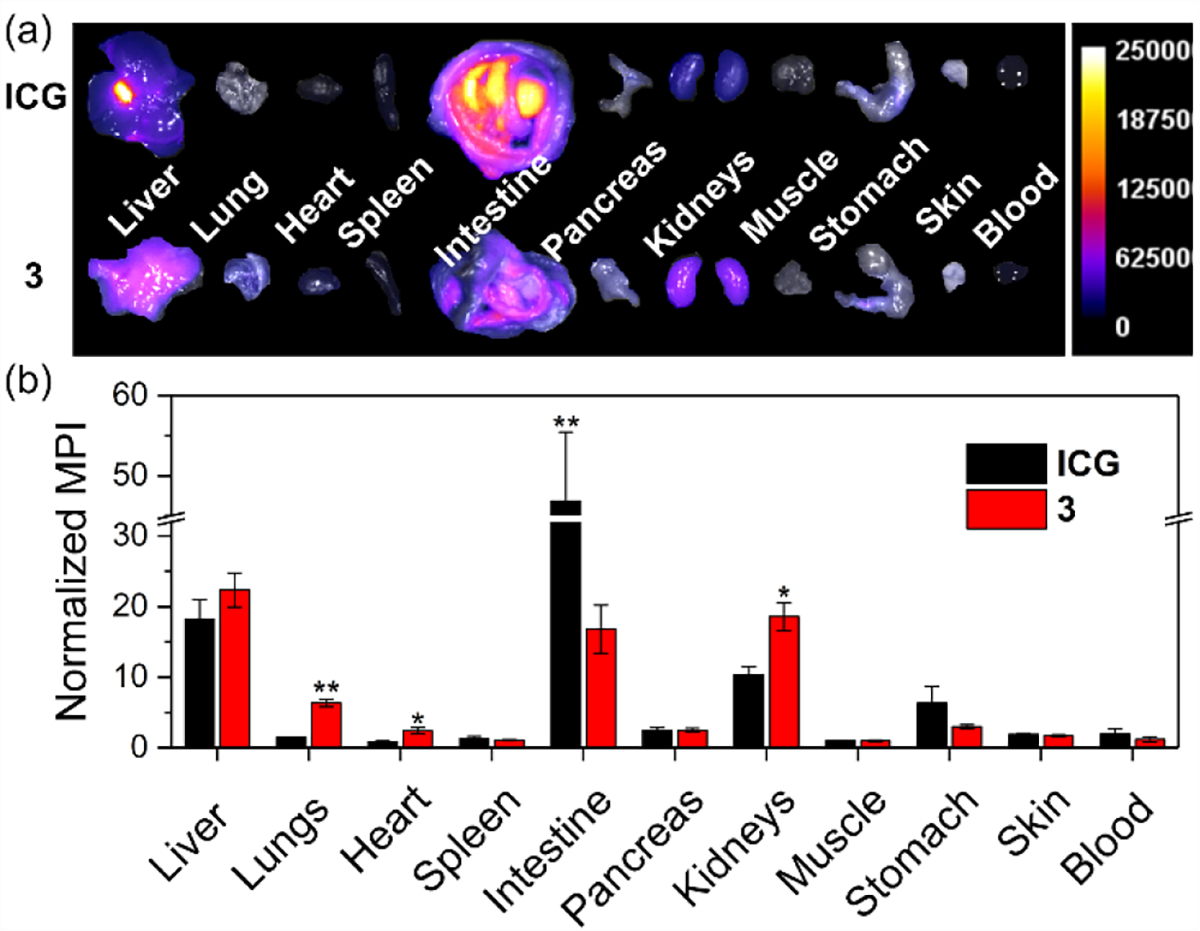
(a) Representative overlaid brightfield and fluorescence ex vivo images of major organs of BALB/c mice sacrificed 2 h after retro-orbital injection of either ICG (top) or 3 (bottom). Scale is in arbitrary fluorescence units. (b) Biodistribution of ICG and 3 in BALB/c mice. Note that this data does not include the large fraction of probe 3 excreted as urine. *p < 0.1, **p < 0.05, otherwise no statistically significant difference.
CONCLUSION
A modular synthetic process enables attachment of two or four shielding arms strategically over the fluorochromes of near-infrared cyanine heptamethine dyes that are electrostatic and spectral analogs of clinically approved ICG. The key synthetic step is a facile substitution reaction that reacts 4’-Cl heptamethine precursors with phenol 6 to give sterically shielded dyes in good yield under mild conditions. A tangible outcome of this work is NIR dye 3 with four shielding arms that greatly enhance chemical and photochemical stability by inhibiting undesired intermolecular reactions with electrophiles (singlet oxygen) or nucleophiles (glutathione). In addition, the shielding arms inhibit dye self-aggregation or dye association with albumin protein which means dye 3 has a much smaller hydrodynamic diameter in blood than the 67 kDa complex of ICG and albumin. Importantly, dye 3 and ICG have essentially the same absorption and fluorescence maxima wavelengths in serum (Table 1, Figures S7 and S17). Thus, dye 3 can be detected using the same cameras and detectors that have been optimized for ICG, and it has great promise as a relatively small, hydrophilic ICG analog for immediate incorporation within Dynamic Contrast Enhanced (DCE) spectroscopy or fluorescence imaging protocols that generate helpful hemodynamic information for a specific patient. Furthermore, the modular synthetic process can be exploited to systematically alter the dye hydrodynamic diameter by changing the number of ethylene glycol units within the appended shielding arms.43,44 Another possibility is to decorate the dye structure with chelators that can strongly bind appropriate metal cations for nuclear imaging or magnetic resonance imaging. Specifically, it should be possible to convert NIR dye 14 into a range of dual modality DCE imaging and spectroscopy agents for characterizing hemodynamics.21 A separate future goal is to develop structural analogs of these shielded NIR dyes with reactive sites for bioconjugation and creation of functional NIR constructs that have very high optical stability.
EXPERIMENTAL SECTION
General.
Column chromatography was performed using Biotage SNAP or Sfär columns. 1H and 13C{1H} NMR spectra were recorded on a Bruker 500 NMR spectrometer. Chemical shifts are presented in ppm and referenced by residual solvent peak. High-resolution mass spectrometry (HRMS) was performed using a time-of-flight (TOF) analyzer with electrospray ionization (ESI). Absorption spectra were recorded on an Evolution 201 UV/vis spectrometer with Thermo Insight software. Fluorescence spectra were collected on a Horiba Fluoromax Plus fluorometer with InGaAs detector and FluoroEssence software. All absorption and fluorescence spectra were collected using quartz cuvettes (1 mL, 1 cm path length).
Synthesis and Characterization.
2,6-Bisazidomethyl-p-cresol 5.
A mixture of 2,6-bisbromomethyl-p-cresol 446 (500 mg, 1.70 mmol, 1 eq) and sodium azide (553 mg, 8.50 mmol, 5 eq) in acetone (10 mL) was stirred under reflux overnight. The reaction was diluted with diethyl ether (30 mL) and filtered through a celite pad. Solvent was removed under reduced pressure and the residue was purified by flash chromatography (SiO2, 0 – 20% DCM in hexanes) to afford the product as a yellow oil (350 mg, 94%, Rf = 0.7 in hexane/ethyl acetate = 4/1) which was stored at – 20 °C. 1H NMR (500 MHz, CDCl3, 25 °C) δ (ppm): 6.99 (s, 2H), 6.15 (s, 1H), 4.42 (s, 4H), 2.29 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3, 25 °C) δ (ppm): 151.4, 130.9, 130.2, 122.5, 51.7, 20.6. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C9H10N6NaO+ 241.0808, found 241.0800.
Compound 6.
A mixture of 5 (500 mg, 2.29 mmol, 1 eq), triethylene glocol monomethyl monopropargyl ether (1.39 g, 6.87 mmol, 3 eq), Cu(MeCN)4PF6 (42.7 mg, 115 μmol, 0.05 eq) and one drop of 2,6-lutidine in DCM (20 mL) was refluxed under argon atmosphere for 24 h. Slovent was removed and the residue was purified by column chromatography (SiO2, 0–5% MeOH in DCM) to afford the product as a yellow oil (1.21 g, 85%, Rf = 0.4 in DCM/MeOH = 20/1). 1H NMR (500 MHz, DMSO-d6, 25 °C) δ (ppm): 9.28 (s, 1H), 7.98 (s, 2H), 6.84 (s, 2H), 5.53 (s, 4H), 4.49 (s, 4H), 3.54 – 3.36 (m, 24H), 3.20 (s, 6H), 2.12 (s, 3H). 13C{1H} NMR (126 MHz, methanol-d4, 25 °C) δ (ppm): 150.9, 131.3, 130.7, 124.0, 71.7, 70.3, 70.1, 69.6, 63.8, 57.9, 49.2, 19.3. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C29H47N6O9+ 623.3399, found 623.3403.
Compound 8.
A mixture of 739 (3.00 g, 14.8 mmol, 1 eq), propargyl bromide (80 wt% in toluene, 9.88 mL, 89.0 mmol, 6 eq), powdered potassium hydroxide (3.33 g, 59.3 mmol, 4 eq) and tetrabutylammonium iodide (548 mg, 1.48 mmol, 0.1 eq) was stirred vigorously at room temperature for 24 h. The mixture was diluted by DCM (20 mL) and filtered through a celite pad. The filtrate was evaporated and the residue was purified by column chromatography (SiO2, 5 – 15% EA in Hexane) to afford 8 as a yellow oil (3.3 g, 80%, Rf = 0.6 in Hexane/ethyl acetate = 4/1). 1H NMR (500 MHz, CDCl3, 25 °C) δ (ppm): 4.13 (d, J = 2.3 Hz, 4H), 3.94 (s, 4H), 3.41 (s, 4H), 2.40 (t, J = 2.3 Hz, 2H), 1.66 – 1.61 (m, 4H), 1.60 – 1.55 (m, 4H). 13C{1H} NMR (126 MHz, CDCl3, 25 °C) δ (ppm): 109.1, 80.4, 74.3, 72.5, 64.3, 58.7, 37.6, 30.5, 27.6. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C16H23O4+ 279.1591, found 279.1570.
Compound 9.
A mixture of 8 (1.00 g, 359 μmol, 1 eq) and TsOH (247 mg, 144 μmol, 0.4 eq) in acetone/water (4/1, 20 mL) was stirred at room temperature for 72 h (the reaction progress was monitored by TLC). The mixture was poured onto saturated sodium hydrogen carbonate solution (50 mL) and extracted with DCM (3 × 50 mL). The organic extracts were dried over anhydrous sodium sulfate, filtered, and then concentrated to give 9 (840 mg, 100%, Rf = 0.4 in Hexane/ethyl acetate = 4/1) as a yellow oil. 1H NMR (500 MHz, CDCl3, 25 °C) δ (ppm): 4.17 (d, J = 2.4 Hz, 4H), 3.51 (s, 4H), 2.43 (t, J = 2.3 Hz, 2H), 2.37 (t, J = 7.0 Hz, 4H), 1.81 (t, J = 7.0 Hz, 4H). 13C{1H} NMR (126 MHz, CDCl3, 25 °C) δ (ppm): 212.7, 80.0, 74.7, 72.6, 58.8, 37.8, 37.1, 29.1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H19O3+ 235.1329, found 235.1322.
Compound 10.
A 100 mL round bottom flask contains DMF (0.397 mL, 5.12 mmol, 4 eq) and DCM (2 mL) was chilled in an ice bath. POCl3 (0.359 mL, 3.84 mmol, 3 eq) was added dropwise. The mixture was stirred vigorously at room temperature for 30 min to afford Vilsmeier intermediate. Compound 9 (300 mg, 1.28 mmol, 1 eq) in DCM (2 mL) was added to the Vilsmeier intermediate. The mixture was stirred at 60 °C for 3 h. DCM was evaporated under reduced pressure then ice-water mixture (~ 50 mL) was added. The mixture was sonicated to suspend the orange oil and kept in fridge (~4 °C) overnight. During this time yellow crystalline needles formed. The solid was collected by filtration, washed with cold water (10 mL) and vacuum dried for 1 h at room temperature to give 10 (300 mg, 76%) which was stored at – 20 °C with hexane. 1H NMR (500 MHz, CDCl3, 25 °C) δ (ppm): 10.24 (s, 1H), 7.56 (br s, 1H), 5.34 (br s, 1 H), 4.11 (d, J = 2.4 Hz, 4H), 3.36 (s, 4H), 2.43 (s, 4H), 2.40 (t, J = 2.4 Hz, 2H). 13C{1H} NMR and MS spectra were not collected due to the poor stability of 10.
Compound 13.
A mixture of 10 (300 mg, 0.972 mmol, 1 eq), 1247 (739 mg, 2.14 mmol, 2.2 eq) and sodium acetate (199 mg, 2.43 mmol, 2.5 eq) in acetic anhydride (2 mL) and ethanol (10 mL) was stirred at 75 °C under argon atmosphere for 5 h. After cooling to room temperature, diethyl ether (100 mL) was added. The solid was collected by filtration and further purified by reverse phase column chromatography (C18, 50 – 75% methanol in water) to afford 13 as a green solid (700 mg, 73%). 1H NMR (500 MHz, methanol-d4, 25 °C) δ (ppm): 8.60 (d, J = 14.2 Hz, 2H), 8.27 (d, J = 8.5 Hz, 2H), 8.03 (d, J = 8.8 Hz, 2H), 8.00 (d, J = 8.5z Hz, 2H), 7.69 (d, J = 8.8 Hz, 2H), 7.65 (dd, J = 8.5, 8.5 Hz, 2H), 7.50 (dd, J = 8.5, 8.5 Hz, 2H), 6.37 (d, J = 14.2 Hz, 2H), 4.36 (t, J = 7.5 Hz, 4H), 4.20 (d, J = 2.4 Hz, 4H), 3.55 (s, 4H), 2.92 (t, J = 7.3 Hz, 4H), 2.87 (t, J = 2.4 Hz, 2H), 2.74 (s, 4H), 2.09 (m, 4H), 2.04 (s, 12H), 1.99 (m, 4H). 13C{1H} NMR (126 MHz, methanol-d4, 25 °C) δ (ppm): 174.4, 174.4, 144.3, 144.2, 144.2, 139.9, 134.1, 132.3, 130.7, 129.9, 128.2, 127.6, 125.0, 122.3, 111.2, 79.8, 75.0, 72.4, 58.4, 51.3, 50.7, 44.1, 37.6, 30.0, 26.7, 26.5, 22.4. HRMS (ESI-TOF) m/z: [M]− calcd for C54H58ClN2O8S2− 961.3329, found 961.3303.
Compound 14.
A solution of the 13 (100 mg, 102 μmol, 1 eq), 6 (75.8 mg, 122 μmol, 1.2 eq) and triethylamine (20.8 μL, 152 μmol, 1.5 eq) in anhydrous DMF (5 mL) was stirred at room temperature under argon atmosphere in the dark for 16 h. After reaction completion, the mixture was directly purified by reverse phase column chromatography (C18, 40 – 55% methanol in water) to afford the product as a green solid (148 mg, 93%). 1H NMR (500 MHz, methanol-d4, 25 °C) δ (ppm): 8.15 (d, J = 8.6 Hz, 2H), 8.05 (d, J = 14.1 Hz, 2H), 8.04 (s, 2H), 7.97 (m, 4H), 7.62 (m, 4H), 7.46 (dd, J = 8.6, 8.6 Hz, 2H), 7.32 (s, 2H), 6.18 (d, J = 14.1 Hz, 2H), 5.79 (s, 4H), 4.60 (s, 4H), 4.27 (t, J = 7.5 Hz, 4H), 4.21 (d, J = 2.3 Hz, 4H), 3.62 – 3.40 (m, 28H), 3.28 (s, 6H), 2.92 – 2.86 (m, 6H), 2.72 (s, 4H), 2.34 (s, 3H), 2.03 (tt, J = 7.5, 7.5 Hz, 4H), 1.93 (tt, J = 7.5, 7.5 Hz, 4H), 1.60 (s, 12H). 13C{1H} NMR (126 MHz, methanol-d4, 25 °C) δ (ppm): 173.8, 163.5, 152.5, 144.9, 141.1, 139.9, 134.9, 134.0, 133.6, 132.2, 130.6, 129.9, 128.2, 127.5, 124.9, 124.8, 124.4, 122.2, 116.9, 111.1, 99.8, 79.8, 75.0, 72.7, 71.6, 70.2, 70.1, 70.1, 70.0, 69.7, 63.9, 58.4, 57.9, 50.9, 50.7, 49.0, 43.9, 37.9, 28.6, 26.9, 26.3, 22.4, 19.4. HRMS (ESI-TOF) m/z: [M]− calcd for C83H103N8O17S2− 1547.6888, found 1547.6851.
Compound 3.
Solutions of copper(II) sulfate (0.5 M in water, 7.63 μL, 3.82 μmol, 0.2 eq), sodium ascorbate (0.5 M in water, 11.5 μL, 5.73 μmol, 0.3 eq) and tris(3-hydroxypropyltriazolylmethyl)amine (0.05 M in water, 76.3 μL, 3.82 μmol, 0.2 eq) were mixed together. The mixture was added to a solution of 14 (30 mg, 19.1 μmol, 1 eq) and 1-azido-2-(2-(2-methoxyethoxy)ethoxy)ethane (14.4 mg, 76.3 μmol, 4 eq) in DMSO (2 mL). The reaction was stirred at room temperature under argon atmosphere in the dark for 8 h. After reaction completion, the mixture was directly purified by column chromatography (C18, 40 – 60% methanol in water) to afford 3 as a green solid (32 mg, 86%). 1H NMR (500 MHz, methanol-d4, 25 °C) δ (ppm): 8.15 (d, J = 8.6 Hz, 2H), 8.12 – 7.95 (m, 10H), 7.66 – 7.59 (m, 4H), 7.47 (dd, J = 8.5, 8.5 Hz, 2H), 7.30 (s, 2H), 6.15 (d, J = 14.1 Hz, 2H), 5.78 (s, 4H), 4.62 (s, 4H), 4.60 – 4.55 (m, 8H), 4.25 (t, J = 7.4 Hz, 4H), 3.86 (t, J = 5.1 Hz, 4H), 3.63 – 3.38 (m, 44H), 3.29 (s, 6H), 3.28 (s, 6H), 2.87 (t, J = 7.5 Hz, 4H), 2.69 (s, 4H), 2.33 (s, 3H), 1.99 (tt, J = 7.5, 7.5 Hz, 4H), 1.92 (tt, J = 7.5, 7.5 Hz, 4H), 1.60 (s, 12H). 13C{1H} NMR (126 MHz, methanol-d4, 25 °C) δ (ppm): 173.8, 163.5, 152.5, 144.9, 144.7, 141.0, 139.9, 134.8, 134.0, 133.6, 132.2, 130.6, 129.9, 128.2, 127.5, 124.9, 124.8, 124.8, 124.4, 122.3, 117.1, 111.2, 99.8, 73.0, 71.7, 71.6, 70.2, 70.1, 70.1, 70.1, 70.0, 69.7, 69.2, 64.2, 63.9, 58.1, 57.9, 57.9, 50.9, 50.7, 50.2, 48.9, 43.9, 38.2, 28.6, 26.9, 26.4, 22.4, 19.4. HRMS (ESI-TOF) m/z: [M]− calcd for C97H133N14O23S2− 1925.9115, found 1925.9105.
IR820.
A mixture of 1141 (145 mg, 0.835 mmol, 1 eq), 1247 (635 mg, 1.84 mmol, 2.2 eq) and sodium acetate (206 mg, 2.50 mmol, 3 eq) in ethanol (10 mL) was stirred at 75 °C under argon atmosphere in the dark for 3 h. The reaction was treated with diethyl ether (100 mL) and filtered. The solid was purified by reverse phase column chromatography (C18, 50 – 70% methanol in water) to afford IR820 as a green solid (620 mg, 87%). 1H NMR (500 MHz, DMSO-d6, 25 °C) δ (ppm): 8.36 (d, J = 14.2 Hz, 2H), 8.29 (d, J = 8.6 Hz, 2H), 8.08 (d, J = 8.6 Hz, 2H), 8.05 (d, J = 8.8 Hz, 2H), 7.81 (d, J = 8.8 Hz, 2H), 7.64 (dd, J = 8.8, 8.8 Hz, 2H), 7.50 (dd, J = 8.6, 8.6 Hz, 2H), 6.40 (d, J = 14.2 Hz, 2H), 4.34 (t, J = 7.7 Hz, 4H), 2.76 (br s, 4H), 2.51 (m, 4H), 1.94 (s, 12H), 1.86 (m, 5H), 1.76 (m, 4H).
Compound 2.
Synthesized from IR820 using the same procedure as 14. Green solid (100 mg of IR820 produced156 mg of 2, i.e., 92% yield). Column chromatography: C18, 40 – 55% methanol in water. 1H NMR (500 MHz, methanol-d4, 25 °C) δ (ppm): 8.16 (d, J = 8.6 Hz, 2H), 8.04 – 7.94 (m, 8H), 7.64 – 7.59 (m, 4H), 7.47 (dd, J = 8.6, 8.6 Hz, 2H), 7.34 (s, 2H), 6.17 (d, J = 14.1 Hz, 2H), 5.81 (s, 4H), 4.59 (s, 4H), 4.26 (t, J = 7.3 Hz, 4H), 3.61 – 3.34 (m, 24H), 3.28 (s, 6H), 2.89 (t, J = 7.3 Hz, 4H), 2.76 (t, J = 6.2 Hz, 4H), 2.35 (s, 3H), 2.09 – 1.89 (m, 10H), 1.60 (s, 12H). 13C{1H} NMR (126 MHz, methanol-d4, 25 °C) δ (ppm): 173.6, 164.4, 152.9, 144.9, 140.0, 139.9, 134.6, 134.0, 133.9, 132.2, 130.6, 129.9, 128.2, 127.5, 124.8, 124.4, 124.3, 122.2, 120.4, 111.1, 99.7, 71.7, 70.2, 70.1, 70.1, 70.0, 69.7, 64.0, 57.9, 50.9, 50.6, 49.1, 43.8, 26.8, 26.3, 24.6, 22.4, 21.0, 19.4. HRMS (ESI-TOF) m/z: [M]− calcd for C75H95N8O15S2− 1411.6364, found 1411.6373.
Compound 1.
Sodium hydroxide (23.5 mg, 589 μmol, 10 eq) and p-cresol (63.6 mg, 589 μmol, 10 eq) were dissolved in water (10 mL). IR820 (50.0 mg, 58.9 μmol, 1 eq) was added and the mixture was stirred at 60 °C under argon atmosphere in the dark. The reaction progress was monitored by absorption spectroscopy. After 2 h, IR820 was totally consumed and the reaction was completed. The mixture was extracted with ethyl acetate (3 × 20 mL), the aqueous phase was concentrated and purified by reverse phase column chromatography (C18, 50 – 70% methanol in water) to afford 1 as a green solid (42 mg, 78%). 1H NMR (500 MHz, methanol-d4, 25 °C) δ (ppm): 8.16 – 8.08 (m, 4H), 7.98 – 7.91 (m, 4H), 7.61 – 7.55 (m, 4H), 7.44 (dd, J = 8.3, 8.3 Hz, 2H), 7.24 (d, J = 8.3 Hz, 2H), 7.08 (d, J = 8.3 Hz, 2H), 6.21 (d, J = 14.3 Hz, 2H), 4.24 (t, J = 7.3 Hz, 4H), 2.89 (t, J = 7.2 Hz, 4H), 2.79 (t, J = 6.2 Hz, 4H), 2.28 (s, 3H), 2.06 (p, J = 6.2 Hz, 2H), 2.03 – 1.89 (m, 8H), 1.66 (s, 12H). 13C{1H} NMR (126 MHz, methanol-d4, 25 °C) δ (ppm): 173.6, 164.2, 158.3, 141.4, 139.9, 133.7, 132.1, 131.9, 130.6, 130.5, 129.9, 128.2, 127.4, 124.7, 122.4, 122.1, 114.6, 110.9, 99.4, 50.9, 50.6, 43.8, 26.6, 26.3, 24.1, 22.4, 21.4, 19.4. HRMS (ESI-TOF) m/z: [M]− calcd for C53H57N2O7S2− 897.3613, found 897.3618.
Fluorescence Quantum Yield Measurements.
Absolute quantum were measured on a Horiba Fluoromax Plus spectrometer with an integrating sphere. Samples were excited at 750 nm with optical density ≤ 0.05. First, all of the photons were recorded with an integrating sphere after the excitation of a blank solvent reference, then the reference was replaced by a sample solution, and the spectrum (740–900 nm) was acquired again. The fluorescence quantum yield (ΦF) was calculated in the FluoroEssence software using the equation below:
where P is the number of photons, F is the fluorescence intensity and E is the intensity at the excitation wavelength, λ is the wavelength. Experiments were conducted in triplicate, with the reported absolute quantum yields corresponding to the mean value ± standard deviation.
Photobleaching Studies.
A solution of dye (2 μM) in PBS buffer, pH 7.4, was placed in a 1 mL (1 cm path length) cuvette that was exposed to air and irradiated, at a distance of 5 cm, by a 150 W Xenon lamp with a 620 nm long-pass filter (0.5 mW/cm2). An absorbance spectrum was recorded every 2 min (Figure S8 – S11). The normalized maximum absorbance of dye was plotted against time and fitted to a non-linear regression, one-phase exponential decay (Table S2).
Chemical Stability Studies.
Monitoring thiol substitution by absorption.
A solution of dye (5 μM) and glutathione (1 mM) in pH 7.4 PBS buffer, pH 7.4, was placed in a 1 mL (1 cm path length) cuvette and capped at room temperature. Absorbance spectrum was recorded every 20 min (Figure S12 – S15).
Characterization of glutathione substitution product.
A solution of CW800-SO3 (20 μM) and GSH (2 mM) in pH 7.4 PBS (20 mL) was stirred at room temperature in the dark. The maxmium absorbance changed from 775 nm to 790 nm after 12 h, indicating reaction completion. The mixture was purified by reverse phase column chromatography (C18, 0 – 10 % MeOH in water) to afford the GSH substitution product of CW800-SO3, whose spectral data are shown in Figure S16.
Albumin Binding Measurements.
The titration experiments measured quenching of bovine serum albumin (BSA) tryptophan fluorescence as dye was added incrementally.26 A fluorescence spectrum (ex: 280 nm, slit width: 3 nm) was acquired after each dye aliquot was added (4 μL of a 1 mM stock solution of ICG or 3). After each aliquot addition, the solution was mixed and allowed to equilibrate for 5 min before spectral acquisition. The relative fluorescence intensity at 335 nm was determined using the following equation, where F0 is the initial fluorescence intensity, F is the fluorescence intensity after each aliquot addition of dye, and the slope of the trend line (m) corresponds to Ka ± SD (Figure S18).
Cell Viability Experiments.
CHO-K1 (Chinese hamster ovary), A549 (lung carcinoma epithelial), U-87 (human glioblastoma) and HT-1080 cells (human fibrosarcoma) were cultured in EMEM medium (supplemented with 10% fetal bovine serum, 0.1 mM non-essential amino acids, 1.5 g/L sodium bicarbonate, 1 mM sodium pyruvate, and 1% penicillin/streptomycin) at 37 °C and 5% CO2 in a humidified incubator. CHO-K1 (Chinese hamster ovary) and A549 (human lung adenocarcinoma) were cultured in F-12K medium (supplemented with 10% fetal bovine serum, and 1% penicillin/streptomycin) at 37 °C and 5% CO2 in a humidified incubator. Cells were then seeded into 96-microwell plates and grown to 80% confluency. The medium was then removed and replaced with either ICG or 3 at various micromolar concentrations (N=3) in their respective medium for 24 h at 37 °C and 5% CO2 in a humidified incubator. After 24 h, the dye was removed and replaced with growth medium containing [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (MTT,1.1mM). After a 4-hour incubation at 37 °C and 5% CO2, SDS-DMSO detergent solution was added to the MTT-growth medium. The samples were incubated overnight, and the absorbance of each well was measured at 590 nm. The readings were then normalized to untreated cells for each dye. All data are the average of triplicate measurements.
Mouse Biodistribution Experiments.
In-vivo experiments were conducted under the approved protocol by the Notre Dame Institutional Animal Care and Use Committee. Three months old healthy female BALB/c mice (N=4) were given a retro-orbital injection of either ICG or 3 (N=2, 100 μL, 10 nmol/mouse). Before the injections, fur covering the abdomen was removed, using a standard Nair procedure, to minimize background fluorescence by the skin. At 2 h, the mice were anesthetized and sacrificed by cervical dislocation followed by the immediate collection of blood from the heart. The skin covering the lower abdomen was removed to expose the abdominal cavity and mice were imaged using a commercial in-vivo imaging station (Ami HT Spectral Imaging). Imaging settings. - ex: 745/20 nm, em: 850/20 nm, exposure: 3 s, percent power: 50%, F-stop: 2, FOV:20, binning: low. For image processing, the mouse body images were pseudo-colored “fire” using ImageJ2 software. After obtaining abdominal cavity images, the major organs were harvested and imaged on a transparent plastic tray using the in-vivo imaging station. Imaging settings. - ex: 745/20 nm, em: 850/20 nm, exposure: 3 s, percent power: 50%, F-stop: 2, FOV:20, binning: low. For the biodistribution analysis of each excised organ, images were imported to ImageJ2, and a manually drawn region of interest was created around each organ. Mean Pixel Intensity (MPI) of each organ was divided by the MPI of the thigh muscle from the same mouse to give a normalized MPI for each dye to compare the fate of the dye post 2 h.
Molecular Modeling.
Molecular modeling employed the semi-empirical method within the MOPAC (2016) program.48 The dielectric constant of the solvent was set at 78.4 for water and 25 °C. The model was generated at the PM7 level.
Supplementary Material
ACKNOWLEDGMENT
The authors are grateful for funding support from the US NIH (R35GM136212).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Supplementary figures, experimental procedures, compound characterization data, copies of NMR and MS spectra, photophysical measurements, absorbance and fluorescence spectra, stability studies, toxicity data, in vivo and ex vivo imaging, computational modeling (PDF)
REFERENCES
- (1).Reinhart MB; Huntington CR; Blair LJ; Heniford BT; Augenstein VA Indocyanine Green :Historical Context, Current Applications, and Future Considerations. Surg. Innov. 2016, 23 (2), 166–175. [DOI] [PubMed] [Google Scholar]
- (2).Kuebler WM; Sckell A; Habler O; Kleen M; Kuhnle GEH; Welte M; Messmer K; Goetz AE Noninvasive Measurement of Regional Cerebral Blood Flow by Near-Infrared Spectroscopy and Indocyanine Green. J. Cereb. Blood Flow Metab. 1998, 18 (4), 445–456. [DOI] [PubMed] [Google Scholar]
- (3).Green MS; Sehgal S; Tariq R Near-Infrared Spectroscopy: The New Must Have Tool in the Intensive Care Unit? Semin. Cardiothorac. Vasc. Anesth. 2016, 20 (3), 213–224. [DOI] [PubMed] [Google Scholar]
- (4).Goncalves LN; van den Hoven P; van Schaik J; Leeuwenburgh L; Hendricks CHF; Verduijn PS; van der Bogt KEA; van Rijswijk CSP; Schepers A; Vahrmeijer AL; Hamming JF; van der Vorst JR Perfusion Parameters in Near-Infrared Fluorescence Imaging with Indocyanine Green: A Systematic Review of the Literature. Life 2021, 11 (5), 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Seeliger B; Agnus V; Mascagni P; Barberio M; Longo F; Lapergola A; Mutter D; Klymchenko AS; Chand M; Marescaux J; Diana M Simultaneous Computer-Assisted Assessment of Mucosal and Serosal Perfusion in a Model of Segmental Colonic Ischemia. Surg. Endosc. 2020, 34 (11), 4818–4827. [DOI] [PubMed] [Google Scholar]
- (6).D’Urso A; Agnus V; Barberio M; Seeliger B; Marchegiani F; Charles AL; Geny B; Marescaux J; Mutter D; Diana M Computer-Assisted Quantification and Visualization of Bowel Perfusion Using Fluorescence-Based Enhanced Reality in Left-Sided Colonic Resections. Surg. Endosc. 2021, 35 (8), 4321–4331. [DOI] [PubMed] [Google Scholar]
- (7).Gitajn IL; Elliott JT; Gunn JR; Ruiz AJ; Henderson ER; Pogue BW; Jiang S Evaluation of Bone Perfusion during Open Orthopedic Surgery Using Quantitative Dynamic Contrast-Enhanced Fluorescence Imaging. Biomed. Opt. Express 2020, 11 (11), 6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Elliott JT; Jiang S; Pogue BW; Gitajn IL Bone-Specific Kinetic Model to Quantify Periosteal and Endosteal Blood Flow Using Indocyanine Green in Fluorescence Guided Orthopedic Surgery. J. Biophotonics 2019, 12 (8), e201800427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sun H; Hu H; Liu C; Sun N; Duan C Methods Used for the Measurement of Blood-Brain Barrier Integrity. Metab. Brain Dis. 2021, 36 (5), 723–735. [DOI] [PubMed] [Google Scholar]
- (10).Milej D; Abdalmalak A; Desjardins L; Ahmed H; Lee TY; Diop M; Lawrence KS Quantification of Blood-Brain Barrier Permeability by Dynamic Contrast-Enhanced NIRS. Sci. Rep. 2017, 7, 1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Forcione M; Chiarelli AM; Davies DJ; Perpetuini D; Sawosz P; Merla A; Belli A Cerebral Perfusion and Blood–Brain Barrier Assessment in Brain Trauma Using Contrast-Enhanced near-Infrared Spectroscopy with Indocyanine Green: A Review. J. Cereb. Blood Flow Metab. 2020, 40 (8), 1586–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hansen ML; Hyttel-Sørensen S; Jakobsen JC; Gluud C; Kooi EMW; Mintzer J; de Boode WP; Fumagalli M; Alarcon A; Alderliesten T; Greisen G Cerebral Near-Infrared Spectroscopy Monitoring (NIRS) in Children and Adults: A Systematic Review with Meta-Analysis. Pediatric Research. 2022. 10.1038/s41390-022-01995-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Forcione M; Yakoub KM; Chiarelli AM; Perpetuini D; Merla A; Sun R; Sawosz P; Belli A; Davies DJ Dynamic Contrast-Enhanced near-Infrared Spectroscopy Using Indocyanine Green on Moderate and Severe Traumatic Brain Injury: A Prospective Observational Study. Quant. Imaging Med. Surg. 2020, 10 (11), 2085–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jagtap J; Sharma G; Parchur AK; Gogineni V; Bergom C; White S; Flister MJ; Joshi A Methods for Detecting Host Genetic Modifiers of Tumor Vascular Function Using Dynamic Near-Infrared Fluorescence Imaging. Biomed. Opt. Express 2018, 9 (2), 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jagtap J; Audi S; Razeghi-Kondelaji MH; Fish BL; Hansen C; Narayan J; Gao F; Sharma G; Parchur AK; Banerjee A; Bergom C A Rapid Dynamic in Vivo Near-Infrared Fluorescence Imaging Assay to Track Lung Vascular Permeability after Acute Radiation Injury. Am. J. Physiol. - Lung Cell. Mol. Physiol. 2021, 320 (3), L436–L450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Jeong H; Kim SR; Kang Y; Kim H; Kim SY; Cho SH; Kim KN Real-Time Longitudinal Evaluation of Tumor Blood Vessels Using a Compact Preclinical Fluorescence Imaging System. Biosensors 2021, 11 (12), 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Liu L; Su X; Mason RP Dynamic Contrast Enhanced Fluorescent Molecular Imaging of Vascular Disruption Induced by Combretastatin-A4P in Tumor Xenografts. J. Biomed. Nanotechnol. 2014, 10 (8), 1545–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).St Lawrence K; Verdecchia K; Elliott J; Tichauer K; Diop M; Hoffman L; Lee TY Kinetic Model Optimization for Characterizing Tumour Physiology by Dynamic Contrast-Enhanced near-Infrared Spectroscopy. Phys. Med. Biol. 2013, 58 (5), 1591–1604. [DOI] [PubMed] [Google Scholar]
- (19).Taruttis A; Morscher S; Burton NC; Razansky D; Ntziachristos V Fast Multispectral Optoacoustic Tomography (MSOT) for Dynamic Imaging of Pharmacokinetics and Biodistribution in Multiple Organs. PLoS One 2012, 7 (1), e30491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lee S; Lim W; Ryu HW; Jo D; Min JJ; Kim HS; Hyun H ZW800–1 for Assessment of Blood-Brain Barrier Disruption in a Photothrombotic Stroke Model. Int. J. Med. Sci. 2017, 14 (13), 1430–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Jennings D; Raghunand N; Gillies RJ Imaging Hemodynamics. Cancer Metastasis Rev. 2008, 27 (4), 589–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Senarathna J; Prasad A; Bhargava A; Gil S; Thakor NV; Pathak AP HemoSYS: A Toolkit for Image-Based Systems Biology of Tumor Hemodynamics. Sci. Rep. 2020, 10, 2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bhargava A; Monteagudo B; Kushwaha P; Senarathna J; Ren Y; Riddle RC; Aggarwal M; Pathak AP VascuViz: A Multimodality and Multiscale Imaging and Visualization Pipeline for Vascular Systems Biology. Nat. Methods 2022, 19 (2), 242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Liang H; Chen X; Jin R; Ke B; Barz M; Ai H; Nie Y Integration of Indocyanine Green Analogs as Near-Infrared Fluorescent Carrier for Precise Imaging-Guided Gene Delivery. Small 2020, 16 (10), 1906538. [DOI] [PubMed] [Google Scholar]
- (25).Teranishi K A Near-Infrared Fluorescent Probe Coated with β-Cyclodextrin Molecules for Real-Time Imaging-Guided Intraoperative Ureteral Identification and Diagnosis. Mol. Pharm. 2020, 17 (7), 2672–2681. [DOI] [PubMed] [Google Scholar]
- (26).Berezin MY; Guo K; Akers W; Livingston J; Solomon M; Lee H; Liang K; Agee A; Achilefu S Rational Approach To Select Small Peptide Molecular Probes Labeled With Fluorescent Cyanine Dyes For In Vivo Optical Imaging. Biochemistry 2011, 50, 2691–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Spa SJ; Hensbergen AW; van der Wal S; Kuil J; van Leeuwen FWB. The Influence of Systematic Structure Alterations on the Photophysical Properties and Conjugation Characteristics of Asymmetric Cyanine 5 Dyes. Dye. Pigment. 2018, 152, 19–28. [Google Scholar]
- (28).Exner RM; Cortezon-Tamarit F; Pascu SI Explorations into the Effect of Meso-Substituents in Tricarbocyanine Dyes: A Path to Diverse Biomolecular Probes and Materials. Angew. Chemie Int. Ed. 2020, 59 (12), 6230–6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Marshall MV; Draney D; Sevick-Muraca EM; Olive DM Single Dose Toxicity Study of IRDye 800CW in Sprague-Dawley Rats. Reporters, Markers, Dye. Nanoparticles, Mol. Probes Biomed. Appl. II 2010, 7576, 61–71. [Google Scholar]
- (30).Cha J; Nani RR; Luciano MP; Kline G; Broch A; Kim K; Namgoong J; Kulkarni RA; Meier JL; Kim P; Schnermann MJ A Chemically Stable Fluorescent Marker of the Ureter. Bioorg. Med. Chem. Lett. 2018, 28 (16), 2741–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hyun H; Owens EA; Narayana L; Wada H; Gravier J; Bao K; Frangioni JV; Choi HS; Henary M Central C-C Bonding Increases Optical and Chemical Stability of NIR Fluorophores. RSC Adv. 2014, 4 (102), 58762–58768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lim SY; Hong KH; Kim D Il; Kwon H; Kim HJ Tunable Heptamethine-Azo Dye Conjugate as an NIR Fluorescent Probe for the Selective Detection of Mitochondrial Glutathione over Cysteine and Homocysteine. J. Am. Chem. Soc. 2014, 136 (19), 7018–7025. [DOI] [PubMed] [Google Scholar]
- (33).Zhu S; Hu Z; Tian R; Yung BC; Yang Q; Zhao S; Kiesewetter DO; Niu G; Sun H; Antaris AL; Chen X Repurposing Cyanine NIR-I Dyes Accelerates Clinical Translation of Near-Infrared-II (NIR-II) Bioimaging. Adv. Mater. 2018, 30, 1802546. [DOI] [PubMed] [Google Scholar]
- (34).Xie B; Stammes MA; van Driel PBAA; Cruz LJ; Knol-Blankevoort VT; Löwik MAM; Mezzanotte L; Que I; Chan A; van den Wijngaard JPHM; Siebes M Necrosis Avid near Infrared Fluorescent Cyanines for Imaging Cell Death and Their Use to Monitor Therapeutic Efficacy in Mouse Tumor Models. Oncotarget 2015, 6 (36), 39036–39049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Stroet MCM; de Blois E; Stuurman DC; de Ridder CMA; Haeck J; Seimbille Y; Mezzanotte L; de Jong M; Löwik CWGM; Panth KM In Vivo Evaluation of Indium-111–Labeled 800CW as a Necrosis-Avid Contrast Agent. Mol. Imaging Biol. 2020, 22 (5), 1333–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Li D-H; Schreiber CL; Smith BD Sterically Shielded Heptamethine Cyanine Dyes for Bioconjugation and High Performance Near-Infrared Fluorescence Imaging. Angew. Chem. Int. Ed. 2020, 59 (29), 12154–12161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Schreiber CL; Li D-H; Smith BD High-Performance Near-Infrared Fluorescent Secondary Antibodies for Immunofluorescence. Anal. Chem. 2021, 93, 3643–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Gamage RS; Li D-H; Schreiber CL; Smith BD Comparison of CRGDfK Peptide Probes with Appended Shielded Heptamethine Cyanine Dye (s775z) for Near Infrared Fluorescence Imaging of Cancer. ACS Omega 2021, 6 (44), 30130–30139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Singh SB; Kaelin DE; Wu J; Miesel L; Tan CM; Meinke PT; Olsen D; Lagrutta A; Bradley P; Lu J; Patel S; Rickert KW; Smith RF; Soisson S; Wei C; Fukuda H; Kishii R; Takei M; Fukuda Y Oxabicyclooctane-Linked Novel Bacterial Topoisomerase Inhibitors as Broad Spectrum Antibacterial Agents. ACS Med. Chem. Lett. 2014, 5 (5), 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Strekowski L; Lipowska M; Patonay G Substitution Reactions of a Nucleofugal Group in Heptamethine Cyanine Dyes. Synthesis of an Isothiocyanato Derivative for Labeling of Proteins with a Near-Infrared Chromophore. J. Org. Chem. 1992, 57 (17), 4578–4580. [Google Scholar]
- (41).Samanta A; Vendrell M; Das R; Chang Y-T Development of Photostable Near-Infrared Cyanine Dyes. Chem. Commun. 2010, 46 (39), 7406–7408. [DOI] [PubMed] [Google Scholar]
- (42).Lin C-M; Usama SM; Burgess K Site-Specific Labeling of Proteins with Near-IR Heptamethine Cyanine Dyes. Molecules 2018, 23 (11), 2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).:Forman HJ; Zhang H; Rinna A Glutathione: Overview of Its Protective Roles, Measurement, and Biosynthesis. Mol. Aspects Med. 2009, 30 (1–2), 1–12.
- (44).Dohmen MPJ; Pereira AM; Timmer JMK; Benes NE; Keurentjes JTF Hydrodynamic Radii of Polyethylene Glycols in Different Solvents Determined from Viscosity Measurements. J. Chem. Eng. Data 2008, 53 (1), 63–65. [Google Scholar]
- (45).Du B; Jiang X; Huang Y; Li S; Lin JC; Yu M; Zheng J Tailoring Kidney Transport of Organic Dyes with Low-Molecular-Weight PEGylation. Bioconjug. Chem. 2019, 31, 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Trost BM; Yeh VSC; Ito H; Bremeyer N Effect of Ligand Structure on the Zinc-Catalyzed Henry Reaction. Asymmetric Syntheses of (−)-Denopamine and (−)-Arbutamine. Org. Lett. 2002, 4 (16), 2621–2623. [DOI] [PubMed] [Google Scholar]
- (47).Li D-H; Smith BD Deuterated Indocyanine Green (ICG) with Extended Aqueous Storage Shelf-Life: Chemical and Clinical Implications. Chem. Eur. J. 2021, 27 (58), 14535–14542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Stewart JJP MOPAC, Colorado Springs, CO: Stewart Computational Chemistry, USA, 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.